
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemoproteomic profiling of itaconations in Salmonella†
Yanling
Zhang
ac,
Wei
Qin
ac,
Dongyang
Liu
ab,
Yuan
Liu
ab and
Chu
Wang
 *abc
*abc
aSynthetic and Functional Biomolecules Center, Beijing National Laboratory for Molecular Sciences, Key Laboratory of Bioorganic Chemistry and Molecular Engineering of Ministry of Education, China
bCollege of Chemistry and Molecular Engineering, China
cPeking-Tsinghua Center for Life Sciences, Peking University, Beijing 100871, China. E-mail: chuwang@pku.edu.cn
First published on 31st March 2021
Abstract
Itaconate is an immunoregulatory and anti-bacterial metabolite, and plays important roles in host–pathogen interactions. Chemoproteomic strategies have been used to explore the anti-inflammatory effects of itaconate on activated macrophages and it has been found that many key proteins in immune pathways were modified; however, how itaconate modulates pathogens was not fully understood. Here, we have designed and synthesized a series of itaconate-based bioorthogonal probes, which enable quantitative and site-specific profiling of itaconated proteins and sites in Salmonella. Among many proteins related to energy metabolism, we identified a key enzyme involved in the glyoxylate cycle, isocitrate lyase (ICL), as the most prominent target. Covalent modification of the active-site cysteine in ICL by itaconate abolishes the enzyme activity and suppresses bacterial growth. Our chemoproteomic study has uncovered the wide array of itaconation targets in Salmonella and provided a comprehensive resource for understanding the anti-bacterial function of this intriguing metabolite.
Introduction
Itaconate has been known as an anti-inflammatory and anti-bacterial metabolite involved in macrophage activation.1,2 It is generated from cis-aconitate by mitochondria-associated enzyme immune responsive gene 1 (IRG1) and its concentration is strikingly upregulated in activated macrophages.3,4 Since the first report of itaconate production in mammalian cells, most of the studies have been devoted to exploring its immunosuppressive function in macrophages.5,6 Itaconate was initially found to reduce cytokine secretion and regulate macrophage metabolism by inhibiting succinate dehydrogenase (SDH).1 Owing to its weak electrophilic property, itaconate can also react with nucleophilic residues, thereby covalently modifying target proteins and affecting their functions. It has been reported that itaconate can exert anti-inflammatory function through the NRF2 and ATF3 signaling pathways by modifying the cysteine sidechains of KEAP1 and GSH, respectively.7,8 It can also modify NLRP3 and block the inflammasome activations.9 Our group has developed a couple of chemoproteomic strategies to globally identify the itaconate modifications (“itaconation”) in activated macrophages and revealed that itaconate can covalently modify protein targets to modulate the glycolysis and necroptosis pathways.10,11 However, the global itaconation landscape in bacteria has not been thoroughly explored.The knowledge of itaconate's anti-bacterial effect could be traced back to as early as 1971 when researchers found that itaconate could inhibit the growth of Salmonella enterica serovar Typhimurium (S. enterica) and Mycobacterium tuberculosis on a non-glucose-based carbon source.12,13 Later on, it was found that knocking out IRG1 in macrophages would lead to the rapid growth of pathogens during invasion. It is generally believed that itaconate can mimic substrate analogues and competitively inhibit the metabolic enzymes in pathogens to interfere with their energy metabolism, including methylisocitrate lyase (MICL) and isocitrate lyase (ICL) in the glyoxylate cycle, and propionyl-CoA carboxylase in the citramalate cycle.14 More recently, a detailed mechanistic study showed that the CoA derivative of itaconate acts as a suicide inhibitor of B12-dependent methylmalonyl-CoA mutase (MCM) in Mycobacterium tuberculosis.15 However, whether itaconate could regulate pathogen functions via direct itaconation is largely unknown.
In this study, we employed quantitative chemoproteomics to survey proteins and sites that are covalently modified by itaconate in S. enterica. With a unique chemical probe that showed specific labeling in bacterial lysates, we were able to identify hundreds of proteins targeted by itaconate. We discovered that itaconate can covalently modify multiple cysteines in ICL including the active-site Cys195. Itaconation results in not only the inhibition of ICL's enzymatic activity but also the destabilization of the enzyme. We believe that the chemoproteomic profiling data would provide a comprehensive resource for studying itaconate's function in pathogenic bacteria and the chemical probe would serve as an enabling tool for ICL inhibitor development in future.
Results and discussion
We recently developed a bioorthogonal itaconate analogue probe named “ITalk” to label and enrich itaconations directly from living macrophages. When the probe was applied to the S. enterica proteome (Fig. 1A), we were surprised to observe that the overall labeling intensity became much lower than that from macrophages (Fig. S1†). To rule out the possibility that the ester linkage in ITalk might be cleaved by bacteria-specific hydrolases, we designed a new probe to replace the ester linkage with an amide. Inspired by a recently reported fumarate analogue probe,16 we also designed two probes with reduced tail lengths from 8 carbons down to 3 carbons (Fig. 1B). We refer to these four probes as “C8E, C8A, C3E and C3A”, with E and A standing for “ester” and “amide”, respectively.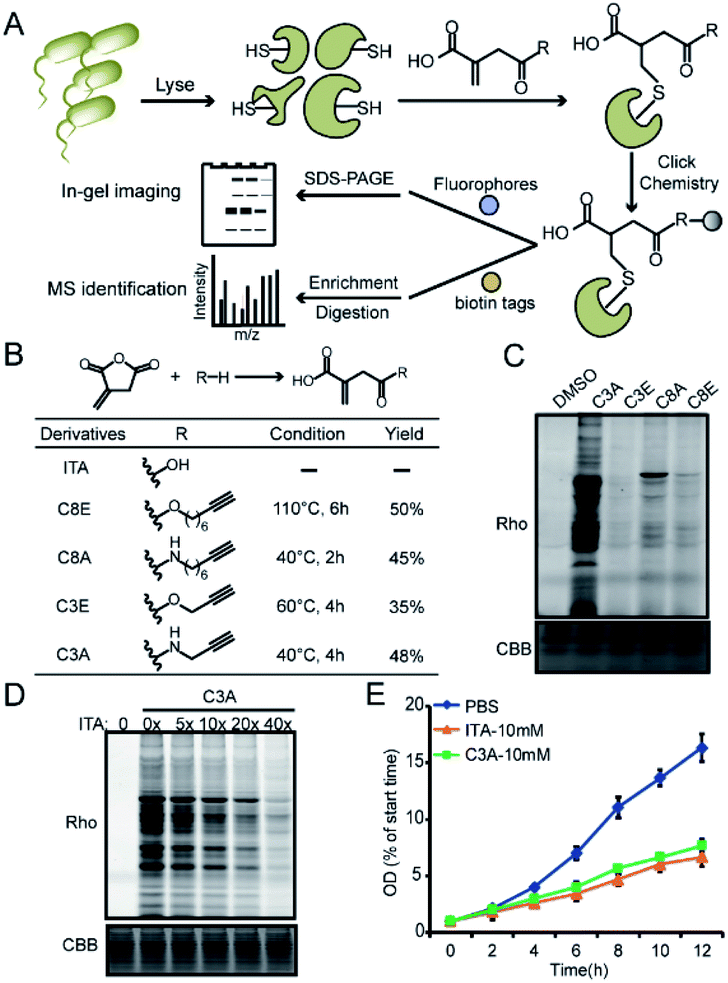 | ||
| Fig. 1 Design and evaluation of the bioorthogonal itaconate-analogue probes. (A) Overall working scheme for profiling itaconation events in S. enterica proteomes. (B) The structures and synthetic yields of itaconate (ITA)-derivative probes including C8E (ITalk), C3A, C3E and C8A. (C) Comparison of the labeling by the four probes in S. enterica lysates. (D) Competitive labeling of C3A by itaconate in a concentration-dependent manner in S. enterica lysates. (E) C3A shows the same inhibition effect as itaconate on the growth of S. enterica. | ||
We synthesized these probes with good yields (Fig. 1B and S2†) and evaluated their reactivities in both macrophages and S. enterica proteomes. Intriguingly, these four probes showed distinguished labeling preference for different proteomes. While C8E maintained the strongest labeling in macrophages, C3A showed the best performance in S. enterica proteomes (Fig. 1C and S3A†). C3A's labeling was clearly concentration-, time-, pH- (Fig. S4A–C†) and activity-dependent (Fig. S3B†), and seemed to be unique to only S. enterica but not E. coli (Fig. S3C†). Furthermore, the proteome reactivity of C3A could be blocked by iodoacetamide, suggesting that the majority of its labeling occurs on cysteines (Fig. S4D†). More importantly, the C3A labeling could be globally competed by itaconate in a concentration-dependent manner but not by methylsuccinate or fumarate (Fig. 4D and S4E†), indicating that C3A shares similar target profiles with itaconate and can be used as a suitable probe for profiling itaconations in S. enterica proteome. Based on a previous report that itaconate could inhibit bacterial growth under glucose limited conditions that many bacteria have to face during infection,4,17 we cultured S. enterica in a minimal medium supplemented with acetate, glycerol or glucose as the unique carbon source and monitored the growth in the presence of C3A, itaconate or methylsuccinate (Fig. 1E and S5†). As expected, both itaconate and C3A showed similar growth inhibition of bacteria in the acetate-only carbon source, while methylsuccinate had little influence, suggesting that the inhibitory effect comes from the electrophilicity of itaconate.
