
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of reaction conditions on the hydrogenolysis of polypropylene and polyethylene into gas and liquid alkanes†
Linxiao
Chen‡
 a,
Yifeng
Zhu‡
b,
Laura C.
Meyer
a,
Lillian V.
Hale
a,
Thuy T.
Le
a,
Abhi
Karkamkar
a,
Johannes A.
Lercher
a,
Oliver Y.
Gutiérrez
*a and
János
Szanyi
*a
a,
Yifeng
Zhu‡
b,
Laura C.
Meyer
a,
Lillian V.
Hale
a,
Thuy T.
Le
a,
Abhi
Karkamkar
a,
Johannes A.
Lercher
a,
Oliver Y.
Gutiérrez
*a and
János
Szanyi
*a
aInstitute for Integrated Catalysis, Pacific Northwest National Laboratory, Richland, WA 99352, USA. E-mail: oliver.gutierrez@pnnl.gov; janos.szanyi@pnnl.gov
bDepartment of Chemistry, Fudan University, 200433, P.R. China
First published on 2nd February 2022
Abstract
Hydrogenolysis of polypropylene (PP) and polyethylene (PE) provides a pathway to convert these plastics into smaller hydrocarbons at relatively low temperature. Among carbon (C)-supported transition metals, ruthenium (Ru) exhibited the highest efficacy, producing mixtures of C1–C38 alkanes. The branching degree of the products depends on the position of the C–C cleavage, which can be tuned by the pressure of H2. Liquid alkanes are produced below 225 °C and 200 °C from PP and PE, respectively, at 30 bar. The C distribution and branching level of the products remain invariant below full conversion of the initial polymer. Increasing H2 pressure favors the hydrogenolysis of internal C–C bonds, reducing methane (CH4) production, and favors linear over branched products. A liquid yield of >57% was achieved with PE under optimum conditions. We reveal the impact of the starting polyolefin structure, reaction conditions, and presence of chlorine on the product distribution and branching degree.
1. Introduction
The annual production of synthetic plastics totaled 367 million metric tons (Mt) in 2020, a nearly 200-fold increase compared to 1950.1 Although this is still less than 5% of all petroleum and gas used in industry,2,3 the use of plastic is foreseen to keep rising to the point of driving oil demand to 2050.4 The concern associated with this trend is that most of the plastic being discarded accumulates in landfills and nature, posing severe threats to the environment.5 Among the 6300 Mt plastic waste generated as of 2015, only 9% had been recycled, 12% was incinerated, and 79% was deposited in landfills.6 As direct combustion is challenging, technologies that enable converting plastic waste into easily accessible energy carriers and other useful products (“upcycle”) are needed.The most common plastics are polyethylene (PE, 32 wt%) and polypropylene (PP, 23 wt%).6 Currently, pyrolysis is the main approach to convert both compounds into smaller hydrocarbons.7–9 Pyrolysis requires high reaction temperatures and provides low selectivity towards higher-value products. The cleavage of C–C bonds is also viable through hydrogenolysis; however, this approach is only beginning to be explored for polymer conversion.10–13 Numerous recent reports are focusing on hydrogenolysis as a conversion strategy (Table 1 (ref. 14–24)). While several platinum (Pt)-group metals are being explored, Ru-based catalysts are attracting most of the attention from the research community.19,20 Typically, the selectivity toward methane has been observed to decrease at high H2 pressure.20 Interestingly, while most studies are focusing on PE as the feedstock, the upcycling of PP, which has an even lower recycling rate than PE, has not been thoroughly explored or compared with PE.
| Feedstock | Average Mw | Catalyst | T (°C) | Major products | Ref. |
|---|---|---|---|---|---|
| PE | ∼35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
Pt/SrTiO3 | 300 | Alkanes | 14 |
| PE | ∼82600 |
mSiO2/Pt/SiO2 | 250 | Alkanes | 15 |
| PE | ∼3500 | Pt/Al2O3 | 280 | Alkylaromatics, alkylnaphthalenes (∼C30) | 16 |
| PE | Not provided | Pt/C or SAPO | ∼25 (plasma) | Gaseous alkanes | 17 |
| PE | ∼120000 |
Pt–Re/SiO2 | 170 | Low chain PE (Mw = 1300) | 18 |
| Aromatic polymers | Not provided | Ru/Nb2O5 | 200 | Arenes | 19 |
| PE | ∼4000 | Ru/C | 200 to 250 | Alkanes | 20 |
| PE | Not provided | Ru/C | 220 to 280 | Alkanes | 21 |
| PE | ∼4000 to ∼50000 |
Ru/CeO2 | 240 to 300 | Alkanes | 22 |
| PP | ∼12000 and ∼250000 |
Ru/TiO2 | 250 | Alkanes | 23 |
| PP | ∼12000 and ∼340000 |
Ru/C | 200 to 250 | Iso-alkanes | 24 |
| PE | ∼4000 | Ru/C | 150 to 250 | Alkanes | This work |
| PP | ∼250000 |
Thus, information on how the polyolefin structure and specific composition (branching, impurities, and the presence of heteroatoms such as chlorine [Cl]) impact activity and selectivity of catalytic conversions is still lacking. These changing chemical and structural compositions are expected to have a very strong impact on the activity of potential metal catalysts, which requires the understanding of fundamental relationships between the catalyst nature and the (mixture) of reacting polymers. In this work we compare the hydrogenolysis of PE and PP on Ru, focusing on the influence of the polymer structure and H2 pressure on product selectivity. We discuss the implications of our results in terms of the reaction mechanism and competitive adsorption of hydrogen, polymer, and alkane products over Ru catalysts.
We address these issues by targeting the hydrogenolytic conversion of PE and PP into liquid alkanes under relatively mild conditions. We report the activity and selectivity for a series of supported metal catalysts, focusing in detail on the most active catalyst, Ru/C. The C1–C38 products were analyzed in detail to establish the carbon distribution and branching level.
2. Experimental
2.1 Chemicals and pre-treatments
PP was purchased from Sigma-Aldrich (isotactic), with average Mw and Mn of ∼250000 g mol−1 and ∼67000, respectively. Before use, the large PP grains were crushed into small particles. PE also was purchased from Sigma-Aldrich as powder, with the average Mw and Mn of ∼4000 g mol−1 and ∼1300, respectively. The transition temperatures (Tg) of PP and PE are 160–165 °C and 106 °C, respectively. Ru/C, Rh/C, Pt/C, palladium (Pd)/C, iridium (Ir)/C, and nickel (Ni)/C at 5 wt% were purchased from Sigma-Aldrich. Prior to the reaction or transmission electron microscopy (TEM) measurements, the catalysts were pre-reduced in 5% H2/nitrogen (N2) flow at 200 °C for 3 h and then passivated in 1% O2/N2 flow. Ethyl acetate (EtOAc) for product extraction was also purchased from Sigma-Aldrich (high-pressure liquid chromatography grade, ≥99.8%).
2.2 Catalytic tests and post-reaction treatments
In a typical procedure, 100 mg of catalyst was mixed with 1000 mg PP/PE in a glass vial (3.7 mL, Fisherbrand). This mixture then was heated to melt the polymer and integrate the catalyst powder in it. The polymer–catalyst mixture then was placed into an autoclave reactor with a total internal volume of 125 mL. The reactor was sealed, and oxygen was removed by pressurizing with H2 and venting in at least five successive cycles. Finally the reactor was pressurized with H2 to the desired pressure, and then heated to the reaction temperature. Upon heating, the pressure increased but then remained relatively invariant during the reaction. We did not use any solvent, and we did not apply stirring. After the reaction, the reactor was quenched in an ice bath. The gas was collected by releasing the gas into a gas-sampling bag (Tedlar®) at room temperature and analyzed by gas chromatography-thermal conductivity detection (GC-TCD) (Inficon Micro GC Fusion gas analyzer with a four-module chassis). The liquids condensed inside the autoclave were dissolved with EtOAc and collected. The post-reaction solid composed of the catalyst and residual oligomers and polymers was further extracted with EtOAc. After filtration, the solution was combined with the liquids and analyzed by gas chromatography-mass spectroscopy (GC-MS) (Agilent 7890A GC, DB-5 column, Agilent 7693 autosampler and Agilent 5975C mass spectrometer) for identification and gas chromatography-flame ionization detection (GC-FID) (Agilent 7890A GC, DB-5 column, Agilent 7693 autosampler) for quantification. The solid residue was dried in an oven at 80 °C, weighed and the resulting mass was corrected for weight of catalyst added.2.3 Product analysis
The solid residue after EtOAc extraction was completely dried and then weighed. The solid conversion, which is not the “polymer conversion”, was calculated as:
Liquid products (C6–C38) were quantified by GC-FID, with butyl-cyclohexane (Sigma-Aldrich, >99%) as the internal standard. The response factor for alkanes of each C number and the retention time of each n-alkanes were obtained with a C7–C30n-alkane standard (Supelco, 1000 μg mL−1 in n-hexane) and a C10–C40 even n-alkane standard (Supelco, 50 μg mL−1 in n-heptane). The response factors in the concentration range of the product solution can be found in Table S2.† All peaks between n-CkH2k+2 and n-Ck−1H2k were assumed as branched Ck alkanes unless GC-MS suggested otherwise. Although peaks from branched C7 products and n-C6H14 overlap with the large EtOAc peak, these products are of low concentration and evaporate to some extent during handling, so they only account for <1% of total C. Therefore, the inability to quantify such products does not affect our analysis nor the C balance in a meaningful way.
The gaseous products (C1–C5) were quantified using a micro-GC-TCD instrument (Inficon Micro GC Fusion gas analyzer) equipped with four columns (12 m Rt®-Q-Bond, 10 m Rxi®-1 ms, 10 m Rt®-Alumina BOND/Na2SO4 and 10 m Rt®-Msieve 5A). Before every analysis run the GC instrument was purged with N2 followed by calibration with a calibrating gas mixture (Matheson) of N2, H2, carbon monoxide, carbon dioxide, methane, ethane, ethylene, propane, propylene, isobutane, n-butane, and n-butene. For the sample analysis, the gas sampling bag was connected to the GC and after flushing the sampling loop, the reaction gas mixture was measured several times until constant signals were obtained. The fraction of each alkane in the mixture was converted to its absolute quantity by the reactor volume (125 mL), and the post-reaction pressure was recorded at room temperature.
Because both PE and PP have high average MW (4000 and 250000 g mol−1, respectively) and large average chain length (285 and 17858 C units, respectively), the stoichiometry of C:H is close to 1:2, which allows us to calculated moles of C in the starting polymers based on their mass. With the information discussed above, the C yield of each phase was calculated as:
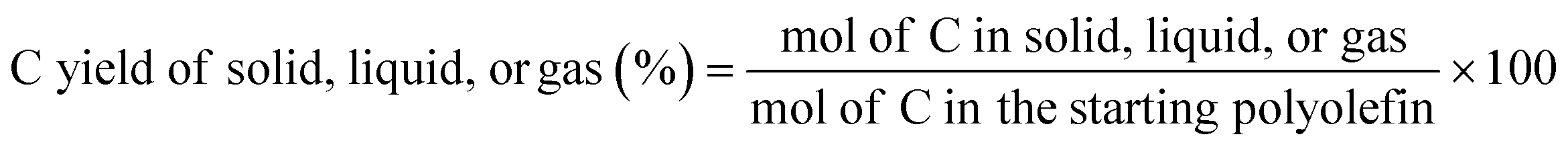
The C selectivity of each phase was calculated as:
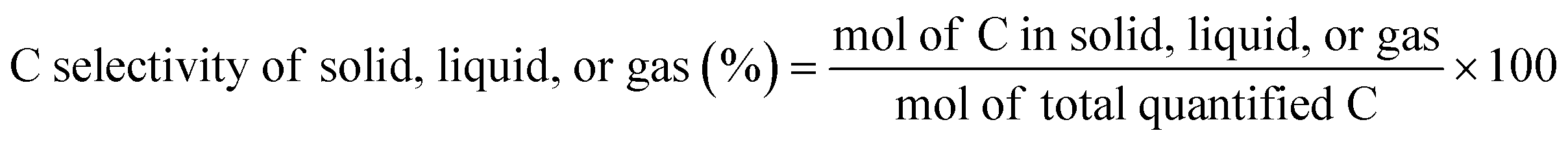
The C yield of each C number from C1 to C38 was calculated as:
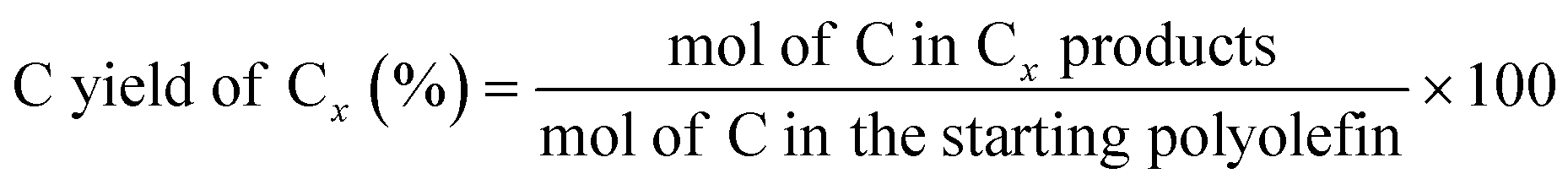
2.4 Catalyst characterization
TEM measurements were performed on an FEI Tecnai F20 electron microscope to determine the metal particle size distribution on all C-supported catalysts. Each catalyst was ground and ultrasonically dispersed in ethanol. Drops of the suspension were applied on a copper grid coated with a carbon film and measurements were carried out with an electron detector with a 200 keV acceleration voltage. The elemental analysis was performed using an EDAX Si (Li) EDS detector and FEI TIA analysis software. Statistical analysis of the metal particle size was realized by counting >300 particles in several places of the mesh. The mean particle size was calculated using the following equation:where dTEM is the volume-area mean diameter of the particle, di is the diameter of the particle, measured from TEM images, and ni is the number of particles with diameter di.
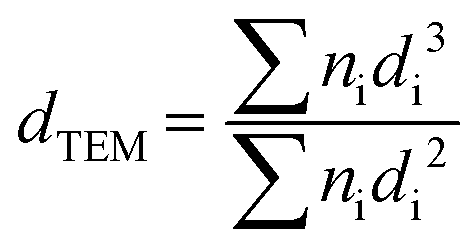
Inductive coupled plasma (ICP) was performed to determine Ru loading on Ru/C. The Ru/C catalyst was digested in concentrated nitric acid in a sealed microwave vessel. After the dissolution of all solids, the solution was analyzed on a Perkin Elmer Optima 7300DV ICP-OES instrument equipped with a cyclonic spray chamber and a Meinhard nebulizer.
H2-Chemisorption was conducted with a Micromeritics ASAP 2020, in chemisorption mode, to determine Ru dispersion on Ru/C. Prior to the measurement, the catalyst was outgassed at room temperature, followed by reduction under H2 at 523 K. The first adsorption isotherm was recorded at room temperature from 0.1 mbar to 600 mbar at 393 K. After evacuation at 393 K for 1 h, a second isotherm set was measured corresponding to physisorbed H2 under the same conditions as the first isotherm. The concentration of chemisorbed H2 was calculated by subtracting the two isotherms and extrapolating to zero H2 pressure. A stoichiometry of 1:1 for metal:hydrogen was assumed to derive the dispersion from the concentration of chemisorbed hydrogen.
N2-Physisorption was conducted to determine the specific surface area and pore volume of Ru/C on the same Micromeritics ASAP 2020 but in physisorption mode at liquid N2 temperature (77 K). The sample was outgassed at 573 K for 2 h prior to the measurements. The textural properties were derived from the Brunauer–Emmett–Teller and Barrett–Joyner–Halenda models applied to the N2 sorption data.
3. Results and discussion
3.1 Comparison of supported metals for polyolefin hydrogenolysis
A series of carbon-supported transition metals (i.e., Ru/C, Rh/C, Pt/C, Pd/C, Ir/C, and Ni/C [all 5 at wt%]) was compared to establish the principal reactivity trend for converting PP and PE by hydrogenolysis (Table 2). Herein “gas products” refers to ≤C5 alkanes, “liquid products” refers to EtOAc-soluble, GC-analyzable alkanes (i.e., C6–C38, which are in the most desirable C range and not necessarily liquid at room temperature), and “solid products” refers to EtOAc-insoluble hydrocarbon oligomers or polymers. Unidentified products accounted for up to 35% of total C. For clarity, they are omitted in the following discussion, but their yield is described in the ESI.†| Catalyst (5 wt%) | Metal particle sizea (nm) | Carbon yieldb (%) | |||||
|---|---|---|---|---|---|---|---|
| PP (MW ≈ 250000) |
PE (MW ≈ 4000) | ||||||
| Solid | Liquid | Gas | Solid | Liquid | Gas | ||
| a Derived from TEM (Fig. S1†) performed after reduction by H2 at 250 °C and passivation by 1% O2. b Reaction conditions: 250 °C, 30 bar H2, 18 h, 1 g PP or PE, 100 mg catalyst. | |||||||
| Blank | N/A | 99.0 | 0 | <0.1 | 96.4 | 0 | 0 |
| Ru/C | 2.4 ± 0.7 | 0 | 0 | 100 | 0 | 0 | 100 |
| Rh/C | 2.3 ± 0.7 | 68.2 | 6.4 | 5.7 | 0 | 35.1 | 17.3 |
| Pt/C | 2.4 ± 0.5 | 94.4 | 0 | 0.2 | 93.8 | 0 | <0.1 |
| Pd/C | 2.8 ± 0.5 | 99.1 | 0 | 0.3 | 93.3 | 0 | <0.1 |
| Ir/C | 2.8 ± 0.5 | 99.0 | 0 | 0.1 | 93.4 | 0 | 0.1 |
| Ni/C | 6 ± 1 | 99.4 | 0 | <0.1 | 94.1 | 0 | <0.1 |
Table 2 shows the carbon yields of solid, liquid, and gas products on each catalyst after reaction for 18 h. Among the catalysts, Ru/C exhibited unique efficacy under the explored conditions, completely converting both PP and PE into gaseous products. Rh/C also produced gas and liquid products under these conditions but showed lower activity compared to Ru/C. While PE was completely converted with Rh/C into gases and liquids, 68% PP remained solid. For Pt/C, Pd/C, Ir/C, and Ni/C, the solid mass loss was <7%, with <1% combined gas and liquid yield, indicating low hydrogenolysis activity under these conditions. Thermogravimetric analysis of fresh and spent Ru/C catalysts did not show signs of coke formation during the reaction.
The superior activity of Ru to other metals for alkane hydrogenolysis has been reported and attributed to the balanced coverage between chemisorbed hydrogen (*H) and chemisorbed hydrocarbon species (*CxHy).20,25–28 The trend among the metals in Table 2 agrees well with the sequence of catalytic activity of metals reported by Rorrer et al. for n-octadecane, i.e., Ru > Rh ≫ Ni/Pt.20 It should be noted in passing that for C2H6 hydrogenolysis, the sequence was slightly different, i.e., Ru > Rh/Ni/Ir ≫ Pt/Pd.25,27 These differences are typical for hydrogenolysis of alkanes varying in size; that is, the structure sensitivity markedly decreases as the molecular size increases.29,30
3.2 Carbon distribution and branching level of liquid products
While the conversion on Ru/C (see Table S1 for ESI† characterization) at 250 °C failed to yield liquid products, lower temperatures improved the selectivity towards liquids (Fig. 1a and b). We repeated selected experiments and found that solid conversions and liquid/gas yields varied within 5% and 3%, respectively. Hydrogenolysis starts as low as 175 °C for PP and 150 °C for PE, with solid conversion increasing with temperature. We note that the lowest temperature (150 °C) at which Ru/C was found to convert PE is much lower than that of Pt catalysts reported in the literature (≥250 °C, Table 1). The conversion of PP requires higher temperature than PE suggesting that under our reaction conditions, PP is less reactive than PE. We cannot, however, discuss the reasons for such a difference because the polymers we investigated have many differences (e.g., MW, viscosity, chemical structure, etc.).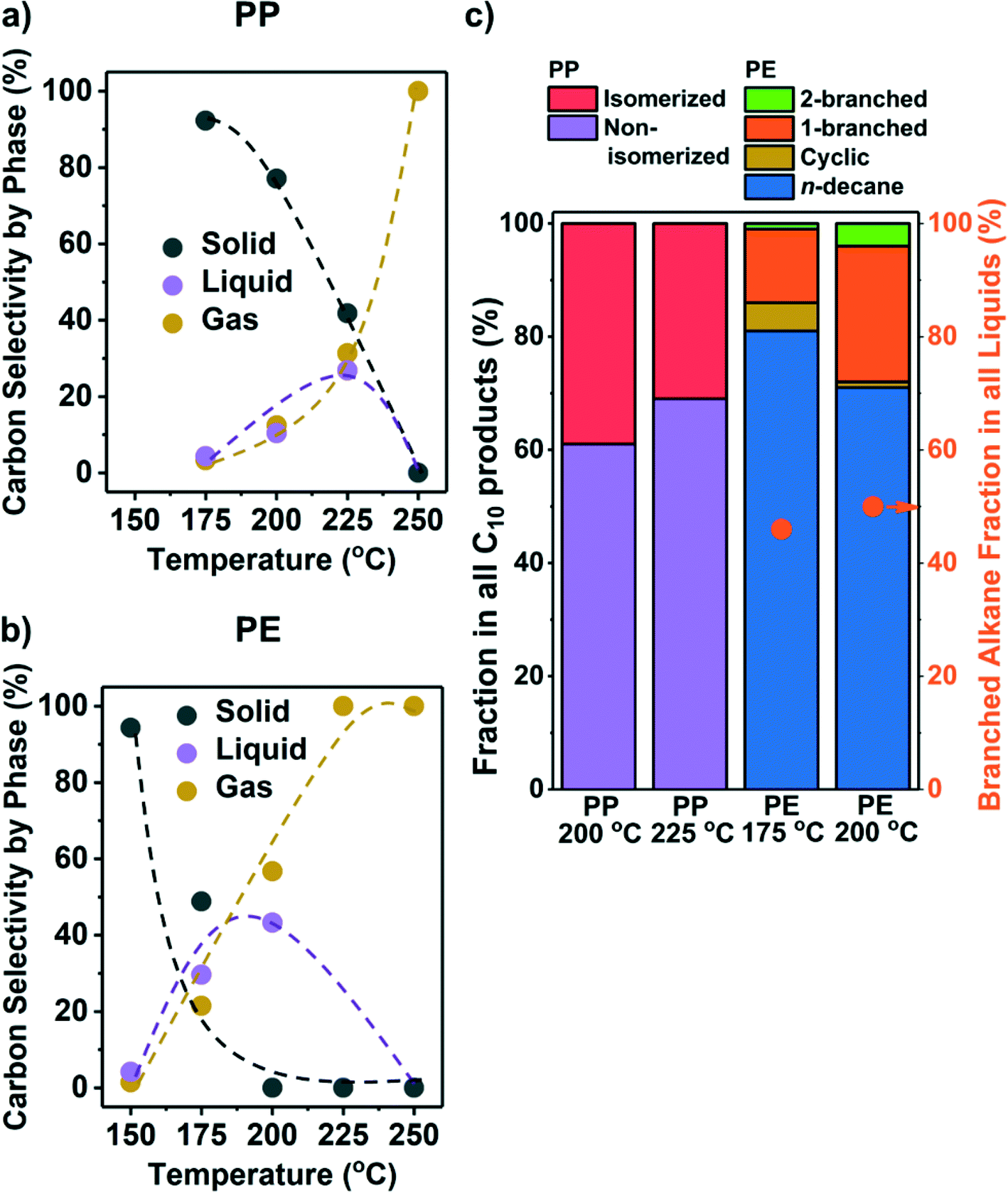 | ||
| Fig. 1 Results from the hydrogenolysis of PP and PE at various temperatures. The C selectivity of solid (black), liquid (purple), and gas (yellow) among all identified C is shown in a) PP and b) PE (see Fig. S8a and b† for C yield including unidentified C, and Fig. S5† for C yield of C1–C38 products by each carbon number). The distribution of C10 products at suitable temperatures for liquid production is shown in c) as stacked bars (see Fig. S4† for GC chromatograms and Table S3† for identities of the products). For PP, purple: non-isomerized alkanes (i.e., 1–4 in Table S3†); red: isomerized alkanes; for PE, blue: n-decane; yellow: branched C10 cyclohexanes; orange: linear C10 alkanes with one branch; green: linear C10 alkanes with two branches. Orange dots in c) show the fraction of branched alkanes in all liquid (C6–C38) products for PE hydrogenolysis. Reaction conditions: 30 bar H2, 18 h, 1 g PP or PE, 100 mg 5 wt% Ru/C. | ||
Liquid alkanes were only produced between 175 °C and 225 °C from PP and 150 °C and 200 °C from PE (Fig. 1a and b). Fig. S5† presents the C yield of C1–C38 products (see Fig. S4† for examples of raw GC-FID data). The figures show that at appropriate temperatures, a mixture of liquid alkanes with wide distribution in molecular weight is produced from both polyolefins. The products of PE (Fig. S5b†) can be divided into the following three categories that are characteristic of the reacting substrate and the sequential processes occurring: 1) CH4 from the hydrogenolysis of terminal C–C bonds, 2) n-alkanes from the hydrogenolysis of internal C–C bonds, and 3) branched alkanes from C–C cleavage at particular positions (see below). PP hydrogenolysis only yields CH4 and branched alkanes without >C3n-alkanes, as the starting PP structure is highly methyl branched. Because CH4 is a low-value product, it is desirable to steer the regioselectivity of hydrogenolysis towards internal C–C bonds. Isomerization may be desirable to increase the product branching level for higher fuel quality,31 particularly with PE as the feedstock. Olefins or aromatics were not observed as products under the present conditions.
Branched alkanes accounted for ∼50% of total C among all liquid products from PE (orange dots in Fig. 1c). All liquid products from PP were branched alkanes. The C10 products were analyzed in detail to gauge the branching level of liquid products and the location, where the C–C bonds are being cleaved. Fig. 1c shows that at temperatures leading to an appreciable liquid yield, products ranged from multiple branched alkanes from PP (Fig. S4b, Table S3†) to n-decane from PE, comprising >60% of total C10. Most branched C10 alkanes from PE contain only one methyl or ethyl side chain (Fig. S4b, Table S3†). The relative absence of branched products with PE compared to PP indicates that metal-catalyzed isomerization is slow compared to C–C cleavage. The PP structure leads, in contrast, to a high degree of branching; the dominating C10 (80 wt%) products from PP hydrogenolysis were 2,4,6-trimethylheptane, 2,4-, 3,5-, and 2,6-dimethyloctane.
3.3 Effects of the reaction time on the product distribution
Let us in the next step establish the sequence of reactions as the conversion changes. Fig. 2a and b show that the conversion was completed in all cases at 36 h. Initially, the liquid and gas amounts produced increased monotonously with reaction time for PE and PP (see Fig. 2 and Table S4†). This indicates that liquid and gaseous alkanes were produced in parallel and that the net consumption of the liquid fraction only started after complete solid conversion (>24 h for PP and >36 h for PE). Fig. S6† shows that the C distribution in the liquid fraction did not change significantly before complete solid conversion (centered around C22 and C18 for PP and PE, respectively), and only started to shift towards shorter alkanes once the polymer was converted. This sequential reaction suggests that in the solvent free system the longer chains of the polyolefins were preferentially adsorbed, blocking secondary hydrogenolysis of intermediately formed hydrocarbons.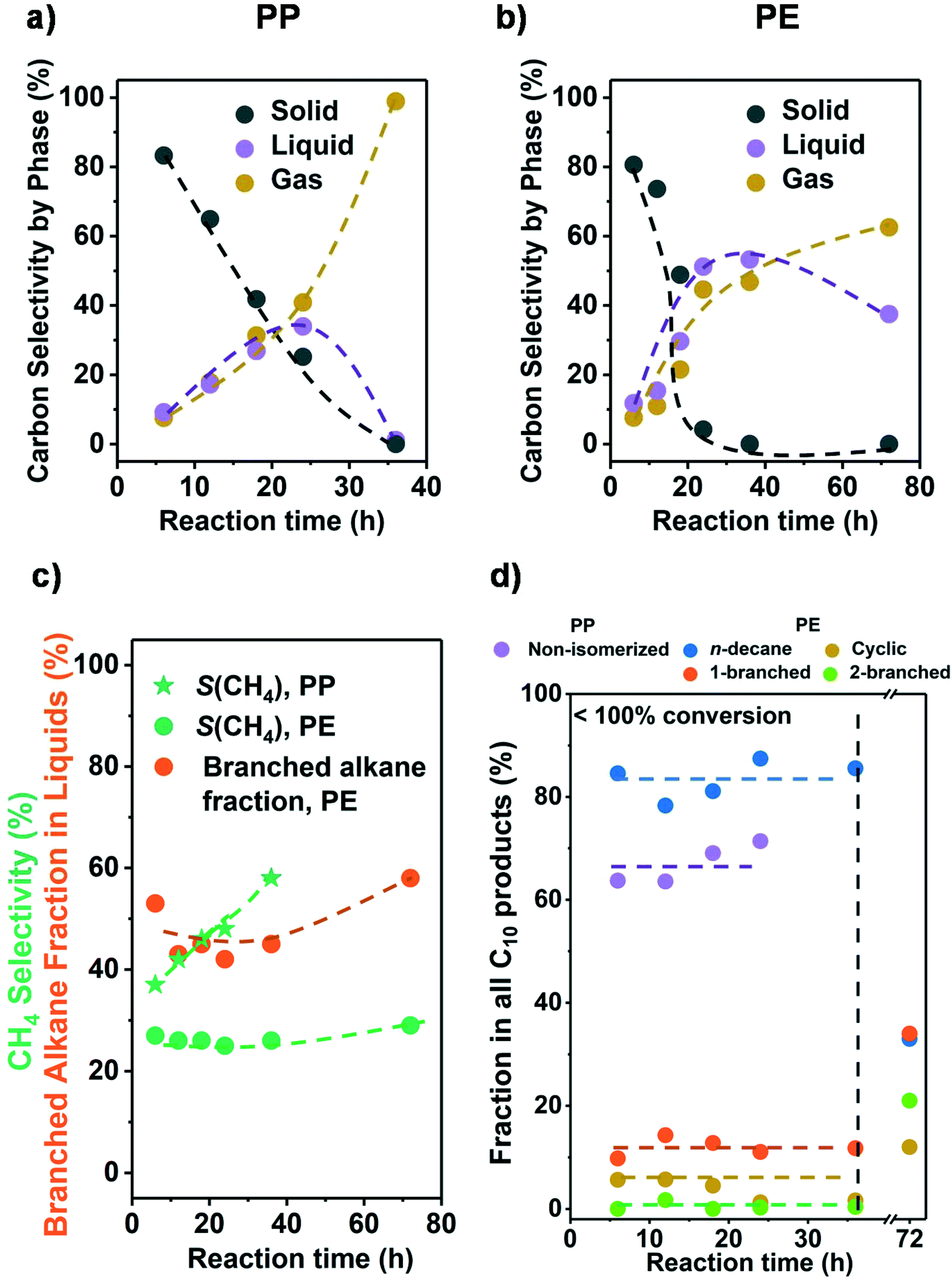 | ||
| Fig. 2 Results from the hydrogenolysis of PP and PE for various time. The C selectivity of solid (black), liquid (purple), and gas (yellow) among all identified C is shown in a) PP and b) PE (see Fig. S7c and d† for C yield including unidentified C, and Fig. S6† for C yield of C1–C38 products by each carbon number). The CH4 selectivity and fraction of branched alkanes in all liquid products from PE are shown in c). The changes in C10 product distribution with time is shown in d) (for PP, purple: non-isomerized alkanes, i.e., 1–4 in Table S3;† red: isomerized alkanes; for PE, blue: n-decane; yellow: branched C10 cyclohexanes; orange: linear C10 alkanes with 1 branch; green: linear C10 alkanes with 2 branches). Reaction conditions: 225 °C for PP and 175 °C for PE, 30 bar H2, 1 g PP or PE, 100 mg 5 wt% Ru/C. | ||
We attribute this effect to the large fraction of carbon atoms contained in the polymer chains. This must increase the probability of a fragment in the polymer, over small hydrocarbon molecules, to interact with the catalyst. Also, the size of such polymer fragments can be significantly larger than hydrocarbon molecules and therefore interact stronger with the catalyst. The dependence of the alkane adsorption energy, and hence hydrogenolysis rate, with chain length has been established with small alkanes and attributed to Van der Waals interactions with metal surfaces as well as entropic factors.32,33
Fig. 2c shows that for a fixed temperature and H2 pressure the selectivity to CH4 did not change until full conversion; that is, intermediately formed products are not able to access the metal surface in the presence of polymer. It should be noted in passing that the CH4 selectivity at full conversion was expectedly higher with PP than with PE. It also should be noted that with incomplete conversion, the selectivity to branched alkanes from PE was constant allowing to estimate the degree of branching (i.e., the products represent the primary cleavage from the polymer). Once the polymer was consumed, excess CH4 and isomers formed. Overall, the results show conclusively that it is possible to produce primary products as long as the product mixture is not exposed to the catalyst after complete conversion of the polymer.
3.4 Effects of the H2 pressure on the reaction rate and selectivity
As the partial pressure of H2 (PH2) has a profound impact on the hydrogenolysis of small alkanes,25,27,34 we investigated its impact on conversion rates and selectivity (Fig. 3). For both polyolefins, the conversion was negligible without the presence of H2 (i.e., only traces of liquid products, mostly alkenes and cyclic alkanes were observed). For PP, the polymer conversion increased monotonously with PH2 between 5 and 75 bar (Fig. 3a). For PE, however, conversion increased until PH2 = 45 bar, and then decreased (Fig. 3b). We attribute this decrease to an excess of H2 competing for binding and reaction sites. This is consistent with the hydrogenolysis of small alkanes, for which negative order in H2 has been reported for transition metals including Ru.25,27,33,34 The subtle differences between PE and PP are attributed to the different reaction temperatures used for the two polymers, causing a lower H coverage with PP.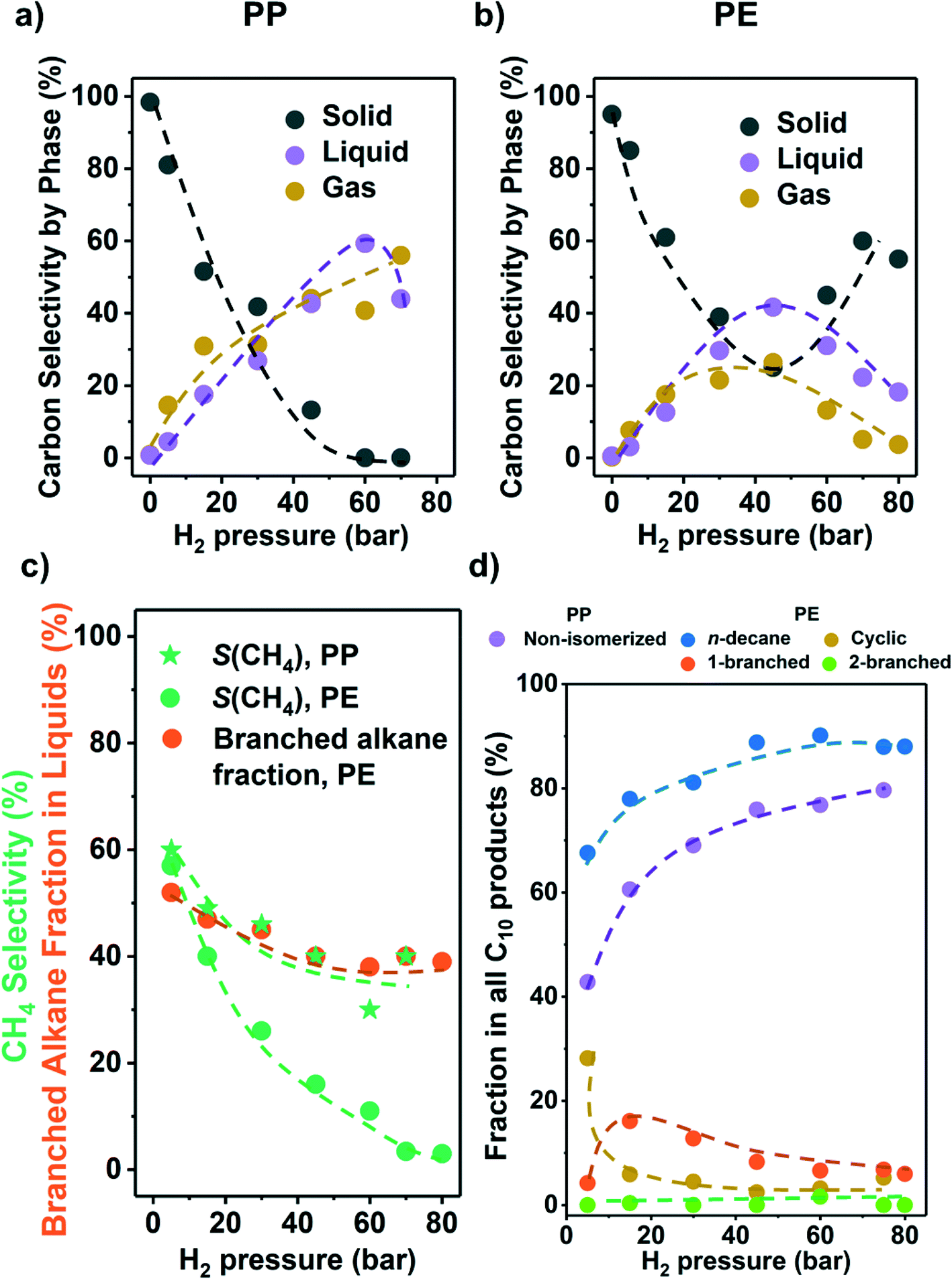 | ||
| Fig. 3 Results from the hydrogenolysis of PP and PE under various H2 pressure. The C selectivity of solid (black), liquid (purple), and gas (yellow) among all quantified C is shown in a) PP and b) PE (see Fig. S7e and f† for C yield including unidentified C, and Fig. S7† for C yield of C1–C38 products by each carbon number). The CH4 selectivity and fraction of branched alkanes in all liquid products from PE are shown in c). The changes in C10 product distribution with PH2 is shown in d) (for PP, purple: non-isomerized alkanes, i.e., 1–4 in Table S3;† red: isomerized alkanes; for PE, blue: n-decane; yellow: branched C10 cyclohexanes; orange: linear C10 alkanes with 1 branch; green: linear C10 alkanes with 2 branches). Reaction conditions: 225 °C for PP and 175 °C for PE, 18 h, 1 g PP or PE, 100 mg 5 wt% Ru/C. | ||
Fig. 3c shows that for both PP and PE, the CH4 selectivity decreased with PH2 below full conversion pointing to differences in the specific binding of the polymer branches with hydrogen pressure.35 This decrease in CH4 selectivity was concluded to be unrelated to varying conversion levels, because under constant PH2, the CH4 selectivity was insensitive to conversion (Fig. 2c). It is interesting to note that that the CH4 selectivity decreased monotonously with increasing PH2 (Fig. 3c), even when the conversion was maintained constant. Thus, we conclude that increasing PH2 leads to preferential hydrogenolysis of internal C–C bonds. We hypothesize that the strands of polymer are hindered to adopt the larger surface ensemble required for terminal C–C bond cleavage. More work to probe this hypothesis is, however, required. The maximum of the molecular weight distribution of the liquid products decreased with increasing PH2 (Fig. S7†) for PP (i.e., more light hydrocarbons being formed). The fact that the maximum was nearly constant for PE suggests that the influence of adsorbed hydrogen is significantly more prominent for branched polymers.
At higher PH2, the fraction of branched alkanes in all liquid products from PE decreased (Fig. 3c), while the fraction of linear C10 in all C10 products from both polyolefins is higher (Fig. 3d). Also, at low PH2 (5 bar), the fraction of cyclic C10 in all C10 products from PE increased significantly (yellow in Fig. 3d). Similar to the CH4 selectivity, these trends in product branching and isomerization level are insensitive to the reaction time, or the solid conversion, under the same PH2 (Fig. 2). Therefore, the changes in selectivity are not associated with the difference in the conversion level. Instead, the results indicate that higher PH2 favors formation of linear products over branched ones.
Mechanistically, hydrogenolysis of the polymer strands on the metal surface occurs at dehydrogenated C–C units.26,36–40 The dehydrogenation process weakens C–C bonds by formation of carbon–metal bonds, increasing the occupancy of antibonding orbitals in C–C bonds.41,42 Intuitively, the formation of the corresponding dehydrogenated transition states requires multiple free adjacent metal sites to accommodate dissociated hydrogen. Therefore, this process is disfavored by relatively high H2 pressures, at least at low reaction temperatures. An example for this is seen in the negative impact of high pressure on the conversion rate of PE at 175 °C.
Such mechanistic picture also helps explain our observations as shown in Fig. 4. For PP, the cleavage can occur between 3C–1C and 3C–2C bonds.
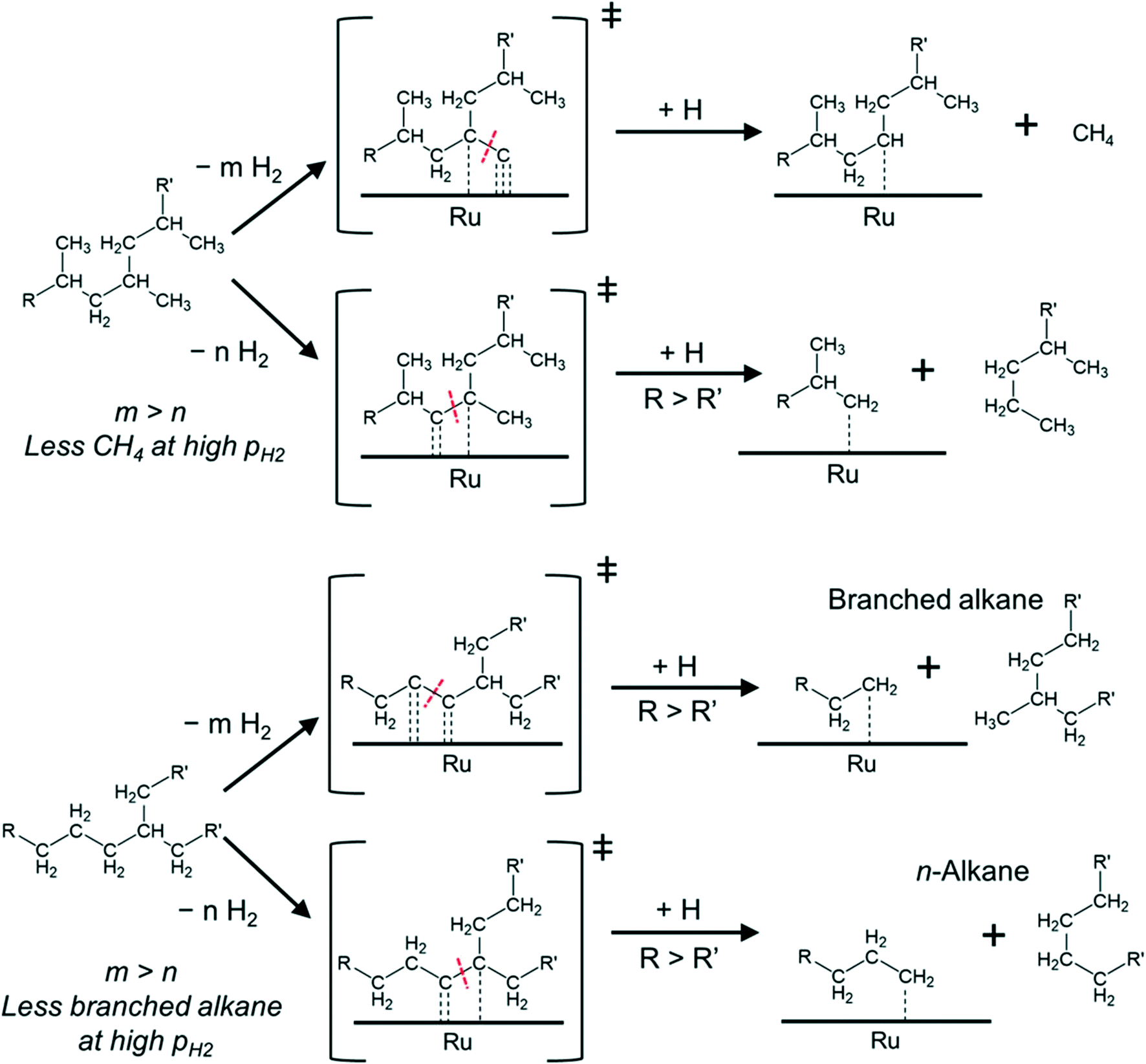 | ||
| Fig. 4 Illustration of the proposed hydrogenolysis pathways for polypropylene (top) and polyethylene (bottom). The first steps of the reaction are adsorption and dehydrogenation, which lead to a hydrogen-depleted intermediate. Upon C–C cleavage, the hydrogenation of the fragments leads to the products. The lighter product (containing R′ in the figure) will desorb preferentially, whereas the surface will remain covered with the longer hydrocarbon chains (the fragment containing R in the figure). | ||
The cleavage of the former is kinetically favored (3C–1C is by far more abundant in PP) and ultimately leads to CH4 upon hydrogenation of the resulting carbide. However, the formation of the corresponding transition states requires removing more hydrogen from the hydrocarbon chain than for the cleavage of a 3C–2C bond (which could lead to long alkane products). Thus, increasing PH2, and the concomitant increase in hydrogen coverage, favors cleavage at 3C–2C positions over 3C–1C. Repulsive interactions between methyl groups and hydrogen at the metal surface could also hinder the oxidative adsorption of 1C with increasing PH2.43
In the case of PE (with one branch every 50–100 C atoms44–46) cleavage can occur between 2C–2C and 3C–2C bonds, in addition to the expected cleavage between xC–1C bonds. If the cleavage occurs at 2C–2C positions, near 3C atoms, subsequent hydrogenation can produce branched alkanes (Fig. 4). In contrast, 3C–2C cleavage has higher probability to produce linear products. We hypothesize that the 2C–2C cleavage requires deeper dehydrogenation than the 3C–2C cleavage. Therefore, increasing PH2 decreases the selectivity to branched products.
Note that in all cases, upon cleavage, the longer and less branched polymer strands have a stronger driving force to remain adsorbed at the surface32 to undergo further reaction. This can lead to multiple xC–1C transition states (particularly for PP), which push the selectivity towards methane.
3.5 High liquid yield under optimal conditions and the effects of Cl
Results indicate that low reaction temperatures (and high PH2) are required to maximize liquid products on Ru/C. Thus, we converted PE at 175 °C and 82 bar H2. Fig. 5 shows the C yield in the three phases and C distribution from C1 to C38. A large fraction of only preliminary identified products forms a part of the liquid phase, i.e., they are EtOAc-soluble, but they have carbon numbers >C38 and therefore beyond detection by GC (see Table S4, Fig. S9,† and related discussion). Hence, Fig. 5 likely underestimates the liquid yield. A high liquid yield of 57 mol. C%, mostly in the diesel range (C12–C20), was achieved after 76 h. As anticipated, the gas yield and CH4 selectivity at 100% solid conversion were only ∼10% and ∼3%, respectively. Beyond full conversion (>48 h, Fig. 5b) the larger alkanes (>C25) were converted to lighter products without much gas production. In contrast, with H2 at 30 bar, a significant fraction of the liquid products was gasified under such conditions (Fig. 2a and b). For PP, it was more difficult to suppress the formation of CH4. By applying high PH2 (60 bar), a maximum of 47% liquid yield at full conversion was achieved but with 30% CH4 selectivity (Fig. S8e, Table S4†).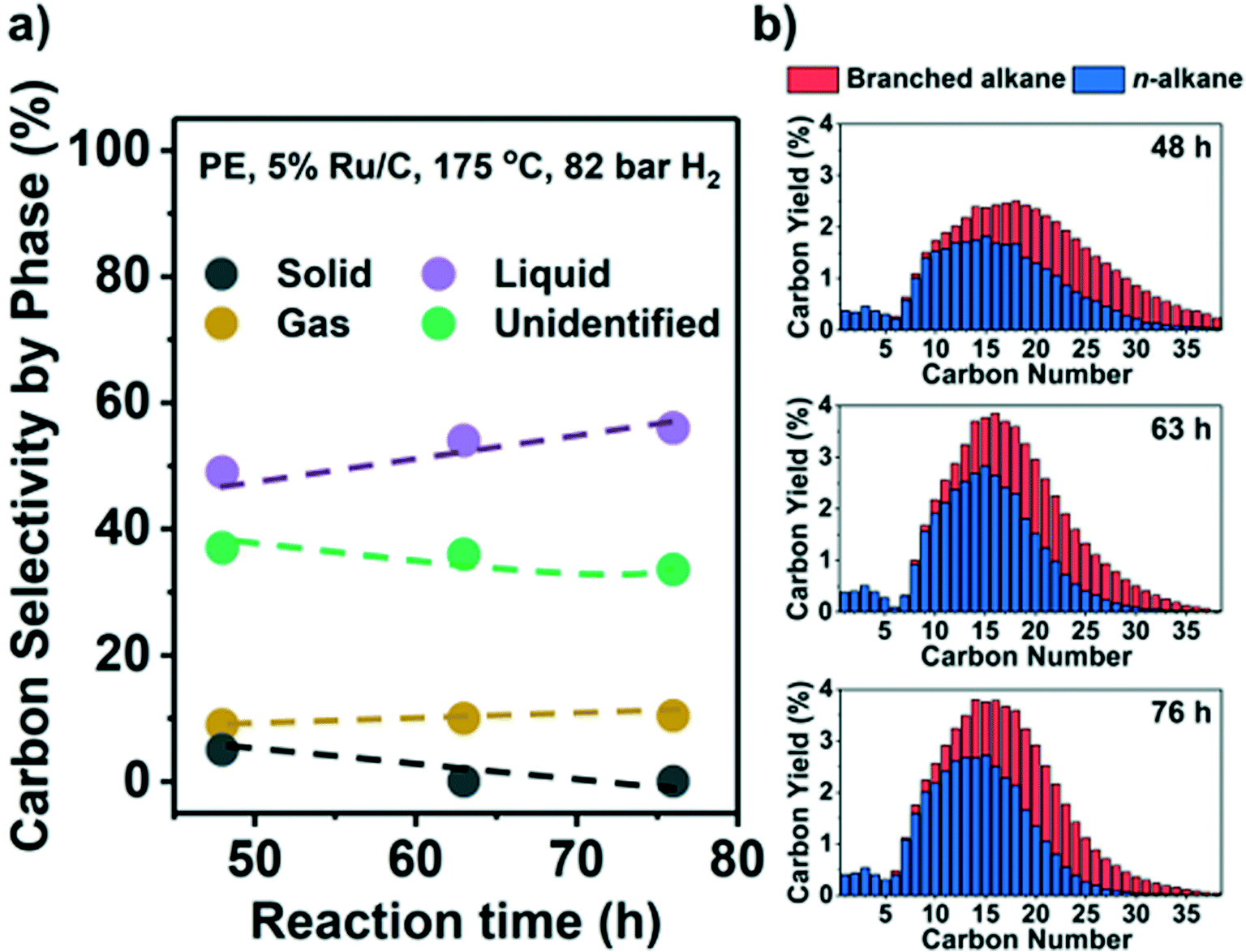 | ||
| Fig. 5 Hydrogenolysis of PE under optimal conditions (175 °C, 82 bar H2) for high liquid yield. The C yield of solid (black), liquid (purple), and gas (yellow), along with unidentified C (green), are shown in a). The C distribution in C1–C38 products is presented in b), with gas products plotted at a one-fifth scale. A liquid yield of >57% was achieved. | ||
A potential challenge faced by hydrogenolysis-based processes is the presence of Cl because PP/PE plastic waste is often mixed with polyvinyl chloride (PVC) even after separation. Experiments in the presence of PVC showed release of HCl above 175 °C in the presence and absence of PP/PE with a strong detrimental effect on catalyst activity. Even at 0.1 wt% PVC (Table S5†), the unconverted fraction of PP increased from 27.6% without PVC, to 87.7%, and further to 96.7% with 1 wt% PVC (225 °C, 30 bar H2). For PE, the solid residue fraction increased from 39.0% without PVC, to 67.6% with 0.1 wt% PVC, and further to 93.2% with 1 wt% PVC (175 °C, 30 bar H2). Control experiments showed that the presence of HCl solution with a Cl content equal to 1 wt% PVC had a similar impact (Table S5†). Therefore, the development of Cl-resistant catalysts or Cl-removal procedures from the feedstock is mandatory. Despite the lower catalytic efficiency, the presence of PVC directs the regioselectivity of hydrogenolysis towards internal C–C bond cleavage and favors branched over linear products (Table S5†). This indicates that Cl at the metal surface particularly hinders the adsorption of 3C (i.e., 3C–1C and 3C–2C cleavage). We characterized the spent catalyst by scanning transmission electron microscopy/energy dispersive X-ray microscopy (STEM/EDX) and by elemental analysis to interrogate the fate of Cl. The signals of Ru and Cl in EDX overlap (Fig. S9b†). Therefore, we could not identify Cl associated with Ru particles. However, we found Cl in areas with few highly disperse Ru particles (Fig. S9c and d†). Thus, we surmise that Cl remains randomly distributed across the used catalyst. According to elemental analysis, a spent sample contains 0.2 wt% Cl, which suggests that 3% of the initial Cl remains in the catalyst. Most of the initial Cl is likely eliminated as HCl, which was detected, but not quantified, after the reaction.
4. Conclusions
The solvent-free hydrogenolysis of polypropylene (PP; MW ∼ 250000) and polyethylene (PE, MW ∼ 4000) using Ru/C catalysts is a low-temperature upcycling approach with high yield to liquid alkanes. Its activity surpasses that of other C-supported Pt-group metals with equal particle size. On Ru/C, C6–C38 alkanes were produced in the temperature range of 175–225 °C from PP and 150–200 °C from PE. PP produces mainly CH4 and branched hydrocarbons, while PE produces CH4 and branched and linear alkanes. Increasing H2 pressure shifts the regioselectivity of hydrogenolysis from terminal C–C towards internal C–C cleavage, thus reducing CH4 production, and favors linear over branched products. These trends in product distribution result from the interplay between 1) the structure of the polymers, 2) the positions along the chain, where C–C units bind to the metal, and 3) the space available at the metal for the dehydrogenation step prior to C–C cleavage. The CH4 selectivity and product distribution in liquid and gas do not change significantly with reaction time or conversion of the initial polymer. This is attributed to the favored adsorption of long-chain polymers over shorter alkanes. Instructed by the findings, we achieved >57% yield of C6–C38 alkanes under optimal conditions. This work demonstrated the feasibility of upcycling both PP and PE by hydrogenolysis and identified Ru/C as a more effective catalyst than the commonly studied Pt. The comparative study on the polyolefin structure, temperature, reaction time, and H2 pressure establishes practical guidelines for improving the product distribution and fundamental knowledge on the reaction mechanism.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
L. C. M., L. V. H., J. A. L., and O. Y. G. were supported by the US Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences (BES), Division of Chemical Sciences, Geosciences and Biosciences (towards a polyolefin-based refinery: understanding and controlling the critical reaction steps, FWP 78459). L. C., T. T. L., A. K., and J. S. were supported by the DOE, Office of Science, BES, Division of Chemical Sciences, Geosciences and Biosciences (impact of catalytically active centers and their environment on rates and thermodynamic states along reaction paths, FWP 47319). We gratefully acknowledge Libor Kovarik for STEM/EDX analysis at Pacific Northwest National Laboratory (PNNL) and Teresa Lemmon and Marie Swita also at PNNL for their help with elemental analysis.References
- I. Tiseo, Annual production of plastics worldwide from 1950 to 2020, Statista, https://www.statista.com/statistics/282732/global-production-of-plastics-since-1950/, (last accessed Aug, 2021) Search PubMed.
-
How much oil is used to make plastic?, U.S. Energy Information Administration Website, https://www.eia.gov/tools/faqs/faq.php?id=34&t=6, (last accessed Aug, 2021) Search PubMed
.
-
How much oil is used to make plastic?, Beston Machinery Website, https://plasticpyrolysisplants.com/much-oil-used-make-plastic/, (last accessed Aug, 2021) Search PubMed
-
A. Ghaddar and R. Bousso, Rising use of plastics to drive oil demand to 2050, IEA, Reuters Website, https://www.reuters.com/article/us-petrochemicals-iea/rising-use-of-plastics-to-drive-oil-demand-to-2050-iea-idUSKCN1ME2QD, (last accessed Aug, 2021) Search PubMed
- D. K. A. Barnes, F. Galgani, R. C. Thompson and M. Barlaz, Philos. Trans. R. Soc., B, 2009, 364, 1985–1998 CrossRef CAS PubMed
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed
- W. Kaminsky, B. Schlesselmann and C. Simon, J. Anal. Appl. Pyrolysis, 1995, 32, 19–27 CrossRef CAS
- R. Bagri and P. T. Williams, J. Anal. Appl. Pyrolysis, 2002, 63, 29–41 CrossRef CAS
- W. Kaminsky and I.-J. N. Zorriqueta, J. Anal. Appl. Pyrolysis, 2007, 79, 368–374 CrossRef CAS
- T. Okui and Y. Ogo, J. Appl. Polym. Sci., 1980, 25, 747–759 CrossRef CAS
- V. Dufaud and J.-M. Basset, Angew. Chem., Int. Ed., 1998, 37, 806–810 CrossRef CAS PubMed
- G. Tosin, C. C. Santini and J.-M. Basset, Top. Catal., 2009, 52, 1203–1210 CrossRef CAS
- X. Jia, C. Qin, T. Friedberger, Z. Guan and Z. Huang, Sci. Adv., 2016, 2, e1501591 CrossRef PubMed
- G. Celik, R. M. Kennedy, R. A. Hackler, M. Ferrandon, A. Tennakoon, S. Patnaik, A. M. LaPointe, S. C. Ammal, A. Heyden, F. A. Perras, M. Pruski, S. L. Scott, K. R. Poeppelmeier, A. D. Sadow and M. Delferro, ACS Cent. Sci., 2019, 5, 1795–1803 CrossRef CAS PubMed
- A. Tennakoon, X. Wu, A. L. Paterson, S. Patnaik, Y. Pei, A. M. LaPointe, S. C. Ammal, R. A. Hackler, A. Heyden, I. I. Slowing, G. W. Coates, M. Delferro, B. Peters, W. Huang, A. D. Sadow and F. A. Perras, Nat. Catal., 2020, 3, 893–901 CrossRef CAS
- F. Zhang, M. Zeng, R. D. Yappert, J. Sun, Y.-H. Lee, A. M. LaPointe, B. Peters, M. M. Abu-Omar and S. L. Scott, Science, 2020, 370, 437–441 CrossRef CAS PubMed
- L. Yao, J. King, D. Wu, S. S. C. Chuang and Z. Peng, Catal. Commun., 2021, 150, 106274 CrossRef CAS
- S. P. Ertem, C. E. Onuoha, H. Wang, M. A. Hillmyer, T. M. Reineke, T. P. Lodge and F. S. Bates, Macromolecules, 2020, 53, 6043–6055 CrossRef CAS
- Y. Jing, Y. Wang, S. Furukawa, J. Xia, C. Sun, M. J. Hülsey, H. Wang, Y. Guo, X. Liu and N. Yan, Angew. Chem., Int. Ed., 2021, 60, 5527–5535 CrossRef CAS PubMed
- J. E. Rorrer, G. T. Beckham and Y. Román-Leshkov, JACS Au, 2021, 1, 8–12 CrossRef CAS PubMed
- C. Jia, S. Xie, W. Zhang, N. N. Intan, J. Sampath, J. Pfaendtner and H. Lin, Chem Catalysis, 2021, 1, 437–455 CrossRef
- Y. Nakaji, M. Tamura, S. Miyaoka, S. Kumagai, M. Tanji, Y. Nakagawa, T. Yoshioka and K. Tomishige, Appl. Catal., A, 2021, 285, 119805 CrossRef CAS
- P. A. Kots, S. Liu, B. C. Vance, C. Wang, J. D. Sheehan and D. G. Vlachos, ACS Catal., 2021, 11, 8104–8115 CrossRef CAS
- J. E. Rorrer, C. Troyano-Valls, G. T. Beckham and Y. Román-Leshkov, ACS Sustainable Chem. Eng., 2021, 9, 11661–11666 CrossRef CAS
- J. H. Sinfelt, Catal. Rev., 1970, 3, 175–205 CrossRef
-
J. H. Sinfelt, in Advances in Catalysis, ed. D. D. Eley, H. Pines and P. B. Weisz, Academic Press, 1973, vol. 23, pp. 91–119 Search PubMed
- J. H. Sinfelt, Catal. Lett., 1991, 9, 159–171 CrossRef CAS
- C. J. Machiels and R. B. Anderson, J. Catal., 1979, 58, 268–275 CrossRef CAS
- H. Shi, O. Y. Gutiérrez, A. Zheng, G. L. Haller and J. A. Lercher, J. Phys. Chem. C, 2014, 118, 20948–20958 CrossRef CAS
- H. Shi, O. Y. Gutiérrez, H. Yang, N. D. Browning, G. L. Haller and J. A. Lercher, ACS Catal., 2013, 3, 328–338 CrossRef CAS
- M. D. Boot, M. Tian, E. J. M. Hensen and S. Mani Sarathy, Prog. Energy Combust. Sci., 2017, 60, 1–25 CrossRef
- D. D. Hibbitts, D. W. Flaherty and E. Iglesia, J. Phys. Chem. C, 2016, 120, 8125–8138 CrossRef CAS
- D. W. Flaherty and E. Iglesia, J. Am. Chem. Soc., 2013, 135, 18586–18599 CrossRef CAS PubMed
- D. W. Flaherty, D. D. Hibbitts and E. Iglesia, J. Am. Chem. Soc., 2014, 136, 9664–9676 CrossRef CAS PubMed
- Y. Nakagawa, S.-I. Oya, D. Kanno, Y. Nakaji, M. Tamura and K. Tomishige, ChemSusChem, 2017, 10, 189–198 CrossRef CAS PubMed
-
F. G. Gault, in Advances in Catalysis, ed. D. D. Eley, H. Pines and P. B. Weisz, Academic Press, 1981, vol. 30, pp. 1–95 Search PubMed
- A. Cimino, M. Boudart and H. Taylor, J. Phys. Chem., 1954, 58, 796–800 CrossRef CAS
- G. Maire, G. Plouidy, J. C. Prudhomme and F. G. Gault, J. Catal., 1965, 4, 556–569 CrossRef CAS
- K. Morikawa, W. S. Benedict and H. S. Taylor, J. Am. Chem. Soc., 1936, 58, 1795–1800 CrossRef CAS
- C. Kemball and H. S. Taylor, J. Am. Chem. Soc., 1948, 70, 345–351 CrossRef CAS
-
I. Chorkendorff and J. W. Niemantsverdriet, Concepts of modern catalysis and kinetics, John Wiley & Sons, New York, 2007 Search PubMed
- J. Kua, F. Faglioni and W. A. Goddard, J. Am. Chem. Soc., 2000, 122, 2309–2321 CrossRef CAS
- H. Shi, O. Y. Gutiérrez, G. L. Haller, D. Mei, R. Rousseau and J. A. Lercher, J. Catal., 2013, 297, 70–78 CrossRef CAS
- Z. Zhou, S. Pesek, J. Klosin, M. S. Rosen, S. Mukhopadhyay, R. Cong, D. Baugh, B. Winniford, H. Brown and K. Xu, Macromolecules, 2018, 51, 8443–8454 CrossRef CAS
- E. Nordmeier, U. Lanver and M. D. Lechner, Macromolecules, 1990, 23, 1072–1076 CrossRef CAS
- M. Jung, Y. Lee, S. Kwak, H. Park, B. Kim, S. Kim, K. H. Lee, H. S. Cho and K. Y. Hwang, Anal. Chem., 2016, 88, 1516–1520 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1re00431j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |