
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceC–H activation and nucleophilic substitution in a photochemically generated high valent iron complex†
Jia Hui
Lim
ab,
Xenia
Engelmann
c,
Sacha
Corby
bd,
Rakesh
Ganguly
 b,
Kallol
Ray
*c and
Han Sen
Soo
*bef
b,
Kallol
Ray
*c and
Han Sen
Soo
*bef
aEnergy Research Institute@NTU (ERI@N), Nanyang Technological University, Interdisciplinary Graduate School, Research Techno Plaza, Singapore 63755
bDivision of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, 21 Nanyang Link, Singapore 637371. E-mail: hansen@ntu.edu.sg
cHumboldt-Universität zu Berlin, Institut für Chemie, Brook-Taylor-Straβe 2, 12489 Berlin, Germany. E-mail: kallol.ray@chemie.hu-berlin.de
dImperial College London, Department of Chemistry, South Kensington Campus, London, SW7 2AZ, UK
eSingapore-Berkeley Research Initiative for Sustainable Energy, 1 Create Way, Singapore 138602
fSolar Fuels Laboratory, Nanyang Technological University, 50 Nanyang Avenue, Singapore 639798
First published on 23rd March 2018
Abstract
The photochemical oxidation of a (TAML)FeIII complex 1 using visible light generated Ru(bpy)33+ produces valence tautomers (TAML)FeIV (1+) and (TAML˙+)FeIII (1-TAML˙+), depending on the exogenous anions. The presence of labile Cl− or Br− results in a ligand-based oxidation and stabilisation of a radical-cationic (TAML˙+)FeIII complex, which subsequently leads to unprecedented C–H activation followed by nucleophilic substitution on the TAML aryl ring. In contrast, exogenous cyanide culminates in metal-based oxidation, yielding the first example of a crystallographically characterised S = 1 [(TAML)FeIV(CN)2]2− species. This is a rare report of an anion-dependent valence tautomerisation in photochemically accessed high valent (TAML)Fe systems with potential applications in the oxidation of pollutants, hydrocarbons, and water. Furthermore, the nucleophilic aromatic halogenation reaction mediated by (TAML˙+)FeIII represents a novel domain for high-valent metal reactivity and highlights the possible intramolecular ligand or substrate modification pathways under highly oxidising conditions. Our findings therefore shine light on high-valent metal oxidants based on TAMLs and other potential non-innocent ligands and open new avenues for oxidation catalyst design.
Introduction
High-valent iron oxo cofactors are key intermediates in carrying out important metabolic reactions such as hydroxylation, epoxidation, and electrophilic aromatic substitution.1–7 Particularly, the heme-containing metalloenzyme cytochrome P450 has generated enormous scientific interest. P450 can utilise O2 to transform into a highly reactive oxo iron(IV)-porphyrin radical-cation, (Porp˙+)FeIV(O) (Compound I; Porp = porphyrin), capable of driving challenging biological oxidations.8,9 Efforts to understand Compound I have thus inspired many metalloporphyrin and porphyrinoid model compounds.10 Through these studies, (Porp˙+)FeIV(O) is now recognised as the reactive species governing the rate-limiting hydrogen abstraction step to give (Porp)FeIV(OH) (Compound II).8,11The redox non-innocent nature of the porphyrin possibly facilitates the stabilisation of a formal FeV centre in Compound I, thereby contributing to its high electrophilic reactivity. Consistent with the observed high reactivity of the (Porp˙+)FeIV(O) core in biological oxidation reactions, a bioinspired (tpfc˙+)MnIV(OH) species showed a five-fold higher electrophilic reactivity compared to its redox-tautomeric form, (tpfc)MnV(O) (tpfc = 5,10,15-tris(pentafluorophenyl)corrole).12 Similarly, Goldberg and co-workers reported an enhanced rate of electrophilic C–H cleavage in a Lewis acid (LA)-induced MnIV(O-LA)(TBP8Cz˙+) (TBP8Cz = octakis(p-tert-butylphenyl)corrolazinato3−) complex.13,14
Concomitantly, non-heme iron complexes bearing ligands such as tetraamido macrocyclic ligands (TAMLs),15 tripodal tris(carbene),16,17 tetracarbene macrocycles,18 N4Py,19,20 and tetramethyl cyclam (TMC)21 have also been investigated as bioinspired and biomimetic counterparts.22 These complexes have provided seminal insights into pivotal biochemical transformations using oxidants such as O2 and H2O2.1,23,24 In particular, the (TAML)Fe system developed by Collins and coworkers is such a potent oxidant that it has been used for pulp bleaching, degradation of recalcitrant pollutants, water oxidation, and even the destruction of explosives in water.15,25–28
In our continuous efforts to develop (photo)chemical solutions for energy research and environmental applications by using only earth-abundant elements in bioinspired systems,29–32 we explored whether (TAML)Fe complex 1 can be activated for novel oxidative chemistry using light as the energy source instead of O2 or H2O2. Nam and Fukuzumi demonstrated that high valent (N4Py)FeIV(O) intermediates can be generated by 2e− oxidation via irradiating (N4Py)FeII in the presence of Ru(bpy)32+ and sacrificial oxidants.33 They have also applied this concept in the asymmetric epoxidation of terminal olefins via the photocatalytic oxidation of a chiral manganese catalyst, (R,R-BQCN)MnII(OTf)2 (BQCN = N,N-dimethyl-N,N-bis(8-quinolyl)cyclohexane-diamine; OTf = CF3SO3).34 In addition, Panda et al. showed photochemical water oxidation by using biuret-modified (TAML)FeIII catalysts with Ru(bpy)32+ as the photosensitiser, via the intermediacy of (TAML)FeV(O).35 Herein, we describe the photochemical generation of bonafide high valent (TAML)Fe using a visible light driven chromophore.
Through a series of transient absorption spectroscopic (TAS) and electrochemical measurements, we show that photoexcited Ru(bpy)32+ can oxidise 1. Ru(bpy)32+ and a sacrificial oxidant are used as the established photosensitiser system in our proof of concept, although the photosensitiser can be replaced with non-noble-metal chromophores and O2 in our idealised future rendition.29,32,36 Interestingly, in the presence of labile chloride or bromide exogenous anions, the (photo)chemically oxidised solution of 1 comprises an equilibrium mixture of the (TAML)FeIV (1+) and (TAML˙+)FeIII (1-TAML˙+) intermediates. In contrast, in the presence of exogenous cyanide, it culminates in a predominantly metal-based oxidation, yielding solely 1+ (Fig. 1).
 | ||
| Fig. 1 Reaction scheme featuring the reactivity of (photo)chemically oxidised 1, depending on the exogenous anions. The presence of exogenous CN− stabilises the high valent FeIV complex (top), whereas Cl− or Br− results in a ligand-based oxidation and stabilisation of a radical-cationic (TAML˙+)FeIII complex, which subsequently leads to C–H activation and nucleophilic substitution on the TAML aryl ring (bottom). | ||
Extensive mechanistic studies and spectroscopic measurements of the (TAML)FeIV and (TAML˙+)FeIII intermediates reveal that the latter reacts via an unusual C–H activation pathway involving a nucleophilic halogen substitution on the TAML aryl ring (Fig. 1). Notably, this nucleophilic aromatic halogenation reaction mediated by (TAML˙+)FeIII represents a novel domain for high-valent metal reactivity and contrasts sharply with the typical halogenation reactions occurring either via a radical (Cl˙) or an electrophilic (Cl+) mechanism at high-valent metal centres. Our work highlights photochemically accessible high valent (TAML)Fe systems with potential applications in the oxidation of pollutants, hydrocarbons, and water, and provides valuable insights into potential side-reactions with exogenous compounds under highly oxidising conditions. This may open new avenues for C–H activation reactions and oxidation catalyst design based on other redox non-innocent ligands.
Results and discussion
Spectroscopic and electrochemical characterisation of 1
We prepared 1 as previously reported.37 The zero-field Mössbauer spectrum of 1 consists of a major doublet (88%, isomer shift δ = 0.195 mm s−1, quadrupole splitting ΔEQ = 3.949 mm s−1) and a minor doublet (12%, δ = 0.208 mm s−1, ΔEQ = 3.158 mm s−1, Fig. S2c in the ESI†). Both species are consistent with intermediate spin FeIII in S = 3/2 ground states. The doublet with larger ΔEQ is tentatively assigned to the five coordinate [(TAML)FeIII(H2O)]− complex and the one with lower ΔEQ corresponds to the six coordinate [(TAML)FeIII(H2O)2]− complex. In solution, the [(TAML)FeIII(H2O)2]− complex represents the major component as evident from the Mössbauer spectrum of 57Fe-labelled 1 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 water:acetonitrile (ACN) mixture (Fig. S2a in the ESI†). This mixed solvent combination was chosen to accommodate the disparate solubilities of 1 (soluble in ACN) and sodium persulfate (Na2S2O8, soluble in water).
:1 water:acetonitrile (ACN) mixture (Fig. S2a in the ESI†). This mixed solvent combination was chosen to accommodate the disparate solubilities of 1 (soluble in ACN) and sodium persulfate (Na2S2O8, soluble in water).
We verified that the axial ligand(s) contained water molecules with an isotope-labelling experiment. The infrared (IR) spectroscopic measurements of 1, after dissolution and heating at 50 °C in D2O for 15 minutes and then drying in vacuo, reveal new IR bands at 2382 and 2445 cm−1 (Fig. S2f in the ESI†). These bands are slightly redshifted from those of D2O (Fig. S2g in the ESI†). We attribute these new bands to the exchange of H by D in the axially bound water molecule(s). Poor electron paramagnetic resonance (EPR) signal to noise ratios were observed for the FeIII in 571 alone, possibly due to aggregation, as observed by Sullivan et al.38 Thus, no EPR spectroscopic data are presented here. We conducted cyclic voltammetry (CV) experiments (Fig. S3 in the ESI†) and verified that the oxidation of 1 by Ru(bpy)33+ (ERu(III)/Ru(II) ≈ +0.85 V)39 should be thermodynamically feasible, in agreement with the redox potential reported previously in the CV of 1.38
Transient absorption measurements
In light of these electrochemical studies, we performed transient absorption spectroscopic (TAS) experiments to verify if 1 can be photochemically oxidised under partially aqueous conditions, while concurrently characterising the absorption features of the oxidised (TAML)Fe intermediates. We probed the characteristic wavelengths (at 365, 470, and 730 nm in absorbance mode, and 600 nm in emission mode) of the photochemically excited Ru(bpy)32+* to observe the fate of Ru(bpy)32+ upon irradiation with 450 nm laser pulses. Some salient results are summarised in Fig. 2 and Table 1, with other details in ESI Table S1 and Fig. S4–S10.† | ||
| Fig. 2 Spectroscopic characterisation data of the reaction between photochemically generated Ru(bpy)33+ and 1. TAS of Ru(bpy)3Cl2, Co(NH3)5Cl3, and 1, featuring spectral changes over time after laser excitation: (a) 0–10 μs and (b) 0–10 μs with an expanded vertical scale. The squares (■) highlight the absorption features of transient oxidised 1. (c) TAS signal of Ru(bpy)3Cl2 and 1 (red), and of Ru(bpy)3Cl2, Co(NH3)5Cl3, and 1 (blue), probed at 365 nm. The long-timescale (0–10 μs) TAS signal is in the inset. The fits to the kinetics are the lines in black. (d) Steady-state spectra of 1 oxidised by Ru(bpy)33+ (blue). The asterisks (*) at 606 and 828 nm are the characteristic bands of oxidised 1. | ||
| Entry | Lifetimes recorded at different probing wavelengths (ns) | |||
|---|---|---|---|---|
| 365 nma | 470 nma | 730 nma | 600 nmb | |
| a Transient absorption signal. b Transient emission signal. c Ru(bpy)3Cl2 (0.020 mM) only. d Ru(bpy)3Cl2 (0.020 mM) + 1 (0.20 mM). e Ru(bpy)3Cl2 (0.020 mM) + Co(NH3)5Cl3 (10 mM). f Ru(bpy)3Cl2 (0.020 mM) + 1 (0.20 mM) + Co(NH3)5Cl3 (10 mM). g More details can be found in Table S1 in the ESI. | ||||
| 1c | 660 ± 20 | 620 ± 20 | 640 ± 20 | 680 ± 30 |
| 2d | 540 ± 20 | 530 ± 20 | 570 ± 20 | 530 ± 20 |
| 3e | 390 ± 20 | 380 ± 20 | 400 ± 20 | 390 ± 20 |
| 4f | τ 1 = 360 ± 20, τ2 = 3010 ± 150 | τ 1 = 390 ± 20, τ2 = 2840 ± 110 | τ 1 = 370 ± 220, τ2 = 3020 ± 120 | 360 ± 20 |
In the presence of 1 and [Co(NH3)5Cl]2+, the photoluminescence of Ru(bpy)32+* at 600 nm decayed with the shortest lifetime, τ1 (Table 1 entry 4), due to both energy transfer to 1 and oxidative quenching by [Co(NH3)5Cl]2+ (Fig. S8 in the ESI†). Concomitantly, a transient oxidised (TAML)Fe species with an absorption band around 365 nm and broad absorption between 450 and 850 nm (Fig. 2b) was observed. A second new, longer-lived time constant, τ2, of around 3010 ns (Table 1 entry 4) was determined from the transient absorption signals at 365 nm (Fig. 2c), which we attribute to electron transfer from 1 to Ru(bpy)33+. Ru(bpy)33+ can oxidise 1, which then absorbs at 365 nm (blue, inset of Fig. 2c) at longer μs timescales after the RuIII has transformed back to the Ru(bpy)32+ ground state. The oxidation of 1 by Ru(bpy)33+ occurs with a near diffusion-controlled bimolecular rate constant of 3.9 × 109 M−1 s−1 (Fig. S10b in the ESI†), likely due to attraction between the cationic Ru(bpy)33+ and anionic 1.
To verify these experiments, we repeated TAS studies with longer timescales up to 4 seconds. From the signals at 600 and 830 nm, oxidised 1 did not decay in 4 seconds (Fig. S11 in the ESI†). We also added 1 equivalent of photochemically generated Ru(bpy)33+ to 1 to independently obtain the steady-state spectra of the oxidised species. The intermediate (Fig. 2d, blue line) has λmax at 606 nm (ε606 ≈ 1200 dm3 mol−1 cm−1) and 828 nm (ε828 ≈ 1700 dm3 mol−1 cm−1), with an estimated lifetime, τ828, of 27 ± 4 min at room temperature (Fig. S12a in the ESI†), suggesting that the stable steady-state intermediate is indeed oxidised 1.
C–H activation and nucleophilic substitution in (photo)chemically oxidised 1
Interestingly, negative-ion mode electrospray ionisation-mass spectrometry (ESI-MS) of the reaction mixture of Ru(bpy)33+ and 1 revealed high resolution ion peaks at mass-to-charge ratios of m/z = 451.9977 and 485.9579 (34 and 68 mass units higher than those of 1), which could have arisen from the nucleophilic aromatic substitution of Cl (from Ru(bpy)3Cl2) into the TAML ligand of 1. We tentatively propose that the photochemical oxidation of 1 is predominantly ligand-based, leading to the stabilisation of radical-cationic (TAML˙+)FeIII (1-TAML˙+), similar to CeIV oxidation in other (TAML)Fe systems.40,41 The oxidation of the TAML results in the formation of a radical-cation (akin to Compound I in P450) that increases its susceptibility to nucleophilic attack, leading to aromatic substitution in the ligand by Cl−. We propose that our observations arose from the use of an outer-sphere oxidant, Ru(bpy)33+, as compared to the more commonly reported inner-sphere oxidants such as (organic) peroxides.To evaluate this hypothesis, we chemically oxidised 1 in the presence of several anions to independently characterise the products. (ArMe)3N˙+ (ArMe = 4-methylphenyl) has been chosen because it is an outer-sphere oxidant (E0′ = 0.40 V vs. Fc+/Fc) suitable for only one electron oxidation of 1.42 In addition, unlike the 1H NMR spectroscopic signals of 2,2′-bipyridyl from Ru(bpy)32+, those of (ArMe)3N do not overlap with the signals of the TAML, therefore enabling the substituted TAML to be monitored by 1H NMR spectroscopy after deligation. Among the common anions inert towards (ArMe)3N˙+, we screened Cl−, Br−, and CN−.
The negative-ion mode ESI-MS data of the reaction of the mixture of 1 oxidised by (ArMe)3N˙+ (4 equivalents) in the presence of Br− (10 equivalents) show mass and isotopic distributions corresponding to those of 1-TAML(Br) and 1-TAML(Br)2 (Fig. 3a), where one and two bromides have substituted for hydrogens on the aromatic ring in 1, respectively. X-ray photoelectron spectroscopy (XPS) measurements also substantiate the presence of aryl bromides, since the binding energy corresponds to Br bound to carbon instead of anionic Br− coordinated to Fe (Fig. 3b).43 To monitor the progress of the substitution reactions, aliquots were withdrawn at specific time intervals, quenched with Na2S2O3 solutions, deligated in pH 7 phosphate buffer solutions, and then analysed by 1H NMR spectroscopy. Within 15 minutes, the signals corresponding to the TAML disappeared (grey dashed line), accompanied by the emergence of a set of new signals (orange dashed lines), which we propose to be mostly from TAML(Br)2 (Fig. 3c). We assigned the major 1H NMR signals (labelled HA, HB, and HC) to those of one TAML(Br)2 isomer, while the remaining signals belong to the minor isomers of TAML(Br)2 with Br substituted at other positions of the aryl ring (Fig. 3c). We managed to isolate 1-TAML(Br)2 by reversed-phase C18 column chromatography (Fig. S14b in the ESI†), but were not successful at growing single crystals of this complex. On the other hand, the aromatic substitution of Cl− proceeded more sluggishly, as evident from the longer reaction time and the moderate conversions of 1-TAML to 1-TAML(Cl) and 1-TAML(Cl)2, based on the ESI-MS data. As a result, the NMR signals resemble the TAML and the minor products cannot be unambiguously assigned (Fig. S16 in the ESI†).
 | ||
| Fig. 3 Characterisation data of 1-TAML(Br)n generated from the reaction involving 1, (ArMe)3N˙+ (4 eq.), and tetraethylammonium bromide (TEABr, 10 eq.). (a) Negative-ion ESI-MS of the crude reaction mixture 30 minutes after the addition of the (ArMe)3N˙+ oxidant. The assigned intermediates are labelled. (b) Br 3d XPS data of 1-TAML(Br)n. The fit (red, binding energy = 70.7 eV) represents the component completely attributable to aryl bromide. (c) 1H NMR spectra of deligated aliquots of the reaction mixture, withdrawn at time intervals 1 (red), 3 (blue), and 15 (green) minutes after the addition of the (ArMe)3N˙+ oxidant. The spectrum of the TAML before oxidation is shown in black. The dashed lines highlight the characteristic 1H NMR signals from the TAML (grey) and the proposed TAML(Br)2 (orange). The 1H NMR signals (labelled HA, HB, and HC) are proposed to arise from the major TAML(Br)2 isomeric product, while the remaining signals (*) likely originate from another isomer of TAML(Br)2 with Br substitution at a different position of the aryl ring. | ||
Having ascertained that the substitution of Cl− or Br− into the TAML had occurred upon the oxidation of 1, we proceeded to investigate the mechanism. In the presence of 10 equivalents of Cl− or Br−, steady-state UV-visible spectral changes were observed (Fig. S17a in the ESI†). The CV measurements of 1 in the presence of 10 equivalents of Cl− or Br− also revealed a small anodic shift in the first redox wave from Br− to Cl− (Fig. S17c†), suggesting that the Cl− interacts more strongly with 1 compared to Br−. We propose that these changes arise from the exchange of the solvent molecule at the axial position with a halide, thus forming 1(X) (X = Cl or Br). Subsequently, the oxidation of 1(X) should give 1+(X) (left, Fig. 4). Apart from our proposed nucleophilic aromatic substitution on 1(X)-TAML˙+ (top, Fig. 4), we also considered the possibility that the coordinated halide on 1+(X) may be electrophilic. Intermolecular (middle, Fig. 4) or intramolecular (bottom, Fig. 4) electrophilic aromatic substitution could then result in 1(X)-TAML(X) products in the presence of excess halides.
 | ||
| Fig. 4 Possible mechanisms that could lead to 1(X)-TAML(X) products following the oxidation of 1(X) in the presence of excess halides. The compounds marked in red arise from a second molecule of X− or 1, instead of the original 1(X) and its oxidised intermediates. | ||
To probe if an intermolecular mechanism is involved, 1,4-dimethoxybenzene (DMOB) has been chosen as an electron-rich aromatic substrate, with straightforward 1H NMR signals (singlets), to facilitate the monitoring of the substitution reaction progress. In the presence of DMOB (10 equivalents), Br− (10 equivalents), and 1,1,2,2-tetrachloroethane as the internal standard, we oxidised 1 with (ArMe)3N˙+ (2 equivalents). The comparison of the 1H NMR spectroscopic and gas chromatography-mass spectrometric (GC-MS) data of the reaction mixtures before and after oxidation revealed that DMOB remained unreacted, and no signals corresponding to brominated DMOB were detected (Fig. S17e†). Similar to previous experiments in the absence of DMOB, the ESI-MS data of the oxidised reaction mixture show 1-TAML(Br)n (Fig. S17f in the ESI†), indicating that the bromide substitution reaction occurs exclusively at the TAML despite the presence of the electron-rich DMOB. Furthermore, we conducted kinetic measurements to evaluate the rate dependence on the concentration of DMOB. We prepared a solution of 1(Br) (generated in situ by mixing 1 with 10 equivalents of Br− for 5 minutes) and added 2 equivalents of (ArMe)3N˙+ with varying concentrations of DMOB. By monitoring the decay of the UV-visible spectral signals of oxidised 1(Br) over time at the absorption maximum of 893 nm (Fig. S17d†), we fit the kinetic data (inset of Fig. S17d†) to obtain the corresponding pseudo-first-order rate constant, kobs. The plot of kobs against the concentration of DMOB does not appear to have a significant positive correlation (Fig. 5a), suggesting that DMOB is not competitive with the reaction between oxidised 1(Br) and exogenous Br−. These results suggest that an intermolecular electrophilic aromatic substitution mechanism is not operational.
 | ||
| Fig. 5 Kinetic experiments performed to probe the mechanism for the formation of 1-TAML(X). Plot of kobs against the concentration of (a) DMOB and (b) Br−. The linear fit of the data in (b) gives a second-order rate constant, k, for the aromatic substitution of oxidised 1(Br). | ||
To explore the feasibility of an intramolecular electrophilic aromatic substitution mechanism, we conducted kinetic measurements with varying concentrations of exogenous bromide. We reacted 1(Br) (generated in situ by mixing 1 with >10 equivalents of Br− for 5 minutes) with 2 equivalents of (ArMe)3N˙+, with varying concentrations of Br−, and obtained the corresponding kobs using the kinetic data collected by UV-visible spectroscopy. If the reaction occurs due to the nucleophilic attack of Br− on the 1(Br)-TAML˙+ radical cation as we proposed, the kobs should increase with increasing Br− concentration. Conversely, the kobs from an intramolecular electrophilic aromatic substitution should be independent of the Br− concentration. The plot of kobs against Br− concentration reveals a positive linear correlation, giving a second-order rate constant k of 9.6 × 10−2 M−1 s−1 for the aromatic substitution of oxidised 1(Br) (Fig. 5b). In a separate experiment, the analysis of ESI-MS data from the reaction of 10 equivalents of Br− with oxidised 1(Cl) (generated in situ by adding 2 equivalents of (ArMe)3N˙+ to a 1 equivalent solution of Cl− and 1) revealed that predominantly 1-TAML(Br)n products are formed instead of 1-TAML(Cl)n products as expected for an intramolecular chloride migration (Fig. S17g in the ESI†). Patently, the subsequently added Br− has substituted on the aromatic ring in 1 ahead of the already coordinated Cl−. These results indicate that the conventional electrophilic halide aromatic substitution pathways are not operating for our system and instead support our proposed atypical mechanism, where 1(X)-TAML˙+ is attacked by an external anion to form 1-TAML(X) products.
Mössbauer spectroscopic analysis on 57Fe-enriched 1 (571) was carried out to obtain further insights into the oxidation process. In the absence of exogenous anions, the oxidation of 571 gives a new species (δ = 0.153 mm s−1, ΔEQ = 3.843 mm s−1, Fig. 6a; blue component), typical of S = 3/2 (TAML)FeIII complexes (Table 2 entry 4). This clearly supports the ligand-based oxidation of 571, forming 571-TAML˙+. In the presence of labile Br−, the Mössbauer spectrum of the oxidised product (1 min after oxidation) shows a mixture of species comprising of 63% of 571(Br)-TAML˙+ and 29% of a new species (δ = −0.161 mm s−1, ΔEQ = 3.232 mm s−1; Table 2 entry 7), which we assign to the formation of S = 1 (TAML)57FeIV(Br) (571+(Br), red component in Fig. 6b). The major 571(Br)-TAML˙+ component undergoes nucleophilic attack by Br− to eventually form 571-TAML(Br)2 (δ = 0.125 mm s−1, ΔEQ = 4.000 mm s−1; Table 2 entry 8 and Fig. S2e in the ESI†) as the sole product in 90 min (bottom, Fig. 7). Similar valence tautomerisation is observed between 571(Cl)-TAML˙+ and 571+(Cl) in the presence of Cl− (Fig. 6c), but the equilibrium markedly shifts to the FeIV571+ form (Table 2 entry 5).
 | ||
| Fig. 6 Mössbauer spectra of (a) 571 and Ru(bpy)33+, (b) 571, Ru(bpy)33+, and Br−, (c) 571, Ru(bpy)33+, and Cl−, and (d) 571+(CN)2. All Ru(bpy)33+ oxidant solutions were generated photochemically from Ru(bpy)3(PF6)2 precursors to minimise interference from the exogenous anions. Samples (a) to (c) were taken 1 minute after the addition of the oxidant. The black lines are the simulations representing >95% of the absorption. Shaded areas are the spectral contributions from 571-TAML˙+ (blue) and 571+ (red). A high-spin FeIII impurity (5–10%) that we attribute to a deligated by-product is observed in all cases. | ||
| Entry | Reaction mixture | ΔEQ (mm s−1) | δ (mm s−1) | Percentagea (%) | Assigned Fe species |
|---|---|---|---|---|---|
|
a There is a small contribution (the remainder of the sample) from 57Fe species with an unusual ΔEQ (0.3–0.7 mm s−1) and δ (0.2–0.5 mm s−1), which we believe to arise from deligated (TAML)Fe (unshaded region of the Mössbauer spectra).
b Samples were dissolved in 1:1 water:acetonitrile.
c Samples were dissolved in acetonitrile.
d Sample was measured as a solid.
|
|||||
| 1 | 571 | 3.071, 3.850 | 0.182, 0.184 | 63, 34 | FeIII |
| 2 | 571 | 3.071, 3.844 | 0.186, 0.186 | 73, 27 | FeIII |
| 3 | 1 | 3.158, 3.949 | 0.208, 0.195 | 12, 88 | FeIII |
| 4 | 571 + RuIII(bpy)33+ (1 eq.)b | 3.843 | 0.153 | 93 | FeIII-TAML˙+ |
| 5 | 571 + RuIII(bpy)33+ (4 eq.) + Cl− (10 eq.), 1 minb | 3.123, 3.542 | −0.075, 0.105 | 71, 29 | FeIV(Cl), FeIII(Cl)-TAML˙+ |
| 6 | 571 + RuIII(bpy)33+ (4 eq.) + Cl− (10 eq.), 30 minb | 3.123, 3.547 | −0.103, 0.183 | 56, 40 | FeIV(Cl), FeIII-TAML(Cl)n |
| 7 | 571 + RuIII(bpy)33+ (4 eq.) + Br− (10 eq.), 1 minb | 3.232, 3.580 | −0.161, 0.152 | 29, 63 | FeIV(Br), FeIII(Br)-TAML˙+ |
| 8 | 571 + RuIII(bpy)33+ (4 eq.) + Br− (10 eq.), 90 minb | 4.000 | 0.125 | 85 | FeIII-TAML(Br)2 |
| 9 | 571+(CN)2 | 2.977 | −0.141 | 80 | FeIV(CN)2 |
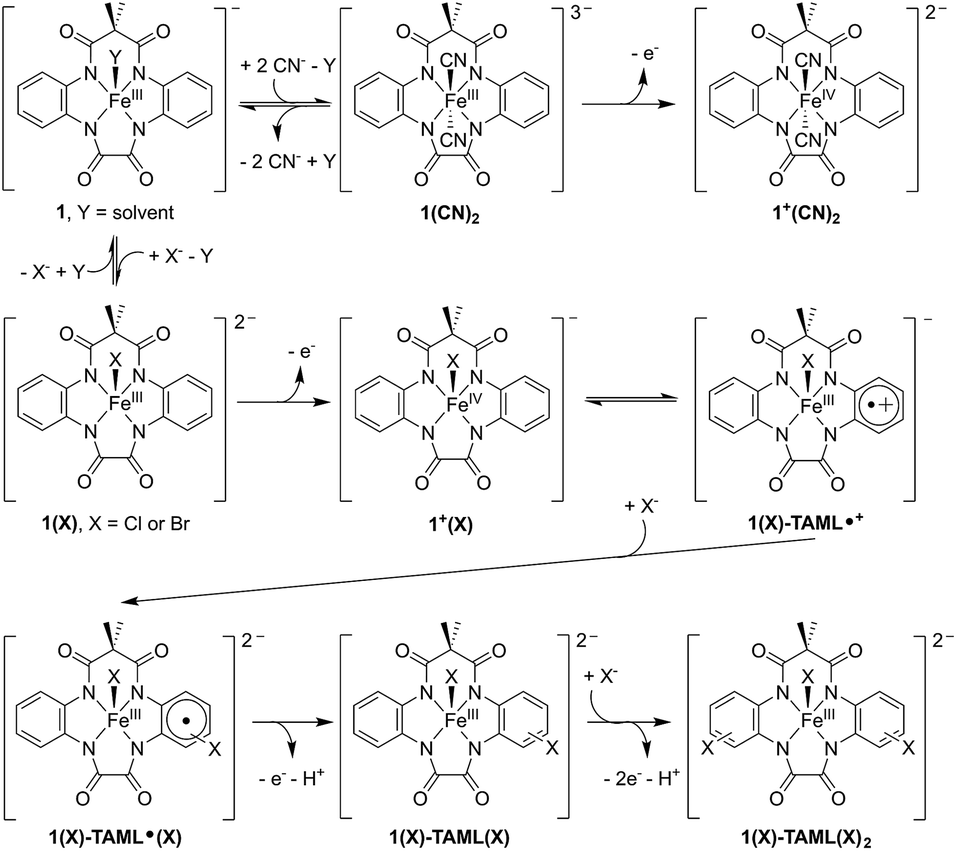 | ||
| Fig. 7 Proposed mechanism for the oxidation of 1, followed by valence tautomerisation between 1(X)-TAML˙+ and FeIV intermediates, depending on the exogenous anion. | ||
Intriguingly, in the presence of excess (10 equivalents) CN−, the UV-visible measurements of 1 reveal a slow formation of a new absorption band with λmax = 440 nm (green, Fig. S17b in the ESI†), which is not observed in the case of Cl− or Br−. The first redox wave in the CV of 1 in the presence of 10 equivalents of CN− becomes more positive compared to the CV of 1 alone, and 1 in the presence of Br− and Cl− (Fig. S17c,† blue). We attribute this observation to the formation of 1(CN)2 (top centre, Fig. 7). The oxidation of 1(CN)2 appeared to solely stabilise one electron-oxidised 1+ as [(TAML)FeIV(CN)2]2− (1+(CN)2, top right, Fig. 7). Negative-ion mode cold-spray ionisation-mass spectrometry (CSI-MS) conducted on the crude reaction mixture at −40 °C revealed that the majority of the FeIV complex was mono-anionic 1+(CN), while a small proportion was identified as the di-anionic complex paired with K+, K[1+(CN)2] (Fig. 8a). Using infrared (IR) spectroscopy (Fig. 8b), we observed a blue-shift in the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretch on going from KCN (2076 cm−1) to [FeIII–CN] (2098 cm−1) to [FeIV–CN] (2127 cm−1), consistent with CN− being coordinated to an increasingly Lewis acidic Fe metal centre, with the highest oxidation state FeIV being most electrophilic. Raman spectroscopic measurements did not give any observable signals for the CN vibration.
N stretch on going from KCN (2076 cm−1) to [FeIII–CN] (2098 cm−1) to [FeIV–CN] (2127 cm−1), consistent with CN− being coordinated to an increasingly Lewis acidic Fe metal centre, with the highest oxidation state FeIV being most electrophilic. Raman spectroscopic measurements did not give any observable signals for the CN vibration.
 | ||
| Fig. 8 Characterisation data of the CN−-stabilised 1+ product generated from the reaction 1 + 5 eq. KCN + 1.05 eq. (ArMe)3N˙+. (a) Negative-ion CSI-MS and the assigned intermediates in the crude reaction mixture containing the dianionic (TAML)FeIV complex [1+(CN)2]2−. (b) Infrared spectra of KCN (black, vmax/cm−1 2076 [KCN]); 1 + 5 eq. KCN (red, vmax/cm−1 2098 [FeIII(CN)]); and 1 + 5 eq. KCN + 1.05 eq. (ArMe)3N˙+ (blue, vmax/cm−1 2127 [FeIV(CN)]). (c) X-ray crystal structure of the dianionic FeIV TAML complex [(1+(CN)2](PPh4)2)], supporting an assignment of the FeIV valency by charge balance. Atom colours: grey for carbon, blue for nitrogen, red for oxygen, yellow for phosphorus, and orange for iron. The hydrogen atoms have been omitted for clarity. The ellipsoids are drawn at 50% probability. Selected bond lengths (Å): Fe1–C20 2.011(6), Fe1–C21 1.993(6), C20–N5 1.137(7), and C21–N6 1.141(7). (d) UV-visible spectrum of a 0.62 mM purified 1+(CN)2 solution. λmax (DCM)/nm 352 (ε/dm3 mol−1 cm−1 1800), 452 (550), 596 (540), 639 (540), 728 (290), and 855 (160). | ||
We isolated the CN−-stabilised FeIV complex (PPh4)2[1+(CN)2] and grew single crystals of the dark blue complex for X-ray crystallography (Fig. 8c). The data were refined to give a disorder model with the TAML occupying two positions, although there was no disorder in the Fe-(CN)2 core (Fig. S19a in the ESI†). Other crystallographic parameters are given in Tables S2–S6 in the ESI.† Mössbauer measurements were performed to confirm the oxidation state of iron in 1+(CN)2. A major doublet (δ = −0.141 mm s−1, ΔEQ = 2.977 mm s−1, Fig. 6d; red component) is consistent with an S = 1 FeIV centre (Table 2, entry 9). To the best of our knowledge, this is the first crystal structure of a high valent cyano-FeIV TAML complex, although Collins and coworkers had spectroscopically characterised a square pyramidal [FeIV(DCB)(CN)]− mono-cyanide before.41,44 Notably, our finding adds to a unique collection of high-valent cyano-FeIV complexes,45,46 with ours being the first di-anionic, octahedral (TAML)FeIV dicyanide complex to be characterised by X-ray crystallography. We also characterised purified 1+(CN)2 using UV-visible spectroscopy (Fig. 8d). Careful scrutiny of the oxidised products of 1 in the presence of different anions (Cl−, Br−, and CN−), therefore, suggests valence tautomerisation between 1+ and 1-TAML˙+ (Fig. 7), governed by the exogenous anion.
From a broader perspective, our findings highlight some of the important aspects for developing photodriven biomimetic oxidation catalysts and an uncommon mechanism for high-valent iron reactivity. Currently, the most commonly used oxidants to obtain high-valent iron species are inner-sphere reagents like peroxides or hypervalent iodine reagents. We report here the generation of (TAML)FeIV (1+) and (TAML˙+)FeIII (1-TAML˙+), by oxidising 1 in the presence of photochemically produced Ru(bpy)33+. The relative proportion of the two tautomers is dependent on the nature of the exogenous anions. The presence of labile Cl− or Br− results in a ligand-based oxidation and stabilisation of a radical-cationic 1-TAML˙+ complex. In contrast, exogenous cyanide culminates in metal-based oxidation to form 1+.
Notably, although valence tautomerisation is observed in some synthetic porphyrinoid counterparts,11,12,14 anion-dependent valence tautomerisation in non-heme iron systems is very uncommon.40,41 The reported radical-cations often show enhanced substrate C–H activation behavior,47 similar to reports on cytochrome P450, and recently on MnIV(O-LA)(TBP8Cz˙+) (LA = Lewis acid, TBP8Cz = octakis(p-tert-butylphenyl)corrolazinato3−) as well.14 However, we show that radical-cations on sterically unprotected ligands, such as, 1-TAML˙+, can undergo unprecedented C–H activation followed by nucleophilic halogen substitution on the TAML aryl ring. The nucleophilic nature of this halogenation reaction contrasts with the more commonly reported mechanisms involving either a halogen radical (X˙) or an electrophilic halogen (X+).40,41,44
Conclusions
In summary, the present work highlights the significance of redox non-innocent ligands under highly oxidising conditions and the possible unexpected reactivity the complexes may have with exogenous compounds, when designing new catalysts for oxidation reactions. These insights should be valuable for future studies and improvements in the designs of biomimetic high valent catalysts. Ligand non-innocence could be an advantage to achieve enhanced oxidation reactions, but the present work highlights the possible alternative reactivity that may be operative for systems involving radical-cations on sterically unprotected ligands. Overcoming the C–H activation followed by nucleophilic halide substitution on the aryl ring will be required to facilitate high turnovers for photocatalytic oxidation reactions involving TAML-based transition metal complexes. Nonetheless, similar nucleophilic reactions could arise in other complexes with redox non-innocent ligands. Whether such a nucleophilic aromatic halogenation reaction mediated by 1-TAML˙+ can also be extended to redox non-innocent ligands such as porphyrinoids is now an intriguing question, which will be investigated in subsequent studies in our laboratories. Moreover, our work also offers a novel approach for intramolecular C–H activation reactions with bona fide high-valent Fe complexes.Experimental
General information
Chemicals were purchased from Sigma-Aldrich, Alfa-Aesar, and Tokyo Chemical Industry (TCI). Unless otherwise stated, the commercial reagents were used as purchased. All reactions involving air- or moisture-sensitive compounds were performed by using standard Schlenk techniques in oven-dried reaction vessels under a nitrogen atmosphere. All UV-visible absorption measurements were carried out on a Shimadzu UV-3600 spectrophotometer, except for kinetic measurements, where an Agilent Cary 8454 spectrophotometer was used instead. Deuterated solvents were purchased from Cambridge Isotope Laboratories and were used as received. The 1H and 13C NMR spectroscopic measurements were recorded on a Bruker AV-500 (500 MHz) NMR spectrometer. The 1H and 13C NMR spectra are reported in parts per million (ppm) downfield referenced to the residual protons of the deuterated solvents.High-resolution mass spectra (HR-MS) were obtained using a Q-TOF Premier LC HR mass spectrometer. Cold-spray ionisation-mass spectrometry (CSI-MS, negative ion mode) was performed using an AccuTOF (JEOL) mass spectrometer equipped with a CSI source. The following conditions were used: needle voltage = −2.0 kV; orifice 1 voltage = 0 V; ring lens voltage = −50 V; and spray temperature = −40 °C. X-ray photoelectron spectroscopy (XPS) data were acquired using a Phoibos 100 spectrometer and a Mg X-ray source (SPECS, Germany). XPS samples were prepared inside a glovebox with a nitrogen atmosphere and SPI double-sided adhesive carbon tape was used to hold the sample on the sample plate. The XPS data were calibrated based on the C 1s position at 284.6 eV and processed using the programme CasaXPS. Elemental analyses were performed with an Elementar vario MICRO cube analyser. Infrared spectroscopic measurements were carried out on a Bruker VERTEX 80 spectrophotometer.
Mössbauer spectra in the absence of magnetic fields were each recorded on a SEECO MS6 spectrometer that consisted of the following instruments: a JANIS CCS-850 cryostat, including a CTI-CRYOGENICS closed-cycle 10 K refrigerator, and a CTI-CRYOGENICS 8200 helium compressor. The cold head and sample mounts are equipped with calibrated DT-670-Cu-1.4L silicon diode temperature probes and heaters. The temperatures are controlled by using a LAKESHORE 335 temperature controller. Spectra are recorded using a LND-45431 Kr gas proportional counter with a beryllium window connected to a SEECO W204 γ-ray spectrometer that includes a high voltage supply, a 10 bit and 5 μs ADC, and two single channel analysers. Motor control and recording of spectra are managed by using a W304 resonant γ-ray spectrometer. For the reported spectra, a RIVERTEC MCO7.114 source (57Co in Rh matrix) with an activity of about 1 GBq was used. The spectra were recorded at 17 K and the data were accumulated for about 1 week for each sample. Mössbauer data were processed and simulated using the WMOSS4 programme (www.wmoss.org). Isomeric shifts are referenced to α-iron at room temperature.
Synthesis of 1
The procedures were adopted and modified from both Sullivan et al. and Ellis et al.38,48 The modified experimental procedures and the characterisation data of the synthetic intermediates can be found in the ESI (Fig. S1 and S20–S23†).Mössbauer spectroscopy
Each Mössbauer sample was prepared by adding appropriate equivalents of the reagent(s) to a 2 mM 1:1 ultrapure water:acetonitrile (ACN) solution of 571. [RuIII(bpy)3(PF6)2]+ (in the absence of chloride) was generated photochemically in the presence of Na2S2O8 using the same procedure described in the section “Oxidation of 1 using independently generated Ru(bpy)33+”. LiCl and LiBr were used as the Cl− and Br− sources respectively, and were dissolved together with 571 before the addition of the oxidant. Upon adding the reagent(s) to the 571 solution, the mixture was shaken vigorously. Subsequently, a ∼0.5 mL aliquot was withdrawn and added to a pre-cooled Mössbauer cup. The half-filled Mössbauer cup was frozen in liquid nitrogen and Mössbauer spectroscopic measurements were conducted on each sample. The Mössbauer data and fits are shown in Fig. 6 and S2 in the ESI,† and their quadrupole splittings (ΔEQ) and isomer shifts (δ) are summarised in Table 2.
Cyclic voltammetry
Cyclic voltammetric measurements were conducted using a Biologic SP-300 potentiostat with 0.10 M n-Bu4NPF6 in anhydrous ACN. A standard three-electrode electrochemical cell was used with a glassy-carbon working electrode (3 mm in diameter from BAS), a Pt wire as the counter electrode, and another Pt wire as the pseudo-reference electrode. The cyclic voltammogram of 1 at various scan rates can be found in Fig. S3 in the ESI.†Nanosecond transient absorption spectroscopic measurements
The transient absorption spectroscopic (TAS) and transient emission spectroscopic (TES) measurements were performed using an Edinburgh Instruments model LP920 transient absorption spectrometer equipped with a pulsed Xe probe lamp in conjunction with a Nd:YAG laser (Continuum model Surelite II-10) as the excitation source. The laser pulse width is 5–8 ns and the repetition rate is 10 Hz. During TAS measurements, the pulses were synchronised with the LP920 system at a frequency of 1 Hz. The pulse energy used was between 4 and 5 mJ per pulse. In all our transient absorption spectra, the detected intensity of the transmitted signals is presented as a logarithm of the ratio of the light intensity from the probe beam after laser excitation to the intensity before laser excitation (ΔOD). Hence, ΔOD refers to the increased absorption (positive ΔOD) or reduced absorption/emission (negative ΔOD) of the transient photoexcited species relative to the ground state.
Pentaamminechlorocobalt(III) chloride (Co(NH3)5Cl3) was added to the sample solution as a solid. Tris(2,2′-bipyridyl)dichlororuthenium(II) hexahydrate (Ru(bpy)3Cl2) was prepared as a 1.0 mM stock solution in a volumetric flask. The stock solution was stored in a refrigerator for a maximum period of one week. The Na salt of 1 was purified by reversed-phase column chromatography and used in all transient absorption measurements. Similarly, a 1.0 mM stock solution of 1 was prepared. A stock solution of 1 was freshly prepared every day, since its stability in ACN is moderate. The quality of 1 may affect the absolute lifetimes of the transient absorption measurements. Therefore, the same batch of 1 was used throughout the whole set of experiments.
The samples were added in the solid form or as stock solutions into a 3.5 mL quartz cuvette (LATECH™, Model: Q-204). Ultrapure water (H2O, Milli-Q Advantage A10, TOC ≤ 5 ppb) and ACN (HPLC grade) were added to make a final volume of 3.0 mL with a ratio of H2O:ACN = 3:1. The cuvette was then fitted with a rubber septum and sealed using Parafilm M®. The cuvette was sonicated to ensure that the solution was homogeneous. Then with a needle fitted through the rubber septum as a gas outlet, argon (Ar) gas was bubbled through the solution for 5 minutes and its transient absorption properties were measured. The detailed composition of each set of experiments is depicted in Table S1 in the ESI.†
Each set of data was fitted to a single exponential or biexponential function to obtain the time constant(s) for the transient absorption signal according to the following two equations respectively:
| y = y0 + A1e−(t − τ0)/τ1 |
| y = y0 + A1e−(t − τ0)/τ1 + A1e−(t − τ0)/τ2 |
The parameters y0, τ0, An, and τn were determined by using a least-squares fitting procedure in Origin. The term y0 corresponds to the vertical intercept at long lifetimes and indicates whether the signal decays to a ‘permanent’ bleach (negative y0) or absorption (positive y0). The term τ0 is the delay time of the excitation pulse from the start of the probe measurement during each photoexcitation cycle. An is the change in optical density after irradiation for the nth exponential term and τn is the corresponding time constant.
The errors corresponding to the fits of the transient signal lifetimes were determined via the principle of error propagation by calculating the root-mean-square deviation from the sum of squares of the uncertainties in each measured value. For each time-resolved measurement, the associated uncertainties included the laser pulse duration, spectrometer time-resolution, mass of samples, and volumes of samples. Hence, the error of the transient signal lifetime, δτ, was calculated according to the following equation:

Transient absorption measurements on a four-second timescale
The same instrumental setup and sample preparations as described in the section “Nanosecond Transient Absorption Spectroscopic Measurements” were used. Instead of the Xe900 xenon lamp, a tungsten halogen lamp (50 W, 12 V) was employed as the probe beam because it would be more stable over a longer timescale.Oxidation of 1 using independently generated Ru(bpy)33+
Ru(bpy)33+ was independently generated using sodium persulfate (Na2S2O8) for stoichiometric oxidation reactions of the Na salt of 1. Co(NH3)5Cl3 has not been used as the sacrificial electron acceptor because both CoII and CoIII are coloured and will interfere with the signals from Ru(bpy)32+, 1, and the product(s) formed. In addition, after Co(NH3)5Cl3 was reduced, a precipitate that was likely to be the poorly soluble Co(OH)2 was formed, which would further complicate UV-visible spectroscopic measurements.The experiment was carried out in a dark room. Stock solutions of 1 and Ru(bpy)3Cl2 were prepared in the same way as those described above in the section “Nanosecond transient absorption spectroscopic measurements”. Ru(bpy)3Cl2 (2.5 mL of a 1.0 mM stock solution) was added into a Schlenk flask connected to a N2 inlet, following which H2O (2.5 mL) was added. Na2S2O8 (6.0 mg) was then added to the solution, giving a composition of [Ru(bpy)3Cl2] = 0.50 mM and [Na2S2O8] = 5.0 mM. The solution was bubbled with N2 for 10 minutes and then irradiated with blue LEDs (450 nm, ∼1 W) for 10 minutes. The solution turned from yellow (Ru(bpy)32+) to green (Ru(bpy)33+). Aliquots (0.20 mL, 1 equivalent or 0.40 mL, 2 equivalents) of the solution were drawn using a micropipette and added into a 1 mL volumetric flask. The Na salt of 1 (0.10 mL) in a 1.0 mM stock solution was measured using a micropipette and added to the Ru(bpy)33+ solution. The solution was diluted with appropriate volumes of ACN and H2O to make up a total volume of 1 mL with a ratio of H2O:ACN = 1:1, giving the final concentrations as [Ru(bpy)33+] = 0.10 mM (1 equivalent) or 0.20 mM (2 equivalents), and [1] = 0.10 mM. The cuvette was then placed in a UV-visible spectrophotometer and its spectrum was recorded at regular time intervals.
Synthesis of tri(p-tolyl)amine ((ArMe)3N)
The synthesis was modified from that described by Goodbrand and Hu (Fig. S13 in the ESI†).49p-Toluidine (2.14 g, 20.0 mmol), 1-iodo-4-methylbenzene (9.16 g, 42.0 mmol), potassium hydroxide (8.75 g, 150 mmol), anhydrous copper(I) chloride (77 mg, 0.78 mmol), and 1,10-phenanthroline (140 mg, 0.80 mmol) were suspended in anhydrous toluene (20 mL). Three freeze–pump–thaw cycles were applied to remove dissolved O2 from the suspension. The mixture was then sealed and heated at 120 °C for 72 hours, and completion was confirmed by TLC. The reaction mixture was diluted with water (100 mL) and extracted with 3 × 150 mL hexane. The organic fractions were combined and rinsed with saturated NaHCO3 solution (100 mL). The combined organic fractions were dried over anhydrous Na2CO3 and filtered. The hexane was removed by rotary evaporation to yield a dark brown precipitate. The precipitate was purified using flash column chromatography and eluted with hexane:DCM = 9:1. Upon solvent removal by rotary evaporation, colourless crystals of (ArMe)3N (4.14 g, 72%) were obtained. 1H NMR (DMSO-d6, 500 MHz, Fig. S24 in the ESI†): δ = 2.24 (s, 9H, Me), 6.84 (d, J = 13.5 Hz, 6H, ArH), 7.06 (d, J = 13.5 Hz, 6H, ArH). The data concur with those reported previously.49
Synthesis of tri(p-tolyl)aminium radical-cations ((ArMe)3N˙+)
This reaction was performed without Schlenk techniques. (ArMe)3N (75 mg, 0.26 mmol) and PbO2 (155 mg, 0.65 mmol) were suspended in ACN (3.0 mL). Hydrofluoroboric acid (HBF4, min. 40%, 57 mL, 0.26 mmol) was added to the suspension, giving a dark blue suspension. The mixture was filtered and the dark blue filtrate containing (ArMe)3N˙+ was used in situ for subsequent reactions.Oxidation of 1 in the presence of bromide anions
This reaction was performed without using any Schlenk techniques. The Na salt of 1 (30 mg, 0.065 mmol), tBuOK (31 mg, 0.27 mmol), and tetraethylammonium bromide (TEABr, 140 mg, 0.65 mmol) were suspended in ACN (30 mL) and sonicated for about 30 seconds until the red solution turned into a green suspension. The (ArMe)3N˙+ (4 equivalents) generated in situ was added dropwise to the suspension, resulting in a black solution. Within 30 minutes, the black solution turned brown, and then red, accompanied by the formation of a white precipitate. Upon the completion of the reaction, as monitored by negative-ion mode ESI-MS, the suspension was filtered and the red filtrate was diluted with diethyl ether (Et2O, 50 mL), giving an orange precipitate. The precipitate was filtered and washed with Et2O (10 mL) and water (5 mL). The residue was dissolved in methanol (MeOH, 5 mL) and filtered. The solvent of the filtrate was removed by rotary evaporation to give the crude TEA[1-TAML(Br)2] product. C18 reversed-phase preparative column chromatography (Merck LiChroprep®RP-18, 40–63 mm) was utilised to further purify the crude product. HR-MS (ESI−, m/z) calculated for C19H1279Br81BrFeN4O4 (1-TAML(Br)2) [M − H]−m/z = 575.8554, found 575.8566. The XPS (Br 3d) binding energy = 70.7 eV, corresponded to Ar–Br and not inorganic Br− (Fig. 3b). Despite many attempts through different solvent combinations, different counter cations (PPh4+ and PPN+), and different temperatures, single crystals of 1-TAML(Br)2 could not be obtained. To gather further structural information of 1-TAML(Br)2, purified 1-TAML(Br)2 was suspended in 1:9 MeOH:pH 5 acetate buffer solution to induce the deligation of 1-TAML(Br)2 to give TAML(Br)2, which was diamagnetic and therefore could be measured by 1H NMR spectroscopy. The 1H NMR spectrum reveals a major product of the symmetrical TAML(Br)2 and other signals, which we believe to be from asymmetrically bromide-substituted TAML(Br)2 or decomposed TAML(Br)2 during acid hydrolysis (Fig. S15 in the ESI†).
Oxidation of 1 in the presence of chloride anions
The same experimental procedures as those for Br− were employed, with TEABr being replaced by tetraethylammonium chloride (TEACl). HR-MS (ESI−, m/z) calculated for C19H13ClFeN4O4 (1-TAML(Cl)) [M − H]−m/z = 451.9975, found 451.9977; and C19H12Cl2FeN4O4 (1-TAML(Cl)2) [M − H]−m/z = 485.9585, found 485.9579. XPS (Cl 2p) measurements revealed two sets of signals: one set has a 2p3/2 binding energy = 197.8 eV (grey) and a 2p1/2 binding energy = 199.3 eV (orange), matching that of inorganic Cl−, while the second set has a 2p3/2 binding energy = 200.4 eV (blue) and a 2p1/2 binding energy = 201.9 eV (green) (Fig. S16b in the ESI†). The new set of signals does not correspond to that of inorganic Cl− but falls within the range of aryl chlorides, giving additional evidence for our proposed formulation of 1-TAML(Cl) and 1-TAML(Cl)2 products.Oxidation of 1 in the presence of cyanide anions
This modified reaction was performed without using any Schlenk techniques. The Na salt of 1 (30 mg, 0.065 mmol), tBuOK (8.0 mg, 0.072 mmol), and potassium cyanide (KCN, 21 mg, 0.32 mmol) were suspended in ACN (AR grade, 5 mL) and sonicated for about 30 seconds until the red solution turned into a green suspension. The suspension was cooled to 0 °C with stirring. (ArMe)3N˙+ (1.05 equivalents) generated in situ was added dropwise to the suspension, resulting in a black solution (moderately stable at room temperature, lifetime, τ837 nm, ≈ 4.4 ± 0.7 hours, Fig. S18a and b in the ESI†). After one minute of stirring, the solvent was removed on a Schlenk line at 0 °C. Once dried, the resulting black-grey solid (stable at room temperature under inert gases), was transferred into a glovebox for subsequent workup. In the glovebox, the solid was suspended in Et2O (20 mL) and filtered. The black residue was washed with Et2O (2 × 10 mL) and dichloromethane (DCM, 10 mL). The black residue was then dissolved in ACN (4 mL) and filtered. Tetraphenylphosphonium chloride (PPh4Cl, 50 mg, 0.13 mmol) was added to the black filtrate, giving a dark blue mixture. The solvent was then evaporated in vacuo. The dried residue was suspended in DCM (5 mL) and filtered to remove KCl. The dark blue filtrate was dried in vacuo. The residue was dissolved in ACN (0.5 mL), and recrystallised via vapor diffusion of Et2O (5 mL) into the ACN solution at −30 °C. After five days, the supernatant was decanted to reveal black crystals of (PPh4)2[1+(CN)2] (20 mg, 27%). Found: C, 71.1; H, 4.9; N, 7.1. Calc. for C69H56FeN6O5P2: C, 71.0; H, 4.8; N, 7.2%.Deligation of 1-TAML(X)n for 1H NMR measurements
The same procedures as those described above in the section “Oxidation of 1 in the presence of bromide/chloride anions” were employed. At specific time intervals, an aliquot (5 mL) was withdrawn, followed by the addition of pH 7 phosphate buffer solution (0.2 M, 1 mL) and aqueous sodium thiosulfate (Na2S2O3, 0.2 M, 1 mL) to the aliquot, resulting in an instantaneous change in the colour of the mixture from black/dark brown (depending on the reaction progress) to orange. The solvent was evaporated in vacuo. The dried residue was suspended in a pH 7 buffer solution (0.2 M, 1 mL) and was allowed to stir for 12 hours, during which the suspension colour changed from orange to beige. The suspension was filtered and the residue was washed with Et2O (2 mL) to remove most of the N(ArMe)3. After removing the Et2O by filtration, the residue was suspended in DMSO-d6. The suspension was filtered and one drop of concentrated hydrochloric acid (HCl, 12 N) was added to the filtrate to ensure that the TAML and the other products are not in their deprotonated forms. The solution was then characterised by 1H NMR spectroscopy.Oxidation of 1(Br) in the presence of DMOB
1,4-Dimethoxybenzene (DMOB, 5.0 mg, 36 μmol), the Na salt of 1 (1.7 mg, 3.6 μmol), and TEABr (7.6 mg, 36 μmol) were dissolved in CD3CN (0.50 mL). The 1H NMR spectrum of the solution was recorded. (ArMe)3N˙+ (3.2 mg, 7.4 μmol) was added to the solution and was characterised by 1H NMR spectroscopy after 5 minutes and after 24 hours with stirring.Kinetic measurements of the reaction of oxidised 1(Br) with varying concentrations of bromide anions
A solution of the Na salt of 1 (0.20 mM) and TEABr (2–14 mM) in ACN (2.0 mL) was allowed to stir for 5 minutes to form 1(Br). (ArMe)3N˙+ (40 μL, 20 mM) was added to the solution and the UV-visible spectrum of the reaction mixture was recorded at 5 second intervals. Each kinetic data set was fitted to a single exponential function and the time constant was determined by using a least-squares fitting procedure in Origin. The corresponding pseudo-first-order rate constant, kobs, was derived from the reciprocal of the time constant. The second-order rate constant, k, for the aromatic substitution of oxidised 1(Br) was determined from the linear fit of the plot of kobs against Br− concentration.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
HSS is supported by the Nanyang Assistant Professorship and MOE Tier 1 grants (M4011611 and M4011791). HSS also gratefully acknowledges the Agency for Science, Technology and Research (A*STAR), AME IRG grants A1783c0002 and A1783c0003, for funding this research. The authors acknowledge support from the Solar Fuels Lab at NTU and the SinBeRISE CREATE. JHL would like to thank Mr. Zonghan Hong (NTU) for the XPS measurements and Ms. Yijie Yang (NTU) for the Raman spectroscopic measurements. KR is funded by UniCat and the Heisenberg Professorship of DFG.Notes and references
- W. Nam, Acc. Chem. Res., 2007, 40, 522–531 CrossRef CAS PubMed.
- J. Hohenberger, K. Ray and K. Meyer, Nat. Commun., 2012, 3, 720 CrossRef PubMed.
- S. Shaik, H. Hirao and D. Kumar, Acc. Chem. Res., 2007, 40, 532–542 CrossRef CAS PubMed.
- C. Krebs, D. Galonić Fujimori, C. T. Walsh and J. M. Bollinger, Acc. Chem. Res., 2007, 40, 484–492 CrossRef CAS PubMed.
- M. H. Sazinsky and S. J. Lippard, in Sustaining Life on Planet Earth: Metalloenzymes Mastering Dioxygen and Other Chewy Gases, ed. P. M. H. Kroneck and M. E. Sosa Torres, Springer International Publishing, Cham, 2015, pp. 205–256, DOI:10.1007/978-3-319-12415-5_6.
- M. Puri and L. Que Jr, Acc. Chem. Res., 2015, 48, 2443–2452 CrossRef CAS PubMed.
- X. Engelmann, I. Monte-Perez and K. Ray, Angew. Chem., Int. Ed., 2016, 55, 7632–7649 CrossRef CAS PubMed.
- J. T. Groves, J. Inorg. Biochem., 2006, 100, 434–447 CrossRef CAS PubMed.
- I. G. Denisov, T. M. Makris, S. G. Sligar and I. Schlichting, Chem. Rev., 2005, 105, 2253–2278 CrossRef CAS PubMed.
- S. R. Bell and J. T. Groves, J. Am. Chem. Soc., 2009, 131, 9640–9641 CrossRef CAS PubMed.
- J. Rittle and M. T. Green, Science, 2010, 330, 933 CrossRef CAS PubMed.
- C. J. Bougher, S. Liu, S. D. Hicks and M. M. Abu-Omar, J. Am. Chem. Soc., 2015, 137, 14481–14487 CrossRef CAS PubMed.
- P. Leeladee, R. A. Baglia, K. A. Prokop, R. Latifi, S. P. de Visser and D. P. Goldberg, J. Am. Chem. Soc., 2012, 134, 10397–10400 CrossRef CAS PubMed.
- R. A. Baglia, C. M. Krest, T. Yang, P. Leeladee and D. P. Goldberg, Inorg. Chem., 2016, 55, 10800–10809 CrossRef CAS PubMed.
- T. J. Collins, Acc. Chem. Res., 2002, 35, 782–790 CrossRef CAS PubMed.
- S. B. Munoz III, W. T. Lee, D. A. Dickie, J. J. Scepaniak, D. Subedi, M. Pink, M. D. Johnson and J. M. Smith, Angew. Chem., Int. Ed., 2015, 54, 10600–10603 CrossRef PubMed.
- G. E. Cutsail III, B. W. Stein, D. Subedi, J. M. Smith, M. L. Kirk and B. M. Hoffman, J. Am. Chem. Soc., 2014, 136, 12323–12336 CrossRef PubMed.
- C. Kupper, J. A. Rees, S. Dechert, S. DeBeer and F. Meyer, J. Am. Chem. Soc., 2016, 138, 7888–7898 CrossRef CAS PubMed.
- L. R. Widger, C. G. Davies, T. Yang, M. A. Siegler, O. Troeppner, G. N. Jameson, I. Ivanovic-Burmazovic and D. P. Goldberg, J. Am. Chem. Soc., 2014, 136, 2699–2702 CrossRef CAS PubMed.
- S. Sahu, B. Zhang, C. J. Pollock, M. Durr, C. G. Davies, A. M. Confer, I. Ivanovic-Burmazovic, M. A. Siegler, G. N. Jameson, C. Krebs and D. P. Goldberg, J. Am. Chem. Soc., 2016, 138, 12791–12802 CrossRef CAS PubMed.
- W. Nam, Y. M. Lee and S. Fukuzumi, Acc. Chem. Res., 2014, 47, 1146–1154 CrossRef CAS PubMed.
- L. Que, Acc. Chem. Res., 2007, 40, 493–500 CrossRef CAS PubMed.
- A. D. Ryabov, in Adv. Inorg. Chem., ed. E. Rudi van and D. H. Colin, Academic Press, 2013, vol. 65, pp. 117–163 Search PubMed.
- J. A. Kovacs, Acc. Chem. Res., 2015, 48, 2744–2753 CrossRef CAS PubMed.
- S. Sen Gupta, M. Stadler, C. A. Noser, A. Ghosh, B. Steinhoff, D. Lenoir, C. P. Horwitz, K. W. Schramm and T. J. Collins, Science, 2002, 296, 326–328 CrossRef CAS PubMed.
- S. K. Khetan and T. J. Collins, Chem. Rev., 2007, 107, 2319–2364 CrossRef CAS PubMed.
- S. Kundu, A. Chanda, S. K. Khetan, A. D. Ryabov and T. J. Collins, Environ. Sci. Technol., 2013, 47, 5319–5326 CrossRef CAS PubMed.
- E. L. Demeter, S. L. Hilburg, N. R. Washburn, T. J. Collins and J. R. Kitchin, J. Am. Chem. Soc., 2014, 136, 5603–5606 CrossRef CAS PubMed.
- J. W. Kee, Y. Y. Ng, S. A. Kulkarni, S. K. Muduli, K. Xu, R. Ganguly, Y. Lu, H. Hirao and H. S. Soo, Inorg. Chem. Front., 2016, 3, 651–662 RSC.
- S. Gazi, W. K. H. Ng, R. Ganguly, A. M. P. Moeljadi, H. Hirao and H. S. Soo, Chem. Sci., 2015, 6, 7130–7142 RSC.
- H. Shao, S. K. Muduli, P. D. Tran and H. S. Soo, Chem. Commun., 2016, 52, 2948–2951 RSC.
- S. K. Muduli, S. Wang, S. Chen, C. F. Ng, C. H. A. Huan, T. C. Sum and H. S. Soo, Beilstein J. Nanotechnol., 2014, 5, 517–523 CrossRef CAS PubMed.
- H. Kotani, T. Suenobu, Y. M. Lee, W. Nam and S. Fukuzumi, J. Am. Chem. Soc., 2011, 133, 3249–3251 CrossRef CAS PubMed.
- D. Shen, C. Saracini, Y. M. Lee, W. Sun, S. Fukuzumi and W. Nam, J. Am. Chem. Soc., 2016, 138, 15857–15860 CrossRef CAS PubMed.
- C. Panda, J. Debgupta, D. Díaz Díaz, K. K. Singh, S. Sen Gupta and B. B. Dhar, J. Am. Chem. Soc., 2014, 136, 12273–12282 CrossRef CAS PubMed.
- H. S. Soo, M. L. Macnaughtan, W. W. Weare, J. Yano and H. M. Frei, J. Phys. Chem. C, 2011, 115, 24893–24905 CAS.
- W. C. Ellis, C. T. Tran, M. A. Denardo, A. Fischer, A. D. Ryabov and T. J. Collins, J. Am. Chem. Soc., 2009, 131, 18052–18053 CrossRef CAS PubMed.
- S. Z. Sullivan, A. Ghosh, A. S. Biris, S. Pulla, A. M. Brezden, S. L. Collom, R. M. Woods, P. Munshi, L. Schnackenberg, B. S. Pierce and G. K. Kannarpady, Chem. Phys. Lett., 2010, 498, 359–365 CrossRef CAS.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. von Zelewsky, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef CAS.
- M. J. Bartos, C. Kidwell, K. E. Kauffmann, S. W. Gordon-Wylie, T. J. Collins, G. C. Clark, E. Münck and S. T. Weintraub, Angew. Chem., Int. Ed., 1995, 34, 1216–1219 CrossRef CAS.
- M. J. Bartos, S. W. Gordon-Wylie, B. G. Fox, L. James Wright, S. T. Weintraub, K. E. Kauffmann, E. Münck, K. L. Kostka, E. S. Uffelman, C. E. F. Rickard, K. R. Noon and T. J. Collins, Coord. Chem. Rev., 1998, 174, 361–390 CrossRef CAS.
- E. Steckhan, in Electrochemistry I, ed. E. Steckhan, Springer Berlin Heidelberg, Berlin, Heidelberg, 1987, pp. 1–69, DOI:10.1007/3-540-17871-6_11.
- Y. P. Zhang, J. H. He, G. Q. Xu and E. S. Tok, J. Phys. Chem. C, 2011, 115, 15496–15501 CAS.
- A. Chanda, D. L. Popescu, F. Tiago de Oliveira, E. L. Bominaar, A. D. Ryabov, E. Munck and T. J. Collins, J. Inorg. Biochem., 2006, 100, 606–619 CrossRef CAS PubMed.
- J. England, E. R. Farquhar, Y. Guo, M. A. Cranswick, K. Ray, E. Munck and L. Que Jr, Inorg. Chem., 2011, 50, 2885–2896 CrossRef CAS PubMed.
- C. C. Cummins and R. R. Schrock, Inorg. Chem., 1994, 33, 395–396 CrossRef CAS.
- S. Hong, X. Lu, Y. M. Lee, M. S. Seo, T. Ohta, T. Ogura, M. Clemancey, P. Maldivi, J. M. Latour, R. Sarangi and W. Nam, J. Am. Chem. Soc., 2017, 139, 14372–14375 CrossRef CAS PubMed.
- W. C. Ellis, C. T. Tran, R. Roy, M. Rusten, A. Fischer, A. D. Ryabov, B. Blumberg and T. J. Collins, J. Am. Chem. Soc., 2010, 132, 9774–9781 CrossRef CAS PubMed.
- H. B. Goodbrand and N.-X. Hu, J. Org. Chem., 1999, 64, 670–674 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedures and characterisation. CCDC 1535587. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc05378a |
| This journal is © The Royal Society of Chemistry 2018 |