
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOn the metal- and bio-catalyzed solvolysis of polyesters and polyurethanes wastes
Francisco G.
Cirujano†
 *a,
Rocio
Villa†
b,
Rebeca
Salas
b,
Miguel
Maireles
a,
Nuria
Martín
a,
Belén
Altava
*a,
Pedro
Lozano
*b and
Eduardo
García Verdugo
*a
*a,
Rocio
Villa†
b,
Rebeca
Salas
b,
Miguel
Maireles
a,
Nuria
Martín
a,
Belén
Altava
*a,
Pedro
Lozano
*b and
Eduardo
García Verdugo
*a
aDepartment of Inorganic and Organic Chemistry, Universitat Jaume I, Av. Vicent Sos Baynat, s/n, 12006, Castelló de la Plana, Castelló, Spain. E-mail: cirujano@uji.es; altava@uji.es; cepeda@uji.es
bDepartamento de Bioquímica y Biología Molecular B e Inmunología, Facultad de Química, Regional Campus of International Excellence “Campus Mare Nostrum”, Universidad de Murcia, 30071 Murcia, Spain. E-mail: plozanor@um.es
First published on 5th September 2024
Abstract
Catalysis is a crucial tool to efficiently address the recycling and upgrading of polymeric waste within the context of a circular economy, providing affordable and selective methods for waste valorization in alignment with the principles of green chemistry. Various catalysts, including metals, metal–organic frameworks, and biocatalysts, have been explored for the degradation of chemical poly(ethylene terephthalate) (PET) and polyurethane (PU) waste through processes like hydrolysis or alcoholysis. This critical review specifically focuses on catalytic tools, examining both homogeneous systems (such as metal salts or coordination organometallic complexes) and heterogeneous systems where the catalysts are immobilized on solids, including metal oxides, layered or porous solids, or inorganic–organic coordination polymers as well as biocatalytic counterparts from 2017 to the present. We provide a comparative analysis of the chemo-catalysts researched, evaluating their performance relative to biocatalysts using a SWOT analysis of both technologies to highlight their strengths and limitations in the context of sustainable waste management practices.
Sustainability spotlightOur consumerist society model is placing an unprecedented demand on resources, resulting in a significant increase in residues and waste generation, which creates fundamental doubts about the sustainability of the model. Among all the industrial activities, the plastic industry has become a cornerstone of modern civilization, while plastic wastes constitute one of the most important problems. The enduring nature of these polymers contributes to widespread environmental pollution, with far-reaching effects not only on land and sea but also on human health due to the pervasive issue of microplastic contamination. The depolymerization of plastic materials, followed by reusing the recovered monomeric products in polymer industry, is an urgent necessity. Our work analyzed how catalysis serves as a crucial tool in efficiently tackling the recycling and upgrading of polymeric waste within the framework of a circular economy, providing affordable and selective methods for waste valorization in alignment with the principles of green chemistry. This work emphasizes the importance of the following UN sustainable development goals, related with: Good health and well-being (SDG 3); Clean water and sanitation (SDG 6); Industry, Innovation and Infrastructure (SDG 9); Responsible consumption and production (SDG 12); Climate action (SDG 13); Life below water (SDG 14); and Life on land (SDG 15). |
1 Introduction
The polymer industry, which took off around the 1950s, has shown impressive growth from its humble beginnings with an annual consumption of 15 million metric tons to a staggering 390.7 million metric tons by 2021.1 This industry has become a cornerstone of modern civilization, influencing a broad range of sectors by underpinning crucial products and applications. Polymers are used in everything from packaging materials and medical devices to consumer goods, automotive components, and construction materials.2,3 Their versatility, durability, and cost-effectiveness have not only revolutionized these industries but also driven innovation and economic growth.4 Moreover, the polymer industry plays a critical role in addressing modern challenges such as sustainability and environmental impact. It has been instrumental in developing lightweight materials that reduce energy consumption in mobility, packing, and construction, as well as components for renewable energy sources like wind turbines.5,6Despite its success, the polymer industry faces significant environmental and social challenges.7 According to Plastics Europe (2022), Europe produced 57.2 Mt of plastics in 2021 and generated 29.5 Mt of plastic waste in 2020 and it is estimated that 65% of this plastic waste was landfilled or incinerated and only 35% was sent to recycling.1,8,9 Moreover, it is predicted that the amount of global plastic waste will increase from 260 to 460 metric tons annually from 2016 to 2030,10 and that more than 500 million metric tons of plastics will be required in 2050.11 Indeed, the impact of plastics on the environment is huge, as one can estimate, on the one hand, by the millions of metric tons of plastics per year delivered to the oceans, and on the other hand, by the large consumption of global petroleum estimated by 2050 (ca. 20%) only for plastics manufacturing. The persistent nature of polymers contributes to widespread environmental pollution, a problem exacerbated in regions with inadequate waste management infrastructures. This has led to significant environmental impact, which is manifested through pollution on both land and sea and the challenge of microplastic contamination. Over 98% of polymers derive from fossil fuels, and prevalent disposal methods like incineration significantly contribute to CO2 emissions. Additionally, although any biodegradable polymer is often considered greener as it does not generate visible permanent wastes, microbial degradation is a biological oxidation process that yields CO2 as final product. Thus, regardless of the origin of the starting materials for the production of polymeric materials, the recovery, depolymerisation and reuse of these materials is the most sustainable way to combat waste generation, where CO2 emission is the most invisible exponent.
The linear economic model of our society, based on the take-make-use-waste should be towards a circular model, where plastic wastes should be considered as chemical feedstocks, achieving the complete recirculation of molecules and materials.12,13 The Circular Economy Action Plan, introduced in 2015 to push the efforts to improve the circularity in Europe,14 responds to escalating environmental degradation and health concerns arising from resource inefficiency and emissions,15 such as the challenging plastic sources.16 The concept of the circular economy replaces the traditional “dispose” step with a “recycle/reuse” step, emphasizing the reuse of materials instead of the current linear product life cycle management processes.17,18 For a sustainable circular economy of polymers and plastics, chemistry should favour decoupling from fossil raw material inputs and contribute to achieving zero CO2 emission by incorporating recycled and biobased feedstock. Only then can we achieve many of the 17 Sustainable Development Goals (SDG) set by the United Nations, 19 such as SDG 3 “Good Health and Well-being”, SDG 8 “Decent Work and Economic Growth”, SDG 9 “industry, innovation, and infrastructure”, SDG 12 “Ensure sustainable consumption and production patterns”, and SDG 13 “Climate Action”. Developing more efficient and economical catalytic methodologies is crucial for achieving sustainable objectives in polymer recycling. Advanced catalytic recycling techniques facilitate the conversion of waste polymers into valuable raw materials that can be reintegrated into the production cycle. This approach not only minimizes environmental impact but also reduces waste production in a sustainable and cost-effective manner.
1.1 Catalysis as the key enabling tool for plastic recycling
This review explores the development of novel chemical and biochemical transformations aimed at the efficient recovery and recycling of polymer materials to mitigate their impact on the environment.19–29 Despite the interest of bio-based polymers (able to be degraded by green solvents, supercritical processes and catalysts),30 special attention will be given to the recycling of polyethylene terephthalate (PET) and polyurethane (PU), as shown in Scheme 1 (top part), which are significant due to their high production volumes and applications ranging from textiles to food and beverage containers.31–35 For recent literature review on polyester depolymerisation please refer to ref. 29c and 29d. The review will cover different recycling processes, particularly focusing on catalytic chemical recycling techniques, which, though less common, are anticipated to play a significant role in future waste management strategies to provide a detailed understanding of the current challenges and innovations within the polymer recycling industry. The need for integrated approaches that consider environmental impacts, technological advancements, and economic factors in developing sustainable polymer lifecycle management strategies will be emphasized.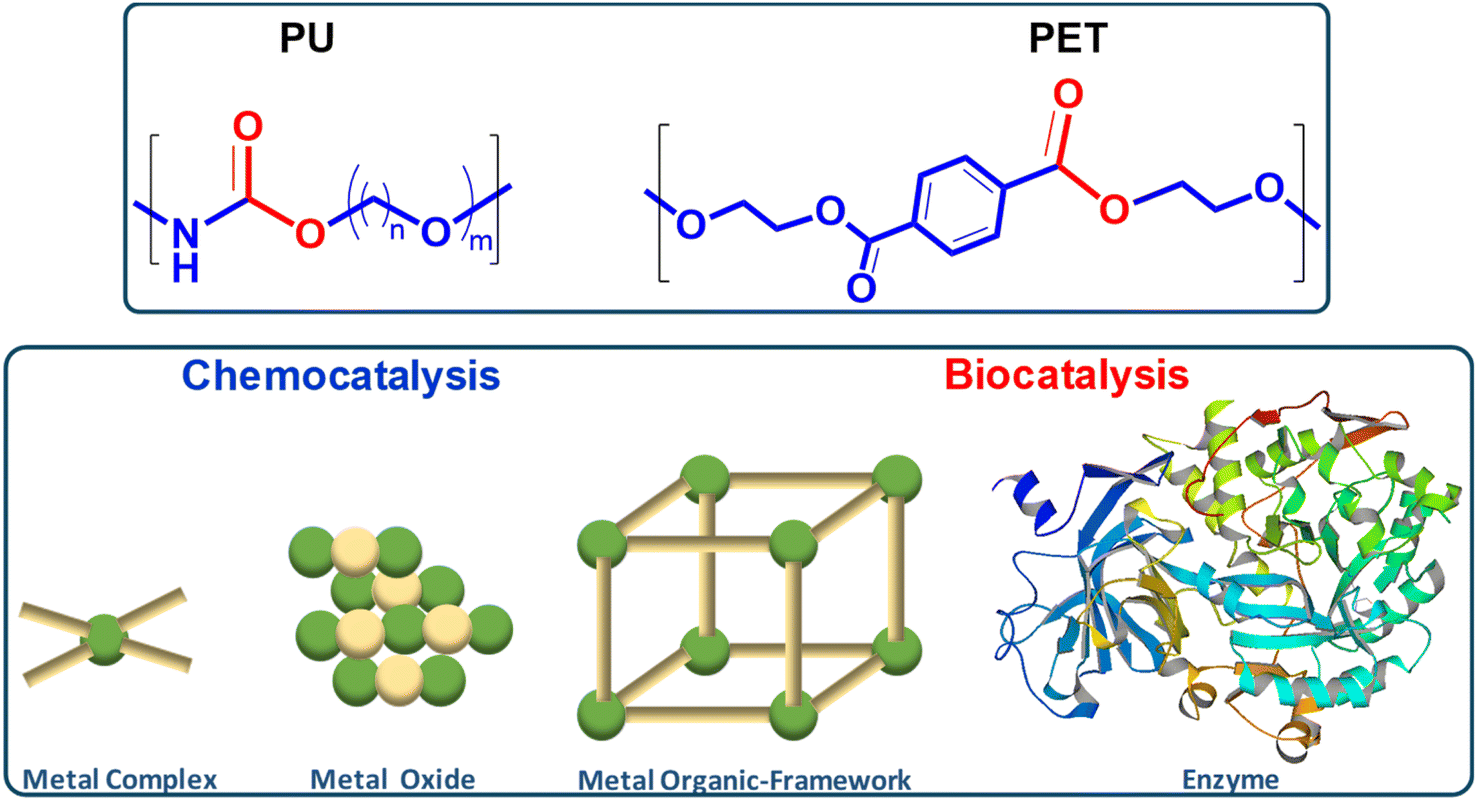 | ||
| Scheme 1 Similarities and differences between PU and PET condensation polymers reviewed here (top). Discrete metal ions, organometallic complexes, metal oxides and porous metal–organic frameworks as chemocatalysts vs. enzyme biocatalysts (bottom). | ||
From a sustainability perspective, the use of non-toxic and abundant first-row transition metals as catalysts is particularly promising. Thus, the chemocatalytic part of the review will be focused mainly on metal catalysis, and the reader is referred to recent works on other approaches such as organocatalysis.36–39 Special attention will be paid to the immobilization of these metals within various frameworks, both organic and inorganic, as depicted in Scheme 1 (bottom part). This immobilization results in robust and reusable heterogeneous catalysts that facilitate recycling without significantly contaminating the polymers or monomers. On the other hand, enzymes represent the most active and selective catalysts operating under mild, non-toxic conditions, making them, from a green chemistry point of view, ideal for polymer, e.g. polyurethane, solvolysis.40 However, the high cost and fragility of enzymes as homogeneous biocatalysts have encouraged interest in developing more versatile, stable heterogeneous catalysts that bridge the gap between traditional inorganic metal catalysts and biological catalysts. In this context, the review also includes recent developments in heterogeneous catalysts such as porous metal oxides or metal–organic frameworks to provide a fresh perspective on the interplay between chemical and biological catalysis.
Table 1 summarizes the different catalytic approaches considered in this work, according to the nature of the active site, either discrete active sites (part 2), sites dispersed in extended dense (part 3) or open porous networks (part 4), or biocatalyst (part 5) approaches for the solvolysis of these type of polymers.
| Active site | Catalyst | Solid | Porous | Polymer |
|---|---|---|---|---|
| Zn | ZnCl2 | No | No | PU/PET |
| Zn(OAc)2 | No | No | PU/PET | |
| ZnO | Yes | Yes | PET | |
| ZnO/SiO2 | Yes | Yes | PET | |
| Zn-MOF | Yes | Yes | PET | |
| Fe | FeCl3 | No | No | PU |
| Fe2O3 | Yes | No | PET | |
| Sn | SnOctoate | No | No | PU |
| Sn/Zn/Al-LDH | Yes | Yes | PU | |
| Ti | TiOBu4 | No | No | PU |
| TiO2–SO4 | Yes | No | PET | |
| His, Asp, Ser | Cutinase | No | No | PET |
| His, Asp, Ser | PETase | No | No | PET |
| Cu, polypeptide | Laccase | No | No | PU |
| His, Asp, Ser | Lipase | No | No | PU |
| His, Asp, Ser | Urethanase | No | No | PU |
2 Discrete metal ions
2.1 Low valence metal catalysts: the special case of Zn(II)-based catalysts
Either rigid and flexible polyurethane (PU) foam was degraded (via glycolysis) into polyol (see Scheme 2), with FeCl3 as the most active catalyst precursor (1 wt%) among different 1st-row transition metals such as Zn, Co, Ni and Fe in the form of metal chloride salts.41 Temperature and reaction time and the type of diol (ethylene glycol (EG) or 1,4-butanediol, being the first more active towards polyol generation than the second) were optimized due to their effect on the solubility of the metal salts. In general, no reaction was observed at temperatures below 160 °C, having an increase in the recovered polyol when increasing the temperature from 160 to 200 °C. Neither longer reaction times nor temperatures higher than 200 °C allowed the recovery of polyol due to the thermal decomposition of such products.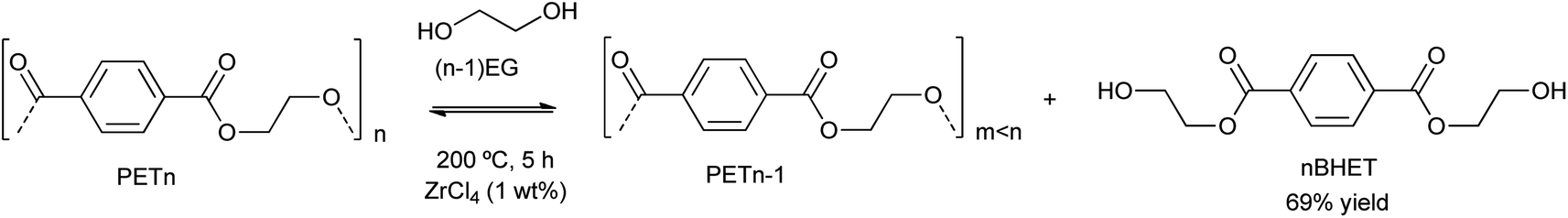 | ||
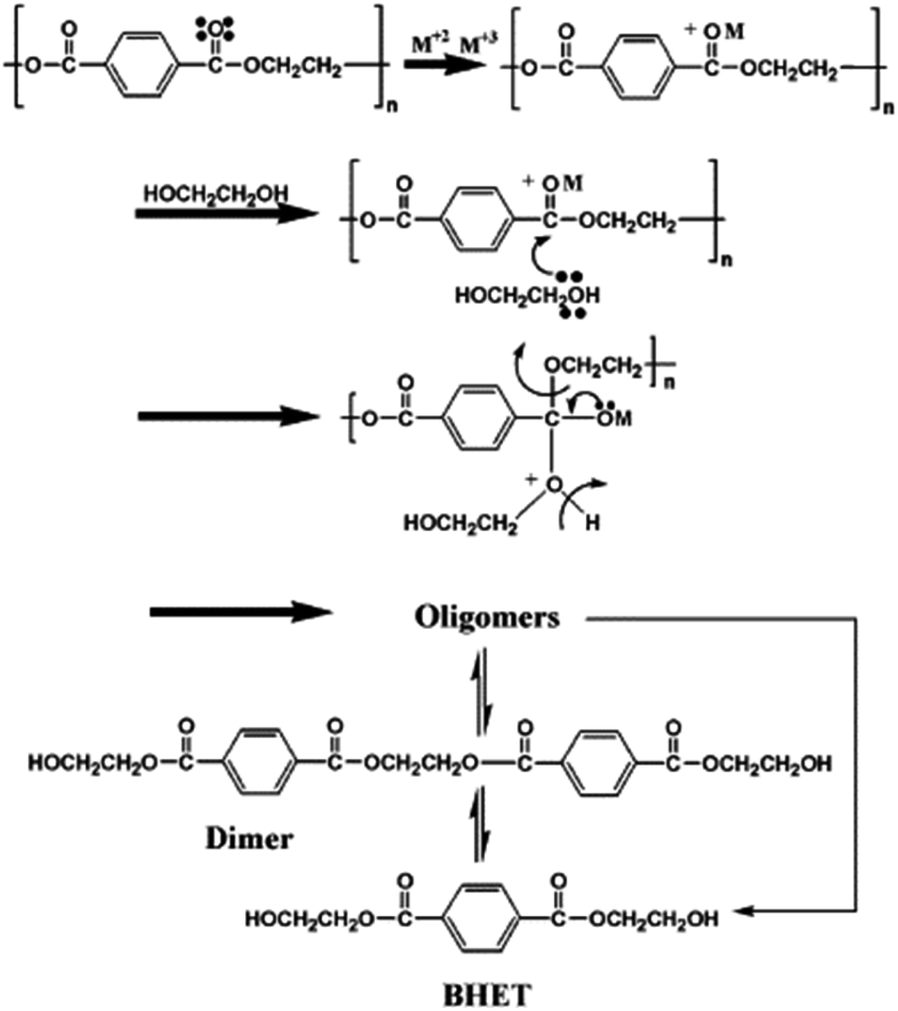
| Scheme 2 PET degradation by glycolysis into BHET using metal chlorides, i.e. ZrCl4 (1% wt), at 200 °C during 5 h. | ||
On the one hand, intermediate trivalent and divalent hard metal cations behaving as Lewis acids were able to degrade rigid PU to more than 90% of polyols. In the case of highly polarizing metal cations, both degradation and re-polymerization processes are favored, thus decreasing the polyol yield. On the other hand, the reactivity of metal chlorides towards rigid PU degradation seems to be related to their corresponding cation charge density and hardness, according to the following order: Fe3+ (44% polyol) > Zn2+ (23% polyol) > Co2+ (44% polyol). In the case of the iron catalyst, the yield increased with the temperature, where the polyol recovery is maximum at 200 °C (see entries 1–3, Table 2). For reaction times larger than 5 h the polyol molecular weight increases because of its re-polymerization.
| Entry | Catalyst | Substrate | Solvent | Temp. (°C) | Time (h) | Monomer yield | Ref. |
|---|---|---|---|---|---|---|---|
| a 5 bar. b 150 bar. c 40 bar. | |||||||
| 1 | FeCl3 (1% wt) | PU | EG | 200 | 5 | 44% | 41 |
| 2 | ZnCl2 (1% wt) | PU | EG | 200 | 5 | 23% | 41 |
| 3 | CoCl2 (1% wt) | PU | EG | 200 | 5 | 44% | 41 |
| 4 | PET | 5 | 67% | 41 | |||
| 5 | ZrCl4 (1% wt) | PET | EG | 200 | 5 | 69% | 41 |
| 6 | ZnCl2 (70% wt) | PU | H2O (14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
140 | 2 | 100% | 42 |
| 7 | Zn-salen (8% wt) | PET | EG (27:1) |
180 | 2 | 50% | 43 |
| 8 | Zn-salen (7% wt) | PET | EG (27:1) |
180 | 1 | 60% | 44 |
| 9 | Zn(OAc)2 (1% mol) | PET | EG (7.6:1) |
196 | 1 | 70% | 45 |
| 10 | Zn(OAc)2 (1% wt) | PU | DEG | 200 | 4 | 48% | 46 |
| 11 | Sn octaoate | PU | EG (15:1) |
190 | 4 | 80% | 47 |
| 12 | ZnO | PET | MeOH | 140 | 2 | 50% | 48 |
| 13 | ZnO | PET | EG (10:1) |
210 | 0.5 | 100% | 49 |
| 14 | Mo/ZnO (1% wt) | PET | EG (4:1) |
189 | 1 | 90% | 50 |
| 15a | ZnMn2O4 (1% wt) | PET | EG (12:1) |
260 | 1 | 93% | 51 |
| 16 | ZnO/Al2O3 (7.5% wt) | PET | Ethanol (sc) | 270 | 1 | 90% | 52 |
| 17 | Ni/Al2O3 (10% wt) | PET | H2O sc | 270 | 1 | 95% | 53 |
| 18b | SO4–TiO2 (10% wt) | PET | H2O (10:1) CO2 sc |
160 | 12 | 99% | 54 |
| 19 | Fe2O3 (2% wt) | PET | EG (13:1) |
220 | 0.5 | 90% | 55 |
| 20 | CeO2 | PET | EG (7:1) |
196 | 0.25 | 90% | 56 |
| 21 | Mg/Zn/Al LDH | PET | EG (10:1) |
196 | 3 h | 80% | 57 |
| 22 | Zn/Sn/Al LDH (1.3% wt) | PU | DEG (5:1) |
190 | 3 | 66% | 58 |
| 23 | e-MnO2 | PET | EG (55:1) |
200 | 0.5 | 100% | 59 |
| 24 | ZnO/SBA-15 (5%) | PET | EG (4:1) |
200 | 1 | 90% | 60 |
| 25 | MAF-6 | PET | EG (6:1) |
180 | 4 | 82% | 61 |
| 26 | Leaf-branch compost cutinase (LCC) | PET | 0.1 M potassium phosphate, pH 8 | 72 | 10 | 90% | 62a |
| 27 | PETase | PET | 50 mM potassium phosphate buffer, pH 7.2 | 30 | 1 | 78% | 63 |
| 28 | Ca2+(1 M)/LCCICCG | PET | 50 mM Tris–HCl buffer, pH 8 | 80 | 12 | 84% | 64 |
| 29 | NusA-IsPETaseMut | PET | 50 mM Gly-NaOH buffer, pH 9 | 30 | 14 days | 19.6% (weight loss) | 65a |
| 30 | S238Y-IsPETaseMut | PET | 50 mM Gly-NaOH buffer, pH 9.4 | 30 | 72 | ∼3% (crystallinity loss) | 65b |
| 31 | RITK-cutinase Mut | PET | 100 mM potassium phosphate buffer, pH 8 | 72 | 24 | 90% | 66 |
| 32c | H2O | PET | H2O | 250 | 1.5 | 85% | 67 |
| 33 | Humicola insolens cutinase | 0.1 M Tris–HCl buffer, pH 7 | 50 | 24 | >99% | ||
| 34 | Trametes versicolor laccase | PU | 0.1 M sodium acetate buffer, pH 4.5 | 37 | 18 days | 17.2% (weight loss) | 68 |
| 35 | Humicola insolens cutinase | PU | 0.1 M potassium phosphate buffer, pH 8 | 50 | 168 | 42% (weight loss) | 69 |
| 36 | Tfcut2 (cutinase) | Dispersed PU | 0.3 M potassium phosphate buffer, pH 8 | 70 | 200 | 0.026 s−1 | 70 |
| 37 | LCC (cutinase) | Solid PU solid | 0.1 M potassium phosphate buffer, pH 8 | 5% | |||
| 38 | Candida rugosa lipase | PU | 0.1 M potassium phosphate buffer, pH 7 | 35 | 1 | 0.12 mg product per L per min | 71 |
| 39 | Tin(II)-2-ethylhexanoate | PU | DEG | 200 | 2 | 99% | 72 |
| 40 | UMG-SP-2 (engineered urethanase) | 0.1 M potassium phosphate buffer, pH 7.5 | 30 | 48 | >99% | ||
The authors proposed two pathways of transesterification reactions occurring during the Lewis acid-catalyzed PU degradation. One is based only on the polymer degradation with no polyol release, and another implies the release and recovery of polyol. The last pathway is disfavored in the case of rigid PU foam with a highly cross-linked structure and highly functionalized polyol products. On the contrary, the use of borderline Lewis acids, such as CoCl2 favors the first pathway of molecular weight decrease with a low polyol release and recovery.
Besides the valorization of PU, the authors employed similar catalysts for the degradation of PET into its bis(2-hydroxyethyl terephthalate) (BHET) monomer (see Scheme 2). However, no clear relation between cation and anion could be established, although the interaction of the metal cation with polyester is key, and therefore a combination of cation hardness, charge density and anion softness will control the catalyst-carbonyl group interaction, promoting its degradation. Indeed, an induction period at the beginning of PET degradation suggested this metal-carbonyl interaction, after which the ethylene glycol attacks the Lewis acid-activated ester groups, resulting in the solubilization/degradation of PET in the alcohol, medium. The highest yields of (BHET) were obtained with ZrCl4 (69%) and CoCl2·6H2O (67%) catalysts at 200 °C, while no yield was observed for the blank reaction (see entries 4–5, Table 2).
Wang et al. proposed the efficient PU degradation, via selective cleavage of C–O/N bonds by coordinatively unsaturated Zn(II) species formed in highly concentrated 70% ZnCl2 aqueous solutions.42 The electrophilic Zn(II) cations behave as Lewis acid for the activation (via coordination) of the oxygen and nitrogen heteroatoms present in the functional groups of PU substrate. This resulted in the eventual cleavage of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O urethane linkages and C–N bonds by the ZnCl2, used in a weight excess of 14:1 with respect to the PU, at 140 °C for 2 h (see entry 6, Table 2). By FTIR and NMR, 2,4-diaminotoluene (DAT), polytetramethylene ether glycol (PTMEG) and 3,3′-dichloro-4,4′-diaminodiphenylmethane (MOCA) were detected as the products of PU degradation (see Scheme 3). However, the C–C and C–O–C bonds were not altered by the Zn(II) of ether linkages as the framework structure remained intact in the degradation process. The mechanism of selective cleavage of C–O and C–N bonds of urethane resulted in the production of carbamic acid (which produced CO and the corresponding amine) and alcohol (PTMEG), which was no further cleavage at the C–O ether bonds.
O urethane linkages and C–N bonds by the ZnCl2, used in a weight excess of 14:1 with respect to the PU, at 140 °C for 2 h (see entry 6, Table 2). By FTIR and NMR, 2,4-diaminotoluene (DAT), polytetramethylene ether glycol (PTMEG) and 3,3′-dichloro-4,4′-diaminodiphenylmethane (MOCA) were detected as the products of PU degradation (see Scheme 3). However, the C–C and C–O–C bonds were not altered by the Zn(II) of ether linkages as the framework structure remained intact in the degradation process. The mechanism of selective cleavage of C–O and C–N bonds of urethane resulted in the production of carbamic acid (which produced CO and the corresponding amine) and alcohol (PTMEG), which was no further cleavage at the C–O ether bonds.
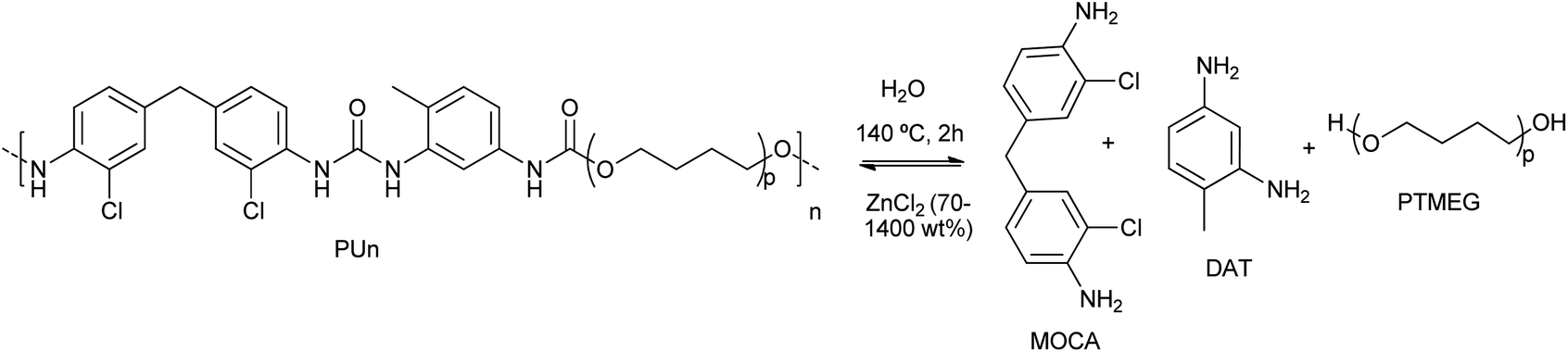 | ||
| Scheme 3 Controllable degradation of PU using ZnCl2 (70–1400 wt%) as a supra-stoichiometric reagent at 140 °C for 2 h. | ||
A similar 70 wt% ZnCl2 aqueous solution was employed for the selective solvolysis of a polyester/polyurethane-coated textile composed of PET fabrics coated with bio-based PU, simplifying the separation of the coating from the fabric for further recycling.73 Suprastoichiometric amounts of ZnCl2 (17.5 times more weight) were employed with respect to the PU-coated polyester under aqueous conditions at 140 °C for 2 h. In this case, the Zn(II) Lewis acid sites are in a cheaper medium with respect to deep eutectic solvents (DES) and ionic liquids (IL). FTIR analysis of the products at the liquid fraction indicated the formation of primary amines and isocyanurates, as well as the polyester polyol (some of them oxidized to aldehydes by the Zn sites). Interestingly, the polyester was not degraded under the conditions employed.
Different salen-type zinc complexes, prepared from amine-aldehyde condensations (see Scheme 4a), were reported for the methanolysis of polylactic acid (PLA) at 80 °C and glycolysis of PET from carbonated drinks bottles to form bis(2-hydroxyethyl) terephthalate (BHET).43 Around 50% yield to colorless (after recrystallization) BHET was obtained after 2 h, working at 180 °C in the presence of 8 wt% loading and 27 equivalents of ethylene glycol (EG) with respect to ester linkages (see entry 7, Table 2).
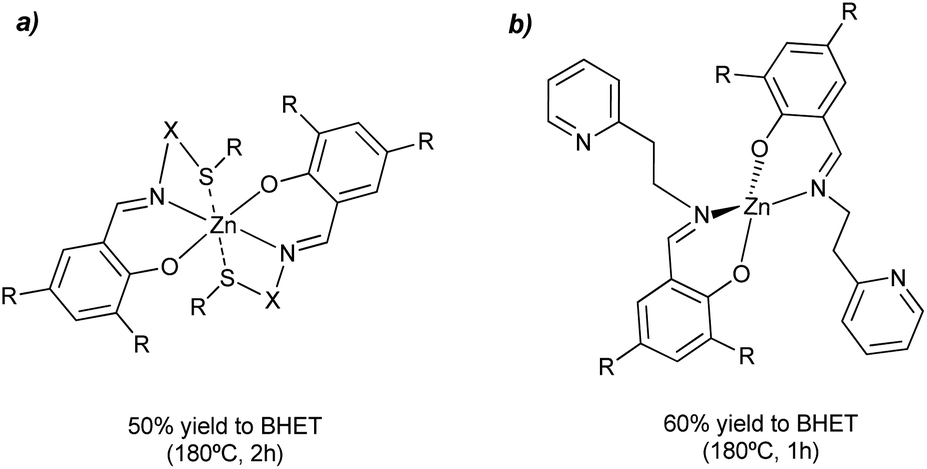 | ||
| Scheme 4 Salen-Zn catalyst (8 wt%), without (a) or with (b) pendant pyridine groups, for the glycolysis of PET into BHET at 180 °C for 1–2 h. | ||
Homoleptic zinc complexes linked by monoanionic phenoxy-imine pyridine moieties were active catalysts in the alcoholysis of PLA and glycolysis of commercial poly(ethylene terephthalate) samples.44 The Zn active site is located in a plane with two (almost perpendicular) bidentate ligands. The salen group chelates the metal while one pendant pyridine points in the other direction to the metal center (see Scheme 4b). The PET sample was contacted with 27.8 equivalents of ethylene glycol in the presence of 0.013 equivalents of Zn (both with respect to the ester functionalities) at 180 °C for 1 h. Under such conditions, up to 95% conversion and 60% yield of BHET was obtained (see entry 8, Table 2). Similar amounts of ZnO required 4 h to convert 72% of the PET, obtaining only 20% yield of BHET. The reported catalyst also outperformed the benchmark zinc acetate, which produced 40% yield of BHET after 4 h while using seven times more catalyst. The zinc sites should work as Lewis acid catalysts that activate the carbonyl group while the pyridine groups might abstract the proton from the alcohol solvent. However, more investigation is needed to understand the catalyst's reaction mechanism and stability under operando conditions. Other authors employed zinc acetate Zn(OAc)2 (1 mol% with respect to PET) as a catalyst for PET granules wastes degradation in EG.45 After 1 h, a BHET yield of 70% was obtained at 196 °C, using an excess of EG:PET molar ratios of 3.8 (see entry 9, Table 2). In contrast, no appreciable yield was detected until 6 h for the blank reaction at 196 °C, even with an EG:PET ratio of 7.6. Alternative catalysts to transition metals, such sodium carbonate, were tested as a depolymerization catalyst, given the lower price and toxicity with respect to Zn. However, higher PET:catalyst molar ratios were needed to achieve similar BHET yields as those obtained with Zn (i.e. PET:Na = 245 vs. PET:Zn = 380). Other sodium salts such as sodium sulfate give poor product yields. This was attributed to the lower solubility with respect to the carbonate. Regarding the mechanism, the formation of Na/Zn-carbonyl intermediates during the transesterification reaction process were proposed. In fact, the metal–oxygen bonds between the catalytic active metal site and the functional groups at the PET substrate are shorter for Zn (2.26 Å) than for Na (2.41 Å). Therefore, a better activation of the ester group by the Zn(II) results in a faster nucleophilic attack by the EG.
Grounded polyurethane foam wastes at 200 °C in diethylene glycol (DEG) was depolymerized (transesterified) in the presence of zinc acetate (1 wt%) as a catalyst (Scheme 5a).46 This zinc-based homogeneous catalyst was more active (48% decrease of the urethane bond area) than organic amine catalysts (see entry 10, Table 2), such as diethylamine (17% decrease of the urethane bond area) or methylamine (25% decrease of the urethane bond area), as authors observed by FTIR. The zinc acetate was also more active than inorganic bases, such as barium or potassium acetates, achieving less than 25% decrease in the urethane bond area. Even the reported tin alkoxide catalyst exhibited lower activity in the PU depolymerization (less than 15% decrease in the urethane bond area). Thus, the divalent Zn ions were key in the activation of the urethane carbonyl group, promoting the nucleophilic attack by the oxygen of the low-weight glycols (see Scheme 5a). On the contrary, the reaction pathway occurring with the tin-based catalysts is different. This is because the alcohol first adds to the tin complex forming an alkoxide intermediate, see for instance their application in split-phase glycolysis.74–77 Indeed, the formation of the active species in the case of the zinc catalyst is ten times higher than the tin complexes (see Scheme 5b).
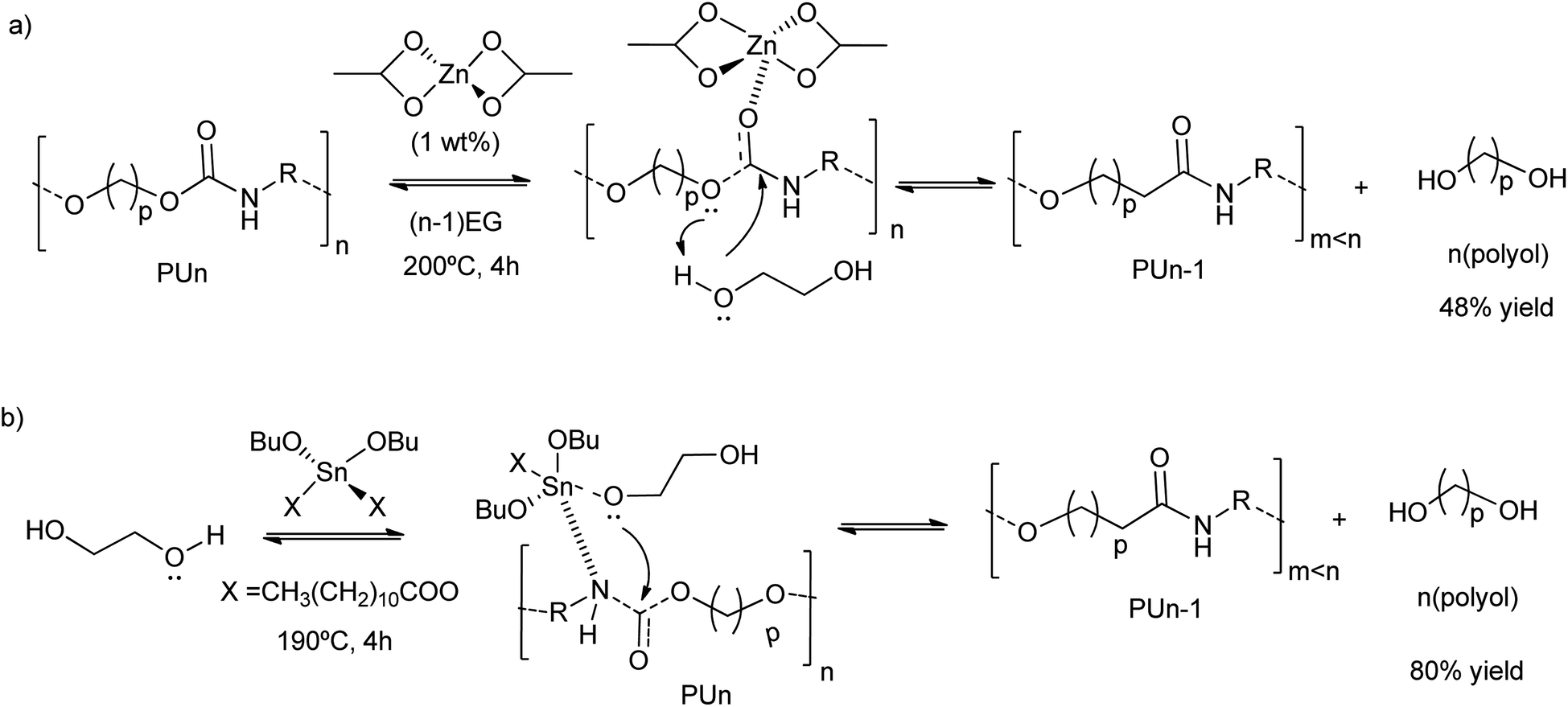 | ||
| Scheme 5 Proposed mechanisms for PU degradation using Zn(OAc)2 (a) or Sn(OBu)2X2 (b) as a catalyst at 190–200 °C for 4 h. | ||
2.2 High valence metal catalysts: tin(IV) and titanium(IV)-based systems
Octoate salts of alkaline or transition metal catalysts (such as Li, Na, K, Sr, Co, Ni, Zn and Sn) were reported as active catalysts for the glycolysis of polyurethane.47,78 On the one hand, scrap foams with a size of 5–25 mm and a composition of polyether polyol poly(propylene oxide-block-ethylene oxide) and toluene diisocyanate (TDI) were employed as substrate. On the other hand, a 15:1 mass ratio of ethylene glycol (EG) with respect to the scrap PU foam and a metal catalyst concentration of 60 mM were reported.47 All catalysts completely degrade the polyurethane chain at 190 °C, as determined by the disappearance of the oligourethanes (see entry 11, Table 2). However, different glycolysis rates were obtained depending on the metal. The best performance catalysts were the Li and Sn octoate salts. The high degradation rate and polyol recovery were attributed to the hard Lewis acid nature of the metal cation.47 Indeed, the interaction of the metal with the hard Lewis base oxygen atom from EG, favors the formation of the metal alkoxide. Note that this intermediate has a non-coordinated hydroxyl group at the glycol that subsequently acts as a nucleophile towards the carbonyl carbon of the urethane group. Besides such first rate-determining step, the transesterification reaction mechanism involves: (i) the formation of a metal alkoxylate, (ii) coordination–insertion of the alkoxide into the urethane group and (iii) transfer from recovered polyol to glycol (see Scheme 6).
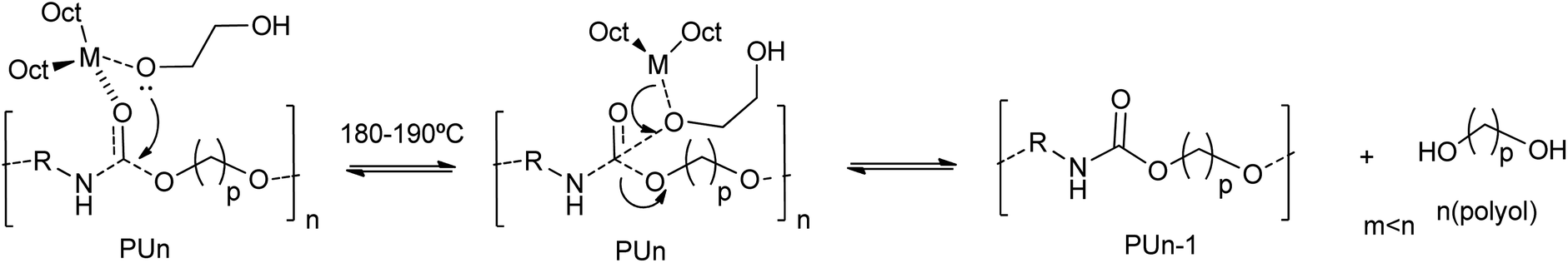 | ||
| Scheme 6 Proposed mechanisms for PU degradation at 180–190 °C using octoate salts of metals (M = Li, Na, K, Sr, Co, Ni, Zn) as a catalyst. | ||
In the case of strontium, which was classified as a soft Lewis acid, the PU degradation rate was slower. For Co, Sn and Zn octoate salts the degradation rates were higher than in the case of Ni salts. Thus, there should be some additional stabilization factor of the Sn-complexation (via coordination-insertion) to the urethane carbonyl group, as well as the not-too-strong (basic alkaline cations)/not-too-weak metal alkoxide bonding resulting from the metal octoate and EG first reaction step. Interestingly, there is no need to separate the stannous octoate from the obtained polyol phase since this same catalyst is employed in PU foaming from the recovered polyol, so no additional purification steps are required when aiming at this application.47 The higher activity of the Zn salt is in line with the above-mentioned high-performance zinc catalysts (zinc acetate and zinc chloride) for both PET and PU degradation, which confirms the similarity between both transesterification reactions.78
Other authors employed titanium(IV) tetrabutoxide for the dissolution of rigid PU foams at 180 °C in dipropylene glycol (DPG).79 Although less active than traditional inorganic Brønsted bases, the titanium catalyst requires less time (45 min) than stannous octoate (143 min) for the PU glycolysis (0.0036 mol cat./DPG 100 g). This strategy allowed the formation of titanium isopropoxide-derived titanium nanodiol catalysts, consisting of a homogenized nanoscale titanium core coated with an alcohol shell.80 This provided a more active catalyst, concerning traditional alkali metal-based catalysts, for the glycolysis of waste polyurethane (PU) pipeline foams. A combination of KOH and the titanium catalyst, even in amounts of only 0.05 wt%, decreased the regenerated polyether polyols viscosity by 10% (Scheme 7). However, little information was provided about PU conversion and reaction mechanisms for the sole titanium catalyst.
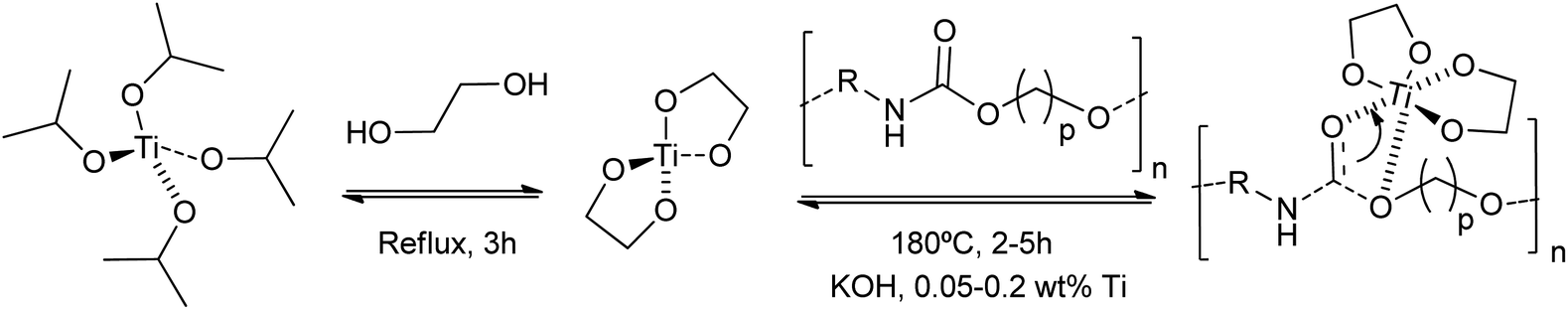 | ||
| Scheme 7 Proposed mechanisms for hard PU degradation at 180 °C for 2–5 h using KOH and 0.05–0.2 wt% Ti-glycol nanocatalysts prepared from titanium isopropoxide and ethyleneglycol. | ||
Finally, we will highlight an example of polyurethane synthesis from PET-based polyols. Three consecutive catalytic steps were carried out to combine the PET degradation and with the PU synthesis from the polyol building blocks obtained. The first step consisted of the glycolysis reaction of PET at high temperatures (230–300 °C) using titanium isopropoxide as catalyst (0.5 wt%).
Crude glycerol was added to the glycolyzed PET oligomeric mixture. The final polyol mixture obtained showed less hydroxyl number and was used as a monomeric mixture for the synthesis of polyurethanes with different physical and chemical properties (i.e. density, compressive strength, etc.).81 However, the use of extreme reaction conditions as well as non-selective catalysts generates uncontrolled reactions and side products.
3 Dense metal oxide architectures
3.1 Low valence metal oxides: the special case of ZnO-based catalysts
ZnO nanoparticles were obtained either from the methanolysis or glycolysis of zinc acetate in a solution of KOH in methanol or ethylene glycol, respectively.48 Methacryloxy and/or amino alkoxysilanes were also employed in the basic alcohol media, probably modulating nanoparticle growth. The colloidal dispersion of ultra-small ZnO nanoparticles (ca. 4 nm) has been employed as a pseudo-homogeneous catalyst (see Scheme 8a). PET methanolysis and glycolysis into dimethylterephthalate (DMT) and bis(2-hydroxyethyl terephthalate) or BHET, respectively, were successfully achieved by such catalyst. The adequate dispersion of the nanoparticles in methanol avoids their aggregation of (<20 nm) favoring their accessibility and diffusion of the PET substrate. Using an alcohol:PET weight ratio of six, a 97% yield to DMT was obtained in less than half an hour. When the temperature was decreased to 140 °C, a 50% yield of DMT was obtained after just 2 h (see entry 12, Table 2). This corresponds to 553 and 117 gPET h−1 gZnO−1, for the methanolysis and glycolysis, respectively. On the other hand, the glycolysis of PET was 4 times slower compared to the methanolysis. The methanolysis occurred from the outside to the inside of the PET, decreasing the particle size of the polymer and increasing its porosity. Key to the mechanism was the activation of the carbonyl bond by the Zn atoms of the nanoparticles, acting as Lewis acid catalysts. Simultaneously, the adjacent oxygen atoms assisted in the deprotonation of the alcohol solvent, which eventually broke the ester linkages generating methyl ester oligomers (see Scheme 8).
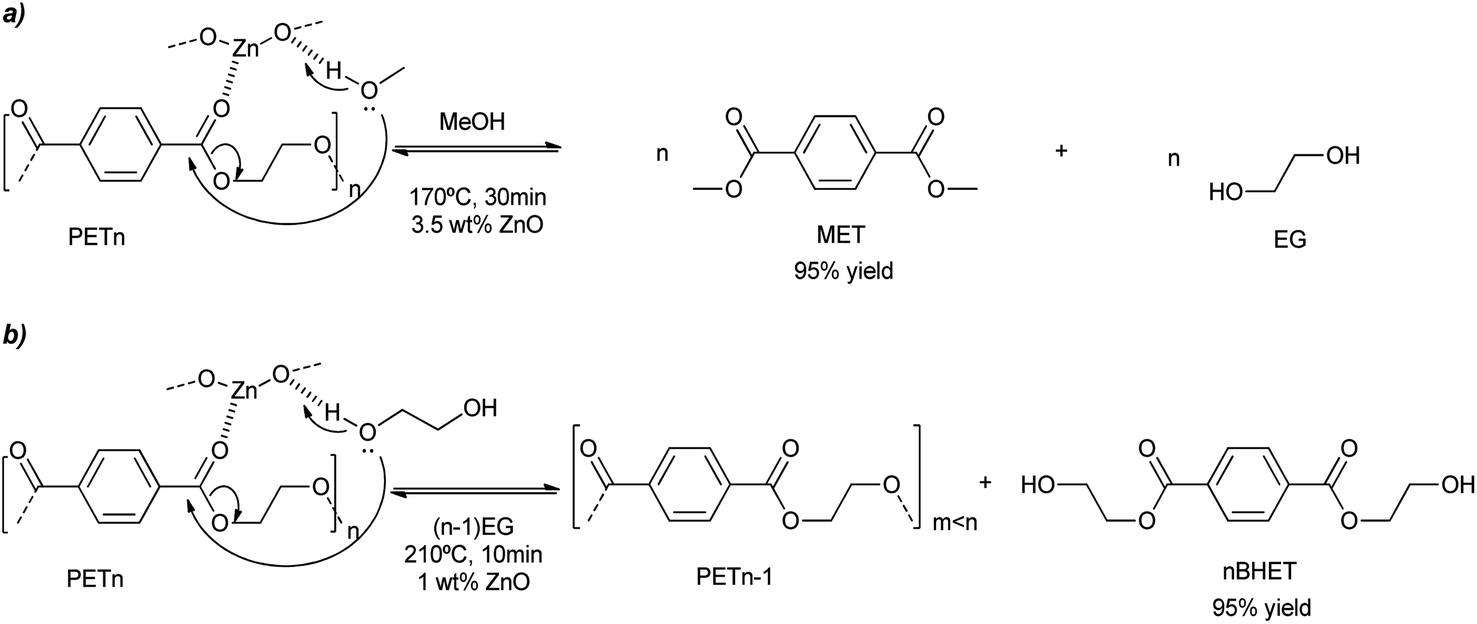 | ||
| Scheme 8 PET degradation mechanism by methanolysis (a) or glycolysis (b) into MET or BHET, respectively, promoted by ZnO nanoparticles (1 wt%) at 210 °C for 10 min. | ||
A solution containing polyvinyl pyrrolidone (PVP), zinc acetate and sodium hydroxide in ethanol or pentanol/ethanol resulted in ZnO nanorods and ZnO nanoplates, respectively.49 Using an EG:PET molar ratio of 10 at 210 °C, a 100% conversion was achieved in half an hour (see entry 13, Table 2). Most importantly, the heterogeneous ZnO could be separated by filtration and recycled in two additional reaction cycles with similar performance as the as-prepared material. At left part of Scheme 9 the authors proposed that the smaller the size of the ZnO nanoparticle (for the same morphology), the higher the catalytic activity (>20% increase in conversion and yield). This was attributed to the large (macromolecular) size of the substrate, favoring its mass transfer to the Zn Lewis acid sites at the small nanoparticles.
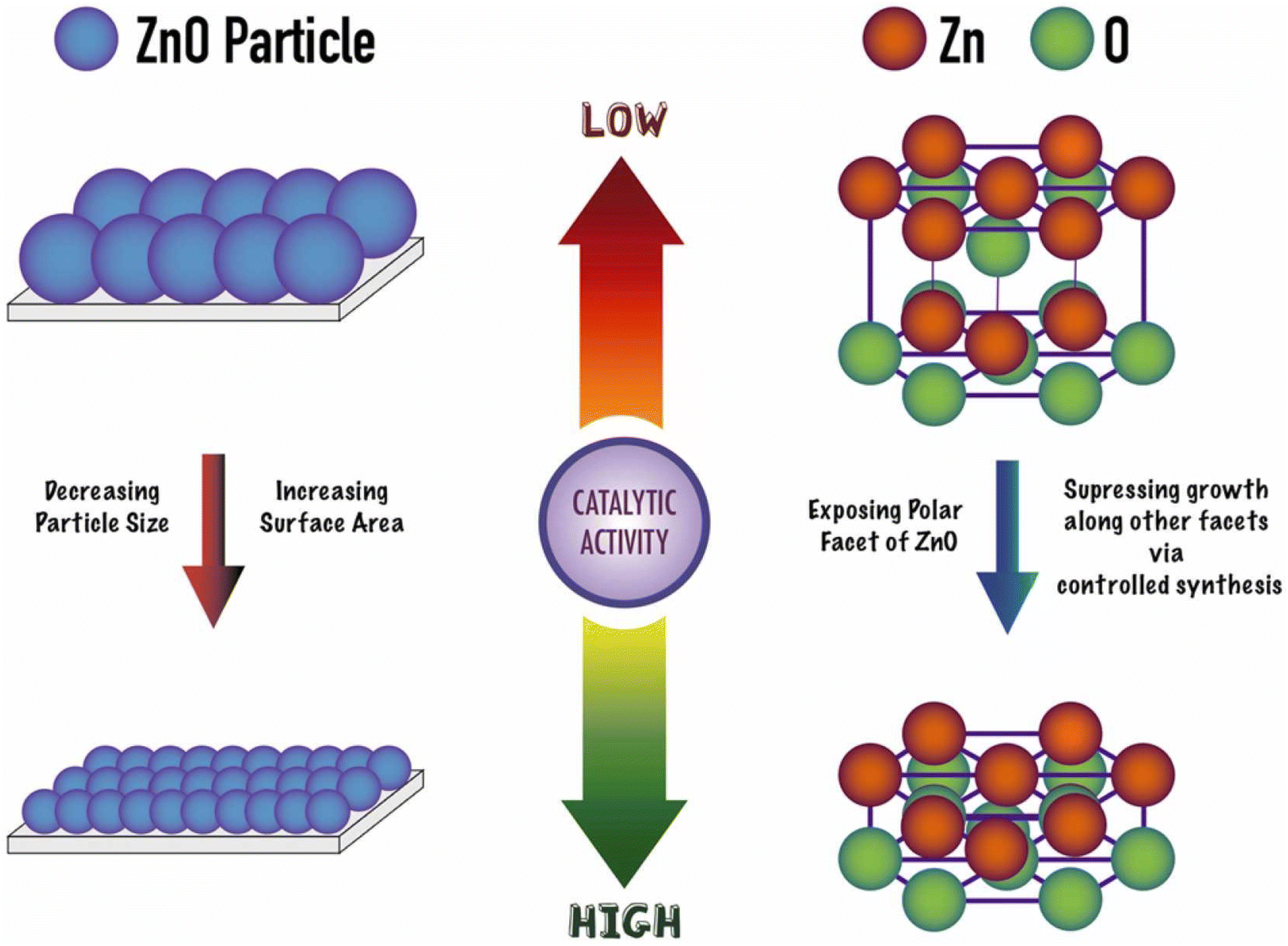 | ||
| Scheme 9 Geometric effects on PET glycolysis activity of ZnO nanoparticles. Reproduced with permission of ref. 49. Copyright (2023) Elsevier. | ||
The authors also point to the important effect of the oxygen atoms adjacent to the Zn sites in withdrawing electron charge from the zinc Lewis acid sites. They also promote hydrogen bonds with ethylene glycol, thus, acting as Lewis base sites with the hydrogen atom from the hydroxyl group of the EG nucleophile (see Scheme 8b). Moreover, (001) facets are more exposed in the case of hexagonal ZnO samples (prisms, disks, and platelets), even for relatively larger crystal size (>250 nm), enhancing its catalytic activity for PET glycolysis (see right part of Scheme 9). Furthermore, the authors point out that microwave heating is superior to conventional heating for PET glycolysis, achieving excellent conversion and yields in short reaction times.49
ZnO nanosheets and nanorods were prepared from thermal decomposition of a zinc/asparagine precipitate in the presence or absence of Mo/Co-sources.50 Only 1 wt% of catalyst was employed for the glycolysis of PET (4:1 EG:PET mass ratio) at 180 °C for 1 h. Less than 60% conversion and BHET monomer yield were obtained with pristine ZnO nanoparticles. On the contrary, those with Co or Mo atoms in its lattice led to quantitative conversions and BHET yields (>90%, see entry 14, Table 2).
The presence of Mo favors the formation of ZnO (100) surfaces with abundance of Zn defects and O vacancies. Atomically dispersed Mo–O and Co–O sites at oxygen bridge bonds of ZnO nanosheets drive the formation of electronically tunable dual Zn–O, Mo–Zn and Co–Zn active sites. XPS analysis of the Mo-doped ZnO suggests electron density migration from Mo to Zn via the O-bridges. Both oxygen vacancies and zinc defects are beneficial for the activation of: (i) the alcohol group (deprotonation) of EG, and (ii) the ester group (carbonylic oxygen) of the PET substrate (see blank inserts in Scheme 10).
 | ||
| Scheme 10 Synthesis of ZnO nanorods and Co/ZnO (a) or Mo/ZnO nanosheets (b), together with the tentative glycolysis pathway in each of them. Adapted with permission of ref. 50. Copyright (2022) American Chemical Society. | ||
The doping of Mo leads to the enhancement of the covalence of the Zn–O bond, which is proposed to lead to electron transfers and the formation of reactive oxygen species at the surface. This species interacts with the OH groups from the EG to produce adsorbed deprotonated EG (*OCH2CH2OH). Therefore, the catalytic synergies of the dual Zn2+–O2− and Mo2+–O2− pairs eventually promote the C–O bond breaking in the PET structure. They also improve the yield of the BHET monomer after subsequent attacks on the ester bonds of PET (Scheme 9b and blank inserts in Scheme 10).
The Mo/ZnO catalyst could be reused in 3 cycles, with only a minor decrease in performance during the first two cycles, which became stronger deactivation in subsequent cycles. However, the catalyst could be regenerated by calcining at 500 °C in air and then at 350 °C under hydrogen. This regenerated the oxygen vacancies and thus maintains its catalytic activity after three additional cycles.50 Ni-doped MgO exhibited similar Ni2+–O2− acid–base pairs and catalytic behavior, being able to decompose PET via glycolysis at 185 °C for 50 min. A 90% yield of BHET monomer was obtained even after 6 reaction cycles, indicating the stable catalytic activity of the composite metal oxide.82
A catalyst with a structure of zinc manganite ZnMn2O4 tetragonal spinel was employed for PET hydrolysis. The Zn-based spinels contain tetrahedrally coordinated Zn ions and octahedrally coordinated Mn ions at the crystal lattice. Quite harsh reaction conditions were employed (260 °C, 5 bars, 1 h) to quantitatively decompose PET into its BHET monomer. Indeed, 12 equivalents of ethylene glycol and 1 wt% catalyst were employed, both with respect to PET.51 The 93% yield of BHET obtained with ZnMn2O4 (see entry 15, Table 2), decreases to 89% and 81% in the case of tetragonal CoMn2O4 and cubic ZnCo2O4 spinels. This was attributed to a lower amount of acid sites and BET areas of the Co-containing catalysts. Based on that the authors proposed that the combination of Zn2+ and Mn3+ cations in the tetragonal ZnMn2O4 spinel possesses a better capability for activation and breaking the CO bonds at the acid sites (Scheme 11). This allows the PET polymer to decompose into oligomers and then into monomers during its degradation pathway. Unfortunately, no additional cycles with the spent catalyst were reported to check its stability.
 | ||
| Scheme 11 PET glycolysis mechanism over double metal oxide catalysts. Adapted with permission of ref. 51. Copyright (2013) Elsevier. | ||
Similar harsh conditions (above the melting point of PET) were employed for the ethanolysis of this condensation polymer when using ZnO (7.5 wt%) on acidic gamma alumina.52 With a catalyst loading of 5 wt% with respect to PET, >90% yield to diethylterephthalate (DET) was obtained after 1 h in supercritical ethanol at 270 °C (Scheme 12a and entry 16, Table 2). Some activity loss was observed since the 5th recycle decreased the DET yield to 75% due to the loss of the ZnO component. Aiming at hydrolysis of PET, nickel was impregnated on alumina, with loadings of 10–15 wt%. Using supercritical water at 260–270 °C, >95% yield of terephthalic acid was obtained after 1 h with the calcined/hydrogenated catalyst (see Scheme 12b and entry 17, Table 2).53 However, the yield decreases to less than 80% after the third cycle. This was probably due to the formation of carbon deposits at the porous support that blocks the metal active sites. Thus, regeneration of the catalyst (calcining at 600 °C to burn the carbon deposits) is necessary, increasing the activity and product yields to 90%.
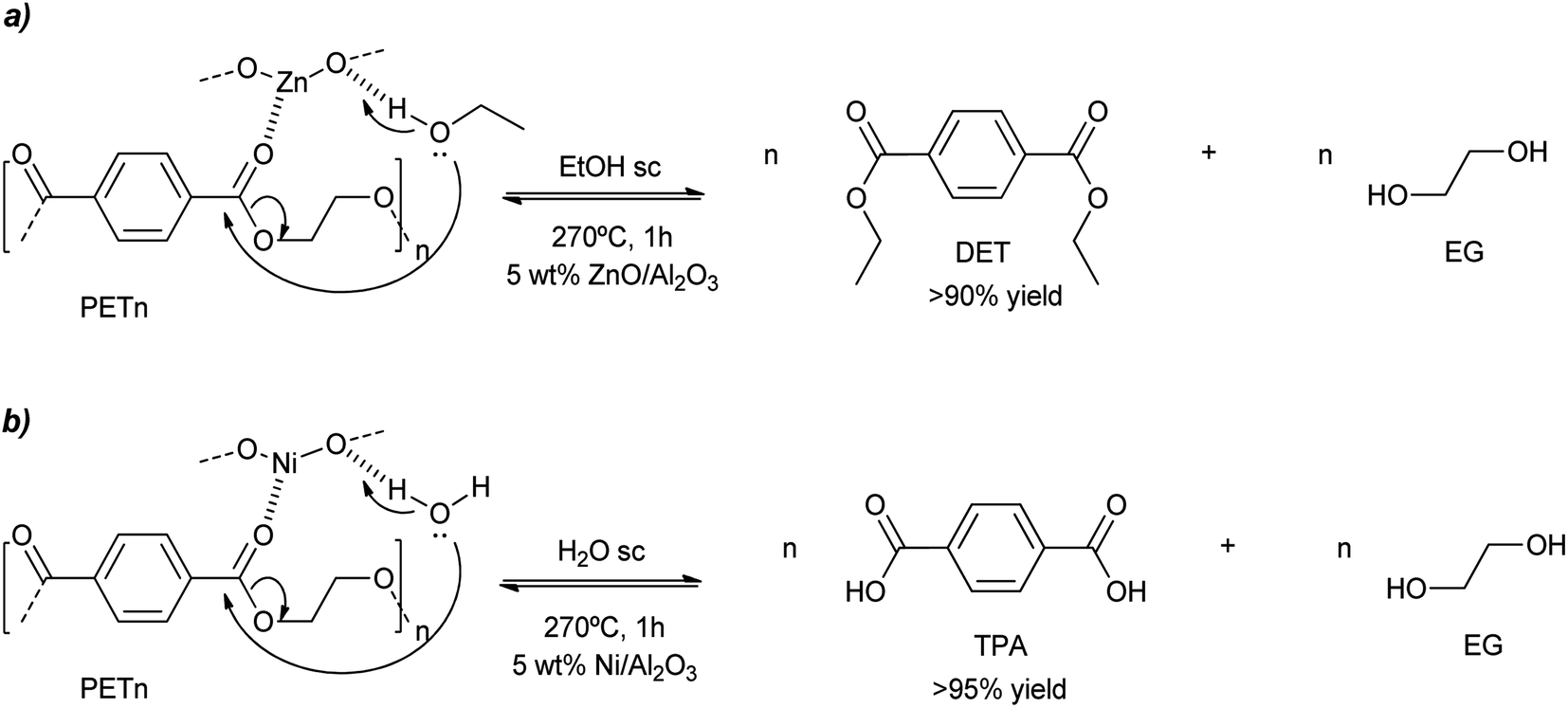 | ||
| Scheme 12 PET degradation mechanism by ethanolysis (a) or hydrolysis (b) into MET or BHET, respectively, promoted by ZnO (a) or Ni (b) doped alumina (5 wt%) at 270 °C for 1 h under supercritical ethanol (a) or water (b). | ||
The interface growth of ZnO and CuO mix metal nanoparticles was reported by bicarbonate assisted co-precipitation of their nitrate precursors.83 The calcination of the oxides allowed for the Cu2+ substitution by Zn2+ in the malachite phase (Cu2(OH)2CO3), and Zn2+ substitution by Cu2+ in the hydrozincite phase (Zn5(OH)6(CO3)2). This fact allowed the tuning of the catalytic activity of the mix-metal oxide for the decomposition of dimethylhexane-1,6-dicarbamate (HDC in Scheme 13a) at 175 °C. Using a ten times mass excess of polyethylene glycol dimethyl ether as solvent, a 70% conversion (after 1 h) of HDC into hexane-1,6-diisocyanate (HDI in Scheme 13a) was obtained. While low amounts of Zn increase the activity of the material, high Cu/Zn ratios decrease both the surface area and catalytic activity. On the other hand, nano-sized Cu2O prepared by hydrolysis/calcination (under an inert atmosphere) steps was an active catalyst for the obtention of methylene di(phenylisocyanate) (MDI). This occurs due to the degradation of methylenediphenyl di(phenylcarbamate) (MDPC) under solvent-free conditions and 210 °C (see Scheme 13b).84 A conversion of 64% based on the MDPC substrate was obtained, with a 48% selectivity to MDI. In comparison, ZnO promoted only a 50% MDPC conversion and 40% selectivity.
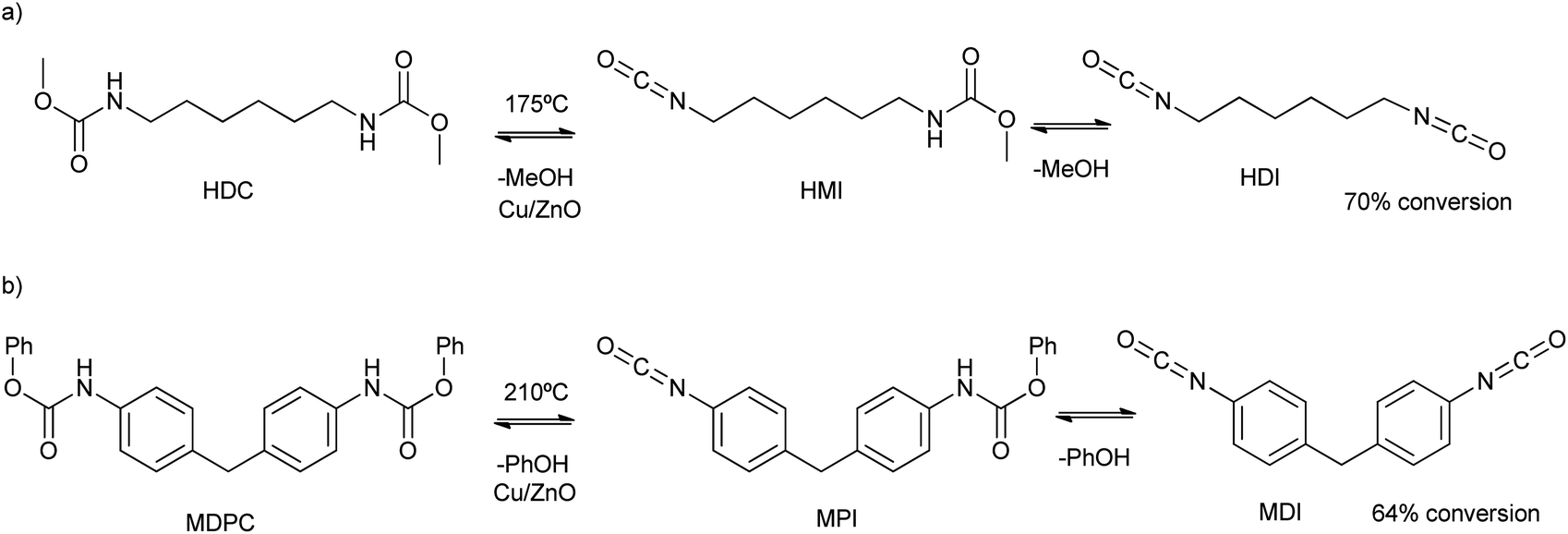 | ||
| Scheme 13 (a) HDC and HMI degradation mechanism into HDI and methanol at 175 °C; (b) MDPC and MPI degradation mechanism into MDI and phenol at 210 °C. | ||
3.2 High valence metal oxide catalysts: Fe(III), Sb(III), Ce(IV) and Ti(IV) oxide-based systems
Sulfated titanium dioxide SO42−/TiO2 was prepared by precipitation of Ti(OH)4 and further H2SO4 treatment and calcination.54 The solid catalyst was employed in the degradation, via hydrolysis, of PET under supercritical CO2 at 160 °C and 15 MPa during 12 h, obtaining 99% yield to terephthalic acid (see entry 18, Table 2). The solid catalyst promoted the formation of Brønsted acid sites that were able to protonate the water solvent. Such water and hydronium ions degrade the PET from the surface and the inside (bulk) promoting the swelling and hydrolysis of the PET. The same authors promoted the same catalytic process but using titania with surface WOx species as acid sites.85 The catalyst preparation, in this case, was hydrothermal and instead of using base-assisted precipitation, a surfactant (CTMABr) was employed. In particular, the number of polytungstate species increases in line with the number of Brønsted sites, which were active in the PET hydrolysis. For that, a PET/catalyst/H2O mass ratio of 1:0.1:10 at 160 °C and 15 MPa was employed during 15 h under supercritical CO2.
Antimony oxide demonstrated a better performance than its antimony acetate precursor in the PET degradation by glycolysis with ethylene glycol (with an EG:PET weight ratio of 5).86 The yield of BHET, using 0.5 wt% of the catalyst, under 200 °C and 2 bar was 98% after 1 h, while only 64% yield was produced with antimony(III) acetate (see Scheme 14). In the case of the reaction in the absence of any catalyst, only 11% yield of BHET was obtained. However, zinc acetate is still a competitive catalyst with respect to Sb2O3, both having a similar performance for PET glycolysis. It is worth mentioning that the monomer containing the antimony catalyst can be employed for further polymerization into PET provided the EG excess is removed by evaporation under vacuum.
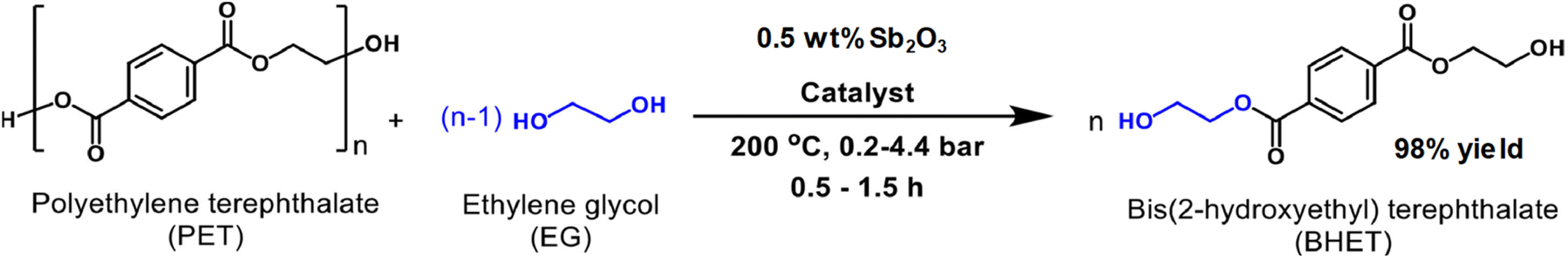 | ||
| Scheme 14 Glycolysis of PET with EG in the presence of antimony(III) oxide as the catalyst. Reproduced with permission of ref. 86. Copyright (2022) Elsevier. | ||
Highly dispersed, ultra-small iron oxide nanoparticles (<10 nm) were prepared from the precipitation of iron chloride in a sodium citrate/ammonia medium, resulting in a Fe3O4 nanodispersion in EG with a 5 wt% solid content.55 The citrate adsorbed on the nanoparticle avoids both the aggregation and oxidation of the nanoparticles, which exhibited Brønsted and Lewis acid sites, and more importantly, BET surface areas >100 m2 g−1. Using a Fe3O4/PET = 2% and an EG/PET = 13, more than 90% yield to BHET was obtained at 210 °C for 30 min (see entry 19, Table 2). The yield was maintained even after three cycles and the iron oxide nanoparticles were magnetically separated and reused after washing (ethanol and water) and redispersing in EG.
Ultra-small (ca. 3 nm) and highly defective CeO2 nanoparticles combined significant BET surface areas (>100 m2 g−1), excellent dispersibility, and a high density of oxygen defects.56 This resulted in efficient PET depolymerization at 196 °C, using an EG:PET weight ratio of 7. The reaction is completed after 0.25 h, obtaining a 99% conversion of PET and a 90% yield of the monomer yield of 90% (see entry 20, Table 2). The small CeO2 nanoparticles, containing Ce3+ ions that promote lattice strain, eventually contain oxygen defects. Those are key active sites in the cleaving of the ester bonds from the PET substrate, leading to oligomers and MHET and BHET through a dynamic equilibrium (see Scheme 15).
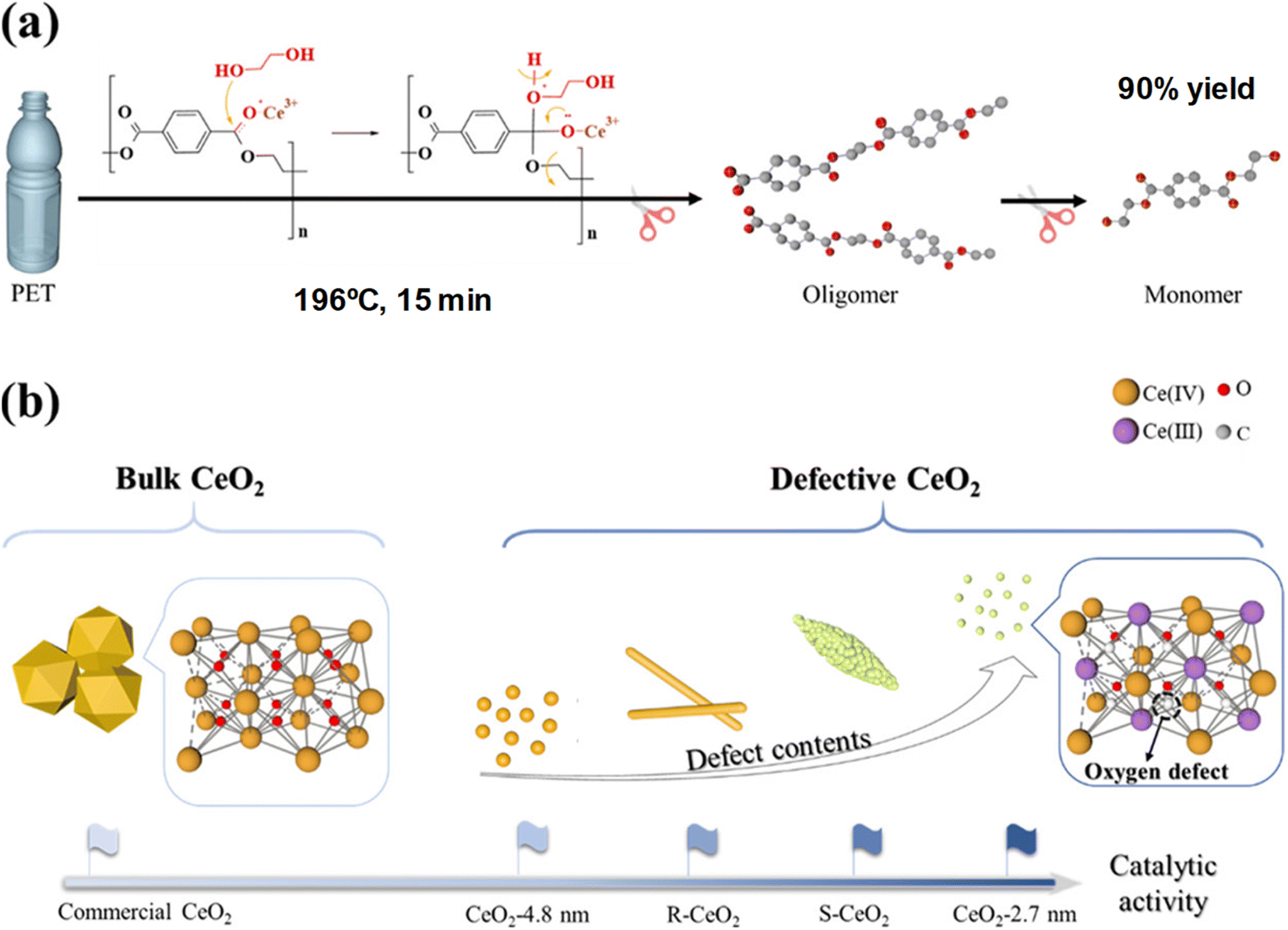 | ||
| Scheme 15 (a) PET glycolysis (196 °C, 15 min) procedure over defective CeO2 NPs. (b) Structures and catalytic activities of CeO2 NPs with different morphologies. Reproduced with permission of ref. 56. Copyright (2022) American Chemical Society. | ||
4 Open 2D/3D metal-containing architectures
4.1 Layered 2D frameworks
Clays, like the smectite clay or montmorillonite K-10 were employed two decades ago as heterogeneous catalysts for the alcoholysis of carbamates.87 The resulting isocyanates are then formed at 180 °C using large amounts of solid catalyst (100 mg with respect to 0.6 mmol of isocyanate). Such high temperatures were necessary to desorb the alcohol product, especially bulkier ones. Yields of the isocyanate products were high in the case of light alcohol leaving groups (e.g. MeO–) and aromatic carbamates (Ph–NHCO–). The authors were able to produce MDI from MDPC (see Scheme 13 b) in decaline with ca. 70% yield after 24 h at 190 °C using a 25 wt% of catalyst. The reaction mechanism proposed requires an optimal number of Brønsted acid sites, such as those present in K-10 solid clay, to promote the alcoholysis avoiding excessive product absorption at the acid sites. In Scheme 16, the authors proposed the carbamate protonation by the Brønsted acid sites of K-10 and subsequent formation of the isocyanate by elimination of the amidic proton and the methoxy group. Both leaving groups are then combined in the form of the methanol byproduct (which is eliminated from the catalyst surface by heating to regenerate the active site).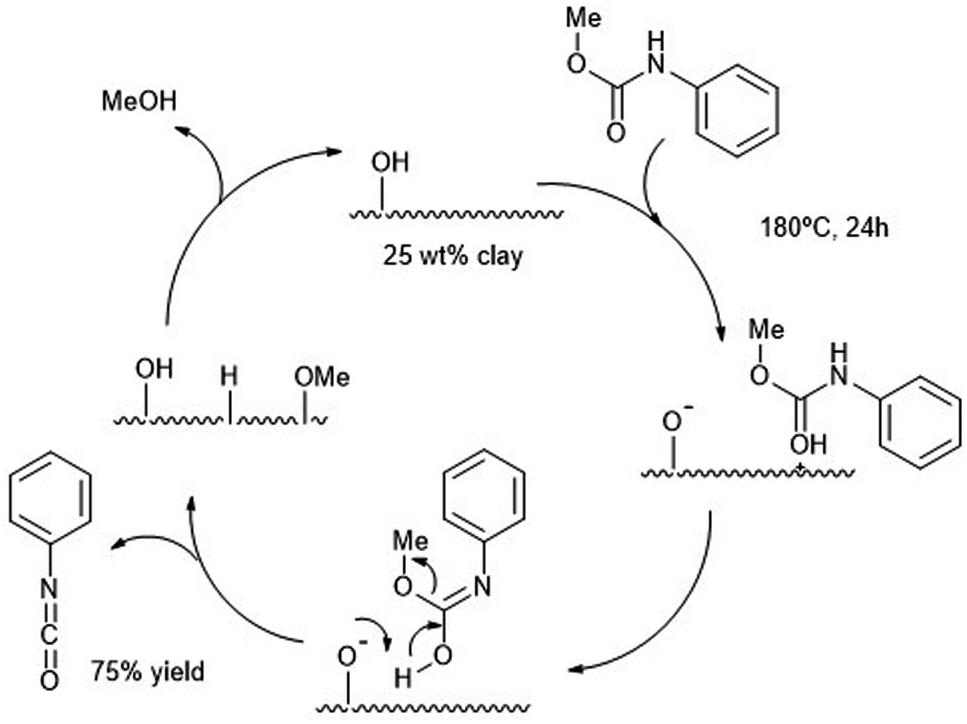 | ||
| Scheme 16 Proposed mechanism for the decomposition of carbamates (180 °C, 24 h) into isocyanates at solid acid K-10 catalyst (25 wt%). | ||
Co-precipitation of Zn, Mg and Al salts in a basic solution of carbonates and hydroxides resulted in the formation of layered double hydroxides.57 The layered of (Mg–Zn)–Al–OH are spaced by 2.4 nm, and named as hydrotalcite or anionic clays (Scheme 17). PET glycolysis was carried out using a 10:1 mass ratio of the alcohol. The catalyst promotes a 95% conversion of PET and 80% selectivity to BHET was obtained after 3 h at 196 °C (see entry 21, Table 2). The active sites were both basic isolated O2− ions, Mg2+–O2− pairs, and OH groups, as well as acid sites such as the Mg2+, Al3+ and Zn2+ cations. On the one hand, the Lewis basic sites are responsible for the activation of the alcohol by interacting with hydroxyl groups. This is key for making the oxygen more nucleophilic for the attack on the carbonyl of the ester group in the PET. On the other hand, the Lewis acid sites withdraw the electron density of the oxygen atom. This makes the carbon atom of the group more electrophilic and susceptible to nucleophilic attack. The catalyst could be reused 4 times without significant loss in activity, the active site was regenerated by washing with water and the structure was maintained. Other authors prepared similar types of hydrotalcites but using Sn instead of Mg, and tested them as heterogeneous catalysts for PU degradation using DEG for 3 h at 190 °C.58 Zn, Sn, Al hydrotalcites (HTCs) were employed in the glycolysis of flexible polyurethane foams. The conditions were: a PUF/DEG mass ratio of 1:5 and HTC/DEG of 0.001 (1.3% catalyst) and 66% of the polyol was recovered (see entry 22, Table 2).
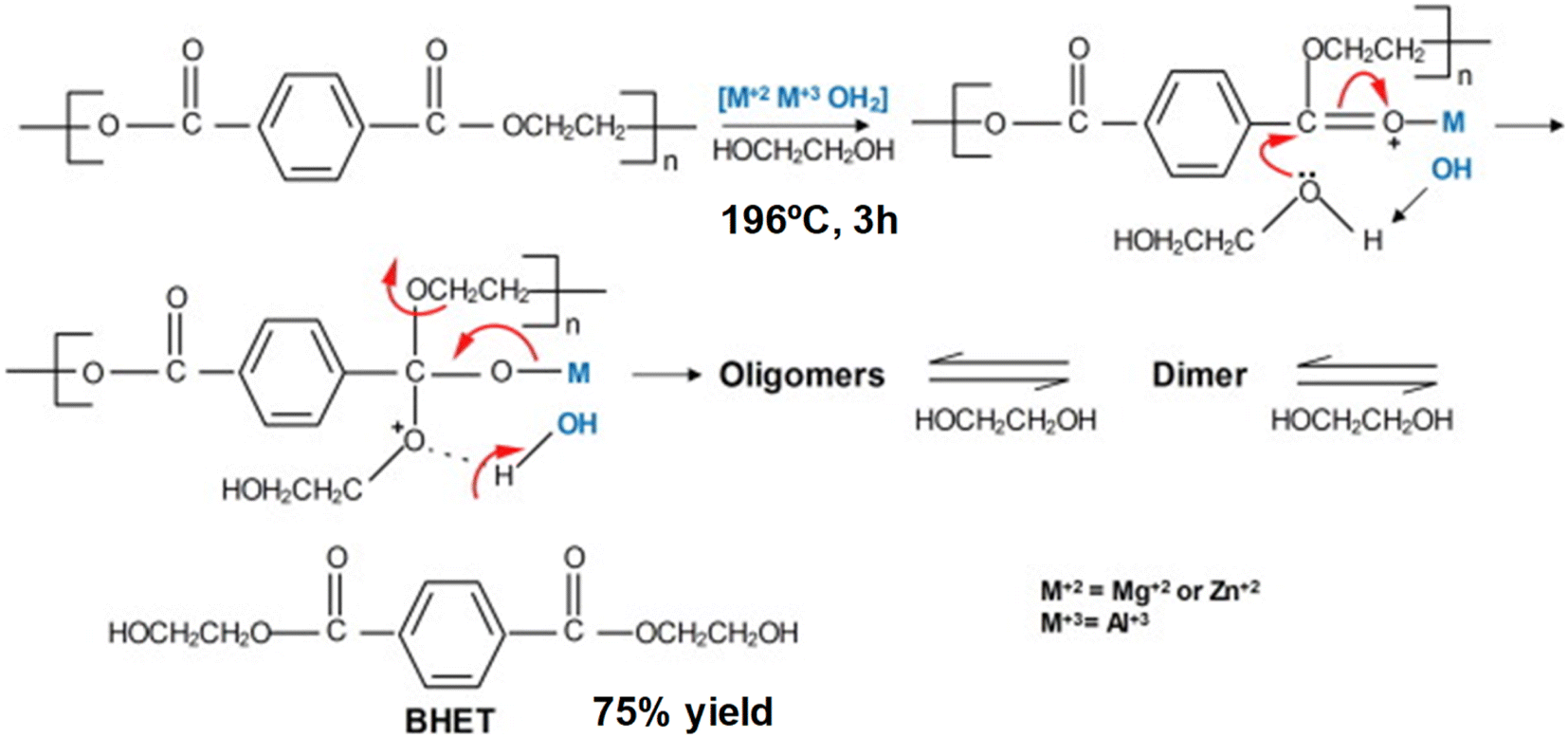 | ||
| Scheme 17 PET glycolysis over Mg/Zn/Al hydrotalcites at 196 °C during 3 h. Reproduced with permission of ref. 57. Copyright (2016) Elsevier. | ||
Two-dimensional (2D) holey and ultrathin MnO2/graphene oxide nanosheets (HGO = holey graphene oxide) were reported as catalysts for PET glycolysis (see Scheme 18).88a The synthetic process consisted of oxidative etching of graphite flakes (the graphene oxide solution) and a self-limiting redox reaction between KMnO4 and Na2SO4 in water (as the MnO2 solution). Both solutions were mixed at 80 °C for half an hour, and the MnO2/HGO was isolated and dried. The optimal EG:PET ratio was 19 and the temperature was 200 °C, resulting in a quantitative BHET yield (100% conversion and yield) after 10 min using the MnO2/HGO catalyst (0.01 wt%). Similar GO–Mn3O4 was prepared with similar oxidation method but under ultrasound conditions, requiring only tens of minutes under ambient conditions. However, very high temperatures (300 °C) were employed for PET glycolysis in this case.88b
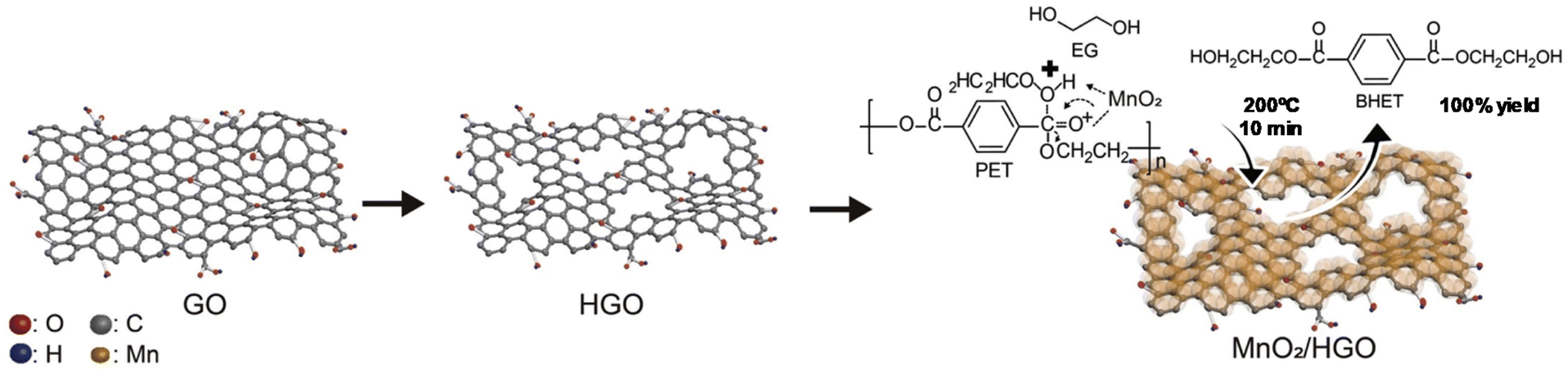 | ||
| Scheme 18 Glycolysis of PET with EG (200 °C, 10 min) in the presence of MnO2/HGO as the catalyst. Reproduced with permission of ref. 88a. Copyright (2021) Elsevier. | ||
A higher surface area was obtained for this material (247 m2 g−1) with respect to MnO2 deposited in GO (69 m2 g−1). This was due to the holey GO support, which was key to its catalytic performance, maintaining its activity after 5 cycles. Even in the absence of the GO, ultrathin-exfoliated MnO2 nanosheets exhibited an optimal performance for the glycolysis of PET. Indeed, 100% yield to BHET concerning bare MnO2 (78% yield) was obtained (see entry 23, Table 2), permitting its use for 5 cycles without apparent loss of activity under reaction conditions (200 °C, 1 h, 0.1 wt% MnO2 and 55:1 EG:PET weight ratio).59 This proved the efficiency of the fluid dynamics-assisted intercalation and exfoliation method in the shear-exfoliation by K+ intercalation between adjacent MnO2 layers. The authors proposed a similar method to produce 2D-Fe nanosheets with high surface area (>200 m2 g−1) obtaining similar activity under similar reaction conditions.89 In this case, the magnetic properties of the catalyst allow its simple separation and recycling five times without apparent loss in activity. It is worth mentioning that only a 10% yield was obtained in the absence of any catalyst. In this case, no active site was available to interact neither with the carbonyl or hydroxyl group from PET and EG, respectively.
4.2 Porous 3D frameworks
A synthesis of the isocyanate HDI by thermal decomposition of the carbonate HDC over Co3O4/ZSM-5 catalyst has been reported (Scheme 13 and 19).90 The metal oxide was incorporated into the microporous medium pore zeolite by: (i) incipient wetness impregnation, (ii) PEG additive, or (iii) deposition precipitation with ammonia evaporation. A 6.5% HDC concentration in chlorobenzene, a 1 wt% Co3O4/ZSM-5 catalyst, under 250 °C temperature, 2.5 h time, 800 ml min−1 nitrogen flow rate and 1.0 MPa pressure was employed. The best performant catalyst was the one prepared with PEG additive (100% HDI yield), followed by one prepared by incipient wetness approach. The one prepared with ammonia was the less active one. The authors showed that catalytic activity is parallel to its acidity (measured by NH3-TPD), surface cobalt content and surface lattice oxygen content. Both acidity and surface oxygen were key in the activation of the carbamate group during its decomposition, as shown in (Scheme 19). The same group reported a similar Zn-doped Co-ZSM-5 catalyst but using dioctyl sebacate as solvent (10 wt% concentration of HDC) at 250 °C for 60 min.91 A yield of 84% of HDI was reported, maintaining its activity for 3 additional cycles. On the other hand, the hydrolysis of dimethyl terephthalate (DMT) was successfully achieved using a ZSM-5 containing niobium oxide sites.92 The Brønsted acidity (in higher number with respect to the Lewis one) and a surface area (higher than 300 m2 g−1) were key to its catalytic activity. The reaction conditions employed were 5 wt% of catalyst with respect to DMT, 5 times more mass of water than DMT and 2 h of reaction. This resulted in a DMT conversion of 99% and a terephthalic acid yield of 94%, maintained during 5 additional reaction cycles.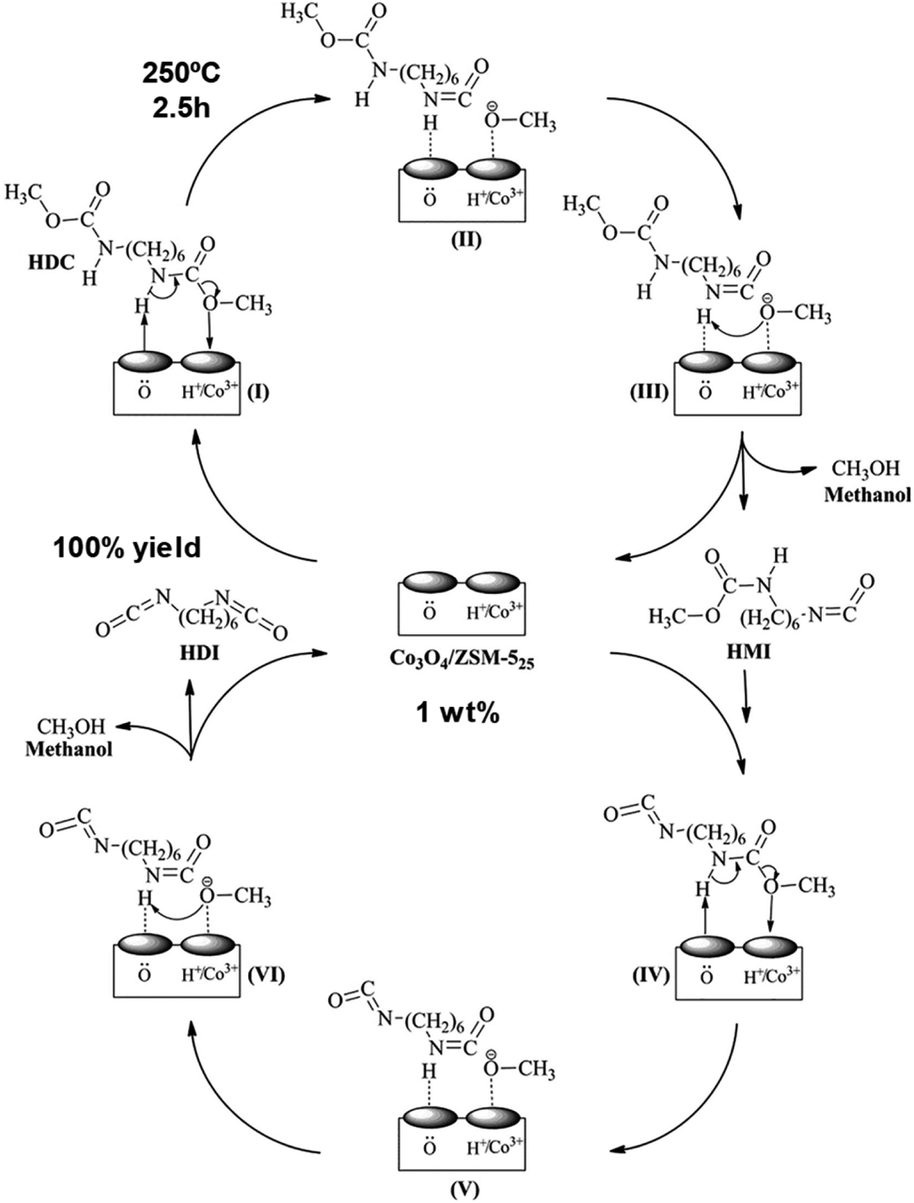 | ||
| Scheme 19 A tentative mechanism for the thermal decomposition (250 °C) of HDC to HDI over Co3O4/ZSM-5 catalyst. Reproduced with permission of ref. 90. Copyright (2017) Elsevier. | ||
Porous solids, such as the mesoporous silica SBA-15 were employed as host (or support) of zinc oxide nanoparticles resulting in active catalysts for PET degradation.60 Using a PET/EG ratio of 1:4 and a catalyst (ZnO)/PET ratio of 0.05, a 100% conversion and 90% yield to BHET was obtained after 1 h at 200 °C (see entry 24, Table 2), similar to the performance of zinc acetate. The heterogeneous catalyst could be recycled 6 times, with a slight decrease in yield after the first 3 cycles (close to 80% BHET yield). However, a more pronounced decrease is observed afterward (down to 60% after the sixth recycle) due to loss of active components.
The zinc–organic framework known as MAF-6 (Metal-Azolate Framework-6) was an active catalyst in the degradation of PET with ethylene glycol. Using a 6:1 weight ratio at 180 °C for 4 h at atmospheric pressure, 92% PET conversion and 82% BHET yield was obtained (see Scheme 20, and entry 25, Table 2).61 The high density of Lewis acid zinc sites, together with the excellent dispersibility of the substrates and catalyst allowed a better performance of the MOF with respect to traditional ZnO (28% PET conversion and 9% BHET yield) and Zn acetate (71% PET conversion and 52% BHET yield) under similar conditions (7.7 mmol Zn). The mechanism proposed assumes that the external surface active sites allow for the PET degradation into oligomers. The oligomers are transformed into dimers that have a sufficiently small size to penetrate the micropores of the MOF and be broken in the inner active sites of the crystal. Similar XRD patterns and low Zn leaching (3.7%) were observed after 5 recycles, maintaining the catalytic activity of the first cycle.
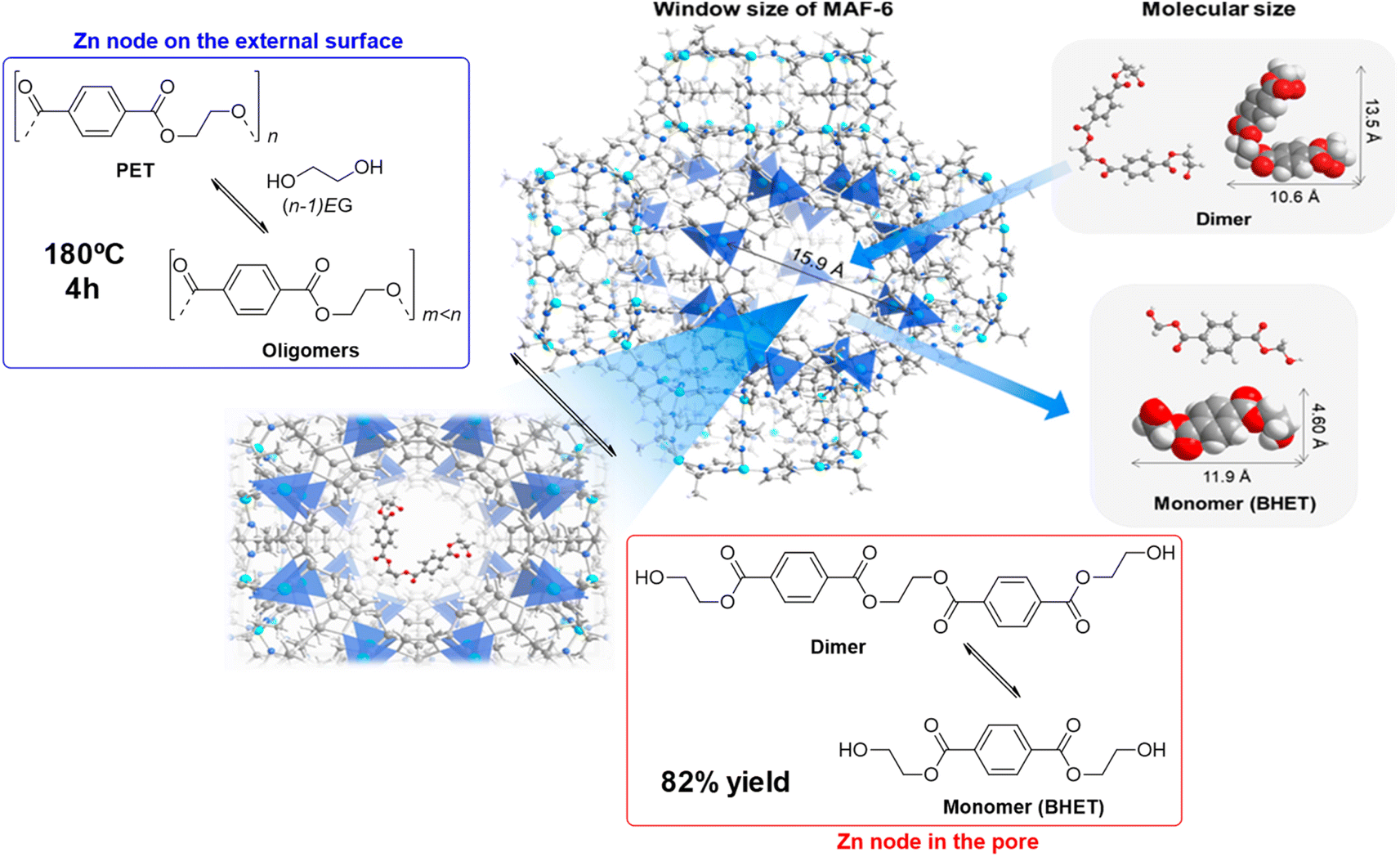 | ||
| Scheme 20 Proposed mechanism for PU degradation at MAF-6 (180 °C, 4 h), together with size of both MAF-6 and, dimer and monomer (BHET). Adapted with permission of ref. 61. Copyright (2021) ACS. | ||
DES@ZIF-8 multifunctional catalyst was prepared from the mixture of the ZnCl2/acetamide (with a molar ratio of 0.4/1) deep eutectic solvent (DES) with the ZIF-8 in a mass ratio of 1:4.93 The XRD of the DES@ZIF-8 is significantly altered by the presence of the DES, although main characteristic peaks of ZIF-8 are still present. The composite was prepared by contacting both MOF and DES in ethanol, stirring at room temperature for 8 h and solvent evaporation. BHET yield was obtained by weighting the reaction mixture after such reaction time at 195 °C and 10 bars (EG:PET mass ratio = 5) and a work-up with hot water. A 100% conversion and 83% BHET selectivity were obtained after 30 min with the best performant Zn-DES@ZIF-8 (maintained during recycling), using only 0.4 wt%. of catalyst with respect to PET. In the case of bare ZIF-8, 73% BHET yield was obtained, which is not so far from the performance of the DES@ZIF-8 composite (only 10% more). The authors proposed that the zinc from the DES interacts with the carbonyl group of the PET. Moreover, the basic amino group of the acetamide does the same with the hydroxyl group of the EG in synergy with the Zn acid sites.
5 Chemo-enzymatic and biocatalytic depolymerization of plastic wastes
5.1 Biocatalytic degradation of PET wastes
The use of biological tools (e.g. microorganisms, enzymes, etc.) for plastic wastes degradation is a sustainable alternative to the chemical processes for eliminating plastic wastes under mild reaction conditions. This green approach is crucial for developing new strategies for plastic recycling/upcycling, via depolymerization of recalcitrant materials into their constituent building blocks overcoming kinetic and thermodynamic limitations.18,30 Recently, it has been reported the suitability of different enzymes for carrying out the degradation of plastic wastes (e.g. polyester, polyethylene, etc.) by using whole cells (e.g. bacterial, fungi, etc.) or enzymes (e.g. laccases, peroxidades, etc.).94 Different hydrolases showed activity to break down bonds of the PET structure, providing terephthalic acid (TA), bis(2-hydroxyethyl)terephthalate (BHET), mono-(2-hydroxyehyl)terephthalic acid (MHET), and ethylene glycol (EG) (Scheme 21).95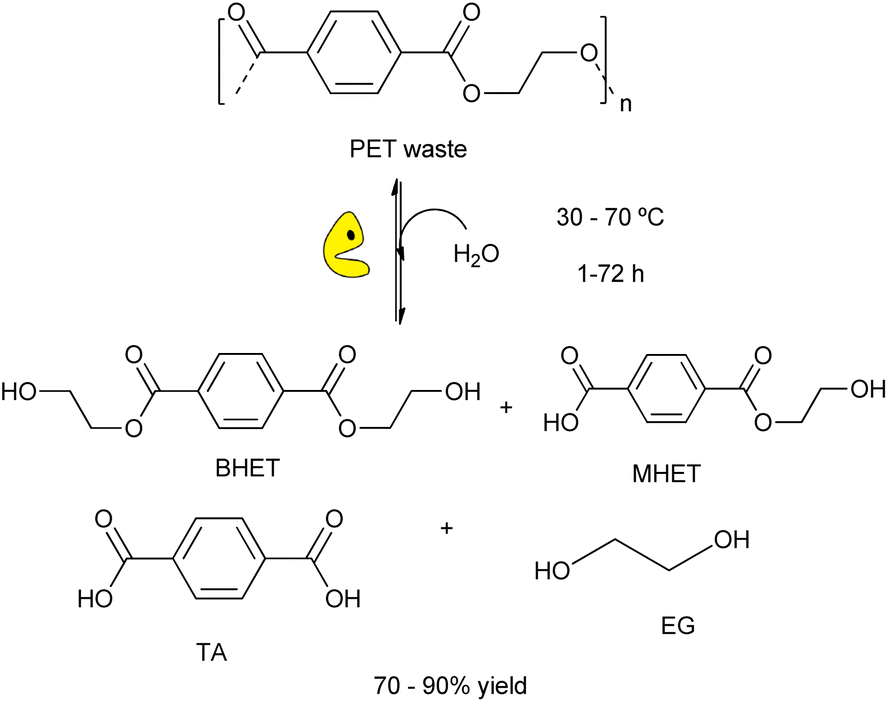 | ||
| Scheme 21 Schematic representation of enzymatic hydrolysis of PET catalysed by engineered biocatalysts under mild conditions (water, RT-70 °C, 1–72 h). | ||
As an alternative, Tournier et al., developed a more sustainable enzymatic depolymerization of PET catalyzed by an engineered cutinase (instead of traditional chemical catalysts), reaching more than 90% postconsumer PET waste degradation after 10 h at 72 °C (entry 26, Table 2). In this study, they successfully demonstrated the reusability of the obtained products (i.e. terephthalic acid monomers) for resynthesizing a new PET under mild reaction conditions.62a It is worth mentioning that the French company CARBIOS is one of the most important examples of a sustainable industrial approach for the depolymerization of PET from waste plastic bottles.62 Taking advantage of protein engineering technology, they have developed a PET depolymerase variant very useful for PET hydrolysis, achieving a productivity rate of 15.5 g L−1 h−1 of terephthalate, which corresponds to 200 g KgPET−1 using 2 mgenzyme gPET. In 2022, CARBIOS also demonstrated remarkable advances in PET depolymerization, providing a 97% depolymerization rate after 16 h at an industrial scale of 1000 L,96 preventing more than 1.8 million tons of plastic foam and a considerable reduction of CO2 emissions. Another important pathway for PET degradation consists in the development of suitable pretreatments of PET wastes to more enzyme-attackable foams which are necessary for PET recycling, demonstrating a remarkable improvement in the substrate accessibility (under microwave irradiation) to obtain PET powders. This approach allows the obtention of more suitable PET-derived constituents (compared to the original untreated PET) for subsequent biocatalytic steps with engineered PETases, achieving 78% yield after 2 h of microwave pretreatment and 1 h of enzymatic reaction at 30 °C (entry 27, Table 2).63 Additionally, a physical pretreatment of PET has been developed to provide a biodegradable form, based on amorphization and micronization towards more accessible substrate for PET hydrolases.64
It should also be noted that the expression and functionality of IsPETaseMut, a variant of the PET-degrading enzyme from Ideonella sakaiensis, has been carried out. By optimizing expression systems in E. coli, two different approaches were recently developed: co-expressing with GroEL/ES chaperones and fusing the enzyme with NusA (entry 29, Table 2). Both strategies significantly increased the yield of soluble IsPETaseMut, with NusA fusion, achieving the highest production at 80 mg L−1, compared to 75 mg L−1 with GroEL/ES co-expression. While GroEL/ES did not affect the catalytic activity of the enzyme, the NusA fusion improved its properties by enhancing PET adsorption. Additionally, the product inhibition effect of TA on IsPETase was notably reduced with NusA-IsPETaseMut, resulting in a weight loss of 19.6% for PET-NP. Despite the initial hydrolysis rate was slower, NusA-IsPETaseMut demonstrated more effective long-term PET degradation, resulting in a 1.4-fold higher adsorption constant toward PET. These results indicate that NusA-IsPETaseMut showed a significant long-term biodegradation behavior compared to IsPETaseMut and IsPETaseMut (GroEL/ES). Moreover, it is demonstrated that optimizing expression systems and fusion partners can significantly enhance enzyme production and performance, opening new opportunities for PET recycling technologies.65a
In the same context, the S238Y mutant, located near the catalytic triad, exhibited a significant enhancement in PET biodegradation, with a 3.3-fold increase in activity compared to the wild-type enzyme.65b In this study, circular PET films were washed with sterile deionized water and dried at room temperature for 48 h before enzymatic treatment. Then, the enzymatic reactions were assayed in buffer (50 mM glycine-sodium hydroxide, pH 9.4) at 30 °C for 72 h (see entry 30, Table 2). Notably, the structural modification improved the activity of the enzyme to break down highly crystallized PET (∼31%), which is commonly found in commercial soft drink bottles. Furthermore, microscopic analysis revealed that mechanical stress on PET surfaces further improved enzyme efficacy by disrupting the crystalline structure (up to 3% loss of crystallinity).
Another strategy for the biocatalytic upcycling consists in the enzymatic depolymerization of PET through the addition of high concentrations of calcium ions (up to 1 M). This allows to carry out the hydrolysis catalysed by the engineered cutinase LCCICCG, exhibiting improved the thermal stability and activity in the presence of calcium cations, providing 84% PET conversion into calcium terephthalate (CaTP·3H2O) at 80 °C for 12 h (entry 28, Table 2). Furthermore, the presence of calcium ions also reduced the amount of base required to maintain optimal pH levels. The recovered building blocks can be further used as raw material for the synthesis of value-added products (i.e. battery electrode production).64
Despite the difficulty of breaking down PET bonds due to its recalcitrant character, promising results for its biocatalytic depolymerization have also emerged using rational design and combinatorial mutagenesis. For instance, the development of a novel ICCG mutant RITK (D53R/R143I/D193T/E208K) cutinase showed excellent (whole-cell) biocatalytic activity, demonstrating the suitability of hydrolytic enzymes for plastic recycling.66 The activity of a PET hydrolase was widely improved, allowing 90% PET depolymerization into 16.7 g L−1 h−1 of terephthalate monomer (for further reuse) using only 3 mgenzyme g−1PET (entry 31, Table 2). Alternatively, Quartinello et al. developed a synergistic chemo-enzymatic hydrolysis of PET from textile waste. In the first place, chemical hydrolysis at 250 °C and 40 bar was performed, reaching 85% conversion into both TA and small oligomers (entry 32, Table 2). Then, a second step of enzymatic hydrolysis of the obtained oligomeric compounds, catalysed by Humicola insolens cutinase (HiC), provided conversions of TA up to 100% (entry 33, Table 2).67 However, one of the main challenges of the biocatalytic approaches is ensuring the operational stability of the biocatalysts under harsh industrial conditions (e.g. high temperatures) required for efficient degradation of large amounts of plastics. In this context, the development of new thermophilic microorganisms is needed for such conditions, although genetic engineering tools for designing these non-conventional host microorganisms are still under development.
5.2 Biocatalytic depolymerization of PU
As described above, PU is a class of synthetic polymer with huge environmental impact, which is obtained from the polycondensation reaction of a diisocyanate (i.e. toluene diisocyanate, TDI) and polyols, providing the characteristic recalcitrant and stable intra-molecular urethane bond.97 Although recycling strategies based on circular chemistry concepts are also a key tool to achieve their depolymerization into valuable secondary materials, traditional chemical PU recycling processes require extreme reaction conditions (180–250 °C, 50 bar). This fact complicates the obtention of pure and useful monomers for further reuse, mainly due to uncontrolled side-products/reactions under these harsh conditions. A sustainable alternative to the traditional chemical hydrolysis of PU is the biocatalytic degradation under milder conditions, reducing the operational costs and the use of toxic compounds. In general, the biocatalytic degradation of PU involves the use of enzymes (i.e. esterases, proteases, ureases, etc.) expressed in fungi and bacteria to produce alcohols, amines, and CO2 (Scheme 22).98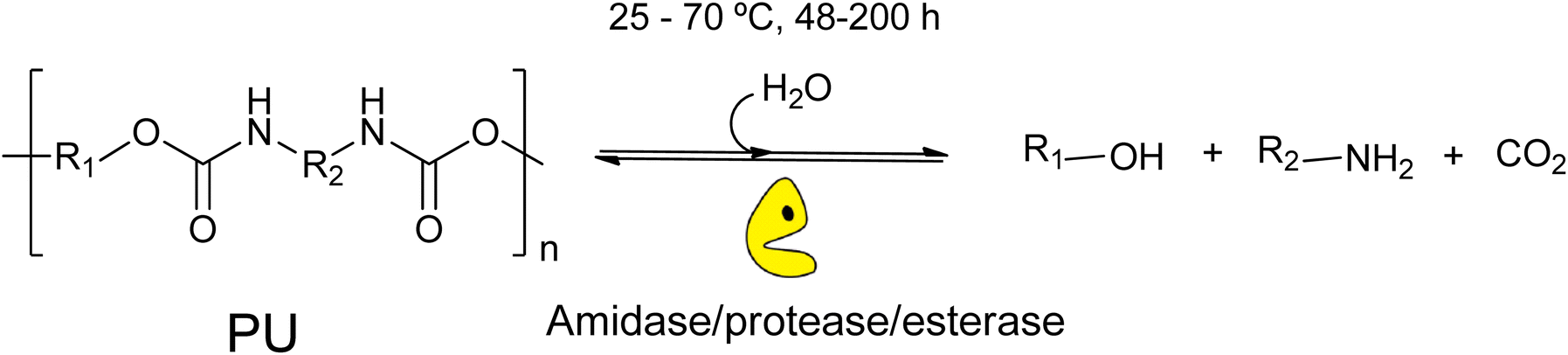 | ||
| Scheme 22 Schematic representation of enzymatic hydrolysis of PU catalysed by amidase, protease, or esterase under mild conditions (water, RT-70 °C, 48–200 h). | ||
Despite the xenobiotic, recalcitrant, and stable nature of PU wastes, few examples of selective biological catalysts have reported for the partial biodegradation of the polyester-polyurethane bonds.99 For instance, the treatment of ultrathin PU using urease and papain enzymes for a long time (1–6 months), achieved a slight change in the PU surface.100 These slight variations and the enormous difficulty of carrying out biocatalytic processes for polyurethane hydrolysis could be explained, not only because of the recalcitrant character and the stability of PU, but also because of the potent inhibitor effect of carbamate and thiocarbamate compounds on the serin proteases and hydrolases. Besides this inhibiting effect, different classes of enzymes have been reported for PU degradation (e.g. esterases, proteases, lipases).101 For example, a Trametes versicolor laccase was evaluated as biocatalysts on four representative polyester- and polyether-based PU wastes, obtaining a remarkable reduction in the molar masses after 18 days of incubation at 37 °C (entry 34, Table 2).68 Alternatively, Humicola insolens cutinase were able to hydrolyse PU-polyester films after 168 h at 50 °C, achieving only 42% yield (entry 35, Table 2).69 In addition, Schmidt et al. studied the degradation of the polyester PU Impranil DLN by the polyester hydrolases LC cutinase (LCC), TfCut2, Tcur1278 and Tcur0390, reaching up to 4.9% weight loss in polyester PU (by turbidimetric assay) after 200 h reaction at 70 °C (entries 36 and 37, Table 2).70
On the other hand, thermostable polyester hydrolases also proved to be suitable biocatalysts for the hydrolysis of polyester PU. For instance, lipases have been an interesting tool to perform the hydrolysis of commercially available solid poly(ester)urethanes. Candida rugosa lipase was used to hydrolyze polyester PU particles in an aqueous medium, which led to valuable ethylene glycol byproduct, with a generation rate of 0.12 mg L−1 min−1 (entry 38, Table 2).71 However, most strategies of biocatalytic hydrolysis of PU require the development of two-step chemo-enzymatic approaches, such as a first step of the depolymerization of polyether-polyurethane using a glycolysis reaction with DEG catalysed by tin(II)-2-ethylhexanoate at high temperature (i.e. >200 °C) (entry 39, Table 2). However, this approach achieved low molecular weight dicarbamates, carbon dioxide, and toluene diamines (TDA). On the contrary, the second biocatalytic hydrolysis step, allowed up to 100% conversion of the obtained dicarbamates, using an engineered urethanase for 48 h at 70 °C (entry 40, Table 2).72
Different enzymes (i.e. lipase, urease, protease, etc.) have been demonstrated as high-performant biocatalysts for the hydrolysis toluene-based urethane model compounds (i.e. bis(2-methoxyethyl) (4-methyl-1,3-phenylene)dicarbamate), using organic solvents (i.e. ethylene glycol or solketal) or, ionic liquids (e.g. 1-butyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide, [C4mim][NTf2]) as a reaction media.102 However, most of the enzymes were unable to catalyse the hydrolysis of the carbamate bonds in pure water. This was due to the insolubility of these di-urethane compounds in water, which was improved (up to 31.6 mU mg−1 for the urease case) in a mixture of solketal:water (90:10, v/v) reaction media. Alternatively, when hydrophobic ILs were used as reaction medium, the urease activity increased twofold (74.1 mU mg−1). The highest specific activity for the hydrolysis of these urethane compounds were obtained by combining lipase and urease biocatalysts in a IL:solketal:H2O (70:25:5, v/v/v) reaction medium after 48 h at 60 °C. These results demonstrated a sustainable biocatalytic strategy for the hydrolytic depolymerization of polyurethane foam wastes.
Another bioprocess for PU degradation is based on the use of abundant microorganisms (i.e. fungi and bacteria) able to degrade synthetic plastics.103–105 For instance, Su et al. demonstrated the use of microbial consortia in the biodegradation of PU wastes, as these microbes were able to use PU as the sole carbon source and accumulate high biomass within 1 week. Extracellular enzyme analyses showed that the consortia secreted esterase and urease, which could be potentially involved in the degradation of PU, although the metabolic pathway of the degradation is still unknown.106 On the other hand, different polyester-polyether urethane degrading yeasts were isolated, resulting in two new strains of Exophilia sp. NS-7 and Rhodotorula sp. NS-12. The results showed that Exophilia sp. NS-7 was esterase, protease, and urease positive, and Rhodotorula sp. NS-12 could produce esterase and urease, and both strains were able to degrade Impranil® as a carbon source, providing a growth rate of 4–6 and 8–12 days, respectively.107 However, the previously mentioned concept of recovery, recycling and reuse (3R) of the compounds does not completely apply, since the microorganisms use PU wastes as nutrients, preventing the concept of a circular economy.108 This drawback can be explained because the structure of the PU residue is not broken down to provide valuable chemical building blocks, or useful constituents of the original polymer to offer promising routes towards life-cycle products. In fact, the microbiological elimination of solid plastic wastes by oxidative metabolic pathway transformations just consists of its full degradation into the most disseminated waste molecule, such as CO2. In this way, the (bio)catalytic breakdown of plastic wastes into their building blocks for further reuse will always be a more sustainable solution than biological digestion.
6 SWOT analysis: chemocatalytic vs. biocatalytic processes for recycling polymeric waste (PET and PUs)
Once the key examples and current tools have been reviewed in the field of catalytic recycling, a Strengths, Weaknesses, Opportunities, Threats (SWOT) analysis provides a comprehensive overview of the relative strengths, weaknesses, opportunities, and threats associated with different catalytic approaches. This analytical framework is crucial for understanding the advantages and limitations of both chemocatalytic and biocatalytic methods, offering insights into their practical impacts. Table 2 shows a compilation of the characteristics of the different catalytic processes analyzed in this work on the depolymerization of polyester and polyurethane waste. Both chemocatalytic and biocatalytic approaches have their unique strengths and challenges. Strategic investments in research and development, as well as thoughtful consideration of environmental impacts, economic factors, and technological capabilities, are essential for advancing sustainable recycling technologies. By assessing the positive aspects and current shortcomings, the SWOT analysis facilitates a critical examination of each approach. Furthermore, it helps identify potential future opportunities and threats, which can guide strategic planning and innovation in the field. This kind of analysis is vital for steering future research directions and improving the technologies involved in polymer recycling, ensuring that they are more efficient, sustainable, and adaptable to varying waste stream conditions. Scheme 23 depict the SWOT analysis for both approaches here evaluated. | ||
| Scheme 23 SWOT analysis of chemo- vs.-bio-catalysis approaches for the degradation of (polyester/polyurethane) plastic waste. | ||
6.1 Chemocatalytic systems
In the first place, the SWOT for the field of chemocatalysis for polymer recycling is presented analysing the examples highlighted through review and current research trends. This comprehensive approach has outlined the current capabilities and future directions of chemocatalytic technologies in addressing the challenges associated with polymer waste.6.2 Strengths
6.3 Weaknesses
6.4 Opportunities
6.5 Threats
6.6 Strengths of biocatalysis
6.7 Weaknesses
6.8 Opportunities
6.9 Threats
7 Technology readiness level of the (bio)catalytic approaches at industrial level
Catalytic and biocatalytic recycling methods for PET and PU waste vary significantly in technological readiness. These technologies generally span Technology Readiness Levels (TRL) 3 to 8, depending on the type of polymer and catalyst used. Catalytic methods for PET and PU recycling are often found in academic settings at lower TRLs (up to TRL 3), focusing on early-stage research and development of the catalyst. In contrast, several examples have progressed to pilot-scale or even commercial deployment, reaching TRLs 8–9. The transition from initial TRLs to full-scale industrial application requires addressing challenges related to efficiency, cost, scalability, and maintaining environmental compliance and product quality.In this context, catalytic glycolysis and hydrolysis of PU have advanced from laboratory research to pilot and industrial-scale applications, showing promising potential for the chemical recovery of polyols from PU waste.109 Companies like Repsol,110 and DOW chemicals,111 operate full-scale plants dedicated to PU recycling, showcasing the commercial viability of these technologies. However, biocatalytic approaches for PU recycling remain at a lower TRL (<4), mainly confined to laboratory-scale demonstrations and early development stages.
On the other hand, both catalytic and biocatalytic processes for PET hydrolysis have reached higher TRLs, bridging the gap from novel academic developments to full-scale industrial applications. Catalytic hydrolysis methods, utilizing zinc or other metal catalysts, are implemented by companies such as Ioniqa Technologies,112 Loop Industries,113 and Indorama Ventures,114 at commercial levels. These methods efficiently depolymerize PET into monomers that can be reused in new production cycles.
Biocatalytic methods, particularly those involving enzymes like PETase, have shown significant promise, reaching pilot and industrial scales. Carbios is a leader in this field, having developed advanced enzymatic recycling techniques that are now operational at industrial levels, paving the way for more sustainable PET recycling processes. Carbios uses engineered enzymes specifically designed to degrade PET, demonstrating the efficiency and scalability of biocatalytic methods, especially in industries like packaging and textiles. This dual approach of catalytic and biocatalytic recycling is setting new standards for circular economies in polymer recycling, highlighting the potential for broader industrial application.115
8 Conclusions
Plastic waste represents a significant challenge in modern society, necessitating a multi-faceted approach to management and recycling. Industries producing polyester and polyurethane must adopt circular chemistry protocols to reintegrate waste back into their production chains, a crucial step for environmental preservation and achieving the Sustainable Development Goals (SDGs) set by the United Nations. The adoption of biodegradable plastics and renewable raw materials marks a strategic advancement in reducing environmental impact. However, the creation of biodegradable waste can lead to a loss of economic value and contribute to CO2 emissions through decomposition processes.This review thoroughly examines catalytic recycling methods, particularly chemocatalytic and biocatalytic strategies for breaking down polyester and polyurethane waste. The analysis reveals a complex balance of strengths, weaknesses, opportunities, and threats associated with these technologies. Chemocatalysis, while robust and efficient, faces environmental and cost challenges. On the other hand, biocatalysis offers an eco-friendlier alternative with greater specificity and lower energy demands, though it struggles with scalability and high operational costs.
Significant advancements in the field underline the need for strategic research and development investments to improve the sustainability and economic viability of recycling technologies. Innovations in catalyst design that can operate under milder conditions and enhance selectivity and stability are particularly promising. These could greatly reduce the environmental impact and improve the economic feasibility of these processes. Traditional metal-based catalysts are well-established, but newer metal–organic hybrids and biocatalysts are still developing within the context of plastic recycling. For instance, Zn-based catalysts are favored for their biocompatibility and stability, with PET frequently used in degradation tests. The review also discusses the high reaction temperatures—often above 150 °C—required in many processes, which lead to significant energy consumption and by-product formation. Efforts to use water as a solvent for polymer hydrolysis under milder conditions are recommended. Biocatalysts, which can operate under less harsh conditions such as temperatures below 80 °C without pressure or organic solvents, present a viable but currently costly option. Future developments may reduce these costs and broaden the industrial applicability of biocatalysts in recycling. The development of hybrid materials (MOF) and advancements in genetic engineering and materials science could significantly advance both chemocatalysis and biocatalysis, making them more adaptable to various industrial applications. Such progress is vital for aligning with global sustainability goals and transitioning towards a circular economy in polymer production and waste management.
Finally, the future of polymer recycling relies heavily on our ability to develop catalytic processes that meet the technical demands of polymer degradation while aligning with environmental regulations and economic realities. Insights from this review are set to guide future research directions and enhance the technologies involved in polymer recycling, ensuring greater efficiency, sustainability, and adaptability to the dynamics of waste stream conditions.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
Francisco G. Cirujano: conceptualization, formal analysis, funding acquisition, investigation, methodology, supervision, writing – original draft, writing – review & editing; Rocio Villa: conceptualization, data curation, formal analysis, investigation, methodology, writing – original draft, writing – review & editing; Rebeca Salas: conceptualization, data curation, formal analysis, investigation, methodology, validation, visualization, writing – original draft, writing – review & editing; Miguel Maireles: investigation, writing – review & editing; Nuria. Martin, data curation, formal analysis, investigation, writing – review & editing; Belen Altava: data curation, funding acquisition, formal analysis, investigation, writing – review & editing; Pedro Lozano: conceptualization; formal analysis, funding acquisition, investigation, methodology, writing – review & editing; Eduardo García-Verdugo: conceptualization; formal analysis, funding acquisition, investigation, methodology, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been partially supported by MICINN-FEDER-AEI 10.13039/501100011033 (PID2021-124695OB-C21/C22, PID2022-142897OA-I00 and PDC2022-133313-C21/C22), MICINN – European Union Next Generation EU-PRTR (TED2021-129626B-C21/C22), and Fundación SENECA (21884/PI/22) grants. F. G. C. and N. M. acknowledge the “Ramon y Cajal” contract with code RYC2020-028681-I and RYC2021-033167-I funded MCIN/AEI/10.13039/501100011033 and by “ESF investing in your future”, “European Union NextGenerationEU/PRTR”. FGC thank Generalitat Valenciana (CISEJI/2023/78). UJI is acknowledged for the project UJI-2023-03.References
- PlasticsEurope, Plastics – the Facts 2022, 2022, accessed 19 Dec 2023, https://plasticseurope.org/wp-content/uploads/2022/10/PE-PLASTICS-THE-FACTS_V7-Tue_19-10-1.pdf.
- X. Yuan, X. Wang, B. Sarkar and Y. S. Ok, Nat. Rev. Earth Environ., 2021, 2, 659–660 CrossRef CAS PubMed.
- A. L. Andrady and M. A. Neal, Philos. Trans. R. Soc., B, 2009, 364, 1977–1984 CrossRef CAS PubMed.
- W. T. Hsu, T. Domenech and W. McDowall, Clean. Environ. Syst., 2021, 2, 100004 CrossRef.
- P. Industry Association: Flexible bag and film division, https://www.plasticsindustry.org/supply-chain/processors/flexible-film-and-bag-division, 08/05/2023.
- L. Mishnaevsky Jr, K. Branner, H. Norgaard Petersen, J. Beauson, M. McGugan and B. F. Sorensen, Materials, 2017, 10, 1285 CrossRef PubMed.
- J. Boucher, G. Billard, E. Simeone and J. Sousa, The Marine Plastic Foot Print: towards a Science-Based Metric for Measuring Marine Plastic Leakage and Increasing the Materiality and Circularity of Plastic, IUCN Publication, Gland, 2020, DOI:10.2305/iucn.ch.2020.01.en.
- OECD, Global Plastics Outlook: Economic Drivers, Environmental Impacts and Policy Options, OECD Publishing, Paris, 2022 Search PubMed.
- E. Butler, G. Devlin and K. McDonnell, Waste Biomass Valorization, 2011, 2, 227–255 CrossRef CAS.
- K. Zheng, Y. Wu, Z. Hu, S. Wang, X. Jiao, J. Zhu, Y. Sun and Y. Xie, Chem. Soc. Rev., 2023, 52, 8–29 RSC.
- (a) H. Sardon and P. Dove Andrew, Science, 2018, 360, 380–381 CrossRef CAS PubMed; (b) R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- R. A. Sheldon and D. Brady, ChemSusChem, 2022, 15, e202102628 CrossRef CAS PubMed.
- P. Lozano and E. García-Verdugo, Green Chem., 2023, 25, 7041–7057 RSC.
- European Commission, The Implementation of the Circular Economy Action Plan, COM, 2019, p. 190 Search PubMed.
- Plastic Europe: Plastics-the Facts 2020. An analysis of European latest plastics production, demand and waste data, 2020, https://www.plasticseurope.org/en/resources/market-data.
- J. Garcia-Martinez, Angew. Chem., Int. Ed., 2021, 60, 4956–4960 CrossRef CAS PubMed.
- Chemistry can help make plastics sustainable — but it isn’t the whole solution, Nature, 2021, vol. 590, pp. 363–364, DOI:10.1038/d41586-021-00391-7.
- H. J. Thamhain, Paper presented at Project Management Institute Annual Seminars & Symposium, Houston, TX Newtown Square, PA, Project Management Institute, https://www.pmi.org/learning/library/phase-gate-processes-promising-complex-547, accessed: December 2023 Search PubMed.
- C. Hussong, J. Langanke and W. Leitner, Green Chem., 2020, 22, 8260–8270 RSC.
- F. R. Wurm, S. Spierling, H. J. Endres and L. Barner, Macromol. Rapid Commun., 2020, 41, 2000351 CrossRef CAS PubMed.
- (a) T. Keijer, V. Bakker and J. C. Slootweg, Nat. Chem., 2019, 11, 190–195 CrossRef CAS PubMed; (b) H. Mutlu and L. Barner, Macromol. Chem. Phys., 2022, 223, 2200111 CrossRef CAS.
- National Science Foundation Centre for Sustainable Polymers, https://csp.umn.edu/sustainable-polymers-101/#1612191883762-e1fcd6cf-bd73, accessed: December 2021.
- Ellen MacArthur Foundation – What is a circular economy, https://ellenmacarthurfoundation.org/topics/circular-economyintroduction/overview, accessed: December 2021.
- I. A. Ignatyev, W. Thielemans and B. Vander Beke, ChemSusChem, 2014, 7, 1579–1593 CrossRef CAS PubMed.
- M. Solis and S. Silveira, Waste Manag., 2020, 105, 128–138 CrossRef CAS PubMed.
- S. M. Al-Salem, P. Lettieri and J. Baeyens, Prog. Energy Combust. Sci., 2010, 36, 103–129 CrossRef CAS.
- C. Baillie, D. Matovic, T. Thamae and S. Vaja, Resour., Conserv. Recycl., 2011, 55, 973–978 CrossRef.
- (a) D. S. Achilias, A. Giannoulis and G. Z. Papageorgiou, Polym. Bull., 2009, 63, 449–465 CrossRef CAS; (b) A. J. Hadi, G. F. Najmuldeen and K. Bin Yusoh, J. Polym. Eng., 2013, 33, 471–481 CAS.
- (a) E. J. Cho, S. Lee, S. J. Oh, M. Han, Y. S. Lee and C. N. Whang, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 52, 16443–16450 CrossRef CAS PubMed; (b) A. L. Andrady, Plastics and the Environment, John Wiley & Sons, New York, 2003 CrossRef; (c) Z. Jia, L. Gao, L. Qin and J. Yin, RSC Sustainability, 2023, 1, 2135–2147 RSC; (d) K. Ghosal and C. Nayak, Mater. Adv., 2022, 3, 1974–1992 RSC; (e) A. Bohre, P. Ram Jadhao, K. Tripathi, K. Kishore Pant, B. Likozar and B. Saha, ChemSusChem, 2023, 16, e202300142 CrossRef CAS PubMed.
- (a) B. Agostinho, A. J. D. Silvestre and A. F. Sousa, Green Chem., 2022, 24, 3115–3119 RSC; (b) M. Imran, B.-K. Kim, M. Han, B. G. Cho and D. H. Kim, Polym. Degrad. Stab., 2010, 95, 1686–1693 CrossRef CAS; (c) Z. Shen, Z. Jia, K. Yu, J. Xie, L. Qin, L. Gao, B. Li, X. Wang and J. Yin, Process Saf. Environ. Prot., 2024, 188, 230–238 CrossRef CAS.
- L. D. Ellis, N. A. Rorrer, K. P. Sullivan, M. Otto, J. E. McGeehan, Y. Roman-Leshkov, N. Wierckx and G. T. Beckham, Nat. Catal., 2021, 4, 539–556 CrossRef CAS.
- M. Chu, Y. Liu, X. Lou, Q. Zhang and J. Chen, ACS Catal., 2022, 12, 4659–4679 CrossRef CAS.
- A. Bohre, P. R. Jadhao, K. Tripathi, K. K. Pant, B. Likozar and B. Saha, ChemSusChem, 2023, 16, e202300142 CrossRef CAS PubMed.
- B. Agostinho, A. J. D. Silvestre, J. A. P. Coutinho and A. F. Sousa, Green Chem., 2023, 25, 13–31 RSC.
- S. C. Kosloski-Oh, Z. A. Wood, Y. Manjarrez, J. P. de los Rios and M. E. Fieser, Mater. Horiz., 2021, 8, 1084–1129 RSC.
- I. Olazabal, E. J. Luna Barrios, S. De Meester, C. Jehanno and H. Sardon, ACS Appl. Polym. Mater., 2024, 6(7), 4226–4232 CrossRef CAS PubMed.
- E. Luna, I. Olazabal, M. Roosen, A. Mueller, C. Jehanno, M. Ximenis, S. de Meester and H. Sardon, Chem. Eng. J., 2024, 482, 148861 CrossRef CAS.
- I. Olazabal, E. Luna, S. De Meester, C. Jehanno and H. Sardon, Polym. Chem., 2023, 14, 2299–2307 RSC.
- I. Olazabal, A. González, S. Vallejos, I. Rivilla, C. Jehanno and H. Sardon, ACS Sustain. Chem. Eng., 2023, 11, 332–342 CrossRef CAS PubMed.
- R. Heiran, A. Ghaderian, A. Reghunadhan, F. Sedaghati, S. Thomas and A. H. Haghighi, J. Polym. Res., 2021, 28, 22 CrossRef CAS.
- R. Esquer and J. J. García, J. Organomet. Chem., 2019, 902, 120972 CrossRef CAS.
- Y. Wang, H. Song, H. Ge, J. Wang, Y. Wang, S. Jia, T. Deng and X. Hou, J. Cleaner Prod., 2018, 176, 873–879 CrossRef CAS.
- J. Stewart, M. Fuchs, J. Payne, O. Driscoll, G. Kociok-Kohn, B. D. Ward, S. Herres-Pawlis and M. D. Jones, RSC Adv., 2022, 12, 1416–1424 RSC.
- S. D'Aniello, S. Laviéville, F. Santulli, M. Simon, M. Sellitto, C. Tedesco, C. M. Thomas and M. Mazzeo, Catal. Sci. Technol., 2022, 12, 6142–6154 RSC.
- R. Lopez-Fonseca, I. Duque-Ingunza, B. de Rivasa, S. Arnaiz and J. I. Gutierrez-Ortiz, Polym. Degrad. Stab., 2010, 95, 1022–1028 CrossRef CAS.
- L. M. dos Santos, C. L. P. Carone, J. Dullius, R. Ligabue and S. Einloft, Polímeros, 2013, 23(5), 608–613 CrossRef CAS.
- C. Molero, A. de Lucas and J. F. Rodriguez, Polym. Degrad. Stab., 2009, 94, 533–539 CrossRef CAS.
- J. T. Du, Q. Sun, X. F. Zeng, D. Wang, J. X. Wang and J. F. Chen, Chem. Eng. Sci., 2020, 220, 115642 CrossRef CAS.
- E. Selvam, Y. Luo, M. Ierapetritou, R. F. Lobo and D. G. Vlachos, Catal. Today, 2023, 418, 114124 CrossRef CAS.
- J. Cao, Y. Lin, W. Jiang, W. Wang, X. Li, T. Zhou, P. Sun, B. Pan, A. Li and Q. Zhang, ACS Sustain. Chem. Eng., 2022, 10, 5476–5488 CrossRef CAS.
- M. Imran, D. H Kim, W. A. Al-Masry, A. Mahmooda, A. Hassan, S. Haider and S. M. Ramay, Polym. Degrad. Stab., 2013, 98, 904–915 CrossRef CAS.
- Y. Yang, F. Chen, T. Shen, A. Pariatamby, X. Wen, M. Yan and E. Kanchanatip, Process Saf. Environ. Prot., 2023, 173, 881–892 CrossRef CAS.
- M. Yan, Y. Yang, F. Chen, D. Hantoko, A. Pariatamby and E. Kanchanatip, Environ. Sci. Pollut. Res., 2023, 30, 102560–102573 CrossRef CAS PubMed.
- X. K. Li, H. Lu, W. Z. Guo, G. P. Cao, H. L. Liu and Y. H. Shi, AIChE J., 2015, 61, 200–214 CrossRef CAS.
- Q. Sun, Y. Y. Zheng, L. X. Yun, H. Wu, R. K. Liu, J. T. Du, Y. H. Gu, Z. G. Shen and J. X. Wang, ACS Sustain. Chem. Eng., 2023, 11, 7586–7595 CrossRef CAS.
- L. X. Yun, H. Wu, Z. G. Shen, J. W. Fu and J. X. Wang, ACS Sustain. Chem. Eng., 2022, 10, 5278–5287 CrossRef CAS.
- G. Eshaq and A. E. ElMetwally, J. Mol. Liq., 2016, 214, 1–6 CrossRef CAS.
- Y. D. Morcillo-Bolanos, W. J. Malule-Herrera, J. C. Ortiz-Arango and A. L. Villa-Holguin, Rev. Fac. Ing. Univ. Antioq., 2018, 87, 77–85 CrossRef CAS.
- S. G. Son, S. B. Jin, S. J. Kim, H. J. Park, J. Shin, T. Ryu, J. M. Jeong and B. G. Choi, FlatChem, 2022, 36, 100430 CrossRef CAS.
- H. Yao, L. Liu, D. Yan, Q. Zhou, J. Xin, X. Lu and S. Zhang, Chem. Eng. Sci., 2022, 248, 117109 CrossRef CAS.
- R. X. Yang, Y. T. Bieh, C. H. Chen, C. Y. Hsu, Y. Kato, H. Yamamoto, C. K. Tsung and K. C. W. Wu, ACS Sustain. Chem. Eng., 2021, 9, 6541–6550 CrossRef CAS.
- (a) V. Tournier, C. M. Topham, A. Gilles, B. David, C. Folgoas, E. Moya-Leclair, E. Kamionka, M. L. Desrousseaux, H. Texier, S. Gavalda, M. Cot, E. Guemard, M. Dalibey, J. Nomme, G. Cioci, S. Barbe, M. Chateau, I. Andre, S. Duquesne and A. Marty, Nature, 2020, 580, 216–219 CrossRef CAS PubMed; (b) https://www.carbios .
- B. Guo, X. Lopez-Lorenzo, Y. Fang, E. Backstrom, A. J. Capezza, S. R. Vanga, I. Furo, M. Hakkarainen and P. Syren, ChemSusChem, 2023, 16, e202300742 CrossRef CAS PubMed.
- R. Xue, C. Qiu, X. Zhou, Y. Cheng, Z. Zhang, Y. Zhang, U. Schroder, U. T. Bornscheuer, W. Dong, R. Wei and M. Jiang, Angew. Chem., Int. Ed., 2023, e202313633 Search PubMed.
- (a) L. Aer, Q. Jiang, I. Gul, Z. Qi, J. Feng and L. Tang, Environ. Res., 2022, 212, 113472 CrossRef CAS PubMed; (b) M. E. Sevilla, M. D. Garcia, Y. Perez-Castillo, V. Armijos-Jaramillo, S. Casado, K. Vizuete, A. Debut and L. Cerda-Mejía, Polymers, 2023, 15, 1779 CrossRef CAS PubMed.
- Y. Fang, K. Chao, J. He, Z. Wang and Z. Chen, 3 Biotech., 2023, 13, 138 CrossRef PubMed.
- F. Quartinello, S. Vajnhandl, J. Volmajer Valh, T. J. Farmer, B. Voncina, A. Lobnik, E. Herrero Acero, A. Pellis and G. M. Guebitz, Microb. Biotechnol., 2017, 10, 1376–1383 CrossRef CAS PubMed.
- A. Magnin, L. Entzmann, E. Pollet and L. Averous, Waste Manag., 2021, 132, 23–30 CrossRef CAS PubMed.
- F. Di Bisceglie, F. Quartinello, R. Vielnascher, G. M. Guebitz and A. Pellis, Polymers, 2022, 14, 411 CrossRef CAS PubMed.
- J. Schmidt, R. Wei, T. Oeser, L. A. D. E. Silva, D. Breite, A. Schulze and W. Zimmermann, Polymers, 2017, 9, 65 CrossRef PubMed.
- R. Gautam, A. S. Bassi and E. K. Yanful, Biotechnol. Lett., 2007, 29, 1081–1825 CrossRef CAS PubMed.
- Y. Branson, S. Soltl, C. Buchmann, R. Wei, L. Schaffert, C. P. S. Badenhorst, L. Reisky, G. Jager and U. T. Bornscheuer, Angew. Chem., 2023, 62, e202216220 CrossRef CAS PubMed.
- D. De Smet, J. Verjans and M. Vanneste, Polymers, 2022, 14, 5452 CrossRef CAS PubMed.
- J. Del Amo, D. Simon, M. J. Ramos, J. F. Rodriguez, A. De Lucas and A. M. Borreguero, J. Mater. Cycles Waste Manage., 2022, 24, 1059–1071 CrossRef CAS.
- D. Simon, A. M. Borreguero, A. De Lucas and J. F. Rodriguez, Waste Manag., 2018, 76, 147–171 CrossRef CAS PubMed.
- J. del Amo, A. M. Borreguero, M. J. Ramos and J. F. Rodriguez, Polymers, 2021, 13, 1418 CrossRef CAS PubMed.
- R. Donadini, C. Boaretti, L. Scopel, A. Lorenzetti and M. Modesti, Chem.–Eur. J., 2023, 30, e202301919 CrossRef PubMed.
- C. Molero, A. de Lucas, F. Romero and J. F. Rodriguez, J. Mater. Cycles Waste Manage., 2009, 11, 130–132 CrossRef CAS.
- M. Murai, M. Sanou, T. Fujimoto and F. Baba, J. Cell. Plast., 2003, 39, 15–27 CrossRef CAS.
- X. Gu, X. Wang, T. Wang, Y. Zhu, X. Guo, S. Liu, S. Zhu and Y. Liu, Polymers, 2022, 14, 5450 CrossRef CAS PubMed.
- X. Luo and Y. Li, J. Polym. Environ., 2014, 22, 318–328 CrossRef CAS.
- Y. Lin, D. Yang, C. Meng, C. Si, Q. Zhang, G. Zeng and W. Jiang, ChemSusChem, 2023, 16, e202300154 CrossRef CAS PubMed.
- C. Jeong, M. J. Hyun and Y. W. Suh, Catal. Commun., 2015, 70, 34–39 CrossRef CAS.
- W. Qingyin, K. Wukui, Z. Yi, Y. Xiangui, Y. Jie, C. Tong and W. Gongying, Chin. J. Catal., 2013, 34, 548–558 CrossRef.
- W. Z. Guo, H. Lu, X. K. Li and G. P. Cao, RSC Adv., 2016, 6, 43171–43184 RSC.
- S. Mohammadi and M. Enayati, Polym. Degrad. Stab., 2022, 206, 110180 CrossRef CAS.
- P. Uriz, M. Serra, P. Salagre, S. Castillon, C. Claver and E. Fernandez, Tetrahedron Lett., 2002, 43, 1673–1676 CrossRef CAS.
- (a) S. B. Jin, J. M. Jeong, S. G. Son, S. H. Park, K. G. Lee and B. G. Choi, Mater. Today Commun., 2021, 26, 101857 CrossRef CAS; (b) G. Park, L. Bartolome, K. G. Lee, S. J. Lee, D. H. Kim and T. J. Park, Nanoscale, 2012, 4, 3879–3885 RSC.
- J. M. Jeong, S. B. Jin, S. G. Son, H. Suh, J. M. Moon and B. G. Choi, React. Chem. Eng., 2021, 6, 297–303 RSC.
- M. Ammar, Y. Cao, P. He, L. Wang, J. Chen and H. Li, Chin. J. Chem. Eng., 2017, 25, 1760–1770 CrossRef.
- Y. Cao, Y. Chi, A. Muhammad, P. He, L. Wang and H. Li, Chin. J. Chem. Eng., 2019, 27, 549–555 CrossRef CAS.
- B. Guo, J. Liu, S. Tang, Y. Liu, Z. Tiana and J. Lva, J. Chem. Technol. Biotechnol., 2022, 97, 1695–1704 CrossRef CAS.
- R. Wang, T. Wang, G. Yu and X. Chen, Polym. Degrad. Stab., 2021, 183, 109463 CrossRef CAS.
- M. E. E. Temporiti, L. Nicola, E. Nielsen and S. Tosi, Microorganisms, 2022, 10, 1180 CrossRef CAS PubMed.
- R. P. Magalhaes, J. M. Cunha and S. F. Sousa, Int. J. Mol. Sci., 2021, 22, 11257 CrossRef CAS PubMed.
- S. Thiyagarajan, E. Maaskant-Reilink, T. A. Ewing, M. K. Julsing and J. van Haveren, RSC Adv., 2022, 12, 947–970 RSC.
- A. Loredo-Trevino, G. Gutierrez-Sanchez, R. Rodriguez-Herrera and C. N. Aguilar, J. Polym. Environ., 2012, 20, 258–265 CrossRef CAS.
- (a) V. Petry do Canto, C. E. Thompson and P. A. Netz, J. Mol. Graphics Modell., 2019, 89, 82–95 CrossRef PubMed; (b) N. Mahajan and P. Gupta, RSC Adv., 2015, 5, 41839–41854 RSC; (c) M. Cregut, M. Bedas, M. J. Durand and G. Thouand, Biotechnol. Adv., 2013, 31, 1634–1647 CrossRef CAS PubMed.
- T. Nakajima-Kambe, Y. Shigeno-Akutsu, N. Nomura, F. Onuma and T. Nakahara, Appl. Microbiol. Biotechnol., 1999, 51, 134–140 CrossRef CAS PubMed.
- S. K. Phua, E. Castillo, J. M. Anderson and A. Hiltner, J. Biomed. Mater. Res., 1987, 21, 231–246 CrossRef CAS PubMed.
- G. Lin, S. Y. Chiou, B. C. Hwu and C. W. Hsieh, Protein J., 2006, 25, 33–43 CrossRef CAS PubMed.
- R. Salas, R. Villa, S. Cano, S. Nieto, E. Garcia-Verdugo and P. Lozano, Catal. Today, 2024, 430, 114516 CrossRef CAS.
- R. Wei and W. Zimmermann, J. Microb. Biotechnol., 2017, 10, 1308–1322 CAS.
- J. Ru, Y. Huo and Y. Yang, Front. Microbiol., 2020, 11, 442 CrossRef PubMed.
- N. Mohanan, Z. Montazer, P. K. Sharma and D. B. Levin, Front. Microbiol., 2020, 11, 580709 CrossRef PubMed.
- T. Su, T. Zhang, P. Liu, J. Bian, Y. Zheng, Y. Yuan, Q. Li, Q. Liang and Q. Qi, Environ. Biotechnol., 2023, 107, 1983–1995 CrossRef CAS PubMed.
- M. Giyahchi and H. Moghimi, Nature, 2023, 13, 5016 CAS.
- A. Magnin, E. Pollet, V. Phalip and L. Averous, Biotechnol. Adv., 2020, 39, 107457 CrossRef CAS PubMed.
- (a) V. Y. Suprun, V. I. Marukha and V. P. Sylovaniuk, Mater. Sci., 2022, 57, 755–764 CrossRef; (b) G. Rossignolo, G. Malucelli and A. Lorenzetti, Green Chem., 2024, 26, 1132–1152 RSC.
- https://www.repsol.com/en/sustainability/sustainability-pillars/environment/circular-economy/our-projects/chemical-recycling-of-polyurethane-foam/index.cshtml .
- https://corporate.dow.com/en-us/purpose-in-action/circular-economy/renuva-program.html .
- https://ioniqa.com/company/ .
- https://www.loopindustries.com/en .
- https://www.indoramaventures.com/en/home .
- https://www.carbios.com/fr/enzymatic-recycling/ .
Footnote |
| † This authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |