
Solution-processed thermally activated delayed fluorescence organic light-emitting diodes using a new polymeric emitter containing non-conjugated cyclohexane units†
Hyung Jong
Kim
,
Chiho
Lee
,
Mallesham
Godumala
,
Suna
Choi
,
Seo Yeon
Park
,
Min Ju
Cho
,
Sungnam
Park
 * and
Dong Hoon
Choi
*
* and
Dong Hoon
Choi
*
Department of Chemistry, Research Institute for Natural Sciences, Korea University, 5 Anam-dong, Sungbuk-gu, Seoul 136-701, Republic of Korea. E-mail: spark8@korea.ac.kr; dhchoi8803@korea.ac.kr
First published on 19th February 2018
Abstract
The importance of thermally activated delayed fluorescence (TADF) materials in organic light-emitting diode (OLED) applications continues to grow as a consequence of their unique properties and excellent performance. Herein, a new green-emitting TADF polymer, P(DMTRZ-Cp), for solution-processable OLEDs was designed and synthesized based on the structure of DMAC-TRZ that shows typical TADF characteristics as a small molecule. The 1,1-diphenylcyclohexane (Cp) moiety introduced into the structure of the polymeric emitter not only acts as a linker between the conjugated monomeric units, but also helps enhancing solubility while disconnecting conjugation along the polymer backbones. P(DMTRZ-Cp) exhibits obvious TADF features such as a small energy gap (0.023 eV) between its lowest singlet and triplet excited states, and obvious delayed photoluminescence (PL) decay behavior. Moreover, this new polymeric emitter exhibits high PL quantum yield over 90% in the film state. By applying solution-processed TADF-OLED devices, P(DMTRZ-Cp) exhibited the maximum external quantum efficiency of up to 15.4% with green emission. As far as we know, this is the first report to introduce a non-conjugated linker, Cp, in the main-chain type polymeric emitter structure that presents TADF characteristics. The Cp linker is considered to be a promising moiety for the development of solution-processable polymeric emitters with high efficiencies.
Introduction
Organic light-emitting diodes (OLEDs) have received much attention over the past few decades due to their applications in full-color flat-panel maximum-resolution displays, wearable display devices, and solid-state lighting.1–3 It is well known that 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratios of singlet and triplet excitons in emissive materials are generated by the recombination of holes and electrons under electrical excitation.4 Conventional fluorescent OLEDs that use singlet excitons alone have a 25% limit on their internal quantum efficiencies (IQEs), with 75% of the triplet excitons wasted through non-radiative pathways. In 1999, phosphorescent organic light-emitting diodes (PhOLEDs) overcame these limitations by using singlet and triplet excitons arising from the strong spin–orbit couplings of heavy metal atoms. However, the scarcity and high cost of specific metals like iridium and platinum have hindered their widespread applications. To address the limitations of fluorescent OLEDs and PhOLEDs, recently a metal-free thermally activated delayed fluorescence (TADF)-OLED with up to 100% IQE has been proposed through the reverse intersystem crossing (RISC) of the triplet to singlet excitons.5–9 Consequently, TADF is now recognized as a next-generation OLED mode.
:3 ratios of singlet and triplet excitons in emissive materials are generated by the recombination of holes and electrons under electrical excitation.4 Conventional fluorescent OLEDs that use singlet excitons alone have a 25% limit on their internal quantum efficiencies (IQEs), with 75% of the triplet excitons wasted through non-radiative pathways. In 1999, phosphorescent organic light-emitting diodes (PhOLEDs) overcame these limitations by using singlet and triplet excitons arising from the strong spin–orbit couplings of heavy metal atoms. However, the scarcity and high cost of specific metals like iridium and platinum have hindered their widespread applications. To address the limitations of fluorescent OLEDs and PhOLEDs, recently a metal-free thermally activated delayed fluorescence (TADF)-OLED with up to 100% IQE has been proposed through the reverse intersystem crossing (RISC) of the triplet to singlet excitons.5–9 Consequently, TADF is now recognized as a next-generation OLED mode.
In general, the structure of a TADF emitter is designed by combining various donor (D) and acceptor (A) units through conjugation. However, TADF materials must have small energy gaps between their lowest singlet and triplet excited states (ΔEST) to efficiently up-convert triplet excitons through RISC following radiative decay. In this regard, the careful selection of D and A units is essential for spatially separating their highest occupied molecular orbitals (HOMOs) and their lowest unoccupied molecular orbitals (LUMOs) to obtain small ΔEST values. Based on this principle, many TADF emitters have been designed and studied to date.
In the respect of device fabrication, high-performance multilayered OLED devices were fabricated by a vacuum deposition technique.10–18 However, this process is relatively expensive, complicated to optimize the conditions, and difficult to apply to flexible substrates for future displays. Parallel strategies to overcome these abovementioned drawbacks include device fabrication using low-cost solution processes, such as spin coating, inkjet printing, or roll-to-roll printing.19–21
Recently, various small molecules and polymeric emitters for implementation in solution-processed TADF-OLED devices have been reported.19–27 Compared to small molecules, polymeric materials with relatively high molecular weights have the advantage that they form good organic layers on substrates in solution processes allowing for the fabrication of laminated OLED devices.28 Thus, the rapid development of solution-processable polymeric emitters is essential for the realization of high-performing TADF-based OLEDs.
Nevertheless, there are some crucial factors that need to be considered when applying polymeric emitters to a TADF device. Among various factors, it is recognized that achieving small ΔEST values while suppressing internal conversion is very difficult in polymer structures. Triplet populations are also effectively quenched by frequent triplet–triplet annihilation (TTA) through intramolecular or intermolecular interactions within the polymer.29
A close look at the literature demonstrating the polymer-based TADF emitters reported so far does not provide clear directions for the design of optimized molecular structures. Despite recognizing the advantages provided by solution processing of polymeric emitters, very few design strategies for improving the disadvantages of polymer-based TADF-OLEDs have been presented to date.30
For instance, Lee et al. synthesized D–A-type alternating copolymers based on benzophenone, which were labelled as “pCzBP” and “pAcBP” and were green and yellow emitters, respectively, with external quantum efficiencies (EQEs) of 8.1% and 9.3%.24 Nikolaenko et al. demonstrated a light-emitting polymer based on D, A and backbone (B) units. This polymer contains host moieties (B–A–B) and TADF emissive cores (D–A–D) and the corresponding device displayed green emission with an EQE of 10.0%.31 A polymer referred to as “PAPTC”, in which the donor unit was in the backbone and the acceptor was bound to the side chain, was reported by Zhu's group. This PAPTC polymer exhibited a green emission with an EQE of 12.6%.32 Wang et al. also reported a conjugated polymer “PFSOTT2” using a similar aforementioned molecular design strategy and this polymer emitter showed nearly 10% EQE with white emission.33 Ren et al. reported a “Copo1” polymeric emitter in which the TADF moiety was bound to the backbone as a pendant group. This polymer shows the highest EQE close to 20% of the reported green polymeric emitters.23 Most recently, Shao et al. demonstrated a novel molecular design for the blue TADF polymer “P-Ac95-TRZ5” based on a non-conjugated polyethylene backbone with a through-space charge transfer effect between the pendant electron-donating acridine unit and electron-accepting triazine unit with a maximum EQE of 12.1%.34
Considering the developmental trends for the previously reported polymeric TADF emitters, progressive improvements in device efficiencies have been demonstrated; despite this, they still suffer from disadvantages associated with complex synthesis procedures, color control issues, and large ΔEST values. Hence, the introduction of non-conjugated linkers which have sp3 bridges minimizes the disadvantages seen in other polymeric emitters by controlling the conjugation length to result in high triplet energies (T1), and small ΔEST values, with an added advantage of enhanced solubility.
In this work, we report a new green polymeric emitter containing non-conjugated linkers in their repeating units that is applicable to OLED devices prepared by solution processes. Using well-known small molecules (i.e.DMAC-TRZ)35,36 and 1,1-diphenylcyclohexane (Cp) as the non-conjugated moiety, a new polymeric emitter (P(DMTRZ-Cp)) was designed and synthesized successfully. The Cp unit is proposed to not only act as a linker between the conjugated monomeric units, but also as a linker capable of disrupting conjugation to retain their essential properties and improving the solubility of the polymer.
The TADF behavior of the newly synthesized polymer was confirmed through investigations of its transient photophysical properties and OLED device performance. By optimizing the device conditions, P(DMTRZ-Cp) as a green polymeric emitter exhibited a maximum EQE of up to 15.4%. The results of this work clearly demonstrated that the use of the 1,1-diphenylcyclohexane unit as a non-conjugated linker is an ideal strategy for use in the development of highly efficient solution-processable polymeric emitters for high-performing TADF-OLEDs.
Experimental
Materials and synthesis
All materials and solvents for the synthesis of the polymeric emitting material were purchased from Alfa Aesar, Sigma-Aldrich, Tokyo Chemical Industry (TCI) and Acros Organics, and were used without further purification. 9,9-Dimethyl-9,10-dihydroacridine,37 10-(4-(4,6-diphenyl-1,3,5-triazin-2-yl)phenyl)-9,9-dimethyl-9,10-dihydroacridine (DMAC-TRZ),35 and 2,2′-(cyclohexane-1,1′-diylbis(4,1-phenylen))bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolane) (Cp)38 were prepared following methods previously reported in the literature.Synthesis of 2,7-dibromo-10-(4-(4,6-diphenyl-1,3,5-triazin-2-yl)phenyl)-9,9-dimethyl-9,10-dihydroacridine (DMTRZ-2br)
10-(4-(4,6-Diphenyl-1,3,5-triazin-2-yl)phenyl)-9,9-dimethyl-9,10-dihydroacridine (DMAC-TRZ) (400 mg, 0.77 mmol) was dissolved in N,N-dimethylformamide (DMF, 30 mL) in a 100 mL two-neck round-bottom flask. N-Bromosuccinimide (NBS) (290 mg, 1.63 mmol) was then added and the reaction mixture was stirred at room temperature for 12 h under nitrogen. After quenching using methanol, the precipitate was collected by filtration and purified by silica-gel column chromatography, with dichloromethane/hexane (1:3) as the eluent, to obtain the required compound in 67% yield (350 mg). 1H NMR (500 MHz, CDCl3): δ (ppm) 9.05 (d, J = 8.55 Hz, 2H), 8.84 (d, J = 7.05 Hz, 4H), 7.60–7.68 (m, 6H), 7.55 (d, J = 2.15 Hz, 2H), 7.53 (d, J = 8.55 Hz, 2H), 7.11 (dd, J1 = 8.85 Hz, J2 = 2.45 Hz, 2H), 6.26 (d, J = 8.85 Hz, 2H), 1.70 (s, 6H). 13C NMR (125 MHz, CDCl3): 171.91, 170.84, 144.28, 139.43, 136.70, 135.95, 132.78, 131.82, 131.76, 131.15, 129.40, 129.01, 128.74, 128.21, 115.88, 113.62, 36.33, 31.01. Mass m/z [(M + H)+] calcd for 674.06, found 675.16. Anal. calcd for C36H26Br2N4: C, 64.11; H, 3.89; N, 8.31. Found: C, 63.80; H, 4.01; N, 8.50.
Synthesis of P(DMTRZ-Cp)
2,7-Dibromo-10-(4-(4,6-diphenyl-1,3,5-triazin-2-yl)phenyl)-9,9-dimethyl-9,10-dihydroacridine (DMTRZ-2br) (150 mg, 0.22 mmol) and 2,2′-(cyclohexane-1,1′-diylbis(4,1-phenylen))bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolane) (Cp) (130 mg, 0.27 mmol) were dissolved in anhydrous toluene (10 mL) in a 100 mL two-neck round-bottom flask. 2.0 M potassium carbonate (2 mL) and a few drops of Aliquat 336 (as a surfactant) were added to the reaction mixture, after which it was degassed with nitrogen. Tris(dibenzylideneacetone)dipalladium(0) (4.3 mg, 4.44 μmol) and tri(o-tolyl)phosphine (5.4 mg, 17.8 μmol) were added to the mixture, after which it was maintained at 100 °C for 18 h under nitrogen. After cooling to room temperature, the reaction mixture was precipitated from methanol (150 mL). The solid was collected by filtration and purified by sequential Soxhlet extraction with acetone, hexane, and dichloromethane. The dichloromethane solution was precipitated into methanol, the solid product was collected and dried under vacuum. P(DMTRZ-Cp) was obtained as a powder in 84% yield (140 mg). Anal. calcd for C54H46N4: C, 86.37; H, 6.17; N, 7.46. Found: C, 86.15; H, 6.32; N, 7.53. GPC: Mn = 4.82 kDa, Mw = 8.58 kDa, PDI = 1.78.Instrumentation
1H and 13C nuclear magnetic resonance spectra were recorded in deuterated chloroform on a Bruker 500 MHz spectrometer. Elemental analyses were conducted using a Thermo Scientific Flash 2000 (Thermo Fisher Scientific) elemental analyzer. The molecular weight of the polymer was measured by gel permeation chromatography (GPC, Agilent 1200 series GPC system, polystyrene standard) using o-dichlorobenzene as an eluent (T = 80 °C).Absorption spectra in solution and film states were recorded using a UV-vis absorption spectrophotometer (HP 8453, photodiode array, λ = 190–1100 nm). The film sample was fabricated by the spin-coating method on a washed glass using a 1.0 wt% toluene solution. Photoluminescence (PL) spectra of solution and film states were obtained using a Hitachi F-7000 fluorescence spectrophotometer at room temperature. Differential scanning calorimetry (DSC, Mettler 821e) was used to identify the thermal properties of the polymer under a nitrogen atmosphere. The electrochemical properties (e.g. oxidation and reduction potentials) were measured by cyclic voltammetry (CV) using a potentiostat (eDAQ, EA161). Ag/AgCl and platinum wire (0.5 mm diameters) were used as the reference and counter electrodes, respectively, and 0.10 M tetrabutylammonium hexafluorophosphate (Bu4NPF6) in distilled acetonitrile was used as the electrolyte solution. A thin polymer film was prepared on a Pt plate using the drop-casting method. Absolute PL quantum yields (PLQYs) were obtained on a JASCO (FP-8600) spectrofluorometer with a xenon lamp excitation source, using an integrating sphere (ILF-835) for solution and film samples.
Transient photoluminescence spectroscopy
A train of 1064 nm pulses with durations of 5 ns was produced at 10 Hz from a Nd:YAG laser (Powerlite Precision II 8000, Continuum). A 355 nm pulse was obtained by the generation of the third harmonic and was used to directly excite the film sample cast on an optical glass. The emission from the film sample was collected by using a lens (focal length = 10 cm), passed through a monochromator and detected by using a photomultiplier tube (PMT) connected to a 500 MHz digital oscilloscope (DSO-X 3054A, Agilent, Inc.). The digital oscilloscope, which was triggered by a TTL signal from the Nd:YAG laser, was used to collect the time-resolved photoluminescence (TRPL) signal from the film sample (25 wt% P(DMTRZ-Cp) doped in the 1,3-bis(N-carbazolyl)benzene (mCP) host) at different emission frequencies. The instrumental response function of our experimental setup was measured to be about 13 ns at 355 nm. The TRPL signal can be measured up to 100 ms in our experimental setup. Temperature-dependent TRPL experiments were carried out by varying the temperature from 100 K to 300 K at intervals of 50 K with a cryostat (VPF-100, Janis). The PL spectra, which were recorded at room temperature and at relatively low temperature (77 K), in the film state were obtained using the same measuring instrumentation equipped with a CCD camera.Computational details
Density functional theory (DFT) calculations at the B3LYP/6-31G(d) level were used to identify the optimized molecular structures, and to determine the electron density distributions and the energies of the highest occupied molecular orbitals (HOMOs) and the lowest unoccupied molecular orbitals (LUMOs).OLED device fabrication
In this study, an ITO-coated glass with a resistance of 10.0 Ω cm−2 (150 nm-thick, AMG Corp.) was used as the anode for the fabrication of OLED devices. The ITO electrode surface was washed sequentially with acetone, deionized water, and isopropyl alcohol for 10 min using ultrasonic treatment.A layer of the poly(3,4-ethylenedioxythiophene):poly(styrene sulfonate) (PEDOT:PSS) film, with a thickness of 40 nm, was coated on the ITO glass after UV-ozone treatment for 20 min (spin coating at 4000 rpm for 30 s followed by baking at 155 °C for 15 min in air). The 30 nm thickness of the poly(9-vinylcarbazole) (PVK) film from a chlorobenzene (CB) solution was spin-coated at 3000 rpm for 30 s on the PEDOT:PSS layer. A blend solution with the host material and the polymeric emitter in toluene was then spin-coated on top of the PVK-treated substrate. Subsequently, a 40 nm-thick 1,3,5-tris(1-phenyl-1H-benzimidazol-2-yl)benzene (TPBi) layer was vacuum-deposited on the previous layer as the electron transport layer. Then LiF and Al cathode layers were vacuum-deposited on the TPBi layer at thicknesses of about 1 nm and 100 nm, respectively. The fabricated hybrid-type devices were characterized using a Keithley SMU 236 instrument and a SpectraScan PR-655 colorimeter to obtain current–voltage–luminance characteristics, current, power, external quantum efficiencies, and the electroluminescence spectra of the polymer-based TADF-OLEDs.
Results and discussion
Design, synthesis and theoretical calculations
In order to design polymeric emitters applicable to TADF-OLED devices, the well-known small molecule, 10-(4-(4,6-diphenyl-1,3,5-triazin-2-yl)phenyl)-9,9-dimethyl-9,10-dihydroacridine (DMAC-TRZ)35,36 was selected as the desired monomeric unit, while 1,1-diphenylcyclohexane (Cp) was chosen as the non-conjugated linker (Fig. 1); Cp was selected since it is easily incorporated synthetically into the polymer backbone, although the conjugation length may be slightly longer than the existing monomer due to the two phenyl moieties.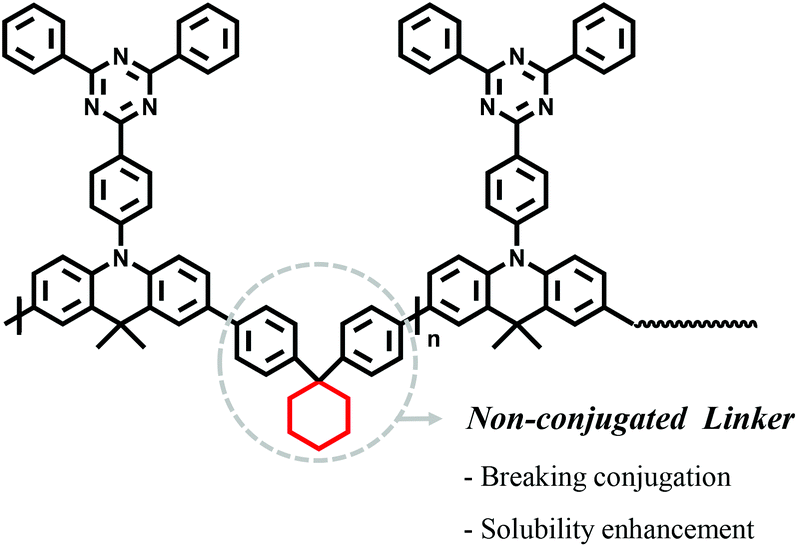 | ||
| Fig. 1 Design strategy and chemical structure of the P(DMTRZ-Cp) polymeric emitter. | ||
According to the literature, various organic semiconducting materials containing cyclohexane units have been reported to be applicable to OLED devices. For example, the well-known 1-bis[4-[N,N-di(4-tolyl)amino]phenyl]cyclohexane (TAPC) is widely used as a hole-transport layer, and many of its derivatives have also been studied.1,39 Recently, the synthesis of host materials based on the cyclohexane unit has been reported; high ET levels were maintained by reducing the conjugation length or by alleviating the effect of conjugation.40 Considering the properties already reported in the literature, alleviating conjugation or disconnecting conjugation along the polymer backbone through the incorporation of sp3-containing units facilitates emissions in the short wavelength range, maintaining TADF characteristics.
In addition, when non-conjugated linkers such as Cp are introduced into the repeating-group design, they are considered to be important factors that help to adjust polymer solubility which is useful in the solution process during device fabrication. Consequently, a new polymeric emitter, P(DMTRZ-Cp) composed of the DMAC-TRZ monomer and cyclohexane linker has been successfully synthesized for solution-processed TADF-OLEDs (Scheme 1).
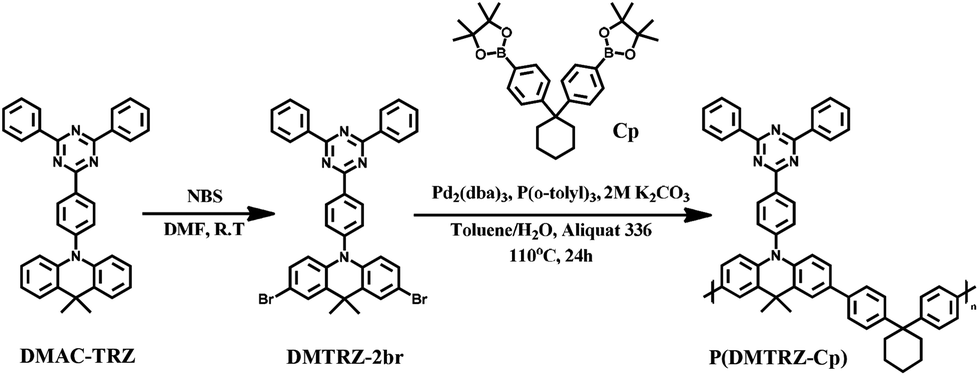 | ||
| Scheme 1 Scheme for the synthesis of the polymeric emitter, P(DMTRZ-Cp). | ||
Following literature reports, the well-known small molecule DMAC-TRZ was synthesized through the Buchwald–Hartwig reaction of 2-(4-bromophenyl)-4,6-diphenyl-1,3,5-triazine and acridine units.35,36 Subsequently, the monomer DMTRZ-2br was prepared by the bromination of DMAC-TRZ, using NBS. The structure of this monomer was confirmed by 1H and 13C NMR spectroscopy techniques and elemental analyses. The final polymer, P(DMTRZ-Cp) was synthesized by Suzuki–Miyaura coupling in the presence of a Pd(0) catalyst using monomers DMTRZ-2br and Cp.38 The resulting polymer was subjected to sequential Soxhlet extraction with acetone, hexane, and dichloromethane, and purified to remove unreacted monomer species as well as the reaction catalyst. The number-average molecular weights (Mns) of P(DMTRZ-Cp) was determined by gel permeation chromatography (GPC) using o-dichlorobenzene as an eluent at 80 °C, and provided the value of 4.8 kDa. The weight-average molecular weights (Mws) and polydispersity index (PDI) of this polymer are shown in Table 1. The new polymeric emitter, P(DMTRZ-Cp), exhibited good solubility in common organic solvents such as dichloromethane, chloroform, and chlorobenzene, which led us to believe that the fabrication of OLED devices by solution processes is possible.
| Polymer emitter | M w/PDI | λ absmax (nm) | λ emmax (nm) | E S/ET/ΔEST (eV) | E optg (eV) |
Φ
PLc (sol/film) |
τ
pd (ns)/τdd (μs) |
HOMOe,f (eV) | LUMOf (eV) |
|---|---|---|---|---|---|---|---|---|---|
| a Solution (toluene). b Film. c Absolute photoluminescence quantum yield (ΦPL) using an integrating sphere in the solution and film states under nitrogen. d PL lifetimes of the prompt (τp) and delayed (τd) decay components for the doped films with mCP. e Obtained from the cyclic voltammograms. f Sample: film on the Pt electrode. LUMO (eV) = HOMO (eV) + Eopt,bg (eV); Eonsetox of ferrocene = 0.35 V. | |||||||||
| P(DMTRZ-Cp) | 8580/1.78 | 339a, 336b | 513b | 2.725/2.702/0.023 | 2.61a, 2.42b | 0.53/0.97 | 12.2/1.8 | −5.37 | −2.95 |
In order to study the HOMO and LUMO distributions theoretically, excited state energies (ES and ET), and the energy differences between the singlet and triplet excited states (ΔEST) of the conjugated unit in the polymer, density functional theory (DFT) calculations at the B3LYP/6-31G* level of theory were performed; these calculations can be used to evaluate the corresponding properties of the polymer applicable to TADF-OLED devices (Fig. 2). For the simplicity of calculations, only the molecular unit that forms the conjugation region was selected as a model structure for use in this calculation. As shown in Fig. 2a and b, the conjugated monomeric unit exhibits large dihedral angles between the central phenyl ring and acridine that are close to 90°; hence, the structure of this unit is severely twisted. These calculations also reveal clearly separated HOMO and LUMO distributions over the acridine and triphenyltriazine moieties, respectively (Fig. 2c). Spatially separated HOMO and LUMO distribution leads to little intramolecular charge transfer between the donor and acceptor entities resulting in a small ΔEST value.30 The computed ΔEST value was found to be small and similar to that of the existing small molecule, DMAC-TRZ (Fig. 2d and Fig. S1†). We confirmed through the calculated values of the computational structure that TADF properties could be retained and it was predicted that this polymeric emitter incorporating Cp linker would be useful for TADF-OLEDs.
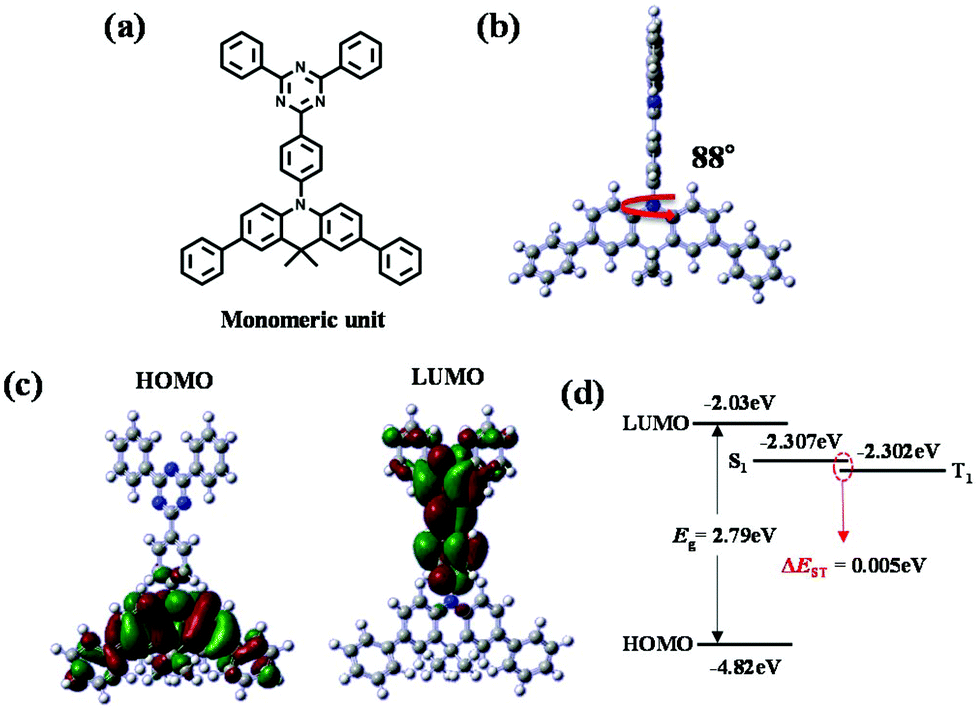 | ||
| Fig. 2 (a) Conjugated monomeric unit in P(DMTRZ-Cp). (b) Optimized geometry, (c) HOMO and LUMO spatial distributions and (d) energy level diagram for the conjugated unit in P(DMTRZ-Cp) calculated at the B3LYP/6-31G (DFT) level of theory with Gaussian 09W. | ||
Photophysical and electrochemical properties
Fig. 3a shows the absorption spectra of the synthesized polymer in solution (solvent: toluene) and film states. In addition, fluorescence spectrum recorded at 300 K and phosphorescence spectrum recorded at 77 K are also shown.21,34 For fluorescence and phosphorescence measurements, the film sample was composed of P(DMTRZ-Cp) mixed in the mCP host which has high triplet energy.41,42 The values related to photophysical properties of the new polymeric emitter measured in this study are summarized in Table 1. The polymeric emitter shows a weak absorption at around 400–450 nm due to its weak intramolecular charge-transfer (ICT) transitions, a consequence of the large dihedral angle between the acridine derivative and the triazine moiety, but strong absorptions are observed at around 300–380 nm, which is attributed to π–π* transitions. The fluorescence spectrum of the polymer film doped into mCP was recorded which exhibited fluorescence maxima at 513 nm for P(DMTRZ-Cp). The polymer exhibits green emission that is red shifted by 10–20 nm due to the phenyl moiety of the non-conjugated linker in addition to intermolecular interaction with mCP host materials, as compared to the emission of the corresponding (reference) small molecule (504 nm for DMAC-TRZ).36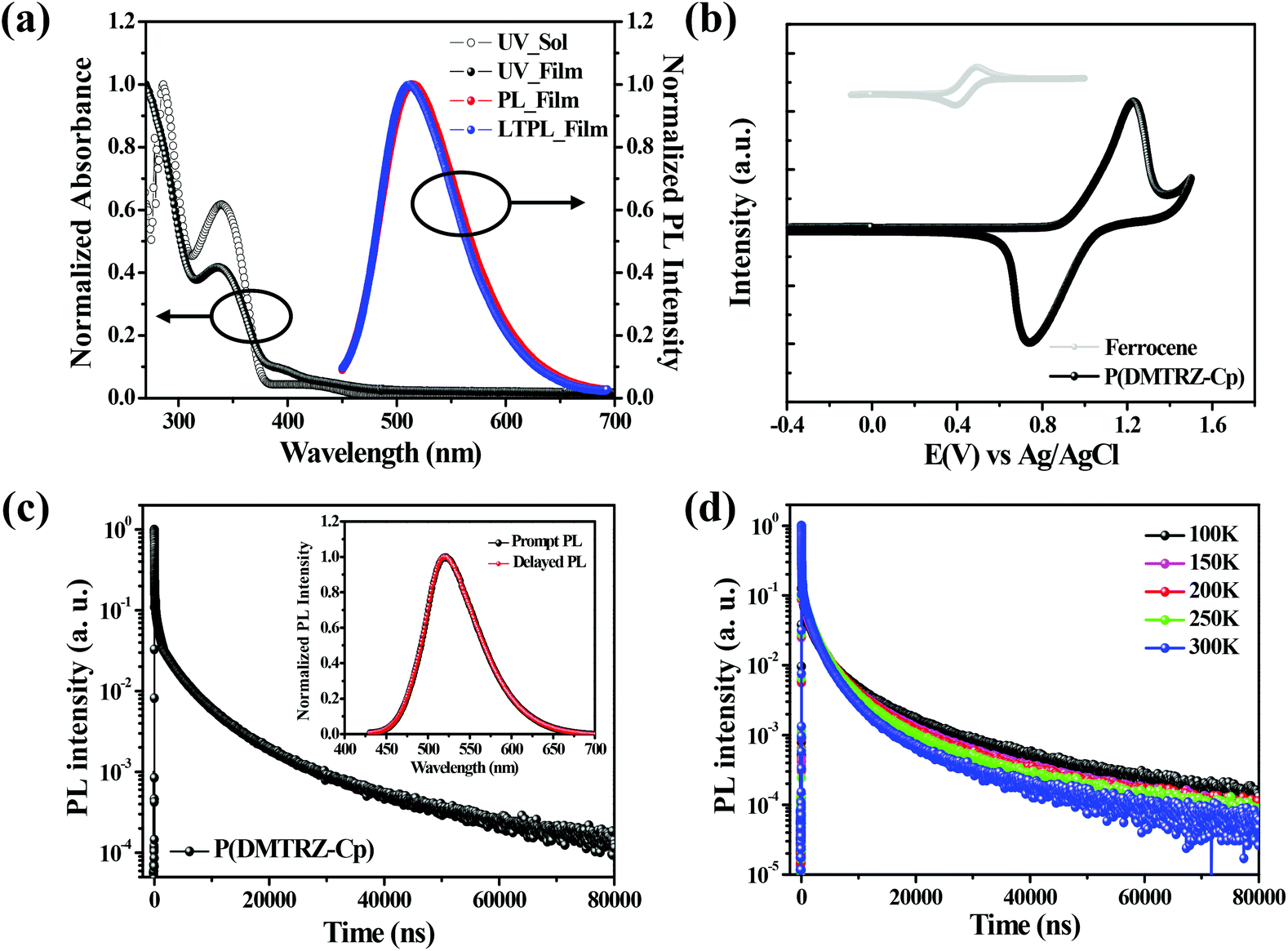 | ||
| Fig. 3 (a) UV-vis absorption spectra, photoluminescence (PL) and low-temperature PL (LTPL) spectra, (b) cyclic voltammogram of the P(DMTRZ-Cp) film. (c) Transient photoluminescence decay curve of the P(DMTRZ-Cp)-doped mCP film at 300 K; the inset is its prompt and delayed emission spectra. (d) Temperature-dependent transient PL decay curves over the 100 to 300 K temperature range for the P(DMTRZ-Cp)-doped mCP film. | ||
To confirm the ICT properties of the excited state of this polymeric emitter for TADF-OLED device applications, fluorescence spectra of the polymer solutions were recorded at room temperature using solvents of differing dipole moments (μ) (i.e., hexane (0.08 D), toluene (0.31 D), chloroform (1.15 D), and tetrahydrofuran (1.75 D)), as shown in Fig. S2.†P(DMTRZ-Cp) exhibits solvatochromic shifts (∼100 nm) as the solvent polarity is increased from hexane to tetrahydrofuran confirming that this polymer exhibits strong ICT characteristics.43–45 A phosphorescence spectrum almost identical to the fluorescence spectrum was also observed for P(DMTRZ-Cp). The energy difference between the singlet and triplet excited states (ΔEST) was determined from the difference in its fluorescence and phosphorescence spectral onset wavelengths. A small ΔEST value of 0.023 eV for P(DMTRZ-Cp) was obtained and it was believed that this polymeric emitter will exhibit TADF behavior.
The PLQY of P(DMTRZ-Cp) was determined to be 0.97 in a mixed film with mCP (Table 1). Compared to the solution state, the PLQY in the film state was enhanced by about a factor of two; this is likely to be due to the suppression of bimolecular deactivation processes and intramolecular nonradiative transitions in the solid state.46–48 An increase in the PLQY of the polymeric emitter was observed under nitrogen, compared to ambient conditions, in both the solution and film states (Table S1†). Through these observations we confirmed that triplet excitons are included in radiative singlet transition processes due to facile up-conversion from the triplet to the singlet state via reverse intersystem crossing (RISC). These phenomena support the conclusion that the new polymeric emitter synthesized in this study has TADF characteristics.20,49 Time-dependent PL measurements also confirmed that its prompt and delayed PL spectra were nearly identical, confirming that the delayed PL originated from a singlet state through RISC and the polymeric emitter has TADF characteristics (inset of Fig. 3c).
The energy levels of the HOMO and LUMO of the polymeric emitter were extracted from cyclic voltammetry (CV) scans shown in Fig. 3b and they were −5.37 eV and −2.95 eV, respectively. The thermal properties of the polymer were further evaluated using DSC (Fig. S3†); only featureless peaks such as glass transition and melting temperature were observed between 40 and 200 °C, indicating that this polymeric emitter has amorphous characteristics.12
For a better understanding of the TADF behavior of the polymeric emitter, we prepared a blended film of P(DMTRZ-Cp) with mCP and its transient photoluminescence spectra were acquired. As can be seen in Fig. 3c, the PL decay occurs over two steps consisting of a very fast prompt PL decay of the order of nanosecond and a delayed fluorescence decay with a microsecond-order lifetime. The lifetime of the prompt PL decay was 12.2 ns, indicating that radiative decay occurs from the S1 to the S0 state. On the other hand, the excitation lifetime for the delayed emission was determined to be 1.8 μs. This emission was confirmed to be radiative decay from the S1 to the S0 states through up-conversion from T1 to S1.50 Using these lifetime values and PL characteristics, the kinetic parameters of the doped film are also obtained and summarized in Table S1.†24,51
The new polymeric emitter, P(DMTRZ-Cp), was found to exhibit TADF behavior on the basis of its transient PL decay curve. In addition, as shown in Fig. 3d, the TADF characteristic was once again verified through PL decay behavior measured in the 100 K–300 K temperature region using the emitter-doped film. The delayed decay of P(DMTRZ-Cp) becomes faster in the microsecond range with increasing the temperature, indicating that the RISC phenomenon from the T1 to S1 excited states by supplying thermal energy is remarkably enhanced.52–54 This observation corroborates that P(DMTRZ-Cp) is a TADF material.
Performance of TADF-OLEDs
To investigate the TADF-OLED performance of the polymeric emitter, multilayered devices were fabricated by a solution process. The optimized device configuration is as follows: ITO/PEDOT:PSS (40 nm)/PVK (30 nm)/P(DMTRZ-Cp):mCP blend (40 nm)/TPBi (40 nm)/LiF (1 nm)/Al (100 nm) (Fig. 4a). | ||
| Fig. 4 (a) Schematic energy level diagram of a device with mCP as the host, (b) current density–voltage–luminance (J–V–L) curves, (c) external quantum efficiency (EQE)–current density (ηext–J) curves and (d) electroluminescence (EL) spectra at a luminance of 1000 cd m−2 of TADF-OLEDs fabricated using the new P(DMTRZ-Cp) polymer emitter. | ||
In these devices, poly(3,4-ethylenedioxythiophene):poly(styrenesulfonic acid) (PEDOT:PSS) and poly(9-vinylcarbazole) (PVK) were used as the hole-injecting layer and hole-transporting layer, respectively, and mCP was used as the host material. In addition, 1,3,5-tris(1-phenyl-1H-benzimidazol-2-yl)benzene (TPBi) was employed as an electron-transporting material.55,56 A schematic energy level diagram of the OLED device is shown in Fig. 4a. The emitting layer was prepared by different doping concentration of P(DMTRZ-Cp) into the mCP host ranging from 5 wt% to 30 wt%. Fig. 4b–d show the performance of TADF-OLED devices fabricated using the polymeric emitter synthesized in this study. These device parameters are summarized in Table 2.
| Polymer emitter | Doping conc. (wt%) |
V
ona (V) |
CEb (cd A−1) | PEc (lm W−1) | EQEd (%) |
λ
peake (nm) |
CIE (x, y)f | ||
|---|---|---|---|---|---|---|---|---|---|
| maximum | At 100 cd m−2 | At 500 cd m−2 | |||||||
| a The driving voltage at a brightness of 1 cd m−2. b Maximum current efficiency. c Maximum power efficiency. d External quantum efficiency at max/100/500 cd m−2. e EL peak wavelength. f The Commission Internationale de L'Eclairage coordinates at a luminance of 1000 cd m−2. | |||||||||
| P(DMTRZ-Cp) | 5 | 6.1 | 14.6 | 5.2 | 4.7 | 4.6 | 3.4 | 520 | (0.30, 0.53) |
| 10 | 5.3 | 19.1 | 7.5 | 6.1 | 6.0 | 5.1 | 520 | (0.31, 0.55) | |
| 15 | 4.1 | 41.2 | 25.9 | 12.5 | 10.4 | 6.8 | 528 | (0.34, 0.57) | |
| 20 | 4.1 | 42.9 | 27.0 | 13.0 | 9.1 | 7.4 | 528 | (0.34, 0.57) | |
| 25 | 4.0 | 50.5 | 31.8 | 15.4 | 9.7 | 6.4 | 528 | (0.35, 0.57) | |
| 30 | 4.0 | 29.7 | 18.7 | 9.0 | 8.2 | 6.4 | 532 | (0.35, 0.57) | |
Fig. 4b and c show current density–voltage–luminance (J–V–L) curves and EQE–current density (ηext–J) curves, respectively, at 20 wt%, 25 wt%, and 30 wt% doping concentrations of P(DMTRZ-Cp). Among the various device conditions (Fig. 4 and Fig. S6†), it showed the best device performance under the doping concentration of 25 wt%. The P(DMTRZ-Cp)-based device (25 wt%) has a low turn-on voltage (at 1 cd m−2) of 4.0 V, a maximum current efficiency of 50.5 cd A−1, Commission Internationale de L'Eclairage (CIE) coordinates of (0.35, 0.57), and a maximum EQE of 15.4%. Polymeric emitter-based OLED devices exhibit green EL spectra similar to those of the corresponding PL spectra of the mCP-doped films (Fig. S4†), confirming that EL is generated by the same origin in radiative decay processes (Fig. 4d).
The EL spectra of the devices using P(DMTRZ-Cp) exhibit the emission maxima at 532 nm. This result clearly shows the complete transfer of energy from the mCP host to the emitter, which is attributed to the confinement of the exciton exclusively in the emissive layer without leakage to the adjacent layers. In addition, the EL spectra of the polymer-based devices are not affected by the changes in the applied voltages over the 5–12 V range, as shown in Fig. S5,† which means that they are not affected by excimer or exciplex emissions and that the devices have excellent operational stability.57,58
Overall, TADF-OLEDs based on P(DMTRZ-Cp) exhibit higher EQE than the theoretical limit of a conventional fluorescent OLED, which is limited to an EQE of 5% based on spin statistics.59 These results, therefore, provide a useful strategy for developing a variety of high-performance polymeric emitters with TADF behavior.
Conclusion
We successfully synthesized P(DMTRZ-Cp) as a green TADF polymeric emitter using a non-conjugated Cp linker. The polymer synthesized using monomers based on the Cp linker, which contains a cyclohexane unit, was found to have sufficient solubility during the solution process while maintaining the TADF characteristics of the existing small molecule (i.e.DMAC-TRZ). The polymeric emitter exhibited fairly a small ΔEST value, which is advantageous for the TADF phenomenon. As a result, OLED devices based on P(DMTRZ-Cp) were effectively fabricated by solution processes and exhibited green EL emission with the maximum EQE value of up to 15.4%. According to the studies on polymeric emitters in TADF-OLEDs reported so far, this is, to the best of our knowledge, the first attempt to apply a cyclohexane unit (i.e., Cp) as a non-conjugated linker in a main-chain polymeric emitter structure. Hence, we believe that the incorporation of the Cp unit with appropriate TADF monomers is a promising strategy for the development of highly efficient solution-processable TADF-polymeric emitters.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Research Foundation of Korea (NRF2015R1A2A1A05001876) and by Key Research Institute Program (NRF20100020209).Notes and references
- C. W. Tang and S. A. Vanslyke, Appl. Phys. Lett., 1987, 51, 913–915 CrossRef CAS.
- J. R. Sheats, H. Antoniadis, M. Hueschen, W. Leonard, J. Miller, R. Moon, D. Roitman and A. Stocking, Science, 1996, 273, 884–888 CAS.
- B. W. D'Andrade and S. R. Forrest, Adv. Mater., 2004, 16, 1585–1595 CrossRef.
- M. A. Baldo, D. F. O'Brien, M. E. Thompson and S. R. Forrest, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 14422–14428 CrossRef CAS.
- A. Endo, K. Sato, K. Yoshimura, T. Kai, A. Kawada, H. Miyazaki and C. Adachi, Appl. Phys. Lett., 2011, 98, 083302 CrossRef.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- Y. Tao, K. Yuan, T. Chen, P. Xu, H. Li, R. Chen, C. Zheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7931–7958 CrossRef CAS PubMed.
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915–1016 RSC.
- M. Y. Wong and E. Z. Colman, Adv. Mater., 2017, 29, 1605444 CrossRef PubMed.
- Y. J. Cho, S. K. Jeon, S. S. Lee, E. Yu and J. Y. Lee, Chem. Mater., 2016, 28, 5400–5405 CrossRef CAS.
- D. Zhang, M. Cai, Z. Bin, Y. Zhang, D. Zhang and L. Duan, Chem. Sci., 2016, 7, 3355–3363 RSC.
- G. Xie, X. Li, D. Chen, Z. Wang, X. Cai, D. Chen, Y. Li, K. Liu, Y. Cao and S. J. Su, Adv. Mater., 2016, 28, 181–187 CrossRef CAS PubMed.
- P. Data, P. Pander, M. Okazaki, Y. Takeda, S. Minakata and A. P. Monkman, Angew. Chem., Int. Ed., 2016, 55, 1–7 CrossRef PubMed.
- R. Komatsu, T. Ohsawa, H. Sasabe, K. Nakao, Y. Hayasaka and J. Kido, ACS Appl. Mater. Interfaces, 2017, 9, 4742–4749 CAS.
- K. Nakao, H. Sasabe, R. Komatsu, Y. Hayasaka, T. Ohsawa and J. Kido, Adv. Opt. Mater., 2017, 5, 1600843 CrossRef.
- J. X. Chen, W. Liu, C. J. Zheng, K. Wang, K. Liang, Y. Z. Shi, X. M. Ou and X. H. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 8848–8854 CAS.
- D. H. Kim, K. Inada, L. Zhao, T. Komino, N. Matsumoto, J. C. Riberre and C. Adachi, J. Mater. Chem. C, 2017, 5, 1216–1223 RSC.
- F. Ni, Z. Wu, Z. Zhu, T. Chen, K. Wu, C. Zhong, K. An, D. Wei, D. Ma and C. Yang, J. Mater. Chem. C, 2017, 5, 1363–1368 RSC.
- Y. J. Cho, K. S. Yook and J. Y. Lee, Adv. Mater., 2014, 26, 6642–6646 CrossRef CAS PubMed.
- Y. J. Cho, B. D. Chin, S. K. Jeon and J. Y. Lee, Adv. Funct. Mater., 2015, 25, 6786–6792 CrossRef CAS.
- K. Albrecht, K. Matsuoka, K. Fujita and K. Yamamoto, Angew. Chem., Int. Ed., 2015, 54, 5677–5682 CrossRef CAS PubMed.
- J. Luo, G. Xie, S. Gong, T. Chen and C. Yang, Chem. Commun., 2016, 52, 2292–2295 RSC.
- Z. Ren, R. S. Nobuyasu, F. B. Dias, A. P. Monkman, S. Yan and M. R. Bryce, Macromolecules, 2016, 49, 5452–5460 CrossRef CAS.
- S. Y. Lee, T. Yasuda, H. Komiyama, J. Lee and C. Adachi, Adv. Mater., 2016, 28, 4019–4024 CrossRef CAS PubMed.
- X. Ban, W. Jiang, K. Sun, B. Lin and Y. Sun, ACS Appl. Mater. Interfaces, 2017, 9, 7339–7346 CAS.
- G. Xie, J. Luo, M. Huang, T. Chen, K. Wu, S. Gong and C. Yang, Adv. Mater., 2017, 29, 1604223 CrossRef PubMed.
- K. Albrecht, K. Matsuoka, D. Yokoyama, Y. Sakai, A. Nakayama, K. Fujita and K. Yamamoto, Chem. Commun., 2017, 53, 2439–2442 RSC.
- S. R. Forrest, Nature, 2004, 428, 911–918 CrossRef CAS PubMed.
- C. Rothe and A. Monkman, J. Chem. Phys., 2005, 123, 244904 CrossRef PubMed.
- Y. Xie and Z. Li, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 575–584 CrossRef CAS.
- A. E. Nikolaenko, M. Cass, F. Bourcet, D. Mohamad and M. Roberts, Adv. Mater., 2015, 27, 7236–7240 CrossRef CAS PubMed.
- Y. Zhu, Y. Zhang, B. Yao, Y. Wang, Z. Zhang, H. Zhan, B. Zhang, Z. Xie, Y. Wang and Y. Cheng, Macromolecules, 2016, 49, 4373–4377 CrossRef CAS.
- Y. Wang, Y. Zhu, G. Xie, H. Zhan, C. Yang and Y. Cheng, J. Mater. Chem. C, 2017, 5, 10715–10720 RSC.
- S. Shao, J. Hu, X. Wang, L. Wang, X. Jing and F. Wang, J. Am. Chem. Soc., 2017, 139, 17739–17742 CrossRef CAS PubMed.
- W. L. Tsai, M. H. Huang, W. K. Lee, Y. J. Hsu, K. C. Pan, Y. H. Huang, H. C. Ting, M. Sarma, Y. Y. Ho, H. C. Hu, C. C. Chen, M. T. Lee, K. T. Wong and C. C. Wu, Chem. Commun., 2015, 51, 13662–13665 RSC.
- T. A. Lin, T. Chatterjee, W. L. Tsai, W. K. Lee, M. J. Wu, M. Jiao, K. C. Pan, C. L. Yi, C. L. Chung, K. T. Wong and C. C. Wu, Adv. Mater., 2016, 28, 6976–6983 CrossRef CAS PubMed.
- S. N. Bagriantsev, K. H. Ang, A. G. Godoy, K. A. Clark, M. R. Arkin, A. R. Renslo and D. L. Minor, ACS Chem. Biol., 2013, 8, 1841–1851 CrossRef CAS PubMed.
- G. E. Park, H. J. Kim, S. Choi, D. H. Lee, M. A. Uddin, H. Y. Woo, M. J. Cho and D. H. Choi, Chem. Commun., 2016, 52, 8873–8876 RSC.
- G. Liaptsis and K. Meerholz, Adv. Funct. Mater., 2013, 23, 359–365 CrossRef CAS.
- I. S. Park, H. Seo, H. Tachibana, J. U. Kim, J. Zhang, S. M. Son and T. Yasuda, ACS Appl. Mater. Interfaces, 2017, 9, 2693–2700 CAS.
- Y. Tao, C. Yang and J. Qin, Chem. Soc. Rev., 2011, 40, 2943–2970 RSC.
- L. Xiao, Z. Chen, B. Qu, J. Luo, S. Kong, Q. Gong and J. Kido, Adv. Mater., 2011, 23, 926–952 CrossRef CAS PubMed.
- R. L. Martin, J. Chem. Phys., 2003, 118, 4775–4777 CrossRef CAS.
- C. Wang, X. Li, Y. Pan, S. Zhang, L. Yao, Q. Bai, W. Li, P. Lu, B. Yang, S. Su and Y. Ma, ACS Appl. Mater. Interfaces, 2016, 8, 3041–3049 CAS.
- G. Xie, D. Chen, X. Li, X. Cai, Y. Li, D. Chen, K. Liu, Q. Zhang, Y. Cao and S. J. Su, ACS Appl. Mater. Interfaces, 2016, 8, 27920–27930 CAS.
- S. Y. Lee, C. Adachi and T. Yasuda, Adv. Mater., 2016, 28, 4626–4631 CrossRef CAS PubMed.
- S. Gan, W. Luo, B. He, L. Chen, H. Nie, R. Hu, A. Qin, Z. Zhao and B. Z. Tang, J. Mater. Chem. C, 2016, 4, 3705–3708 RSC.
- P. Rajamalli, V. Thangaraji, N. Senthilkumar, C. C. R. Wu, H. W. Lin and C. H. Cheng, J. Mater. Chem. C, 2017, 5, 2919–2926 RSC.
- S. Y. Lee, T. Yasuda, Y. S. Yang, Q. Zhang and C. Adachi, Angew. Chem., 2014, 126, 6520–6524 CrossRef.
- T. Nakagawa, S. Y. Ku, K. T. Wong and C. Adachi, Chem. Commun., 2012, 48, 9580–9582 RSC.
- J. S. Kang, T. R. Hong, H. J. Kim, Y. H. Son, R. Lampande, B. Y. Kang, C. Lee, J. K. Bin, B. S. Lee, J. H. Yang, J. W. Kim, S. Park, M. J. Cho, J. H. Kwon and D. H. Choi, J. Mater. Chem. C, 2016, 4, 4512–4520 RSC.
- W. Liu, J. X. Chen, C. J. Zheng, K. Wang, D. Y. Chen, F. Li, Y. P. Dong, C. S. Lee, X. M. Ou and X. H. Zhang, Adv. Funct. Mater., 2016, 26, 2002–2008 CrossRef CAS.
- D. Y. Chen, W. Liu, C. J. Zheng, K. Wang, F. Li, S. L. Tao, X. M. Ou and X. H. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 16791–16798 CAS.
- J. X. Chen, W. Liu, C. J. Zheng, K. Wang, K. Liang, Y. Z. Shi, X. M. Ou and X. H. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 8848–8854 CAS.
- G. Hughes and M. R. Bryce, J. Mater. Chem., 2005, 15, 94–107 RSC.
- F. Wang, J. Hu, X. Cao, T. Yang, Y. Tao, L. Mei, X. Zhang and W. Huang, J. Mater. Chem. C, 2015, 3, 5533–5540 RSC.
- J. Y. Hu, Y. J. Pu, Y. Yamashita, F. Satoh, S. Kawata, H. Katagiri, H. Sasabe and J. Kido, J. Mater. Chem. C, 2013, 1, 3871–3878 RSC.
- L. Zhao, S. Wang, S. Shao, J. Ding, L. Wang, X. Jing and F. Wang, J. Mater. Chem. C, 2015, 3, 8895–8903 RSC.
- C. Mayr, S. Y. Lee, T. D. Schmidt, T. Yasuda, C. Adachi and W. Brutting, Adv. Funct. Mater., 2014, 24, 5232–5239 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: DSC, PL & EL spectra, J–V–L curves etc. based on a non-conjugated polymeric emitter. See DOI: 10.1039/c7py02113e |
| This journal is © The Royal Society of Chemistry 2018 |