DOI:
10.1039/D3SC06462J
(Edge Article)
Chem. Sci., 2024,
15, 3578-3587
Crystal property engineering using molecular–supramolecular equivalence: mechanical property alteration in hydrogen bonded systems†‡
Received
2nd December 2023
, Accepted 24th January 2024
First published on 26th January 2024
Abstract
Most crystal engineering strategies exercised until now mainly rely on the alteration of weak non-covalent interactions to design structures and thus properties. Examples of mechanical property alteration for a given structure type are rare with only a few halogen bonded cases. The modular nature of halogen bonds with interaction strength tunability makes the task straightforward to obtain property differentiated crystals. However, the design of such crystals using hydrogen bond interactions has proven to be non-trivial, because of its relatively higher difference in bonding energies, and more importantly, disparate geometries of the functional groups. In the present crystal property engineering exercise, with the support of CSD analysis, we replaced a supramolecular precursor that leads to plastically bendable crystals, with a molecular equivalent, and obtained an equivalent crystal structure. As a result, the new structure, with comparable hydrogen bonding chains, produces elastically bendable single crystals (as opposed to plastically bendable crystals). In addition, the crystals show multidirectional (here two) elastic bending as well as rare elastic twisting. The occurrence of multiple isostructural examples, including a solid solution, with identical properties further demonstrates the general applicability of the proposed model. Crystals cannot display the concerned mechanical property in the absence of the desired structure type and fracture in a brittle manner on application of an external stress. Nanomechanical experiments and energy framework calculations also complement our results. To the best of our knowledge, this is the first example of a rational crystal engineering exercise using solely hydrogen bond interactions to obtain property differentiated crystals. This strategy namely molecular–supramolecular equivalence has been unexplored till now to tune mechanical properties, and hence is useful for crystal property engineering.
Introduction
Studying the mechanical properties of molecular crystals has been an overreaching goal in recent years in the area of crystal engineering.1 Among many other mechanical responses, say thermosalient, photosalient, chemosalient, and self-healing, the analysis and understanding of flexible molecular crystals has been the focus of vast research activity.2–5 Flexible crystals possess the potential for a variety of applications that range from smart materials to biological systems.6–15 Mechanically bendable or twistable crystals are generally divided into two major categories, elastic and plastic.1 Elastic crystals exhibit reversible deformation characteristics, whereas plastic crystals show an irreversible or permanent deformation of crystals.
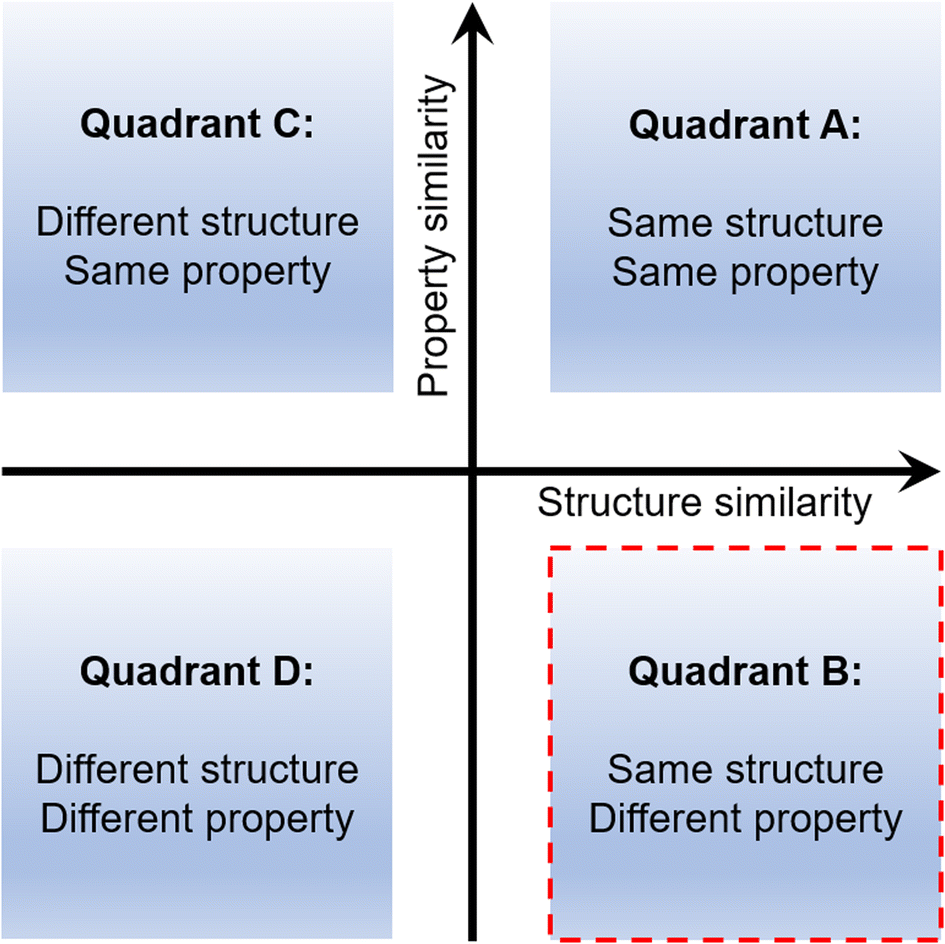
Although many mechanically soft molecular crystals are known today,10–29 the rational design30,31 of such systems still remains a formidable challenge. Many unanswered questions still remain. How can one obtain a crystal with desired mechanical properties? What should be the starting point in such an exercise? Can we alter a property within a given structure type in a predictable manner? An attempt to address such queries needs an in-depth understanding of the structure–property relationship. It is generally observed that systems with the same or comparable structures display similar properties (Quadrant A in Fig. 1), which is unsurprising.30,32 And the opposite relation, i.e., different structures possess dissimilar properties, is also similarly true (Quadrant D).33–36 These two quadrants belong to the conventional structure–property relation types. The third is the same property emerging from a different set of structures (i.e., Quadrant C). Reports for Quadrant C are known for both proteins and small molecule systems.37,38 And for the last one, equivalent/similar structures result in entirely different responses (i.e., Quadrant B).39,40 Quadrants B and C, which attract significant attention, are the parts of a non-classical structure–property paradigm and are rarely explored in crystal engineering. Exploring such systems can unravel new and effective crystal engineering methodologies.
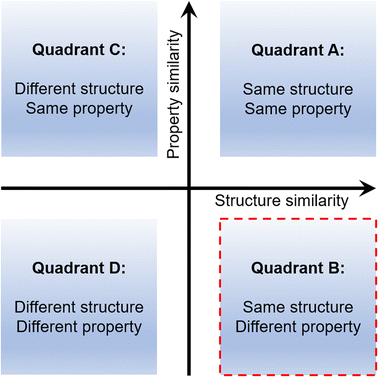 |
| | Fig. 1 Different quadrants in the crystal property engineering exercise. | |
Recently, distinctly different properties, namely elastic and plastic deformation, have been achieved starting from a given structure type by using different halogenated aromatic esters.39 A similar observation was made in some halogenated phenols too.40 In all these cases, a drastic change in macroscopic properties was achieved by altering halogen bond strength via replacing higher halogens with lower halogens or vice versa. Hence, the modular nature of halogen bonded synthons (say I⋯I → Br⋯Br → Cl⋯Cl) makes it relatively straightforward to design crystals by a gradual chemical change (here interaction strength), within a given structure type. However, to the best of our knowledge, such an exercise in hydrogen bonded systems has not been done so far, which meets the criteria of Quadrant B. This is perhaps because of the challenge posed by the relatively higher difference in bonding energies and more importantly disparate geometries of the groups (e.g., –OH, –NH2, and –COOH) and the synthons formed. Hence, it is not trivial to tune interaction strength significantly by replacing one hydrogen bonding group with another while maintaining the structural equivalence, so that property differentiated crystals evolve. Such a methodology is generally proven to be challenging for common hydrogen bonded systems. Does this need a different strategical approach? Here, based on the previous experience in this series, we converted a plastically bendable crystal type into an elastically bendable crystal type using the molecular–supramolecular mimicry, which remained unexplored in crystal property engineering. The general applicability and effectiveness of the design strategy are tested using multiple examples.
Results and discussion
Crystal engineering background
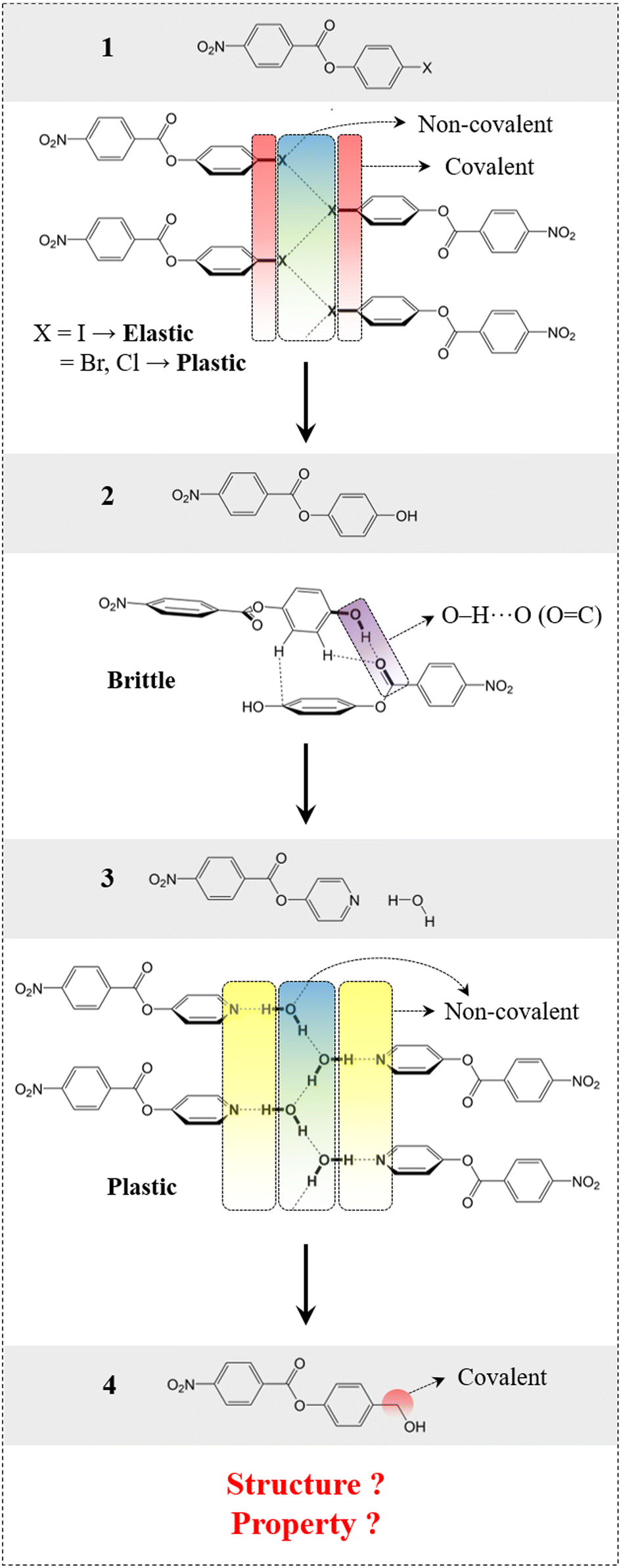
First, we present here the background for our crystal engineering that enabled us to achieve a hydrogen bonded system with an unusual behaviour (the same structure, but different properties: Quadrant B). We identified a rational design strategy to develop a desired molecular system, considering some of our previously reported molecules as a starting point (compounds 1–3, Fig. 2).39,41 The structural pattern of compound 1 (4-halophenyl 4-nitrobenzoate) was considered as a prototype (say structure type 1) that includes (a) bi-directional molecular arrangement of π-stacked columns involving C–H⋯O (NO2) contacts and (b) a halogen bonded catemer with interaction strength tunability, which is a crucial property guiding factor.39 Stronger halogen bonds formed by heavier halogens, like iodine(I), give rise to elastic deformation whereas weaker halogen bonds formed by bromine (Br) and chlorine (Cl) result in plastic bending in the crystals with comparable structures.39 In a related study, a strategy was used to obtain a hydrogen bonded equivalent (i.e. compound 2, 4-hydroxyphenyl 4-nitrobenzoate) (say structure type 2) by replacing the halogen bond synthons. However, this led to an undesired structure as well as a property (i.e. brittleness).41 This study helped us to introduce a retrosynthetic design strategy to functionally modify 2 and obtain compound 3 (4-pyridinyl 4-nitrobenzoate hydrate). This in turn helped to understand the key criteria to obtain the desired structure type (say structure type 3) with an O–H⋯O–H⋯O–H hydrogen bonded catemer, mimicking the halogen bonding pattern in structure type 1. To form such a hydrogen bonded catemer, the hydrogen atom of the O–H group must orient out of the aromatic ring plane, which is not the case in 2 due to the extended conjugation of the phenolic oxygen with the aromatic ring.41 To make such an arrangement, the (aryl)C–O covalent bond needed to be disconnected by replacing with a supramolecular equivalent (pyridine)N⋯H–O–H to obtain 3, which then formed the desired structure. The presence of three non-covalent units involving dynamic hydrogen bonds, parallel to the bending faces, made this supramolecular region significantly mechanically weak, resulting in permanent molecular migrations in the crystals of 3 leading to plastic deformation, via breaking/reformation of intermolecular interactions formed by the water molecule. Now, if one can transform this hydrogen bonded, plastically bendable crystal of 3 into an elastic one within an equivalent structure, then the current goal of property alteration should be achieved.
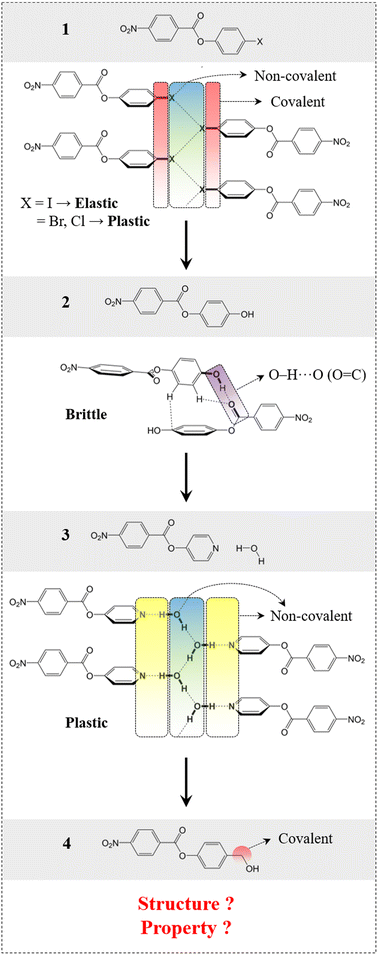 |
| | Fig. 2 Rational design of compound 4, starting from compound 1via a systematic crystal engineering approach. Schematic presentation of intermolecular interactions involved in compounds 1–3. | |
Methodology
To achieve elasticity, one needs to convert the weak supramolecular interaction region in 3 (plastic) into a relatively stronger one within an equivalent structure (structure type 3).24,41,42 This basically means monitoring of chemical factors within a given geometrical factor. For this to be done, the (pyridine)N⋯H–O–H non-covalent units were thought to be replaced with covalent molecular units while maintaining the core structure guided by O–H⋯O–H⋯O–H chains. The covalent molecular unit should be such that the O–H group can avoid conjugation with the aromatic ring, favouring the out-of-plane orientation and the formation of similar catemeric hydrogen bonded chains. For this purpose, we introduced a –CH2− group as a covalent molecular unit between the aryl ring and the O–H group, inhibiting any kind of aromatic –O–H conjugation that can lead to the undesired in-plane orientation. This has resulted in compound 4 [4-(hydroxymethyl)phenyl 4-nitrobenzoate], and we explored to see whether this can fulfill the criteria to adopt for the desired structure type in order to achieve the property under consideration.
CSD study and initial verification of the structural model
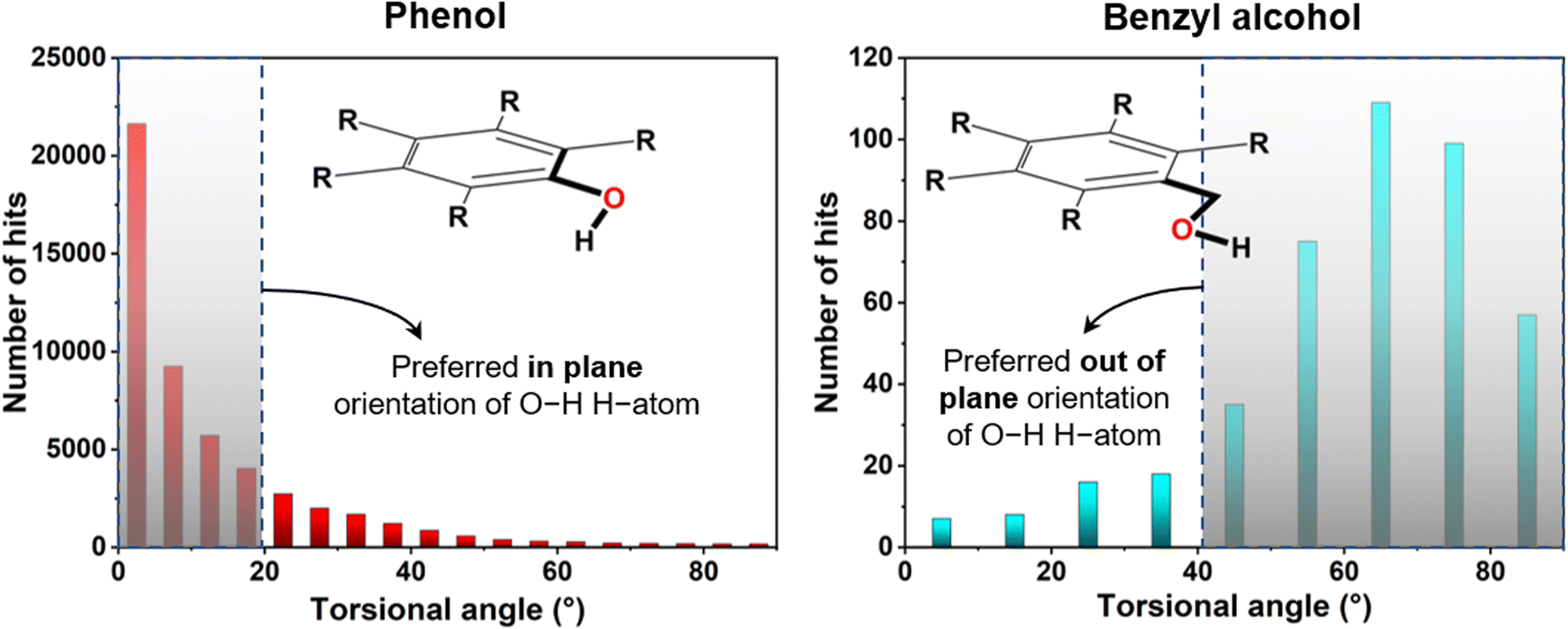
For initial verification of the structural model, a CSD study was carried out for the benzyl alcohol unit of compound 4 in comparison to the phenolic part (Fig. 3). Here, we searched the torsional angular distribution for the phenol and benzyl alcoholic hydroxyl hydrogen atom to obtain the preferred orientation. In phenol, the hydrogen atom almost always is in the aromatic ring's plane, maintaining a lower torsional angle (<20°) so that the molecule can attain highly stabilizing aromatic C–O conjugation. On the other hand, benzyl alcohol showed a completely opposite trend in that the concerned hydrogen atom prefers to be out of the aromatic ring plane and maintains a higher torsional angle (>40°). In the latter case, the in-plane steric crowding factor prevails in the absence of any conjugation. This thus signifies the usefulness of using the benzyl alcohol unit in obtaining the concerned structure type involving compound 4.
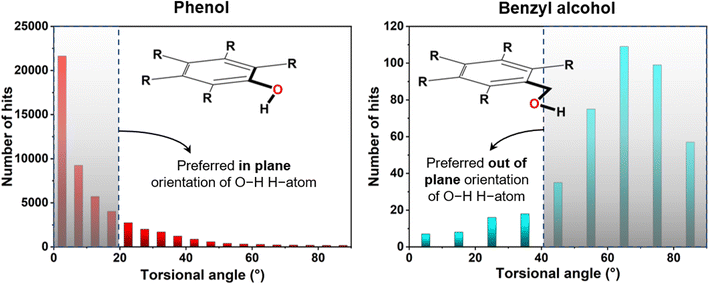 |
| | Fig. 3 Torsional angle distribution for the hydroxyl H-atom of phenol and benzyl alcohol as found from CSD analysis (version: 5.42 updated till September 2021). | |
Crystal structure analysis
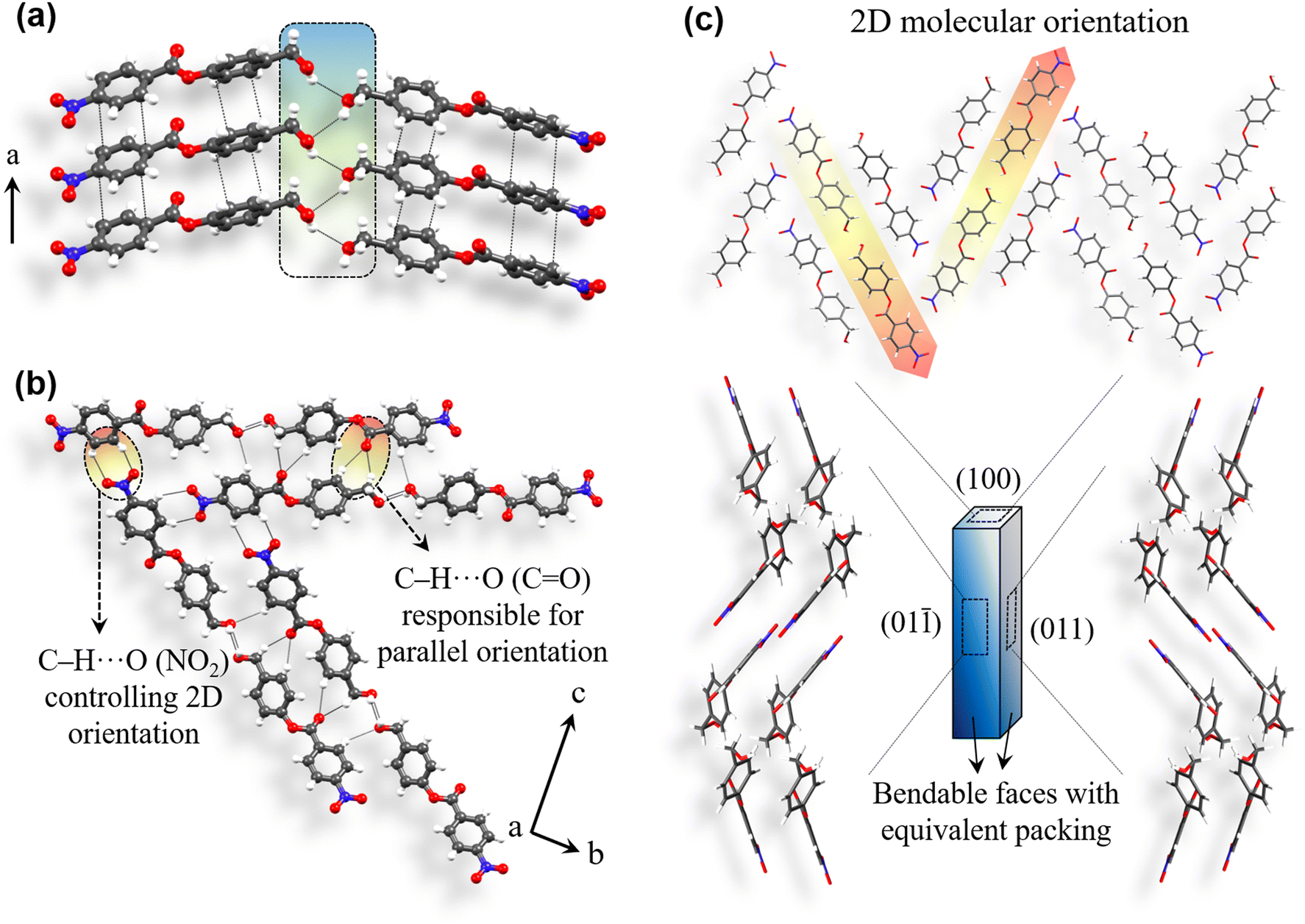
Compound 4 was synthesized using 4-nitrobenzoyl chloride and 4-hydroxybenzyl alcohol (ESI S1‡). After purification, slow evaporation crystallization experiments were performed by dissolving compound 4 in different solvents. Significantly good, diffraction quality crystals were obtained from dichloromethane (DCM) solution (ESI S2‡). Crystals of 4 were then subjected to single crystal X-ray diffraction (SCXRD) experiments (ESI S3‡) to determine the structure and intermolecular interactions involved. Compound 4 crystallizes in an orthorhombic space group P212121 with Z′ = 1. Molecules form π-stacking interactions (3.5 Å, 3.41 Å) along the crystal needle direction, i.e. a-axis (Fig. 4a). Here, the Ar–CH2–OH site is disordered. However, it shows that the –CH2− linker could still maintain the desired out-of-plane orientation of the O–H group as well as hydroxyl hydrogen atom. Parallel stacked columns are then involved in the anticipated O–H⋯O–H⋯O–H (D/Å, d/Å, θ/°: 3.15 Å, 2.44 Å, 145.8°) hydrogen bonded catemer formation. C–H⋯O (3.41 Å, 2.52 Å, 160.4°; 3.72 Å, 2.85 Å, 149.2°; 3.37 Å, 2.61 Å, 139.7°) interactions from the ester carbonyl group and alcoholic O-atom further orient these hydrogen bonded units in parallel, along the c-axis. The packing attains a two directional (2D) molecular arrangement in the bc-plane owing to the formation of C–H⋯O (3.34 Å, 2.53 Å, 144.9°; 3.64 Å, 2.85 Å, 143.6°) contacts involving the NO2 group (Fig. 4b). Such 2D orientation has led to two pairs of equivalent major faces (011)/(0![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) and (01)/(01). The structure of 4 (Fig. 4c) is geometrically equivalent and isostructural to that of 3 (see ESI S18‡), as anticipated.
) and (01)/(01). The structure of 4 (Fig. 4c) is geometrically equivalent and isostructural to that of 3 (see ESI S18‡), as anticipated.
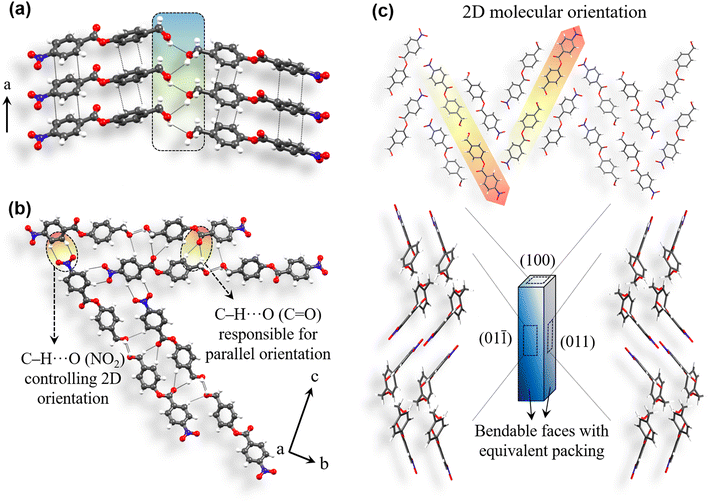 |
| | Fig. 4 Crystal packing of compound 4. (a) π-stacked columns are connected via an O–H⋯O–H⋯O–H hydrogen bonded catemer involving an out of plane –CH2–OH group. Disordered Ar–CH2–OH site is presented in a simplified way for better understanding of the interaction pattern. (b) NO2 group forms C–H⋯O contacts, helping in 2D molecular orientation in the bc-plane (c). (c) Crystal packing shows equivalent arrangement for (011)/(0) and (01)/(01) faces. | |
Qualitative mechanical assessment: deformation experiments
We performed mechanical testing by holding a long pristine crystal with a pair of forceps and poking it at the middle with a metallic needle. The crystal went through a progressive macroscopic bending deformation and finally attained a smooth semicircular bent shape (Fig. 5a, ESI Movie S1‡). Interestingly, unlike 3, on withdrawal of the external mechanical stress, the crystal regained its initial pristine shape completely. This process was repeated many times, which confirmed the elastic bending nature of the crystal (ESI S9, Fig. S2a and Movie S2‡). Crystal breaks beyond a threshold limit of ∼3% strain (Fig. 5a4) (see ESI S10‡).
 |
| | Fig. 5 (a) Snapshots show a progressive elastic bending of a crystal of 4. Crystal breaks beyond a threshold limit (a4). (b1) A straight hanging crystal is bent along two equivalent directions: (b2) out of plane and (b3) in-plane deformation. (c) Reversible elastic twisting. | |
The equivalent packing with respect to the major faces (011)/(0) and (01)/(01) has further prompted us to study the mechanical responses along different directions.11c It is generally difficult to perform manual bending of bare single crystals on each specific face, as the crystals flip or rotate, creating difficulties in identifying faces. Hence, we first fixed one end of the crystal with glue at the edge of a glass slide (cantilever-like geometry) (Fig. 5b1). We applied an external load on the hanging side of the crystal vertically from the bottom, as shown in Fig. 5b2 and ESI Movie S3.‡ Subsequently, the crystal was bent upward smoothly (out of plane), and it reverted back to the initial straight position on unloading. Following this, a horizontal load was then applied on the same crystal. Notably, the crystal showed similar elastic bending (Fig. 5b3 and ESI Movie S3‡). Here, it is an ‘in plane’ elastic bending, but on a different face. The same experiments, repeated on multiple crystals, also displayed the same phenomena, confirming the multi-directional (here, two) reversible (elastic) bendability of the crystals of 4.
In parallel, the existence of the two directional packing with multi-directional elastic flexibility has further encouraged us to test the current crystal for its ability to twist. For this, we have taken a good quality long crystal in paratone oil on a glass plate. The translational movement of the crystal was restricted by holding one end of the crystal with the help of a pair of forceps. The other end was kept free. Then an external torque was applied along the needle direction of the crystal using a metallic needle. The crystal here could take a twisted geometry (Fig. 5c, ESI S9, Fig. S2b and Movie S4‡). And when the external torque was released, the crystal regained the initial straight geometry. The process is repeatable. This is an example of elastic twisting and possibly the first case of elastic mechanical twisting observed in molecular crystals. Note that several examples of crystals that can be twisted plastically using external mechanical stress and light/thermally induced twisting are known in the literature, but elastic twisting induced by mechanical stress has not been observed.2c,3b,39
Quantitative mechanical assessment: nanoindentation experiments
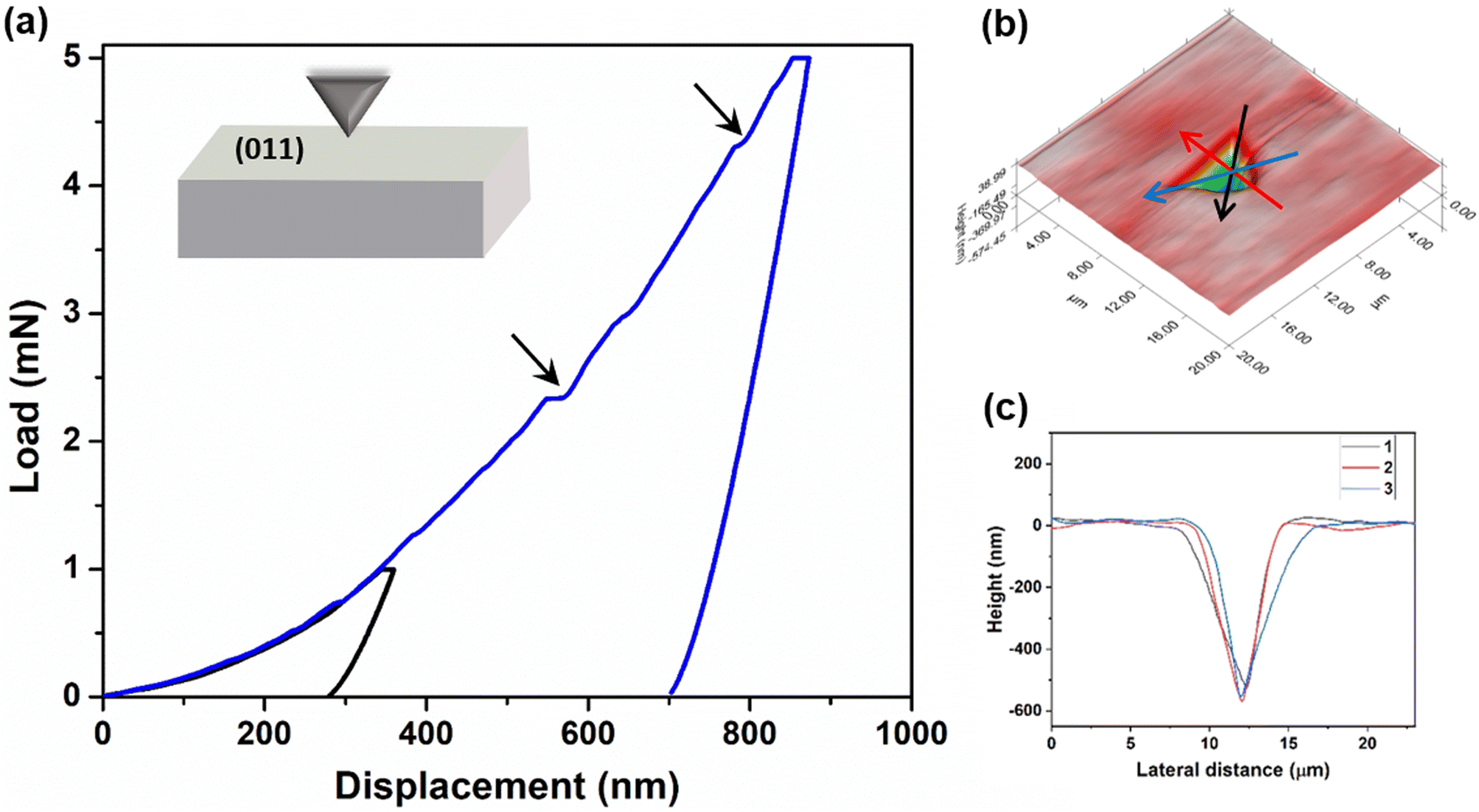
In addition to qualitative assessment, quantitative mechanical characterization of crystals of 4 was carried out using the state-of-the-art nanoindentation technique.43 Indentation experiments were performed on a major face (011) at two different peak loads 1 mN and 5 mN with a loading rate of 0.2 mN s−1 (Fig. 6a and ESI S11‡). The elastic modulus (E) and the hardness (H) were obtained applying the standard Oliver–Pharr method.44 The experimental E and H are 7.98 ± 0.33 GPa and 0.27 ± 0.02 GPa, respectively, at 5 mN load. Here, in the P–h curves, the sudden displacement of the indenter tip is observed at constant load. These are generally known as pop-ins, which occur due to sudden breakage of compressed molecular layers when the force applied by the tip exceeds their resistive force. The generation of pop-ins signifies the restriction that may forbid long range molecular movement under compression. These crystals seem to be compressible in nature as no significant pile-up is observed (Fig. 6b and c). Crystals of 4 are considerably soft in nature, indicating the ease of molecular reorganization locally to a certain extent. This might be helping in the reversibility of deformation in the crystals, thus resulting in elastic bending.
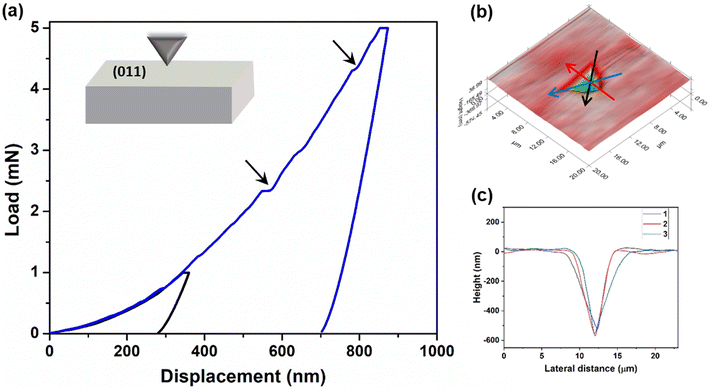 |
| | Fig. 6 Nanoindentation studies on crystals of 4. (a) Representative load–depth (P–h) curves obtained using two different loads 1 and 5 mN on the (011) face. The presence of pop-ins is shown by arrows. (b) 3D mapped indentation surface. (c) Height profile of the residual indent impression on the sample, obtained from the scanning probe microscopy (SPM) image, along different lines red, blue and black. | |
Energy framework calculations: analysis of interaction energy distribution
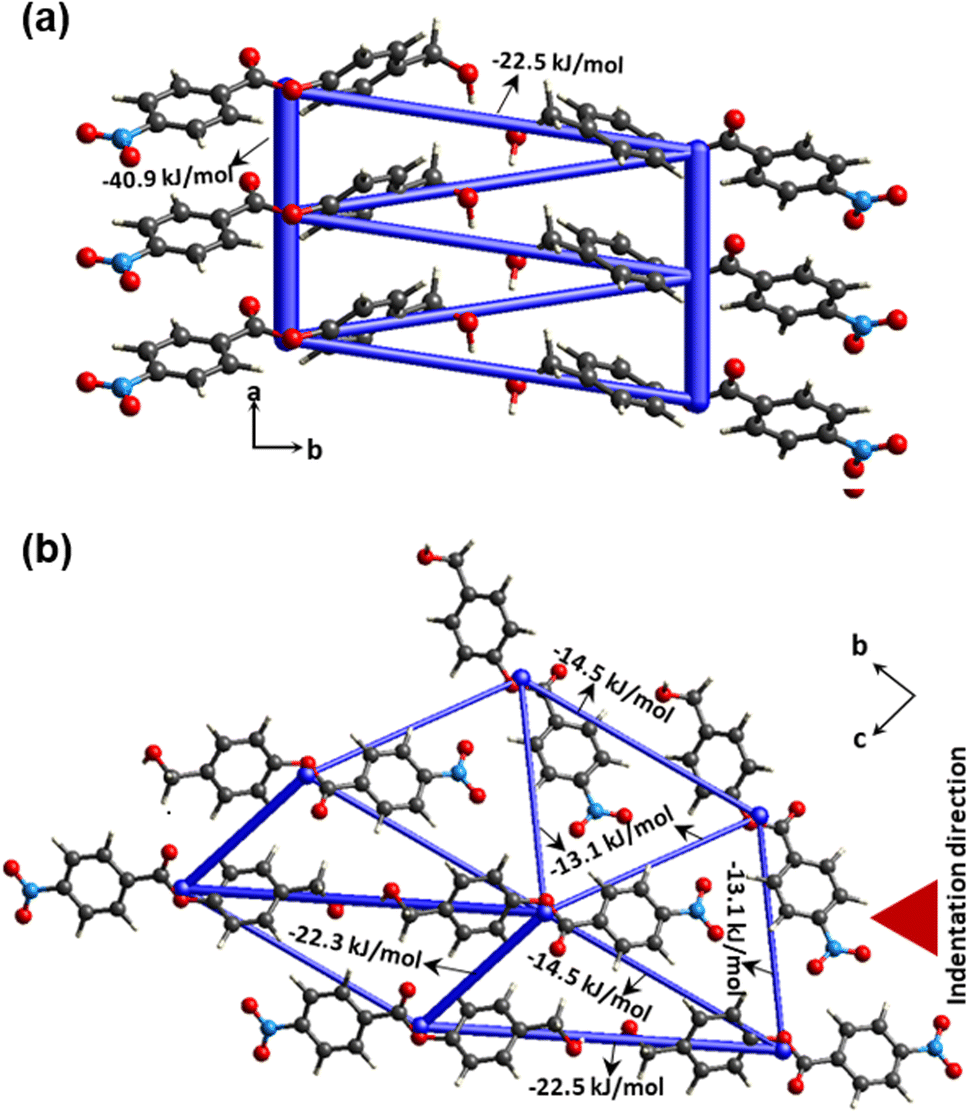
In order to access the information on intermolecular interaction energy distribution around the molecules in crystal 4, we have performed energy framework calculations using the software suite Crystal-Explorer17.45 We extracted atomic coordinate information from experimental crystallographic data to use as an input for the computation procedure. The molecular wave function B3LYP-D2/6-31G (d,p) was applied to run the DFT calculation. The pairwise total intermolecular interaction energies (ETot) thus obtained from energy framework calculations show that π-stacking interactions dominate in the present structure (ETot = –40.9 kJ mol−1) (Fig. 7a). Two π-stacked columns are connected through –22.5 kJ mol−1. In the bc-plane, molecules are connected via C–H⋅⋅⋅O with each other and designate similar framework energy distribution along different directions (ETot = −13.1 kJ mol−1; −14.5 kJ mol−1 and −22.3 kJ mol−1) (Fig. 7b). All these interactions contribute more or less similarly along different directions. The crystal has a near-isotropic packing, which is generally the case in most elastically bendable crystals with a good or a reasonably high elastic limit.
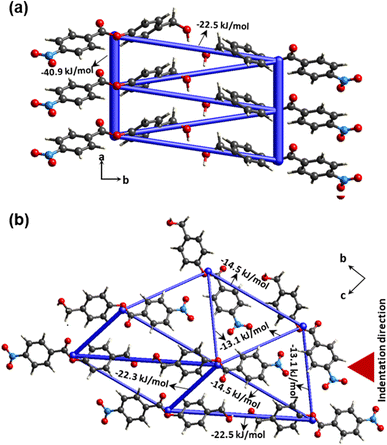 |
| | Fig. 7 Energy framework calculations were performed for crystal 4, (a) along the a-axis for π-stacked units and (b) in the bc-plane. | |
Generalization of the design strategy
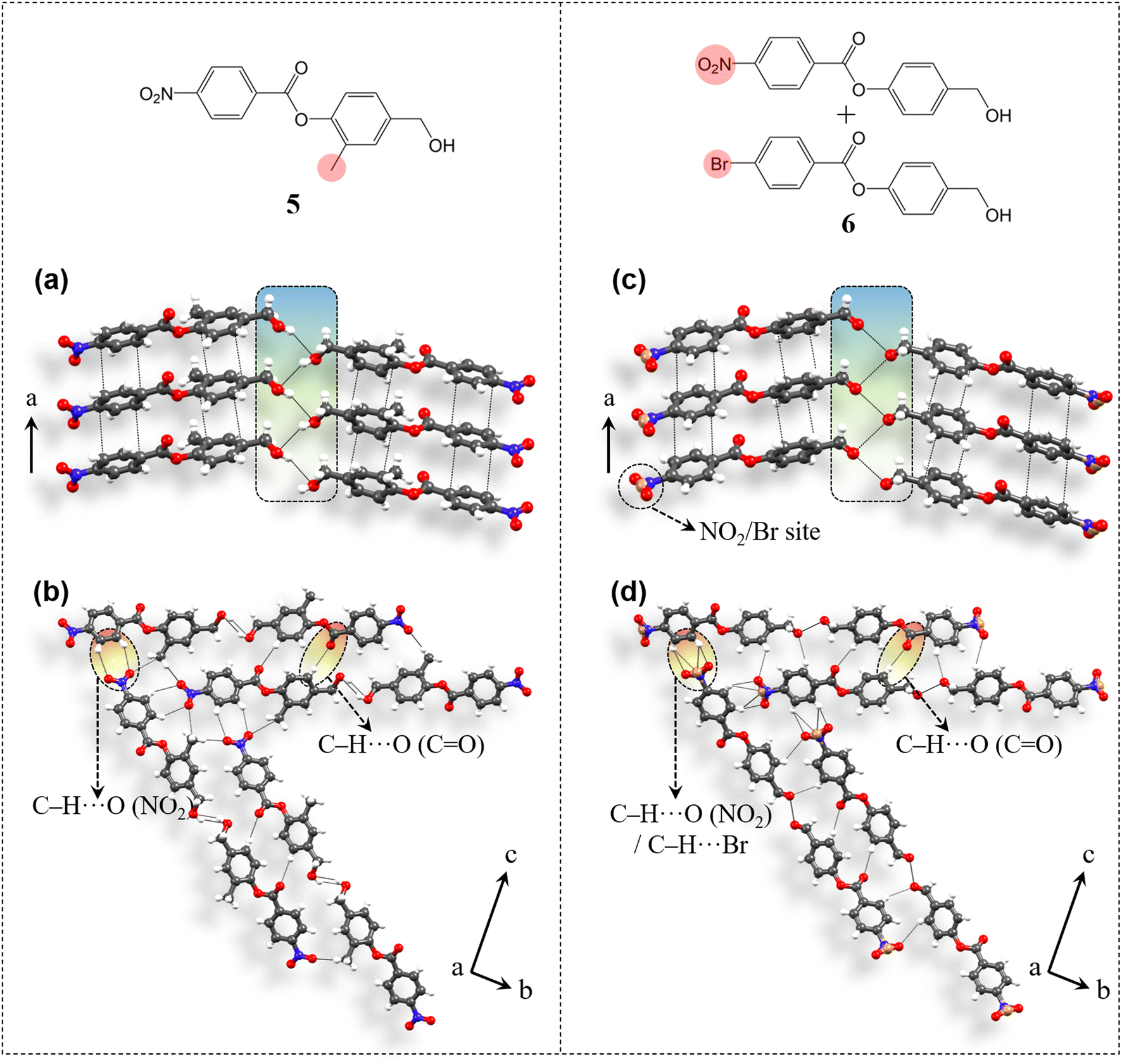
The applicability of the design strategy to different systems proves its reliability. To understand the generality and robustness of the proposed design strategy, we have further explored additional examples with closely related molecular structures. The presence of chemically innocent o-hydrogens in the phenolic ring of 4 has prompted us to replace one of them with a non-perturbing substituent, which here is a methyl group and thus have obtained compound 5 [4-(hydroxymethyl)-2-methylphenyl 4-nitrobenzoate] (Fig. 8). 5 was synthesized from 4-nitrobenzoyl chloride and 4-(hydroxymethyl)-2-methylphenol (see ESI S1‡). Diffraction quality crystals were obtained through crystallization from DCM solution. Crystal structure analysis shows that 5 adopts the P212121 space group with Z′ = 1 and is isostructural to 4. Molecules are π-stacked (3.61 Å, 3.41 Å) along the unit cell short axis a (Fig. 8a). Disordered –CH2–OH groups are then involved in the desired O–H⋯O–H⋯O–H (3.6 Å, 2.87 Å, 149.96°) catemer synthon formation. Such parallel units are connected through C–H⋯O (ester) contacts (3.54 Å, 2.61 Å, 173.2°) along the c-axis. NO2 groups orient the molecules along two different directions in the bc-plane via C–H⋯O formation (3.61 Å, 2.8 Å, 146.5°) in obtaining 2D packing (Fig. 8b), resulting in equivalent major faces (011)/(0) and (01)/(01) (ESI S14‡). Methyl groups further stabilize the packing via C–H⋯O contacts (3.55 Å, 2.63 Å, 161.5°).
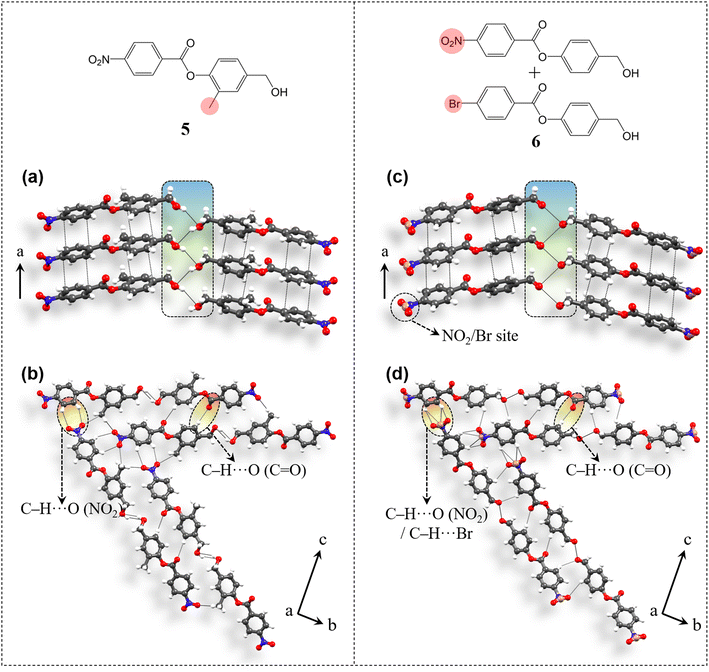 |
| | Fig. 8 Crystal packing and intermolecular interactions involved in compounds 5 and 6. 5 exhibits (a) π-stacked columns with the O–H⋯O–H⋯O–H catemer and (b) C–H⋯O (NO2) mediated 2D molecular orientation. (c and d) Similar structural pattern was observed for 6 too. Disordered sites are presented in a simplified manner for better understanding of the interaction pattern. | |
Further, extending the generality of the design approach, we have synthesized a multicomponent system, which here is a binary solid solution (molecular alloy, 6) of 4-(hydroxymethyl)phenyl 4-nitrobenzoate (compound 4) and 4-(hydroxymethyl)phenyl 4-bromobenzoate (molecule A) (see ESI S2‡ for details). Solid solutions gain importance as each molecular component brings its own characteristics, allowing property tuning by the variation of the composition in multifunctional crystalline materials, without necessarily perturbing the parent structure type. However, the targeted synthesis of solid solutions is challenging as it requires a very delicate balance between the geometrical and chemical factors of the concerning functional groups. The existence of NO2/halo isostructurality46 has prompted us to synthesize a solid solution here involving molecule 4 with NO2 functionality and molecule A with the bromo group (in a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 ratio), thus forming solid solution crystal 6 (Fig. 8). Crystal structure (space group P212121, Z′ = 1) determination revealed that both the molecules are present, however in a ratio of 90:10 with NO2 and Br occupying the same crystallographic site. Again 6 is isostructural to 4. Molecules are π-stacked (3.5 Å, 3.43 Å) along the short axis a (Fig. 8c). Disordered –CH2–OH groups then form the anticipated O–H⋯O–H⋯O–H (3.19 Å) hydrogen bonded catemer units. Such parallel units are stabilized by C–H⋯O (ester) (3.46 Å, 2.53 Å, 175.7°) interactions and are further 2D oriented in the bc-plane using C–H⋯O(NO2)/Br (3.35 Å, 2.54 Å, 145.7°; 3.66 Å, 2.88 Å, 142.8°; 3.72 Å, 3.08 Å, 127.6°; 3.74 Å, 3.14 Å, 124.2°) contacts (Fig. 8d) to develop equivalent major faces (ESI S14‡) similar to 4 and 5.
:50 ratio), thus forming solid solution crystal 6 (Fig. 8). Crystal structure (space group P212121, Z′ = 1) determination revealed that both the molecules are present, however in a ratio of 90:10 with NO2 and Br occupying the same crystallographic site. Again 6 is isostructural to 4. Molecules are π-stacked (3.5 Å, 3.43 Å) along the short axis a (Fig. 8c). Disordered –CH2–OH groups then form the anticipated O–H⋯O–H⋯O–H (3.19 Å) hydrogen bonded catemer units. Such parallel units are stabilized by C–H⋯O (ester) (3.46 Å, 2.53 Å, 175.7°) interactions and are further 2D oriented in the bc-plane using C–H⋯O(NO2)/Br (3.35 Å, 2.54 Å, 145.7°; 3.66 Å, 2.88 Å, 142.8°; 3.72 Å, 3.08 Å, 127.6°; 3.74 Å, 3.14 Å, 124.2°) contacts (Fig. 8d) to develop equivalent major faces (ESI S14‡) similar to 4 and 5.
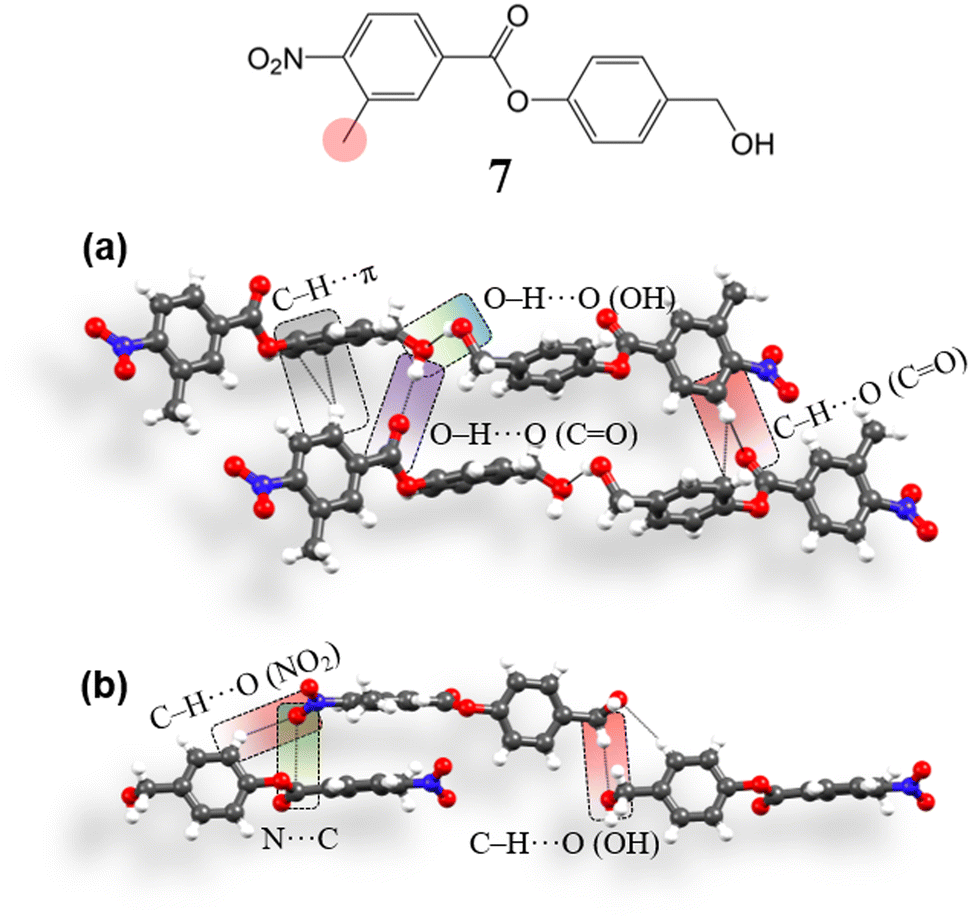
To compare and verify the effectiveness of the structural model, we have then tried to obtain a crystal structure deviating from the required type (i.e. opposed to 4, 5 and 6), using a system with a similar molecular structure. To obtain such a structure, we replaced one of the m-hydrogens by a methyl group in the carboxylic containing ring with an aim to introduce a geometrical perturbation at the site of the C–H⋯O (NO2) bifurcated synthon and thus have obtained compound 7 [4-(hydroxymethyl)phenyl 3-methyl-4-nitrobenzoate] (ESI S1‡). 7 crystallizes in the space group P1 (Z′ = 2). Aromatic rings are engaged in the formation of C–H⋯π (3.49 Å, 2.64 Å, 158.3°, 136.8°; 3.57 Å, 2.72 Å, 143.9°) interactions, perturbing the continuous stacking of rings (Fig. 9a). Further, the formation of the –CH2–OH mediated O–H⋯O–H⋯O–H catemer is perturbed. Here, two OH groups form an O–H⋯O (OH) (2.82 Å, 1.97 Å, 159.8°) hydrogen bond in that the second free hydrogen of the OH group interacts with C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, forming O–H⋯O (CO) (2.85 Å, 1.98 Å, 177.8°) interaction. The NO2 group in the structure interacts with phenolic site ring hydrogens forming C–H⋯O (NO2, ester) (3.5 Å, 2.72 Å, 139.5°; 3.42 Å, 2.6 Å, 144.3°; 3.41 Å, 2.61 Å, 137.9°; 3.48 Å, 2.66 Å, 144.2°; 3.31 Å, 2.66 Å, 126.5°) interactions. NO2 groups are also involved in orthogonal interactions (3.03 Å, 3.17 Å) (Fig. 9b), stabilizing the 3D interlocked packing. In summary, although the molecular structure of 7 is very similar to that of 4, 5 and 6, its crystal structure deviates from that of the latter (4, 5, and 6).