We next applied C3A to profile itaconate modified proteins in S. enterica proteomes (Fig. 2A). Bacterial lysates were incubated with PBS or 5 mM itaconate for 3 hours, respectively, and then labeled with 500 μM C3A for 12 hours. We also included a third sample without C3A labeling as the negative control. After eliminating the unreacted probes, the C3A-labeled proteins were conjugated with an azide-biotin tag and enriched by streptavidin beads. After trypsin digestion, the peptides were further labeled with a triplex dimethylation reagent (light for the non-probe control, medium for the C3A-labeled sample and heavy for the itaconate-competition sample) and subjected to quantitative proteomics analysis (Fig. 2A). By averaging the corresponding peptide ratios resulting from the dimethyl labeling, we got two quantitative ratios for each protein – the medium/light ratio is the “enrichment” ratio and the medium/heavy ratio is the “competition” ratio. We generated four biological replicates and obtained a total of 1230 C3A-labeled proteins (Fig. 2B, Table S1†). By further applying an enrichment ratio cutoff of 10 and a competition ratio cutoff of 1.5, we finally selected 197 proteins as prominent itaconate-modified targets (Fig. 2C). Functional survey of these proteins by PANTHER18 revealed that many of them are enzymes related to energy metabolism, and majority of them are cytoplasmic proteins (Fig. 2D). The list is, to the best of our knowledge, the first database for itaconate modified proteins in S. enterica, which provides a rich resource for the community to explore the anti-bacterial properties of itaconate.
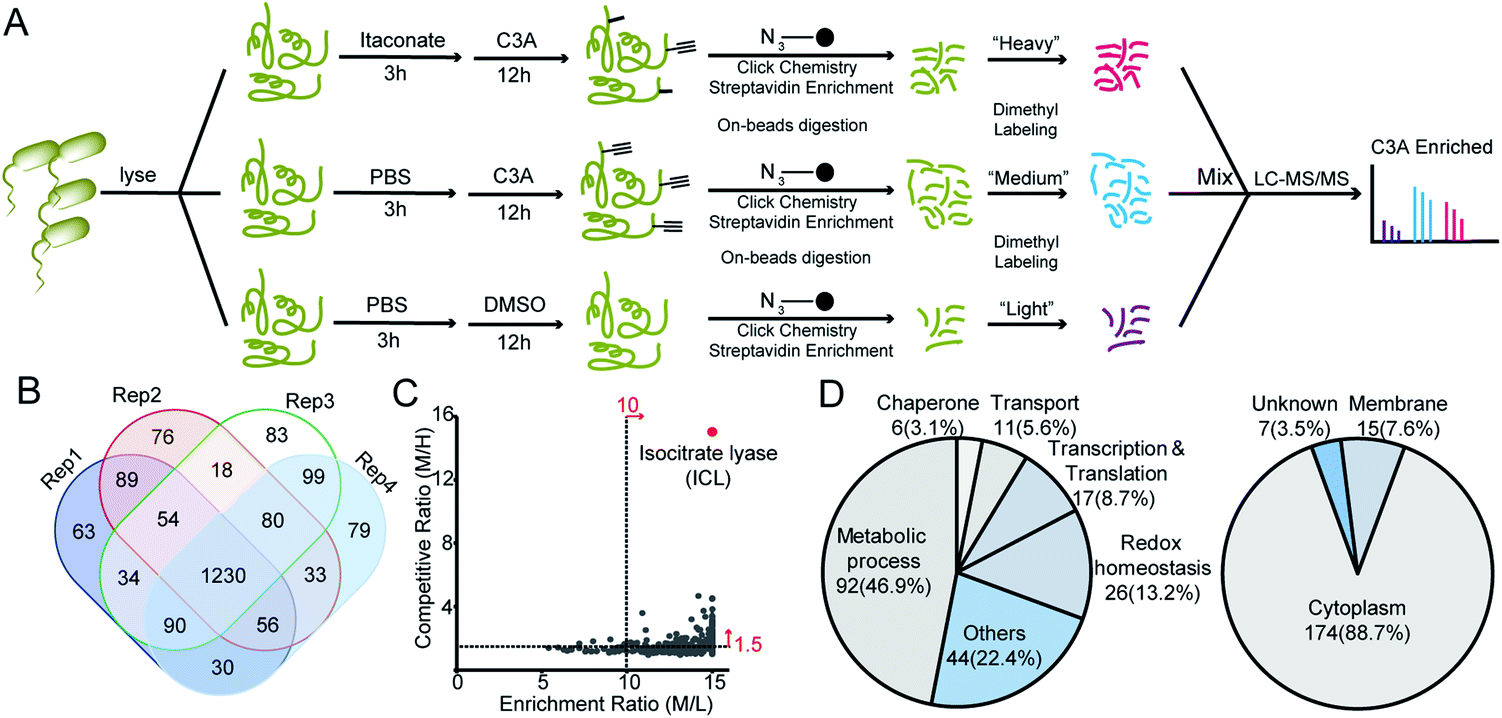 | ||
| Fig. 2 Quantitative chemoproteomic profiling of itaconated proteins by C3A in S. enterica lysates. (A) Workflow for the triplex dimethylation-based quantitative chemoproteomic profiling of itaconated proteins in S. enterica by C3A. (B) Overlap of the itaconated proteins identified by C3A in four biological replicates. (C) Distribution of the averaged enrichment and competition ratios of the C3A-labeled proteins. (D) Bioinformatics analysis of the prominent 197 itaconated protein targets in terms of functional annotation and cellular location. | ||
We next performed the TOP-ABPP19 experiment in order to identify itaconation sites (Fig. 3A). We used an acid cleavable azide-biotin tag to enrich and release the modified peptides for LC-MS/MS analysis, and from two biological repeats, 1319 cysteine sites from 781 proteins were identified (Fig. 3B, Table S2†). We cross-checked these itaconation sites with the above-mentioned 197 prominent target proteins with strong competition ratios, and found that 129 proteins have itaconation sites identified (Table S3†). Interestingly, 66 proteins were identified to contain more than 1 itaconation site and 35 of them have more than 2 sites (Fig. 3C). No other amino acid sites were detected beyond cysteines, suggesting the specific reactivity of C3A in the S. enterica proteome. We biochemically confirmed the itaconation sites on some of the targets including enolase (ENO), oligoribonuclease (ORN), ATP synthase gamma chain (ATPG), probable transcriptional regulatory protein (YEBC), putative oxidoreductase (YIEF), and putative intracellular proteinase (YHBO). C3A's labeling on these proteins was completely abolished by mutation of the corresponding cysteines to serines (Fig. 3D).
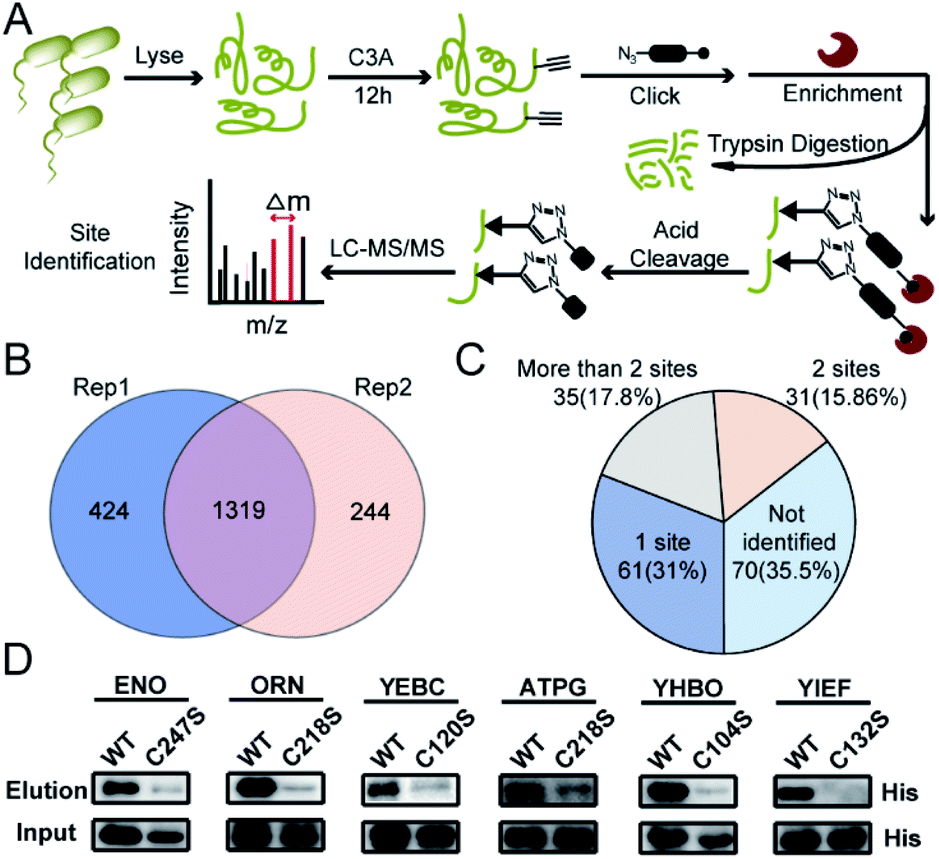 | ||
| Fig. 3 Site-specific profiling of itaconated cysteines by C3A in S. enterica lysates. (A) The TOP-ABPP workflow for chemoproteomic profiling of itaconated sites in S. enterica lysates. (B) Overlap of the itaconated cysteines identified by C3A in two biological replicates. (C) Distribution of the itaconation targets in terms of the number of modification sites per protein. (D) Biochemical validation of the novel itaconation sites in ENO, ORN, YEBC, ATPG, YHBO, and YIEF by comparing the C3A labeling between the wild-type (WT) proteins and their cysteine-to-serine mutants. | ||
Among all the itaconated proteins identified by our chemoproteomic profiling, the most prominent one is isocitrate lyase (ICL, also known as ACEA) which has maximum ratios in both the enrichment and competition profiling experiments (Fig. 2B). ICL is a Mg2+-dependent enzyme involved in the glyoxylate cycle that catalyzes the reversible conversion of isocitrate to succinate and glyoxylate20 (Fig. S6A†). This catalysis bypasses two oxidative steps in the TCA cycle to synthesize succinate. Thus, the glyoxylate cycle is more carbon conserving and can help maintain an adequate supply of TCA intermediates under glucose limited conditions.21 Given its absence in vertebrates and conservation among most prokaryotes, ICL is considered as an ideal drug target candidate.22 Notably, a recent study independently discovered via the approaches of structural biology and medicinal chemistry that itaconate could serve as a covalent inhibitor for the two ICL isoforms from Mycobacterium tuberculosis by targeting their active-site cysteines.23 Consistent with this finding, our chemoproteomic profiling identified 5 itaconation sites in the ICL of S. enterica including the catalytic Cys195 (Table S3†).
We recombinantly overexpressed the wild-type ICL and each of the 5 cysteine-to-serine single mutants with the N-terminal 6xHis tag in S. enterica. The C195S mutant almost lost all the labeling signal, indicating that it is the main itaconation site in ICL (Fig. 4A). We next purified each of the ICL variants (Fig. S6B†) and incubated the purified protein with itaconate to confirm the modifications on five cysteine sites by LC-MS/MS (Fig. 4B and S6C†). We then measured the enzymatic activity of the wild-type and mutant ICLs following a previously reported assay.24 Consistent with the knowledge that C195 serves as a catalytic residue to aid the product formation of succinate, the activity of C195S was completely abolished, while that of C318S was partially lost (Fig. 4C). We also observed a dose-dependent inhibition by itaconate of the activity of wild-type ICL with an IC50 value around 2.6 mM (Fig. 4D). Lastly, we knocked out the endogenous ICL from S. enterica and complemented back with either the wild-type protein or each of the five mutants (Fig. 4E). The growth defects of these strains under the limited carbon source were highly consistent with the observed loss of the enzymatic activity, with the C195S and C318S strains showing complete and partial growth inhibition, respectively. These data collectively suggested that Cys195 is a functionally critical site for ICL and covalent modification of this specific site by itaconate mediated the metabolite's anti-bacterial activity.
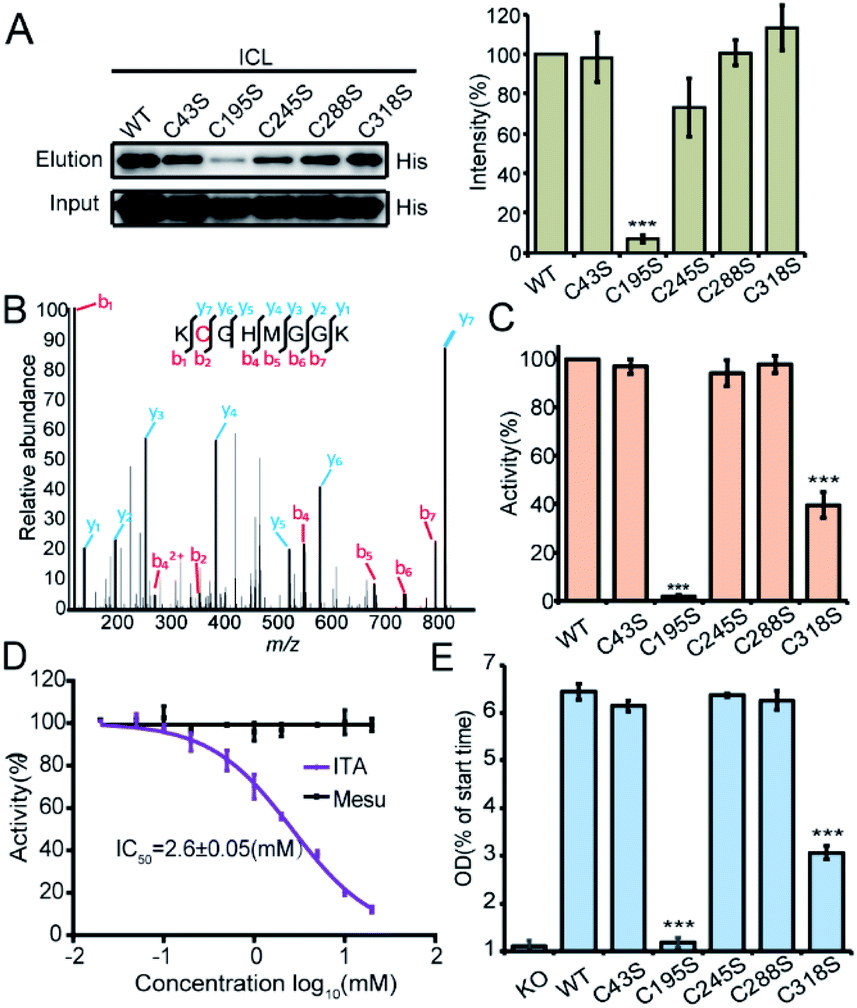 | ||
| Fig. 4 Functional characterization of the itaconations on ICL. (A) Mutation of Cys195 abolished the C3A labeling on ICL (left). The quantification (right) of the labeling intensities was averaged from three repeats. (B) MS/MS spectrum of the peptide of ICL containing the itaconated Cys195. The purified ICL protein was incubated with 1 mM itaconate for 2 hours, and then the labeled proteins were digested by trypsin and analyzed by LC-MS/MS. (C) Enzyme activity of the purified wild-type (WT) ICL and the five cysteine-to-serine mutants. (D) Itaconate inhibits the enzyme activity of ICL with an IC50 of 2.6 mM. (E) Strains with overexpression of certain ICL mutants (C195S or C318S) showed growth defects. | ||
Conclusions
In this study, we developed a novel bioorthogonal probe, C3A, for quantitative and site-specific chemoproteomic profiling of itaconate modifications in S. enterica. Compared to the ITalk probe used for profiling in living macrophages, C3A has an amide linkage and a shorter tail, which resulted in dramatically different labeling performance in bacterial lysates. While the exact reason for the enhanced and unique labeling in the S. enterica proteome by C3A remains elusive, the probe has enabled the first global profiling of itaconation events in bacteria, which will provide a rich resource for investigating the anti-bacterial role of the metabolite. In future, the probe's permeability should be further improved for profiling in living pathogens. On a related note, a recent study showed that itaconate and its derivatives have different impacts on downstream pathways,25 warranting the need to develop new chemoproteomic tools to map endogenous itaconation in both hosts and pathogens.Itaconate has long been considered to inhibit ICL by competitive binding as a product analogue. An elegant study has recently provided unambiguous structural and biochemical evidence that itaconate indeed covalently modifies the active-site cysteine of ICL in Mycobacterium tuberculosis to inhibit the enzyme's activity,23 which justifies the rationale of developing covalent inhibitors for this functionally important enzyme. Our profiling study independently discovered and confirmed such a covalent modification event. Since C3A showed specific labeling on the active-site Cys195 in the purified enzyme, we envision that the probe should be highly compatible with high-throughput screening platforms, such as Fluopol-ABPP,26 to obtain more potent covalent inhibitors for the enzyme.
It is interesting to observe that the C318S mutant of ICL also loses partially the enzymatic activity although the site is distant to the catalytic center. Our preliminary data revealed that the mutation might affect the enzyme's stability (Fig. S7A†), but a structural biology approach would ultimately provide an answer for this mystery. Since Cys318 is also subjected to itaconate's modification as evidenced by the tandem mass spectra, it could in principle serve as another site for allosteric inhibitor development. Lastly, with probes available for monitoring itaconation in both macrophages and bacteria, the diversified roles of itaconate could be further explored in the context of host–pathogen interface in future.
Author contributions
Chu Wang, Yanling Zhang and Wei Qin conceived the project. Yanling Zhang conducted most of the experiments unless specified otherwise. Dongyang Liu carried out the C8A synthesis. Yuan Liu helped perform MS data analysis. Yanling Zhang and Chu Wang analyzed the data and wrote the manuscript with input from all the authors.Conflicts of interest
The authors declare no competing interests.Acknowledgements
We thank the Computing Platform of the Center for Life Science for supporting the LC-MS/MS data analysis. This work is supported by grants from Ministry of Science and Technology of People's Republic of China (No. 2016YFA0501500) and the National Natural Science Foundation of China (No. 91953109, 21778004 and 21925701) to C. W.Notes and references
- V. Lampropoulou, A. Sergushichev, M. Bambouskova, S. Nair, E. E. Vincent, E. Loginicheva, L. Cervantes-Barragan, X. Ma, S. C. Huang, T. Griss, C. J. Weinheimer, S. Khader, G. J. Randolph, E. J. Pearce, R. G. Jones, A. Diwan, M. S. Diamond and M. N. Artyomov, Cell Metab., 2016, 24, 158–166 CrossRef CAS PubMed.
- L. A. J. O'Neill and M. N. Artyomov, Nat. Rev. Immunol., 2019, 19, 273–281 CrossRef PubMed.
- C. L. Strelko, W. Lu, F. J. Dufort, T. N. Seyfried, T. C. Chiles, J. D. Rabinowitz and M. F. Roberts, J. Am. Chem. Soc., 2011, 133, 16386–16389 CrossRef CAS PubMed.
- A. Michelucci, T. Cordes, J. Ghelfi, A. Pailot, N. Reiling, O. Goldmann, T. Binz, A. Wegner, A. Tallam, A. Rausell, M. Buttini, C. L. Linster, E. Medina, R. Balling and K. Hiller, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7820–7825 CrossRef CAS PubMed.
- Z. Zasłona and L. A. J. O’Neill, Mol. Cell, 2020, 78(5), 814–823 CrossRef PubMed.
- T. Cordes, A. Michelucci and K. Hiller, Annu. Rev. Nutr., 2015, 35, 451–473 CrossRef CAS PubMed.
- E. L. Mills, D. G. Ryan, H. A. Prag, D. Dikovskaya, D. Menon, Z. Zaslona, M. P. Jedrychowski, A. S. H. Costa, M. Higgins, E. Hams, J. Szpyt, M. C. Runtsch, M. S. King, J. F. McGouran, R. Fischer, B. M. Kessler, A. F. McGettrick, M. M. Hughes, R. G. Carroll, L. M. Booty, E. V. Knatko, P. J. Meakin, M. L. J. Ashford, L. K. Modis, G. Brunori, D. C. Sévin, P. G. Fallon, S. T. Caldwell, E. R. S. Kunji, E. T. Chouchani, C. Frezza, A. T. Dinkova-Kostova, R. C. Hartley, M. P. Murphy and L. A. O'Neill, Nature, 2018, 556, 113–117 CrossRef CAS PubMed.
- M. Bambouskova, L. Gorvel, V. Lampropoulou, A. Sergushichev, E. Loginicheva, K. Johnson, D. Korenfeld, M. E. Mathyer, H. Kim, L. H. Huang, D. Duncan, H. Bregman, A. Keskin, A. Santeford, R. S. Apte, R. Sehgal, B. Johnson, G. K. Amarasinghe, M. P. Soares, T. Satoh, S. Akira, T. Hai, C. de Guzman Strong, K. Auclair, T. P. Roddy, S. A. Biller, M. Jovanovic, E. Klechevsky, K. M. Stewart, G. J. Randolph and M. N. Artyomov, Nature, 2018, 556, 501–504 CrossRef CAS PubMed.
- A. Hooftman, S. Angiari, S. Hester, S. E. Corcoran, M. C. Runtsch, C. Ling, M. C. Ruzek, P. F. Ruzek, A. F. McGettrick, K. Banahan, M. M. Hughes, A. D. Irvine, R. Fischer and L. A. J. O'Neill, Cell Metab., 2020, 32, 468–478 CrossRef CAS PubMed.
- W. Qin, K. Qin, Y. Zhang, W. Jia, Y. Chen, B. Cheng, L. Peng, N. Chen, Y. Liu, W. Zhou, Y. L. Wang, X. Chen and C. Wang, Nat. Chem. Biol., 2019, 15, 983–991 CrossRef CAS PubMed.
- W. Qin, Y. Zhang, H. Tang, D. Liu, Y. Chen, Y. Liu and C. Wang, J. Am. Chem. Soc., 2020, 142, 10894–10898 CrossRef CAS PubMed.
- J. O. Williams, T. E. Roche and B. A. McFadden, Biochemistry, 1971, 10, 1384–1390 CrossRef CAS PubMed.
- B. A. McFadden and S. Purohit, J. Bacteriol., 1977, 131, 136–144 CrossRef CAS PubMed.
- I. A. Berg, L. V. Filatova and R. N. Ivanovsky, FEMS Microbiol. Lett., 2002, 216, 49–54 CrossRef CAS PubMed.
- M. Ruetz, G. C. Campanello, M. Purchal, H. Shen, L. McDevitt, H. Gouda, S. Wakabayashi, J. Zhu, E. J. Rubin, K. Warncke, V. K. Mootha, M. Koutmos and R. Banerjee, Science, 2019, 366, 589–593 CrossRef CAS PubMed.
- R. A. Kulkarni, D. W. Bak, D. Wei, S. E. Bergholtz, C. A. Briney, J. H. Shrimp, A. Alpsoy, A. L. Thorpe, A. E. Bavari, D. R. Crooks, M. Levy, L. Florens, M. P. Washburn, N. Frizzell, E. C. Dykhuizen, E. Weerapana, W. M. Linehan and J. L. Meier, Nat. Chem. Biol., 2019, 15, 391–400 CrossRef CAS PubMed.
- J. D. McKinney, K. H. zu Bentrup, E. J. Muñoz-Elías, A. Miczak, B. Chen, W.-T. Chan, D. Swenson, J. C. Sacchettini, W. R. Jacobs and D. G. Russell, Nature, 2000, 406, 735–738 CrossRef CAS PubMed.
- H. Mi, A. Muruganujan, X. Huang, D. Ebert, C. Mills, X. Guo and P. D. Thomas, Nat. Protoc., 2019, 14, 703–721 CrossRef CAS PubMed.
- E. Weerapana, A. E. Speers and B. F. Cravatt, Nat. Protoc., 2007, 2, 1414–1425 CrossRef CAS PubMed.
- S. Hillier and W. T. Charnetzky, J. Bacteriol., 1981, 145, 452–458 CrossRef CAS PubMed.
- F. C. Fang, S. J. Libby, M. E. Castor and A. M. Fung, Infect. Immun., 2005, 73, 2547–2549 CrossRef CAS PubMed.
- D. G. Russell, Nat. Rev. Mol. Cell Biol., 2001, 2, 569–577 CrossRef CAS PubMed.
- B. X. C. Kwai, A. J. Collins, M. J. Middleditch, J. Sperry, G. Bashiri and I. K. H. Leung, RSC Med. Chem., 2021, 12(1), 57–61 RSC.
- E. M. el-Mansi, C. MacKintosh, K. Duncan, W. H. Holms and H. G. Nimmo, Biochem. J., 1987, 242, 661–665 CrossRef CAS PubMed.
- A. Swain, M. Bambouskova, H. Kim, P. S. Andhey, D. Duncan, K. Auclair, V. Chubukov, D. M. Simons, T. P. Roddy, K. M. Stewart and M. N. Artyomov, Nat. Metab., 2020, 2, 594–602 CrossRef CAS PubMed.
- D. A. Bachovchin, S. J. Brown, H. Rosen and B. F. Cravatt, Nat. Biotechnol., 2009, 27, 387–394 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, synthetic procedures, characterization data for reaction products and additional figures. See DOI: 10.1039/d1sc00660f |
| This journal is © The Royal Society of Chemistry 2021 |