
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Single-crystalline WSe2 nanoflakes as efficient electrocatalysts†
Veronika
Brune
 ,
Fabian
Hartl
,
Thomas
Fischer
,
Ziyaad
Aytuna
,
David
Patrun
,
David
Hillebrand
and
Sanjay
Mathur
*
,
Fabian
Hartl
,
Thomas
Fischer
,
Ziyaad
Aytuna
,
David
Patrun
,
David
Hillebrand
and
Sanjay
Mathur
*
University of Cologne, Institute of Inorganic Chemistry, Greinstraße 6, Köln 50939, Germany. E-mail: sanjay.mathur@uni-koeln.de
First published on 13th March 2024
Abstract
This study presents a novel single molecular source with covalently bonded ligand systems, featuring pre-existing W–Se bonds, for direct synthesis of single crystalline flakes of 2D tungsten diselenide (WSe2). Synthesis and structural characterization of [W{(SeC2H4)2NMe}2] expand the class of single- source precursors for metal sulfides and selenides of general formula [MIV{(XC2H4)2NMe}2] (MIV = Ti, Zr, Hf, Mo, W, Sn; X = S, Se). Comprehensive analysis including NMR, elemental analysis, and single crystal X-ray diffraction, provide insights into structural characteristics of [W{(SeC2H4)2NMe}2] in solution and solid state. Microwave-assisted solvothermal decomposition at 300 °C for 15 minutes yielded highly crystalline WSe2 flakes, confirmed by XRD, SEM, EDS, and TEM analyses. Reduced reaction time under mild conditions enables reproducible generation of layered 2D WSe2 materials for large-scale applications. Electrochemical studies reveal increased electrocatalytic activity, attributed to enhanced electrochemical surface area (ECSA, 67.3 cm2) observed in the as-prepared WSe2 flower-like nanostructures.
Introduction
Two-dimensional (2D) semiconductors, such as transition metal dichalcogenides (TMDCs; MX2, M = transition metal, X = S, Se) have attracted recent attention for their visible band gap, unlike graphene.1 Their van der Waals-layered structure offers unique chemical and physical traits, making them sought-after materials in optoelectronics and catalysis.2–4 Typically comprising three-atom-thick layers (X–M–X), few-layered two-dimensional TMDCs exhibit different physical properties compared to bulk counterparts, with direct and enlarged band gaps.5 Bulk MX2 materials have indirect bandgaps around 1 eV (MoS2: 1.2 eV, MoSe2: 1.1 eV, WS2: 1.0 eV, WSe2: 0.9 eV),5,6 while few-layered TMDC possess a direct and increased band gap (MoS2: 1.9 eV, MoSe2: 1.6 eV, WS2: 1.8 eV, WSe2: 1.5 eV).7,8 Tungsten based dichalcogenides, which are known for low toxicity, natural abundance and cost-effectiveness, hold scientific interest due to their superior optoelectronic properties9 particularly in applications like LEDs,10,11 solar energy harvesting,12–16 photodetectors,17,18 energy storage, and water splitting catalysts.19–23Despite extensive research on WSe2, its thin film production remains relatively limited, primarily due to reliance on complex precursors and vacuum-based deposition techniques. Unlike sulfur-based TMDCs such as MoS2 and WS2, there are fewer reports on WSe2 thin films, typically using precursors such as W(CO)6, WCl6 or WO3 in combination with a selenium source (H2Se, selenium powder). However, these methods often result in the incorporation of hetero-elements and formation of secondary phases.24–26 To address challenges such as stoichiometry control and impurity reduction our study introduces a novel single-source precursor (SSP) with preformed tungsten-selenide bonds. This molecular precursor was targeted for mild and efficient decomposition, bypassing issues linked to multiple precursors and ensuring controlled phase formation with specific stoichiometry, oxidation states, and purity.24–26 The optimal coordination of selenium-containing ligands around tungsten in the precursor stabilizes tungsten in the desired tetravalent (W(IV)) state, facilitating WSe2 formation in a single decomposition step under mild conditions.
This study presents the synthesis of flower-like WSe2 microspheres via microwave assisted solvothermal synthesis from a single molecular source. The W–Se precursor belongs to a precursor class containing a chelating tridentate, dianionic chalcogenolate-ligand [(−XC2H4)2NMe] (X = S, Se) in combination with M2+ or M4+ centers ([MIV{(XC2H4)2NMe}2] or [MII(XC2H4)2NMe], respectively with X = S, Se and MIV = Ti, Zr, Hf, Mo, W, Sn, MII = Sn).27–29 This unique precursor class, known for a well-defined decomposition pathway and good solubility in organic solvents, offers a new synthetic access to phase-pure crystalline WSe2 from a single molecular source.9,30 The results emphasize the need for carefully designed molecular precursors and optimized decomposition parameters to achieve high-quality, well-defined structures of WSe2 for harnessing the anisotropic properties of layered TMDCs.31 Despite various efforts on chemical synthesis of metal chalcogenides, few single molecular precursors exist for TMDCs, including WSe2. Notably, [(Et4N)2WSe4] and [Cat]2[WSe4] have been reported as single-source precursors for solvothermal decomposition (300 °C, 12 h) as well as through two-step dry thermal decomposition process (1st step: 300 °C at 450 sccm Ar flow for 30 min; 2nd step: annealing at 600 °C for 30 min at 450 sccm Ar flow) to yield particles and films, respectively.30 Moreover, [(Et4N)2WSe4] has been used in a heating-up method to produce 1T’WSe2 sheets on different substrates.32 However, the limited availability of stable and soluble molecular precursors continues to be a bottleneck in the chemically-controlled processing of WSe2 materials with high purity and crystallinity.
This report describes a novel molecular precursor [W{(SeC2H4)2NMe}2], demonstrating excellent solubility and stability in organic solvents, which can be efficiently transformed into WSe2 flakes through microwave-assisted solvothermal processing.
Results and discussion
This work is motivated by the need of swift and reproducible synthesis of phase pure WSe2 material via solution-based decomposition of a single molecular source. The molecular complex reported here is synthesized by the reductive cleavage of the diselenide bonds in cyclic selenium-containing ligand [{(SeC2H4)2Me}2]. A subsequent reaction with WCl6, resulted in the formation of the desired complex bis(2,2′-(methylazanediyl)bis(ethane-1-selenolate)) tungsten(IV) [W{(SeC2H4)2Me}2] (Scheme 1). The initial selenium containing ligand was synthesized following an optimized synthesis route established by Hartl et al. to isolate the air stable crystalline ligand [{(SeC2H4)2NMe}2].28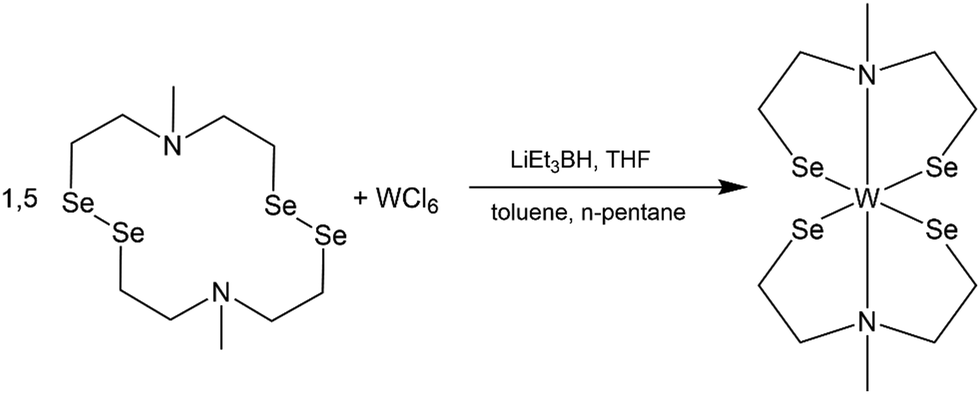 | ||
| Scheme 1 Reaction of cyclic selenium containing ligand [{(SeC2H4)2NMe}2] with tungsten hexa-chloride (WCl6) to the desired complex [W{(SeC2H4)2NMe}2]. | ||
The synthesis of metal complexes containing selenium-based organic ligands involves the intermediary step of reductive cleavage of Se–Se bond due to the instability of unsaturated selenols.33 The reduction of diselenide rings to corresponding selenols enable their in situ reaction with metal centers, which represents a well-known synthetic strategy.34 To ensure controlled transformations, all reaction steps were conducted under an inert atmosphere, preventing the uncontrolled oxidation of the tungsten center or the uncontrolled re-oxidation of the reduced ligand system.
The resulting dark product was analyzed using multinuclear NMR spectroscopy analysis including 1D and 2D NMR experiments (1H, 13C, 1H, 1H COSY, 1H, 13C HSQC, 1H, 13C HMBC). The spectral data confirmed the coordination of two dianionic tridentate ligands to the same tungsten center. The proton NMR spectrum exhibited eight inequivalent proton signals in an integrative ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2:6:2:2:4:2 corresponding to five inequivalent carbon signals suggesting a rigid tungsten complex in solution (Fig. S1, ESI†). The split proton signals, result from the fixed orientation of the protons towards (endo protons, highlighted in red) or away (exo protons, highlighted in green) from the metal center, which allowed the identification of 22 proton (Fig. 1 and 2b) signals but only five non-equivalent carbon signals, due to the symmetrical arrangement of the ligands. A similar non-fluxional behavior in solution has been reported previously within this precursor class.27,29 The methyl-group protons appeared as a singlet signal at 3.50 ppm, correlated with the carbon signal at 59.1 ppm (Fig. S1, ESI†). The CH2-protons exhibit complex multiplets, mirroring the comparable structure observed in both solution and the solid-state as shown in the molecular structure presented in Fig. 1. Notably, the CH2-protons appearing at 3.13–3.18 ppm and 2.65–2.75 ppm, correlated with the high-field carbon signal at 39.4 ppm. The CH2-protons with an integrative proton ratio of four at 2.45 ppm correlate with the carbon signal observed at 66.8 ppm. The multiplets with a chemical shift of 4.08–4.18 ppm and 3.50–3.58 ppm relate to the 13C chemical shift of 74.5 ppm. The carbon signal displaying the most down-field shift corresponds to the CH2-protons at chemical shift of 4.45–4.52 ppm and 2.18–2.29 ppm.
:2:2:6:2:2:4:2 corresponding to five inequivalent carbon signals suggesting a rigid tungsten complex in solution (Fig. S1, ESI†). The split proton signals, result from the fixed orientation of the protons towards (endo protons, highlighted in red) or away (exo protons, highlighted in green) from the metal center, which allowed the identification of 22 proton (Fig. 1 and 2b) signals but only five non-equivalent carbon signals, due to the symmetrical arrangement of the ligands. A similar non-fluxional behavior in solution has been reported previously within this precursor class.27,29 The methyl-group protons appeared as a singlet signal at 3.50 ppm, correlated with the carbon signal at 59.1 ppm (Fig. S1, ESI†). The CH2-protons exhibit complex multiplets, mirroring the comparable structure observed in both solution and the solid-state as shown in the molecular structure presented in Fig. 1. Notably, the CH2-protons appearing at 3.13–3.18 ppm and 2.65–2.75 ppm, correlated with the high-field carbon signal at 39.4 ppm. The CH2-protons with an integrative proton ratio of four at 2.45 ppm correlate with the carbon signal observed at 66.8 ppm. The multiplets with a chemical shift of 4.08–4.18 ppm and 3.50–3.58 ppm relate to the 13C chemical shift of 74.5 ppm. The carbon signal displaying the most down-field shift corresponds to the CH2-protons at chemical shift of 4.45–4.52 ppm and 2.18–2.29 ppm.
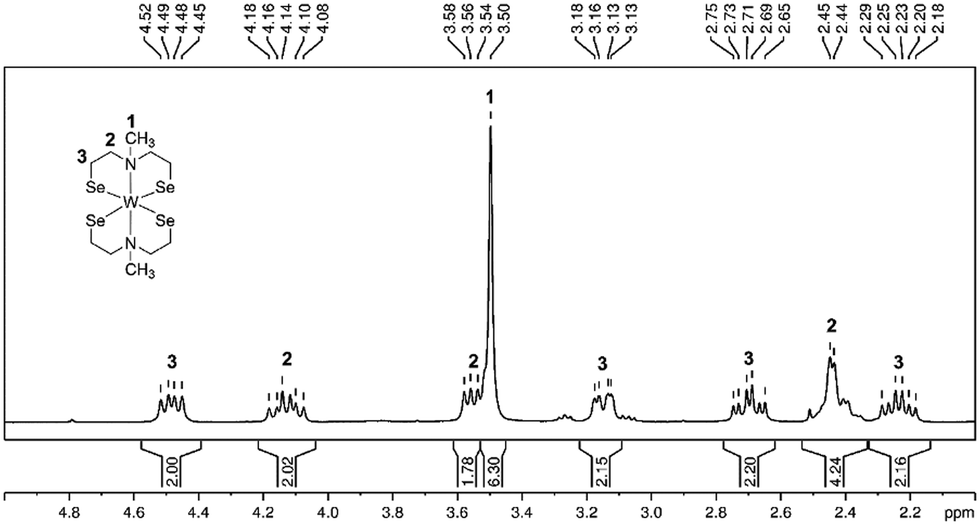 | ||
| Fig. 1 1H NMR spectrum (CDCl3, rt) of complex [W{(SeC2H4)2NMe}2] with proton integration and assignment of protons to the molecular structure. | ||
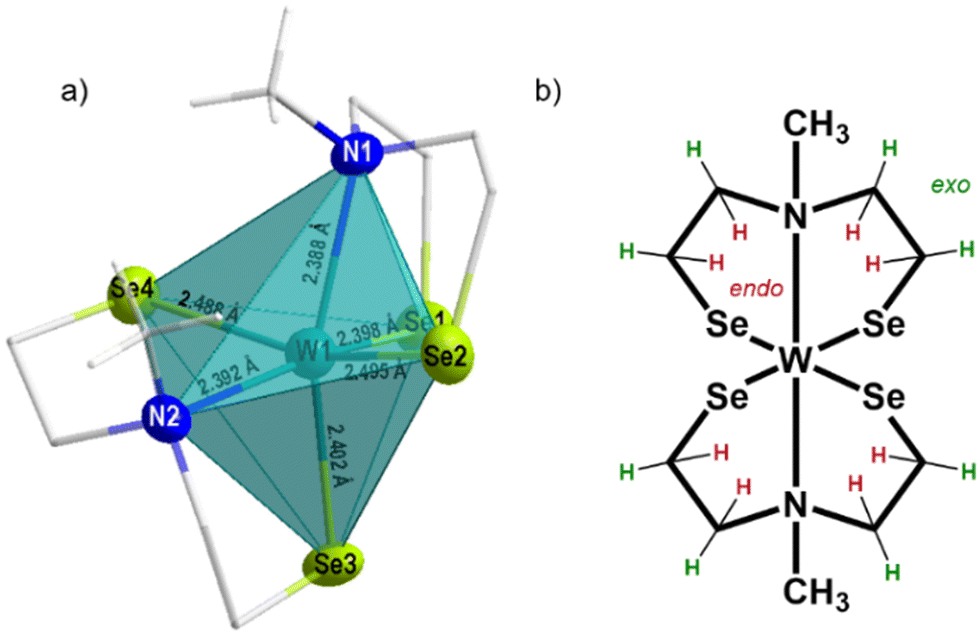 | ||
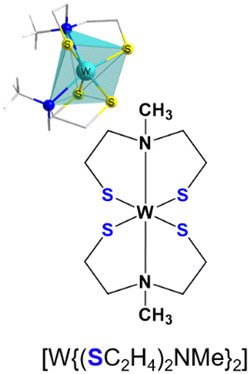
| Fig. 2 Molecular structure of [W{(SeC2H4)2NMe}2], (a) showing the distorted octahedral coordination and bond length around the tungsten center, (b) precursor chemical structure showing in total 22 protons, highlighted are the exo (green) and end (red) oriented protons as identified by NMR analysis. | ||
The elemental analysis of the tungsten precursor exhibited close agreement between the calculated and measured values for C, H and N contents. Furthermore, the solid-state structure was elucidated by single crystal X-ray structure analysis of black crystals grown from a chloroform/n-pentane solution revealing a triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , with similar cell parameters as the isotypical compounds ([M{(XC2H4)2NMe}2]; M = TiIV, ZrIV, HfIV, WIV, MoIV, SnIV, X = S, Se). The molecular structure as shown in Fig. 2a confirmed the coordination of two chelating dianionic ligands to the same metal center in a tridentate fashion, resulting in a distorted octahedral coordination, consequently stabilizing tetravalent oxidation state for the tungsten center.
, with similar cell parameters as the isotypical compounds ([M{(XC2H4)2NMe}2]; M = TiIV, ZrIV, HfIV, WIV, MoIV, SnIV, X = S, Se). The molecular structure as shown in Fig. 2a confirmed the coordination of two chelating dianionic ligands to the same metal center in a tridentate fashion, resulting in a distorted octahedral coordination, consequently stabilizing tetravalent oxidation state for the tungsten center.
The methyl groups of the coordinated ligands point towards each other, due to the steric demand of the electron lone pairs present on the selenium atoms. This particular arrangement of the two coordinated ligands contributes to the rigidity of the molecular framework in solution (see NMR analysis) and the solid-state (Fig. 2). The Se–W bond length, ranging from 2.4–2.5 Å, differs from analogous sulfur compound [W{(SC2H4)2NMe}2] (2.2–2.4 Å)29,35–37 however they are in accordance with other reported W–Se bond lengths.31 The W–N bond lengths (2.388(5) and 2.391(6) Å) are within the range of literature-reported values (1.92–2.49 Å)38,39 and less elongated, when compared to the sulfur containing structure (2.375(6) and 2.377(6) Å) (Table 1). The triangular planes formed by the tridentate coordination of each ligand (Fig. S2a and b, ESI†) display a twist of 73.5°, slightly larger compared to the sulfur containing compound due to the increased repulsion of sterically demanding electron lone pairs at selenium. To investigate the thermal decomposition behavior of the molecular precursor, TG-DSC measurements were conducted (Fig. S6, ESI†), revealing a multistep decomposition process. The theoretical mass loss of approximately 50%, indicative of the formation of desired WSe2 material was observed at a temperature of around 410 °C, as evidenced by an endothermic peak in the DSC curve. However, no plateau formation was observed in the TG curve of the molecular precursor, indicating its limited stability at higher temperatures.
| Cell parameter | Complex | |
|---|---|---|
| [W{(SeC2H4)2NMe}2] | [W{(SC2H4)2NMe}2]29 | |
| Empirical formulas | WSe4C10H22N2 | WS4C10H22N2 |
| Formula weight | 669.98 g mol−1 | 482.38 g mol−1 |
| Crystal system | Triclinic | |
| Space group |
P |
|
| R (int) | 0.0353 | 0.1026 |
| Unit cell dimensions | a = 7.9788 (3) Å | a = 7.8311 (7) Å |
| b = 9.7801 (4) Å | b = 9.6503 (8) Å | |
| c = 11.6660 (5) Å | c = 11.2664 (1) Å | |
| α = 80.387 (3)° | α = 82.455 (7)° | |
| β = 82.715 (3)° | β = 84.507 (7)° | |
| γ = 65.910 (3)° | γ = 66.593 (6)° | |
| Volume | 817.64(6) Å3 | 762.1(2) Å3 |
| Z | 2 | 2 |
|
|
|
The complex [W{(SeC2H4)2NMe}2] was tested as a precursor to obtain crystalline WSe2 structures through microwave-assisted solvothermal decomposition. Dissolving the precursor in dry NMP (N-methyl-2-pyrrolidone) produced a dark brown solution that was solvothermally treated for 15 min at 300 °C, yielding a black precipitate which was separated from soluble byproducts by centrifugation. The precipitated product was washed with chloroform and ethanol in several subsequent dispersion-centrifugation cycles, followed by drying at reduced pressure (50 °C for 30 min). The XRD diffraction data confirmed the formation of WSe2 (ref. ICDD 00-038-1388; Fig. 3a, cyan pattern) with low crystallinity. Further, calcination at 500 °C for 30 min under reduced pressure (Fig. 3a, black pattern) and 700 °C for 7 h under N2 (Fig. 3a, blue pattern) enhanced the crystallinity of WSe2 flakes (Fig. 3a). Microscopic analysis validated the production of interconnected thin flake-like WSe2 structures (Fig. 3b and 4b).
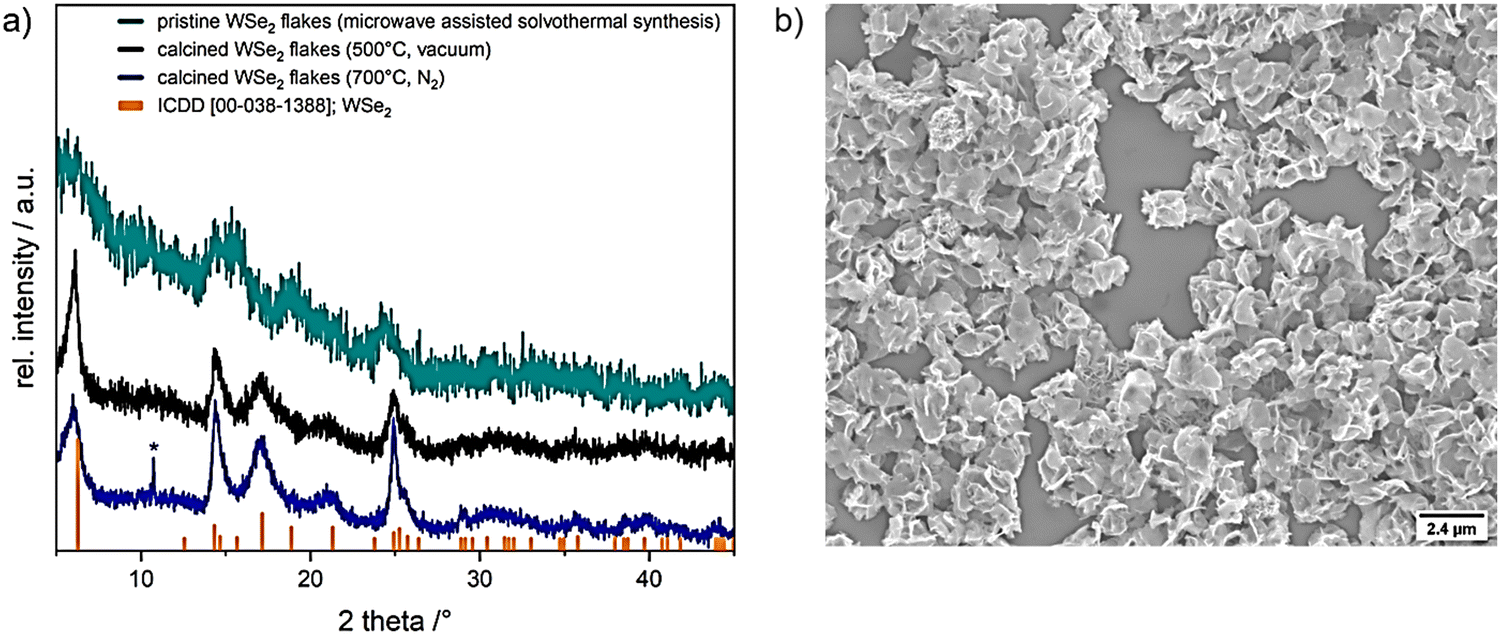 | ||
| Fig. 3 (a) XRD patterns of microwave assisted solvothermal decomposition of [W{(SeC2H4)2NMe}2] precursor in NMP (cyan) and calcined WSe2 flakes in vacuum at 500 °C (for 30 min (black)) and under nitrogen atmosphere at 700 °C (for 7 h (blue)), reference WSe2 (ICDD 00-038-1388) is shown in orange, *showing a peak which could assume the formation of WO2 (ICDD 00-082-0728, WO2; Fig. S5, ESI†). (b) SEM micrograph of resulting in WSe2 microstructures. | ||
 | ||
| Fig. 4 (a) SEM image of WSe2 with flower-like arrangement of 2D flakes, (b) SEM image of dispersed WSe2 flakes, (c)–(f) elemental mapping showing homogeneous distribution of tungsten and selenium located on as-prepared WSe2 structures. | ||
The X-ray diffractogram of the product obtained from microwave-assisted solvothermal decomposition of [W{(SeC2H4)2NMe}2] showed the incipient crystallization of WSe2 (ICDD collection code: 00-038-1388, Fig. 3a, cyan pattern). To improve the crystallinity of as-obtained WSe2 flakes, calcination was performed in vacuum (at 500 °C) and under N2 atmosphere (at 700 °C). The broad diffraction peaks and average full width at half maximum (FWHM) indicate small crystallite sizes, estimated to be about 10 nm using the Scherrer formula.40 A small amount of tungsten oxide phase possibly formed by the thermal oxidation of the selenide phase during calcination was detected with minor intensity in the XRD pattern after calcination at 700 °C (blue pattern, Fig. 3a). The partial oxidation of as-synthesized WSe2 flakes could not be avoided under the given experimental conditions and sample transfer set-up. This limited stability at higher temperatures of decomposition product from molecular precursor have been observed in the previous reports on isotypic complexes of zirconium and hafnium.27 Further structural analysis and an in-depth microscopic analysis of pristine WSe2 flakes (SEM and HRTEM) confirmed the formation of nanocrystalline flakes of regular morphology. The 2D flake-like structure observed in this study has been reported for WSe2 prepared by solvothermal syntheses.41–43
As shown in Fig. 5, the isolated WSe2 flakes comprises a few-layered material, packed in a disordered manner with minimal stacking.41,42 Using EDS analysis, confirmation of the correct stoichiometry of tungsten and selenium was established by capturing low-energy electrons emitted from the sample's surface following the interaction with the primary electron beam, utilizing secondary electron (SE) detectors. The measurement reveals the expected metal–ligand ratio of 1:2, confirming the formation of stoichiometric WSe2 (Fig. 4c and d, Fig. S3, ESI†) across a wide area of the sample. Point EDS-spectra further supported the specific compositional ratio between tungsten and selenium to be 1:2 (WSe2, Fig. 4c and d, (Fig. 4e and f showing the main O and C contamination resulting from the substrate) Fig. S3 and S4, ESI†).
 | ||
| Fig. 5 (a) HRTEM image of pristine WSe2 flower like structures with interplanar distance of 0.67 nm (a) corresponding to the (002) plane. Fast Fourier transformation (FFT) of selected area yields highly ordered hexagonal pattern (inset) corresponding to the (100) (II) and (110) plane (III), respectively. (b) Hexagonal SAED pattern confirms the high crystallinity of WSe2 flakes. | ||
HRTEM analysis of WSe2 nanostructures provided detailed insight into morphology and crystallinity of individual flakes. The results evidently confirm the layered structure of WSe2 and its hexagonal lattice (Fig. 5). Multilayers reveal an interlayer distance of 0.67 nm, corresponding to the (002) lattice plane (d = 0.648 nm). Additionally, layers oriented perpendicular to the optical axis (highlighted area in Fig. 5a) exhibit a hexagonal lattice arrangement, validated by fast Fourier transformation (FFT) (Fig. 5a inset). The pattern compares well with the (001) and (110) lattice plane distances of WSe2 (II: 0.28 nm and III: 0.16 nm). Moreover, selected area electron diffraction (SAED) of isolated flakes confirm the single crystalline nature of WSe2 platelets (Fig. 5b), with the diffraction pattern correlating with the WSe2 structure.
The catalytic potential of WSe2 flakes in the oxygen evolution reaction (OER) was evaluated utilizing a nickel foam (1 cm x 1 cm) coated with a mixture of pristine WSe2 flakes, Super P®Li (a high purity and structured carbon black powder) and Nafion (acting as a binder) as anode in the water-splitting reaction. Typically, OER experiences a high overpotential and sluggish kinetics due to oxygen-containing intermediate species that bind onto the active catalytic surface.44 Using linear sweep voltammetry (LSV) at a scan rate of 2 mV s−1 (Fig. 6a), a bare nickel foam with a platinum counter electrode against calomel and anode without WSe2 catalyst (only Super P®Li and Nafion on nickel foam) served as the references. The overpotential, calculated at a current density of 10 mA cm−2, revealed the performance of WSe2 composite catalyst against the two references demonstrating higher current density and superior electrochemical performance (Fig. 6a). The WSe2 catalyst showed an overpotential of 210 mV, which was 79 mV lower than that of bare nickel foam (Ni-foam, 289 mV),45,46 nickel foam containing Super P®Li and Nafion (Ni-foam-SN) and in comparison to values reported in the literature (Fig. 6a).14,16 These results greatly demonstrate the faster OER kinetics of prepared WSe2 nanocrystals.
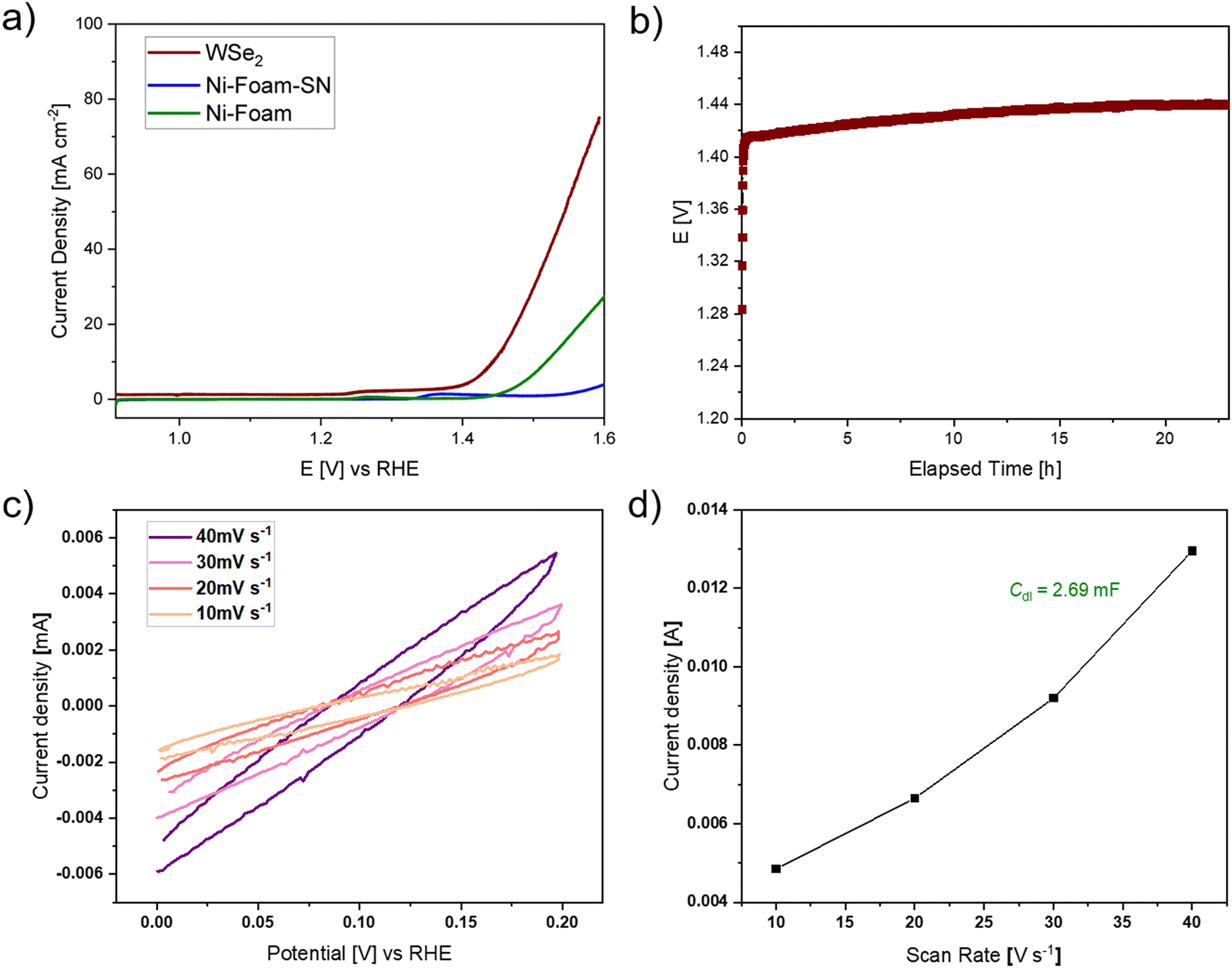 | ||
| Fig. 6 (a) Linear sweep voltammetry of WSe2 flower-like structures with Super P®Li and Nafion on nickel foam (WSe2, red curve) in comparison to only Super P®Li and Nafion on nickel foam (Ni-foam-SN, blue curve) and bare nickel foam (Ni-foam, green curve) used as references (b) chronopotentiometry of WSe2 at a current density of 10 mA cm−2, (c) cyclic voltammetry curves at different scan rates from 10–40 mV s−1 and (d) current density vs. scan rate for calculation of the double-layer capacitance (Cdl = 2.69 mF). | ||
The durability of the WSe2 catalyst under alkaline conditions was investigated using chronopotentiometry measurements for >22 hours. The overpotential of 210 mV remained stable during this period (Fig. 6b) showing a good stability over time. Electrochemical active surface area (ECSA [cm2]), characterizing the electroactive surface area of the catalyst, was determined by cyclic voltammetry measurements in a voltage range of 0 to 0.2 V vs. RHE at varying scan rates (10–40 mV s−1). Fig. 6c illustrates the obtained CV curves, from which the current density was extracted at a potential of 0.1 V vs. RHE. The ECSA is calculated from the specific capacitance (Cs = 0.04 mF cm−2) of used alkaline system and the double-layer capacitance (Cdl, which is calculated from measured CV curves, Fig. 6d). The ECSA of the WSe2 electrocatalyst was found to be 67.3 cm2, showing a higher active surface area compared to reported values (34.86 cm2, 41.65 cm2) for WSe2,44,47 which facilitates the binding of electroactive oxygen-based intermediate species on the catalyst surface.44,48
Conclusions
Easy availability of high-purity nanophase materials exhibiting specific properties, tailored shape and microstructure is essential for transforming their scientific potential into innovative advanced functional materials and efficient devices. Extensive research in the synthesis and processing of inorganic nanomaterials have resulted in a wealth of data, however, synthetic protocols to validate reproducible scaled-up production of ordered nanostructures, are still elusive. Application of tailored molecular precursors offer potential for addressing the prevailing challenges of energy turnaround of materials processing methods, achieving high atom-economy for resource-efficient use of raw materials and reduction of consequent purification and post-synthesis treatment steps.49–53 The approach reported here for the fabrication of high purity WSe2 nanostructures by low-temperature transformation of a single molecular precursor emphasizes the power of synthesis in controlled growth of nanostructured materials. Leveraging the solubility in common organic solvents and stability of this precursor, microwave-assisted solvothermal decomposition enabled a rapid and reproducible material processing of crystalline WSe2 flakes, as verified by chemical and structural analysis. Through the systematic expansion of the molecular library of metal-chalcogenide complexes reliable and efficient chemical pathway for molecule-based materials is feasible, which however demands synthesis and characterization of new precursors for accessing kinetically stabilized compositions.54,55 Nanoflakes of single crystalline WSe2 exhibited notably enhanced electrochemical performance in terms of overpotential, active surface area, and stability, when compared to prior studies.Experimental
All manipulations of air- and moisture-sensitive materials were carried out under a nitrogen atmosphere using Stock-type all-glass assemblies. All solvents and reactants were used without further purification except when specified. Tungsten hexachloride (99.9%) was purchased from ABCR Chemicals, potassium selenocyanate (98.5%) was purchased from Alfa Aesar and LiEt3BH (1 M in THF) was purchased from Merck, N-methyl-2-pyrrolidone (NMP) was purchased from TCI. Super P®Li was purchased from TIMCAL and Nafion D-521 Dispersion (5 w/w% H2O/iso-propanol) was used as a binder and purchased from Alfa Aesar. Toluene, n-heptane, n-pentane, and tetrahydrofuran (THF), which were used for the syntheses of metal complexes, were dried by standard methods with appropriate desiccating reagents and distilled prior to their use.56 Microwave-assisted thermal decomposition experiments have been performed with a Discover 2.0 microwave system from CEM using a fixed power program. Typically, 30 mg of molecular precursor were dissolved in 3.5 mL NMP and flushed with argon. The reaction mixture was rapidly heated to 300 °C and maintained for 15 min. The black precipitate was removed from the supernatant by centrifugation. The precipitate was washed three times by dispersing it in chloroform, sonicating the dispersion, and removing the supernatant by either decantation or centrifugation. Pristine WSe2 flakes were dried under reduced pressure (10−2 Torr) at 50 °C for 0.5 h using a vacuum oven.Data collection for single crystal X-ray structural elucidation was performed on STOE IPDS II/2T diffractometer using graphite-monochromatized Mo Kα radiation (0.71073 Å). The data were corrected for Lorentz and polarization effects. A numerical absorption correction based on crystal-shape optimization was applied for all data. The programs used in this work are STOE's X- Area, including X-RED and X-Shape for data reduction and absorption correction, SIR-92 and SHELXL-2014 for structure solution, and SHELXL for structure refinement.57–62 CCDC 2307367 contain the supplementary crystallographic data for [W{(SeC2H4)2NMe}2].† NMR spectra were recorded either on a Bruker Avance II 300 spectrometer; chemical shifts are quoted in part per million relative to external TMS (1H and 13C). Powder X-ray diffraction (XRD) patterns were measured on a Stoe Stadi-P diffractometer equipped with a Mythen 1 K detector using Mo Kα radiation (λ = 0.71073 Å). Samples were measured in sealed glass capillaries with a diameter of 0.3 mm. The size and morphology of deposited films were analyzed using a Zeiss Sigma 300 VP RISE, a field emission scanning electron microscope (SEM) with integrated energy-dispersive X-ray spectroscopy (EDS) measurement instruments from Oxford Instruments Xplore 30, and a JEM-2200FS transition electron microscope (TEM), operated at 200 kV with an UHR pole piece and in-column filter. The acquisition of scanning electron microscopy (SEM) images is enhanced by the utilization of both Inlens and secondary electron (SE) detectors, offering distinct advantages in imaging and analytical capabilities. Inlens detectors excel in acquiring high-quality, detailed images with improved contrast and resolution, especially when imaging topographical features and surface morphology.
Synthesis of 5,12-dimethyl-1,2,8,9-tetraselena-5,12-diazacyclotetradecane, [{(SeC2H4)2NMe}2]
Caution! N,N-Bis(chloroethyl)methylamine hydrochloride is toxic and carcinogenic. Resorption of CN-formed during the reaction can be deadly. Exposure, including skin contact, must be avoided. This substance and any contaminated surfaces and glassware must be handled with the necessary precautions and safety standards.In a three-necked round bottom flask, under nitrogen atmosphere, KSeCN (3.11 g, 21.6 mmol) was dissolved in MeOH (30 mL). N,N-bis(chloroethyl)methylamine (1.99 g, 10.2 mmol) dissolved in MeOH (20 mL) was added slowly through a dropping funnel under continuous stirring. The red suspension was stirred at 80 °C for 5 hours. After that point, the reaction mixture was not sensitive against oxygen or ambient moisture. The grey suspension was cooled down to room temperature and KOH (5.81 g, 103.6 mmol) was added portion-wise under water bath cooling. The reaction mixture was found to turn yellowish and was stirred at room temperature for 16 h. Chloroform (60 mL) and water (60 mL) were added to the suspension, forming a yellow layer. The layers were separated, and the aqueous layer was extracted with chloroform. The combined organic phases were washed with water and dried over MgSO4. The excess of organic solvent was removed under reduced pressure. In a 100 mL round bottom flask, the yellow crude product was dissolved in minimum amount of CHCl3 and the remaining volume was made up by adding methanol. The flask was stored overnight at −30 °C. After decantation, selenium containing ligand (1.51 g, 3.11 mmol, 61%) was obtained as a light-yellow crystalline product.
1H NMR (300 MHz, CDCl3) δ [ppm]: 3.11 (t, 8H, J = 8.2 Hz, –N–CH2–CH2–Se–), 2.78 (t, 8H, J = 8.2 Hz, –N–CH2–CH2–Se–), 2.31 (s, 6H, –N(CH3)).
13C NMR (75 MHz, CDCl3) δ [ppm]: 58.2 (–N–CH2–CH2–Se–), 43.7 (–N(CH3)), 27.9 (–N–CH2–CH2–Se–).
Synthesis of bis(2,2′-(methylazanediyl)bis(ethane-1-selenolate)) tungsten(IV), [W{(SeC2H4)2NMe}2]
The selenium containing ligand (441 mg, 0.908 mmol) was dissolved in dry THF (15–20 mL) under N2 atmosphere. To the resulting yellow solution, LiHBEt3 (3.61 mL, 1 M in THF) was added dropwise until full discoloration occurred and gas evolution seized. Following that WCl6 (240 mg, 0.605 mmol) was added, when the colorless solution turned immediately into a deep red-brownish suspension. The wine-red reaction mixture was stirred at room temperature for 60 minutes and the solvent was removed under reduced pressure. The solid residue was extracted with CHCl3 (2 × 10 mL) and decantated into a separate flask. The volume of the solvent was reduced by 50% and n-pentane (50 mL) was added to the dark precursor solution. A dark brown precipitate was formed. The flask was stored in the freezer overnight to complete the precipitation of the product. After decantation, the residue was washed twice with n-pentane. After drying under reduced pressure, the final compound (269 mg, 401 mmol, 66%) was obtained as a black powder. Calc. for [W{(SeC2H4)2NMe}2] (MW 670): C, 17.9, H, 3.3, N, 4.1. Found: C, 19.2, H, 3.5, N, 3.6.1H NMR (300 MHz, CDCl3) δ [ppm]: 4.48 and 2.23 (m, 2H and 2H, –N–CH2–CH2–Se–), 4.13 and 3.55 (m, 2H and 2H, –N–CH2–CH2–Se–), 3.49 (s, 6H, –N(CH3)), 3.16 and 2.69 (m, 2H and 2H, –N–CH2–CH2–Se–), 2.49 (m, 4H, –N–CH2–CH2–Se–).
13C NMR (75 MHz, CDCl3) δ [ppm]: 100.6 (–N–CH2–CH2–Se–), 74.0 (–N–CH2–CH2–Se–), 66.6 (–N–CH2–CH2–Se–), 59.1 (CH3), 39.0 (–N–CH2–CH2–Se–).
Electrochemical testing
Pristine WSe2 flakes were used in the electrochemical measurements reported here. All electrochemical studies were performed using a three-electrode setup containing a platinum coil as a counter electrode and a calomel electrode (Hg/Hg2Cl2, 3 M KCl) in 1 M KOH electrolyte as the reference. The electrode preparation was performed by homogeneous dispersion of 4.0 mg of as-prepared WSe2 flakes, 0.5 mg Super P®Li (high purity and structured carbon black powder with a moderate surface area) as conductive carbon and 100 μl 5 wt% Nafion solution (5 w/w% H2O and iPrOH 1:1) drop-coated onto the nickel foam functioning as current collector, resulting in an active mass loading of 4 mg cm−2 in the prepared electrode. Prior to electrochemical investigation, cyclic voltammetry was measured for 5 cycles in a voltage range of 0–0.7 V with a scan rate of 0.05 V s−1.
Author contributions
Special thanks are due to Ms Dr Corina Hegemann for help with the single crystal X-ray diffraction studies and the structure refinement. Mr Dirk Pullem is acknowledged for performing elemental analysis, Ms Silke Kremer for single-crystal analysis, Mr Daniel Moog for X-ray powder diffraction analysis and Mrs Sophie Lüggert for her synthetic support.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors gratefully acknowledge the financial and infrastructural support provided by the University of Cologne. The authors thank the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG) for the generous financial support of the scientific personnel and support through large equipment grant for the Transmission Electron Microscope (grant no. 259830545) and Raman Imaging Scanning Electron Microscope (grant no. 450610390).References
- Y. Guo and J. Robertson, Appl. Phys. Lett., 2016, 108, 233104 CrossRef
.
- H. Jiang, J. Phys. Chem. C, 2012, 116, 7664–7671 CrossRef CAS
- K. Kadiwala, E. Butanovs, A. Ogurcovs, M. Zubkins and B. Polyakov, J. Cryst. Growth, 2022, 593, 126764 CrossRef CAS
- S. Chandrasekaran, L. Yao, L. Deng, C. Bowen, Y. Zhang, S. Chen, Z. Lin, F. Peng and P. Zhang, Chem. Soc. Rev., 2019, 48, 4178–4280 RSC
- C. R. Ryder, J. D. Wood, S. A. Wells and M. C. Hersam, ACS Nano, 2016, 10, 3900–3917 CrossRef CAS PubMed
- S. Tongay, J. Zhou, C. Ataca, K. Lo, T. S. Matthews, J. Li, J. C. Grossman and J. Wu, Nano Lett., 2012, 12, 5576–5580 CrossRef CAS PubMed
- D. Muoi, N. N. Hieu, H. T. T. Phung, H. V. Phuc, B. Amin, B. D. Hoi, N. V. Hieu, L. C. Nhan, C. V. Nguyen and P. T. T. Le, Chem. Phys., 2019, 519, 69–73 CrossRef CAS
- J. Gusakova, X. Wang, L. L. Shiau, A. Krivosheeva, V. Shaposhnikov, V. Borisenko, V. Gusakov and B. K. Tay, Phys. Status Solidi A, 2017, 214, 1–7 CrossRef
- V. Sethi, D. Runacres, V. Greenacre, L. Shao, A. L. Hector, W. Levason, C. H. de Groot, G. Reid and R. Huang, J. Mater. Chem. A, 2023, 11, 9635–9645 RSC
- O. Del Pozo-Zamudio, A. Genco, S. Schwarz, F. Withers, P. M. Walker, T. Godde, R. C. Schofield, A. P. Rooney, E. Prestat, K. Watanabe, T. Taniguchi, C. Clark, S. J. Haigh, D. N. Krizhanovskii, K. S. Novoselov and A. I. Tartakovskii, 2D Mater., 2020, 7, 031006 CrossRef CAS
- C. Wang, F. Yang and Y. Gao, Nanoscale Adv., 2020, 2, 4323–4340 RSC
- F. Bozheyev and K. Ellmer, J. Mater. Chem. A, 2022, 10, 9327–9347 RSC
- M. D. Haque, M. H. Ali and A. Z. M. T. Islam, Sol. Energy, 2021, 230, 528–537 CrossRef CAS
- C.-H. Chiang, Y.-C. Yang, J.-W. Lin, Y.-C. Lin, P.-T. Chen, C.-L. Dong, H.-M. Lin, K. M. Chan, Y.-T. Kao, K. Suenaga, P.-W. Chiu and C.-W. Chen, ACS Nano, 2022, 16, 18274–18283 CrossRef CAS PubMed
- Y. Wang, B. Ren, J. Zhen Ou, K. Xu, C. Yang, Y. Li and H. Zhang, Sci. Bull., 2021, 66, 1228–1252 CrossRef CAS PubMed
- P. M. Pataniya, X. Yang, B. Li, D. Kannichankandy and C. K. Sumesh, Int. J. Energy Res., 2022, 46, 12073–12081 CrossRef CAS
- S. Ghosh, A. Varghese, K. Thakar, S. Dhara and S. Lodha, Nat. Commun., 2021, 12, 3336 CrossRef CAS PubMed
- Y. Zhang, L. Wang, Y. Lei, B. Wang, Y. Lu, Y. Yao, N. Zhang, D. Lin, Z. Jiang, H. Guo, J. Zhang and H. Hu, ACS Nano, 2022, 16, 20937–20945 CrossRef CAS PubMed
- L. Wei, Y. Su, Y. Ma, Y. Gu, Y. Qin, X. Wu, Y. He, X. Li, Y. Peng and Z. Deng, Chem. Eng. J., 2022, 448, 137591 CrossRef CAS
- J. Hu, J. Li, Z. Wei and S. Zhang, J. Solid State Electrochem., 2022, 26, 1605–1612 CrossRef CAS
- C. Zhang, B. Fei, D. Yang, H. Zhan, J. Wang, J. Diao, J. Li, G. Henkelman, D. Cai, J. J. Biendicho, J. R. Morante and A. Cabot, Adv. Funct. Mater., 2022, 32, 1–13 Search PubMed
- R. Konar and G. D. Nessim, Mater. Adv., 2022, 3, 4471–4488 RSC
- K. Share, J. Lewis, L. Oakes, R. E. Carter, A. P. Cohn and C. L. Pint, RSC Adv., 2015, 5, 101262–101267 RSC
- R. Dziobek-Garrett, S. Hilliard, S. Sriramineni, O. Ambrozaite, Y. Zhu, B. M. Hudak, T. H. Brintlinger, T. Chowdhury and T. J. Kempa, ACS Nanosci. Au, 2023, 3, 441–450 CrossRef CAS PubMed
- X. Wang, X. Shi, C. Gu, Q. Guo, H. Liu, X. Li and Y. Wu, APL Mater., 2021, 9, 071109 CrossRef CAS
- Y. Xuan, A. Jain, S. Zafar, R. Lotfi, N. Nayir, Y. Wang, T. H. Choudhury, S. Wright, J. Feraca, L. Rosenbaum, J. M. Redwing, V. Crespi and A. C. T. van Duin, J. Cryst. Growth, 2019, 527, 125247 CrossRef CAS
- V. Brune, C. Hegemann, M. Wilhelm, N. Ates and S. Mathur, Z. Anorg. Allg. Chem., 2022, 648, e20220004 CrossRef
- F. Hartl, V. Brune, S. Lüggert, C. Hegemann, D. van Gerven, M. Wilhelm, S. Ji, H. Choi and S. Mathur, Inorg. Chem., 2023, 62, 6274–6287 CrossRef CAS PubMed
- V. Brune, C. Hegemann and S. Mathur, Inorg. Chem., 2019, 58, 9922–9934 CrossRef CAS PubMed
- J. Kim, Y. R. Lim, Y. Yoon, W. Song, B. K. Park, J. Lim, T.-M. Chung and C. G. Kim, RSC Adv., 2019, 9, 6169–6176 RSC
- V. K. Greenacre, A. L. Hector, R. Huang, W. Levason, V. Sethi and G. Reid, Dalton Trans., 2022, 51, 2400–2412 RSC
- Y. J. Park, H.-S. So, H. Hwang, D. S. Jeong, H. J. Lee, J. Lim, C. G. Kim and H. S. Shin, ACS Nano, 2022, 16, 11059–11065 CrossRef CAS PubMed
-
T. Kataoka and S. Watanabe, Comprehensive Organic Functional Group Transformations II, Elsevier, Oxford, 2005, pp. 237–254 Search PubMed
- M. Bühner and H. Sund, Eur. J. Biochem., 1969, 11, 73–79 CrossRef PubMed
- T. E. Burrow, A. J. Lough, R. L. Richards and R. H. Morris, Inorg. Chim. Acta, 1997, 259, 125–135 CrossRef CAS
- M. Bhattacharyya, R. Prakash, C. Nandi, M. G. Chowdhury, B. Raghavendra, T. Roisnel and S. Ghosh, J. Chem. Sci., 2019, 131, 123 CrossRef CAS
- C. O. Kienitz, C. Thöne and P. G. Jones, Inorg. Chem., 1996, 35, 3990–3997 CrossRef CAS PubMed
- U. Jayarathne, J. T. Mague and J. P. Donahue, Polyhedron, 2013, 58, 13–17 CrossRef CAS
- N. Bartalucci, M. Bortoluzzi, T. Funaioli, F. Marchetti, G. Pampaloni and S. Zacchini, Dalton Trans., 2017, 46, 12780–12784 RSC
- A. Monshi, M. R. Foroughi and M. R. Monshi, World J. Nano Sci. Eng., 2012, 02, 154–160 CrossRef
- K. Xiang, S. Li, J. Chen, Y. Wu, F. Yang, Y. Li, W. Dai, J. Wang and K. Shen, Langmuir, 2022, 38, 8585–8594 CrossRef CAS PubMed
- P. Martínez-Merino, E. Sani, L. Mercatelli, R. Alcántara and J. Navas, ACS Sustainable Chem. Eng., 2020, 8, 1627–1636 CrossRef
- M. S. Sokolikova, P. C. Sherrell, P. Palczynski, V. L. Bemmer and C. Mattevi, Nat. Commun., 2019, 10, 712 CrossRef CAS PubMed
- M. Velpandian, G. Ummethala, S. K. Malladi and P. Meduri, J. Colloid Interface Sci., 2022, 623, 561–573 CrossRef CAS PubMed
- J. Wang, L. Li, L. Meng, L. Wang, Y. Liu, W. Li, W. Sun and G. Li, RSC Adv., 2018, 8, 35131–35138 RSC
- H. Wu, T. Zhu, X. Lu and G. W. Ho, J. Mater. Chem. A, 2017, 5, 24153–24158 RSC
- S. B. Kale, A. Bhardwaj, V. C. Lokhande, D.-M. Lee, S.-H. Kang, J.-H. Kim and C. D. Lokhande, Chem. Eng. J., 2021, 405, 126993 CrossRef CAS
- T. Singh, R. Müller, J. Singh and S. Mathur, Appl. Surf. Sci., 2015, 347, 448–453 CrossRef CAS
- T. Lehnen, M. Valldor, D. Nižňanský and S. Mathur, J. Mater. Chem. A, 2014, 2, 1862–1868 RSC
- M. Veith, S. Mathur and V. Huch, J. Am. Chem. Soc., 1996, 118, 903–904 CrossRef CAS
- S. Mathur, M. Veith, T. Ruegamer, E. Hemmer and H. Shen, Chem. Mater., 2004, 16, 1304–1312 CrossRef CAS
- U. Atamtürk, E. Jung, T. Fischer and S. Mathur, Chem. Mater., 2022, 34, 7344–7356 CrossRef
- U. Atamtürk, V. Brune, S. Mishra and S. Mathur, Molecules, 2021, 26, 5367 CrossRef PubMed
- I. Muliuoliene, S. Mathur, D. Jasaitis, H. Shen, V. Sivakov, R. Rapalaviciute, A. Beganskiene and A. Kareiva, Opt. Mater., 2003, 22, 241–250 CrossRef CAS
- S. Mathur, H. Shen, R. Rapalaviciute, A. Kareiva and N. Donia, J. Mater. Chem., 2004, 14, 3259 RSC
-
S. Hünig, P. Kreitmeier, G. Märkl and J. Sauer, Arbeitsmethoden in Der Organischen Chemie, Verlag Lehmanns, Berlin, 2006 Search PubMed
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2007, 64, 112–122 CrossRef PubMed
- STOE X-SHAPE, 1999.
- Stoe & Ci GmbH, STOE Win XPOW, Stoe & Ci GmbH, Darmstadt, Germany, 2010 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed
- STOE X-RED32, 2002.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2307367. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ma01100c |
| This journal is © The Royal Society of Chemistry 2024 |