O, forming O–H⋯O (CO) (2.85 Å, 1.98 Å, 177.8°) interaction. The NO2 group in the structure interacts with phenolic site ring hydrogens forming C–H⋯O (NO2, ester) (3.5 Å, 2.72 Å, 139.5°; 3.42 Å, 2.6 Å, 144.3°; 3.41 Å, 2.61 Å, 137.9°; 3.48 Å, 2.66 Å, 144.2°; 3.31 Å, 2.66 Å, 126.5°) interactions. NO2 groups are also involved in orthogonal interactions (3.03 Å, 3.17 Å) (Fig. 9b), stabilizing the 3D interlocked packing. In summary, although the molecular structure of 7 is very similar to that of 4, 5 and 6, its crystal structure deviates from that of the latter (4, 5, and 6).
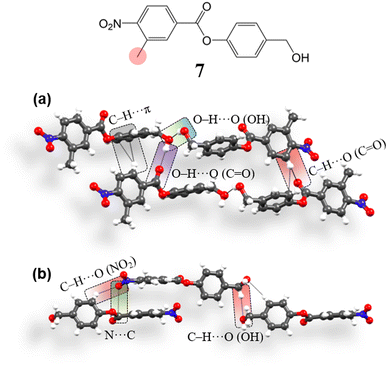 |
| | Fig. 9 (a and b) Intermolecular interactions involved in the crystal packing of 7. | |
Now to evaluate the mechanical behaviour, we performed mechanical testing on long acicular crystals of 5, 6 and 7 and compared with 4. The crystals of both 5 and 6 which are isostructural to 4 have undergone smooth elastic bending in forming a semicircular arc upon application of external stress (ESI S15 and Movie S5‡ for crystal 5). On the other hand, the crystals of 7 broke apart without showing any visible elastic/plastic deformation (ESI S15‡). Perhaps the rigid structure type with undesired packing/interactions in 7 does not allow stress dissipation (or structural buffering) as required for mechanical flexibility. The mechanical properties of crystals 5–7 are again quantitatively probed with the nanoindentation technique (ESI S5 and S16‡). Both 5 (E = 8.58 ± 0.45 GPa and H = 0.27 ± 0.02 GPa) and 6 (E = 8.10 ± 0.46 GPa and H = 0.21 ± 0.01 GPa) display similar behaviour to 4, whereas 7 (E = 12.70 ± 1.09 GPa and H = 0.37 ± 0.03 GPa) differs noticeably. These examples clearly signify the effectiveness of the demonstrated design strategy in transforming plastic crystals of 3 into elastic crystals using isostructural systems 4, 5 and 6, and thus proving the generality of our crystal engineering approach for fine tuning of mechanical properties.
Conclusions
The present work represents an example of a rational supramolecular synthesis, similar to molecular synthesis. Here, a mechanical property, namely plastic deformation in the given precursor crystal, is transformed into another distinct mechanical response, namely elastic deformation, within the equivalent structure/packing. Supramolecular synthons in the given structure are replaced by a molecular equivalent of a non-covalent bond in a way so that it can still maintain the parent structure type, i.e. geometrically similar but with altered chemical factors, which play a crucial role in deciding the mechanical properties outcome. The crystal thus obtained is 2D elastically bendable as well as elastically twistable. To the best of our knowledge, this is the first example of a systematic crystal engineering exercise using solely hydrogen bond interactions to obtain crystals with distinct mechanical properties. At the same time, there are no reports showing crystals with elastic twisting using mechanical stress. The generality of the design strategy was tested using multiple examples, including a multicomponent system. Crystals do not exhibit the desired property when the structural packing and interactions deviate from the required type, indicating the significance of the discussed design strategy/model. Crystals were quantitatively probed with the nanoindentation technique to find out their hardness, elastic modulus, and other characteristic features. It shows that these crystals are soft in nature and internally compressible to a certain extent which can further help in molecular adjustment during structural buffering for elastic deformation within isotropic packing. The present work shifts from the conventional structure–property paradigm of a similar structure with the same properties to a rare/unique case of a similar structure but with distinct macroscopic mechanical responses (i.e. Quadrant B) involving fully hydrogen bonded systems. Our work shows how one can use the molecular–supramolecular equivalent strategy as a designing tool to aim a particular property in crystal engineering practices.
Data availability
All experimental and characterization data in this article are available in the ESI‡.
Author contributions
S. M. performed all the experiments, computational calculations and assisted in the project design. S. S., C. M. R, and S. M. analyzed the results and co-wrote the manuscript. C. M. R. and S. S. designed the project. S. S. coordinated the project and prepared the initial draft.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
We are thankful to Prof. Horst Puschmann for helping to refine the crystal structures and Mr Manik Lal Maity for fruitful discussion during the synthesis of the molecules. S. M. thanks CSIR India for the fellowship (File No. 09/921(0183)/2017-EMR-I). C. M. R. thanks SERB for funding (No. CRG/2021/004992).
Notes and references
- S. Saha, M. K. Mishra, C. M. Reddy and G. R. Desiraju, Acc. Chem. Res., 2018, 51, 2957–2967 CrossRef CAS PubMed.
-
(a) P. Naumov, S. Chizhik, M. K. Panda, N. K. Nath and E. Boldyreva, Chem. Rev., 2015, 115, 12440–12490 CrossRef CAS PubMed;
(b) W. M. Awad, D. W. Davies, D. Kitagawa, J. M. Halabi, M. B. Al-Handawi, I. Tahir, F. Tong, G. Campillo-Alvarado, A. G. Shtukenberg, T. Alkhidir, Y. Hagiwara, M. Almehairbi, L. Lan, S. Hasebe, D. P. Karothu, S. Mohamed, H. Koshima, S. Kobatake, Y. Diao, R. Chandrasekar, H. Zhang, C. C. Sun, C. Bardeen, R. O. Al-Kaysi, B. Kahr and P. Naumov, Chem. Soc. Rev., 2023, 52, 3098–3169 RSC;
(c) R. Rai, B. P. Krishnan and K. M. Sureshan, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 2896–2901 CrossRef CAS PubMed.
-
(a) B. B. Rath and J. J. Vittal, Acc. Chem. Res., 2022, 55, 1445–1455 CrossRef CAS PubMed;
(b) L. Zhu, R. O. Al-Kaysi and C. J. Bardeen, J. Am. Chem. Soc., 2011, 133, 12569–12575 CrossRef CAS PubMed.
- Z. Lu, Y. Zhang, H. Liu, K. Ye, W. Liu and H. Zhang, Angew. Chem., Int. Ed., 2020, 59, 4299–4303 CrossRef CAS PubMed.
-
(a) S. Bhunia, S. Chandel, S. K. Karan, S. Dey, A. Tiwari, S. Das, N. Kumar, R. Chowdhury, S. Mondal, I. Ghosh, A. Mondal, B. B. Khatua, N. Ghosh and C. M. Reddy, Science, 2021, 373, 321–327 CrossRef CAS PubMed;
(b) S. Mondal, P. Tanari, S. Roy, S. Bhunia, R. Chowdhury, A. K. Pal, A. Datta, B. Pal and C. M. Reddy, Nat. Commun., 2023, 14, 6589 CrossRef CAS PubMed.
- Y. Cao and H. Li, Nat. Nanotechnol., 2008, 3, 512–516 CrossRef CAS PubMed.
- M. A. Garcia-Garibay, Angew. Chem., Int. Ed., 2007, 46, 8945–8947 CrossRef CAS PubMed.
- A. Facchetti, Chem. Mater., 2011, 23, 733–758 CrossRef CAS.
- S. Lv, D. M. Dudek, Y. Cao, M. M. Balamurali, J. Gosline and H. Li, Nature, 2010, 465, 69–73 CrossRef CAS PubMed.
-
(a) R. Jada, T. Feiler, A. Mondal, A. A. L. Michalchuk, C. M. Reddy, B. Bhattacharya, F. Emmerling and R. Chandrasekar, Adv. Opt. Mater., 2022, 2201518 Search PubMed;
(b) M. Annadhasan, D. P. Karothu, R. Chinnasamy, L. Catalano, E. Ahmed, S. Ghosh, P. Naumov and R. Chandrasekar, Angew. Chem., Int. Ed., 2020, 59, 13821 CrossRef CAS PubMed.
-
(a) H. Liu, Z. Bian, Q. Cheng, L. Lan, Y. Wang and H. Zhang, Chem. Sci., 2019, 10, 227–232 RSC;
(b) L. Lan, L. Li, P. Naumov and H. Zhang, Chem. Mater., 2023, 35, 7363–7385 CrossRef CAS;
(c) B. Tang, S. Tang, C. Qu, K. Ye, Z. Zhang and H. Zhang, CCS Chem., 2023, 5, 2348–2357 CrossRef CAS.
- S. Hayashi and T. Koizumi, Angew. Chem., Int. Ed., 2016, 55, 2701–2704 CrossRef CAS PubMed.
- S. Hayashi, F. Ishiwari, T. Fukushima, S. Mikage, Y. Imamura, M. Tashiro and M. Katouda, Angew. Chem., Int. Ed., 2020, 59, 16195–16201 CrossRef CAS PubMed.
- Y. Chen, Z. Chang, J. Zhang and J. Gong, Angew. Chem., Int. Ed., 2021, 60, 22424–22431 CrossRef CAS PubMed.
- R. Samanta, S. Das, S. Mondal, T. Alkhidir, S. Mohamed, S. P. Senanayak and C. M. Reddy, Chem. Sci., 2023, 14, 1363–1371 RSC.
- S. Das, A. Mondal and C. M. Reddy, Chem. Soc. Rev., 2020, 49, 8878–8896 RSC.
- B. Bhattacharya, A. A. L. Michalchuk, D. Silbernagl, N. Yasuda, T. Feiler, H. Sturm and F. Emmerling, Chem. Sci., 2023, 14, 3441–3450 RSC.
- K. Yadava, X. Qin, X. Liu and J. J. Vittal, Chem. Commun., 2019, 55, 14749–14752 RSC.
- S. Takamizawa and Y. Miyamoto, Superelastic organic crystals, Angew. Chem., Int. Ed., 2014, 53, 6970–6973 CrossRef CAS PubMed.
- S. Ghosh and M. K. Mishra, Cryst. Growth Des., 2021, 21, 2566–2580 CrossRef CAS.
- K. Chen, J. Wang, Y. Feng, H. Liu, X. Zhang, Y. Hao, T. Wang, X. Huang and H. Hao, J. Mater. Chem. C, 2021, 9, 16762–16770 RSC.
- I. S. Divya, S. Kandasamy, S. Hasebe, T. Sasaki, H. Koshima, K. Woźniak and S. Varughese, Chem. Sci., 2022, 13, 8989–9003 RSC.
- J. Lin, Y. Cao, Y. Liu, M. Li, Y. Chen, J. Zhou, S. Wu and J. Gong, Chem. Commun., 2023, 59, 619–622 RSC.
- S. Hu, M. K. Mishra and C. C. Sun, Chem. Mater., 2019, 31, 3818–3822 CrossRef CAS.
-
(a) M. Dakovic, M. Borovina, M. Pisacic, C. B. Aakeroy, Z. Soldin, B. M. Kukovec and I. Kodrin, Angew. Chem., Int. Ed., 2018, 57, 14801–14805 CrossRef CAS PubMed;
(b) M. Pisačić, I. Kodrin, A. Trninić and M. Đaković, Chem. Mater., 2022, 34, 2439–2448 CrossRef PubMed;
(c) S. P. Thomas, A. Worthy, E. Z. Eikeland, A. J. Thompson, A. Grosjean, K. Tolborg, L. Krause, K. Sugimoto, M. A. Spackman, J. C. McMurtrie, J. K. Clegg and B. Iversen, Chem. Mater., 2023, 35, 2495–2502 CrossRef CAS.
- C. M. Reddy, IUCrJ, 2019, 5, 505–506 CrossRef PubMed.
- M. K. Panda, S. Ghosh, N. Yasuda, T. Moriwaki, G. D. Mukherjee, C. M. Reddy and P. Naumov, Nat. Chem., 2015, 7, 65–72 CrossRef CAS PubMed.
- S. Bhandary, A. J. Thompson, J. C. McMurtrie, J. K. Clegg, P. Ghosh, S. R. N. K. Mangalampalli, S. Takamizawa and D. Chopra, Chem. Commun., 2020, 56, 12841–12844 RSC.
- R. Devarapalli, S. B. Kadambi, C. T. Chen, G. R. Krishna, B. R. Kammari, M. J. Buehler, U. Ramamurty and C. M. Reddy, Chem. Mater., 2019, 31, 1391–1402 CrossRef CAS.
- S. Saha and G. R. Desiraju, Chem. Commun., 2016, 52, 7676–7679 RSC.
- G. R. Krishna, R. Devarapalli, G. Lal and C. M. Reddy, J. Am. Chem. Soc., 2016, 138, 13561–13567 CrossRef CAS PubMed.
- A. Kidera, Y. Konishi, T. Ooi and H. A. Scheraga, J. Protein Chem., 1985, 4, 265–297 CrossRef CAS.
- S. Saha and G. R. Desiraju, J. Am. Chem. Soc., 2018, 140, 6361–6373 CrossRef CAS PubMed.
- S. Saha and G. R. Desiraju, Chem. Commun., 2018, 54, 6348–6351 RSC.
- G. M. J. Schmidt, Pure Appl. Chem., 1971, 27, 647–648 CrossRef CAS.
- E. J. Stollar and D. P. Smith, Essays Biochem., 2020, 64(4), 649–680 CrossRef CAS PubMed.
- S. K. Panigrahi and G. R. Desiraju, Natl. Acad. Sci. Lett., 2004, 27, 1–11 Search PubMed.
- S. Saha and G. R. Desiraju, Chem.–Eur. J., 2017, 23, 4936–4943 CrossRef CAS PubMed.
- S. Saha and G. R. Desiraju, J. Am. Chem. Soc., 2017, 139, 1975–1983 CrossRef CAS PubMed.
- A. Mukherjee and G. R. Desiraju, IUCrJ, 2014, 1, 49–60 CrossRef CAS PubMed.
- S. Saha and G. R. Desiraju, Chem. Commun., 2017, 53, 6371–6374 RSC.
-
(a) U. B. R. Khandavilli, M. Lusi and P. J. Frawley, IUCrJ, 2019, 6, 630–634 CrossRef CAS PubMed;
(b) C. C. Sun and D. J. W. Grant, Pharm. Res., 2004, 21, 382–386 CrossRef CAS PubMed;
(c) B. Pal, G. Raj, R. Jana, T. Moriwaki, G. D. Mukherjee, B. Mukhopadhyay and P. Naumov, Cryst. Growth Des., 2017, 17, 1759–1765 CrossRef;
(d) C. M. Reddy, IUCrJ, 2019, 6, 505–506 CrossRef CAS PubMed;
(e) S. Saha and G. R. Desiraju, J. Am. Chem. Soc., 2018, 140, 6361–6373 CrossRef CAS PubMed see the structure of 6B with the O–H⋯N–H⋯O–H catemer that produces the elastic crystal (Refcode: LIDPUM)..
- S. Varughese, M. S. R. N. Kiran, U. Ramamurty and G. R. Desiraju, Angew. Chem., Int. Ed., 2013, 52, 2701–2712 CrossRef CAS PubMed.
- W. C. Oliver and G. M. Pharr, J. Mater. Res., 2004, 19, 3–20 CrossRef CAS.
-
M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, P. R. Spackman, D. Jayatilaka and M. A. Spackman, CrystalExplorer17, University of Western Australia, 2017, http://hirshfeldsurface.net Search PubMed.
-
(a) S. Saha and G. R. Desiraju, Chem. Commun., 2021, 57, 4976–4978 RSC;
(b)
A. I. Kitaigorodskii, Mixed Crystals, Springer, Berlin, 1984 CrossRef;
(c) S. Ranjan, R. Devarapalli, S. Kundu, S. Saha, S. Deolka, V. R. Vangala and C. M. Reddy, IUCrJ, 2020, 7, 173–183 CrossRef CAS PubMed.
Footnotes |
| † This paper is dedicated to Prof. Gautam R. Desiraju. |
| ‡ Electronic supplementary information (ESI) available: Experimental details, computation, and movies. CCDC 2269655, 2311705–2311707. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc06462j |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
C Malla
Reddy
a,
C Malla
Reddy