
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A bifunctional iminophosphorane squaramide catalyzed enantioselective synthesis of hydroquinazolines via intramolecular aza-Michael reaction to α,β-unsaturated esters†
Guanglong
Su
 a,
Connor J.
Thomson‡
a,
Ken
Yamazaki‡
ab,
Daniel
Rozsar
a,
Kirsten E.
Christensen
a,
Trevor A.
Hamlin
*b and
Darren J.
Dixon
*a
a,
Connor J.
Thomson‡
a,
Ken
Yamazaki‡
ab,
Daniel
Rozsar
a,
Kirsten E.
Christensen
a,
Trevor A.
Hamlin
*b and
Darren J.
Dixon
*a
aDepartment of Chemistry, Chemistry Research Laboratory, University of Oxford, Mansfield Road, Oxford OX1 3TA, UK. E-mail: darren.dixon@chem.ox.ac.uk
bDepartment of Theoretical Chemistry, Amsterdam Institute of Molecular and Life Sciences (AIMMS), Amsterdam Center for Multiscale Modeling (ACMM), Vrije Universiteit Amsterdam, De Boelelaan 1083, 1081 HV, Amsterdam, The Netherlands. E-mail: t.a.hamlin@vu.nl
First published on 18th March 2021
Abstract
An efficient synthesis of enantioenriched hydroquinazoline cores via a novel bifunctional iminophosphorane squaramide catalyzed intramolecular aza-Michael reaction of urea-linked α,β-unsaturated esters is described. The methodology exhibits a high degree of functional group tolerance around the forming hydroquinazoline aryl core and wide structural variance on the nucleophilic N atom of the urea moiety. Excellent yields (up to 99%) and high enantioselectivities (up to 97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 er) using both aromatic and less acidic aliphatic ureas were realized. The potential industrial applicability of the transformation was demonstrated in a 20 mmol scale-up experiment using an adjusted catalyst loading of 2 mol%. The origin of enantioselectivity and reactivity enhancement provided by the squaramide motif has been uncovered computationally using density functional theory (DFT) calculations, combined with the activation strain model (ASM) and energy decomposition analysis (EDA).
:3 er) using both aromatic and less acidic aliphatic ureas were realized. The potential industrial applicability of the transformation was demonstrated in a 20 mmol scale-up experiment using an adjusted catalyst loading of 2 mol%. The origin of enantioselectivity and reactivity enhancement provided by the squaramide motif has been uncovered computationally using density functional theory (DFT) calculations, combined with the activation strain model (ASM) and energy decomposition analysis (EDA).
Introduction
Heterocyclic organic compounds containing a hydroquinazoline core are commonplace amongst various natural products and potent drug substances used in the clinic.1 These include, for instance, DPC 963, a second-generation non-nucleoside reverse transcriptase inhibitor (NNRTI) for HIV treatment,2 fungicidal 2-azolyl-3,4-dihydroquinazolines compounds3 and the anti-human cytomegalovirus drug, letermovir (Fig. 1).4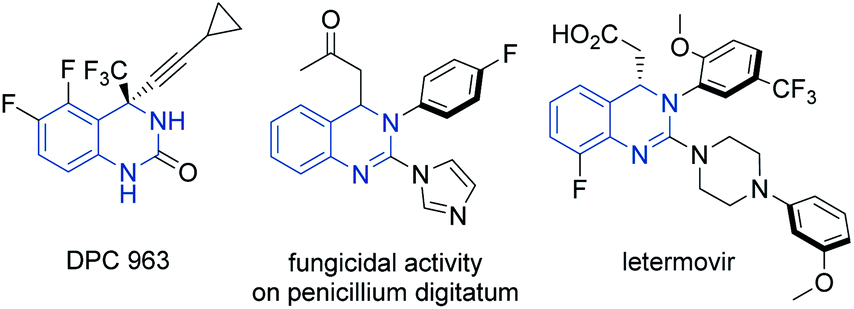 | ||
| Fig. 1 Representative pharmaceutically active compounds containing a hydroquinazoline core. | ||
Although much effort has been directed towards the synthesis of hydroquinazoline compounds,5 highly enantioselective catalytic methods are still relatively uncommon, especially for unbiased/unactivated systems (Scheme 1). In 2015, the Mashima group developed an enantioselective hydrogenation of quinazolinium salts to yield chiral tetrahydroquinazolines with excellent enantioselectivity under chiral iridium catalysis.6 A palladium-catalyzed enantioselective allylic C–H amination to generate the chiral hydroquinazoline core in good yield and high enantioselectivity was later described by Gong and coworkers.7
 | ||
| Scheme 1 Previous enantioselective syntheses of hydroquinazoline. | ||
Specifically, for dihydroquinazolines bearing a trifluoromethyl group attached to a newly generated quaternary carbon center, an extensive range of metal and metal-free catalyzed enantioselective addition reactions to reactive cyclic ketimines using alkyne,8 ketone,9 nitroalkane,10 β-keto acid,11 nitrile,12 alcohol13 and isocyanoacetate14 nucleophiles, have been developed.15
Enantioselective aza-Michael reactions enabled by metal-free catalysts are other powerful and promising approaches to access such pharmaceutically relevant N-heterocycles.16 However, in this field, catalyst promoted addition of pronucleophilic ureas to tethered β-substituted α,β-unsaturated esters remains largely unsolved due to the high pKa of the urea and low electrophilicity of the Michael acceptor.17,18 To our knowledge only two reports describe the synthesis of the chiral hydroquinazoline core in such a way. In 2016, a single moderately enantioselective phase-transfer-catalyzed intramolecular aza-Michael reaction (IAMR) was described by Tschaen and coworkers en route to letermovir.19 In 2017, Ruck and co-workers then developed the enantioselective IAMR reaction of related guanidine containing substrates.20 However, only N-aryl nucleophiles were compatible and transformation of the guanidine IAMR product to drug molecules bearing urea motifs – such as in DPC 963 (shown in Fig. 1) – was not feasible. Against this backdrop, we envisaged that the enhanced Brønsted basicity and broadly tunable structure of the bifunctional iminophosphorane (BIMP) superbase catalyst system developed in our group21 could provide the solution to the challenging pKa related reactivity and modest stereocontrol in the IAMR, and herein we wish to report our findings.
Results and discussion
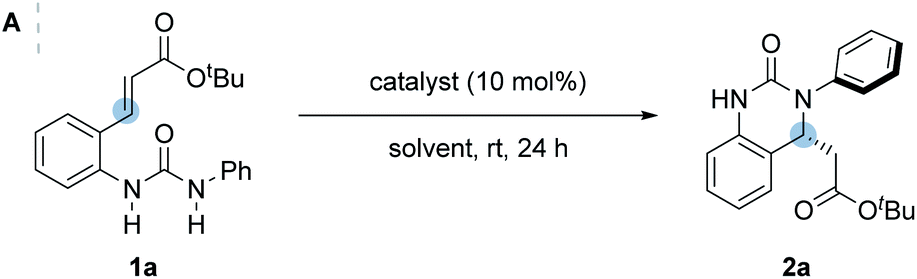
Urea 1a bearing an α,β-unsaturated tert-butyl ester was chosen as the model substrate for the IAMR reaction. An initial reactivity study of various bifunctional organocatalysts revealed that moderately Brønsted basic cinchona-derived bifunctional catalyst A failed to promote any detectable reaction in Et2O at room temperature after 24 hours (Table 1, entry 1). In contrast, catalyst B bearing a superbasic iminophosphorane motif smoothly gave the desired product 2a in 96% isolated yield and 68.5:31.5 er under identical conditions (Table 1, entry 2). With excellent reactivity identified, a series of modifications to the BIMP catalyst structure was then performed to optimise the IAMR reaction. Changing the H-bond donor from a urea to the more acidic thiourea improved the enantioselectivity to 74:26 er but lowered the isolated yield to 73% (Table 1, entry 3). The introduction of a second stereogenic center adjacent to the thiourea motif in the catalyst allowed for rapid library generation and solved the issue of poor reactivity (Table 1, entries 4–6). Variation of the chiral backbone and optimization of reaction conditions revealed that 10 mol% catalyst F in 0.025 M toluene at room temperature gave the desired product in almost quantitative yield and 78.5:21.5 er (Table 1, entry 7).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Concentration (M) | Yield (%)a | erb |
| a Yields of isolated products. b Determined by HPLC analysis on chiral stationary phase. c 12 days reaction time. d 4 hours reaction time. e 10 hours reaction time. N.D. = not determined. | |||||
| 1c | A | Et2O | 0.1 | <1 | N.D. |
| 2 | B | Et2O | 0.1 | 96 | 68.5:31.5 |
| 3 | C | Et2O | 0.1 | 73 | 74:26 |
| 4 | D | Et2O | 0.1 | 99 | 60:40 |
| 5 | E | Et2O | 0.1 | 96 | 74.5:25.5 |
| 6 | F | Et2O | 0.1 | >99 | 75:25 |
| 7 | F | Toluene | 0.025 | >99 | 78.5:21.5 |
| 8d | G | Toluene | 0.025 | 53 | 67.5:32.5 |
| 9d | H | Toluene | 0.025 | 11 | 65.5:34.5 |
| 10e | I | Toluene | 0.025 | 76 | 79:21 |
| 11e | K | Toluene | 0.025 | 71 | 80.5:19.5 |
| 12 | L | Toluene | 0.025 | >99 |
94.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :5.5 :5.5
|

|
|||||
A third stereogenic center adjacent to the amide motif was then incorporated and enantioselectivity increased to 80.5:19.5 er with catalyst J slightly outcompeting diastereomeric catalyst I (Table 1, entries 10 & 11). Excitingly, a squaramide substitution for the thiourea (catalyst K) boosted the enantiocontrol to 94.5:5.5 er.
The major enhancement in selectivity likely arises from the higher acidity/H-bond donor ability of the squaramide and/or the modified 3D structure resulting from the differing bond angles at the squaramide.22 Additional catalyst structure-performance studies gave no further improvement (see ESI† for optimization details).
With the optimal conditions in hand, the scope of the enantioselective IAMR reaction was then explored (Scheme 2A). Notably, the IAMR reactions were found to typically have very clean reaction profiles and no effort was made to exclude moisture or air from the scoping experiments. Varying the substituents on the quinazolinone aryl core gave rise to minimal fluctuation in enantioselectivity and compatible functionalities varied from electron-donating groups to electron-withdrawing groups. Elevated temperatures of up to 80 °C were required to ensure solubility of the substrates in some cases (1b and 1d). A pyridine-based substrate (1i) was also found to be well-tolerated affording the desired product in excellent yield and enantioselectivity under the standard reaction conditions. The substituent effect on the N-aryl ring was then examined. Substrates possessing single iodine, bromine and fluorine atoms at various ring positions as well as a 3,5-dichloro example, performed typically well providing the desired hydroquinazoline core in excellent yield and good er (1j to 1p). The rates of the cyclization reactions were found to decrease with increasing electron-richness of the N-aryl rings. For substrates 1q to 1y, extra reaction time or heating to 50 °C was required to maintain the high yield without compromising enantioselectivity. However, the positional effect of the substituents on reaction enantioselectivity was negligible (1t to 1v). Interestingly, ortho substituents (such as thiomethyl, tert-butyl and ethynyl in 1w–1y) gave rise to a slight uplift in enantioselectivity (96:4–97:3 er). The methodology was also applicable to less activated, higher pKa, alkyl-substituted ureas. The high Brønsted basicity of the BIMP catalyst system indeed smoothly provided N-allyl and N-benzyl substituted hydroquinazolines in almost quantitative yield and good er (85:15). Even less activated ureas (1ab to 1ad) demanded harsher reaction conditions to deliver the cyclized product in moderate to excellent yield and good er. Finally, the methyl ester acceptor (1ae) also proved to be a good substrate. After 48 hours hydroquinazoline product 2ae was obtained in almost quantitative yield in high enantioselectivity (92:8 er). Other conjugate acceptors including phenyl esters, enones and α,β-unsaturated amides were also examined, however satisfactory enantioselectivities were not obtained (see ESI† for details).
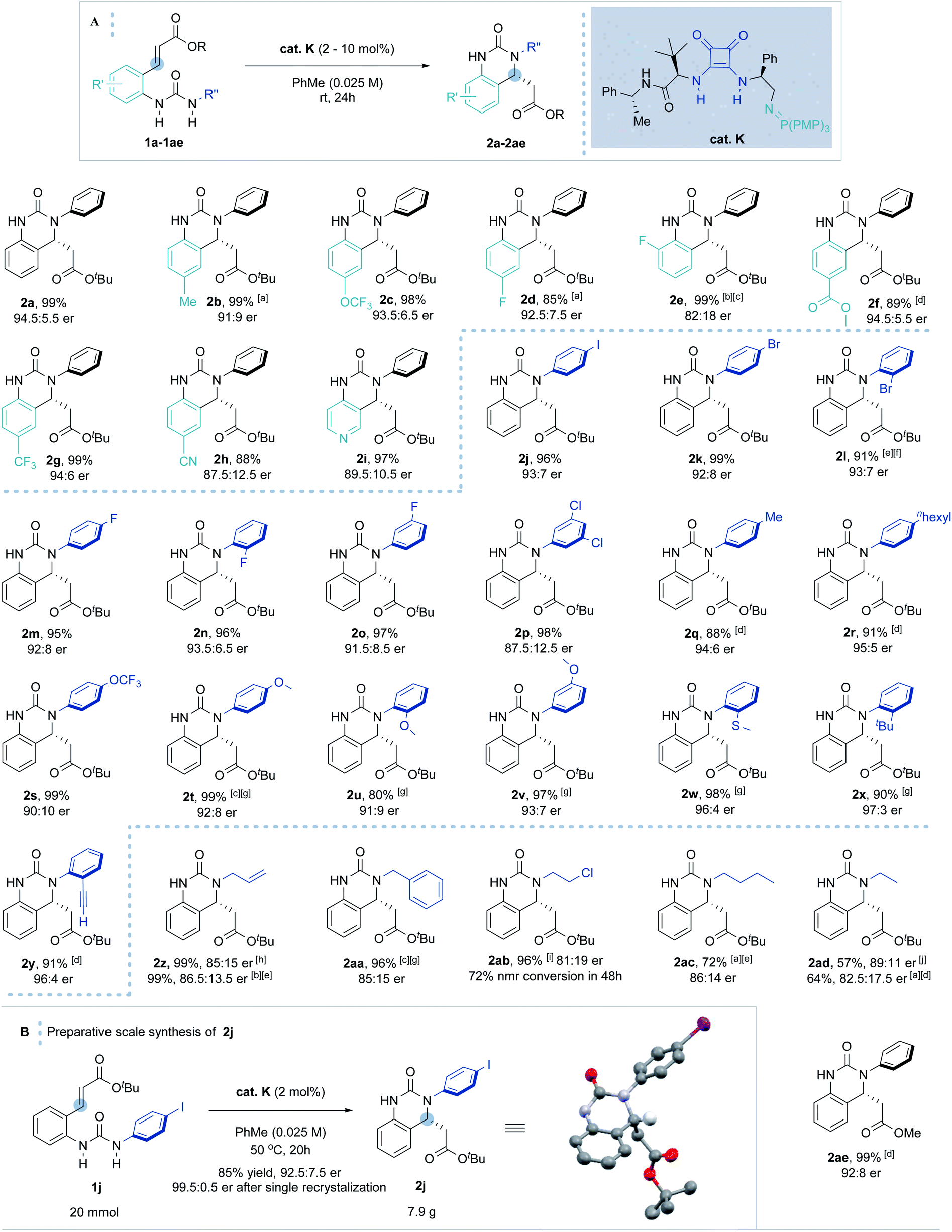 | ||
| Scheme 2 (A) Scope of the BIMP-catalyzed intramolecular aza-Michael reaction to α,β-unsaturated ester. [a] Reaction carried out at 80 °C. [b] Reaction carried out at 40 °C. [c] 30 hours reaction time. [d] 48 hours reaction time. [e] 72 hours reaction time. [f] 5 mol% cat. K was used. [g] Reaction carried out at 50 °C. [h] Reaction carried out at 60 °C. [i] 120 hours reaction time. [j] 216 hours reaction time. (B) Preparative scale synthesis of 2j. Stereochemical configuration was assigned by analogy with (R)-2j (determined by single crystal X-ray diffraction studies).33 | ||
Increasing the reaction scale 100 fold (to 20 mmol) and decreasing the catalyst loading to 2 mol% delivered the desired product in good yield (7.9 g, 85%) without compromising enantioselectivity (92.5:7.5 er). Pleasingly, only a single recrystallization was required to afford essentially enantiopure 2j (Scheme 2B). Furthermore, and to demonstrate potential industrial applicability of the chemistry, various derivatizations of this product were carried out (Scheme 3). For example, removal of the tert-butyl carboxylate ester with TFA, activation as the acid chloride, and subsequent treatment with benzyl amine and methanol gave the methyl ester (3) and amide (4) in excellent to moderate yield, respectively. Suzuki coupling with an N-methyl substituted pyrazole boronic acid and Sonogashira coupling with erlotinib successfully installed various functionalities in the para-position of the N-aryl ring.
In order to paint a mechanistic picture, density functional theory calculations on the aza-Michael reaction step were performed. All calculations reported in this paper were performed using the Amsterdam Density Functional (ADF) software.23 Equilibrium structures and transition structure geometries were optimized using the BLYP functional24,25 and the DZP basis set.26 Solvent effects of toluene were accounted for using the conductor-like screening model (COSMO) of solvation.27 Dispersion interactions were included using Grimme's DFT-D3 correction with Becke–Johnson damping.28 The zeroth-order regular approximation (ZORA) was used to account for scalar relativistic effects.29 This level is referred to as COSMO(toluene)-ZORA-BLYP-D3(BJ)/DZP. All stationary points have been verified, through vibrational analysis, to be minima (zero imaginary frequencies) or transition structures (one imaginary frequency). The character of the normal mode associated with the imaginary frequency has been analyzed to ensure it resembles the reaction under consideration. Optimized structures were illustrated using CYLview20.30 Potential energies were refined by means of single point calculations using the M06-2X functional31 and the TZ2P basis set.26 This level is denoted COSMO(toluene)-ZORA-M06-2X/TZ2P//COSMO(toluene)-ZORA BLYP-D3(BJ)/DZP. The reported Gibbs free energies in solution are calculated by adding thermal corrections computed at 298 K from vibrational frequencies obtained through numerical differentiation of the analytical gradient at COSMO(toluene)-ZORA-BLYP-D3(BJ)/DZP and a standard concentration (1 mol L−1) to the total electronic energy at COSMO(toluene)-ZORA-M06-2X/TZ2P//COSMO(toluene)-ZORA-BLYP-D3(BJ)/DZP.
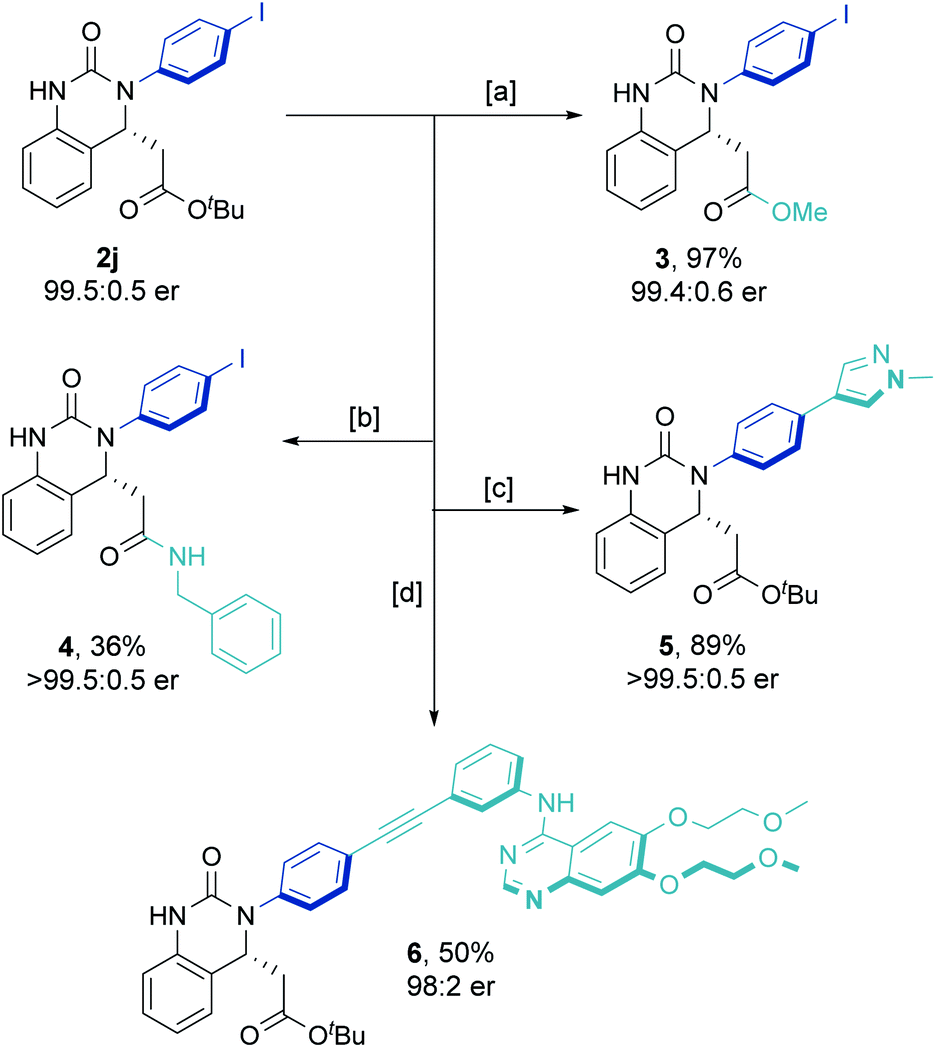 | ||
| Scheme 3 Derivatization of enantioenriched 2j. [a] (i) TFA, CH2Cl2, 0 °C to RT, 5 h. (ii) SOCl2, MeOH, 20 h. [b] (i) TFA, CH2Cl2, 0 °C to RT, 5 h. (ii) (COCl)2, DMF (cat.), CH2Cl2, 2 h. (iii) BnNH2, Et3N, CH2Cl2, 18 h. [c] 1-Methyl-1H-pyrazole-3-boronic acid pinacol ester, Pd(dppf)Cl2CH2Cl2, Cs2CO3, 1,4-dioxane, H2O. [d] Erlotinib (HCl complex), PdCl2(PPh3)2, CuI, PPh3, Et3N. | ||
To elucidate the origin of stereocontrol in the novel BIMP squaramide catalyzed IAMR reaction, we performed a state-of-the-art DFT study. Due to the conformational freedom and the existence of two potential activation modes of the BIMP catalyst, we computed and compared all the possible TSs for the enantio-determining Michael reaction step involving substrate 1ae (see the ESI† for additional details).32 The most energetically preferred transition structures that lead to either (R)- or (S)-product are shown in Scheme 4. The TS–ModeA–LA1–RA1–R that forms the (R)-product was found to be favoured by 1.2 kcal mol−1, which agrees with the experimentally confirmed absolute stereochemical configuration by single crystal X-ray diffraction studies. Pleasingly, our computational approach predicted the enantioselectivity for the formation of product 2ae in 88:12 er, which was in excellent agreement with the experimental selectivity of 92:8 er. The energetically preferred TS conformation in TS–ModeA–LA1–RA1–R engages in several weak stabilizing interactions. The squaramide moiety interacts with the ester carbonyl group by hydrogen bonding and with the urea carbonyl group by CO–π interaction to activate both the electrophile and the nucleophile. The “left arm” of the BIMP catalyst bearing the amide group additionally interacts with the aromatic scaffold in the substrate by both CH–π, and CO–π interactions without significant steric repulsion. The “right arm” of the BIMP catalyst bearing the iminophosphorane moiety activates the nucleophilic urea by both hydrogen bonding and through CH–π interactions between the PPh3 and the aromatic ring on the N atom in the case of the N-aryl substrates. In addition to these catalyst/substrate interactions, the hydrogen bonding and the CH–π interactions within the catalyst also provides the rigidity of this particular lowest energy transition structure.34 This conformation creates an ideal-fit pocket within which the substrate can perfectly fit that maximizes stabilizing interactions and minimizes steric repulsion during the C–N bond forming step of the Michael reaction. Analysis of non-covalent interaction (NCI) plots allows one to qualitatively visualize these weak interactions between the catalyst and the substrate (Scheme S5 and S6†).35 Therefore, the TS that has a catalyst conformation and coordination mode of the substrate that both reduces steric repulsion and maximizes interactions is energetically preferred in this reaction.
In order to obtain deeper insight into the origin of the catalytic activity imparted by the squaramide motif of the BIMP catalyst, an activation strain analysis (ASA) and an energy decomposition analysis (EDA) were carried out on archetypal model systems. The ASM involves decomposing the electronic energy ΔE into the strain energy ΔEstrain associated with the structural deformation of the hydrogen bond donor (HB) and methyl acrylate (MA) from their equilibrium geometry and the interaction energy ΔEint between the deformed reactants [eqn (1)].36 The EDA separates the interaction energy (ΔEint) into the following three chemically meaningful energy terms: classical electrostatic interaction (ΔVelstat), Pauli repulsion (ΔEPauli) between closed-shell orbitals which is responsible for steric repulsion, and stabilizing orbital interaction (ΔEoi) that accounts, among others, for HOMO–LUMO interactions [eqn (2)].37
| ΔE = ΔEstrain + ΔEint | (1) |
| ΔEint = ΔVelstat + ΔEPauli + ΔEoi | (2) |
First, we analyzed the interaction between HB (urea, thiourea, and squaramide) and MA in the formation of complexes U–MA, TU–MA, SQ–MA (Scheme 5A). The interaction becomes more stabilizing from U–MA, TU–MA, SQ–MA (ΔEint = −9.0 to −10.1 to −11.5 kcal mol−1) mainly due to the more stabilizing ΔVelstat term as a result of the electrostatic nature of hydrogen bonds. The ΔEoi term is also very important and involves significant charge transfer from the lone pair of the oxygen atom of MA and the two 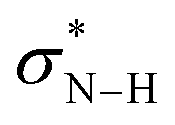 orbitals on the HB. This flow of charge out of the substrate to the catalyst induces a polarization of the π–MO away from the C
orbitals on the HB. This flow of charge out of the substrate to the catalyst induces a polarization of the π–MO away from the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond (the importance of which is explained below). The stabilizing ΔVelstat and ΔEoi interactions play a significant role in the hydrogen bonding stabilization by the squaramide catalyst that can be seen by the decreased H⋯OC bond length in SQ–MA compared to U–MA and TU–MA.
C bond (the importance of which is explained below). The stabilizing ΔVelstat and ΔEoi interactions play a significant role in the hydrogen bonding stabilization by the squaramide catalyst that can be seen by the decreased H⋯OC bond length in SQ–MA compared to U–MA and TU–MA.
 | ||
| Scheme 4 (A) Lowest energy TS structure for the formation of (R)-product and (B) lowest energy TS structure for the formation of (S)-product of the BIMP squaramide-catalyzed intramolecular aza-Michael reaction computed at COSMO(toluene)-ZORA-M06-2X/TZ2P//COSMO(toluene)-ZORA-BLYP-D3(BJ)/DZP. Energies (kcal mol−1) and forming bond lengths (Å) of TS geometries are provided in the insert. | ||
 | ||
| Scheme 5 (A) Interaction energies and the energy decomposition analysis (EDA) of the hydrogen bond donor–methyl acrylate complexes (HB–MA). (B) Energy barriers of the aza-Michael reaction transition structures and the activation strain analysis (ASA) and the energy decomposition analysis (EDA). (C) Molecular orbital diagram and the most significant occupied orbital overlaps computed at COSMO(toluene)-ZORA-M06-2X/TZ2P//COSMO(toluene)-ZORA-BLYP-D3(BJ)/DZP. Energies (kcal mol−1) are provided in the insert. | ||
We then analyzed the transition structures and the energy barriers for the aza-Michael reaction (Scheme 5B). The uncatalyzed reaction goes with the highest reaction barrier (ΔG‡ = 16.3 kcal mol−1). The urea, thiourea, and squaramide catalyzed reactions go with barriers of 12.0, 11.2, and 8.9 kcal mol−1, respectively. In order to elucidate the trend in the reactivity, we performed the ASA on the transition structures. The trend in Gibbs free energy activation barriers is the same as for the electronic activation energy barriers. The lower, more favourable, barrier for the squaramide catalyzed reaction compared to the uncatalyzed one (ΔE‡ = −8.8 vs. 2.9 kcal mol−1) originates from a more stabilizing interaction energy (ΔE‡int = −29.2 vs. −20.0 kcal mol−1). The differences in the ΔE‡strain also contribute to the trend but are less decisive for the overall reactivity trends (ΔE‡strain = 20.4 vs. 23.0 kcal mol−1). Next, using the EDA method, the trend in the more stabilizing ΔE‡int was analyzed. This successfully identified the role of a reduction in Pauli repulsion between the reactants being the reason for the more enhanced reactivity of the catalyzed reactions.37 The origin of the less destabilizing Pauli repulsion (ΔE‡Pauli) for the transformation was quantified by performing a Kohn–Sham molecular orbital (KS–MO) analysis (Scheme 5C). The occupied π–MOHB–MA (2p atomic orbitals on the reacting CC double bonds) contributes to the trend in the Pauli repulsion. The computed orbital overlap S between the π–MOHB–MA and the lone pair of the nucleophile HOMO−1Nu decreases from 0.141 for TSuncatalyzed to 0.106 for TSSQ–MA, which is caused by the aforementioned polarization of the π–MOHB–MA away from the reactive carbon center of the CC bond due to the charge transfer interaction with the hydrogen bond donor catalyst. The squaramide catalyst emerges as the best of our studied bifunctional iminophosphorane squaramide catalysts as it is able to reduce the destabilizing Pauli repulsion between the reactants and thereby impart the greatest reactivity enhancement of our intramolecular aza-Michael reaction.38 These systematic computational analyzes explain the origin of reactivity and enantioselectivity in this BIMP squaramide catalyzed aza-Michael reaction.
Conclusions
In summary, an efficient and highly enantioselective BIMP-catalyzed intramolecular aza-Michael reaction affording decorated hydroquinazoline structures with excellent yields and enantiomeric ratios has been developed. A novel BIMP squaramide system was found to be effective in activating both aromatic and aliphatic ureas. DFT calculations uncovered the fact that the optimal catalyst conformation creates a pocket-like binding site for the substrate to impart enantiofacial selectivity, whilst the squaramide motif demonstrates advantages over urea and thiourea H-bond donor groups on decreasing the destabilizing Pauli repulsion between the reactants (combined ASM and EDA). The catalytic ring formation strategy demonstrated broad functional group tolerance including of esters, nitriles, heterocycles, alkenes, and alkynes, and catalyst loading can be lowered down to 2 mol% in a multi-gram scale synthesis. The hydroquinazoline aza-Michael reaction products were stable towards a series of late-stage structural derivatizations thus demonstrating relevance to pharmaceutical development.Author contributions
D. J. D., G. S. and C. J. T. designed the experiments. G. S. performed all the experiments. T. A. H. designed and guided the computational work. K. Y. performed the computations. K. E. C. performed the crystallographic study. G. S., K. Y. and T. A. H. prepared the manuscript and all other authors contributed to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. J. T. thanks Bayer AG for generous financial support. K. Y. thanks the Honjo International Scholarship Foundation for funding. D. R. thanks the EPSRC Centre for Doctoral Training in Synthesis for Biology and Medicine (EP/L015838/1) for studentships, generously supported by AstraZeneca, Diamond Light Source, Defence Science and Technology Laboratory, Evotec, GlaxoSmithKline, Janssen, Novartis, Pfizer, Syngenta, Takeda, UCB and Vertex. T. A. H. thanks The Netherlands Organization for Scientific Research (NWO) for financial support. All DFT calculations were carried out on the Dutch national e-infrastructure with the support of SURF Cooperative.Notes and references
- (a) W. Shagufta and I. Ahmad, Med. Chem. Commun., 2017, 8, 871–885 RSC; (b) B. K. Tiwary, K. Pradhan, A. K. Nanda and R. Chakraborty, J. Chem. Biol. Ther., 2015, 1, 104 Search PubMed; (c) S. Patterson, M. S. Alphey, D. C. Jones, E. J. Shanks, I. P. Street, J. A. Frearson, P. G. Wyatt, I. H. Gilbert and A. H. Fairlamb, J. Med. Chem., 2011, 54, 6514–6530 CrossRef CAS; (d) K. Hemalatha and G. Madhumitha, Eur. J. Med. Chem., 2016, 123, 596–630 CrossRef CAS PubMed; (e) P. Thanigaimalai, K.-C. Lee, S.-C. Bang, J.-H. Lee, C.-Y. Yun, E. Roh, B.-Y. Hwang, Y. Kim and S.-H. Jung, Bioorg. Med. Chem., 2010, 18, 1555–1562 CrossRef CAS; (f) P. Thanigaimalai, V. K. Sharma, K.-C. Lee, C.-Y. Yun, Y. Kim and S.-H. Jung, Bioorg. Med. Chem. Lett., 2010, 20, 4771–4773 CrossRef CAS PubMed; (g) H. Hasegawa, M. Muraoka, K. Matsui and A. Kojima, Bioorg. Med. Chem. Lett., 2003, 13, 3471–3475 CrossRef CAS; (h) P. Bernardelli, E. Lorthiois, F. Vergne, C. Oliveira, A.-K. Mafroud, E. Proust, N. Pham, P. Ducrot, F. Moreau, M. Idrissi, A. Tertre, B. Bertin, M. Coupe, E. Chevalier, A. Descours, F. Berlioz-Seux, P. Berna and M. Li, Bioorg. Med. Chem. Lett., 2004, 14, 4627–4631 CrossRef CAS PubMed; (i) N. M. A. Gawad, H. H. Georgey, R. M. Youssef and N. A. El-Sayed, Eur. J. Med. Chem., 2010, 45, 6058–6067 CrossRef; (j) J. S. Byun, J. M. Sohn, D. G. Leem, B. Park, J. H. Nam, D. H. Shin, J. S. Shin, H. J. Kim, K.-T. Lee and J. Y. Lee, Bioorg. Med. Chem. Lett., 2016, 26, 1073–1079 CrossRef CAS PubMed; (k) S. Patterson, M. S. Alphey, D. C. Jones, E. J. Shanks, I. P. Street, J. A. Frearson, P. G. Wyatt, I. H. Gilbert and A. H. Fairlamb, J. Med. Chem., 2011, 54, 6514–6530 CrossRef CAS PubMed; (l) K.-A. S. Schlegel, Z.-Q. Yang, T. S. Reger, Y. Shu, R. Cube, K. E. Rittle, P. Bondiskey, M. G. Bock, G. D. Hartman, C. Tang, J. Ballard, Y. Kuo, T. Prueksaritanont, C. E. Nuss, S. M. Doran, S. V. Fox, S. L. Garson, R. L. Kraus, Y. Li, V. N. Uebele, J. J. Renger and J. C. Barrow, Bioorg. Med. Chem. Lett., 2010, 20, 5147–5152 CrossRef CAS PubMed.
- (a) R. W. King, R. M. Klabe, C. D. Reid and S. K. Erickson-Viitanen, Antimicrob. Agents Chemother., 2002, 46, 1640–1646 CrossRef CAS PubMed; (b) J. W. Corbett, S. S. Ko, J. D. Rodgers, S. Jeffrey, L. T. Bacheler, R. M. Klabe, S. Diamond, C.-M. Lai, S. R. Rabel, J. A. Saye, S. P. Adams, G. L. Trainor, P. S. Anderson and S. K. Erickson-Viitanen, Antimicrob. Agents Chemother., 1999, 43, 2893–2897 CrossRef CAS PubMed; (c) J. W. Corbett, S. S. Ko, J. D. Rodgers, L. A. Gearhart, N. A. Magnus, L. T. Bacheler, S. Diamond, S. Jeffrey, R. M. Klabe, B. C. Cordova, S. Garber, K. Logue, G. L. Trainor, P. S. Anderson and S. K. Erickson-Viitanen, J. Med. Chem., 2000, 43, 2019–2030 CrossRef CAS PubMed.
- W. J. Li, Q. Li, D. L. Liu and M. W. Ding, J. Agric. Food Chem., 2013, 61, 1419–1426 CrossRef CAS PubMed.
- (a) P. S. Verghese and M. R. Schleiss, Drugs Future, 2013, 38, 291–298 CrossRef PubMed; (b) T. Goldner, G. Hewlett, N. Ettischer, H. Ruebsamen-Schaeff, H. Zimmermann and P. Lischka, J. Virol., 2011, 85, 10884–10893 CrossRef CAS PubMed.
- (a) V. K. Pandey, M. A. Kumar and N. Trivedi, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2008, 47B, 1910–1914 CAS; (b) W. Seitz, H. Geneste, G. Backfisch, J. Delzer, C. Graef, W. Hornberger, A. Kling, T. Subkowski and N. Zimmermann, Bioorg. Med. Chem. Lett., 2008, 18, 527–531 CrossRef CAS PubMed; (c) H. Rhim, Y. S. Lee, S. J. Park, B. Y. Chung and J. Y. Lee, Bioorg. Med. Chem. Lett., 2005, 15, 283–286 CrossRef CAS PubMed; (d) T. Xie, Y. Xiao, S. Zhao, X.-Q. Hu and P.-F. Xu, J. Org. Chem., 2016, 81, 10499–10505 CrossRef CAS PubMed; (e) J. Willwacher, S. Rakshit and F. Glorius, Org. Biomol. Chem., 2011, 9, 4736–4740 RSC; (f) Z. Xin, Z. Pei, T. V. Geldem and M. Jirousek, Tetrahedron Lett., 2000, 41, 1147–1150 CrossRef CAS; (g) Y. S. Lee, B. H. Lee, S. J. Park, S. B. Kang, H. Rhim, J.-Y. Park, J.-H. Lee, S.-W. Jeong and J. Y. Lee, Bioorg. Med. Chem. Lett., 2004, 14, 3379–3384 CrossRef CAS PubMed; (h) A. V. Ivachtchenko, S. M. Kovalenko and O. G. Drushlyak, J. Comb. Chem., 2003, 5, 775–788 CrossRef CAS; (i) S. Fukamachi, H. Konishi and K. Kobayashi, Synthesis, 2010, 10, 1593–1598 Search PubMed; (j) K. Kobayashi, Y. Yokoi and H. Konishi, Synthesis, 2011, 10, 1526–1528 CrossRef; (k) B. Kaur and R. Kaur, ARKIVOC, 2007, 15, 315–323 Search PubMed.
- Y. Kita, K. Higashida, K. Yamaji, A. Iimuro and K. Mashima, Chem. Commun., 2015, 51, 4380–4382 RSC.
- P. Wang, M. Shen, T. Wang, H. Lin and L. Gong, Angew. Chem., Int. Ed., 2017, 56, 16032–16036 ( Angew.Chem. , 2017 , 129 , 16248–16252 ) CrossRef CAS PubMed.
- (a) N. A. Magnus, P. N. Confalone and L. Storace, Tetrahedron Lett., 2000, 41, 3015–3019 CrossRef CAS; (b) G. S. Kauffman, G. D. Harris, R. L. Dorow, B. R. P. Stone, R. L. Parsons, J. A. Pesti, N. A. Magnus, J. M. Fortunak, P. N. Confalone and W. A. Nugent, Org. Lett., 2000, 2, 3119–3121 CrossRef CAS PubMed; (c) B. Jiang and Y. G. Si, Angew. Chem., Int. Ed., 2004, 43, 216–218 CrossRef CAS PubMed; (d) F.-G. Zhang, H. Ma, J. Nie, Y. Zheng, Q. Gao and J. A. Ma, Adv. Synth. Catal., 2012, 354, 1422–1428 CrossRef CAS.
- B. Jiang, J. J. Dong, Y. G. Si, X. L. Zhao, Z. G. Huang and M. Xu, Adv. Synth. Catal., 2008, 350, 1360–1366 CrossRef CAS.
- H. Xie, Y. Zhang, S. Zhang, X. Chen and W. Wang, Angew. Chem., Int. Ed., 2011, 50, 11773–11776 CrossRef CAS PubMed.
- (a) H. N. Yuan, S. Wang, J. Nie, W. Meng, Q. Yao and J. A. Ma, Angew. Chem., Int. Ed., 2013, 52, 3869–3873 CrossRef CAS PubMed; (b) H. N. Yuan, S. Li, J. Nie, Y. Zheng and J. A. Ma, Chem. Eur. J., 2013, 19, 15856–15860 CrossRef CAS.
- F. G. Zhang, X. Y. Zhu, S. Li, J. Nie and J. A. Ma, Chem. Commun., 2012, 48, 11552–11554 RSC.
- D. Zhou, X. Yu, J. Zhang, W. Wang and H. Xie, Org. Biomol. Chem., 2016, 14, 6193–6196 RSC.
- M. X. Zhao, H. L. Bi, R. H. Jiang, X. W. Xu and M. Shi, Org. Lett., 2014, 16, 4566–4569 CrossRef CAS PubMed.
- S. Li and J. Ma, Chem. Soc. Rev., 2015, 44, 7439–7448 RSC.
- For reviews on enantioselective aza-Michael reactions, see: (a) M. G. Vinogradov, O. V. Turova and S. G. Zlotin, Org. Biomol. Chem., 2019, 17, 3670–3708 RSC; (b) M. Sánchez-Rosellò, J. L. Aceña, A. Simón-Fuentesa and C. del Pozo, Chem. Soc. Rev., 2014, 43, 7430–7453 RSC; (c) S. Nayak, P. Panda, S. Bhakta, S. K. Mishra and S. Mohapatra, RSC Adv., 2016, 6, 96154–96175 RSC; (d) C. Bhanja, S. Jena, S. Nayak and S. Mohapatra, Beilstein J. Org. Chem., 2012, 8, 1668–1694 CrossRef CAS PubMed; (e) D. Enders, C. Wang and J. X. Liebich, Chem. Eur. J., 2009, 15, 11058–11076 CrossRef CAS PubMed; (f) J. Wang, P. Li, P. Y. Choy, A. S. C. Chan and F. Y. Kwong, ChemCatChem, 2012, 4, 917–925 CrossRef CAS; (g) P. R. Krishna, A. Sreeshailam and R. Srinivas, Tetrahedron, 2009, 65, 9657–9672 CrossRef CAS; (h) E. Reyes, M. Fernández, U. Uría, J. L. Vicario, D. Badía and L. Carrillo, Curr. Org. Chem., 2012, 16, 521–546 CrossRef CAS; (i) L. Xu and C. Xia, Eur. J. Org. Chem., 2005, 4, 633–639 CrossRef.
- D. S. Allgäuer, H. Jangra, H. Asahara, Z. Li, Q. Chen, H. Zipse, A. R. Ofial and H. Mayr, J. Am. Chem. Soc., 2017, 139, 13318–13329 CrossRef.
- For intermolecular examples, see: (a) G. Sundararajan and N. Prabagaran, Org. Lett., 2001, 3, 389–392 CrossRef CAS PubMed; (b) Y. Lin, W. J. Hirschi, A. Kunadia, A. Paul, I. Ghiviriga, K. A. Abboud, R. W. Karugu, M. J. Vetticatt, J. S. Hirschi and D. Seidel, J. Am. Chem. Soc., 2020, 142, 5627–5635 CrossRef CAS PubMed.
- G. R. Humphrey, S. M. Dalby, T. Andreani, B. Xiang, M. R. Luzung, Z. J. Song, M. Shevlin, M. Christensen, K. M. Belyk and D. M. Tschaen, Org. Process Res. Dev., 2016, 20, 1097–1103 CrossRef CAS.
- C. K. Chung, Z. Liu, K. W. Lexa, T. Andreani, Y. Xu, Y. Ji, D. A. DiRocco, G. R. Humphrey and R. T. Ruck, J. Am. Chem. Soc., 2017, 139, 10637–10640 CrossRef CAS.
- M. Formica, D. Rozsar, G. Su, A. J. M. Farley and D. J. Dixon, Acc. Chem. Res., 2020, 53, 2235–2247 CrossRef CAS PubMed and references cited therein.
- (a) J. P. Malerich, K. Hagihara and V. H. Rawal, J. Am. Chem. Soc., 2008, 130, 14416–14417 CrossRef CAS PubMed; (b) H. Konishi, T. Y. Lam, J. P. Malerich and V. H. Rawal, Org. Lett., 2010, 12, 2028–2031 CrossRef CAS PubMed; (c) K. S. Yang, A. E. Nibbs, Y. E. Türkmen and V. H. Rawal, J. Am. Chem. Soc., 2013, 135, 16050–16053 CrossRef CAS PubMed; (d) S. M. Banik, A. Levina, A. M. Hyde and E. N. Jacobsen, Science, 2017, 358, 761–764 CrossRef CAS PubMed; (e) A. E. Wendlandt, P. Vangal and E. N. Jacobsen, Nature, 2018, 556, 447–451 CrossRef CAS PubMed; (f) G. Kardos and T. Soós, Eur. J. Org. Chem., 2013, 21, 4490–4494 CrossRef.
- (a) G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS; (b) C. Fonseca Guerra, J. G. Snijders, G. te Velde and E. J. Baerends, Theor. Chem. Acc., 1998, 99, 391–403 Search PubMed; (c) ADF2018.105, SCM Theoretical Chemistry, Vrije Universiteit Amsterdam, The Netherlands, 2018, http://www.scm.com Search PubMed.
- (a) J. C. Slater, Quantum Theory of Molecules and Solids, McGraw-Hill, New York, 1974 Search PubMed; (b) A. D. Becke, J. Chem. Phys., 1986, 84, 4524–4529 CrossRef CAS; (c) A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- E. van Lenthe and E. J. Baerends, J. Comput. Chem., 2003, 24, 1142–1156 CrossRef CAS PubMed.
- (a) A. Klamt and G. Schüürmann, J. Chem. Soc. Perkin Trans., 1993, 2, 799–805 RSC; (b) A. Klamt, J. Phys. Chem., 1995, 99, 2224–2235 CrossRef CAS; (c) A. Klamt and V. Jonas, J. Chem. Phys., 1996, 105, 9972–9981 CrossRef CAS; (d) C. C. Pye and T. Ziegler, Theor. Chem. Acc., 1999, 101, 396–408 Search PubMed.
- (a) S. Grimme, J. Antony, S. Ehrlich and S. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef; (b) A. D. Becke and E. R. Johnson, J. Chem. Phys., 2005, 123, 154101 CrossRef PubMed.
- (a) E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1993, 99, 4597–4610 CrossRef CAS; (b) E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1994, 101, 9783–9792 CrossRef CAS.
- C. Y. Legault, CYLview20, Unversité de Sherbrooke, Sherbrooke, 2020, http://www.cylview.org Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- (a) J. C. Golec, E. M. Carter, J. W. Ward, W. G. Whittingham, L. Simon, R. S. Paton and D. J. Dixon, Angew. Chem., Int. Ed., 2020, 59, 17417–17422 CrossRef CAS PubMed; (b) M. Formica, G. Sorin, A. J. M. Farley, J. Díaz, R. S. Paton and D. J. Dixon, Chem. Sci., 2018, 9, 6969–6974 RSC; (c) H. Shi, I. N. Michaelides, B. Darses, P. Jakubec, Q. N. N. Nguyen, R. S. Paton and D. J. Dixon, J. Am. Chem. Soc., 2017, 139, 17755–17758 CrossRef CAS PubMed; (d) B. Kótai, G. Kardos, A. Hamza, V. Farkas, I. Pápai and T. Soós, Chem. Eur. J., 2014, 20, 5631–5639 CrossRef PubMed.
- Low temperature single crystal X-ray diffraction data for 2j was collected using a Rigaku Oxford SuperNova diffractometer. Raw frame data were reduced using CrysAlisPro and the structures were solved using ‘Superflip’ [L. Palatinus and G. Chapuis, J. Appl. Cryst., 2007, 40, 786–790.] before refinement with CRYSTALS. (a) P. Parois, R. I. Cooper and A. L. Thompson, Chem. Cent. J., 2015, 9, 30 CrossRef PubMed; (b) R. I. Cooper, A. L. Thompson and D. J. Watkin, J. Appl. Cryst., 2010, 43, 1100–1107 CrossRef CAS ; as per the ESI† (CIF).
- (a) S. C. C. van der Lubbe and C. F. Guerra, Chem.–Asian J., 2019, 14, 2760–2769 CAS; (b) S. Tsuzuki, K. Honda, T. Uchimaru, M. Mikami and K. Tanabe, J. Am. Chem. Soc., 2000, 122, 3746–3753 CrossRef CAS; (c) M. Nishio, Phys. Chem. Chem. Phys., 2011, 13, 13873–13900 RSC.
- (a) E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed; (b) J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef PubMed.
- (a) P. Vermeeren, S. C. C. van der Lubbe, C. Fonseca Guerra, F. M. Bickelhaupt and T. A. Hamlin, Nat. Protoc., 2020, 15, 649–667 CrossRef CAS PubMed; (b) F. M. Bickelhaupt and K. N. Houk, Angew. Chem., Int. Ed., 2017, 56, 10070–10086 CrossRef CAS PubMed; (c) L. P. Wolters and F. M. Bickelhaupt, WIRES Comput. Mol. Sci., 2015, 5, 324–343 CrossRef CAS PubMed; (d) I. Fernández and F. M. Bickelhaupt, Chem. Soc. Rev., 2014, 43, 4953–4967 RSC; (e) W.-J. van Zeist and F. M. Bickelhaupt, Org. Biomol. Chem., 2010, 8, 3118–3127 RSC.
- F. M. Bickelhaupt and E. J. Baerends, in Reviews in Computational Chemistry, ed. K. B. Lipkowitz and D. B. Boyd, Wiley, Hoboken, 2000, pp. 1–86 Search PubMed.
- (a) T. A. Hamlin, I. Fernandez and F. M. Bickelhaupt, Angew. Chem., Int. Ed., 2019, 58, 8922–8926 CrossRef CAS PubMed; (b) P. Vermeeren, T. A. Hamlin, I. Fernandez and F. M. Bickelhaupt, Angew. Chem., Int. Ed., 2020, 59, 6201–6206 CrossRef CAS; (c) P. Vermeeren, F. Brinkhuis, T. A. Hamlin and F. M. Bickelhaupt, Chem.–Asian J., 2020, 15, 1167–1174 CrossRef CAS PubMed; (d) P. Vermeeren, T. A. Hamlin, I. Fernandez and F. M. Bickelhaupt, Chem. Eur J., DOI:10.1002/chem.202004496; (e) P. Vermeeren, M. Dalla Tiezza, M. van Dongen, I. Fernandez, F. M. Bickelhaupt and T. A. Hamlin, Chem. Eur J., DOI:10.1002/chem.202100522R2.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2054508. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc00856k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |