
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIron-catalyzed urea synthesis: dehydrogenative coupling of methanol and amines†
Elizabeth M.
Lane
a,
Nilay
Hazari
 b and
Wesley H.
Bernskoetter
*c
b and
Wesley H.
Bernskoetter
*c
aDepartment of Chemistry, Brown University, Providence, RI 02912, USA
bDepartment of Chemistry, Yale University, New Haven, CT 06511, USA
cDepartment of Chemistry, The University of Missouri, Columbia, MO 65201, USA. E-mail: bernskoetterwh@missouri.edu
First published on 9th April 2018
Abstract
Substituted ureas have numerous applications but their synthesis typically requires the use of highly toxic starting materials. Herein we describe the first base-metal catalyst for the selective synthesis of symmetric ureas via the dehydrogenative coupling of methanol with primary amines. Using a pincer supported iron catalyst, a range of ureas was generated with isolated yields of up to 80% (corresponding to a catalytic turnover of up to 160) and with H2 as the sole byproduct. Mechanistic studies indicate a stepwise pathway beginning with methanol dehydrogenation to give formaldehyde, which is trapped by amine to afford a formamide. The formamide is then dehydrogenated to produce a transient isocyanate, which reacts with another equivalent of amine to form a urea. These mechanistic insights enabled the development of an iron-catalyzed method for the synthesis of unsymmetric ureas from amides and amines.
Introduction
Ureas are frequent intermediates in organic synthesis and appear in resin precursors, dyes, and agrochemicals.1,2 However, their most prominent role is in pharmaceuticals, where they are key functional groups in a host of medicinal compounds3 such as antibacterial,4 antiatherosclerotic,5 antidepressant,6 and anticancer7 drugs. The most prevalent metal-free methods for synthesizing ureas use highly toxic or dangerous starting materials, such as phosgene and its derivatives, isocyanates, azides,8–10 or carbon monoxide (CO).9,11 Carbon dioxide (CO2) can be used as a non-toxic urea precursor, but these reactions typically require high temperatures and pressures, expensive dehydrating agents, or strong bases.9 Additionally, all current metal-free methods produce stoichiometric amounts of inorganic salts as waste.Transition metal catalysis is a potential strategy for improving the preparation of ureas. Of the most highly explored methods, the synthesis of symmetric ureas via the metal-catalyzed oxidative carbonylation of amines requires high catalyst loadings, generally gives low yields, and often forms significant amounts of side products (such as oxamides, carbamate esters, or CO2). Furthermore, elevated pressures of CO and harsh oxidizing conditions are needed.12–16 Urea generation through the metal-catalyzed reaction of CO2 and substituted amines requires high temperatures and pressures and base additives, and the reaction yields and scope are poor.17–21 Metal-catalyzed dehydrogenative coupling approaches for urea synthesis avoid concerns about high pressures and have the advantage of producing dihydrogen (H2) as the only byproduct. Unfortunately, the few examples of dehydrogenative coupling of formamides and amines22–25 and amide cross-coupling26,27 require high catalyst loadings (2–5 mol%) and most also need a stoichiometric peroxide additive. In addition, the preparation of the formamide starting materials is often tedious and typically employs harsh formylating reagents.30 A desirable alternative is to instead dehydrogenatively couple methanol with an amine to form a formamide intermediate. Subsequent dehydrogenative coupling of the formamide with another equivalent of amine generates a urea (and H2; Scheme 1). The paucity of catalysts suitable for even the first step of this transformation is demonstrated by the fact that while numerous metal catalysts are capable of dehydrogenative amidation,28 only four exhibit any activity with respect to coupling methanol and amines to formamides in the absence of harsh oxidative conditions,29–32 and only one of those does so with turnover numbers (TONs) greater than 50.32 The incorporation of primary amines into dehydrogenative amidation and ureation reactions faces the additional challenge of potential imine formation via dehydration from a plausible hemiaminal intermediate.33 While imines can be valuable and dedicated dehydrogenative imination catalysts exist,34–36 in the case of ureation they represent undesirable side products that detrimentally affect selectivity. As primary amines are required for dehydrogenative ureations that proceed through isocyanate intermediates, the minimization or elimination of imination further restricts the suite of suitable catalysts.
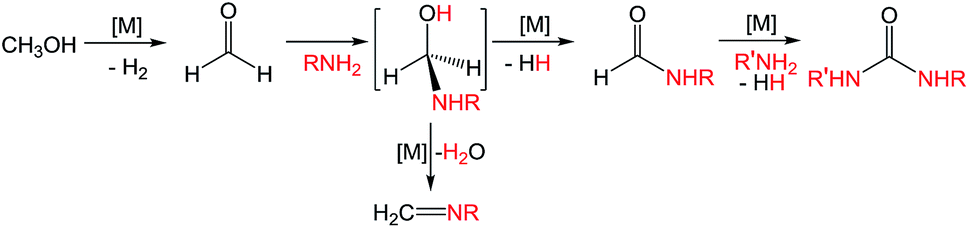 | ||
| Scheme 1 Metal-promoted pathway for dehydrogenative urea formation. | ||
Recently, Kim and Hong described the sole example of catalytic urea formation directly from methanol and amines (Fig. 1).37 Their precious metal ruthenium catalyst exhibits high selectivities and excellent yields for the production of symmetric and unsymmetric ureas, with TONs of up to 190 and 15, respectively, and its applicability toward reversible38 hydrogen storage has been demonstrated.39 However, the synthesis of unsymmetric ureas requires high catalyst loadings and a complicated two-step process. To date, no base-metal catalysts for the production of ureas from methanol and amines have been described, with the most closely related examples instead relying on formamide starting materials24 or isocyanate reagents.40 Base-metal catalysts for dehydrogenative urea synthesis are particularly desirable for pharmaceutical applications where there are stringent requirements regarding product separation from toxic metals.41 Previous work in our laboratories has shown the catalytic ability of iron-pincer complexes of the type RPNPFe(H)(CO) (RPNP = N[CH2CH2(PR2)]2, R = iPr, Cy) for both methanol dehydrogenation42 and the dehydrogenative amidation of alcohols with secondary amines,32 suggesting the applicability of this class of compounds toward ureation. Herein we report the use of iPrPNPFe(H)(CO) (1, Fig. 1) in the first base-metal catalyzed dehydrogenative coupling of methanol and primary amines to selectively form ureas. This system exhibits excellent activities in the absence of any additives and has been employed to isolate ureas on a scale of several hundred milligrams, corresponding to TONs of up to 160 for symmetric ureas and 176 for unsymmetric ureas.
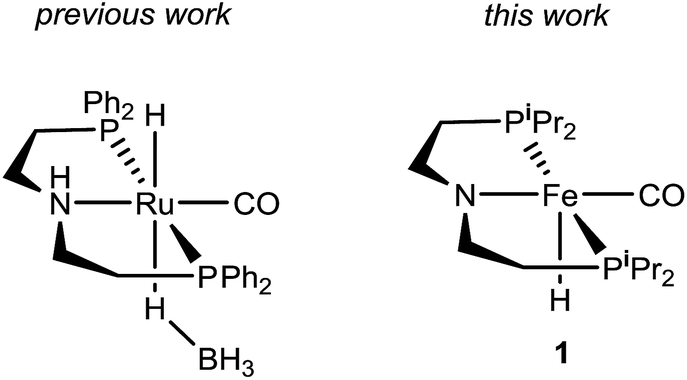 | ||
| Fig. 1 Ruthenium and iron catalysts for symmetric and unsymmetric urea formation from dehydrogenative coupling. | ||
Results and discussion
Catalytic studies
Due to the relative insolubility of 1,3-dicyclohexylurea in most organic solvents and the corresponding ease of post-reaction isolation, initial investigations into the ability of 1 to catalyze the formation of symmetric ureas employed methanol and cyclohexylamine as the substrates. A promising TON of 23 for the formation of 1,3-dicyclohexylurea was observed using a one to four molar ratio of methanol to amine and 0.5 mol% of 1 in 5 mL tetrahydrofuran (THF) at 80 °C for 8 hours. Empirical optimization of the reaction resulted in minimal improvements (Tables S1–S5†) except for increasing the reaction temperature, which nearly tripled the TON for urea formation to 66 (Table 1, entry 12). While the total yield of urea is modest (ca. 33%), sterics may play a limiting kinetic role given the need to attach two amine substrates to the methanol-derived carbonyl carbon of the urea.|
|
|||
|---|---|---|---|
| Entry | Amine | TONc | Yield (%) |
a Reaction conditions: 3 mmol methanol, 12 mmol amine, 0.5 mol% 1, 5 mL THF at 120 °C for 8 hours. Each entry is an average of two trials unless otherwise indicated.
b Average of three trials.
c Based on yield of isolated urea of >99% purity (as determined by 1H NMR spectroscopy) unless otherwise indicated.
d >98% purity.
e >97% purity.
f Mixture of isomers, ∼75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25 cis:trans. :25 cis:trans.
|
|||
| 1 |
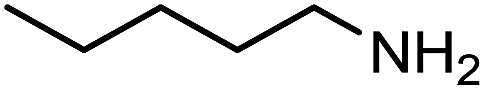
|
160 | 80% |
| 2 |
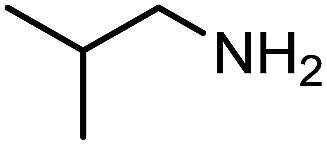
|
150 | 75% |
| 3 |
|
147 | 74% |
| 4 |
|
144 | 72% |
| 5 |
|
12d | 6% |
| 6b |
|
156e | 78% |
| 7 |
|
140 | 70% |
| 8 |
|
126 | 63% |
| 9b |
|
90 | 45% |
| 10 |
|
123 | 62% |
| 11 |
|
116 | 58% |
| 12b |
|
66 | 33% |
| 13 |
|
22e,f | 11% |
| 14 |
|
0 | — |
| 15 |
|
0 | — |
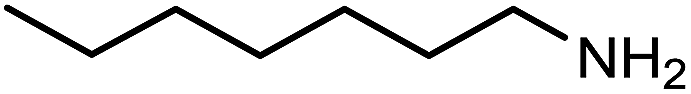
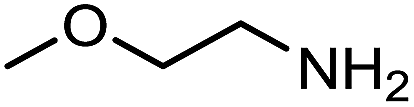
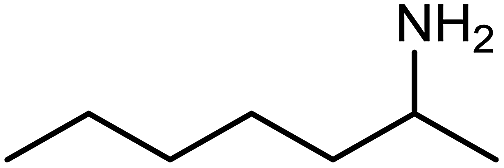
The encouraging preliminary results prompted examination of the substrate scope for dehydrogenative ureation (Table 1). The most productive substrates were terminal alkylamines, with pentylamine giving the highest TON of 160 (80% yield, Table 1, entry 1). Small steric changes such as using a branched alkylamine (isobutylamine, entry 2) or elongating the alkyl chain (heptylamine, entry 3) did not significantly alter catalyst performance. Likewise, capping the alkylamine chain with a methoxy group (2-methoxyethylamine, entry 4) afforded good yields. However, switching from a terminal amine (entry 3) to an internal amine (2-aminoheptane, entry 5) significantly decreased the TON. While initial amidation proceeded favorably (TON = 53 for the formamide), it is possible that the sterically hindered internal amine significantly increased the barrier either for attack by a second equivalent of amine or for the dehydrogenation of intermediates necessary to form the corresponding symmetric urea.
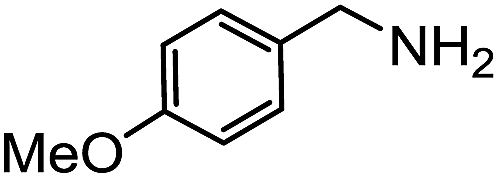
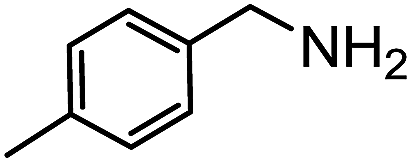
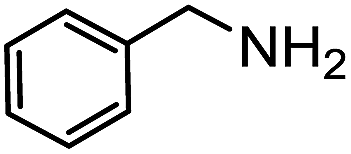
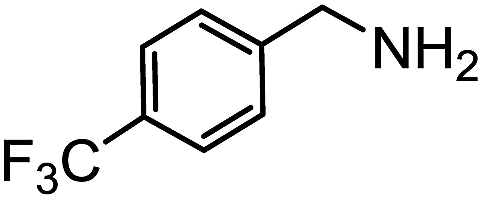
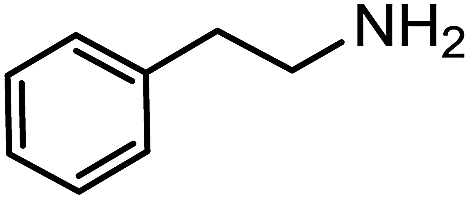
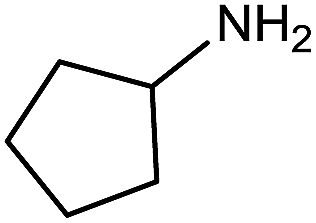
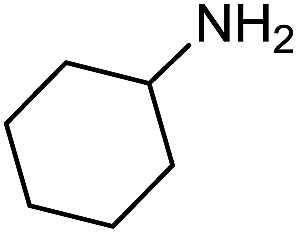
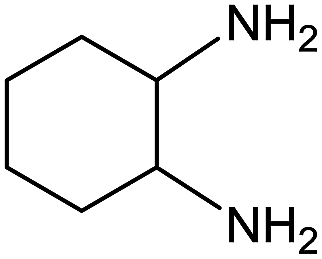
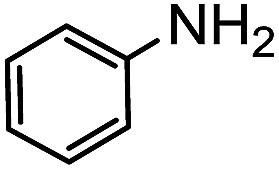
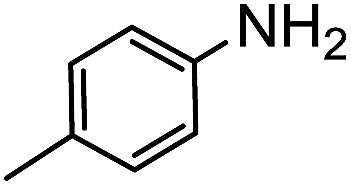
Electronic influences were probed using a series of benzylamine derivatives (entries 6–10) and it was found that the presence of electron-donating substituents such as methoxy and methyl in the para position of the benzyl moiety enhanced the TON (entries 6 and 7, respectively) compared to unsubstituted benzylamine (entry 8). An electron-withdrawing substituent such as a trifluoromethyl group slightly decreased the TON (entry 9). These substituent changes affect the nucleophilicity of the amine substrate which likely explains this trend in TON. Attempts to decrease steric hindrance by switching from benzylamine to 2-phenethylamine had little effect (entries 8 versus 10, respectively), although it is possible that the increased flexibility in the short carbon chain could counteract some of the desired steric relief. Entries 11 and 12 demonstrate that while smaller ring structures can still perform reasonably well, there is significant steric hindrance that increases rapidly with size, as the change of a cyclopentyl group to a cyclohexyl group dropped the TON by nearly half. The attempted synthesis of a cyclic urea (entry 13) from a diamine was successful, although it gave a poor yield compared to its monoamine counterpart (entry 12). This could be partially due to the cis/trans mixture of the starting amine (∼60:40), as formation of the strained trans product is less favorable compared to its cis counterpart (though both cis and trans products were observed), thereby detracting from catalytic performance. However, analogous experiments by Kim and Hong displayed a similar reduced activity toward diamines that was attributed to amine coordination effects, which could also be involved here.37 Finally, aniline and its methyl derivative (entries 14 and 15) reacted poorly under these conditions, giving very little conversion to corresponding formamides and no detectable urea. This is attributed to a lack of nucleophilicity from the poor electron donation of the aryl substituent, which has been a common obstacle for catalytic dehydrogenative coupling reactions using amines.43 Addition of a co-catalytic amount of exogenous base did little to overcome these limitations (Table S6†). Overall, 1 represents one of only two examples of metal complexes capable of catalyzing urea synthesis from alcohols and primary amines. It has the advantage of containing a cheaper, more abundant base metal while still displaying good yields in the production of symmetric ureas from a range of substrates, without the formation of imines. Furthermore, in several cases 1 affords isolated pure urea product on a synthetically useful scale.
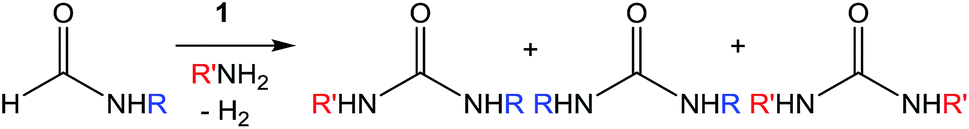
Encouraged by the performance of 1 in the synthesis of symmetric ureas, further investigations addressed the preparation of unsymmetric ureas,44 which are most prevalent as key functional groups in pharmaceuticals.6 While unsymmetric ureas can be acquired via the transamidation of ureas, either by metal-free9 or metal-catalyzed44 means, these methods have limited scope, can require base or reductant additives, and suffer from the same toxicity issues in generating the urea starting materials. Kim and Hong accessed these products from methanol and amines using a one-pot, two-step method that, while effective, required significant and sequential catalyst loadings (6 mol% total, added in two portions) and was restricted to benzylamine as a substrate. The direct reaction of methanol with two different primary amines has been shown to result in a distribution of symmetric and unsymmetric ureas,37 so our approach focused on selectively forming unsymmetric ureas from the reactions of formamides and amines (Table 2). As previously mentioned, the few metal-catalyzed examples of this process exhibit very low TONs (<50).23–25 Re-optimization of the reaction conditions for producing unsymmetric rather than symmetric ureas using 1 resulted in a change of the ratio of starting materials (to 1:1 formamide:amine) and in reaction time from 8 hours to 16 hours (Tables S7–S9†). Initial experiments involving benzylformamide and cyclohexylamine gave excellent yields of the desired unsymmetric urea (Table 2, entry 1) with high selectivity. However, it was found that some scrambling of the starting formamide or the unsymmetric product (or both) had occurred to give trace amounts of both symmetric ureas (Scheme 2). Further experiments under catalytic conditions showed that scrambling of the starting formamide can occur in small amounts in the absence of catalyst, but suggested an iron-catalyzed enhancement of this process (Table S10†). NMR experiments also indicated that while hydrogenation of the unsymmetric urea product to the opposing formamide and amine pair could occur, it was a very minor process (ca. 1.5% conversion) under the conditions studied (10 mol% 1, THF-d8, 120 °C, 16 hours, 1 atm H2; Fig. S1†). Additional NMR experiments revealed that the urea product can undergo further reactions with both amines and formamides in solution, providing yet another pathway to the observed scrambling (Fig. S2–S4†). While insightful, these experiments did not distinguish whether scrambling of the starting formamide or of the product urea was the predominant process for producing the undesirable symmetric ureas (Fig. S5†). Catalytic trials involving the opposing reactant pair of cyclohexylformamide and benzylamine (Table 2, entry 3) revealed a significant decrease in selectivity, presumably due to a corresponding increase in the rate of scrambling compared to unsymmetric ureation.
|
|
|||||
|---|---|---|---|---|---|
| Entry | R | R′ | Yieldb (%) | Conv.c (%) | Sel.d (%) |
| a Reaction conditions: 3 mmol formamide, 3 mmol amine, 0.5 mol% 1, 5 mL THF at 120 °C for 16 hours. Each entry is an average of two trials. b Isolated yield. c Based on formamide consumption. d Selectivity: percentage unsymmetric urea (compared to symmetric ureas) in final product. | |||||
| 1 |
|
|
85% | 89% | 96% |
| 2 |
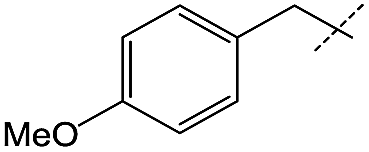
|

|
86% | 93% | 92% |
| 3 |
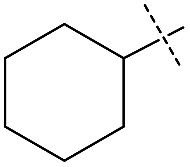
|
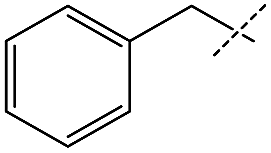
|
49% | 79% | 68% |
| 4 |
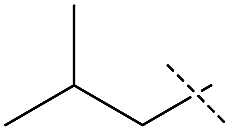
|
|
85% | 86% | 82% |
| 5 |
|

|
11% | 28% | 78% |
| 6 |
|
|
0% | 16% | — |
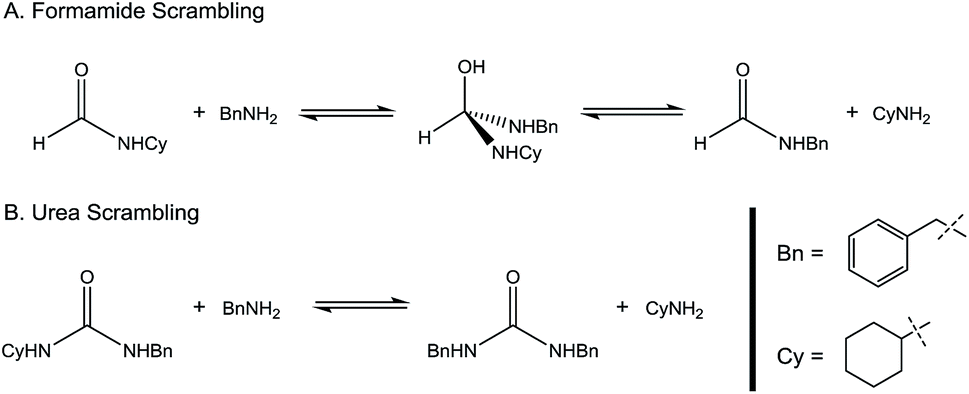 | ||
| Scheme 2 Potential scrambling pathways in unsymmetric urea formation. | ||
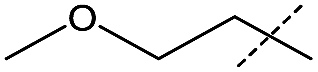
Efforts to enhance yield through minor substituent manipulation did not have a significant effect. While 4-methoxybenzylamine and pentylamine were both high-performing substrates for the formation of symmetric ureas, there was little advantage to the N-(4-methoxybenzyl)formamide and pentylamine combination over benzylformamide and cyclohexylamine in either TON or selectivity (Table 2, entry 2). This indicated that starting from combinations of formamides and amines that minimized scrambling (for example, benzylformamide or its derivatives rather than cyclohexylformamide) was far more important than small electronic or steric changes in substituents. As a result, other experiments with respect to unsymmetric urea production focused on species that were reminiscent of medicinally-relevant ureas (entries 4–6).6
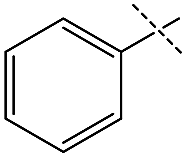
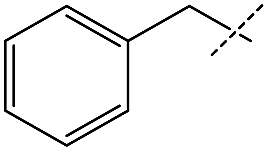
Although the initial reaction of isobutylformamide and ethanolamine did not yield urea, changing the –OH group on the amine to a methoxy group (entry 4) gave excellent turnover, albeit with poorer selectivity than for formamides containing benzyl derivatives. The failure of ethanolamine was attributed to its preference for forming an iron-alkoxide species with the catalyst.32 The production of unsymmetric ureas containing an aniline moiety on one side is highly desirable in a variety of medical applications including for antitumor and anticonvulsant agents,4,7,45 however, formanilide was a poor substrate for dehydrogenative coupling and scrambling to form the more active pentylformamide was observed as the primary process (entry 5). A similar lack of nucleophilicity is reflected in the amine of entry 6 as only formamide scrambling was observed with benzylformamide (a known active substrate). Overall, while the possibility of obtaining unsymmetric ureas in high yields with good selectivities was demonstrated, it was found that kinetic control was far more important than qualitative changes in sterics or electronics. As a result, a full substrate scope would not be as informative and was not performed. Regardless, 1 exhibits TONs over three times greater than the existing catalysts for this method of obtaining unsymmetric ureas23–25 and the one-pot two-step method starting from alcohol and amine,37 and has the added benefits of being both base-metal-catalyzed and additive free.
Mechanistic studies
There are two main pathways proposed for urea formation, as shown in Scheme 3. Kim and Hong postulated that after initial dehydrogenative amidation (that can proceed via either a hemiaminal or hemiacetal intermediate), their ruthenium-catalyzed ureation followed path (a) to generate an aminal, followed by metal-catalyzed dehydrogenation to yield the urea product.37 Alternatively, isocyanate intermediates (usually metal-bound) have been implicated in other metal-mediated urea formation reactions starting from amides,23,27 as well as in the majority of metal-free ureation cases.8,9 These examples would suggest path (b), whereby urea generation stems from metal-catalyzed dehydrogenation of a formamide to an isocyanate, followed by nucleophilic attack of an amine. Mechanistic investigations were therefore performed using 1 to elucidate whether iron-catalyzed urea formation proceeds via an aminal or an isocyanate intermediate. Our previous attempts to make a tetrasubstituted species, such as 1,3-dimorphylurea, were unsuccessful (Fig. S6†). This could be due to the inability of the secondary amide intermediate to undergo dehydrogenation to the isocyanate, although steric hindrance in generating a putative aminal intermediate cannot be ruled out. Therefore, a similar analysis was performed using two synthetic routes for the preparation of 1,1,3-tripentylurea (Scheme 4). While the NMR-scale reaction of pentylformamide and dipentylamine displayed approximately 80% conversion to the desired urea,46 the alternative combination of dipentylformamide and pentylamine did not produce any coupling product (Fig. S7–S8†). The main difference between these two routes is the ability of the starting formamide to form an isocyanate intermediate, as the sterics of the final product are identical. Further evidence for an isocyanate intermediate was obtained through the reaction of cyclohexylformamide with 1 in the absence of other reagents. In this reaction the iron-dihydride species (presumably generated from formamide dehydrogenation) was observed by NMR spectroscopy, along with cyclohexyl isocyanate (confirmed by GC analysis) as the sole organic product (Fig. S9†). These results indicate that iron-catalyzed dehydrogenative ureation most likely proceeds through an isocyanate intermediate as shown in Scheme 3b.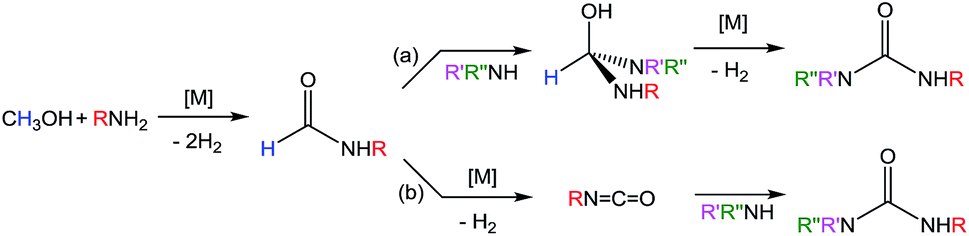 | ||
| Scheme 3 Proposed pathways for metal-catalyzed urea formation from methanol and primary amines. | ||
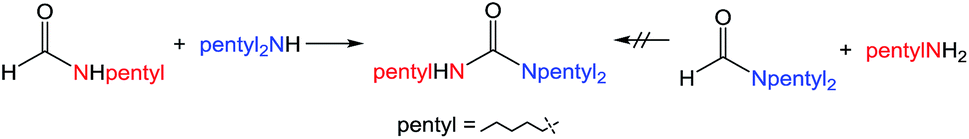 | ||
| Scheme 4 Formamide-dependent synthesis of 1,1,3-tripentylurea. | ||
Conclusions
In conclusion, complex 1 displays excellent catalytic ability for urea formation via the dehydrogenative coupling of methanol and amines. While most precious metal catalysts for the coupling of alcohols and primary amines stop at the formamide, this base metal species has a rare propensity to undergo further coupling to the urea. The performance of 1 in the synthesis of symmetric ureas from methanol and primary amines rivals that of the only other (ruthenium catalyzed) example, and its TONs in the synthesis of unsymmetric ureas from formamides and primary amines are several times better than those of any existing catalysts. In addition, 1 performs these dehydrogenative reactions without the use of an exogenous base or a hydrogen acceptor, enhancing its environmental and economic profile. In contrast to the proposed behavior of the ruthenium catalyst, mechanistic studies suggest that in this case the final ureation step proceeds via an isocyanate intermediate. Trapping the transient isocyanate species with other reagents (such as alcohols to give urethanes)47 may provide alternative synthetic pathways to a suite of desirable compounds starting from dehydrogenative coupling. As previously observed in amidation reactions, however, 1 is highly sensitive to steric hindrance,32 which will necessitate further manipulation of the ancillary ligand environment to ensure its widespread utilization. Furthermore, the occurrence of reagent and product scrambling in the generation of unsymmetric ureas is a drawback to this application of catalyst 1. This issue is not unique to iron and further understanding of ways to control scrambling outside of reagent selection will greatly expand the substrate scope and applicability of metal-mediated unsymmetric urea formation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, Catalysis Science Program, under Award DE-SC0018222 and the Curators of the University of Missouri.References
- A. Abad, J. Loveras and A. Michelena, Field Crops Res., 2004, 87, 257 CrossRef
.
- B. Kocyigit-Kaymakcioglu, A. O. Celen, N. Tabanca, A. Ali, S. I. Khan, I. A. Khan and D. E. Wedge, Molecules, 2013, 18, 3562 CrossRef CAS PubMed
- P. Y. S. Lam, P. K. Jadhav, C. J. Eyermann, C. N. Hodge, Y. Ru, L. T. Bacheler, J. L. Meek, M. J. Otto, M. M. Rayner, Y. N. Wong, C.-W. Chang, P. C. Weber, D. A. Jackson, T. R. Sharpe and S. Erickson-Viitanen, Science, 1994, 263, 380 CAS
- P. Sikka, J. K. Sahu, A. K. Mishra and S. R. Hashim, Med. Chem., 2015, 5, 479 Search PubMed
- K. Matsuda, Med. Res. Rev., 1994, 14, 271 CrossRef CAS PubMed
- I. Gallou, Org. Prep. Proced. Int., 2007, 39, 355 CrossRef CAS
- D. Bankston, J. Dumas, R. Natero, B. Riedl, M.-K. Monahan and R. Sibley, Org. Process Res. Dev., 2002, 6, 777 CrossRef CAS
- H. Babad and A. G. Zeiler, Chem. Rev., 1973, 73, 75 CrossRef CAS
- F. Bigi, R. Maggi and G. Sartori, Green Chem., 2000, 2, 140 RSC
- A. R. Kulkarni, S. Garai and G. A. Thakur, J. Org. Chem., 2017, 82, 992 CrossRef CAS PubMed
- S. C. Srivastava, A. K. Shrimal and A. Srivastava, J. Organomet. Chem., 1991, 414, 65 CrossRef CAS
- P. Giannoccaro, C. F. Nobile, P. Mastrorilli and N. Ravasio, J. Organomet. Chem., 1991, 419, 251 CrossRef CAS
- H. Alper, G. Vasapollo, V. Hartstock and M. Miekuz, Organometallics, 1987, 6, 2391 CrossRef CAS
- P. Giannoccaro, J. Organomet. Chem., 1987, 336, 271 CrossRef CAS
- E. S. Smirnova, J. M. Muñoz Molina, A. Johnson, N. A. G. Bandeira, C. Bo and A. M. Echavarren, Angew. Chem., Int. Ed., 2016, 55, 7487 CrossRef CAS PubMed
- F. Qian, J. McCusker, Y. Zhang, A. D. Main, M. Chlebowski, M. Kokka and L. McElwee-White, J. Org. Chem., 2002, 67, 4086 CrossRef CAS PubMed
- M. T. Zoeckler and R. M. Laine, J. Org. Chem., 1983, 48, 2539 CrossRef CAS
- F. Shi, Q. Zhang, Y. Ma, Y. He and Y. Deng, J. Am. Chem. Soc., 2005, 127, 4182 CrossRef CAS PubMed
- A. Ion, V. Parvulescu, P. Jacobs and D. D. Vos, Green Chem., 2007, 9, 158 RSC
- M. Tamura, K. Ito, Y. Nakagawa and K. Tomishige, J. Catal., 2016, 343, 75 CrossRef CAS
- M. Xu, A. R. Jupp and D. W. Stephan, Angew. Chem., Int. Ed., 2017, 56, 14277 CrossRef CAS PubMed
- S. Kotachi, Y. Tsuji, T. Kondo and Y. Watanabe, J. Chem. Soc., Chem. Commun., 1990, 549 RSC
- T. Kondo, S. Kotachi, Y. Tsuji, Y. Watanabe and T. Mitsudo, Organometallics, 1997, 16, 2562 CrossRef CAS
- G. S. Kumar, R. A. Kumar, P. S. Kumar, N. V. Reddy, K. V. Kumar, M. L. Kantam, S. Prabhakar and K. R. Reddy, Chem. Commun., 2013, 49, 6686 RSC
- V. Krishnakumar, B. Chatterjee and C. Gunanathan, Inorg. Chem., 2017, 56, 7278 CrossRef CAS PubMed
- H. Jiang, A. Lin, C. Zhu and Y. Cheng, Chem. Commun., 2013, 49, 819 RSC
- X. Li, B. Li, J. You and J. Lan, Org. Biomol. Chem., 2013, 11, 1925 CAS
- For a representative example, see: C. Gunanathan, Y. Ben-David and D. Milstein, Science, 2007, 317, 790 CrossRef CAS PubMed
- N. Ortega, C. Richter and F. Glorius, Org. Lett., 2013, 15, 1776 CrossRef CAS PubMed
- B. Kang and S. H. Hong, Adv. Synth. Catal., 2015, 357, 837 Search PubMed
- S. Chakraborty, U. Gellrich, Y. Diskin-Posner, G. Leitus, L. Avram and D. Milstein, Angew. Chem., Int. Ed., 2017, 56, 4229 CrossRef CAS PubMed
- E. M. Lane, K. B. Uttley, N. Hazari and W. H. Bernskoetter, Organometallics, 2017, 36, 2020 CrossRef CAS
- A. Kumar, N. A. Espinosa-Jalapa, G. Leitus, Y. Diskin-Posner, L. Avram and D. Milstein, Angew. Chem., Int. Ed., 2017, 56, 14992 CrossRef CAS PubMed
- B. Gnanaprakasam, J. Zhang and D. Milstein, Angew. Chem., Int. Ed., 2010, 49, 1468 CrossRef CAS PubMed
- A. Mukherjee, A. Nerush, G. Leitus, L. J. W. Shimon, Y. B. David, N. A. Espinosa-Jalapa and D. Milstein, J. Am. Chem. Soc., 2016, 138, 4298 CrossRef CAS PubMed
- B. Saha, S. M. Wahidur Rahaman, P. Daw, G. Sengupta and J. K. Bera, Chem.–Eur. J., 2014, 20, 6542 CrossRef CAS PubMed
- S. K. Kim and S. H. Hong, Org. Lett., 2016, 18, 212 CrossRef CAS PubMed
- E. Balaraman, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2011, 50, 11702 CrossRef CAS PubMed
- J. Kothandaraman, S. Kar, R. Sen, A. Goeppert, G. A. Olah and G. A. S. Prakash, J. Am. Chem. Soc., 2017, 139, 2549 CrossRef CAS PubMed
- X. Huang, T. Zhuang, P. A. Kates, H. Gao, X. Chen and J. T. Groves, J. Am. Chem. Soc., 2017, 139, 15407 CrossRef CAS PubMed
- A. M. Thayer, Chem. Eng. News, 2013, 91, 10 Search PubMed
- S. Chakraborty, P. O. Lagaditis, M. Förster, E. A. Bielinski, N. Hazari, M. C. Holthausen, W. D. Jones and S. Schneider, ACS Catal., 2014, 4, 3994 CrossRef CAS
- For a few examples of catalytic dehydrogenative coupling reactions using aniline as a substrate, see:
(a)
Ref. 28
;
(b) L. U. Nordstrøm, H. Vogt and R. Madsen, J. Am. Chem. Soc., 2008, 130, 17672 CrossRef PubMed
- M. Zhang, S. Imm, S. Bähn, L. Neubert, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2012, 51, 3905 CrossRef CAS PubMed
- J. A. Bergman, K. Woan, P. Perez-Villarroel, A. Villagra, E. M. Sotomayor and A. P. Kozikowski, J. Med. Chem., 2012, 55, 9891 CrossRef CAS PubMed
- See ESI† for more details.
- H. Alper and F. W. Hartstock, J. Chem. Soc., Chem. Commun., 1985, 1141 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization data, and select NMR spectra. See DOI: 10.1039/c8sc00775f |
| This journal is © The Royal Society of Chemistry 2018 |