
Self-healing elastomers based on conjugated diolefins: a review
Prasanta Kumar
Behera
 ,
Subhra
Mohanty
and
Virendra Kumar
Gupta
*
,
Subhra
Mohanty
and
Virendra Kumar
Gupta
*
Polymer Synthesis & Catalysis Group, Reliance Research and Development Center, Reliance Industries Limited, Navi Mumbai 400701, India. E-mail: Virendrakumar.Gupta@ril.com
First published on 1st March 2021
Abstract
The formation of microcracks is a common problem in elastomeric materials. The propagation of microcracks to bigger cracks results in the failure of the elastomeric material, which needs to be replaced by a new material, thus creating a huge elastomer waste. The introduction of self-healing property into the elastomeric material is an important way to repair microcracks and to stop the crack propagation process. This review summarizes the progress in self-healing elastomers (polybutadiene rubber, styrene–butadiene rubber, polyisoprene rubber, butyl rubber, nitrile butadiene rubber, chloroprene rubber, and silicon rubber) based on conjugated diolefins. The use of various self-healing mechanisms and their effect on the self-healing property as well as reprocessability has been discussed. The challenges and future research opportunities are highlighted.
 Prasanta Kumar Behera | Dr Prasanta Kumar Behera received his MTech degree in Rubber Technology and PhD in Polymer Chemistry from Indian Institute of Technology Kharagpur, India. He is currently working as Research Scientist in Research & Development, Reliance Industries Limited. His area of research primarily focuses on the synthesis of functional polymers and their applications in designing self-healing/shape-memory materials and superhydrophobic coatings. |
 Subhra Mohanty | Dr Subhra Mohanty is currently working as Asst. Vice President in Research and Development of Reliance Industries. She acquired her PhD degree in Rubber Technology from Indian Institute of Technology, Kharagpur, India. She has about 25 peer reviewed publications and 15 patents to her credit. Her research interests are mostly on various functional polymer synthesis and the development of new applications. |
 Virendra Kumar Gupta | Dr Virendra Kumar Gupta is currently Head, R&D Polymer, Reliance Industries Limited. He received his Ph.D. in Chemistry from Banaras Hindu University, Varanasi. His current research interests include material science, circular economy, and sustainability. He has 160 patents, 75 research publications and 80 presentations in conferences. He has successfully commercialized 25 technologies in the area of polymers. |
1. Introduction
In today's society, rubbers (elastomers) have been used in various fields such as tires, shoes, seals, and many elastomeric products.1–5 The rise in the demand for rubber products drives the increased production of rubber materials. When the rubber material gets damaged, it needs to be replaced and reprocessed.6–8 As rubber materials are permanently crosslinked, it is difficult to reprocess and decompose thermally at high temperatures (400–700 °C).9–11 The growing demand for rubber products and the problem in their reprocessability cause huge rubber wastes.12 Thus, it is a big challenge for the research community to come up with a solution to this problem.The solution to the above problem can be achieved in two ways. The first solution is to make the crosslinked rubber reprocessable and the second one is to improve the application life by making them self-healable.13,14 Making the rubber materials self-healable is a better way to overcome the hurdle as the damaged rubber article does not need to be replaced and remoulded (reprocessed). In recent years, the significant effort by the research community for the development of self-healing rubber gives a new hope to overcome the technological and environmental issues associated with crosslinked rubber wastes.15 Making rubber self-healable will improve its application life and hence reduce rubber waste. When a crack is generated in the self-healing rubber, it repairs the crack by itself or by application of external stimuli.16,17 Thus, there is no need to replace the self-healing rubber again and again.
Most rubber-based wastes are generated from tire and automotive applications.18 Conjugated diolefin based elastomers such as polybutadiene (PB, BR, butadiene rubber), styrene–butadiene rubber (SBR), polyisoprene rubber (IR) (natural rubber, NR), butyl rubber (IIR), nitrile butadiene rubber (NBR), and chloroprene rubber (CR) are widely used in tire, tube, and other automotive applications.19–22 The lifetime of tire and other rubber articles can be improved by making these rubbers self-healable. Most self-healing polymeric systems are based on dynamic covalent networks or supramolecular interactions.23,24 The presence of a dynamic network further helps in the reprocessing of rubber materials. Thus, it is important to review the self-healing systems based on PB, SBR, NR, IIR, NBR, and CR, which will help in the design of rubber-based products with interesting self-healing and reprocessable properties. In this review, various self-healing approaches used in the conjugated diolefin based elastomeric system are discussed. The effect of different self-healing mechanisms on the self-healing efficiency and recyclability have been described.
2. Various types of self-healing approaches
The self-healing approaches in polymers can be broadly classified into two categories, i.e., extrinsic and intrinsic approaches (Fig. 1).25 In the extrinsic self-healing approach, the polymer itself does not exhibit self-healing property. Thus, external healing agents are incorporated to induce the self-healing property in the polymer.26–28 In the intrinsic self-healing system, the polymer does not need any external healing agent. The reversible interaction between the functional groups present in the polymer is the reason behind the self-healing property.29–31 The functional groups in intrinsic self-healing polymers interact through reversible non-covalent interactions (hydrogen bonding,32,33 ionic interactions,34–36 π–π interactions,37,38 coordination bond,39 and host–guest interactions40) or reversible (dynamic) covalent bond formation (e.g., Diels Alder & retro-DA reaction,41–49 Alder–ene reaction,50,51 reversible electrophilic substitution,52 reversible cyclodimerization,53–55 disulfide metathesis reaction,56–58 and boronate ester bearing B–N dative bond59). Depending on the nature of the healing agent or the functional moieties present in it, the extrinsic and intrinsic approaches may be autonomous or non-autonomous (Fig. 2).60–62 Autonomous self-healing means it does not need an external stimuli, while the non-autonomous self-healing system needs external stimuli to show the self-healing property.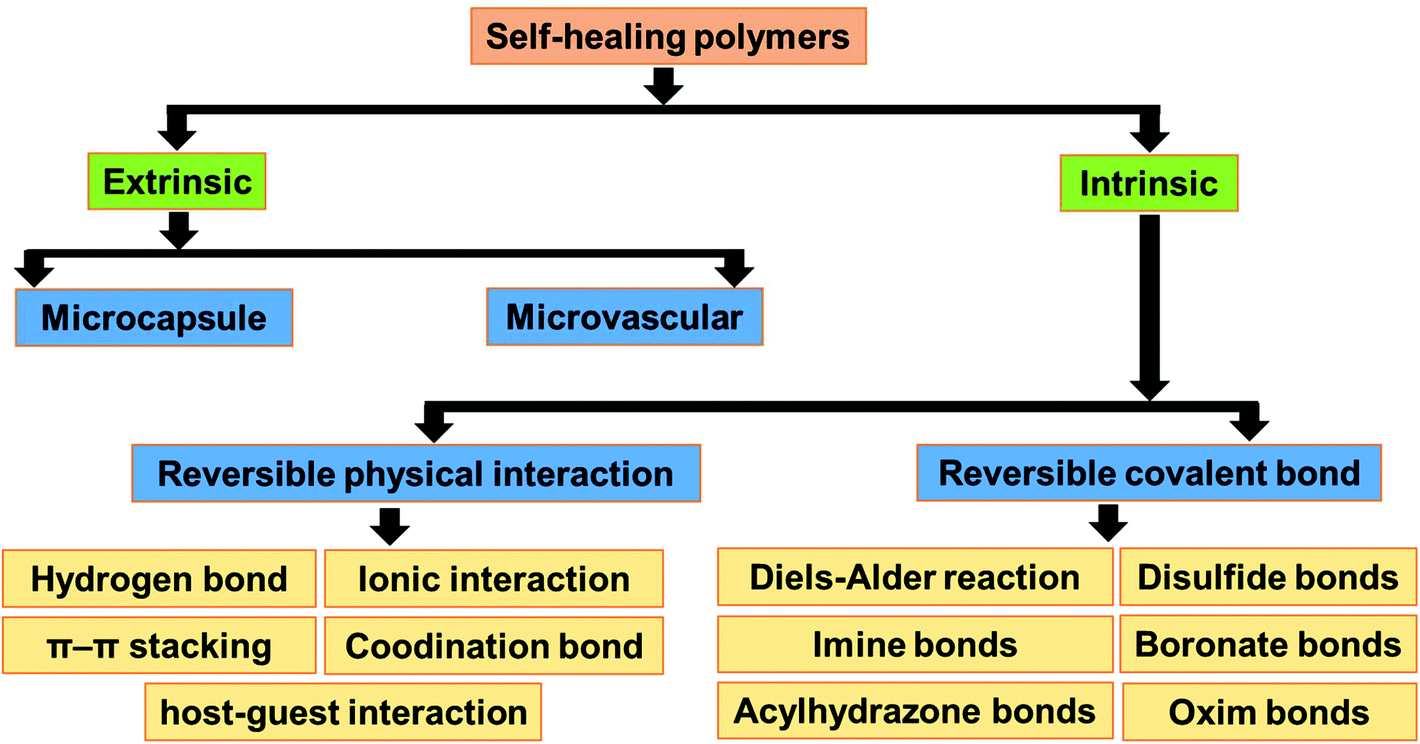 | ||
| Fig. 1 Classification of self-healing polymers based on the mechanism of the healing process. | ||
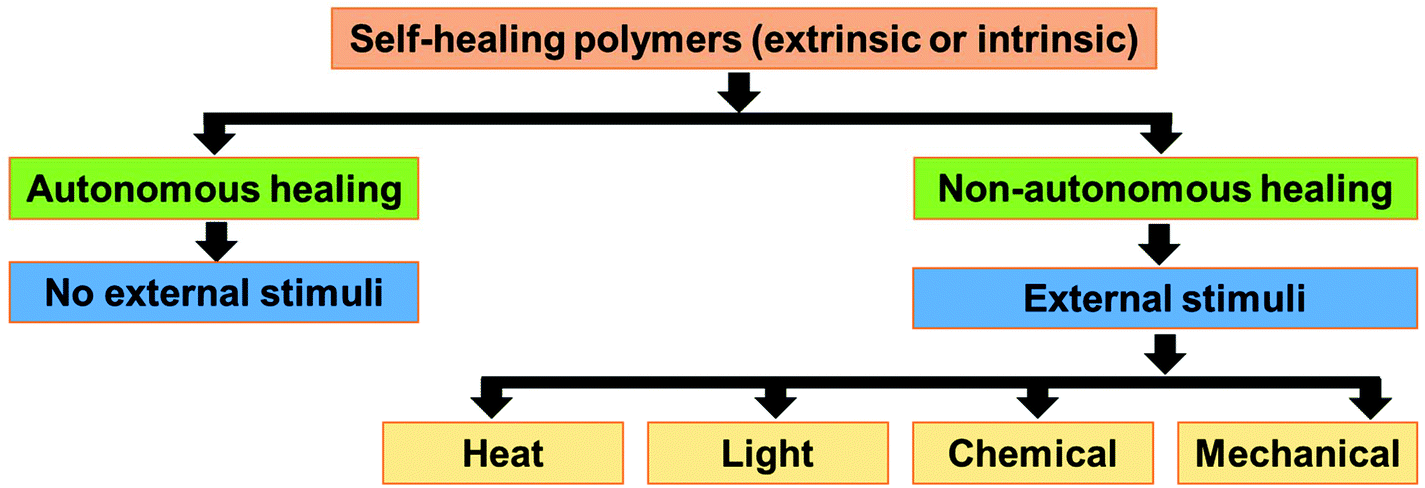 | ||
| Fig. 2 Classification of self-healing polymers based on the stimuli required for the healing process. | ||
3. Self-healing polybutadiene (PB)
The presence of the main chain and pendant double bonds in PB makes it a suitable rubber for further modification. Different functional groups are incorporated into the PB rubber via reaction with the pendant as well as the main chain double bond.63–66 The grafting of suitable functional groups onto PB may help in showing its self-healing property.3.1. Self-healing PB via Diels–Alder (DA) and retro Diels–Alder (DA) reactions
DA and rDA reactions are the most versatile mechanisms to develop self-healing polymers (Fig. 3). A diene and a dienophile are needed to prepare the self-healing PB via DA and rDA approach. Dienes or dienophiles are grafted onto the PB chain and then the crosslinking reaction is performed via the DA reaction. The covalent crosslinks formed via the DA reaction break at high temperature (110–160 °C) and the formation of DA adducts takes place when the temperature is reduced to 55–75 °C. When cracks appear, the rubber is heated above the rDA reaction temperature, which leads to the breaking of DA adducts. The further reduction of the temperature to 55–75 °C results in the formation of DA adducts and heals the cracks. | ||
| Fig. 3 Schematic representation of Diels–Alder (DA) and retro Diels–Alder (rDA) reaction in furan and maleimide systems. | ||
Polybutadienes can be easily functionalized via the thiol–ene reaction using furfuryl thiol. In comparison to the cis/trans units, the vinyl groups are more reactive toward the thiol–ene reaction.67 Trovatti et al.68 reported the preparation of a crosslinked PB rubber using the DA reaction and claimed that this strategy can be utilized to develop a recyclable tire. First, the furan-functionalized PB is prepared via the thiol–ene reaction and then crosslinked via the DA reaction using bismaleimide as a dienophile. On heating above 110 °C, the crosslinked PB undergoes decrosslinking via the rDA reaction. Bai et al.69 studied the recyclability and self-healing property of PB rubber prepared by the DA reaction between furan-functionalized PB and bismaleimide (Fig. 4). The crosslinked elastomer exhibits good recyclability and can be thermally remoulded to produce a new sample without a significant impact on the mechanical properties. The crack in the crosslinked rubber starts to decrease at 90 °C and completely disappears when the temperature reaches above 150 °C. This indicates that the crosslinked PB rubber shows self-healing behavior at high temperature.
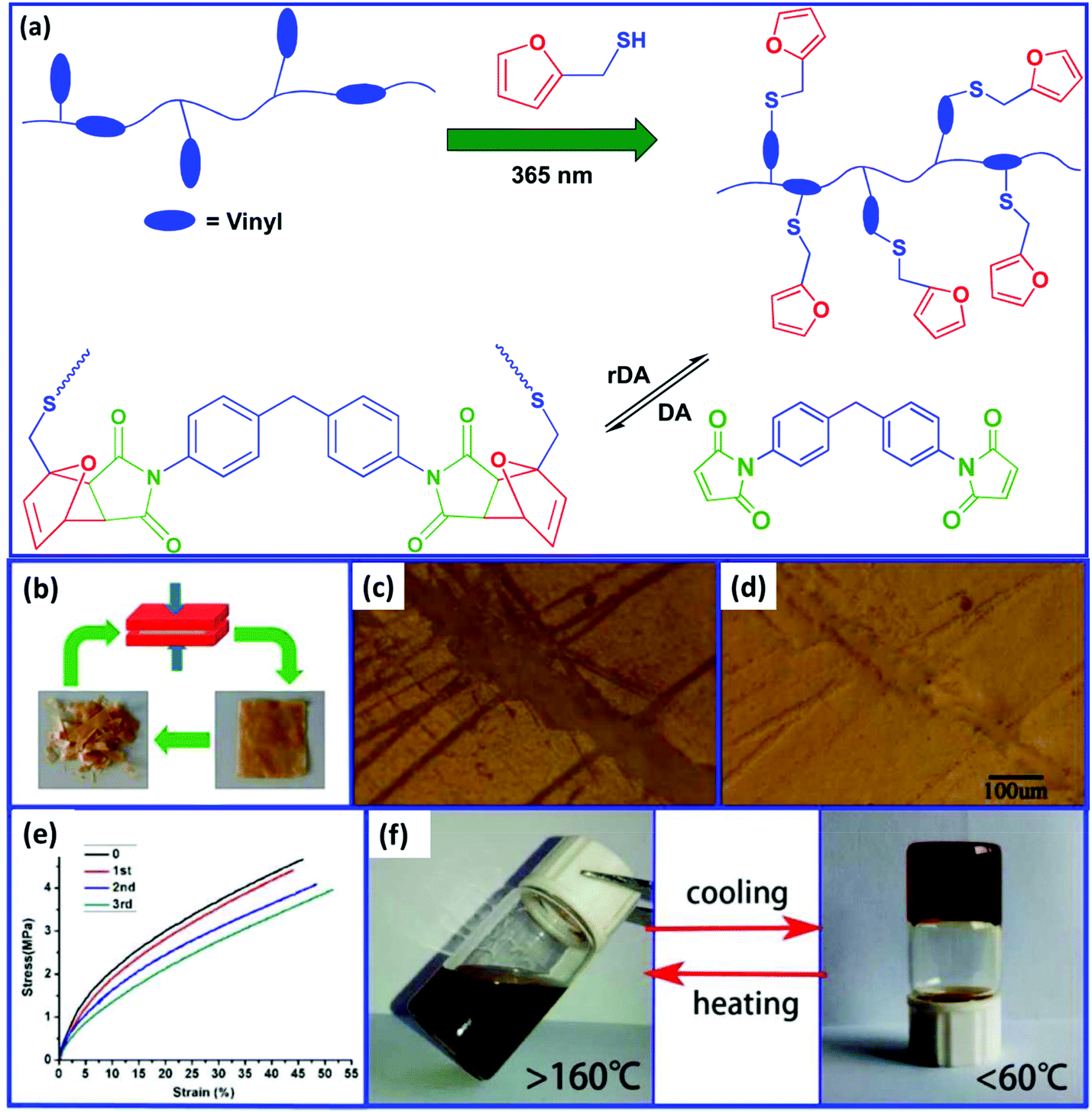 | ||
| Fig. 4 (a) Schematic representation for the preparation of the recyclable PB rubber, (b) recyclability study, (c) the photograph of the scratched film at room temperature, (d) the photograph of the film after healing at 150 °C, (e) stress–strain curves of the pristine and the recycled samples, and (f) the sol–gel process in dichlorobenzene at different temperatures.69 Reproduced from ref. 69 with permission from American Chemical Society, copyright 2015. | ||
Besides the thiol–ene reaction, other alternative methods have also been reported to incorporate DA moieties into PB rubber. Berto et al.70 reported a three-step method to prepare a tetrafuran telechelic cis-1,4-PB rubber (Fig. 5). They first prepared the epoxidized PB rubber via the epoxidation of PB using m-CPBA (meta-chloroperoxybenzoic acid), which on treatment with periodic acid gave the aldehyde telechelic PB rubber with different chain lengths (low molecular weight). The aldehyde telechelic PB is then functionalized with diethanolamine (DEA) to prepare tetrahydroxyl telechelic PB, which on further reaction with furfuryl isocyanate, give a tetrafuran-functionalized telechelic PB. The reaction of the liquid-like furan-functionalized PB with bismaleimide via DA chemistry results in the formation of elastomeric networks. The tensile strength of the DA crosslinked product is highly dependent on the molecular weight of the furan telechelic PB. Low molecular weight oligomers exhibit higher tensile strength and lower elongation at break, which is due to the increased crosslink density. Interestingly, these materials can be recycled over five times without a loss in their mechanical properties.
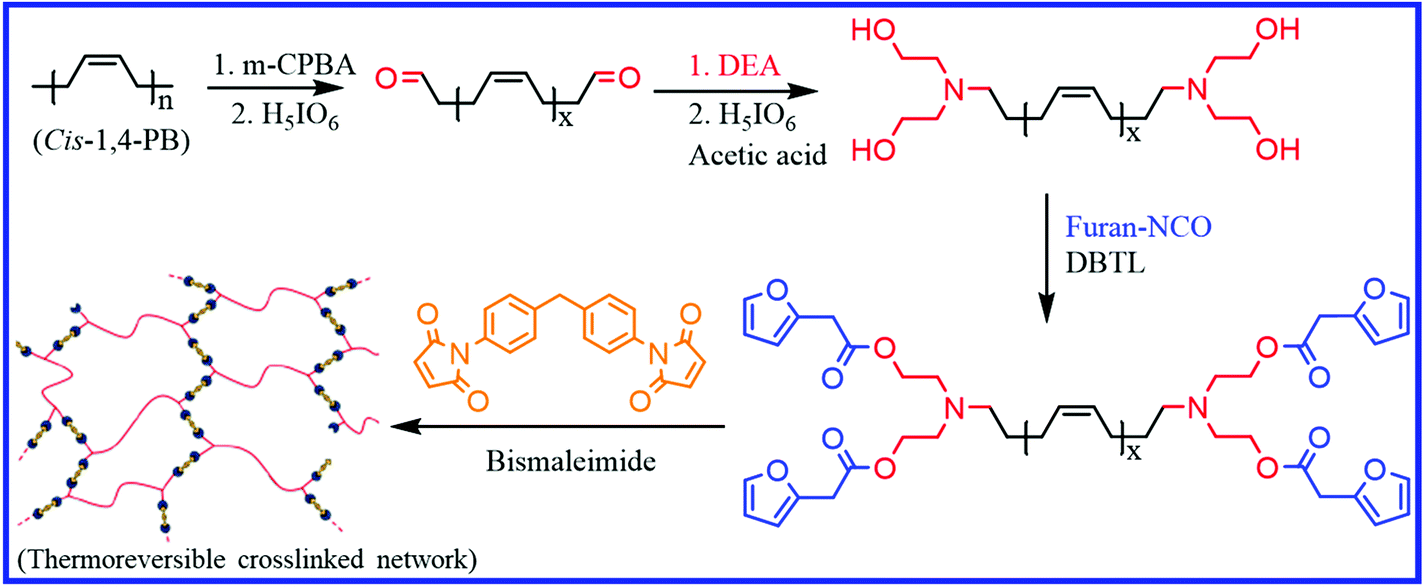 | ||
| Fig. 5 Schematic representation of the preparation of furan telechelic PB rubber and its DA adduct.70 Reproduced from ref. 70 with permission from American Chemical Society, copyright 2018. | ||
3.2. Self-healing PB via disulfide metathesis reaction
Similar to DA and rDA click reactions, the disulfide metathesis reaction is also based on a dynamic covalent bond, as shown in Fig. 6. The disulfide moieties present in rubber can undergo disulfide metathesis reaction at room temperature or under external stimuli (temperature or UV light). The exchange of polymer segments via the disulfide metathesis reaction results in the self-healing property.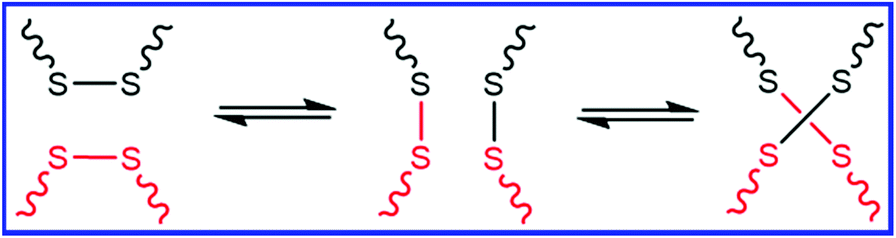 | ||
| Fig. 6 Schematic representation of the disulfide metathesis reaction. | ||
Xiang et al.71 reported the preparation of a self-healable and reprocessable PB rubber by thiol–ene photo-click polymerization between the double bonds in PB and thiols in polysulfide (Fig. 7). Under UV irradiation, the rearrangement of the crosslinked network takes place via the disulfide metathesis reaction, which induces the self-healing property in the rubber. The healing efficiency increases with an increase in the content of disulfide bonds. The increase in the intensity of the UV light and the time of irradiation also improves the healing efficiency. The conventional sulfur-crosslinked PB rubber can also be made self-healable and recyclable by triggering the disulfide rearrangement reaction with a suitable catalyst. Xiang et al.72 reported the use of CuCl2 to catalyze the disulfide metathesis reaction in sulfur-crosslinked PB rubber. The catalyst is incorporated in the rubber during compounding, which accelerates the disulfide exchange reaction. The sulfur-crosslinked PB with 0.1 phr of CuCl2 shows a healing efficiency of 75% when healed at 110 °C for 12 h, whereas the sulfur-crosslinked PB without CuCl2 exhibits a healing efficiency of only 12%. This indicates that CuCl2 catalyzes disulfide metathesis through a circulated crossover reaction between the disulfide and polysulfide bonds. Interestingly, the incorporation of CuCl2 also helps in the reprocessing of sulfur-crosslinked rubber.
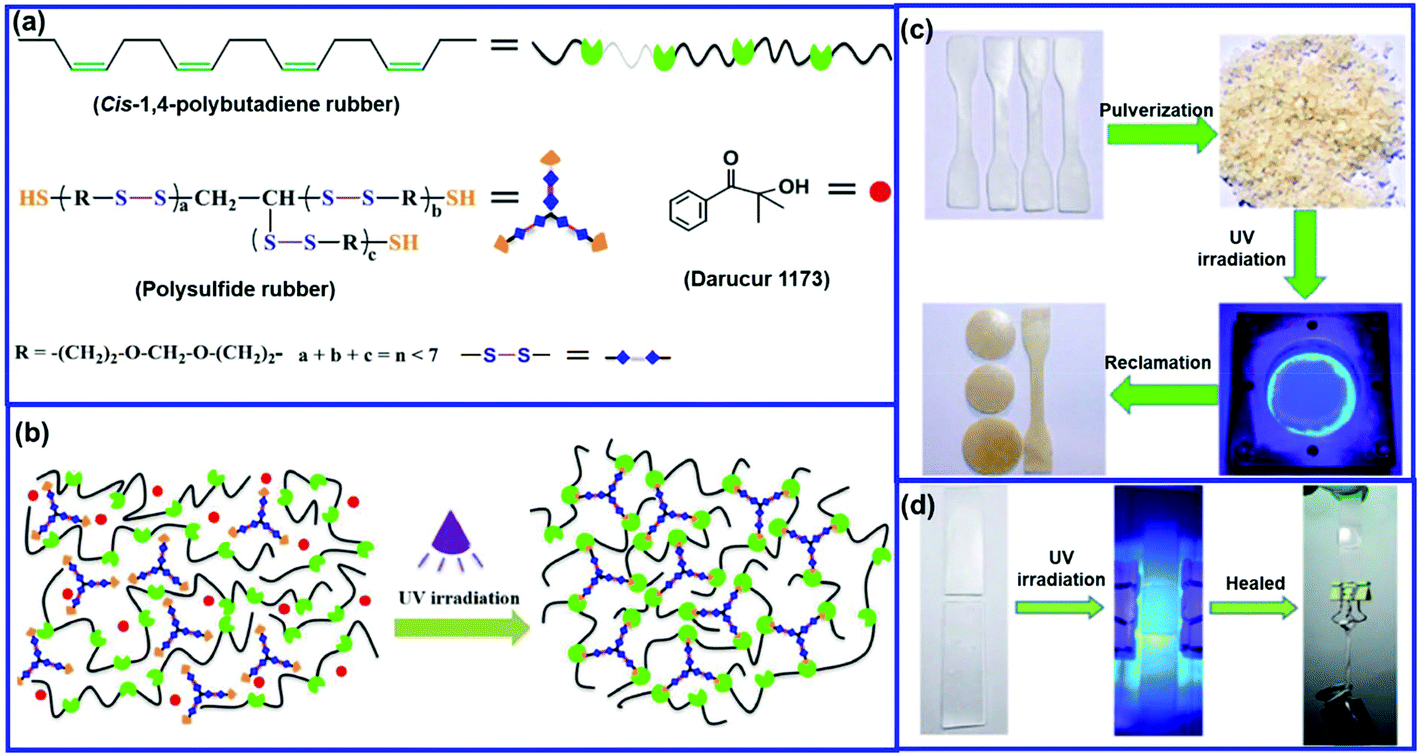 | ||
| Fig. 7 (a) Chemical structures of the main components of UV-curable PB rubber, (b) photocrosslinking process in the UV-curable rubber, (c) reprocessing of the cured rubber, and (d) photographs illustrating the healing processes of the UV-cured rubbers.71 Reproduced from ref. 71 with permission from Elsevier, copyright 2019. | ||
3.3. Self-healing PB via hydrogen bonding, ionic interaction, and crystallization
Self-healing PB rubber can be prepared by the introduction of functional groups that undergo reversible ionic association, hydrogen bonding, or crystallization. Wang et al.73 studied the effect of the combination of covalent and non-covalent crosslinks on the self-healing, shape-memory, and mechanical properties of PB rubber. They prepared the self-healing PB elastomer via the self-assembly of PB oligomers having carboxylic acid and amine groups, followed by covalent crosslinking via the thiol–ene reaction (using a tri-functional thiol compound) (Fig. 8). The interaction between the carboxylic acid and the amine groups results in the formation of dynamic ionic-hydrogen bonds and induced the self-healing property in the elastomer. At low covalent crosslinking density, these PB show self-healing properties at room temperature. The increase in the covalent crosslink density improves the shape-memory and mechanical properties but reduces the self-healing efficiency. Without a crosslinker, the healing efficiency is about 93% and decreased to 77% and 33% with the increase in the crosslinker concentration to 2% and 6%, respectively. The presence of irreversible covalent crosslinks reduces the chain mobility, which in turn reduces the healing rate and healing efficiency.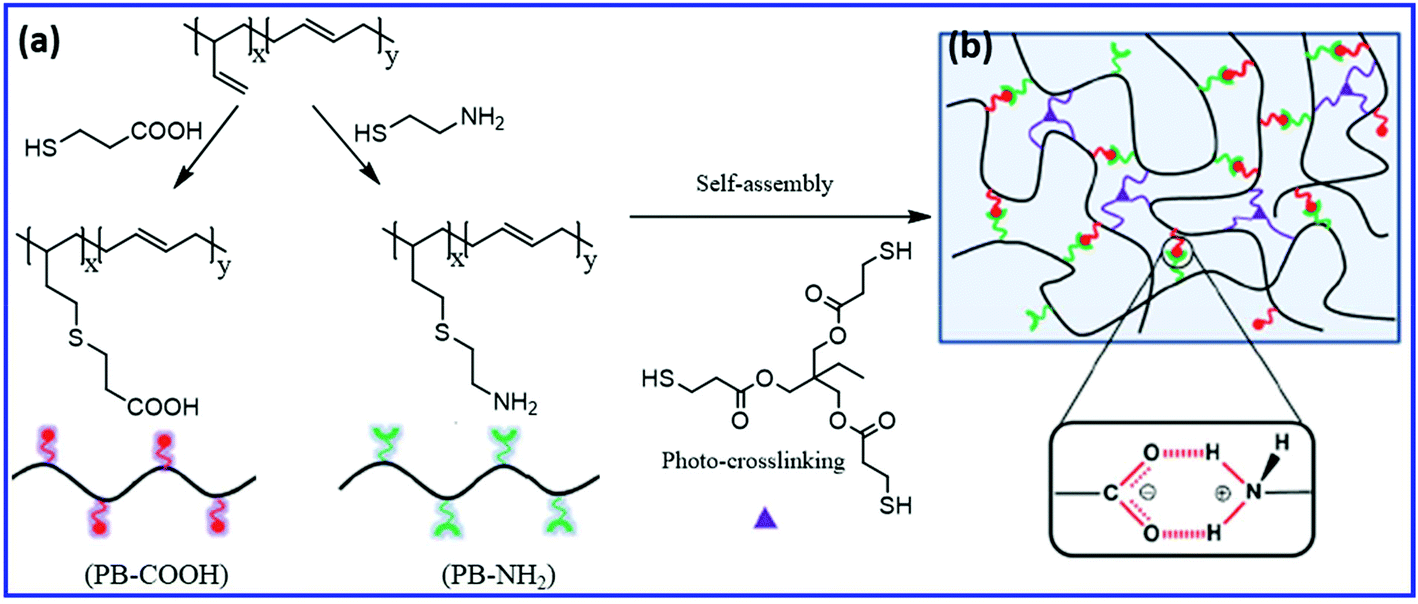 | ||
| Fig. 8 (a) Schematic representation of the preparation of –COOH and –NH2 functionalized PB via the thiol–ene reaction. (b) The polymer network having both ionic hydrogen bonds and covalent cross-links.73 Reproduced from ref. 73 with permission from Royal Society of Chemistry, copyright 2015. | ||
Jasra et al.74 introduced the concept of hydrogen bonding in the preparation of a thermoreversible and self-healing PB rubber blend. They prepared the self-healing PB via blending PB rubber with maleic anhydride-grafted polypropylene, followed by the ring-opening of the maleic anhydride group using triazole derivatives such as 3-amino-1,2,4-triazole and 4-amino-1,2,4-triazole. The carbonyl groups involved in the formation of thermo-reversible hydrogen bonding with amine and carboxylic acid protons which help the blend to show self-healing properties in the temperature range of 40–85 °C.
The grafting of crystallizable hydrocarbons onto PB can impart self-healing property. The crystallizable hydrocarbon side chains in PB will undergo association to form crystalline domains. These crystalline domains will melt on heating and again recrystallize on reducing the temperature. The melting and reformation of crystallites will act as a reversible network and induce self-healing property in the elastomer. Xu et al.75 reported the preparation of thermoresponsive self-healing PB coatings by the grafting of 1H,1H,2H,2H-perfluorodecanethiol (PFDT) onto vinyl pendant groups of 1,2-PB via thiol–ene reaction. PB-g-PFDT exhibits a glass transition at 44 °C and a melting transition at 68 °C. When the scratched coating surface of PB-g-PFDT is healed at 50 °C for 50 min, no significant change in the scratch is observed. Although the chain mobility of PB-g-PFDT is improved above the Tg, the crystallites of PFDT still restrict the migration of the polymer chains to the scratched area. Interestingly, the scratch on this coating completely disappeared when healing was performed at 80 °C for 5 min, followed by cooling down to room temperature. At 80 °C, the melting of the PFDT crystallites occurs and further reduction in temperature results in the reformation of the crystallites, which leads to the complete healing of the scratch. This PB-g-PFDT coating also exhibits anti-smudge property and various liquids (with a wide range of surface tensions) readily slide off the coating surfaces without leaving any residue. In addition to other flat surfaces, PB-g-PFDT can also be used on cotton fabrics to prepare superamphiphobic fabrics that are capable of repetitively healing the damage.
Self-healing properties are of utmost importance in PB rubber as these are mostly used in the sidewall of a tire. In the event of any cut/puncture in the sidewall, the tire is discarded. The inbuilt self-healing properties of PBR is one of the most desired properties in the automotive segment. In this section, self-healing PB has been discussed using various mechanisms such as the DA/rDA reaction, disulfide metathesis reaction, hydrogen bonding, ionic interaction, and side-chain crystallization. Semicrystalline polycaprolactone oligomers can also be grafted onto PB rubber and their impact on the self-healing property could be evaluated. From the multiple literature studies of various methodologies, it is concluded that the functionalization of PB rubber is more suitable from the process prospective, which imparts self-healing characteristics under specific conditions.
4. Self-healing styrene–butadiene rubber (SBR)
SBR is a synthetic rubber comprising styrene (10–25%) and butadiene (75–90%) monomers. The butadiene unit in SBR is approximately composed of 60 to 70% trans-1,4; 15 to 20% cis-1,4; and 15 to 20% 1,2 configurations at 50 °C. High styrene and high vinyl grades are also available for special applications.76 Carboxylated SBR (XSBR) is a modified grade of SBR, which contained pendant –COOH groups.77 The –COOH groups in XSBR can be easily utilized to design self-healing rubber.78The introduction of non-covalent supramolecular networks is an important way to design self-healable and recyclable rubber materials. In most cases, the non-covalent supramolecular networks do not impart good mechanical properties and make them inferior for certain applications. To overcome this difficulty, Xu et al.79 prepared a self-healing carboxylated SBR via the reaction between carboxyl groups of carboxylated SBR (XSBR) and zinc oxide (ZnO), where Zn2+ acts as a crosslinker between the XSBR molecules (Fig. 9). Secondary ionic networks are also formed via further self-aggregation of the ion pairs of Zn2+ salts, whose rearrangements results in good recycling ability. Interestingly, pristine XSBR with 5 wt% zinc oxide exhibits a tensile strength of 6.7 MPa (increased to 10.3 MPa after 3 recycles), which is far higher than that of the most reported non-covalent supramolecular rubbers.
 | ||
| Fig. 9 (a) Schematic representation of network formation by the reaction between carboxyl groups of XNBR and zinc oxide. (b) Schematic for ion multiplet, ion cluster, and ionic crosslinked network.79 Reproduced from ref. 79 with permission from American Chemical Society, copyright 2016. | ||
The functional groups on nanoparticles also interact with the –COOH group of XSBR and result in the self-healing property. Xu et al.80 prepared self-healing carboxylated styrene–butadiene rubber (XSBR)/nano-chitosan (NCS) composites via the salt-forming reaction, in which NCS is responsible for constructing the supramolecular hybrid network (Fig. 10). The –NH2 group of NCS interacts with the –COOH group of XSBR, which results in the formation of carboxylate ammonium salt ([COO−][NH3+]). The carboxylate ammonium groups further aggregate to form week ionic clusters, which are responsible for the self-healing property. In XSBR/NCS composites, NCS acts as a multifunctional crosslinker and improves the mechanical properties without sacrificing the self-healing property. With 20 wt% NCS loading, the tensile strength of the XSBR/NCS composite increases twice that of pristine XSBR and the healing efficiency is found to be 92% after healing at room temperature for 24 h. The NCS loading strongly affects the healing efficiency of the XSBR/NCS composites. The self-healing efficiency continuously increases up to 20 wt% of NCS loading, which is due to the increase in the ionic bond crosslinks in the supramolecular hybrid network. A further increase in the NCS loading (30% and 40%) significantly reduces the healing efficiency. The decrease in the healing efficiency at higher filler loading is due to the formation of the filler-filler network, which hinders the mobility and the interfacial interpenetration of the XSBR molecular chains, and reconstitution of ionic bond crosslinks.
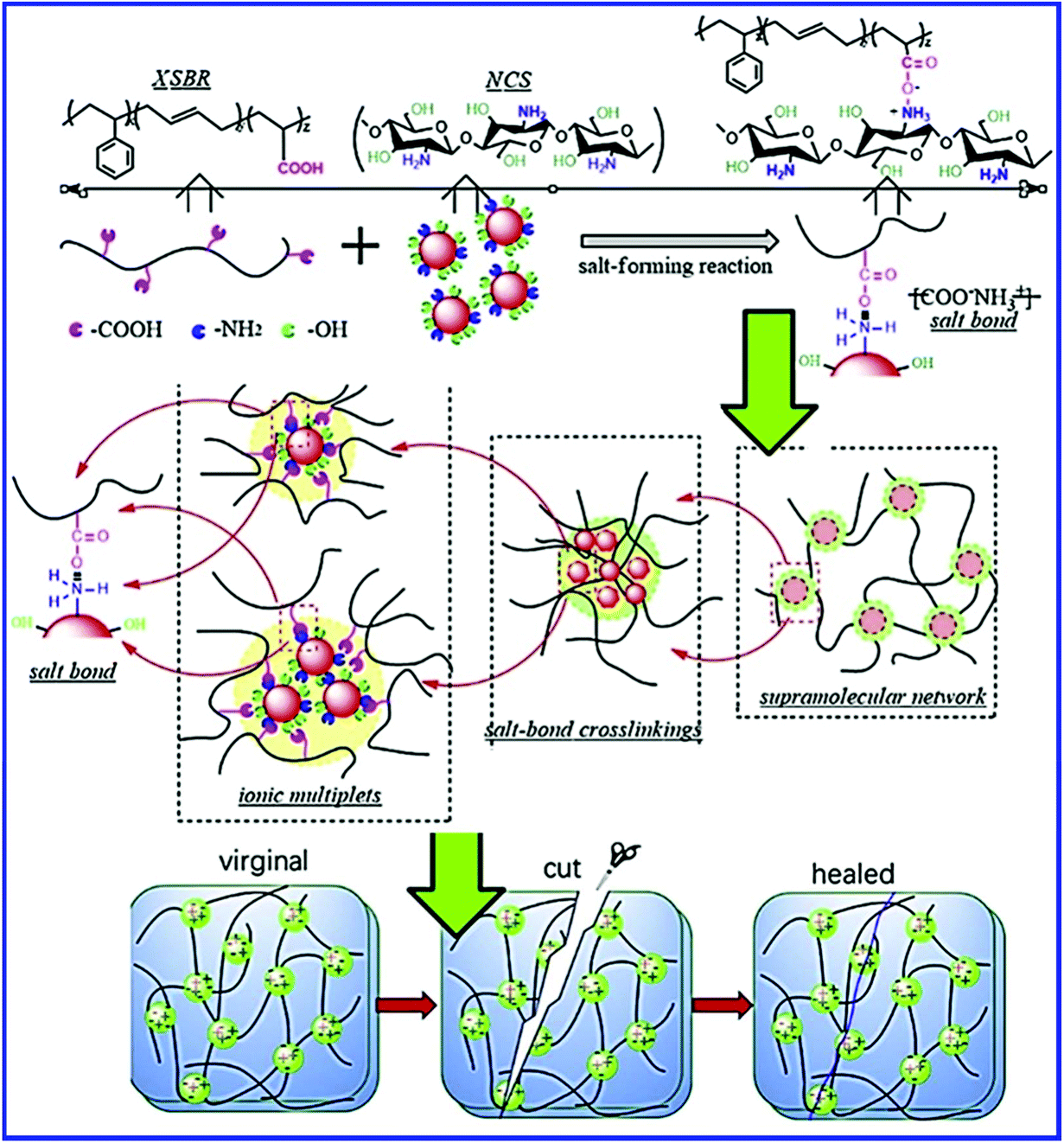 | ||
| Fig. 10 Schematic representation of salt-forming reaction between XSBR and NCS, the evolution of the supramolecular hybrid network in the XSBR/NCS composites, and self-healing by the reestablishment of ionic bond crosslinking in the supramolecular XSBR network.80 Reproduced from ref. 80 with permission from Elsevier, copyright 2019. | ||
Besides chitosan, multiwall carbon nanotube (MWCNT) also have a great impact on the self-healing and mechanical properties of SBR. Kuang et al.81 reported the preparation of self-healing SBR/CNT nanocomposites via the DA and rDA approaches. In this case, the furfuryl-grafted SBR (SBR-FS) and furfuryl-terminated MWCNT (MWCNT-FA) react with bismaleimide (BM) to form covalently bonded and reversibly cross–linked rubber composites (Fig. 11). The Young's modulus of the SBR-FS/BM/MWCNT-FA increases by 200–300% with just 5 wt% loading of MWCNT-FA. The tremendous increase in the modulus is because of the increase in the crosslink density as well as the reinforcing effect of the MWCNT-FA. The increase in MWCNT-FA further improves the healing efficiency. Thus, MWCNT-FA plays a dual role in improving the healing efficiency and mechanical properties via the formation of DA adducts with BM as well by acting as the reinforcing filler. The effect of the MWCNT-FA content on the healing efficiency is highly dependent on the furan/maleimide ratio. The incorporation of 5 wt% MWCNT-FA in SBR-FS/BM (with furan/maleimide ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) improves the healing efficiency from 37% to 90% when heated at 100 °C for 5 h. In SBR-FS/BM, where the furan/maleimide ratio is 1:1, the healing efficiency decreases with the addition of 5 wt% MWCNT-FA. The decrease in the healing efficiency is due to the increased crosslink density, which reduces the mobility of the polymer chains. The healing efficiency of the MWCNT-FA based composite is much higher than that of the MWCNT-COOH (–COOH functionalized MWCNT) because the –COOH functional groups in MWCNT-COOH do not take part in the DA and rDA reaction. The crosslinked SBR-FS/MWCNT-FA/BM nanocomposites are reprocessable similar to thermoplastic elastomers on heating to about 130–150 °C.
:1) improves the healing efficiency from 37% to 90% when heated at 100 °C for 5 h. In SBR-FS/BM, where the furan/maleimide ratio is 1:1, the healing efficiency decreases with the addition of 5 wt% MWCNT-FA. The decrease in the healing efficiency is due to the increased crosslink density, which reduces the mobility of the polymer chains. The healing efficiency of the MWCNT-FA based composite is much higher than that of the MWCNT-COOH (–COOH functionalized MWCNT) because the –COOH functional groups in MWCNT-COOH do not take part in the DA and rDA reaction. The crosslinked SBR-FS/MWCNT-FA/BM nanocomposites are reprocessable similar to thermoplastic elastomers on heating to about 130–150 °C.
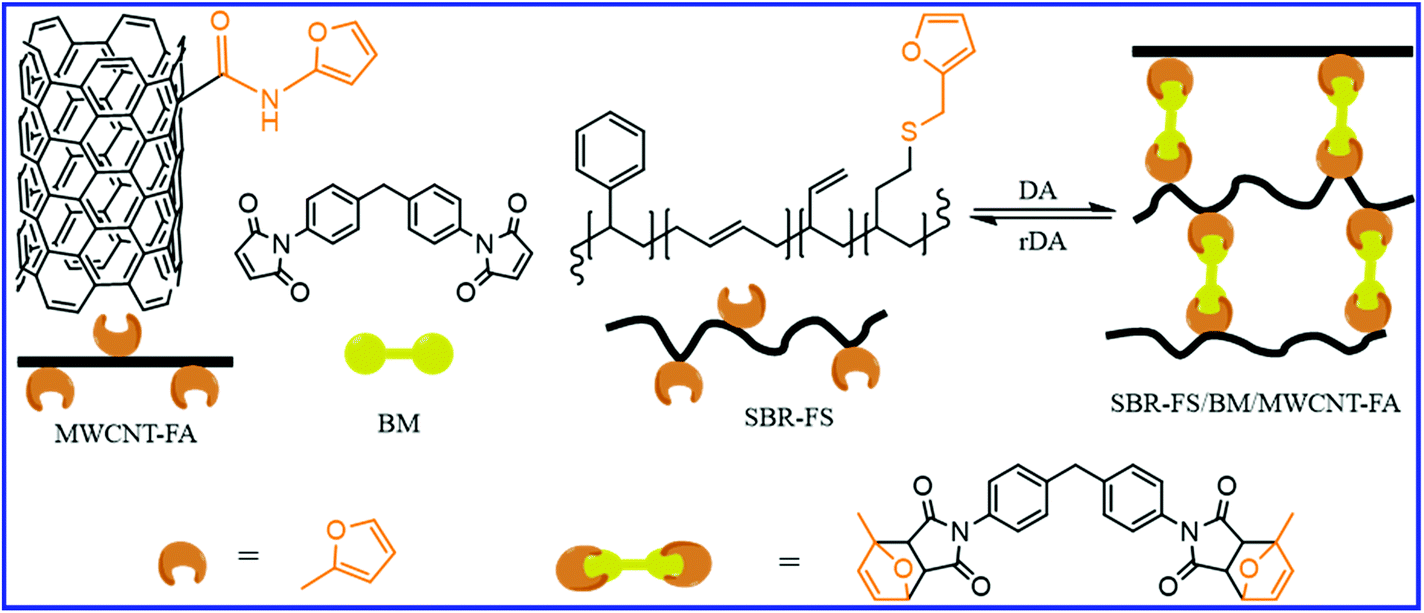 | ||
| Fig. 11 Schematic representation for the preparation self-healable SBR-FS/BM/MWCNT-FA nanocomposites via DA reaction of the furan group of rubber and CNT with bismaleimide.81 | ||
A few works have been reported in the area of self-healing SBR, which is having the single largest synthetic rubber application in the rubber industry. High styrene content SBR or hydrogenated SBR with self-healing property will further widen the application areas of SBR.
5. Self-healing polyisoprene rubber (IR) (Natural rubber-NR and synthetic IR)
Natural polyisoprene rubber (NR) and synthetic polyisoprene rubber (IR) are composed of cis-1,4-polyisoprene.82 The cis-1,4 isoprene content in NR is higher than that of IR.83,845.1. Self-healing IR via the DA and rDA reaction
Natural polyisoprene rubber (NR) is one of nature's most exceptional materials, which is predominantly used in the tire industry. NR can be made self-healable by grafting a suitable functional moiety on its backbone. Tanasi et al.85 prepared a furan-functionalized NR (NR-g-furan) by grafting furfuryl amine molecules onto the maleic anhydride-functionalized NR (NR-g-MA) (Fig. 12). The DA reaction between the furan groups of furan-functionalized NR and the maleimide groups of bismaleimide gives a crosslinked NR (NR-DA). The crosslinked NR is thermoreversible and shows self-healing property when heated to the rDA and DA temperature. These samples show a healing efficiency of 38–85% after healing at 130 °C for 4 h, followed by another 3–7 h at 40 °C.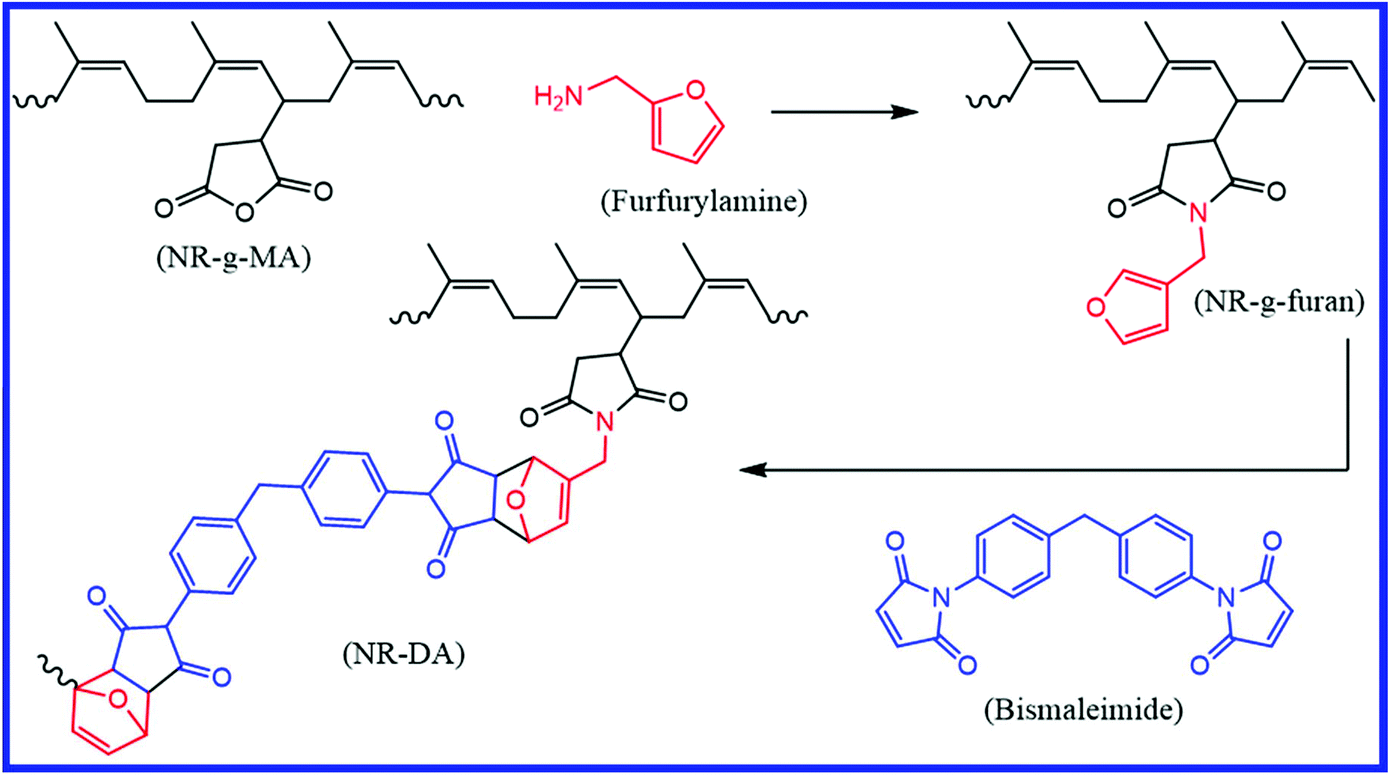 | ||
| Fig. 12 Schematic representation of the preparation of furan-functionalized NR and the crosslinking of furan-functionalized NR via the DA reaction using bismaleimide.85 Reproduced from ref. 85 with permission from Elsevier, copyright 2019. | ||
5.2. Self-healing IR via ionic network
Introducing ionic interactions is one of the important ways to design self-healing isoprene rubbers. Miwa et al.86 reported the preparation of self-healing IR by introducing carboxyl groups to 1.7% of the repeating units, followed by neutralization of 24–90% of the carboxyl groups with sodium hydroxide (Fig. 13a). The association of the ionic group forms a network structure where the ionic moieties undergo continuous hopping between the ionic aggregates at room temperature. This ionic hopping makes the network dynamic and results in self-healing property at room temperature (28 °C). The rate of ionic-hoping of the ionomers increases with decreasing neutralization level, which in turn improves the healing rate. Carboxylated cis-IR with 55% neutralization shows ∼80% recovery after 6 h at room temperature, whereas more than 40 h is required for carboxylated IR with 90% neutralization. At the same neutralization levels, the self-healing rate decreases with the increase in the molecular weight, which is due to the reduction in the diffusion and randomization of the polymer chains between the damaged faces.87 Interestingly, the introduction of ionic moieties to trans-IR (via 90% neutralization of carboxyl groups) does not result in the self-healing property at room temperature. The blending of ionically-modified trans-IR with ionically-modified cis-IR improves the modulus with a reduction in the self-healing efficiency.88 Yang et al.89 reported a facile method for the preparation of the self-healing NR ionomer by reacting brominated NR with histidine (Fig. 13b). The strong ionic interaction between the ionic moieties results in a physically crosslinked elastomer with faster healing time (∼10 s) and a significant self-healing efficiency (93.98%).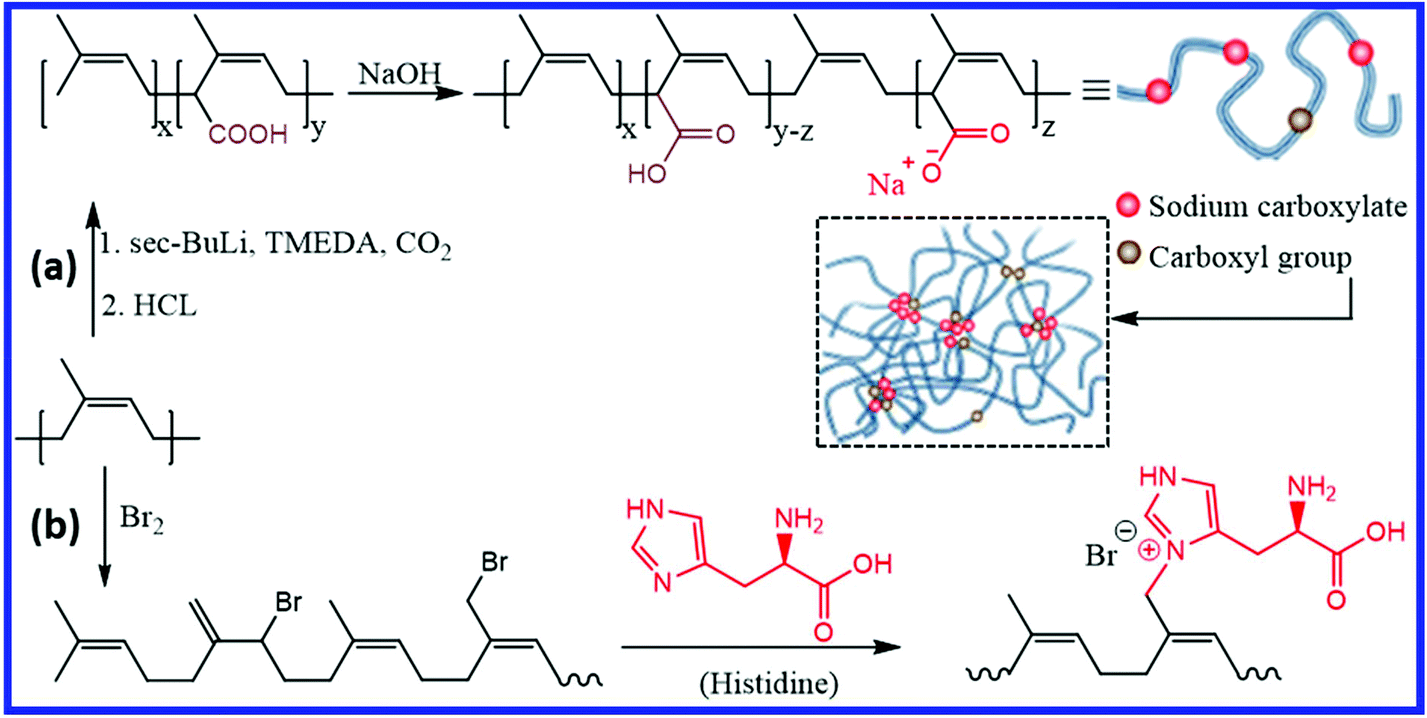 | ||
| Fig. 13 (a) Schematic illustration of the preparation of sodium carboxylate-functionalized IR and ionic association in it.86 (b) Schematic representation of the preparation of self-healing NR ionomer from brominated NR.89 Reproduced from ref. 86 with permission from Springer Nature, copyright 2019. Reproduced from ref. 89 with permission from Elsevier, copyright 2020. | ||
The strength of the rubbers based on an ionic cross-linking network is quite limited and their long-term dimensional stability is questionable. Thus, it is important to incorporate both ionic and covalent crosslinks to develop self-healing rubber with good mechanical properties. Xu et al.90 reported the preparation of a potential self-healing material via controlled peroxide-induced vulcanization to generate massive ionic crosslinks via the polymerization of zinc dimethacrylate (ZDMA) in NR. They controlled the vulcanization process to generate reversible supramolecular networks via ionic crosslinks by retarding the formation of covalent crosslinks. The ionic crosslinks can be easily rearranged between themselves and result in the self-healing property. A fully cut and mended sample nearly gains its original strength when healed at the ambient temperature. In comparison to ZDMA, the use of excess zinc oxide (ZnO) and methacrylic acid (MAA) in NR, where the in situ formation of ZDMA takes place during the peroxide curing process, results in better mechanical properties.91 The improved mechanical properties are due to the presence of residual nano ZnO, which acts as a reinforcing filler and helps in the formation of the new ionic supramolecular hybrid network in the NR/MAA/ZnO compounds. Interestingly, the self-healing efficiency of the NR/MAA/ZnO compounds decreases with an increase in the healing temperature from 23 °C to 80 °C. Although a higher temperature accelerates the mobility of the NR chains across the damaged surface, the seriously deteriorated ionic associations are unable to construct powerful new ionic crosslinks during healing. The ratio of ZnO/MAA in the NR/MAA/ZnO compound also has a strong impact on the mechanical properties and the rate of healing. An increase in the ZnO/MAA ratio improves the mechanical properties and healing efficiency but reduces the rate of healing. To improve the self-healing and mechanical properties, Liu et al.92 incorporated a dual ionic network into NR (Fig. 14). The reaction of lithiated NR with maleic anhydride (MA) generates lithium carboxylate (–COOLi)-based ionic network (NR-MA), which on further reaction with zinc methacrylate (ZDMA) and peroxide results in a second ionic network (NR-MA-ZDMA). As compared to the ZDMA-crosslinked NR, the combination of lithium carboxylate-based network and ZDMA network provides better mechanical and self-healing properties.
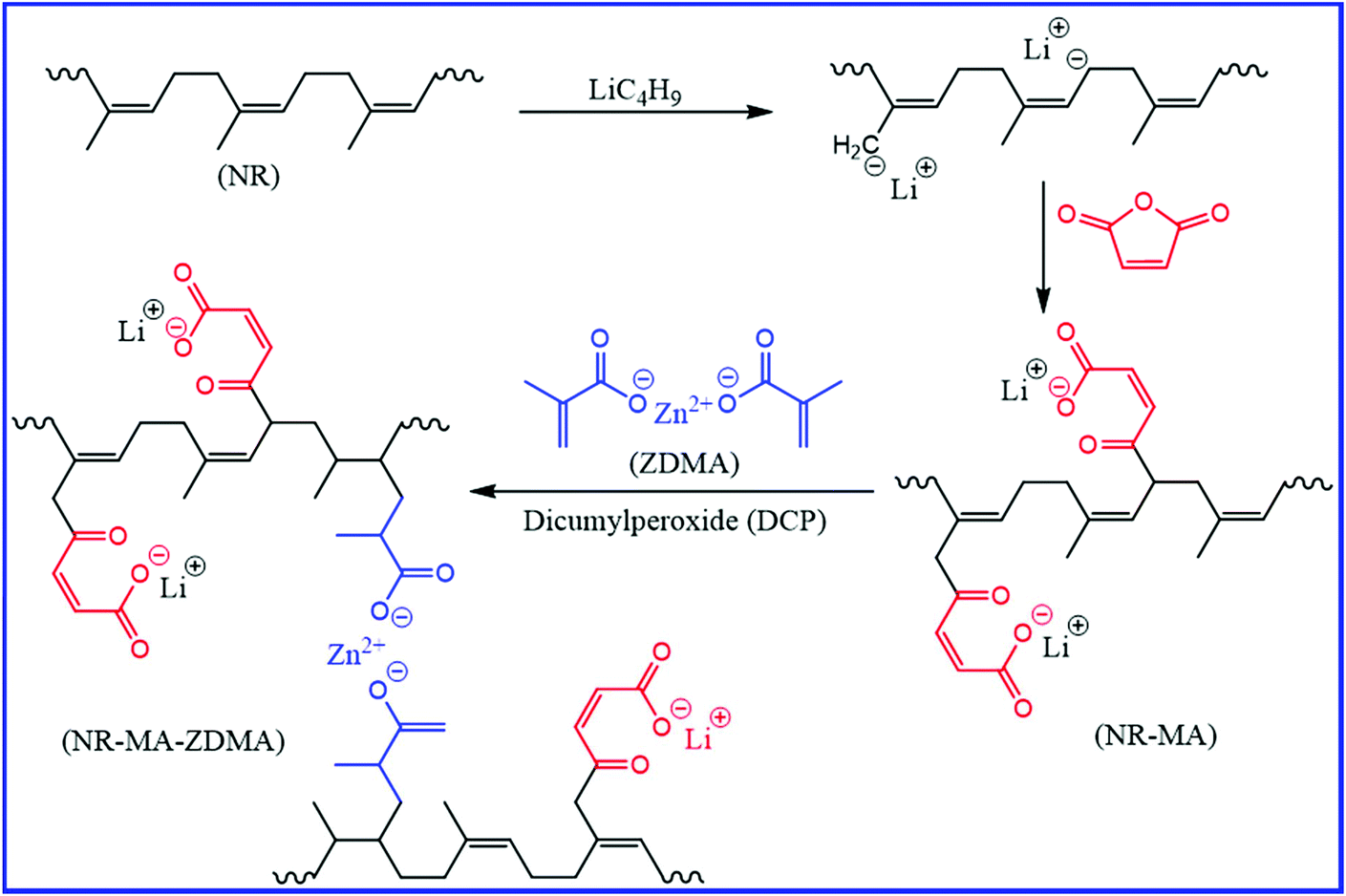 | ||
| Fig. 14 Schematic representation of the preparation of self-healing NR having a dual ionic network.92 Reproduced from ref. 92 with permission from American Chemical Society, copyright 2020. | ||
Rahman et al.93 reported that at high temperatures, epoxidized natural rubber (ENR) shows the self-repairing property due to the interdiffusion of rubber chains and the strong interaction between epoxy groups. Thus, the incorporation of ZDMA into ENR, followed by crosslinking with peroxide, can further improve the self-healing and mechanical properties of ENR. Liu et al.94 reported the preparation of self-healing rubber via controlled peroxide vulcanization of ZDMA (30 phr) filled ENR (ENR/ZDMA). Controlled peroxide vulcanization is performed at a somewhat lower temperature (140 °C) than conventional curing (150 °C), which suppresses the covalent cross-linking of ENR and facilitates the self-polymerization and grafting of ZDMA molecules on to ENR. The self-polymerized and grafted ZDMA form a reversible ionic cluster and induce the self-healing property. The percentage of epoxidation in ENR also affects the self-healing property of the ENR/ZDMA compounds. ENR25/ZDMA (ENR with 25% epoxidation) shows self-healing properties at a lower temperature than that of ENR40/ZDMA (40% epoxidation). After healing at 80 °C for 1 h, the tensile strength of the ENR40/ZDMA composites is recovered to more than 80% of the standard sample, whereas the tensile strength of the ENR25/ZDMA samples is restored to about 70% after healing at room temperature for 50 min. The self-healing property of ENR can also be tuned by blending with a suitable ionomer. Rahman et al.95 studied the self-healing property of the ENR/ionomer blend, where the ionomer is a partially neutralized product of poly(ethylene-co-methacrylic acid) (EM, 15% acid content) with sodium (EMNa) or zinc (EMZn). The ENR/EMNa blend shows complete self-healing, whereas partial self-repairing is observed for the ENR/EMZn blend.
5.3. Self-healing IR based on reversible coordination bond and hydrogen bonding
Self-healing natural rubber can be prepared via the introduction of reversible coordinate bonds. ENR and modified ENR with a suitable functional group can form a reversible coordinate bond with different metal atoms. Han et al.96 developed a facile and effective approach to fabricate self-healing NR via metal–ligand coordinate bond formation. They modified the commercially available ENR by polydopamine (PDA) and then cross-linked it via reversible catechol-Fe3+ coordination bonds (Fig. 15a). The resulting Fe3+/PDA-ENR elastomer exhibits good tensile strength (2 MPa) with a self-healing efficiency of 89.3 ± 3.6% after healing at 50 °C for 12 h. Interestingly, even at low temperature (−10 °C), the Fe3+/PDA@ENR elastomer shows a healing efficiency of ∼80% after 12 h of healing. Liu et al.97 reported the preparation of a self-healing blend based on ZnCl2-decorated ENR (ENR-Zn) and histidine (His)-modified cellulose nanocrystal (His-CNC) (Fig. 15b). Histidine and Zn2+ form supramolecular metal–ligand coordination, which results in excellent self-healing property with an efficiency of 107.17%, 95.43%, and 88.65% for the first, second, and third cycle, respectively. The ENR-Zn/His-CNC blend exhibits a tensile strength of 1 MPa, which is far lower than that of the Fe3+/PDA-ENR. Tang et al.98 incorporated Fe3+-intercalated graphene nanosheet (Fe3+-GN) into the gelatin-ENR (GA-ENR)-based emulsion to obtain a self-healing elastomer via reversible coordination between Fe3+ and gelatin (GA) (Fig. 15c). The Fe3+-GN/GA-ENR composite exhibit low tensile strength (1.6 MPa) with a healing efficiency of 94%. It is always challenging to achieve higher self-healing efficiency along with good mechanical properties via a reversible coordination mechanism.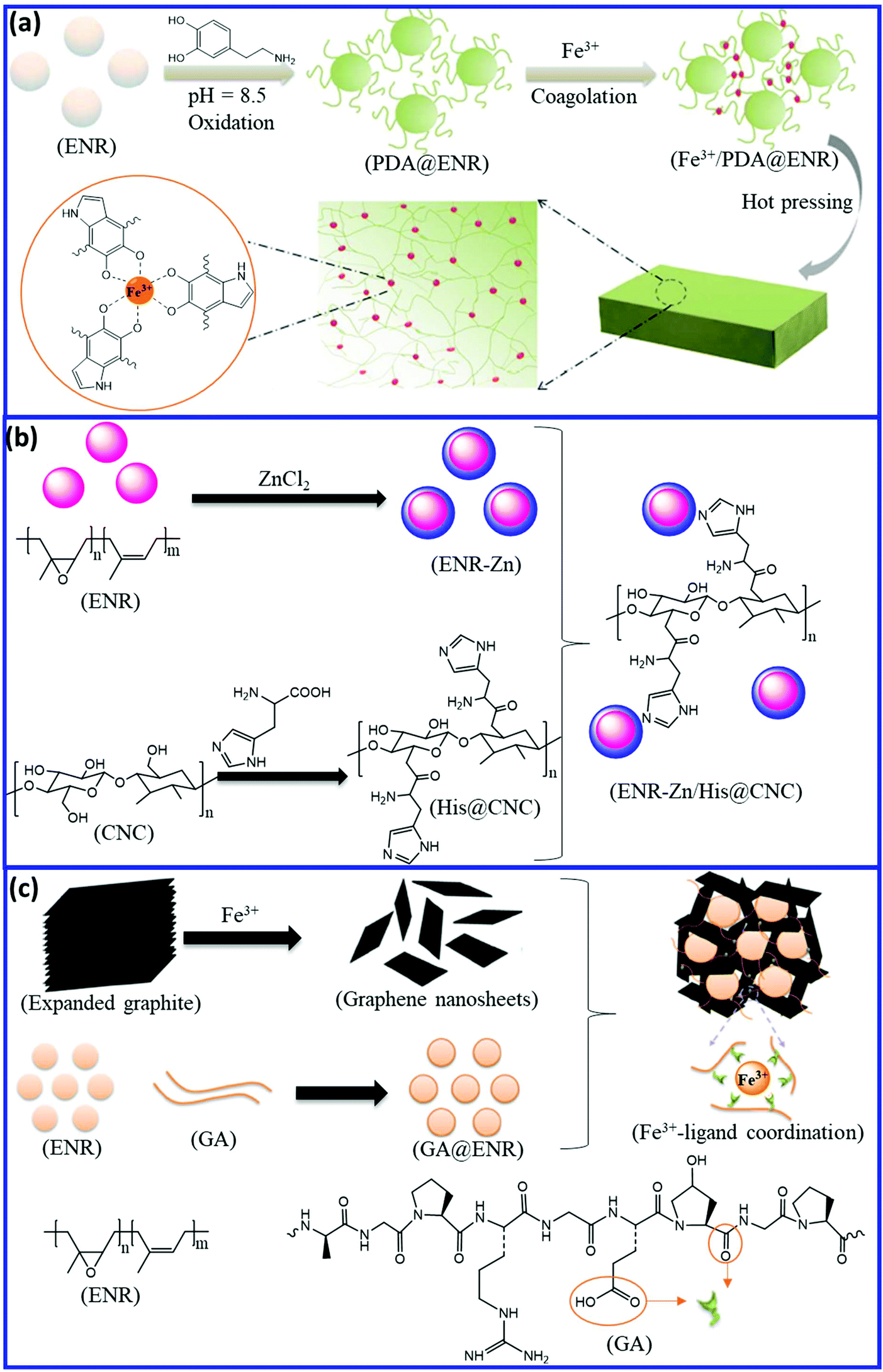 | ||
| Fig. 15 Schematic representation of the preparation of self-healing ENR via reversible coordinate bonds; (a) catechol-Fe3+, (b) histidine-Zn2+, and (c) gelatine-Fe3+.96–98 Reproduced from ref. 96 with permission from American Chemical Society, copyright 2017. Reproduced from ref. 98 with permission from American Chemical Society, copyright 2019. | ||
The mechanical and self-healing properties of IR and ENR can be improved by introducing a dual dynamic network.99 Liu et al.100 reported a self-healing polyisoprene via the dual-dynamic network by introducing weaker multiple hydrogen bonds and stronger Zn–triazole coordinate bond into the unvulcanized cis-1,4-polyisoprene matrix (Fig. 16a). The dual dynamic system results in excellent recovery in the mechanical properties of about 74% (tensile strength before and after healing are 21 MPa and 15.5 MPa, respectively) when it is healed at 80 °C for 24 h. Chen et al.101 introduced coordination as well as boronic ester bonds into ENR to develop a dual dynamic self-healing elastomer. They prepared the dynamic dual crosslinked elastomer by crosslinking ENR using a dithiol-bearing boronic ester crosslinker, followed by the coordination of the epoxy group with Zn2+ (Fig. 16b). The network rearrangement through boronic ester transesterifications and reversible Zn2+–O coordination provide excellent healing efficiency (60–100%) and good mechanical properties (1.60 to 14.63 MPa).
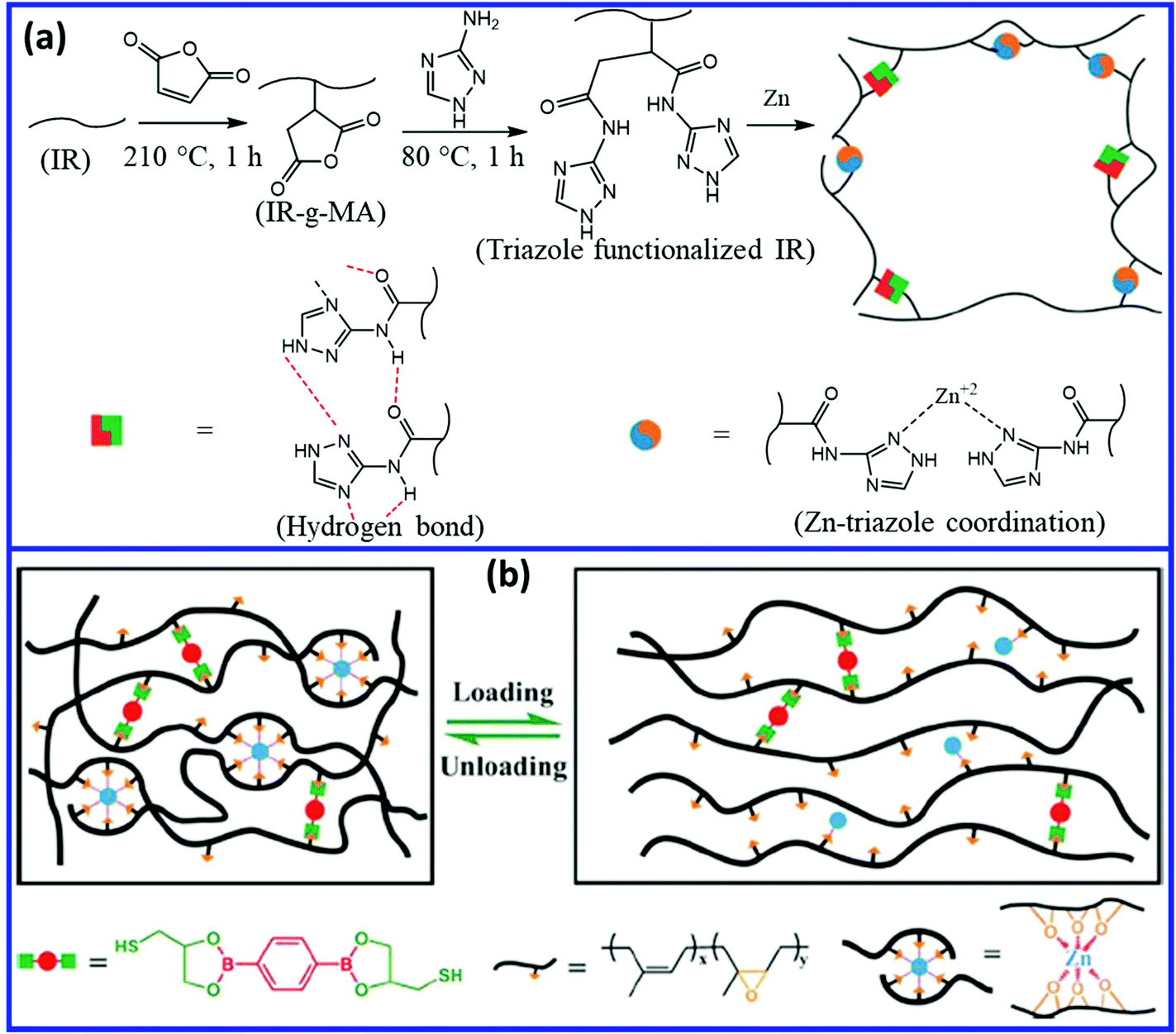 | ||
| Fig. 16 (a) Schematic representation of the preparation of self-healing IR via a dual dynamic network based on reversible hydrogen and coordinate bond.100 (b) Self-healing ENR networks with dual dynamic crosslinks of boronic ester bonds and non-covalent Zn2+–O coordinate bonds.101 Reproduced from ref. 100 with permission from Royal Society of Chemistry, copyright 2019. Reproduced from ref. 101 with permission from American Chemical Society, copyright 2019. | ||
5.4. Self-healing IR and epoxy-functionalized IR based on dynamic disulfide metathesis
Polydiene rubbers including NR (IR) are mostly cured using the sulfur vulcanization process. The sulfur vulcanization process creates sulfur crosslinks of different lengths (mono-, di-, and poly-sulfide bonds), pendant side groups, and cyclic sulfides. Depending on the accelerator/sulfur (a/s) ratio, the sulfur vulcanizations are generally classified as conventional (CV, a/s = 0.1–0.6), semi-efficient (semi-EV, a/s = 0.7–2.5), and efficient (EV, a/s = 2.5–12).102–104 The CV system, where the a/s ratio is low, forms more disulfide and polysulfide linkages, whereas the EV system results in more monosulfide linkages. The disulfide and polysulfide bonds can undergo disulfide exchange reactions and induce the self-healing property in the elastomer. Hernández et al.105 prepared self-healing NR using the conventional sulfur curing system with a low a/s ratio (0.2). At a constant a/s ratio (0.2), the increase in the sulfur content (from 0.7 phr to 2.5 phr) reduces the healing efficiency, which is due to the increased crosslink density of the system. The increased crosslink density reduces the chain mobility, which retards the disulfide exchange reaction and hence the healing efficiency. Further, an increase in the exposure time of the cut surfaces to the open atmosphere results in a decrease in the healing efficiency of the mended samples.Epoxidized polyisoprene (epoxidized natural rubber, ENR) contains two reactive functional moieties (epoxy group and unsaturated double bond) and can be easily modified with various crosslinkers and organic molecules to develop self-healing ENR. Imbernon et al.106 introduced disulfide functionality into the ENR using dithiodibutyric acid (DTDB) as a crosslinker (Fig. 17a). DTDB-cured ENR exhibits strong elastomeric properties and behaves like a standard natural rubber until 100 °C. The disulfide exchange reaction makes DTDB-cured ENR reprocessable when heated above 150 °C. After adhering two specimens for 30 min at 180 °C, ENR with disulfide bond (DTDB-cured ENR) shows superior lap-shear strength (20 N) than that without the disulfide bond (5 N). The incorporation of both disulfide functionality and hydrogen bonding into ENR can further improve the self-healing property. Cheng et al.107 prepared a dual cross-linked self-healing ENR based on dynamic disulfide metathesis and thermoreversible hydrogen bonding (Fig. 17b). Two aromatic disulfides (Dithiodibenzoic acid, DTSA, and 4,4′-dithiodianiline, DTDA) along with sulfur (S) are used to improve the self-healing property. The increase in the sulfur content in the DSTA/DDTA/S curing system reduces the healing efficiency as it results in the formation of more permanent crosslinks, which reduce the mobility of the polymer chains. The presence of carbonyl, epoxy, and hydroxyl groups in the dual crosslinked system results in the formation of thermoreversible hydrogen bonds, which further helps in the improvement of mechanical and self-healing properties. The dual cross-linked ENR exhibits good self-healing efficiency (up to 98%) when healed at 120 °C for 24 h.
 | ||
| Fig. 17 (a) Schematic representation of the preparation of self-healing ENR having aliphatic disulfide functionality.106 (b) Self-healing ENR having aromatic disulfide functionality and hydrogen bonding.107 Reproduced from ref. 106 with permission from Royal Society of Chemistry, copyright 2015. Reproduced from ref. 107 with permission from American Chemical Society, copyright 2019. | ||
5.5. Self-healing nanocomposites of epoxy-functionalized IR
The introduction of suitable nanofillers into the ENR can also induce the self-healing property in the nanocomposites. Nie et al.108 and Chen et al.109 studied the self-healing property of ENR/chitin nanocrystals (ENR/CNCs) composites where the supramolecular networks are formed through hydrogen bonding between the CNCs and the epoxy groups of ENR. The supramolecular network improves the mechanical properties and induces the self-healing property in the composite. With 20 wt% CNCs loading, the tensile strength of the ENR/CNCs composites increases twice that of the original strength of neat ENR. The ENR/CNCs composite with 20 wt% filler loading shows good self-healing efficiency (83%). The healing behavior is strongly affected by the CNCs contents, healing temperature, and healing time. Nie et al.110 further studied the effect of the combination of various fillers on the self-healing and mechanical properties of the ENR composites. They dispersed the chitin nanocrystals (CNCs), carboxymethyl chitosan (CMCS), and carboxyl functional multi-walled carbon nanotube (CNT) into ENR in the latex state. The formation of multiple hydrogen bonds between the functional groups of the fillers and ENR result in a healing efficiency of 91%. Interestingly, the tensile strength, as well as the elongation at break of the ENR/CNCs/CMCS/CNT composite, is much higher than that of the pristine ENR.Cao et al.111 prepared the self-healing nanocomposites based on the ENR/cellulose nanocrystals via the dispersion of cellulose nanocrystals in ENR in the latex state. The formation of supramolecular networks via dynamic hydrogen bond between the epoxy oxygen of ENR and hydroxyl groups on the cellulose nanocrystals induces the self-healing property in the composite. Supramolecular hydrogen bonding also improves the mechanical property of the composite. The self-healing property is strongly affected by the cellulose nanocrystals’ content and healing time. Cao et al.112 further reported the preparation of recyclable and self-healable elastomers composites based on ENR and TOCNs (TEMPO-oxidized cellulose nanocrystals) having a large number of surface carboxyl groups (Fig. 18). The formation of interfacial ester bonds by the reaction between the carboxyl groups of TOCNs and the epoxy groups of ENR results in a covalently crosslinked network structure, which improves the mechanical property of the composite. Interestingly, the network with exchangeable β-hydroxyl ester bonds at the ENR-TOCN interface can change the network topology via the transesterification reactions. This transesterification reaction plays an important role in the recycling and self-healing process and results in a healing efficiency of ∼80%.
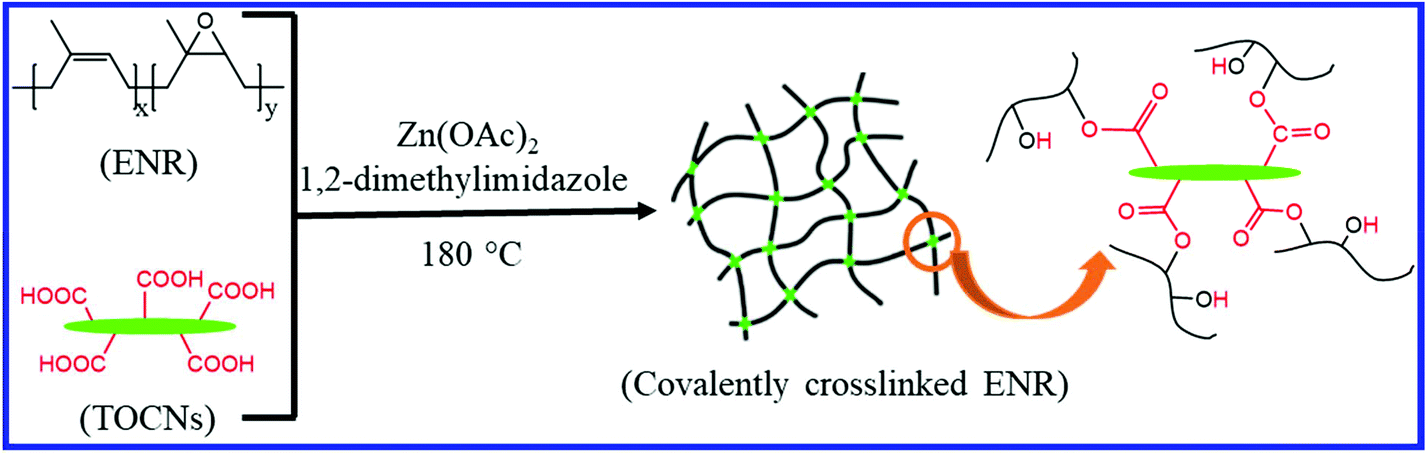 | ||
| Fig. 18 Schematic representation of the preparation of TOCNs cross-linked ENR via the epoxy-acid reaction112 Reproduced from ref. 112 with permission from Royal Society of Chemistry, copyright 2019. | ||
Other fillers such as silica and graphene oxide (having –OH functionality) can induce the self-healing property via the formation of hydrogen bonding with NR and ENR. Sattar et al.113 reported the preparation of self-healing NR-silica (NR/SiO2) composites via Mg2+-assisted heteroaggregation of NR (bearing proteins and phospholipids) and silica to produce a reversible supramolecular network mainly constructed by ion-dipole, electrostatic, and hydrogen bonding (Fig. 19a). The Mg2+ ions interact with the negatively charged colloids of NR and SiO2 particles and drive the mutual assembly via reversible electrostatic interaction and hydrogen bonding. The hydrogen bond is formed between the silanol groups on the silica surface and the lipid groups of NR. The combination of hydrogen bonding and ionic interaction in the NR-silica composites result in the self-healing property. Interestingly, the NR/SiO2 composites prepared via the wet compounding method (mixing in latex state) possess higher healing efficiency (79%) than that prepared via the dry mixing process (51.6%). Utrera-Barrios et al.114 prepared self-healing composites based on ENR and thermally-reduced graphene oxide (TRGO) having surface functional groups (hydroxyl, carboxyl, and epoxy) (Fig. 19b). During vulcanization, the hydroxyl and carboxyl group of TRGO reacts with the epoxy group of ENR, which generates covalent linkage (C–O–C) between the TRGO and ENR, which provides good mechanical properties. The ring-opening of the epoxide also generates the –OH functionality in ENR. The –OH and epoxy functionality of ENR are involved in the formation of reversible hydrogen bonding with the functional group of TRGO, which induces the self-healing property in the ENR/TRGO composite. Guo et al.115 developed a self-healing composite by incorporating serine-modified Ti3C2MXenes (s-MXenes) into serine-modified ENR (s-ENR) (Fig. 19c). The supramolecular hydrogen bonding interfaces between the s-MXenes and s-ENR results in excellent self-healing property with a healing efficiency of 99.3% after healing at room temperature for 90 min.
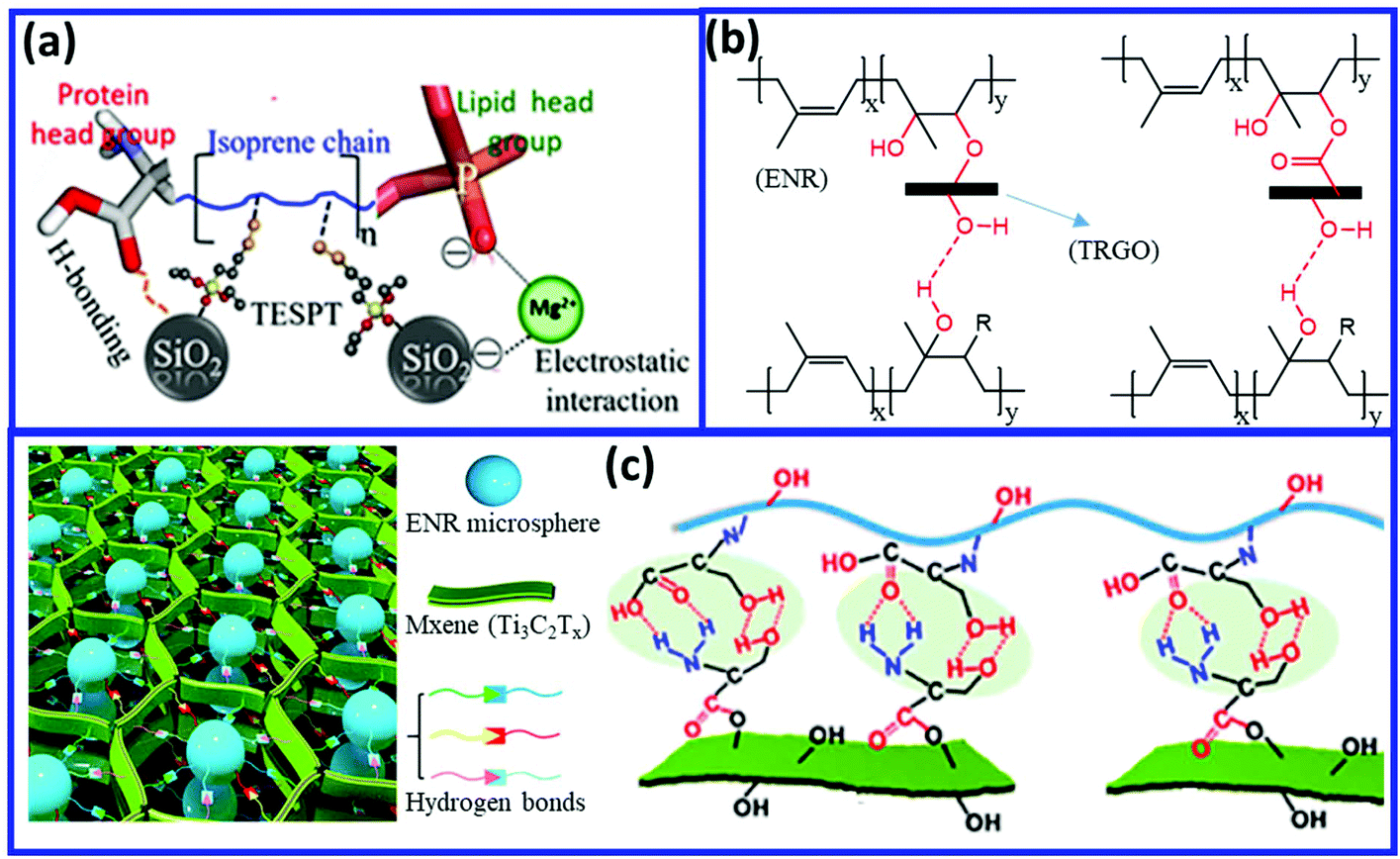 | ||
| Fig. 19 (a) Schematic representation of the electrostatic interaction and hydrogen bonding in the NR/SiO2 composites.113 (b and c) Hydrogen bonding in the ENR/TRGO and serine-modified ENR/Mxene composites.114,115 Reproduced from ref. 113 with permission from American Chemical Society, copyright 2019. Reproduced from ref. 114 with permission from American Chemical Society, copyright 2020. Reproduced from ref. 115 with permission from American Chemical Society, copyright 2020. | ||
Various mechanisms such as DA-rDA reaction, ionic interaction, reversible coordination, hydrogen bonding, disulfide metathesis, and transesterification have been utilized to prepare self-healing IR. Self-healing IR based on transesterification needs very high temperature (160 °C) to show the self-healing property, whereas the DA-rDA-based system exhibits self-healing property at 130 °C. The DA-rDA-based IR further needs to be maintained at 40 °C for a longer time to achieve suitable mechanical property through the formation of DA adducts. Thus, compared to the transesterification reaction, the DA-rDA-based system needs more time to show better healing efficiency. IR based on ionic interaction, reversible coordination, hydrogen bonding, and disulfide metathesis exhibits self-healing property in between 25 °C to 120 °C. Different functional fillers like chitin nanocrystals (CNC), carboxymethyl chitosan (CMCS), carboxyl functional multi-walled carbon nanotube (CNT), cellulose nanocrystals, silica, thermally-reduced graphene oxide (TRGO), and serine-modified Ti3C2MXenes have been explored to develop self-healing ENR composites via H-bonding. Serine-modified ENR/Ti3C2MXene composite shows superior self-healing property over the ENR/CNC, ENR/CMCS, ENR/CNT, and ENR/TRGO composites.
6. Self-healing isoprene isobutyl rubber (butyl rubber, IIR)
Butyl rubber is a random copolymer of isobutylene and isoprene, which contains 1–3% of isoprene units.116 The presence of the double bond and the methyl group in the isoprene unit make the IIR suitable for various modification and crosslinking processes. Bromobutyl rubber (BIIR) is an important grade of IIR, which is prepared via the bromination of IIR.117,118 The allylic bromide unit in BIIR can be easily modified with amine, phosphine, and imidazole derivatives to form ionomers.119–1246.1. Self-healing IIR via ionic interaction and hydrogen bonding
The introduction of ionic moieties is an important way to convert commercial IIR to a self-healing material.125 Das et al.126 reported the self-healing property of ionomeric butyl rubber (IIIR) prepared by the modification of bromobutyl rubber (BIIR) with butylimidazole. The reaction between the allyl bromide group of BIIR with butylimidazole results in the formation of ionic imidazolium groups having associated bromide anion. The interaction between the imidazolium ionic moieties results in the formation of reversible ionic associates those act as physical crosslinks (Fig. 20a). These ionic association break with an increase in the temperature and again, the association of ionic moieties takes place with a further reduction in the temperature. The association, dissociation, and rearrangement of the ionic groups induce the self-healing property in the rubber (Fig. 20b). The ionically-modified BIIR shows superior mechanical properties (elastic modulus, tensile strength, ductility, and hysteresis loss) over those of conventionally sulfur-cured BIIR. The structure of the alkyl imidazole (modifier) also has a great impact on the self-healing property of BIIR. Suckow et al.127 studied the effect of various alkyl imidazole such as 1-methylimidazole, 1-butylimidazole, 1-hexylimidazole, 1-nonylimidazole, and 1-(6-chlorohexyl)-1H-imidazole on the self-healing property of ionically-modified BIIR. The self-healing property mainly depends on the nature of the ionic cluster. The strong ionic cluster reduces the chain dynamics, which in turn improves the mechanical properties but reduces the self-healing efficiency. Imidazole with short aliphatic substituents (1-methylimidazole) forms strong clusters and results in poor self-healing property, whereas imidazole with medium alkyl chain length (e.g. butyl, hexyl, and nonyl) reduces the cluster strength and improves the chain dynamics and hence the self-healing efficiency. The weaker cluster strength results in poor mechanical properties. Thus, a balance between the mechanical or self-healing property is needed to get the optimum value of both. BIIR modified with 1-hexylimidazole shows the optimum properties with a healing efficiency of 74% (based on the tensile strength) and 98% (based on the elongation at break) and tensile strength and elongation at break of 10.7 MPa and 1040%, respectively. Mordvinkin et al.128 reported that imidazole (methyl-, butyl-, hexyl, or nonylimidazole)-modified BIIR form ionic clusters of about 20 ionic groups. This indicates that the overall structure is governed by the spacing of the ionic groups along the chain and by their strong aggregation tendency in all the cases. The fraction of free ionic groups is small in all the imidazole-modified BIIR. The amplitude of the internal motion of the ionic clusters increases with an increase in the length of the alkyl group of alkylimidazolium. | ||
| Fig. 20 (a) Schematic representation of the formation of the ionic cluster in the imidazole-modified BIIR.127 (b) Dissociation and association ionic groups during the self-healing process.126 Reproduced from ref. 126 with permission from American Chemical Society, copyright 2015. Reproduced from ref. 127 with permission from American Chemical Society, copyright 2017. | ||
Stein et al.129 studied the effect of the combination of ionic cluster and hydrogen bonding on the self-healing and reprocessability of bromobutyl rubber. In this case, BIIR is reacted with modifiers having one uracil and one imidazole moiety. The reaction of the allyl bromide units of BIIR with the imidazole moiety results in the formation of imidazolium bromide units (ionic groups), which undergo association to form ionic clusters. The supramolecular physical crosslink is formed via triple hydrogen bonding between the uracil groups with a bifunctional linker having two diamidopyridyl moieties. The presence of dual dynamic crosslinks does not help to show the improved self-healing property and the healing efficiency is lower than that of the hexyl imidazole-based ionomer.
6.2. Self-healing IIR-ionomer nanocomposites
The addition of the filler also affects the self-healing property of the ionically-modified BIIR.130 Le et al.131,132 reported the preparation of self-healing butyl rubber (IIR) by the modification of bromobutyl (BIIR) with imidazole. Further, they loaded the imidazole-modified BIIR with carbon nanotubes (CNTs) to reinforce the matrix and to induce the electrical conductivity. The cation–π interaction between the imidazolium group of butyl rubber and the CNT improves the rubber-CNT interaction and results in better mechanical performance and higher electrical conductivity. The electrically conductive nature of the composite helps to accelerate the healing process just by applying an electrical current across the damaged surface. Sallat et al.133 studied the effect of addition of silica nanoparticles on the self-healing and mechanical properties of ionically-modified BIIR (modified using 1-butylimidazole). In addition to pristine (precipitated) silica, they also used silanized silica nanoparticles with aliphatic or imidazolium functional groups. The interaction between the ionic group of ionically-modified BIIR and the functional groups of silica nanoparticles strongly improves the mechanical properties but with a reduction in the self-healing efficiency. The tensile strength of ionically-modified BIIR is 10.2 MPa and increased to 12 MPa, 18.9 MPa, and 18.9 MPa, respectively, due to its composites with pristine, alkyl-functionalized, and imidazolium-functionalized silica nanoparticles. No improvement in the tensile strength is observed when the modification of silica is performed via the in situ process during compounding. This may be because of the presence of unreacted modifier molecules that are not successfully grafted over the silica nanoparticles. The pristine ionically-modified BIIR shows a healing efficiency of 87 ± 50%, which reduces to 32–43% for its silica nanocomposites. Interestingly, the tensile stress and elongation at break of the composites after healing are comparable with those of the pristine ionomer.Most of the reported literature on self-healing BIIR is based on imidazolium-based ionic moiety. Phosphonium, ammonium, and pyridinium based ionic groups can also be incorporated into BIIR via reaction with phosphine, amine, and pyridine-based nucleophiles. The effect of the phosphonium, ammonium, and pyridinium-based ionic moieties on the self-healing property of BIIR should be evaluated and compared with the imidazolium-based ionic moiety.
7. Self-healing nitrile butadiene rubber (NBR)
7.1. Self-healing NBR based on coordination bond
NBR contains a double bond as well as a nitrile functionality. The nitrile group in NBR acts as a ligand and forms complexes with metal salts. Nitrile rubber is also crosslinked using metal salts, where the crosslinks are formed via the coordination reaction between the nitrile group and the metal atom.134 If the coordination bond between the nitrile group and the metal is reversible, then the coordination crosslinked NBR can be easily reprocessed. The functional group of the functionalized NBR can also be utilized to form a reversible coordinate bond with suitable metal ions. Cheng et al.135 reported the preparation of recyclable NBR by the functionalization of epoxidized NBR with dopamine, followed by the formation of coordination crosslinks between the catechol groups of dopamine and Fe3+ ion. The introduction of crosslinks via reversible coordination chemistry can also induce the self-healing property in NBR. Zhang et al.136 reported the preparation of self-healable and reprocessable coordination-crosslinked NBR using cobalt neocaprate (cobalt(II) decanoate) as a crosslinker (Fig. 21). When the healing is performed at 190 °C for 10 min, the coordination-crosslinked NBR shows a healing efficiency of 72%, whereas it is only 26% in sulfur-crosslinked NBR. NBR loaded with 30 phr of cobalt neocaprate exhibits superior tensile strength and higher elongation at break than that of the sulfur (1.5 phr)-cured NBR sample. The cobalt neocaprate-crosslinked NBR exhibits higher modulus and tensile strength after recycling. The nature of the metal–ligand bond also has a strong influence on the self-healing efficiency. Das et al.137 studied the effect of various metal salts (Co+2, Ni+2, and Zn+2 based metal salts) on the self-healing property of 2,6-diaminopyridine-functionalized carboxylated nitrile butadiene rubber (DAP-XNBR). Zn+2-coordinated DAP-XNBR exhibits better self-healing efficiency than that of Co+2 and Ni+2, whereas the tensile strength of the Co+2 and Ni+2-coordinated DAP-XNBR is much higher than that of Zn+2. The higher healing efficiency in Zn+2 coordinated DAP-XNBR is due to the dynamic nature of the metal–ligand bond, whereas the stronger metal–ligand bond in Ni+2-coordinated DAP-XNBR reduces the healing efficiency. The healing efficiency Co+2, Ni+2, and Zn+2-coordinated DAP-XNBR increases with an increase in the healing temperature.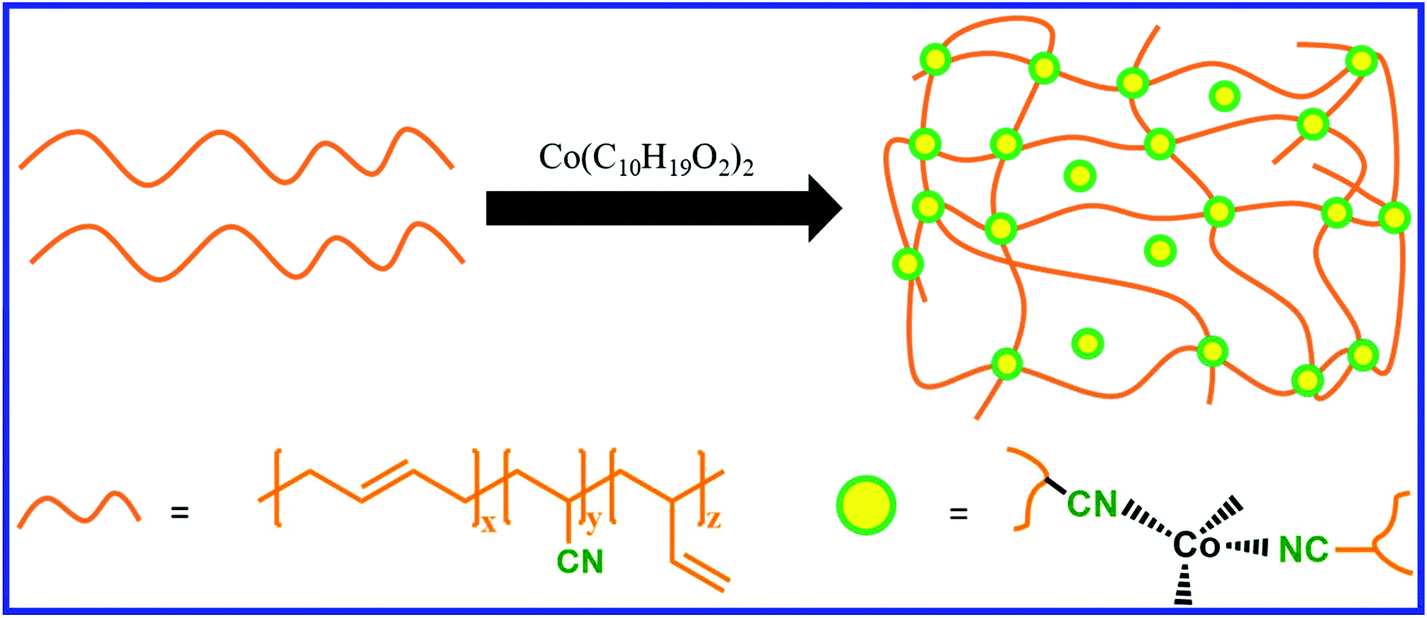 | ||
| Fig. 21 Schematic representation of the preparation of coordination crosslinked NBR.136 | ||
7.2. Self-healing NBR via blending with other polymers
NBR can also be made self-healable by blending with suitable polymeric materials. Schüssele et al.138 studied the self-healing property of NBR by blending with hyperbranched polyethyleneimine and their urea derivatives. Neat NBR shows a healing efficiency of 4% when the cut samples are healed at 100 °C for 12 h. However, the blending of 12 phr of hyperbranched polyethyleneimine (PEI) with NBR results in a healing efficiency of 27–44%. The blending of high molecular weight PEI (2000 g mol−1) with NBR shows better healing efficiency (44%) than that of low molecular weight PEI (800 g mol−1). The blending of poly(acrylic acid)-grafted ground tire rubber (PAA-g-GTR) also has a great impact on the self-healing property of NBR. Barrios et al.139 compared the self-healing property of the XNBR/ZnO compound with the PAA-g-GTR-added XNBR/ZnO (XNBR/ZnO/PAA-g-GTR). In XNBR/ZnO, Zn2+ form ionic crosslinks with the acid group of XNBR, and the association and dissociation of ionic groups results in the self-healing property. The addition of PAA-g-GTR into the XNBR/ZnO compound further improves the healing efficiency, which is due to the formation of additional ionic clusters via interaction between the acid groups of PAA-g-GTR and Zn2+. The healing efficiency of the XNBR/ZnO compound is 15%, which increased to 70% for the XNBR-ZnO-PAA-g-GTR compound. NBR as an additive also improves the self-healing efficiency of other self-healing polymeric systems. Feng et al.140 reported that the self-healing efficiency of epoxy resins (based on the Diels–Alder reaction) can be improved by introducing NBR into it. The blending of 0, 5, 10, and 15 phr of NBR to the self-healing epoxy system results in a healing efficiency of 70.0%, 81.5%, 84.9%, and 88.6%, respectively.7.3. Self-healing NBR via DA/rDA reaction and disulfide metathesis reaction
The self-healing property of NBR can be improved via the introduction of dual dynamic covalent bonds. Sarkar et al.141 reported the preparation of self-healing XNBR based on dynamic photo-triggered disulfide metathesis and thermoreversible Diels–Alder (DA) “click” chemistry (Fig. 22). They first prepared a furan-functionalized XNBR (FXNBR) via the reaction of XNBR with furfuryl amine, followed by crosslinking with a dual functional crosslinker (BMDS) having maleimide as well as disulfide functionalities that can participate in the DA reaction and disulfide metathesis reaction. The BM-DS crosslinked FXNBR (FXNBR-BMDS) shows a healing efficiency of 60–91% when healed at 125 °C for 4 h, followed by cooling down to RT, and this temperature was maintained for another 12 h. The increased BM-DS content in the FXNBR-BMDS results in an increase in the tensile strength but reduces the self-healing efficiency. The improved tensile strength is due to the increase in the crosslink density, which in turn reduces the chain mobility and self-healing efficiency. The FXNBR-BMDS also shows self-healing property under UV irradiation (for 4 h), which is due to the disulfide metathesis reaction. The FXNBR-BMDS sample with similar BMDS content (furan:BMDS = 1:0.07) shows a healing efficiency of 74% and 78% under thermal and UV irradiation, respectively. The authors studied the individual effect of thermal and UV irradiation on the self-healing property. The study of the effect of a combined dose of thermal and UV irradiation on the self-healing property would have been more interesting.
 | ||
| Fig. 22 Schematic representation of the preparation of self-healing XNBR having disulfide moiety and DA bond.141 | ||
8. Self-healing chloroprene rubber (CR)
Sulfur-crosslinked CR can be made self-healable and reprocessable by the incorporation of a suitable catalyst that can cause a disulfide metathesis reaction. Xiang et al.142 reported the use of organic complex copper(II) methacrylate (MA-Cu) as a novel catalyst to trigger disulfide metathesis at elevated temperatures. When the catalyst is incorporated into the vulcanized CR, the reshuffling of inherent sulfur crosslinks is enabled typically at 120 °C, while the microstructure of the material remains unchanged at a lower temperature (typically lower than 100 °C). The presence of the catalyst activates the disulfide bond, as a result of which the sulfur bonds are reversibly exchanged inside the vulcanized chloroprene rubber as well as across the surfaces at 120 °C. The exchange of disulfide bonds via the metathesis reaction results in repeated reshaping, self-healing, and recycling of the crosslinked CR. The disulfide metathesis reaction is only possible for disulfide and polysulfide linkages. However, the formation of more monosulfide linkages during vulcanization may act as permanent crosslinks and retard the self-healing process.9. Self-healing diene-functionalized silicone rubber
Silicone rubber (polysiloxane) differs from other elastomers in the structure of its backbone, which consists of Si–O–Si units. The wide applications of silicone rubber in biomedical devices and seals have attracted significant attention for the development of self-healing silicone rubber that can improve the application life. Besides a large number of pieces of literature on self-healing silicone rubber,143–154 only a few have been reported on the field of diene-functionalized self-healing silicone rubber. Zhao et al.155 reported a healable polysiloxane elastomer via the crosslinking of polydimethylsiloxane-bearing maleimide pendants (AMS-DMAS-M) with furan–end functionalized siloxane (APMS-F) via the Diels–Alder (DA) reaction (Fig. 23). For comparison purposes, they also prepared a crosslinked polysiloxane sample without the DA bonds. The DA-crosslinked polysiloxane elastomers show the self-healing property for thermal treatment at 140 °C for 3 h and at 80 °C for different times. At 140 °C, the cleavage of the crosslinks (DA adduct) takes place via retro Diels–Alder reaction, which generates the furan (diene) and maleimide (dienophile) functionality on the polysiloxane elastomers. These furan and maleimide groups are again recombined at 80 °C and result in the formation of DA crosslinks. The increase in the thermal treatment time at 80 °C results in an increase in the healing efficiency, which is due to the formation of more crosslinks via the DA reaction. DA-crosslinked polysiloxane elastomers show a healing efficiency of 29%, 59%, 89%, and 95% on respective thermal treatment for 1 h, 6 h, 12 h, and 24 h at 80 °C. The content of DA adducts and the molecular weight of the polysiloxane elastomer has a strong impact on the self-healing efficiency. An increase in the content of DA adducts and a decrease in the molecular weight of the polysiloxane elastomer results in an improvement in the healing efficiency. The higher healing efficiency of the low molecular weight polysiloxane elastomer is due to the better chain mobility than that of the high molecular weight counterpart. Interestingly, the DA-crosslinked polysiloxane elastomer shows similar healing efficiency even after remolding via hot compression at 140 °C (for 1.5 h under a pressure of 20 MPa), followed by cooling down to 80 °C and remaining at this temperature for 24 h. This indicates that the DA-crosslinked polysiloxane elastomer is reprocessable similar to thermoplastic elastomers. In contrast, the crosslinked polysiloxane sample without DA bonds does not exhibit self-healing and reprocessable properties. The incorporation of nanoparticles into the DA-based self-healing polysiloxane system also affects the mechanical and self-healing properties. Zhao et al.156 studied the effect of graphene nanosheets on self-healable and mechanical properties of polysiloxane/graphene nanocomposites. The pristine DA crosslinked polysiloxane elastomer shows a tensile strength of 0.059 MPa and a self-healing efficiency of >90% (healing condition: 80 °C for 12 h) even after three successive healing cycles. The incorporation of 35 wt% graphene nanosheets results in an 18-fold increase in the tensile strength without compromising the self-healing efficiency.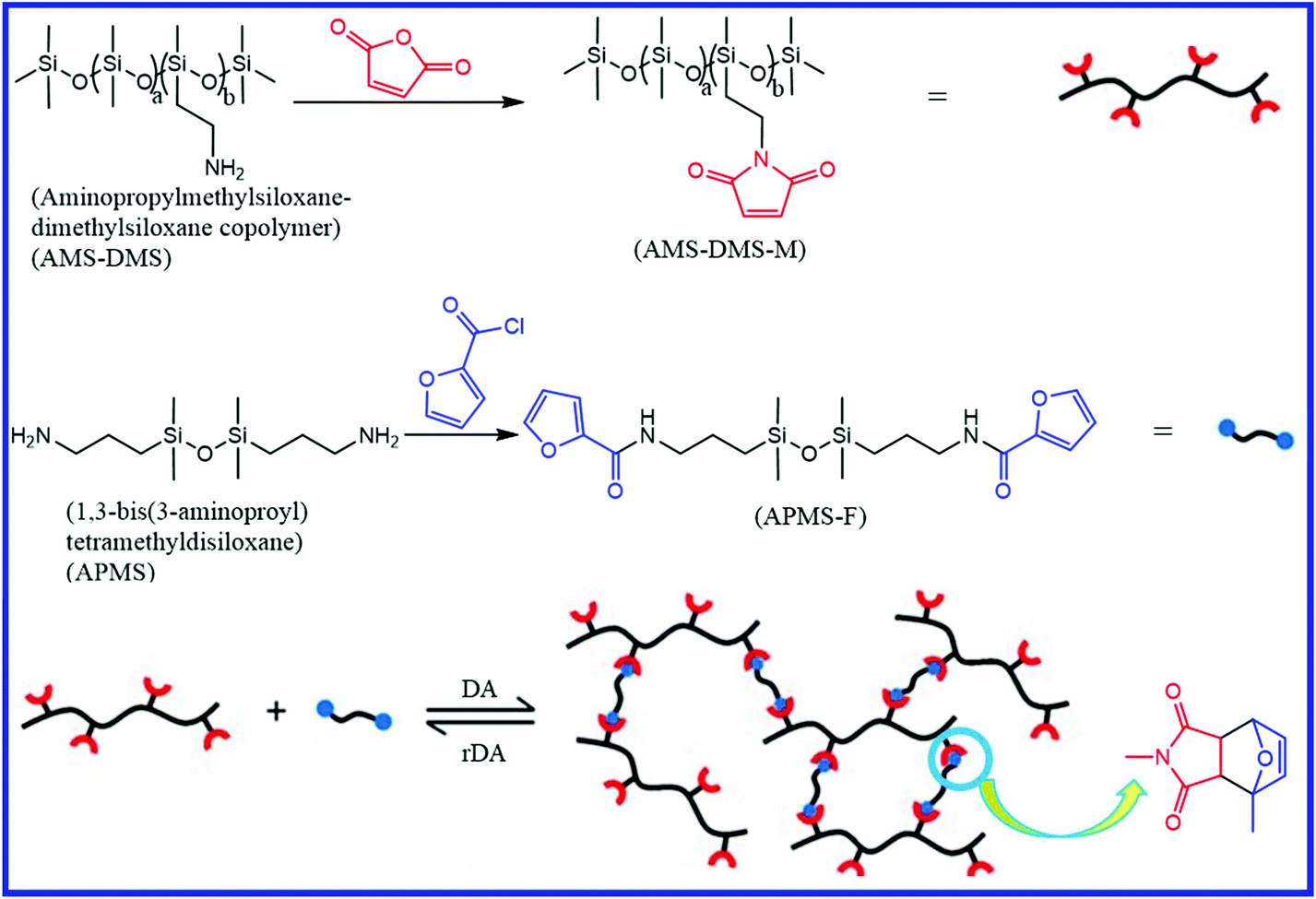 | ||
| Fig. 23 Schematic representation of the preparation of the self-healing polysiloxane elastomer with DA bonds.155 Reproduced from ref. 155 with permission from Royal Society of Chemistry, copyright 2016. | ||
10. Summary
The mechanism of self-healing, healing efficiency, healing conditions, recyclability, and recyclability conditions of the different diolefin-based elastomeric systems is summarized in Table 1.| Rubber/modified rubber/other chemicals | Healing mechanism | Healing conditions | Healing efficiency (%) | Recyclability | Ref. |
|---|---|---|---|---|---|
| Furan functionalized PB & bismaleimide | DA & rDA | — | — | Recyclable at 110 °C | 68 |
| Furan functionalized PB & bismaleimide | DA & rDA | 150 °C & 60 °C | — | Recyclable at 160 °C | 69 |
| PB/polysulfide | Disulfide metathesis | UV irradiation | 93–97 | Recyclable under UV light | 71 |
| S-crosslinked PB/CuCl2 | Disulfide exchange | 110 °C 12 h | 75 | Recyclable at 110 °C | 72 |
| PB oligomers having carboxylic acid and amine groups/tri-thiol crosslinker | Ionic hydrogen bonds | RT, 1 h | 33–93 | — | 73 |
| PB/1H,1H,2H,2H-perfluorodecanethiol | Crystallite melting | 80 °C | — | — | 75 |
| XSBR/NCS composites (NCS 20 wt%) | Supramolecular hybrid network | RT, 24 h | 92 | — | 80 |
| Furan functionalized SBR/MWCNT & bismaleimide | DA & rDA | 100 °C, 5 h | 90 | Recyclable at 130–150 °C | 81 |
| Furan functionalized NR & bismaleimide | DA & rDA | 100 °C, 4 h & 40 °C, 3–7 h | 38–85 | Recyclable | 85 |
| Carboxylated IR & NaOH | Ionic interaction | 28 °C | 80 | — | 86 |
| NR/methacrylic acid/ZnO | Ionic interaction | RT, 20 min | 96–100 | — | 91 |
| Dopamine modified ENR/Fe3+ | Reversible coordinate bond | 50 °C, 12–24 h | 89–93 | — | 96 |
| ZnCl-decorated ENR/histidine modified CNC | Reversible coordinate bond | RT, 2 min | 107 | — | 97 |
| Maleic anhydride grafted NR/3-amino-1,2,4-triazole/ZnCl2 | Hydrogen bond & reversible coordinate bond | 80 °C, 12–24 h | 74 | — | 100 |
| ENR/dithiol-bearing boronic ester/ZnCl2 | boronic ester transesterifications & reversible coordinate bond | 80 °C, 24 h | 60–100 | Recyclable at 160 °C | 101 |
| NR/S/accelerator (CBS) a/s = 0.2 | Disulfide exchange | 70 °C, 7 h | 3–80 | — | 105 |
| ENR/dithiodibenzoic acid/4,4′-dithiodianiline/S | Disulfide metathesis and hydrogen bonding | 120 °C, 5 h | 98 | Recyclable at 155 °C | 107 |
| ENR/chitin nanocrystals (20 phr) | Hydrogen bonding | RT, 24 h | 83 | — | 108 |
| ENR/cellulose nanocrystals (20 phr) | Hydrogen bonding | RT, 24 h | 86 | — | 111 |
| ENR/TEMPO-oxidized cellulose nanocrystals | Transesterification reaction | 160 °C, 3 h | 80 | Recyclable at 160 °C | 112 |
| NR/SiO2/MgSO4 (activator and sulfur curing agents) | Hydrogen bonding and ionic interaction | 50 °C, 24 h | 51–79 | — | 114 |
| Serine modified Ti3C2MXenes/serine modified ENR | Hydrogen bonding | RT, 90 min | 99.3 | — | 115 |
| BIIR/1-butylimidazole | Ionic interaction | RT–100 °C, 1–192 h | — | Recyclable in solvent or above 130 °C | 126 |
| BIIR/1-methylimidazole | Ionic interaction | 70 °C, 16 h (pressed together with a compression of 0.1 mm) | 18 ± 13 | 127 | |
| BIIR/1-butylimidazole | 25 ± 16 | ||||
| BIIR/1-hexylimidazole | 74 ± 43 | ||||
| BIIR/1-nonylimidazole | 57 ± 39 | ||||
| BIIR/1-butylimidazole | Ionic interaction and acid–base interaction | 70 °C, 16 h (under slight pressure) | 87 ± 50 | 133 | |
| BIIR/1-butylimidazole/pristine silica | 40 ± 25 | ||||
| BIIR/1-butylimidazole/alkyl functionalized silica | 43 ± 23 | ||||
| BIIR/1-butylimidazole/imidazolium functionalized silica | 32 ± 16 | ||||
| NBR/cobalt neocaprate | Coordination | 190 °C, 10 min | 72 | Recyclable | 136 |
| NBR/sulfur | — | 26 | — | ||
| NBR | 100 °C, 12 h | 4 | — | 138 | |
| NBR/PEI | 27–44 | — | |||
| Furan functionalized NBR & bismaleimide with disulfide moiety | DA, rDA and Disulfide metathesis | 125 °C for 4 h, cooling down to RT and held for 12 h | 60–75–91 | Recyclable at 130 °C | 141 |
| UV irradiation for 4 h | 78 | ||||
| Furan and maleimide functionalized polysiloxane | DA & rDA | 140 °C for 3 h and 80 °C for 1–24 h | 29–95 | Recyclable at 140 °C | 155 |
11. Conclusions, challenges, and future research opportunities
Diels–Alder reaction, disulfide metathesis reaction, ionic interaction, reversible coordinate bond, and reversible hydrogen bonding are important mechanisms that induce self-healing property in elastomeric materials. Each mechanism has its own merits and demerits. Self-healing elastomers based on the Diels–Alder reaction show good mechanical property but need high temperatures to show self-healing property. Though the Diels–Alder reaction-based self-healing systems are covalently crosslinked, these can be easily reprocessed by heating above the retro Diels–Alder reaction temperature. The disulfide exchange mechanism, ionic interaction, hydrogen bonding, and reversible coordinate bond induce the self-healing property at somewhat lower temperatures (also depend upon the crosslink density and elastomeric system) but do not provide good mechanical properties. Depending on the elastomeric system, the disulfide mechanism shows self-healing property under thermal or UV irradiation or even at room temperature. The longer exposure of the cut surfaces of disulfide-based self-healing elastomers to the open atmosphere reduces the healing efficiency due to the deactivation of the sulfide radicals. In the case of ionically-modified elastomers, the healing efficiency strongly depends on the nature of the ionic moiety. Very weak ionic interaction reduces the healing efficiency and the mechanical properties. The formation of a very strong ionic cluster provides good mechanical properties but reduces the self-healing efficiency. Hence, an optimized ionic interaction is crucial for achieving better self-healing property. The introduction of both permanent and dynamic crosslinks improves the mechanical property but the permanent network reduces the chain dynamics and hence the self-healing property. Thus, it is quite a challenging task to design a self-healing elastomer with good mechanical as well as self-healing properties.The self-healing elastomers based on multi-dynamic networks need to be explored to achieve excellent self-healing and good mechanical properties. For example, aromatic disulfide, ionic interaction, and DA adducts can be introduced to a single elastomeric system. Aromatic disulfide can show self-healing property at room temperature and the ionic group can be manipulated to show the self-healing property at 60–100 °C; furthermore, the DA adduct will show self-healing property at 120–160 °C. In this kind of multi-mechanism healing system, the healing of micro-cracks will occur at room temperature due to disulfide metathesis reaction, whereas the ionic interaction (physical crosslinks) and DA adducts (covalent crosslinks) will provide good mechanical properties. For a crack of intermediate intensity, the material can be heated above the dissociation temperature of the ionic crosslinks so that the material can heal via a dual mechanism, i.e., the involvement of both ionic and disulfide exchange mechanisms. In case of a bigger crack, the elastomer can be heated above the rDA reaction temperature, which will improve the chain mobility and the healing will take place through the combination of disulfide metathesis, reversible ionic interaction, and reversible DA/rDA reaction. Multi-stimuli (heat, light, chemical, mechanical, etc.) responsive self-healing elastomers can also be explored by introducing various functional moieties into a single elastomeric system. A detailed study of the individual and combined effects of the stimuli on the self-healing efficiency would be more interesting.
There are few reports in the area of self-healing SBR, which has the single largest synthetic rubber application in the rubber industry. High styrene content SBR or hydrogenated SBR with self-healing property will further expand the application areas of SBR. More work should also be done on the development of self-healing CR, which has not been explored much.
Elastomers are mostly used by compounding with different fillers. Conventional fillers hinder the interaction between the functional groups of the self-healing elastomers, resulting in reduced healing efficiency. Fillers with a suitable functional moiety that can reversibly interact with the functional groups of the self-healing elastomers should be explored to achieve optimum self-healing and mechanical properties. Without in-depth studies of self-healing elastomer composites, commercialization is nearly an impossible target. Hence, maximum effort should be given to the development of self-healing elastomeric composite rather than pristine rubber.
Despite the great efforts in the development of self-healing elastomers, there are serious limitations that need to be resolved. Evaluating the self-healing efficiency through tensile measurement or monitoring superficial cracks is insufficient to ensure the behavior in the service of self-healing elastomeric materials. The impact of external environmental conditions on the self-healing property of the elastomers and elastomer composites also needs to be evaluated for real applications.
Elastomers such as PB, SBR, NR, and IIR are mostly used in the tire industry and always perform under cyclic thermal/mechanical stress. Thus, future research should also be focused on mechano-responsive and thermoresponsive self-healing elastomers that can exhibit self-healing property under mechanical/thermal stress. IIR-based rubbers having self-healing properties will help in puncture-proof tires during the application itself. These special PB/SBR/NR used in tire tread/sidewall will increase the life cycle of the tire. Hence, this self-healing concept development and commercialization will have utmost importance and the need of the hour for next-generation automatic vehicles. The application research of these new materials is to be explored in the automotive segment. Another important upcoming application area of elastomers is in the flexible electronic segment. Electronic items, being costly, a higher life cycle is the most desired. If self-healing elastomers can be used in this area, the life cycle of the products can be enhanced. This will in turn reduce the cost of gadgets as well as the desired reduction in electronic wastes.
The increasing demand for elastomeric materials is resulting in huge elastomeric wastes. This review has described the recent advances in smart elastomeric materials, which could be a potential alternative to reduce the elastomeric waste by extending their application life and making them suitable for reprocessing. We hope that this review will serve as an inspiration for the research community to take this field ahead and to make the sense of self-healing elastomers in real life for a sustainable society.
Conflicts of interest
The authors declare no conflict of interest.References
- E. McDonel, K. Baranwal and J. Andries, in Polymer blends, Elsevier, 1978, pp. 263–292 Search PubMed.
- D. Barnard, C. Baker and I. Wallace, Rubber Chem. Technol., 1985, 58, 740–750 CrossRef CAS.
- B.-K. Kwak, Elastomers Compos., 1974, 9, 5–13 Search PubMed.
- K. Hashimoto, A. Maeda, K. Hosoya and Y. Todani, Rubber Chem. Technol., 1998, 71, 449–519 CrossRef CAS.
- T. S. Mroczkowski, US Pat., 5162409, 1992 Search PubMed.
- H. Schnecko, in Rubber recycling, Macromolecular Symposia, Wiley Online Library, 1998, pp. 327–343 Search PubMed.
- B. Adhikari, D. De and S. Maiti, Prog. Polym. Sci., 2000, 25, 909–948 CrossRef CAS.
- L. Bockstal, T. Berchem, Q. Schmetz and A. Richel, J. Cleaner Prod., 2019, 236, 117574 CrossRef CAS.
- J. E. Morin, D. E. Williams and R. J. Farris, Rubber Chem. Technol., 2002, 75, 955–968 CrossRef CAS.
- C. Berrueco, E. Esperanza, F. Mastral, J. Ceamanos and P. García-Bacaicoa, J. Anal. Appl. Pyrolysis, 2005, 74, 245–253 CrossRef CAS.
- S. Mohanty, P. Mukunda and G. B. Nando, Polym. Degrad. Stab., 1995, 50, 21–28 CrossRef CAS.
- L. Imbernon and S. Norvez, Eur. Polym. J., 2016, 82, 347–376 CrossRef CAS.
- A. Jones and H. Dutta, Mech. Mater., 2010, 42, 481–490 CrossRef.
- H. Ur Rehman, Y. Chen, M. S. Hedenqvist, H. Li, W. Xue, Y. Guo, Y. Guo, H. Duan and H. Liu, Adv. Funct. Mater., 2018, 28, 1704109 CrossRef.
- A. M. Wemyss, C. Bowen, C. Plesse, C. Vancaeyzeele, G. T. Nguyen, F. Vidal and C. Wan, Mater. Sci. Eng., R, 2020, 141, 100561 CrossRef.
- J. L. Wietor and R. P. Sijbesma, Angew. Chem., Int. Ed., 2008, 47, 8161–8163 CrossRef CAS PubMed.
- S. R. White, B. J. Blaiszik, S. L. Kramer, S. C. Olugebefola, J. S. Moore and N. R. Sottos, Am. Sci., 2011, 99, 392–399 CrossRef.
- P. Jacob, P. Kashyap, T. Suparat and C. Visvanathan, Waste Manage. Res., 2014, 32, 918–926 CrossRef PubMed.
- Z. Bao, J. Tao and C. Flanigan, Polym. Compos., 2017, 38, 918–926 CrossRef CAS.
- G. Hermenegildo, E. Bischoff, R. Mauler, M. Giovanela, L. Carli and J. Crespo, J. Elastomers Plast., 2017, 49, 47–61 CrossRef CAS.
- T. Shimizu, T. Tsuboi, T. Endo, K. Kobayashi and M. Daitoku, US Pat., 20160311259A1, 2016 Search PubMed.
- S. Mohanty, G. B. Nando, K. Vijayan and N. Neelakanthan, Polymer, 1996, 37, 5387–5394 CrossRef CAS.
- K. Imato and H. Otsuka, Dynamic Covalent Chemistry: Principles, Reactions, and Applications, Wiley Online Library, 2017, pp. 359–387 Search PubMed.
- Y. Yang and M. W. Urban, Adv. Mater. Interfaces, 2018, 5, 1800384 CrossRef.
- K. Urdl, A. Kandelbauer, W. Kern, U. Müller, M. Thebault and E. Zikulnig-Rusch, Prog. Org. Coat., 2017, 104, 232–249 CrossRef CAS.
- S. R. White, N. R. Sottos, P. H. Geubelle, J. S. Moore, M. R. Kessler, S. Sriram, E. N. Brown and S. Viswanathan, Nature, 2001, 409, 794–797 CrossRef CAS PubMed.
- D. Y. Zhu, M. Z. Rong and M. Q. Zhang, Prog. Polym. Sci., 2015, 49, 175–220 CrossRef.
- S. An, M. W. Lee, A. L. Yarin and S. S. Yoon, Chem. Eng. J., 2018, 344, 206–220 CrossRef CAS.
- S. J. Garcia, Eur. Polym. J., 2014, 53, 118–125 CrossRef CAS.
- N. Zhong and W. Post, Composites, Part A, 2015, 69, 226–239 CrossRef CAS.
- Z. Wang, X. Lu, S. Sun, C. Yu and H. Xia, J. Mater. Chem. B, 2019, 7, 4876–4926 RSC.
- P. Cordier, F. Tournilhac, C. Soulié-Ziakovic and L. Leibler, Nature, 2008, 451, 977–980 CrossRef CAS PubMed.
- F. Herbst, D. Döhler, P. Michael and W. H. Binder, Macromol. Rapid Commun., 2013, 34, 203–220 CrossRef CAS PubMed.
- N. Duan, Z. Sun, Y. Ren, Z. Liu, L. Liu and F. Yan, Polym. Chem., 2020, 11, 867–875 RSC.
- P. K. Behera, P. Mondal and N. K. Singha, Polym. Chem., 2018, 9, 4205–4217 RSC.
- S. L. Banerjee, S. Samanta, S. Sarkar and N. K. Singha, J. Mater. Chem. B, 2020, 8, 226–243 RSC.
- S. Burattini, H. M. Colquhoun, B. W. Greenland and W. Hayes, Faraday Discuss., 2009, 143, 251–264 RSC.
- S. Burattini, H. M. Colquhoun, J. D. Fox, D. Friedmann, B. W. Greenland, P. J. Harris, W. Hayes, M. E. Mackay and S. J. Rowan, Chem. Commun., 2009, 6717–6719 RSC.
- Z. Wang, C. Xie, C. Yu, G. Fei, Z. Wang and H. Xia, Macromol. Rapid Commun., 2018, 39, 1700678 CrossRef PubMed.
- M. Nakahata, Y. Takashima and A. Harada, Macromol. Rapid Commun., 2016, 37, 86–92 CrossRef CAS PubMed.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698–1702 CrossRef CAS PubMed.
- A. A. Kavitha and N. K. Singha, Macromolecules, 2010, 43, 3193–3205 CrossRef CAS.
- N. B. Pramanik, G. B. Nando and N. K. Singha, Polymer, 2015, 69, 349–356 CrossRef CAS.
- A. A. Kavitha and N. K. Singha, ACS Appl. Mater. Interfaces, 2009, 1, 1427–1436 CrossRef CAS PubMed.
- P. K. Behera, P. Mondal and N. K. Singha, Macromolecules, 2018, 51, 4770–4781 CrossRef CAS.
- P. Mondal, P. K. Behera, B. Voit, F. Böhme and N. K. Singha, Macromol. Mater. Eng., 2020, 2000142 CrossRef CAS.
- P. Mondal, G. Jana, P. K. Behera, P. K. Chattaraj and N. K. Singha, Polym. Chem., 2019, 10, 5070–5079 RSC.
- S. Banerjee, B. V. Tawade and B. Améduri, Polym. Chem., 2019, 10, 1993–1997 RSC.
- S. K. Raut, P. K. Behera, T. S. Pal, P. Mondal, K. Naskar and N. K. Singha, Chem. Commun., 2021, 57, 1149–1152 RSC.
- P. Mondal, P. K. Behera and N. K. Singha, Chem. Commun., 2017, 53, 8715–8718 RSC.
- P. Mondal, P. K. Behera and N. K. Singha, Prog. Polym. Sci., 2021, 113, 101343 CrossRef CAS.
- P. Mondal, G. Jana, P. K. Behera, P. K. Chattaraj and N. K. Singha, Macromolecules, 2020, 53, 8313–8323 CrossRef CAS.
- C.-M. Chung, Y.-S. Roh, S.-Y. Cho and J.-G. Kim, Chem. Mater., 2004, 16, 3982–3984 CrossRef CAS.
- S. Banerjee, R. Tripathy, D. Cozzens, T. Nagy, S. Keki, M. Zsuga and R. Faust, ACS Appl. Mater. Interfaces, 2015, 7, 2064–2072 CrossRef CAS PubMed.
- M. Bag, S. Banerjee, R. Faust and D. Venkataraman, Sol. Energy Mater. Sol. Cells, 2016, 145, 418–422 CrossRef CAS.
- A. Rekondo, R. Martin, A. R. de Luzuriaga, G. Cabañero, H. J. Grande and I. Odriozola, Mater. Horiz., 2014, 1, 237–240 RSC.
- R. Martin, A. Rekondo, A. R. de Luzuriaga, G. Cabañero, H. J. Grande and I. Odriozola, J. Mater. Chem. A, 2014, 2, 5710–5715 RSC.
- P. K. Behera, S. K. Raut, P. Mondal, S. Sarkar and N. K. Singha, ACS Appl. Polym. Mater., 2021, 3, 847–856 CrossRef CAS.
- C. Kim, H. Ejima and N. Yoshie, J. Mater. Chem. A, 2018, 6, 19643–19652 RSC.
- C.-H. Li, C. Wang, C. Keplinger, J.-L. Zuo, L. Jin, Y. Sun, P. Zheng, Y. Cao, F. Lissel and C. Linder, Nat. Chem., 2016, 8, 618–624 CrossRef CAS PubMed.
- B. Jony, M. Thapa, S. Mulani and S. Roy, In Proceedings of the American Society for Composites—Thirty-third Technical Conference, 2018.
- S. García, H. Fischer and S. Van Der Zwaag, Prog. Org. Coat., 2011, 72, 211–221 CrossRef.
- L. Dai, X. Wang, Z. Bu, B. G. Li and S. Jie, J. Appl. Polym. Sci., 2019, 136, 46934 CrossRef.
- C. M. Geiselhart, J. T. Offenloch, H. Mutlu and C. Barner-Kowollik, ACS Macro Lett., 2016, 5, 1146–1151 CrossRef CAS.
- L. Xu, B.-G. Li, S. Jie, Z. Li and Z. Bu, Ind. Eng. Chem. Res., 2019, 58, 13085–13092 CrossRef CAS.
- P. K. Behera, P. Mandal, M. Maiti, R. V. Jasra and N. K. Singha, Rubber Chem. Technol., 2016, 89, 335–348 CrossRef CAS.
- O. Türünç and M. A. Meier, Macromol. Rapid Commun., 2010, 31, 1822–1826 CrossRef PubMed.
- E. Trovatti, T. M. Lacerda, A. J. Carvalho and A. Gandini, Adv. Mater., 2015, 27, 2242–2245 CrossRef CAS PubMed.
- J. Bai, H. Li, Z. Shi and J. Yin, Macromolecules, 2015, 48, 3539–3546 CrossRef CAS.
- P. Berto, A. Pointet, C. Le Coz, S. Grelier and F. Peruch, Macromolecules, 2018, 51, 651–659 CrossRef CAS.
- H. Xiang, J. Yin, G. Lin, X. Liu, M. Rong and M. Zhang, Chem. Eng. J., 2019, 358, 878–890 CrossRef CAS.
- H. Xiang, H. Qian, Z. Lu, M. Rong and M. Zhang, Green Chem., 2015, 17, 4315–4325 RSC.
- D. Wang, J. Guo, H. Zhang, B. Cheng, H. Shen, N. Zhao and J. Xu, J. Mater. Chem. A, 2015, 3, 12864–12872 RSC.
- R. Jasra, M. Maiti and V. Srivastava, US Pat., 20150045496A1, 2015 Search PubMed.
- F. Xu, X. Li, D. Weng, Y. Li and J. Sun, J. Mater. Chem. A, 2019, 7, 2812–2820 RSC.
- A. F. Halasa, J. Prentis, B. Hsu and C. Jasiunas, Polymer, 2005, 46, 4166–4174 CrossRef CAS.
- M. Alimardani and F. Abbassi-Sourki, J. Compos. Mater., 2015, 49, 1267–1282 CrossRef CAS.
- S. Mukhopadhyay, P. Sahu, H. Bhajiwala, S. Mohanty, V. Gupta and A. K. Bhowmick, J. Mater. Sci., 2019, 54, 14986–14999 CrossRef CAS.
- C. Xu, X. Huang, C. Li, Y. Chen, B. Lin and X. Liang, ACS Sustainable Chem. Eng., 2016, 4, 6981–6990 CrossRef CAS.
- C. Xu, J. Nie, W. Wu, L. Fu and B. Lin, Carbohydr. Polym., 2019, 205, 410–419 CrossRef CAS PubMed.
- X. Kuang, G. Liu, X. Dong and D. Wang, Macromol. Mater. Eng., 2016, 301, 535–541 CrossRef CAS.
- N. Candau, L. Chazeau, J.-M. Chenal, C. Gauthier and E. Munch, Phys. Chem. Chem. Phys., 2016, 18, 3472–3481 RSC.
- S. Toki and B. S. Hsiao, Macromolecules, 2003, 36, 5915–5917 CrossRef CAS.
- M. Bruzzone, G. Corradini and F. Amato, Rubber Chem. Technol., 1966, 39, 1593–1607 CrossRef.
- P. Tanasi, M. H. Santana, J. Carretero-González, R. Verdejo and M. A. López-Manchado, Polymer, 2019, 175, 15–24 CrossRef CAS.
- Y. Miwa, J. Kurachi, Y. Kohbara and S. Kutsumizu, Commun. Chem., 2018, 1, 1–8 CrossRef CAS.
- Y. Miwa, J. Kurachi, Y. Sugino, T. Udagawa and S. Kutsumizu, Soft Matter, 2020, 16, 3384–3394 RSC.
- Y. Miwa, M. Yamada, Y. Shinke and S. Kutsumizu, Polym. Chem., 2020, 11, 6549–6558 RSC.
- X. Yang, J. Liu, D. Fan, J. Cao, X. Huang, Z. Zheng and X. Zhang, Chem. Eng. J., 2020, 389, 124448 CrossRef.
- C. Xu, L. Cao, B. Lin, X. Liang and Y. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 17728–17737 CrossRef CAS PubMed.
- C. Xu, L. Cao, X. Huang, Y. Chen, B. Lin and L. Fu, ACS Appl. Mater. Interfaces, 2017, 9, 29363–29373 CrossRef CAS PubMed.
- J. Liu, C. Xiao, J. Tang, Y. Liu and J. Hua, Ind. Eng. Chem. Res., 2020, 59, 12755–12765 CrossRef CAS.
- M. A. Rahman, L. Sartore, F. Bignotti and L. D. Landro, ACS Appl. Mater. Interfaces, 2013, 5, 1494–1502 CrossRef CAS PubMed.
- Y. Liu, Z. Li, R. Liu, Z. Liang, J. Yang, R. Zhang, Z. Zhou and Y. Nie, Ind. Eng. Chem. Res., 2019, 58, 14848–14858 CrossRef CAS.
- M. A. Rahman, M. Penco, I. Peroni, G. Ramorino, A. M. Grande and L. D. Landro, ACS Appl. Mater. Interfaces, 2011, 3, 4865–4874 CrossRef CAS PubMed.
- Y. Han, X. Wu, X. Zhang and C. Lu, ACS Appl. Mater. Interfaces, 2017, 9, 20106–20114 CrossRef CAS PubMed.
- X. Liu, G. Su, Q. Guo, C. Lu, T. Zhou, C. Zhou and X. Zhang, Adv. Funct. Mater., 2018, 28, 1706658 CrossRef.
- Y. Tang, Q. Guo, Z. Chen, X. Zhang, C. Lu, J. Cao and Z. Zheng, ACS Appl. Mater. Interfaces, 2019, 11, 23527–23534 CrossRef CAS PubMed.
- Q. Guo, B. Huang, C. Lu, T. Zhou, G. Su, L. Jia and X. Zhang, Mater. Horiz., 2019, 6, 996–1004 RSC.
- J. Liu, J. Liu, S. Wang, J. Huang, S. Wu, Z. Tang, B. Guo and L. Zhang, J. Mater. Chem. A, 2017, 5, 25660–25671 RSC.
- Y. Chen, Z. Tang, Y. Liu, S. Wu and B. Guo, Macromolecules, 2019, 52, 3805–3812 CrossRef CAS.
- S. Ghorai, A. K. Jalan, M. Roy, A. Das and D. De, Polym. Test., 2018, 69, 133–145 CrossRef CAS.
- A. Birley, Br. Polym. J., 1989, 21, 362–362 CrossRef.
- N. H. H. Shuhaimi, N. S. Ishak, N. Othman, H. Ismail and S. Sasidharan, J. Elastomers Plast., 2014, 46, 747–764 CrossRef CAS.
- M. Hernández, A. M. Grande, W. Dierkes, J. Bijleveld, S. Van Der Zwaag and S. J. García, ACS Sustainable Chem. Eng., 2016, 4, 5776–5784 CrossRef.
- L. Imbernon, E. Oikonomou, S. Norvez and L. Leibler, Polym. Chem., 2015, 6, 4271–4278 RSC.
- B. Cheng, X. Lu, J. Zhou, R. Qin and Y. Yang, ACS Sustainable Chem. Eng., 2019, 7, 4443–4455 CrossRef CAS.
- J. Nie, W. Mou, J. Ding and Y. Chen, Composites, Part B, 2019, 172, 152–160 CrossRef CAS.
- Y. Chen, J. Nie, C. Xu, W. Wu and Z. Zheng, ACS Sustainable Chem. Eng., 2019, 8, 1285–1294 CrossRef.
- J. Nie, J. Huang, J. Fan, L. Cao, C. Xu and Y. Chen, ACS Sustainable Chem. Eng., 2020, 8, 13724–13733 CrossRef.
- L. Cao, D. Yuan, C. Xu and Y. Chen, Nanoscale, 2017, 9, 15696–15706 RSC.
- L. Cao, J. Fan, J. Huang and Y. Chen, J. Mater. Chem. A, 2019, 7, 4922–4933 RSC.
- M. A. Sattar, S. Gangadharan and A. Patnaik, ACS Omega, 2019, 4, 10939–10949 CrossRef PubMed.
- S. Utrera-Barrios, M. H. Santana, R. Verdejo and M. A. López-Manchado, ACS Omega, 2020, 5, 1902–1910 CrossRef CAS PubMed.
- Q. Guo, X. Zhang, F. Zhao, Q. Song, G. Su, Y. Tan, Q. Tao, T. Zhou, Y. Yu and Z. Zhou, ACS Nano, 2020, 14, 2788–2797 CrossRef CAS PubMed.
- R. N. Webb, T. D. Shaffer and A. H. Tsou, Kirk–Othmer Encyclopedia of Chemical Technology, 2000 Search PubMed.
- R. Cao, X. Zhao, X. Zhao, X. Wu, X. Li and L. Zhang, Ind. Eng. Chem. Res., 2019, 58, 16645–16653 CrossRef CAS.
- P. Xie, K. Wang, J. Zhang, Y. Hu and G. Luo, Ind. Eng. Chem. Res., 2018, 57, 10883–10892 CrossRef CAS.
- J. S. Parent, G. D. White, R. A. Whitney and W. Hopkins, Macromolecules, 2002, 35, 3374–3379 CrossRef CAS.
- J. S. Parent, A. Penciu, S. A. Guillén-Castellanos, A. Liskova and R. A. Whitney, Macromolecules, 2004, 37, 7477–7483 CrossRef CAS.
- G. Davidson, D. Adkinson, S. Malmberg, L. Ferrari, C. Siegers and S. Chadder, US Pat., 9388258B2, 2016 Search PubMed.
- L. Ferrari, N. Suhan, C. Siegers and T. B. Carmichael, US Pat., 9969824B2, 2018 Search PubMed.
- J. S. Parent, A. M. Porter, M. R. Kleczek and R. A. Whitney, Polymer, 2011, 52, 5410–5418 CrossRef CAS.
- A. Ozvald, J. S. Parent and R. A. Whitney, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2438–2444 CrossRef CAS.
- A. Das, A. Sallat, F. Böhme, E. Sarlin, J. Vuorinen, N. Vennemann, G. Heinrich and K. W. Stöckelhuber, Polymers, 2018, 10, 94 CrossRef PubMed.
- A. Das, A. Sallat, F. Böhme, M. Suckow, D. Basu, S. Wießner, K. W. Stöckelhuber, B. Voit and G. Heinrich, ACS Appl. Mater. Interfaces, 2015, 7, 20623–20630 CrossRef CAS PubMed.
- M. Suckow, A. Mordvinkin, M. Roy, N. K. Singha, G. Heinrich, B. Voit, K. Saalwächter and F. Böhme, Macromolecules, 2017, 51, 468–479 CrossRef.
- A. Mordvinkin, M. Suckow, F. Böhme, R. H. Colby, C. Creton and K. Saalwächter, Macromolecules, 2019, 52, 4169–4184 CrossRef CAS.
- S. Stein, A. Mordvinkin, B. Voit, H. Komber, K. Saalwächter and F. Böhme, Polym. Chem., 2020, 11, 1188–1197 RSC.
- H. Kim, A. L. Yarin and M. W. Lee, Composites, Part B, 2020, 182, 107598 CrossRef CAS.
- H. H. Le, F. Böhme, A. Sallat, S. Wießner, M. auf der Landwehr, U. Reuter, K. W. Stöckelhuber, G. Heinrich, H. J. Radusch and A. Das, Macromol. Mater. Eng., 2017, 302, 1600385 CrossRef.
- H. Le, S. Hait, A. Das, S. Wiessner, K. Stöckelhuber, F. Böhme, U. Reuter, K. Naskar, G. Heinrich and H.-J. Radusch, eXPRESS Polym. Lett., 2017, 11, 230–242 CrossRef CAS.
- A. Sallat, A. Das, J. Schaber, U. Scheler, E. S. Bhagavatheswaran, K. W. Stöckelhuber, G. Heinrich, B. Voit and F. Böhme, RSC Adv., 2018, 8, 26793–26803 RSC.
- H. Mou, F. Shen, Q. Shi, Y. Liu, C. Wu and W. Guo, Eur. Polym. J., 2012, 48, 857–865 CrossRef CAS.
- Z. Cheng, M. Yan, L. Cao, J. Huang, X. Cao, D. Yuan and Y. Chen, Ind. Eng. Chem. Res., 2019, 58, 3912–3920 CrossRef CAS.
- Z. F. Zhang, X. T. Liu, K. Yang and S. G. Zhao, Macromol. Res., 2019, 27, 803–810 CrossRef CAS.
- M. Das, S. Pal and K. Naskar, eXPRESS Polym. Lett., 2020, 14, 860–880 CrossRef CAS.
- A. C. Schüssele, F. Nübling, Y. Thomann, O. Carstensen, G. Bauer, T. Speck and R. Mülhaupt, Macromol. Mater. Eng., 2012, 297, 411–419 CrossRef.
- S. Utrera-Barrios, J. Araujo-Morera, L. P. de Los Reyes, R. V. Manzanares, R. Verdejo, M. Á. López-Manchado and M. H. Santana, Eur. Polym. J., 2020, 110032 CrossRef CAS.
- L. Feng, H. Zhao, X. He, Y. Zhao, L. Gou and Y. Wang, Polym. Eng. Sci., 2019, 59, 1603–1610 CrossRef CAS.
- S. Sarkar, S. L. Banerjee and N. K. Singha, Macromol. Mater. Eng., 2021 DOI:10.1002/mame.202000626.
- H. P. Xiang, M. Z. Rong and M. Q. Zhang, ACS Sustainable Chem. Eng., 2016, 4, 2715–2724 CrossRef CAS.
- S. H. Cho, H. M. Andersson, S. R. White, N. R. Sottos and P. V. Braun, Adv. Mater., 2006, 18, 997–1000 CrossRef CAS.
- S. H. Cho, S. R. White and P. V. Braun, Chem. Mater., 2012, 24, 4209–4214 CrossRef CAS.
- H. Lu and S. Feng, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 903–911 CrossRef CAS.
- Y. Wang, L. Ding, C. Zhao, S. Wang, S. Xuan, H. Jiang and X. Gong, Compos. Sci. Technol., 2018, 168, 303–311 CrossRef CAS.
- Y. Liu, K. Zhang, J. Sun, J. Yuan, Z. Yang, C. Gao and Y. Wu, Ind. Eng. Chem. Res., 2019, 58, 21452–21458 CrossRef CAS.
- Y. Miwa, K. Taira, J. Kurachi, T. Udagawa and S. Kutsumizu, Nat. Commun., 2019, 10, 1–6 CrossRef CAS.
- A. Strąkowska, A. Kosmalska, M. Masłowski, T. Szmechtyk, K. Strzelec and M. Zaborski, Polym. Bull., 2019, 76, 3387–3402 CrossRef.
- X. Zhao and J. Zhang, React. Funct. Polym., 2019, 138, 114–121 CrossRef CAS.
- H. Sun, X. Liu, S. Liu, B. Yu, N. Ning, M. Tian and L. Zhang, Chem. Eng. J., 2020, 384, 123242 CrossRef CAS.
- L. Zhao, X. Shi, Y. Yin, B. Jiang and Y. Huang, Compos. Sci. Technol., 2020, 186, 107919 CrossRef CAS.
- H. Yan, S. Dai, Y. Chen and J. Ding, ChemistrySelect, 2019, 4, 10719–10725 CrossRef CAS.
- K. V. Deriabin, N. A. Ignatova, S. O. Kirichenko, A. S. Novikov and R. M. Islamova, Polymer, 2021, 212, 123119 CrossRef CAS.
- J. Zhao, R. Xu, G. Luo, J. Wu and H. Xia, J. Mater. Chem. B, 2016, 4, 982–989 RSC.
- L. Zhao, B. Jiang and Y. Huang, J. Mater. Sci., 2019, 54, 5472–5483 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |