Recent advances in C–H fluorination
Aijun
Lin†
,
C. Bryan
Huehls†
and
Jiong
Yang
*
Department of Chemistry, Texas A&M University, College Station, Texas 77843-3255, USA. E-mail: yang@mail.chem.tamu.edu; Tel: (+1)9798452889
First published on 7th March 2014
Abstract
Incorporation of fluorine atom(s) into organic compounds is often desirable in the discovery of new pharmaceuticals, agrochemicals, and materials. However, development of transformations to incorporate fluorine is challenging because of its highly electronegative nature. Recent advances in C–H functionalization have allowed new approaches to C–F bonds. Herein we highlight progress in C–H fluorination, which is arguably the most efficient approach to incorporate fluorine since it obviates the need of pre-functionalization of organic compounds.
 Aijun Lin, C. Bryan Huehls and Jiong Yang | Dr. Aijun Lin was born in Jintan, China. He received his Ph.D. from Nanjing University in 2011 for his work on asymmetric organocatalysis under the guidance of Prof. Chengjian Zhu. After a one-year postdoctoral stint at the same group, he joined the research group of Prof. Jiong Yang at Texas A&M University in 2012 as a postdoctoral research associate. His current research focuses on transition metal catalysis, especially those based on iron, cobalt, and nickel. C. Bryan Huehls attended Ball State University where he first began research in synthetic methodology under the guidance of Prof. Robert E. Sammelson. After receiving his B.A. in chemistry in 2009, he started his graduate study at Texas A&M University in Spring 2010 and joined the research group of Prof. Jiong Yang. His doctoral research focused on developing new methods for synthesis of indolenines and exploring iron-catalyzed transformations. He attended the ACS Summer School of Green Chemistry and Sustainable Energy, and he was a BASF-TAMU Graduate Student Symposium Finalist in 2013. Prof. Jiong Yang received his Ph.D. from The Ohio State University for his work on total synthesis of natural products under the tutelage of Prof. Leo A. Paquette. He pursued postdoctoral research in chemical genetics in the laboratories of Prof. Peter G. Schultz (The Scripps Research Institute) and then Prof. Stuart L. Schreiber (Harvard University/Broad Institute of MIT and Harvard) as a NIH postdoctoral fellow before starting his independent career at Texas A&M University. He is interested in developing new synthetic methods and strategies for synthesis of bioactive compounds, with the goal of bringing the power of modern synthetic organic chemistry to bear on biomedical problems. |
The past decade witnessed the rapid development of transition metal-catalyzed C–H functionalization as a powerful tool for organic synthesis, with heteroatom directed C–H activation as a commonly used strategy for regioselection. In 2006, Sanford and co-workers described the first Pd-catalyzed aryl and benzyl C–H fluorination of 2-phenylpyridine and 8-methylquinoline derivatives with N-fluoropyridinium tetrafluoroborate or its trimethyl congener as the electrophilic fluorine source under microwave conditions (Scheme 1, eqn (1)).8 For C(sp2)–H fluorination, 2,6-difluorinated products were formed except when ortho- or meta-substituted 2-phenylpyridines were used, which led to monofluorinated products. While quinoline and pyridine were used as the directing group in this pioneering study, other N-heterocycles, such as quinoxaline, pyrazole, benzo[d]oxazole, and pyrazine, were found to be equally effective for directed ortho-fluorination under similar conditions.9 Building upon their success on electrophilic C–H fluorination, Sanford and co-workers also described the first Pd-catalyzed nucleophilic C–H fluorination in which 8-methylquinoline derivatives were fluorinated at the benzyl position by Pd(OAc)2 and PhI(OPiv)2, with AgF as the source of the nucleophilic fluoride (Scheme 1, eqn (2)).10
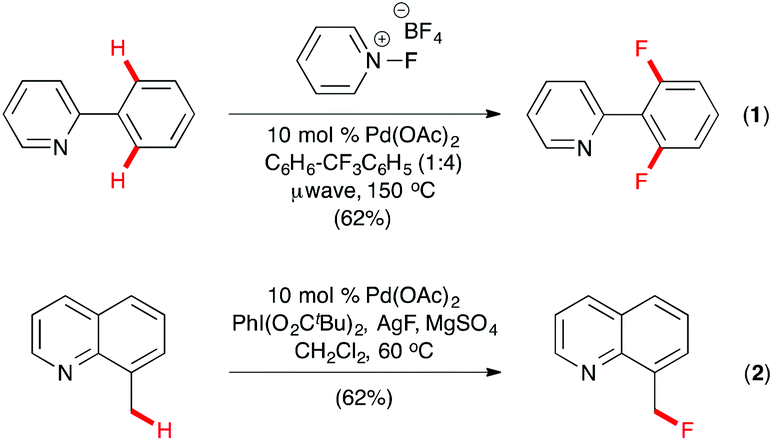 | ||
| Scheme 1 Pd-catalyzed C(sp2)–H and C(sp3)–H fluorination by Sanford. | ||
Yu and co-workers described ortho-C–H fluorination of triflamide-protected benzylamines with N-fluoro-2,4,6-trimethylpyridinium triflate using Pd(OTf)2 as the catalyst and N-methylpyrrolidinone (NMP) as an essential additive.11 2,6-Difluorinated products were formed unless the ortho- or meta-position of the substrates had been substituted (Scheme 2, eqn (3)). Hypothesizing that the slow displacement of the monofluorinated products from the PdII center caused difluorination of the substrates, Yu and co-workers successfully developed a method for C–H monofluorination using the amide derivatives of benzoic acid and 2,3,5,6-tetrafluoro-4-(trifluoromethyl) aniline as the substrates (Scheme 2, eqn (4)).12 The monoselectivity of the reaction was attributed to the weaker coordination of the L-type acidic amide directing group, which allowed rapid dissociation of the monofluorinated products.
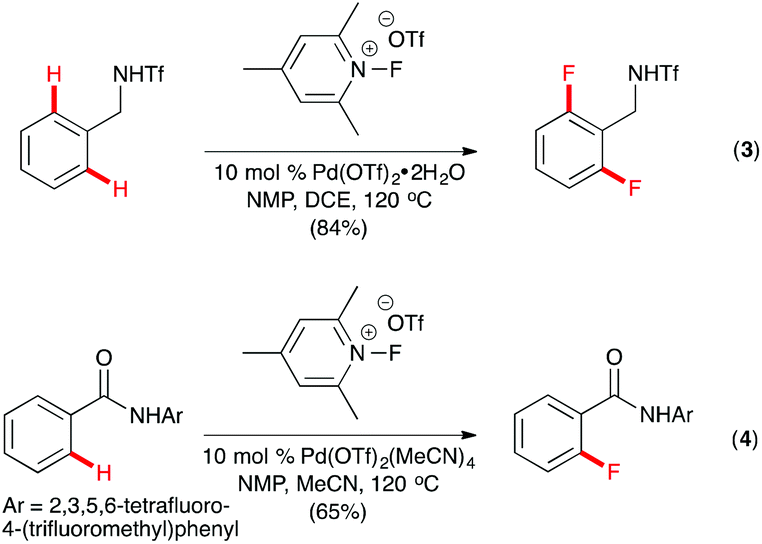 | ||
| Scheme 2 Pd-catalyzed C(sp2)–H fluorination by Yu. | ||
While a mechanistic pathway involving reductive elimination of PdIV(R)(F) intermediates appears to be operative in the Pd-catalyzed C–H fluorination reactions,13 Daugulis and co-workers hypothesized that the amide derivative of 8-aminoquinoline, which they introduced as a directing group for Pd-catalyzed C–H arylation,14 might promote Cu-catalyzed C–H fluorination through CuIII intermediates.15 Indeed, ortho-fluorination of 8-aminoquinoline benzamide was effected with AgF and NMO using CuI as the catalyst (Scheme 3, eqn (5)). Selective mono- or difluorination could be achieved by adjusting the loading of the reagents and the reaction time.16
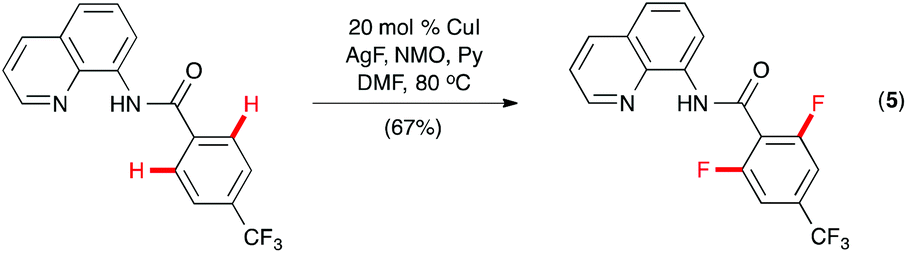 | ||
| Scheme 3 Cu-catalyzed C(sp2)–H fluorination by Daugulis. | ||
The heteroatom-directed C–H functionalization provided a powerful approach for fluorination of organic compounds, but the limitations of this approach are also obvious. For example, in addition to requiring the presence of a directing group, the Pd-catalyzed C–H fluorination of 2-phenylpyridine incorporated fluorine into the phenyl only (vide supra) and fluorination of pyridine could not be achieved. Because of the important role of fluorinated heterocycles in pharmaceuticals and agrochemicals, there is an urgent need of new approaches to fluorinate heterocycles. Inspired by the classic Chichibabin reaction of pyridine and NaNH2 to form 2-aminopyridine, Fier and Hartwig developed a practical approach for C–H monofluorination of pyridine by AgF2 with exclusive site selectivity at the C–H bonds adjacent to nitrogen under mild conditions (Scheme 4, eqn (6)).17 For example, reaction of 2-phenylpyridine with AgF2 in MeCN gave 2-fluoro-6-phenylpyridine as the only fluorinated product in 88% yield. This excellent site-selectivity was rationalized by a mechanism of initial N-coordination of AgF2 with pyridine followed by addition of the Ag–F bond across the π system to give an amido-silver(II)-fluoride intermediate, and abstraction of hydrogen by a second equivalent of AgF2 to give the product. The reaction also was found to be effective for C–H monofluorination of other 6-membered N-heterocycles, such as quinolines, pyrazines, pyrimidines, and pyridazines.
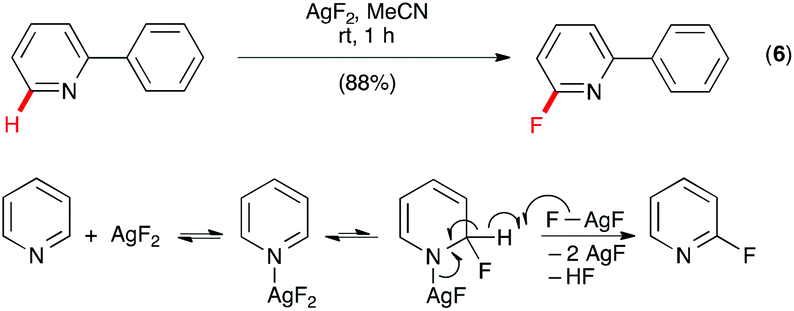 | ||
| Scheme 4 Monofluorination of pyridine by AgF2. | ||
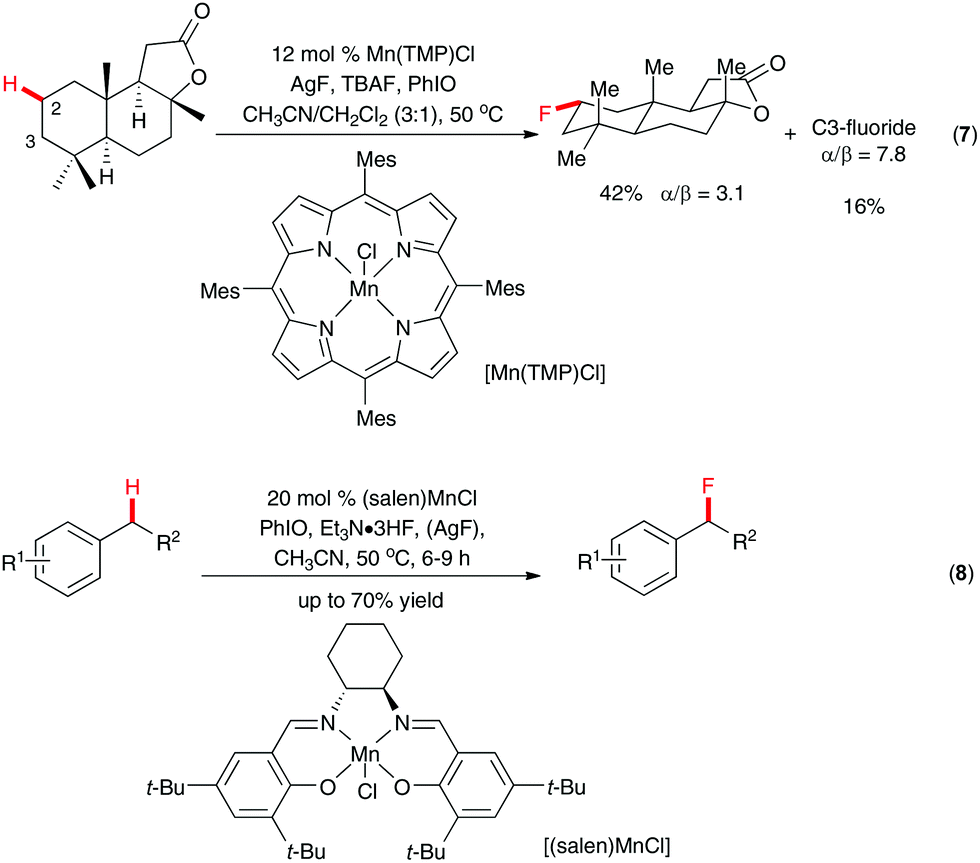
The challenge of selective C(sp3)–H fluorination without a directing group lies in identifying a catalytic system that is both sufficiently reactive and predictably selective. In an elegant study, Groves and co-workers discovered that the manganese porphyrin system Mn(TMP)Cl could be used for regioselective C(sp3)–H fluorination by silver fluoride/tetrabutylammonium fluoride trihydrate with iodosylbenzene as the oxo-transfer agent.18 The unique reactivity of the system was demonstrated in the fluoridation of a number of polycyclic natural/unnatural compounds. For example, among the 26 unactivated C(sp3)–H bonds of sclareolide, only those at C2 and C3 reacted to give methylene-fluorinated products (Scheme 5, eqn (7)). The proposed reaction mechanism involved abstraction of hydrogen by the in situ formed oxomanganese species O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) MnV(TMP) and capture of the resulting C-centered radical with fluorine by a trans-MnIV(TMP)F2 species. A related manganese–salen system was also reported by the same research group for selective benzyl C–H fluorination with the inexpensive triethylamine trihydrofluoride as the source of nucleophilic fluoride (Scheme 5, eqn (8)).19 Even though less efficient, potassium fluoride in the presence of 18-crown-6 could also be used as the source of fluorine.
MnV(TMP) and capture of the resulting C-centered radical with fluorine by a trans-MnIV(TMP)F2 species. A related manganese–salen system was also reported by the same research group for selective benzyl C–H fluorination with the inexpensive triethylamine trihydrofluoride as the source of nucleophilic fluoride (Scheme 5, eqn (8)).19 Even though less efficient, potassium fluoride in the presence of 18-crown-6 could also be used as the source of fluorine.
 | ||
| Scheme 5 Mn-catalyzed C(sp3)–H fluorination. | ||
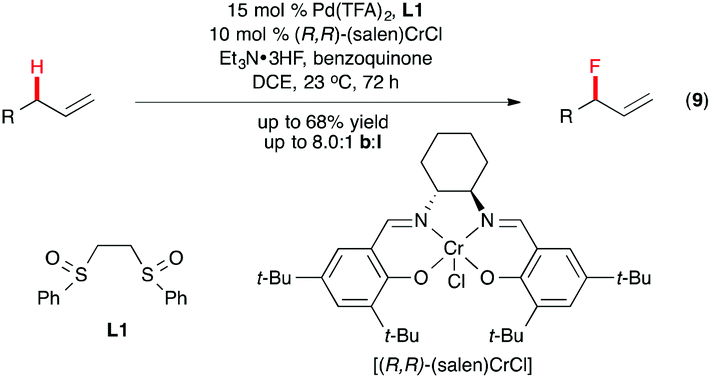
The electrophilic PdII–sulfoxide system developed by White and co-workers is a powerful tool for C(sp3)–H functionalization.20 Recently, Braun and Doyle demonstrated this catalytic system to be effective for allylic C–H fluorination as well.21 Their optimized reaction conditions include Pd(OTf)2-1,2-bis(phenylsulfinyl)ethane (L1), a Lewis acidic co-catalyst (salen)CrCl, and benzoquinone as the oxidant, with triethylamine trihydrofluoride as the nucleophilic fluoride source (Scheme 6). The allyl fluoride product was formed with high branch![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :linear regioselectivity, and in good yields.
:linear regioselectivity, and in good yields.
 | ||
| Scheme 6 Pd-catalyzed allylic C(sp3)–H fluorination. | ||
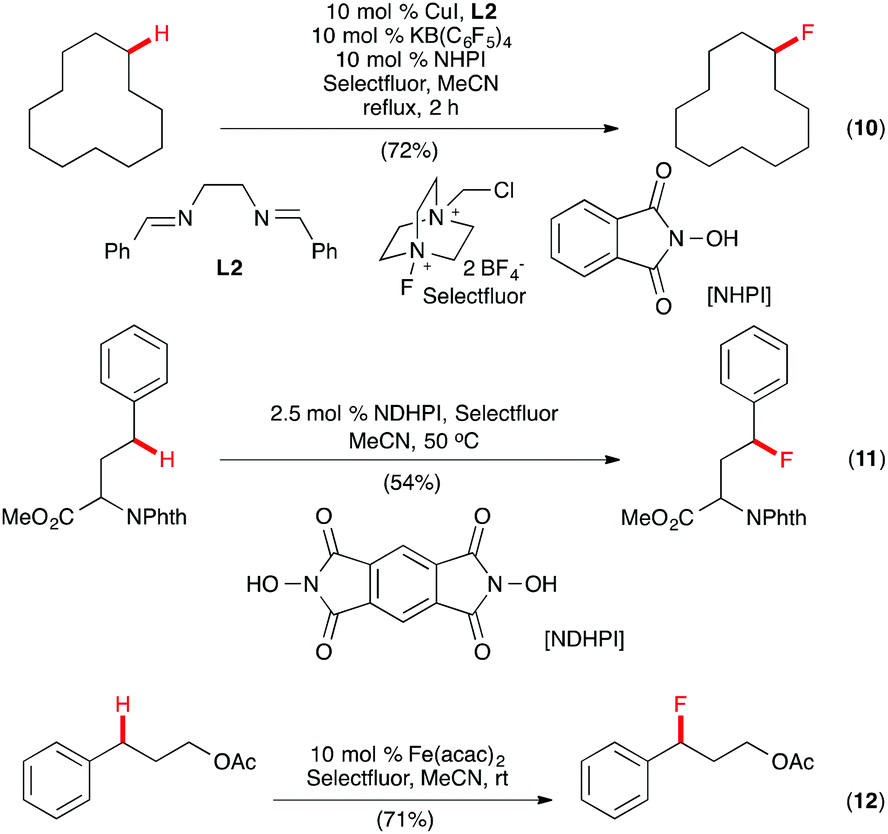
A multicomponent system of CuI-bis(imine) (L2) complex, KB(C6F5)4, KI, and N-hydroxyphthalimide (NHPI) was reported by Lectka and co-workers for aliphatic, allylic, and benzylic monofluorination with Selectfluor (Scheme 7, eqn (10)).22 A radical mechanism was proposed with NHPI, known to form the phthalimide N-oxygen radical in situ in the presence of redox active metals, serving as a radical initiator. Evidence to support this hypothesis included reaction inhibition by the radical trapping agent 2,2,6,6-tetramethylpiperidin-1-yloxyl (TEMPO). Interestingly, in a related study, Inoue and co-workers found that NHPI alone was capable of initiating benzylic C–H fluorination with Selectfluor, but synthetically useful yields for the benzylic and aliphatic C–H fluorination required N,N-dihydroxypyromellitimide (NDHPI) to be used (Scheme 7, eqn (11)).23 A recent report by Lectka and co-workers showed that benzylic C(sp3)–H monofluorination could also be effected with Selectfluor using Fe(acac)2 as the catalyst (Scheme 7, eqn (12)).24
 | ||
| Scheme 7 Some radical C(sp3)–H fluorination. | ||
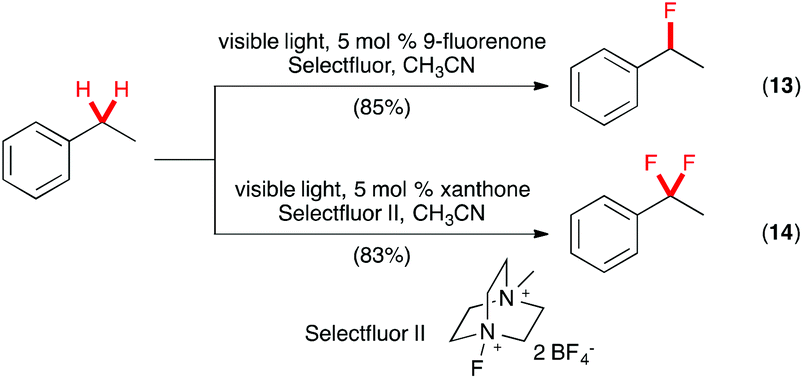
A transition metal-free photochemical method also was described recently for benzylic C(sp3)–H fluorination.25 In this elegant study, Chen and co-workers reported that a wide range of substrates underwent benzylic C(sp3)–H fluorination with Selectfluor using 9-fluorenone as the catalyst (Scheme 8, eqn (13)). No special equipment was necessary as the reaction could be carried out in regular glassware with household CFL as the source of visible light. Under similar conditions, the challenging C(sp3)–H benzylic difluorination could be effected with Selectfluor II using xanthone as the catalyst (eqn (14)).
 | ||
| Scheme 8 Photolytic benzylic C(sp3)–H fluorination. | ||
Chemists have gone a long way developing new methods for synthesis of C–F bonds. Particularly, recent advances in transition metal-catalysis and radical reactions have allowed innovative approaches for C–H fluorination. However, significant challenges remain. Some of these challenges are inherent in C–H activation, such as developing new directing groups for mild and regioselective C–H activation, new catalytic systems with unique selectivity, and new reaction conditions with broader substrate scope. Further challenges have to be met for effective C–H fluorination, such as developing economical electrophilic fluorine sources, fluorination processes based on nucleophilic fluoride, and identifying operationally simple reaction conditions to address the special needs in synthesizing 18F PET imaging agents, which would benefit from a rapid reaction (due to the short, 110 min half-life of 18F) with dilute aqueous [18F]fluoride (produced by proton bombardment of 18O-enriched water).7d,26 These challenges are expected to stimulate further efforts to invent innovative approaches to incorporate fluorine into organic compounds.
Acknowledgements
We thank the National Science Foundation (CHE-1150606) and the Robert A. Welch Foundation (A-1700) for financial support.Notes and references
- C. Dong, F. Huang, H. Deng, C. Schaffrath, J. B. Spencer, D. O'Hagan and J. H. Naismith, Nature, 2004, 427, 561 CrossRef CAS PubMed ; and references cited therein.
- (a) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed; (b) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359 CrossRef CAS PubMed; (c) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; (d) H.-J. Böhm, D. Banner, S. Bendels, M. Kansy, B. Kuhn, K. Müller, U. Obst-Sander and M. Stahl, ChemBioChem, 2004, 5, 637 CrossRef PubMed.
- (a) P. Jeschke, ChemBioChem, 2004, 5, 570 CrossRef CAS PubMed; (b) P. Maienfisch and R. G. Hall, Chimia, 2004, 58, 93 CrossRef CAS.
- R. Berger, G. Resnati, P. Metrangolo, E. Weber and J. Hulliger, Chem. Soc. Rev., 2011, 40, 3496 RSC.
- S. M. Ametamey, M. Honer and P. A. Schubiger, Chem. Rev., 2008, 108, 1501 CrossRef CAS PubMed.
- (a) D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308 RSC; (b) J. Emsley, Chem. Soc. Rev., 1980, 9, 91 RSC.
- For some reviews: (a) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214 CrossRef CAS PubMed; (b) M. P. Sibi and Y. Landais, Angew. Chem., Int. Ed., 2013, 52, 3570 CrossRef CAS PubMed; (c) C. Hollingworth and V. Gouverneur, Chem. Commun., 2012, 48, 2929 RSC; (d) M. Tredwell and V. Gouverneur, Angew. Chem., Int. Ed., 2012, 51, 11426 CrossRef CAS PubMed; (e) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470 CrossRef CAS PubMed; (f) T. Furuya, J. E. M. N. Klein and T. Ritter, Synthesis, 2010, 1804 CAS; (g) S. Lectard, Y. Hamashima and M. Sodeoka, Adv. Synth. Catal., 2010, 352, 2708 CrossRef CAS; (h) D. Cahard, X. Xu, S. Couve-Bonnaire and X. Pannecoucke, Chem. Soc. Rev., 2010, 39, 558 RSC; (i) M. C. Pacheco, S. Purser and V. Gouverneur, Chem. Rev., 2008, 108, 1942 CrossRef PubMed; (j) V. V. Grushin, Acc. Chem. Res., 2010, 43, 160 CrossRef CAS PubMed; (k) K. L. Kirk, Org. Process Res. Dev., 2008, 12, 305 CrossRef CAS; (l) T. Furuya, C. A. Kuttruff and T. Ritter, Curr. Opin. Drug Discovery. Dev., 2008, 11, 803 CAS; (m) J.-A. Ma and D. Cahard, Chem. Rev., 2008, 108, PR1 CrossRef CAS; (n) M. Shimizu and T. Hiyama, Angew. Chem., Int. Ed., 2005, 44, 214 CrossRef CAS PubMed; (o) J.-A. Ma and D. Cahard, Chem. Rev., 2004, 104, 6119 CrossRef CAS PubMed.
- K. L. Hull, W. Q. Anani and M. S. Sanford, J. Am. Chem. Soc., 2006, 128, 7134 CrossRef CAS PubMed.
- S.-J. Lou, D.-Q. Xu, A.-B. Xia, Y.-F. Wang, Y.-K. Liu, X.-H. Du and Z. Y. Xu, Chem. Commun., 2013, 49, 6218 RSC.
- K. B. McMurtrey, J. M. Racowski and M. S. Sanford, Org. Lett., 2012, 14, 4094 CrossRef CAS PubMed.
- X. Wang, T.-S. Mei and J.-Q. Yu, J. Am. Chem. Soc., 2009, 131, 7520 CrossRef CAS PubMed.
- K. S. L. Chan, M. Wasa, X. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2011, 50, 9081 CrossRef CAS PubMed.
- For a review: (a) K. M. Engle, T.-S. Mei, X. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2011, 50, 1478 CrossRef CAS PubMed For some reports: (b) N. D. Ball and M. S. Sanford, J. Am. Chem. Soc., 2009, 131, 3796 CrossRef CAS PubMed; (c) T. Furuya and T. Ritter, J. Am. Chem. Soc., 2008, 130, 10060 CrossRef CAS PubMed; (d) T. Furuya, D. Benitez, E. Tkatchouk, A. E. Strom, P. Tang, W. A. Goddard III and T. Ritter, J. Am. Chem. Soc., 2010, 132, 3793 CrossRef CAS PubMed; (e) J. M. Racowski, J. B. Gary and M. S. Sanford, Angew. Chem., Int. Ed., 2012, 51, 3414 CrossRef CAS PubMed; (f) J. R. Brandt, E. Lee, G. B. Boursalian and T. Ritter, Chem. Sci., 2014, 5, 169 RSC.
- (a) V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154 CrossRef CAS PubMed For recent reviews: (b) M. Corbet and F. De Campo, Angew. Chem., Int. Ed., 2013, 52, 9896 CrossRef CAS PubMed; (c) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726 CrossRef CAS PubMed.
- A. J. Hickman and M. S. Sanford, Nature, 2012, 484, 177 CrossRef CAS PubMed.
- T. Truong, K. Klimovica and O. Daugulis, J. Am. Chem. Soc., 2013, 135, 9342 CrossRef CAS PubMed.
- P. S. Fier and J. F. Hartwig, Science, 2013, 342, 956 CrossRef CAS PubMed.
- W. Liu, X. Huang, M.-J. Cheng, R. J. Nielsen, W. A. Goddard III and J. T. Groves, Science, 2012, 337, 1322 CrossRef CAS PubMed.
- W. Liu and J. T. Groves, Angew. Chem., Int. Ed., 2013, 52, 6024 CrossRef CAS PubMed.
- M. S. Chen and M. C. White, J. Am. Chem. Soc., 2004, 126, 1346 CrossRef CAS PubMed.
- M.-G. Braun and A. G. Doyle, J. Am. Chem. Soc., 2013, 135, 12990 CrossRef CAS PubMed.
- S. Bloom, C. R. Pitts, D. C. Miller, N. Haselton, M. G. Holl, E. Urheim and T. Lectka, Angew. Chem., Int. Ed., 2012, 51, 10580 CrossRef CAS PubMed.
- Y. Amaoka, M. Nagatomo and M. Inoue, Org. Lett., 2013, 15, 2160 CrossRef CAS PubMed.
- S. Bloom, C. R. Pitts, R. Woltornist, A. Griswold, M. G. Holl and T. Lectka, Org. Lett., 2013, 15, 1722 CrossRef CAS PubMed.
- J.-B. Xia, C. Zhu and C. Chen, J. Am. Chem. Soc., 2013, 135, 17494 CrossRef CAS PubMed.
- L. Cai, S. Lu and V. W. Pike, Eur. J. Org. Chem., 2008, 2853 CrossRef CAS.
Footnote |
| † These authors contributed equally. |
| This journal is © the Partner Organisations 2014 |