
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLinker engineering in metal–organic frameworks for dark photocatalysis†
Yating
Pan‡
a,
Jingxue
Wang‡
a,
Shengyi
Chen
b,
Weijie
Yang
 b,
Chunmei
Ding
c,
Amir
Waseem
d and
Hai-Long
Jiang
*a
b,
Chunmei
Ding
c,
Amir
Waseem
d and
Hai-Long
Jiang
*a
aDepartment of Chemistry, University of Science and Technology of China Hefei, Anhui 230026, P. R. China. E-mail: jianglab@ustc.edu.cn
bSchool of Energy and Power Engineering, North China Electric Power University, Baoding 071003, P. R. China
cDalian National Laboratory for Clean Energy, State Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, P. R. China
dDepartment of Chemistry, Quaid-i-Azam University, Islamabad 45320, I. R. Pakistan
First published on 9th May 2022
Abstract
Dark reactions featuring continuous activity under light off conditions play a critical role in natural photosynthesis. However, most artificial photocatalysts are inactive upon the removal of the light source, and the artificial photocatalysts with dark photocatalysis abilities have been rarely explored. Herein, we report a Ti-based metal–organic framework (MOF), MIL-125, exhibiting the capability of dark photocatalytic hydrogen production. Remarkably, the introduction of different functional groups onto the linkers enables distinctly different activities of the resulting MOFs (MIL-125-X, X = NH2, NO2, Br). Dynamic and thermodynamic investigations indicate that the production and lifetime of the Ti3+ intermediate are the key factors, due to the electron-donating/-withdrawing effect of the functional groups. As far as we know, this is the first report on dark photocatalysis over MOFs, providing new insights into the storage of irradiation energy and demonstrating their great potential in dark photocatalysis due to the great MOF diversity.
Introduction
Energy storage and conversion are important solutions toward global sustainable development.1–3 Natural photosynthesis refers to the solar energy capture and storage through a series of donor and acceptor cofactors as charge transfer chains. These charges captured by cofactors demonstrate charge separation in the light reaction and possess a long lifetime capable of driving reactions in the dark.4 Specifically, solar energy is captured by the photosensitizers to produce carbohydrates from CO2 and H2O, where water oxidation generates O2 and protons in light reactions, and the protons react with CO2 to afford carbohydrates in dark reactions. Inspired by this, artificial photocatalytic systems have been developed for converting solar energy into chemical energy and meeting current energy and environmental challenges.5–7 Unfortunately, compared with natural photocatalytic systems, most artificial photocatalytic materials display activity under light irradiation only, and lack the ability to store electrons in the dark to achieve further reaction like the natural photocatalytic system; accordingly, the reports on photocatalysis operated in the dark are limited.8–15 Therefore, it is very attractive to mimic the natural plant cells and develop artificial photocatalysts with a dark photocatalysis capability, which are able to store reducing/oxidizing energy to drive reactions in the dark and would be candidates for energy storage.8–15Photosensitive unit and energy storage substance are the two important components to achieve dark photocatalysis in photocatalysts.11 Therefore, the relevant investigations mainly focus on the catalysts composed of these two components, such as Cu2O/TiO2,12 C3N4/carbon nanotube/graphene,13 and g-C3N4@Au@SrAl2O4: Eu2+, Dy3+,14etc. The structural complexity of composites causes the difficulty of accurately understanding the structure–property relationship. Unfortunately, the study on single-component photocatalysts with dark photocatalysis behavior has remained extremely rare thus far.8,10,15 It is highly desired to explore photocatalysts with well-defined and readily tailorable structures for unveiling the underlying mechanism and achieving rational regulation of dark photocatalysis.
As a class of crystalline porous materials featuring well-defined and tunable structures, metal–organic frameworks (MOFs) possess semiconductor-like behavior and great potential in fundamental photocatalysis.16–39 The diversity of organic linkers and metal-oxo clusters enables MOFs to be promising materials for dark photocatalysis, as they integrate the photosensitive unit (organic linkers) with the energy storage substance (metal-oxo clusters). Moreover, the atomically precise and tailorable structures of MOFs provide congenital conditions for activity regulation by structural alteration in photocatalysis, including light absorption, charge transfer kinetics, etc.40–43 It is assumed that the understanding of the structure–activity relationship toward dark photocatalysis would be better achieved by a systematic structural regulation of MOFs.
With the above thoughts in mind, herein, a representative Ti-MOF, MIL-125,44 constructed of cyclic octamers of TiO5(OH) linked by 1,4-benzenedicarboxylate (BDC), featuring an evident color change upon light irradiation by converting Ti4+ to Ti3+ as per previous reports,24,45 has been found to exhibit dark photocatalysis behavior. It is demonstrated that such color change could be maintained for a long time after the light is off, indicating the electron storage ability due to the long-lived Ti3+ electronic state. As a result, dark photocatalytic hydrogen production over MIL-125 has been successfully achieved in the presence of a Pt co-catalyst. Significantly, thanks to the tunable MOF structure, the introduction of different functional groups on the BDC linker greatly affects the generation and lifetime of Ti3+, resulting in a distinctly different dark photocatalysis activity (Scheme 1). To our knowledge, this is unprecedented work on dark photocatalysis over MOFs.
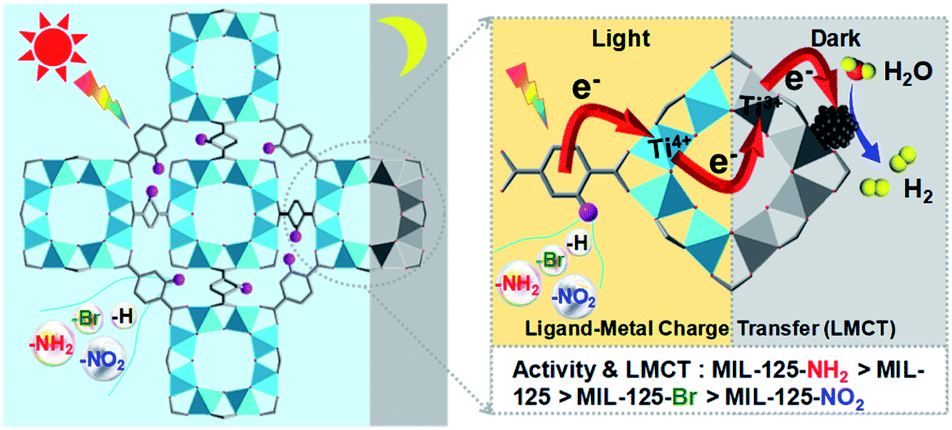 | ||
| Scheme 1 Illustration showing the dark photocatalysis over MIL-125 and MIL-125 with different functional groups, MIL-125-X (X = NH2, NO2, Br). | ||
Results and discussion
Typically, MIL-125 and MIL-125-NH2 were synthesized directly with the Ti precursor and the linkers via a solvothermal method. Unfortunately, it is difficult to obtain MIL-125-NO2 and MIL-125-Br with good crystallinity by the sole or mixed linker strategy under solvothermal conditions. Alternatively, they were fabricated by linker-exchange of MIL-125,46 and the maximum exchange extent is ∼50% (Fig. S1†). Powder X-ray diffraction patterns (XRD) indicate that all MIL-125-X (X = NH2, NO2, Br) feature the same structure as MIL-125 (Fig. S2†). Scanning electron microscopy (SEM) images show that MIL-125-X have a similar morphology and size to MIL-125 (Fig. S3†). Moreover, infrared (IR) spectra manifest the successful introduction of the functional groups in MIL-125-X (Fig. S4†). The functional groups on the linkers extend the light absorption of MIL-125 (Fig. S5 and S6†).On light irradiation, the color of MIL-125 changes from white to blue by using methanol (MeOH) as the electron donor in an oxygen-free environment (Fig. 1), similar to that observed in the previous report on MIL-125 photocatalysis.21,24 The color change is ascribed to the generation of Ti3+ based on the electron spin resonance (ESR) results (Fig. S7†). Compared to MIL-125 in the dark, new ESR peaks at g = 1.945, 2.002 and 2.025 under light irradiation are assignable to Ti3+ and O2˙− adsorbed onto Ti-oxo clusters (the residual O2 molecules accept electrons from electron-trapped Ti-oxo cluster),35,47 manifesting that Ti4+ in MIL-125 accepts electrons and is converted to a Ti3+ intermediate via the common ligand–metal charge transfer (LMCT) process. Upon turning off the light, the catalyst slowly fades for a very long time (43 hours, Fig. 1), possibly due to the acceptance of electrons by the inevitably leaked air (O2), during which negligible hydrogen is detected (Fig. S8†). This phenomenon indicates that MIL-125 can produce a long-lived intermediate that gives rise to the MOF discoloration under light irradiation. This long-lived intermediate can also be evidenced by the maintained ESR signal after turning off the light for a long time (Fig. S7†). In sharp contrast, upon light off, the introduction of Pt nanoparticles (NPs, ∼3 nm, Fig. S9, Table S1†) into the reaction solution resulted in the blue color gradually disappearing and hydrogen being produced accordingly is in the dark. This suggests that the electrons stored in the long-lived intermediate can be released upon introducing Pt NPs to achieve the dark photocatalytic hydrogen production, which is similar to the dark reaction of natural photosynthesis. By optimizing the amount of sacrificial agent (methanol), the dark photocatalytic hydrogen production can last up to 1.5 hours and reach a plateau (Fig. 2a). The rate of MIL-125 is quite fast (∼270.2 μmol g−1 h−1) in the first 30 min, and the average rate is 116.7 μmol g−1 h−1 over 90 min. This indicates that the stored electrons are rapidly released to produce hydrogen by introducing the Pt co-catalyst upon light off, then the number of electrons is reduced and the rate of releasing electrons to hydrogen is slowed down, and finally, the electrons are completely consumed. The elevated temperature can accelerate the H2 release (Fig. S10†). Moreover, it is found that the methanol concentration plays an important role in the dark photocatalysis with acetonitrile as a solvent. As the methanol concentration increases, the hydrogen production activity increases at first and then decreases, manifesting a volcanic trend (Fig. S11†). In fact, similar results can be observed when TEOA or TEA is adopted as a sacrificial agent instead (Fig. S12†).
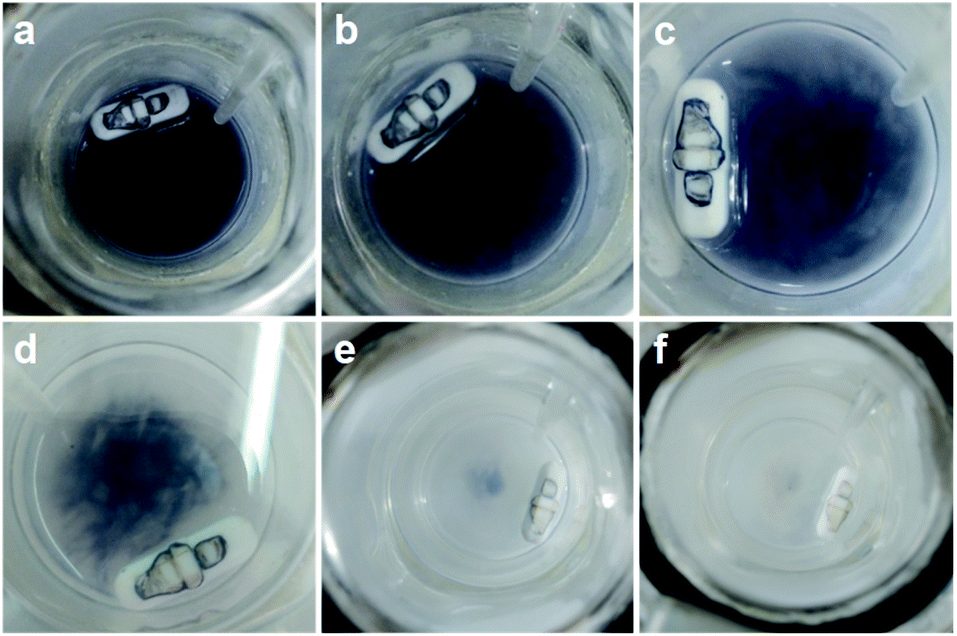 | ||
| Fig. 1 Color change of MIL-125 in the dark along with time: (a) 0 h; (b) 1 h; (c) 5 h; (d) 30 h; (e) 32 h; (f) 43 h. | ||
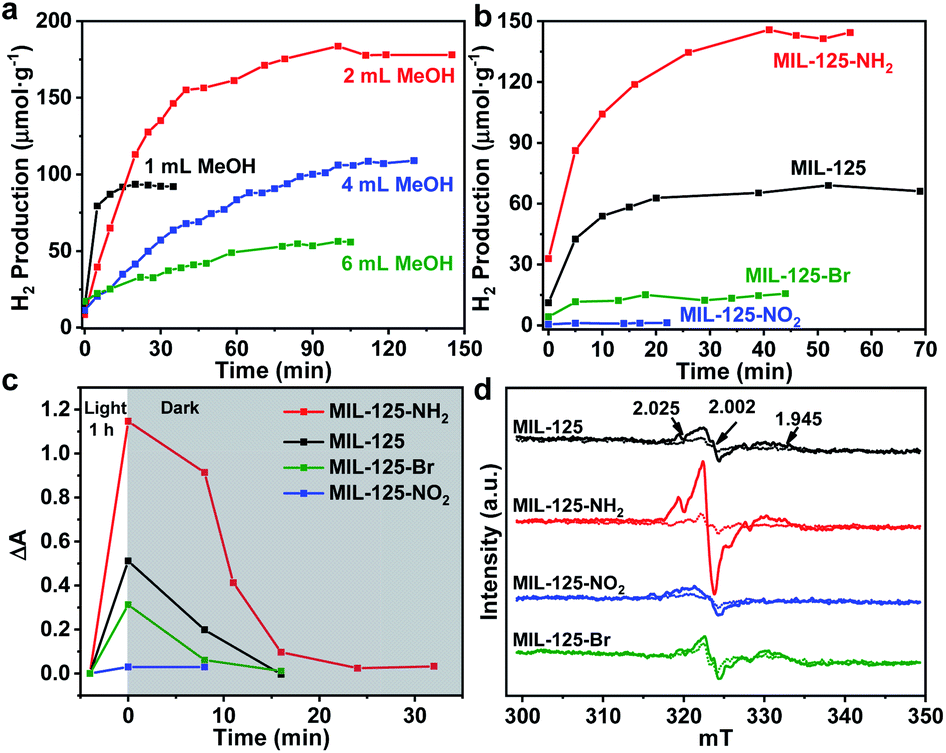 | ||
| Fig. 2 (a) Dark photocatalytic hydrogen production activity of MIL-125 with different amounts of methanol at 25 °C. (b) Dark photocatalytic hydrogen production activity comparison of MIL-125 and MIL-125-X (X = NH2, NO2, Br) with 0.5 mL TEOA as a sacrificial agent at 25 °C. (c) Decay profiles of UV-Vis absorption at 530 nm (raw data in Fig. S18†). (d) ESR spectra (solid and dotted lines represent signals in the dark and when light is on, respectively) for MIL-125 and MIL-125-X (X = NH2, NO2, Br). | ||
To explore whether the dark photocatalytic activity would be influenced by a minor structural modulation of the MOF, MIL-125-X (X = NH2, NO2, Br) featuring different functional groups tethered on its organic linker (BDC) has been examined in the presence of TEOA as the sacrificial agent (TEOA is efficient for the hole removal of MIL-125-X compared with MeOH, Fig. S13†) and Pt NPs as electron acceptors (Fig. S9 and S14†). As mentioned above, the introduction of functional groups onto the BDC linker extends the light absorption of MIL-125 to some extent (Fig. S6†). Intuitively, MIL-125-X may showcase improved dark photocatalytic hydrogen production activity.24,46 However, the results are beyond our expectations. In comparison with MIL-125, the dark photocatalytic activity and maintained time length of MIL-125-NH2 are significantly increased, while the activity and time length of MIL-125-Br are much reduced; MIL-125-NO2 almost gives no activity (Fig. 2b and S15†). The further synthesized MIL-125-0.5NH2 with 50% –NH2 group also presents the expected activity improvement in the dark reaction (Fig. S16†). The hydrogen source has been also identified by an isotopically labelled D2O experiment (Fig. S17†).
In terms of the three key aspects of photocatalysis, light absorption, charge separation and transfer, and redox ability, it is evident that the light absorption influence on the activity can be largely ruled out based on the above experimental results. Given that the same redox reaction is conducted over these MOFs, the remaining dominant factor should be charge separation and transfer ability. To disclose this, the reactive intermediates that accept and store photogenerated electrons for dark photocatalysis have been characterized. A new broad peak in the UV-Vis absorption spectra in the range of 450–800 nm, assignable to the Ti3+ absorption, can be observed under vacuum and light irradiation;24 the peak gradually decreases along with prolonged time in the dark (Fig. 2c and S18†). This observation further supports the formation of the Ti3+ intermediate, as mentioned above (Fig. S7†). Given that different functional groups possess discriminative electron-donating/-withdrawing abilities, the Ti3+ ESR and the UV-Vis absorption signal present different intensities for MIL-125-X (X = NH2, NO2, Br) under light irradiation (Fig. 2c and d). MIL-125-NH2 produces a significantly higher ESR and UV-Vis absorption signal than MIL-125, whereas MIL-125-Br and MIL-125-NO2 have much lower signal intensity. The results reveal that the electron-donating –NH2 group is beneficial to the Ti3+ production, while the –Br and –NO2 groups featuring electron-withdrawing behavior are detrimental to Ti3+ generation. Consistent with this, the UV-Vis absorption decay profile of the Ti3+ signal along with the dark time indicates that the Ti3+ intermediate produced by MIL-125-NH2 with an electron-donating group possesses a longer lifetime, whereas MIL-125-X (X = Br, NO2) with electron-withdrawing groups exhibits shorter life spans (Fig. 2c and S18†).
Photoelectrochemical characterizations are adopted to further investigate the influence of different functional groups on the lifetime of the intermediate produced by the LMCT process. Compared with MIL-125, the photocurrent and electrochemical impedance spectroscopy (EIS) measurements of MIL-125-NH2 with an electron-donating group demonstrate an enhanced photocurrent response and a reduced Nernst radius (Fig. 3a and b). On the contrary, MIL-125-X with electron-withdrawing groups (X = Br, NO2) manifests a lower photocurrent response and larger radii. The results indicate that the charge separation and transfer, i.e. LMCT process, can be promoted by an electron-donating group. The open-circuit potential (OCP) response reveals a long decay time of MIL-125-NH2, while MIL-125-Br and MIL-125-NO2 give a reduced decay time (Fig. S19†), which are supportive and in line with the above results on the promoting effect of dark photocatalysis by an electron-donating group. Given the electron-donating/-withdrawing effect of diverse groups on the linkers, it is infeasible to distinguish the charge separation ability by means of photoluminescence (PL) intensity which reflects the concentration of charge carriers (Fig. 3c). The time-resolved PL spectra demonstrate a prolonged lifetime of photogenerated carriers in MIL-125-NH2 involving the electron-donating group (Fig. 3d and Table S2†), indicating the inhibited recombination in MIL-125-NH2 compared with MIL-125, and the favorable charge separation and electron storage by introducing an electron-donating group. The steady-state PL is almost quenched when introducing TEOA in MeCN; the time-resolved PL under this condition gives a similar trend to that without TEOA (Fig. S20,† and 3d), where the reduced intensity and lifetime might be due to the consumed holes by TEOA. Combining all these results, it can be deduced that the dark photocatalytic hydrogen production largely depends on the yield and lifetime of the Ti3+ electron-storage intermediate influenced by charge separation (LMCT process) under light irradiation, and different functional groups of MOF linkers play significant roles on the intermediate production.
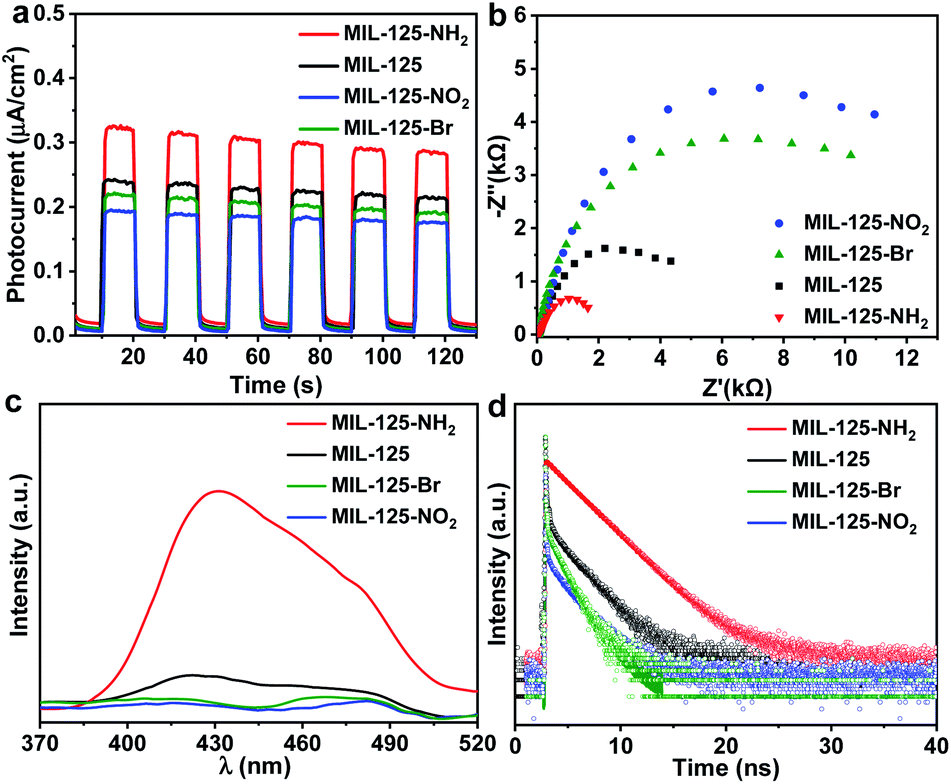 | ||
| Fig. 3 (a) Photocurrent responses, (b) EIS Nyquist plots, (c) photoluminescence (PL) spectra (excited at 250 nm), and (d) time resolved PL spectra for MIL-125 and MIL-125-X (X = NH2, NO2, Br) in MeCN. | ||
A time-dependent density-functional theory (TD-DFT) calculation has been adopted to simulate the influence of the functional group on the LMCT process and charge separation (Fig. 4 and S21†). The charge density distribution diagram of the MIL-125 excited state reveals that the electrons are mainly distributed on the Ti-oxo cluster and the holes are left on the linker (Fig. 4a). The charge density distribution changes after grafting the linkers with different functional groups. The electron-donating group (-NH2) shows more electron density distribution on the cluster and a longer distance between electrons and holes (Fig. 4b and d), which indicates the improved charge separation. By contrast, the electron density is lower on the MOF cluster with the electron-withdrawing group (–NO2) and there is significant overlap in the distribution between electrons and holes (Fig. 4c), implying the possible quick recombination of e–h pairs. The overlapping percentage of electrons and holes can be used to describe the degree of charge separation (Fig. 4d). The calculation results indicate that the introduction of an electron-donating group is beneficial, while an electron-withdrawing group is detrimental to the charge separation and electron storage in the MIL-125 system, thereby resulting in their differentiated activity in dark photocatalysis.
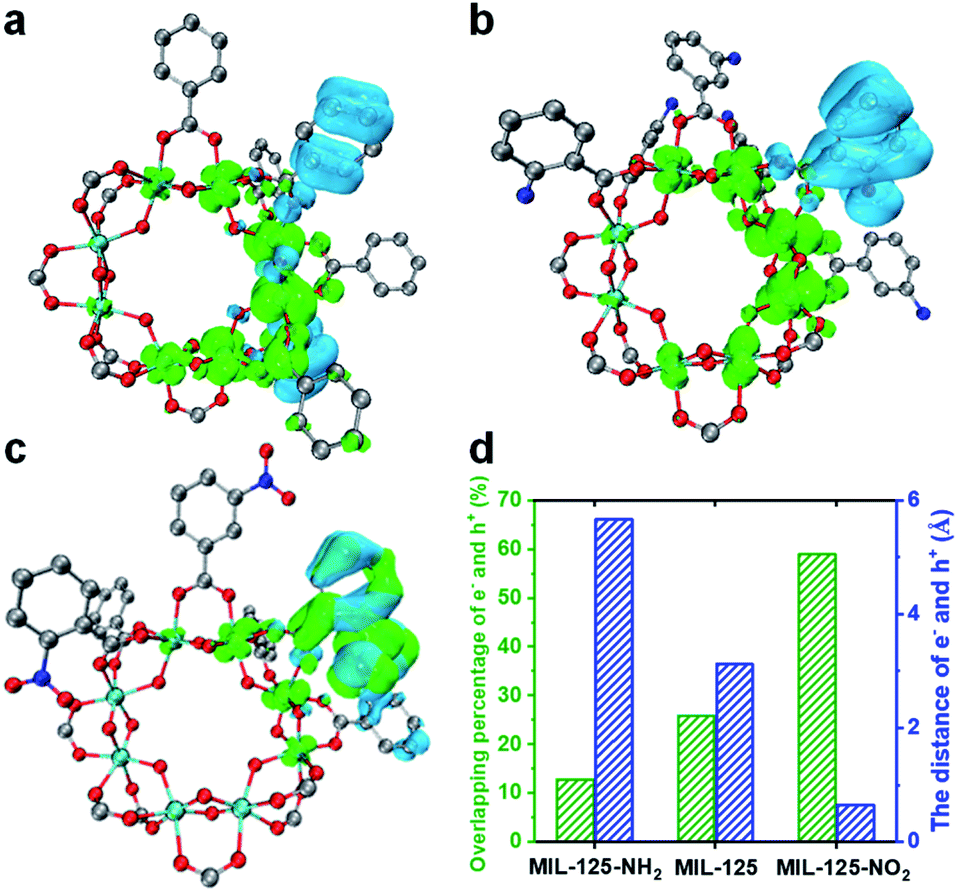 | ||
| Fig. 4 Electron–hole distribution diagrams of (a) MIL-125, (b) MIL-125-NH2 and (c) MIL-125-NO2 in their excited states (red: O; sky blue: Ti; blue: N; gray: C; olive color: electrons; cyan color: holes). (d) Overlapping percentage and distance between electrons and holes in MIL-125 and MIL-125-X (X = NH2, NO2). | ||
According to the above results, it should be safe to conclude that the yield and lifetime of the Ti3+ intermediate that store and relay electrons are largely responsible for the dark photocatalytic activity of the MIL-125 series. The functional groups on the MIL-125 linker provide different effects on the charge separation, where the better separated electrons allow the longer yield and lifetime of Ti3+ electron-storage intermediates, accounting for their distinctly different dark photocatalytic activity. The generated Ti3+ percentage in the light harvesting step and the percentage of these Ti3+ intermediates converted into H2 in the dark reaction are roughly estimated to be 8.6% and 55.3% in MIL-125-NH2, respectively, based on the consumed TEOA and the generated H2 amount (Table S3†).
In addition, the redox ability of the catalyst may be another significant factor influencing the activity of dark photocatalysis. The lowest unoccupied molecular orbital (LUMO) energy potential of MIL-125-NH2, MIL-125, MIL-125-Br and MIL-125-NO2 is at −0.92, −0.60, −0.41 and −0.31 V (vs. NHE), respectively, based on the Mott–Schottky analysis (Fig. S22†), which is similar to the trend of the DFT calculation (Fig. S21†). In combination with the bandgap energy (Eg) (Fig. S23†), the overall band structure positions can be calculated (Fig. S24a†). Their LUMO energy potential levels are all higher than the standard redox potential for hydrogen production. The introduction of electron-donating groups induces the more negative LUMO energy potential, giving rise to the easier proton reduction. This is further supported by the linear sweep voltammetry (LSV) curves, which show that MIL-125-NH2 has a lower hydrogen production overpotential than MIL-125 (Fig. S24b†), while MIL-125-Br and MIL-125-NO2 give much higher overpotentials. These results demonstrate that the introduction of functional groups will affect the redox ability of MOFs, which also contribute to the differentiated dark photocatalytic activity.
Conclusions
In summary, the formation of a long-lived Ti3+ intermediate under light irradiation enables MIL-125 with electron storage abilities to achieve dark photocatalysis. The electron-donating/-withdrawing group on the BDC linker greatly affects the redox ability and e–h separation (LMCT process) in the resulting isoreticular MIL-125-X (X = NH2, NO2, Br). The altered charge separation capability plays a crucial role and gives rise to the differentiated electron storage ability and lifetime of the Ti3+ intermediate generated by the LMCT process in these MOFs under light irradiation. Upon light off, the storage electrons in the Ti3+ intermediate can be released for proton reduction, and accordingly, the MOFs involving diverse groups exhibit distinctly different dark photocatalytic activities, among which MIL-125-NH2 possesses superior activity to the other counterparts. Given the huge structural diversity, it is believed that MOFs are very promising in dark photocatalysis. Moreover, in addition to dark photocatalytic hydrogen production, the reaction should be extendable, such as benzyl alcohol oxidation (Fig. S25†). As far as we know, this is the first study on the systematic regulation of dark photocatalytic hydrogen production over MOF-based materials. This work opens a new avenue to the lifetime modulation of electron relay toward enhanced dark photocatalysis.Experimental section
Preparation of MIL-125
MIL-125 was synthesized with some modifications based on a previous report.44 Typically, 0.6 g of terephthalic acid (BDC) was ultrasonically dispersed in 9.0 mL of extra dry N,N-dimethylformamide (DMF) in a 20 mL Teflon-lined stainless steel autoclave. Then, 1.2 mL of extra dry methanol and 0.312 mL of tetrabutyltitanate were added and stirred for 5 min. The mixture reacted in an oven at 130 °C for 15 h. The product was filtered and then washed with DMF and methanol several times, followed by drying in an oven at 85 °C overnight.Preparation of MIL-125-X (X= NH2, NO2, Br) and MIL-125-0.5NH2
Generally, the synthesis of MIL-125-NH2 was the same as that of MIL-125, except for the replacement of BDC with the same molar amount of 2-aminoterephthalic acid (ATA). For MIL-125-NO2, MIL-125-Br and MIL-125-0.5NH2, typically, 196.0 mg of BDC-NO2, 506.7 mg of BDC-Br and 90.6 mg of ATA were added to a mixture of 56.2 mg of MIL-125, 9.0 mL of extra dry N,N-dimethylformamide (DMF) and 1.0 mL of extra dry methanol, respectively. Then the mixture was heated in an oven at 130 °C for 15 h. The rest of the procedure was the same as for MIL-125.Dark photocatalytic hydrogen production experiments
Typically, 10 mg of photocatalyst was dispersed in a suspension of different solvents and sacrificial agents (the reaction temperature was 25 °C unless otherwise stated):(a) MeOH was used as the solvent and sacrificial agent for the photocatalytic hydrogen production activity of MIL-125 (4 mL of MeOH) and MIL-125-NH2 (2 mL of MeOH) under light on and off conditions (Fig. S8 and S13†);
(b) MeOH (2 mL) was used as the solvent and sacrificial agent for the photocatalytic hydrogen production over MIL-125 under different reaction temperatures (25 °C and 35 °C) (Fig. S10†);
(c) Different amounts of MeOH, TEA and TEOA as sacrificial agents and MeCN as a solvent were applied for studying the effect of sacrificial agent concentration on dark photocatalysis (Fig. 3a, S11 and S12†);
(d) TEOA (0.5 mL) as a sacrificial agent and MeCN (3.5 mL) as a solvent were applied in the investigation of different functional groups on MIL-125-X on dark photocatalysis.
The suspension was transferred into an optical reaction vessel (160 mL) and purged with nitrogen for 15 min to remove air. The reaction solution was irradiated for 1 h using a 300 W Xe lamp (LX-300F, Japan) equipped with a UV cut-off filter (200–800 nm). Then, 0.25 mL of a pre-synthesized Pt nanoparticle (NPs, ∼3 nm) solution (0.5 mg mL−1) was introduced into the reaction system once the light was turned off. The size and added amount of Pt NPs were fixed in this work. Hydrogen gas was measured after certain intervals of time by gas chromatography (Shimadzu GC-2014) with a thermal conductivity detector (TCD).
Isotopically labelled D2O experiment
Typically, 10 mg of MIL-125 were dispersed in 0.5 mL of TEOA and 3.5 mL of MeCN. The isotopically labelled D2O experiment was conducted by replacing the 0.25 mL H2O solution containing Pt NPs with a 0.25 mL D2O solution containing Pt NPs.Dark photocatalytic benzyl alcohol oxidation experiment
Generally, 10 mg of MIL-125 were dispersed in 0.5 mL of benzyl alcohol and 3.5 mL of anhydrous MeCN, and the suspension was transferred into an optical reaction vessel (160 mL) with a tee knob of an oxygen balloon (the tee knob was first disconnected) and purged with nitrogen for 15 min to remove air. The reaction solution was irradiated by the 300 W Xe lamp (LX-300F, Japan) equipped with a UV cut-off filter (200–800 nm). One hour later, the light was turned off and the tee knob of the oxygen balloon was connected. The product was measured and analysed by GC (Shimadzu GC-2010 Plus) equipped with an FID detector.Estimated percentages of excited and converted Ti3+ species
Generally, 10 mg of MIL-125-NH2 were dispersed in 3.5 mL of MeCN and 0.5 mL of TEOA. The suspension was transferred into an optical reaction vessel (160 mL) and purged with nitrogen for 15 min to remove air. The reaction solution was irradiated by the 300 W Xe lamp (LX-300F, Japan) equipped with a UV cut-off filter (200–800 nm). One hour later, the light was turned off. The consumed TEOA was analysed by GC (Shimadzu GC-2010 Plus) equipped with a FID detector, so that the excited Ti3+ in the light-harvesting step could be evaluated. The generated H2 amount was measured by GC (Shimadzu GC-2014) with a TCD to estimate the converted Ti3+.Computational methods
Density functional theory (DFT) was carried out to elucidate the electronic properties of the titled MOFs in their excitation states. In this work, the MIL-125 and MIL-125-NH2 models were constructed based on the structural formula Ti8O8(OH)4(BDC)6 and Ti8O8(OH)4(BDC-NH2)6, respectively.46 MIL-125-NO2 was achieved by replacing three BDC (total of six BDC units) with BDC-NO2. The structural optimization was performed by the PBE1PBE density functional method and the def2-SVP basic set.48,49 Grimme's DFT-D3 method was used for the correction of dispersion interaction via the Becke–Johnson damping function.50,51 The calculation of the excited states was performed with time-dependent density-functional theory (TDDFT). Due to the large computational cost, only the first singlet transitions were calculated. The hole and electron distributions, which represent the difference between the electron density of the first singlet excited state and the ground state, were calculated and analyzed by the wave function analysis software Multiwfn 3.8, and the results were visualized by the visualization program VMD 1.9.1.52–54To better understand the electron excitation process, some indices describing electron holes are defined as follows:
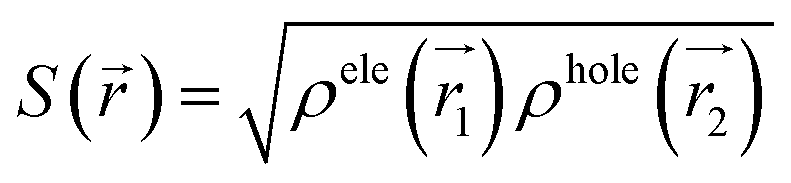
![[r with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0072_20d1.gif) 1) ρhole(2) are defined as the density of the hole and the electron, respectively. Subsequently, Sr, which represents the overlap of holes and electrons, is defined as follows:
1) ρhole(2) are defined as the density of the hole and the electron, respectively. Subsequently, Sr, which represents the overlap of holes and electrons, is defined as follows: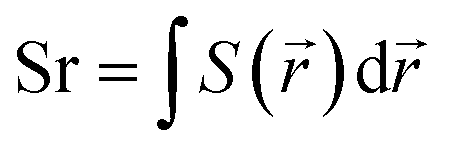
The center of mass of the electron (hole) can be calculated by the following formula:
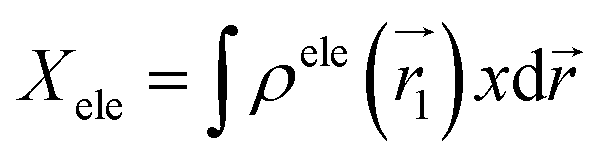
| μele = (Xele,Yele,Zele) |
![[D with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0044_20d1.gif) = |μele − μhole| = |μele − μhole| |
| D = || |
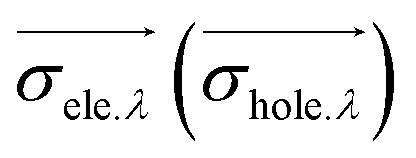 indicates the distribution breadth of electrons (holes) in the corresponding direction:
indicates the distribution breadth of electrons (holes) in the corresponding direction:where λ represents the three directions of coordinates (x, y, z).
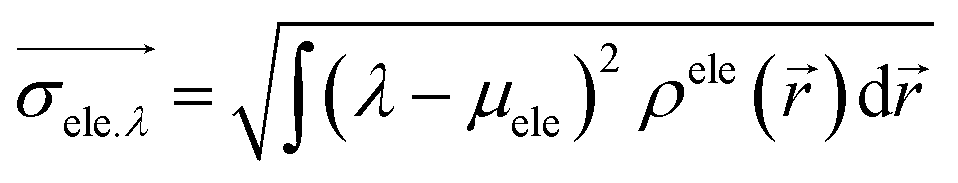
H
λ
measures the average extent of holes and electrons in the λ direction:
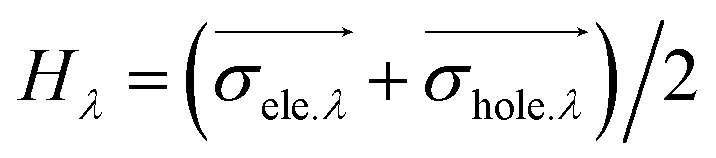
H
CT measures the average extent of holes and electrons in the direction of charge transition:
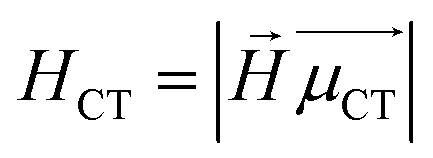
![[H with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0048_20d1.gif) is the vector written together with Hx, Hy, and Hy, and
is the vector written together with Hx, Hy, and Hy, and 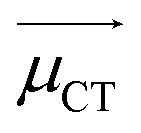 is the unit vector in the direction of charge transition, which can be directly obtained by using the position of the center of mass of electrons and holes.
is the unit vector in the direction of charge transition, which can be directly obtained by using the position of the center of mass of electrons and holes.
The H index reflects the overall average distribution breadth of electrons and holes:
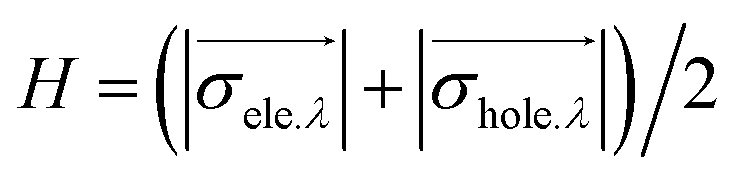
Finally, we define the t index that is used to measure whether electrons and holes are significantly separated:
| t = D − HCT |
If the t index is larger than 0, this implies that the separation of holes and electrons is sufficient, because the centroids of holes and electrons are far away, and their average degree of extension in this direction is relatively not so high.
Photoelectrochemical measurements
The photoelectrochemical measurements were performed on a CHI 760E electrochemical workstation (Chenhua Instrument, Shanghai, China) with a standard three-electrode system: a working electrode (the photocatalyst-coated ITO plate), a counter electrode (Pt plate) and a reference electrode (Ag/AgCl), with a 300 W Xenon lamp (LX-300F, Japan) as the light source and a 0.1 M Na2SO4 solution as the electrolyte. A suspension (200 μL, prepared by 2 mg of catalyst, 10 μL of Nafion and 1 mL of ethanol) was dropped onto the ITO plate surface with an exposed area of 1.0 × 1.0 cm2 as the working electrode. A bias potential of +0.5 V was applied in this measurement process.Electrochemical impedance spectroscopy
Electrochemical impedance spectroscopy was performed on a Zahner Zennium electrochemical workstation in a standard three-electrode system (photocatalyst-coated glassy carbon as the working electrode, a Pt plate as the counter electrode, and Ag/AgCl as the reference electrode), with a 0.1 M Na2SO4 aqueous solution as the electrolyte. The working electrode was prepared by dropping 30 μL of the suspension (2 mg of catalyst was dispersed into a mixed solution of 10 μL of 5 wt% Nafion and 1 mL of ethanol) onto the surface of the glassy carbon electrode. Then, the EIS measurements were conducted with a bias potential of −1.7 V in the dark.Linear sweep voltammetry (LSV) curves
LSV curves were also collected on a CHI 760E electrochemical workstation with a standard three-electrode system, the same as that in photoelectrochemical measurements. However, the working electrodes were prepared by dropping 100 μL of suspension to the surface of an ITO plate with a 2.0 × 2.0 cm2 exposed area. A bias potential of −0.6 V to −0.05 V was applied in this measurement process.Open-circuit potential (OCP) response test
On a CHI 760E electrochemical workstation (Chenhua Instrument, Shanghai, China) with a standard three-electrode system of a photocatalyst-coated ITO working electrode, a Pt counter electrode and an Ag/AgCl reference electrode, the OCP measurements were carried out in a 0.1 M Na2SO4 electrolytic solution. The ITO working electrode with a 2.0 × 2.0 cm2 photocatalyst-coated area was prepared by a 100 μL suspension (2 mg of catalyst, 10 μL of 5 wt% Nafion and 1 mL of ethanol). For the first 50 s, the OCP was measured under light irradiation and the rest of the data were collected in the dark.Data availability
All experimental and computation data and methods related to this study can be found in the test and ESI.†Author contributions
H.-L. J. conceived the idea and supervised the project. Y. P. and J. W. performed the experiments and collected the data. S. C. and W. J. conducted the DFT calculations. H.-L. J. and Y. P. co-wrote the manuscript. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2021YFA1500400), the NSFC (21725101 and 22161142001), the Dalian National Laboratory Cooperation Fund, CAS (DNL201911), the Fundamental Research Funds for the Central Universities (WK3450000007 and WK2060000038) and the Supercomputing Center of USTC.References
- J. Ran, J. Zhang, J. Yu, M. Jaroniecc and S. Z. Qiao, Chem. Soc. Rev., 2014, 43, 7787–7812 RSC.
- Q. Wang and K. Domen, Chem. Rev., 2020, 120, 919–985 CrossRef CAS PubMed.
- X.-B. Li, Z.-K. Xin, S.-G. Xia, X.-Y. Gao, C.-H. Tung and L.-Z. Wu, Chem. Soc. Rev., 2020, 49, 9028–9056 RSC.
- A. Magnuson, M. Anderlund, O. Johansson, P. Lindblad, R. Lomoth, T. Polivka, S. Ott, K. Stensjö, S. Styring, V. Sundström and L. Hammarström, Acc. Chem. Res., 2009, 42, 1899–1909 CrossRef CAS PubMed.
- J. Kim, D. Hansora, P. Sharma, J. Jang and J. Lee, Chem. Soc. Rev., 2019, 48, 1908–1971 RSC.
- Y. Wang, A. Vogel, M. Sachs, R. Sprick, L. Wilbraham, S. Moniz, R. Godin, M. Zwijnenburg, J. R. Durrant, A. Cooper and J. Tang, Nat. Energy, 2019, 4, 746–760 CrossRef CAS.
- Y. Zhao, C. Ding, J. Zhu, W. Qin, X. Tao, F. Fan, R. Li and C. Li, Angew. Chem., Int. Ed., 2020, 59, 9653–9658 CrossRef CAS PubMed.
- G. Vincent, W.-h. Lau, D. Klose, H. Kasap, F. Podjaski, M.-C. Pigni, E. Reisner, G. Jeschke and B. V. Lotsch, Angew. Chem., Int. Ed., 2017, 56, 510–514 CrossRef PubMed.
- H. Zhang, J. Ming, J. Zhao, Q. Gu, C. Xu, Z. Ding, R. Yuan, Z. Zhang, H. Lin, X. Wang and J. Long, Angew. Chem., Int. Ed., 2019, 58, 7718–7722 CrossRef CAS PubMed.
- G. Cui, X. Yang, Y. Zhang, Y. Fan, P. Chen, H. Cui, Y. Liu, X. Shi, Q. Shang and B. Tang, Angew. Chem., Int. Ed., 2019, 58, 1340–1344 CrossRef CAS PubMed.
- M. Sakar, C.-C. Nguyen, M.-H. Vu and T.-O. Do, ChemSusChem, 2018, 11, 809–820 CrossRef CAS PubMed.
- L. Liu, W. Yang, Q. Li, S. Gao and J. K. Shang, ACS Appl. Mater. Interfaces, 2014, 6, 5629–5639 CrossRef CAS PubMed.
- Q. Zhang, H. Wang, Z. Li, C. Geng and J. Leng, ACS Appl. Mater. Interfaces, 2017, 9, 21738–21746 CrossRef CAS PubMed.
- X. Liu, X. Chen, Y. Li, B. Wu, X. Luo, S. Ouyang, S. Luo, A. A. A. Kheraifd and J. Lin, J. Mater. Chem. A, 2019, 7, 19173–19186 RSC.
- H. Schlomberg, J. Kröger, G. Savasci, M. W. Terban, S. Bette, I. Moudrakovski, V. Duppel, F. Podjaski, R. Siegel, J. Senker, R. E. Dinnebier, C. Ochsenfeld and B. V. Lotsch, Chem. Mater., 2019, 31, 7478–7486 CrossRef CAS PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- H.-C. Zhou and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5415–5418 RSC.
- B. Li, H.-M. Wen, Y. Cui, W. Zhou, G. Qian and B. Chen, Adv. Mater., 2016, 28, 8819–8860 CrossRef CAS PubMed.
- L. Jiao, Y. Wang, H.-L. Jiang and Q. Xu, Adv. Mater., 2018, 30, 1703663 CrossRef PubMed.
- A. Dhakshinamoorthy, Z. Li and H. Garcia, Chem. Soc. Rev., 2018, 47, 8134–8172 RSC.
- C. T. Saouma, S. Richard, S. Smolders, M. F. Delley, R. Ameloot, F. Vermoortele, D. E. D. Vos and J. M. Mayer, J. Am. Chem. Soc., 2018, 140, 16184–16189 CrossRef CAS PubMed.
- X. Zhang, Z. Chen, X. Liu, S. K. Hanna, X. Wang, R. Taheri-Ledari, A. Maleki, P. Li and O. K. Farha, Chem. Soc. Rev., 2020, 49, 7406–7427 RSC.
- W.-H. Li, W.-H. Deng, G.-E. Wang and G. Xu, EnergyChem, 2020, 2, 100029 CrossRef.
- Y. Fu, D. Sun, Y. Chen, R. Huang, Z. Ding, X. Fu and Z. Li, Angew. Chem., Int. Ed., 2012, 51, 3364–3367 CrossRef CAS PubMed.
- H.-Q. Xu, J. Hu, D. Wang, Z. Li, Q. Zhang, Y. Luo, S.-H. Yu and H.-L. Jiang, J. Am. Chem. Soc., 2015, 137, 13440–13443 CrossRef CAS PubMed.
- H. Zhang, J. Wei, J. Dong, G. Liu, L. Shi, P. An, G. Zhao, J. Kong, X. Wang, X. Meng, J. Zhang and J. Ye, Angew. Chem., Int. Ed., 2016, 55, 14310–14314 CrossRef CAS PubMed.
- Y. Wang, N.-Yu. Huang, J.-Q. Shen, P.-Q. Liao, X.-M. Chen and J.-P. Zhang, J. Am. Chem. Soc., 2018, 140, 38–41 CrossRef CAS PubMed.
- J. Wang, L. Qiao, H. Nie, H. Huang, Y. Li, S. Yao, M. Liu, Z. Zhang, Z. Kang and T. Lu, Nat. Commun., 2021, 12, 813 CrossRef CAS PubMed.
- N. Li, J. Liu, J.-J. Liu, L.-Z. Dong, Z.-F. Xin, Y.-L. Teng and Y.-Q. Lan, Angew. Chem., Int. Ed., 2019, 58, 5226–5230 CrossRef CAS PubMed.
- Z.-B. Fang, T.-T. Liu, J. Liu, S. Jin, X.-P. Wu, X.-Q. Gong, K. Wang, Q. Yin, T.-F. Liu, R. Cao and H.-C. Zhou, J. Am. Chem. Soc., 2020, 142, 12515–12523 CrossRef CAS PubMed.
- T. Zhou, Y. Du, A. Borgna, J. Hong, Y. Wang, J. Han, W. Zhang and R. Xu, Energy Environ. Sci., 2013, 6, 3229–3234 RSC.
- D. Kim, D. R. Whang and S. Y. Park, J. Am. Chem. Soc., 2016, 138, 8698–8701 CrossRef CAS PubMed.
- T. Zhang and W. Lin, Chem. Soc. Rev., 2014, 43, 5982–5993 RSC.
- Y. An, Y. Liu, P. An, J. Dong, B. Xu, Y. Dai, X. Qin, X. Zhang, M.-H. Whangbo and B. Huang, Angew. Chem., Int. Ed., 2017, 56, 3036–3040 CrossRef CAS PubMed.
- J.-D. Xiao, L. Han, J. Luo, S.-H. Yu and H.-L. Jiang, Angew. Chem., Int. Ed., 2018, 57, 1103–1107 CrossRef CAS PubMed.
- S. Z. Yang, B. Pattengale, E. L. Kovrigin and J. Huang, ACS Energy Lett, 2017, 2, 75–80 CrossRef CAS.
- Z. Xia, C. He, X. Wang and C. Duan, Nat. Commun., 2017, 8, 361 CrossRef PubMed.
- Y. Zhang, J. Guo, L. Shi, Y. Zhu, K. Hou, Y. Zheng and Z. Tang, Sci. Adv., 2017, 3, e1701162 CrossRef PubMed.
- C. Xu, Y. Pan, G. Wan, H. Liu, L. Wang, H. Zhou, S.-H. Yu and H.-L. Jiang, J. Am. Chem. Soc., 2019, 141, 19110–19117 CrossRef CAS PubMed.
- C. H. Hendon, D. Tiana, M. Fontecave, C. Sanchez, L. D’arras, C. Sassoye, L. Rozes, C. Mellot-Draznieks and A. Walsh, J. Am. Chem. Soc., 2013, 135, 10942–10945 CrossRef CAS PubMed.
- M. B. Chambers, X. Wang, L. Ellezam, O. Ersen, M. Fontecave, C. Sanchez, L. Rozes and C. Mellot-Draznieks, J. Am. Chem. Soc., 2017, 139, 8222–8228 CrossRef CAS PubMed.
- Y.-P. Wei, Y. Liu, F. Guo, X.-Y. Dao and W. Y. Sun, Dalton Trans., 2019, 48, 8221–8226 RSC.
- X.-P. Wu, L. Gagliardi and D. G. Truhlar, J. Am. Chem. Soc., 2018, 140, 7904–7912 CrossRef CAS PubMed.
- M. Dan-Hardi, C. Serre, T. Frot, L. Rozes, G. Maurin, C. Sanchez and G. Férey, J. Am. Chem. Soc., 2009, 131, 10857–10859 CrossRef CAS PubMed.
- M. A. Nasalevich, C. H. Hendon, J. G. Santaclara, K. Svane, B. v. d. Linden, S. L. Veber, M. V. Fedin, A. J. Houtepen, M. A. v. d. Veen, F. Kapteijn, A. Walsh and J. Gascon, Sci. Rep., 2016, 6, 23676 CrossRef CAS PubMed.
- S.-Y. Han, D.-L. Pan, H. Chen, X.-B. Bu, Y.-X. Gao, H. Gao, Y. Tian, G.-S. Li, G. Wang, S.-L. Cao, C.-Q. Wan and G.-C. Guo, Angew. Chem., Int. Ed., 2018, 57, 9864–9869 CrossRef CAS PubMed.
- S. Mohajernia, P. Andryskova, G. Zoppellaro, S. Hejazi, S. Kment, R. Zboril, J. Schmidt and P. Schmuki, J. Mater. Chem. A, 2020, 8, 1432–1442 RSC.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- S. Grimme, J. Antony and S. Ehrlich, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- Z. Liu, T. Lu and Q. Chen, Carbon, 2020, 165, 461–467 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graph., 1996, 14, 33–38 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials and instrumentation, supplemental figures and table. See https://doi.org/10.1039/d1sc06785k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |