
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural engineering of catalysts for ammonia electrosynthesis from nitrate: recent advances and challenges
Wenxi
Qiu†
a,
Yuanting
Liu†
a,
Minghao
Xie
b,
Zhaoyu
Jin
 c,
Panpan
Li
*a and
Guihua
Yu
*b
c,
Panpan
Li
*a and
Guihua
Yu
*b
aCollege of Materials Science and Engineering, Sichuan University, Chengdu 610065, P. R. China. E-mail: panpanli@scu.edu.cn
bMaterials Science and Engineering Program and Walker Department of Mechanical Engineering, The University of Texas at Austin, Austin 78712, USA. E-mail: ghyu@austin.utexas.edu
cInstitute of Fundamental and Frontier Sciences, University of Electronic Science and Technology of China, Chengdu 610054, P. R. China
First published on 26th September 2023
Abstract
Ammonia (NH3) is an indispensable industrial chemical used in fertilizer production and energy carriers. However, its production through the Haber–Bosch process requires high temperature and high pressure, consuming significant energy and releasing large amounts of CO2, rendering it unsustainable. As a result, sustainable approaches for ammonia synthesis powered by renewable electricity have gained significant attention, such as the electrocatalytic N2 reduction reaction (N2RR) and nitrate reduction reaction (NitRR). This review summarizes recent advancements in the design strategies of electrocatalysts for the NitRR, highlighting synthetic methods such as doping, alloying, single-atom engineering, nanoconfinement, size-regulation, and tandem catalysis. These strategies aim to tune the adsorption of reactants and intermediates or enhance proton–electron transfer. Future studies could explore new electrocatalysts for efficient NitRR based on the strategies summarized in this review to improve nitrate pollution removal efficiency and ammonia production rates. Furthermore, the challenging questions raised at the end of the paper, such as optimizing the reaction kinetics of the NitRR and improving catalyst selectivity and stability, can provide new directions and insights for future catalyst design.
 Panpan Li | Panpan Li is a professor in the College of Materials Science and Engineering in Sichuan University. She obtained her Bachelor's degree in Chemistry in 2013 and her Doctor's degree in Environmental Science in 2019, both from Sichuan University. After graduation, she conducted her postdoctoral research in Prof. Guihua Yu's Lab at the University of Texas at Austin. Her research interests are focused on the design of hydrogel materials for electrocatalysis, as well as innovative electrochemical technologies for environmental and energy sustainability. |
 Guihua Yu | Guihua Yu is the Temple Foundation Endowed Professor of Materials Science and Mechanical Engineering at the University of Texas at Austin. He received his BS degree with the highest honor from USTC, PhD from Harvard University, followed by postdoc at Stanford University. His research is focused on creative synthesis of nanoarchitected polymeric materials and hybrid organic–inorganic nanomaterials, the fundamental understanding of their chemical and physical properties, and exploration of their technologically important applications in the fields of environment, energy, and sustainability. |
Broader contextThe production of ammonia (NH3) under mild conditions utilizing renewable energy sources without CO2 emissions is a promising solution to the energy crisis and environmental concerns. NH3 electrosynthesis powered by electricity has emerged as an alternative to the Haber–Bosch process, which is energy-consuming and environmentally unfriendly. However, the electrocatalytic nitrate reduction reaction (NitRR) involved in this process is complex and involves a nine-proton and eight-electron transfer process, lowering the overall kinetic rate, lifting the overpotentials, and resulting in unexpected byproducts. Additionally, the competing hydrogen evolution reaction (HER) leads to low faradaic efficiency. Therefore, designing catalysts with good activity and selectivity is crucial. To ensure high performance, research focuses on advancing catalyst design strategies. This review presents the recent advances in designing strategies to construct improved electrocatalysts for the NitRR, highlighting alloy, doping, single-atom engineering, nanoconfinement, size-regulation, and tandem catalysis. These strategies aim to tailor the adsorption of reactants and intermediates or strengthen proton–electron transfer. Although the strategies have shown potential to boost NitRR catalytic activity and efficiency, there are still challenges in the future development of this field, as illustrated at the end of the review. |
Introduction
Nitrogen is an essential element for life in living organisms. The nitrogen cycle plays a crucial role in maintaining the balance of elements in the biosphere and has a direct impact on human life.1,2 Unfortunately, anthropogenic activities are severely damaging the nitrogen cycle.3 For instance, the excessive release of nitrate into the environment poses a significant threat to both the ecological environment and public health.4 The accumulation of nitrate in rivers and lakes can cause water eutrophication, which is detrimental to aquatic life.5 In addition, the presence of nitrate in drinking water causes damage to the human endocrine system and leads to severe diseases.6,7 Moreover, nitrite, produced from nitrate, is a carcinogenic compound that can cause cancer.8 Therefore, removing nitrate from water is of utmost importance.9,10 Traditional biological, physical, and chemical methods have been used to remove nitrate from water, but they generate secondary pollution and require post-treatment, which limits their widespread use.11–14 Compared with these technologies, the electrocatalytic nitrate reduction reaction (NitRR) is a promising alternative approach as it uses renewable electricity and operates under mild conditions without producing harmful residues.4,15–17Electrocatalytic nitrate reduction is a promising approach for mitigating and upcycling nitrate pollution by producing ammonia (NH3), a valuable chemical commodity.18,19 NH3 is not only a crucial synthetic chemical for nitrogen-containing compound production but is also recognized as a carbon-free energy carrier for hydrogen storage.20,21 Currently, large-scale ammonia production is achieved via the energy-intensive Haber–Bosch (H–B) method, which converts N2 and H2 to NH3 under high pressure and temperature. However, the H–B process consumes 1–2% of the world's annual fossil energy and results in roughly 1% of the global annual CO2 emissions.22 The electrochemical synthesis of ammonia is a promising alternative that can reduce energy consumption and carbon runoff during industrial production. However, the low solubility of nitrogen in aqueous electrolytes, the difficulties in breaking the triple bond of N2 (941 kJ mol−1), and the suppression of the hydrogen evolution reaction (HER) lead to poor efficiency and limit the further reliable application of the electrochemical nitrogen reduction reaction (NRR). Nitrogen oxides,23–25 including NO3−, NO2−, NO, and N2O, are considered more potential ammonia synthesis pathways due to the lower dissociation energies of the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond (204 kJ mol−1) and/or N–O bond (176 kJ mol−1). However, nitrogen oxide concentrations are generally lower than those that have been reported. For example, the concentrations of nitrate in industrial wastewater, textile wastewater, and polluted groundwater were only 41.6 mM, 7.4 mM, and 0.9–1.2 mm, respectively, whereas the other nitrogen oxide concentrations were lower. Consequently, potential solutions include concentrating diluted nitrogen-containing reagents prior to reduction and designing highly selective catalysts suitable for treating low-concentration wastewater. In addition, with the consideration of disturbing species and complex components in real wastewater, the design of electrocatalysts with high anti-interference is more conducive to its practical application. As the major nitrogen pollutant in surface and groundwater, converting NO3− into more valuable ammonia is an effective way to manage the disturbed nitrogen cycle, which has attracted much attention in past decades. However, the NitRR involves the transfer of nine protons coupled with eight electrons, which is a kinetically sluggish process.26 Moreover, undesired by-products, such as NO2−, N2H4, and N2, generated during the NitRR can affect the selectivity of ammonia production.27 Additionally, the hydrogen evolution reaction (HER) competes with the NitRR at high overpotential, leading to low faradaic efficiency. To address these challenges, researchers have been focusing on designing efficient electrocatalysts with high selectivity for producing NH3.28
O bond (204 kJ mol−1) and/or N–O bond (176 kJ mol−1). However, nitrogen oxide concentrations are generally lower than those that have been reported. For example, the concentrations of nitrate in industrial wastewater, textile wastewater, and polluted groundwater were only 41.6 mM, 7.4 mM, and 0.9–1.2 mm, respectively, whereas the other nitrogen oxide concentrations were lower. Consequently, potential solutions include concentrating diluted nitrogen-containing reagents prior to reduction and designing highly selective catalysts suitable for treating low-concentration wastewater. In addition, with the consideration of disturbing species and complex components in real wastewater, the design of electrocatalysts with high anti-interference is more conducive to its practical application. As the major nitrogen pollutant in surface and groundwater, converting NO3− into more valuable ammonia is an effective way to manage the disturbed nitrogen cycle, which has attracted much attention in past decades. However, the NitRR involves the transfer of nine protons coupled with eight electrons, which is a kinetically sluggish process.26 Moreover, undesired by-products, such as NO2−, N2H4, and N2, generated during the NitRR can affect the selectivity of ammonia production.27 Additionally, the hydrogen evolution reaction (HER) competes with the NitRR at high overpotential, leading to low faradaic efficiency. To address these challenges, researchers have been focusing on designing efficient electrocatalysts with high selectivity for producing NH3.28
A large number of catalytic materials have been intensively studied as potential electrocatalysts for the NitRR, including noble metals,29 transition metals,30–32 and metal-free catalysts.33 Recent advances in catalysts have emphasized the importance of catalysts’ structure and composition at the nanoscale or even at the atomic level. In this review, we summarize the latest developments in the NitRR for NH3 production and structural engineering strategies such as electronic structure optimization and mass transport regulation to improve the NitRR activity and selectivity. First, we discuss the possible reaction pathways for nitrate electroreduction to NH3. We then categorize a series of design strategies into six sections, alloying, doping, single-atom engineering, size regulation, nanoconfinement, and tandem catalysis as shown in Fig. 1 and Table 1. These strategies aim to either tune the adsorption of reactants and intermediates or enhance proton–electron transfer. Hence, it is necessary to understand in-depth the reaction pathways through theoretical calculations and advanced characterization, which can guide the design of enhanced electrocatalysts for the NitRR. Finally, we offer an overview of the current challenges and prospects associated with the design strategies for NitRR catalysts.
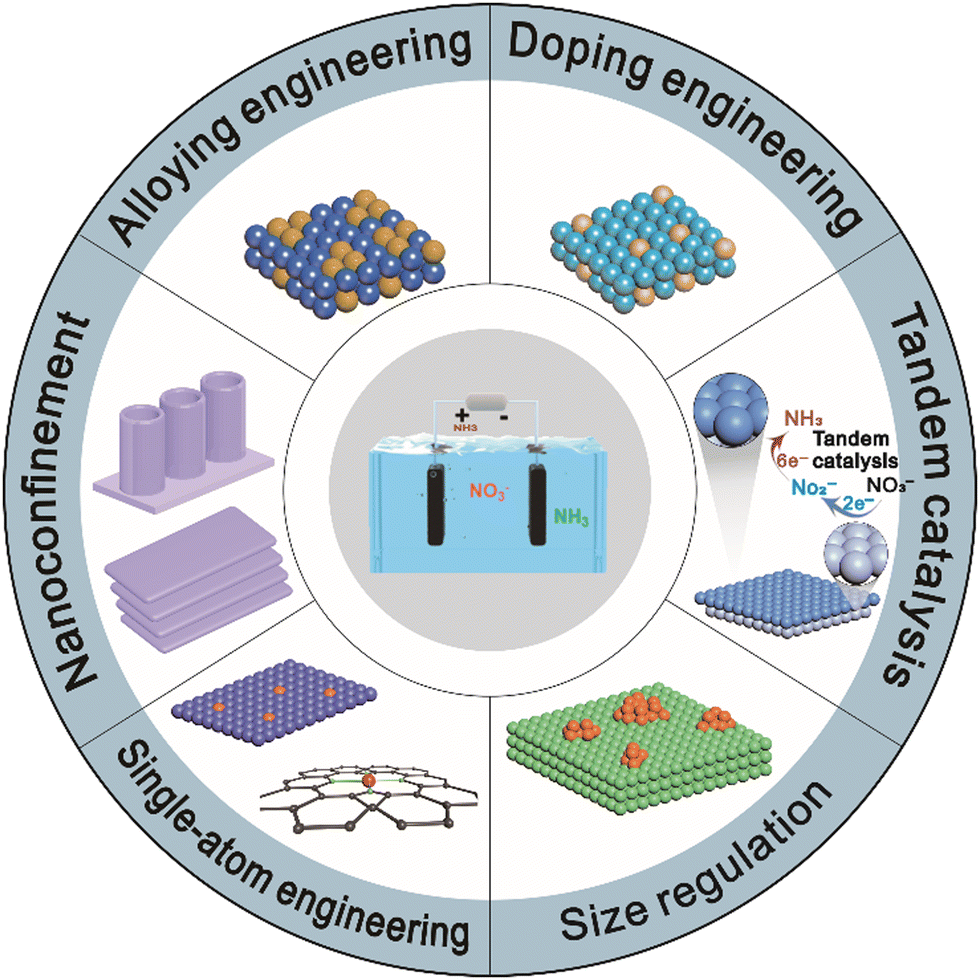 | ||
| Fig. 1 The schematic illustration of electrocatalyst design strategies for promoting the NitRR performance. | ||
| Catalyst | Electrolyte | Faradaic efficiency | Ammonia yield rate | Ref. | |
|---|---|---|---|---|---|
| Alloying | CuNi alloys | 1.0 M KOH + 0.1 M KNO3 | 99.0 ± 1% at −0.15 V vs. RHE | N.A. | 31 |
| CuNi Ns/CF | 1.0 M KOH + 44.3 g L−1 NO3− | 97.03% at −0.48 V vs. RHE | 94.57 mg h−1 cm−2 at −0.48 V vs. RHE | 85 | |
| PdCu/Cu2O hybrids | 0.5 M Na2SO4 + 100 ppm NO3−–N | 94.32% at −0.8 V vs. RHE | 0.19 mmol h−1 cm−2 at −0.8 V vs. RHE | 86 | |
| Doping | N–C-1000 | 0.1 M KOH + 0.1 M KNO3 | 95% at −0.7 V vs. RHE | 1.30 mg h−1 cm−2 at −0.7 V vs. RHE | 85 |
| B–MoS2/CC | 0.5 M Na2SO4 + 0.1 M NaNO3 | 92.3% at −0.7 V vs. RHE | 10.8 mg h−1 cm−2 at −0.7 V vs. RHE | 95 | |
| Fe/Ni2P | 0.2 M K2SO4 + 0.05 M NO3− | 94.3% at −0.4 V vs. RHE | 4.17 mg h−1 cm−2 at −0.4 V vs. RHE | 96 | |
| Single-atom catalysts | Fe–PPy SACs | 0.1 M KOH + 0.1 M KNO3 | ∼100.0% at −0.30 V vs. RHE | 2.75 mg h−1 cm−2 at −0.7 V vs. RHE | 78 |
| Fe SAC | 0.25 M K2SO4 + 0.50 M KNO3 | 75% at −0.66 V vs. RHE | 20.0 mg h−1 cm−2 at −0.85 V vs. RHE | 79 | |
| Cu-cis-N2O2 | 0.5 M Na2SO4 + 1000 ppm KNO3–N | N.A. | 27.84 mg h−1 cm−2 at −1.6 V vs. RHE | 111 | |
| GdSA-D-NiO400 | 1.0 M KOH + 1.0 M KNO3 | 97% at −0.1 V vs. RHE | 0.628 mg mgcat−1 h−1 at −0.1 V vs. RHE | 77 | |
| Ru1–TiOx/Ti | 1.0 M KOH + 0.5 M NaNO3 | 87.6% at −0.3 V vs. RHE | 2.2 mol g−1 h−1 at −0.3 V vs. RHE | 124 | |
| VCu-Au1Cu SAAs | 1.0 M KOH + 0.1 M KNO3 | 98.7% at −0.2 V vs. RHE | 0.55 mg h−1 cm−2 at −0.2 V vs. RHE | 125 | |
| Size regulation | Ru NCs/TiO2 NTs | 0.05 M Na2SO4 + 100 ppm KNO3–N | >90% from −0.1 to −0.4 V vs. RHE | 600 μg h−1 cm−2 at −0.4 V vs. RHE | 132 |
| Rh@Cu-0.6% | 0.1 M Na2SO4 + 0.1 M KNO3 | 93.0% at −0.20 V vs. RHE | 1.27 mmol h−1 cm−2 at −0.4 V vs. RHE | 133 | |
| Nanoconfinement | TiO2 NTS/CuOx | 0.5 M Na2SO4 + 100 ppm KNO3–N | 92.23% at −0.75 V vs. RHE | 1.24 mg NH3 cm−2 h−1 at −0.75 V vs. RHE | 138 |
| Cu SAG | 1.0 M PBS + 20 mM NO3− | 78% at −0.8 V vs. RHE | 440 μg cm−2 h−1 at −0.8 V vs. RHE | 144 | |
| PdCu–H | 0.1 M KOH + 0.1 M KNO3 | 87.3% at −0.3 V vs. RHE | 0.551 mmol h−1 mg−1 at −0.3 V vs. RHE | 140 | |
| Tandem catalysis | CuCoSP | 0.1 M KOH + 0.1 M KNO3 | 93.3% at −0.175 V vs. RHE | 19.89 mg NH3 cm−2 h−1 at −0.175 V vs. RHE | 142 |
| Cu nanosheets | 1.0 M KOH + 0.2 M KNO3 | 95% at −0.36 V vs. RHE | 1.41 mmol h−1 cm−2 at −0.59 V vs. RHE | 143 | |
| FeB2 | 0.1 M KOH + 0.1 M KNO3 | 96.8% at −0.6 V vs. RHE | 25.5 mg h−1 cm−2 at −0.6 V vs. RHE | 158 | |
Reaction mechanisms
Pathways and mechanisms
Understanding the electrocatalytic nitrate reduction reaction mechanism is essential for designing efficient electrocatalysts. This complex process involves the transport of multiple electrons and protons, with various nitrogen oxidation states ranging from −3 to +5. Among them, N2 and NH3 are the most thermodynamically stable forms,6,34 with N2 being harmless and environmentally friendly. From the perspective of “upcycling waste to value”, NH3 is the desirable product. These species are obtained from the following Reactions (1) and (2), respectively. Nitrate electroreduction is divided into two parts, including the indirect autocatalytic reduction pathway and the indirect electrocatalytic reduction pathway.35 The pathway without nitrate participation is called indirect autocatalytic reduction.36 Direct nitrate electroreduction involves the regulation of active adsorbed hydrogen atoms (Had.) and the electron reduction from the cathode. In the adsorbed-hydrogen-mediated pathway, H2O is adsorbed on the electrode surface and then reduced to produce Had.. Then, Had. and nitrate are gradually converted into NH4+via intermediates such as NO2−ad., NOad., Nad., NHad., and NH2ad., among others.37 It is worth noting that two Nad. species combine to form N2. The migration barrier of Nad. (0.75 eV) is significantly higher than that of Had. (0.10 eV). Therefore, the N–N bond is generated less kinetically than the N–H bond.38| NO3− + 9H+ + 8e− → NH3 + 3H2O E0 = −0.12 V vs. SHE | (1) |
| 2NO3− + 12H+ + 10e− → N2 + 6H2O E0 = 1.17 V vs. SHE | (2) |
Fig. 2 displays the electron-mediated pathway of nitrate electroreduction. Initially, nitrate is transformed into nitrite.39–41 The first step of the NitRR is the adsorption of nitrate on the active sites, which is considered to be the rate-determining step.42,43 Adsorption energy plays a pivotal role in the initiation of the reaction and the adsorption efficiency is related to the nitrate concentration, cathode materials, and the mass transfer rate of reactants.44,45 After the absorption step, the electroreduction of NO3−ad. to nitrite is depicted as Reaction (3), which comprises a proton-coupled double-electron transfer and another proton coupling process successively. This three-step reaction follows an electrochemical–chemical–electrochemical (ECE) mechanism and is known as one of the rate-determining steps during the overall reaction. The first electron transfer reaction (NO3−ad. to NO32−ad.) occurs at a significant negative potential with estimated Tafel slopes of 120 mV dec−1.44 Through the initial electrochemical reduction, a short-lived (≈20 μs) NO32−ad. is generated according to Reaction (4).39,46 Then, with further homogenous chemical reduction, NO32−ad. is hydrolyzed to a nitrogen dioxide radical (Reaction (5)).47,48 Consequently, the second electron transfer reaction converts NO2˙ to NO2− as shown in Reaction (6).49
| NO3− + 2H+ + 2e− → NO2− + H2O E0 = 0.93 V vs. SHE | (3) |
| NO3−ad. + e− → NO32−ad. E0 = −0.89 V vs. SHE | (4) |
| NO32−ad. + 2H+ → NO2˙ + H2O k = 5.5 × 104 s−1 | (5) |
| NO2˙ad. + e− → NO2−ad. E0 = 1.04 V vs. SHE | (6) |
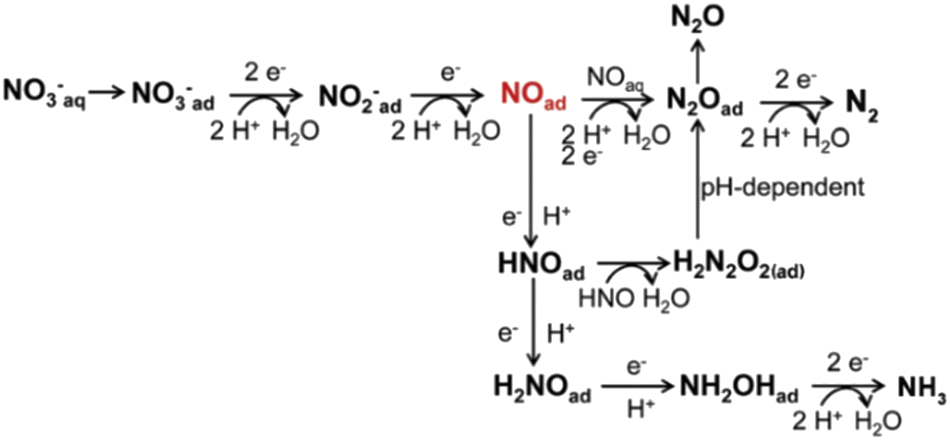 | ||
| Fig. 2 Reaction pathway diagram for nitrate electroreduction. | ||
Moreover, nitrite can be reduced to NO by direct electron transfer and a deoxidation process.42 This step is a divergent center for generating dinitrogen or ammonia. The selectivity of the subsequent pathway is determined by nitric oxide.50 There are multiple pathways to form N2, while the NOad. intermediate undergoes a continuous charge-transfer path to produce ammonia. One pathway involves the reaction of NOad. with aqueous NO to generate the N2Oad. intermediate.51–53 N2Oad. can be reduced to N2 instead of desorbing from the cathode surface.54,55 The hydrogenation of NOad. produces HNOad. as per Reaction (7). The produced HNOad. obtains an electron and yields H2NOad. according to Reaction (8). Next, the reduction of H2NOad. occurs (Reaction (9)), releasing hydroxylamine (NH2OHad.). Finally, the absorbed hydroxylamine is rapidly deoxidized to ammonia by Reaction (10).56 Many simulations support the large energy barrier in the step of NOad. to NHOad., making this step the rate-determining step. Another reaction for HNOad. involves rapid dimerization to produce hyponitrous acid (H2N2O2).57,58 However, the monoanionic form (HN2O2−) is unstable and easily decomposes to give N2O in a pH-dependent process. Consequently, N2O can be reduced to N2.59
| NOad. + H+ + e− → HNOad. | (7) |
| HNOad. + H+ + e− → H2NOad. | (8) |
| H2NOad. + H+ + e−→ NH2OHad. | (9) |
| NH2OHad. + 2H+ + 2e−→ NH3 + H2O | (10) |
Catalyst chemical composition
The selectivity of the electrocatalytic reduction-to-nitrate reaction highly depends on the chemical composition of catalysts. Several studies have summarized the activity trends and selectivity of the NitRR on various metals, providing guidelines for catalyst design.22,52–55 Experimental results have demonstrated the NitRR performance on noble metals in sulfuric acid solutions. Rh has the highest NitRR activity, with the activity decreasing in the order of Rh > Ru > Ir > Pt ≈ Pd.27,60 The high coverage of hydrogen on the active sites of the catalyst could hinder the adsorption of NO3− and its reduced intermediates, resulting in a low FE for the NitRR. Therefore, noble metals are not desirable catalysts for this reaction. Pt has low activity for the NitRR because of the weak adsorption for NO3−.61–63 Yet, NH3 FE on the Pd surface is low because of a serious competing HER and complicated intermediates, such as NOx, N2H4, and NH2OH produced during the pathways from NO3− to NH3.64 However, Ru shows a significant increase in NH3 FE from 0 V vs. RHE.65 To optimize the activity for the NitRR, the surface of noble metals can be modified with adatoms or alloys and the synergistic effect is beneficial for nitrate reduction in a tandem catalysis process, such as simultaneous acceleration of sequential NO3−-to-NO2− and NO2− reduced to N2, NH3 or other products.55,66,67The premise for achieving high conversion of nitrate to NH3 is the moderate H adsorption ability of active sites. Several transition metals are preferred, such as Cu-, Ti-, Co- and Ni-based materials. Among these, Cu dominates and presents the highest activity toward the nitrate reduction reaction, as it can manage the competing HER better than others, including Ag, Au, Ni, Zn, Al, Sn and Bi.59,68–70 However, undesirable byproducts, such as nitrite, are usually generated during the NitRR when using Cu as the catalyst. The efficiency of Cu-based materials can be improved by modifying or alloying other metals. For instance, Li et al. reported a metasequoia-like nanocrystalline Cu49Fe1 catalyst for the NitRR in a neutral medium. Cu49Fe1 achieved a faradaic efficiency of up to 94.5% and a good NH3 selectivity of 86.8% due to the enhanced adsorption of the intermediates via a Fe-shifted Cu d-band center.71
Electronic structure
The electronic structure of the catalyst determines the adsorption strength of NO3− and other reaction intermediates, which considerably influence the NitRR catalytic activity. We could change the electronic structure of catalysts based on various design strategies. With the introduction of other metals,72–74 elements,33,75,76 or a single atom,77–79 the electronic structure of the catalyst surface and the outermost orbital electronic properties can be regulated due to the different properties of dopants and host atoms. These changes include valence, electronegativity, charge density and polarization, which can strengthen orbital hybridization between the electrocatalyst and nitrate or intermediates, thereby improving its adsorption strength.80 In single-atom catalysts, the surrounding matrix is an effective parameter for modulating the electronic states of the single metal atom due to strong interaction with the coordinated elements. Similarly, regulation of the size of the catalyst to clusters can tune the electronic states of active sites, leading to better activity.Catalyst geometry
In addition to chemical composition, geometry is another crucial factor in designing high-activity catalysts, as it closely correlates with reactant delivery and gas diffusion during the catalytic reaction. The electrocatalytic nitrate reduction reaction involves charge transfer and liquid–solid phase mass transport. The development of catalysts with two-dimensional (2D) or three-dimensional (3D) structures is expected to provide liquid channels and gas channels. Pak et al.81 reported that the Ti plate with TiO2 nanotube arrays showed improved nitrate reduction efficiency. The nanotube array geometric structure helps the diffusion of nitrate and intermediates. The arrays of vertically aligned metal nanowires leads to high electrocatalytic activity due to their geometry. Napolskii et al.82 prepared highly ordered rhodium nanowire arrays with an average diameter of 55 nm and a length of 1.5–11 μm via a template method and used them for the nitrate reduction reaction. The much-increased current density was achieved due to the enhanced mass transport in the shortest Rh nanowires catalyst. To evaluate the effect of layer morphology on transport, Cattarin et al.83 reported that a spongy Cu–Ni alloy layer with bimodal porosity was deposited from vigorous hydrogen evolution using a large current. The Cu–Ni alloy featured in macroscopic pores tens of micrometers in diameter. The porous Cu–Ni alloy electrode showed a higher current due to the enhanced transport.Design strategies for electrocatalysts
The dependency of NitRR on the composition and geometry of the electrode has been reported, and therefore the catalyst design strategies must take these critical factors into account for achieving excellent selectivity and efficiency. In this section, we aim to provide a summary of recent advances in the investigation of nitrate reduction electrocatalysts, with a focus on design strategies that consider cross-scale interactions over the surface and interface. Our goal is to categorize the design strategies for promoting the intrinsic activity of catalysts based on changes in the local electronic environment of the active centers. In the sections below, we discuss different strategies and provide insights into the structure–activity relationship of electrocatalysts.Alloying engineering
Alloy catalysts with metal elements can provide different and active centers,84 while the multiple compositions of alloy catalysts can tune the d-band center and surface potential,87–89 influencing the adsorption of reactants and intermediates. In the NitRR field, the alloying catalyst is also a promising design strategy.72,73,85,90–93 Wang et al.74 synthesized a series of CuNi alloys by adjusting the concentration ratio of Cu and Ni. When Cu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ni = 50:50, the Cu50Ni50 exhibited the highest NitRR catalytic activity. The d-band center of CuNi alloys shifts 0.14, 0.28, and 0.32 eV towards the Fermi level with the increase of Ni composition ratio, enhancing the adsorption energy of intermediates (Fig. 3a). From Fig. 3b, the volcanic-type relationship between NO3− adsorption energy and experimental overpotential on all CuNi alloys indicates the introduction of Ni atoms leads to a higher adsorption energy of NO3−. However, with increasing Ni concentration, the Cu site decreases, and the adsorption energy of *NH2 intermediates exceeds the optimal value, resulting in a decline in the NitRR performance. Notably, the ratio of two metals in copper–nickel alloy has an important effect on the catalytic performance. To explore the relationship between the proportion of copper–nickel metal and catalytic performance, a series of copper–nickel alloys with different proportions have been reported with carbon substrate. For instance, Dong et al.94 fabricated a series of CuNi alloy nanoparticles embedded in a nitrogen-doped carbon matrix (CuNi/NC) with different Cu–Ni ratios; Liu et al.93 reported a series of Cu–Ni catalysts with component-controllable CuxNiy nanoparticles encapsulated in N-doped carbon film (CuxNiy/NC).
:Ni = 50:50, the Cu50Ni50 exhibited the highest NitRR catalytic activity. The d-band center of CuNi alloys shifts 0.14, 0.28, and 0.32 eV towards the Fermi level with the increase of Ni composition ratio, enhancing the adsorption energy of intermediates (Fig. 3a). From Fig. 3b, the volcanic-type relationship between NO3− adsorption energy and experimental overpotential on all CuNi alloys indicates the introduction of Ni atoms leads to a higher adsorption energy of NO3−. However, with increasing Ni concentration, the Cu site decreases, and the adsorption energy of *NH2 intermediates exceeds the optimal value, resulting in a decline in the NitRR performance. Notably, the ratio of two metals in copper–nickel alloy has an important effect on the catalytic performance. To explore the relationship between the proportion of copper–nickel metal and catalytic performance, a series of copper–nickel alloys with different proportions have been reported with carbon substrate. For instance, Dong et al.94 fabricated a series of CuNi alloy nanoparticles embedded in a nitrogen-doped carbon matrix (CuNi/NC) with different Cu–Ni ratios; Liu et al.93 reported a series of Cu–Ni catalysts with component-controllable CuxNiy nanoparticles encapsulated in N-doped carbon film (CuxNiy/NC).
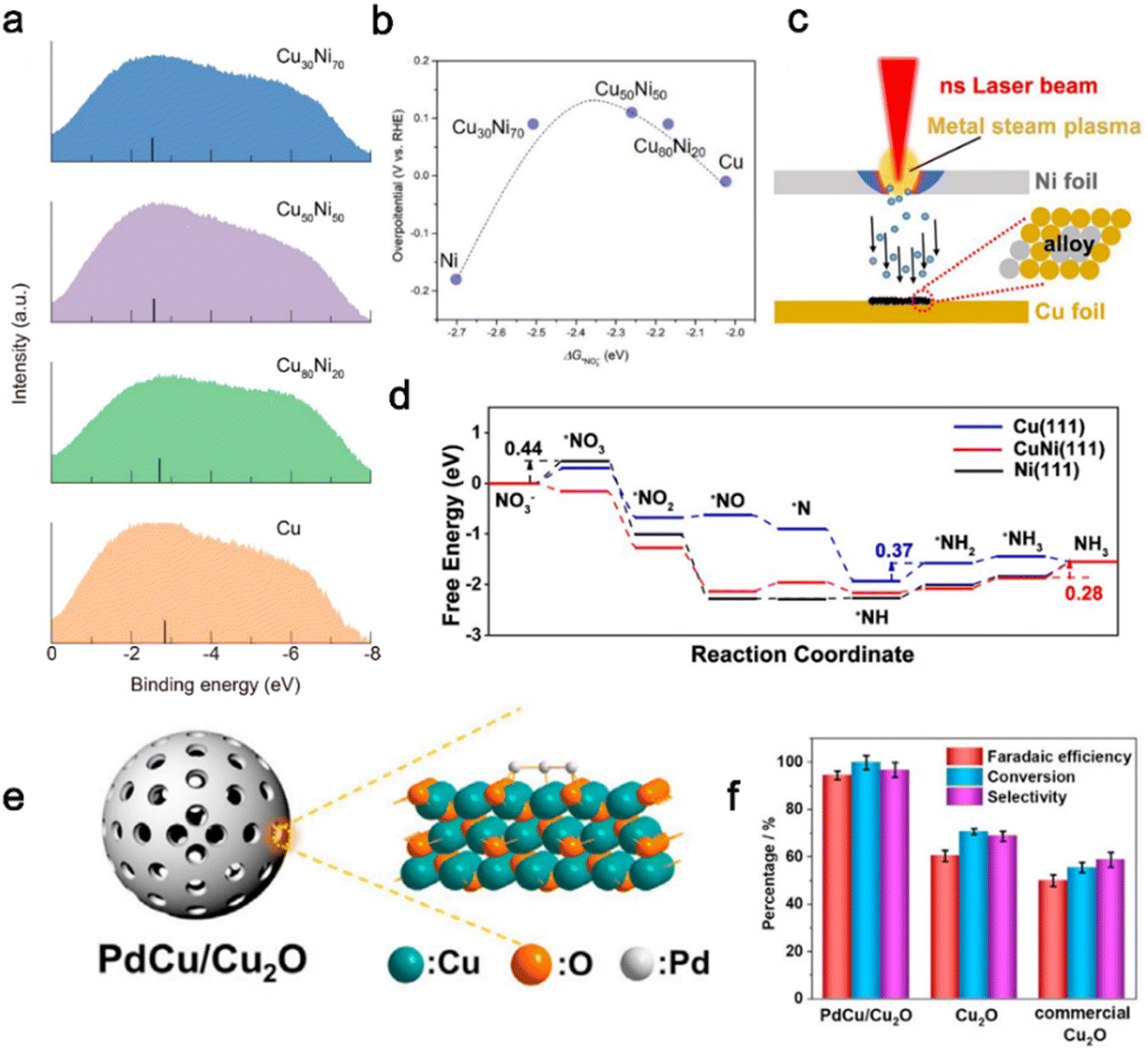 | ||
| Fig. 3 Alloy engineering. (a) UPS spectra of various CuNi alloys and pure Cu catalysts. (b) The volcano-type relationship between overpotentials of electrocatalytic nitrate reduction and adsorption energies of *NO3− on all CuNi alloys.74 Copyright 2020, American Chemistry Society. (c) Mechanism illustration of the laser irradiation process. (d) Free energies for electrocatalytic reduction of nitrate into NH3 on different catalyst surfaces, respectively.85 Copyright 2023, The Royal Society of Chemistry. (e) Schematic illustration of mesoporous hollow sphere PdCu/Cu2O hybrid structures. (f) Faradaic efficiency and selectivity of ammonia, and the conversion rate of nitrate over different samples.86 Copyright 2021, Cell Press. | ||
Except for traditional chemical synthesis, the laser irradiation method is also a fast and facile strategy, which is conducive to the formation of alloys due to the characteristics of rapid heating and cooling during the preparation. Yu et al.85 synthesized CuNi alloy nanoparticles (CuNi NPs/CF) using a laser irradiation method. As shown in Fig. 3c, when the Ni foil located in the upper layer was irradiated by the laser beam, the Ni atoms on its surface absorbed the laser energy and instantly evaporated to produce Ni plasma gas. After the laser treatment, a black CuNi alloy was formed in the corresponding region of the Cu foil surface in the lower layer. CuNi NPs/CF showed excellent NitRR activity with a maximum FE of 97.03% and a highest yield rate of 94.57 mg h−1 cm−2. By comparing the free energies of NO3− reduction on CuNi(111), Ni(111), and Cu(111) (Fig. 3d), the energy barrier of the hydrogenation reduction step of the intermediate *NH was significantly reduced after introducing Ni atoms, which could be attributed to the surrounding Ni sites providing sufficient H* to Cu sites. Besides, the Gibbs free energy change (ΔG) of the potential-determining step (PDS) on CuNi(111) was 0.28 eV, while the ΔG of PDS on Ni(111) and Cu(111) was 0.44 eV and 0.37 eV, respectively (Fig. 3d), implying a more favorable reaction path on CuNi(111).
Alloying has presented a unique strategy to tailor the electronic structure of catalysts. Noble metals such as Pd, Pt, Rh and Ag show high activity for nitrate reduction, and many noble metal alloys for the NitRR have been reported, such as PdAg alloys, PtRh alloys, PtIr alloys and PdN alloys. However, due to the high cost and resource scarcity of precious metals, the choice of non-metallic elements is considered more promising. Alloying precious metals with non-metals is an effective option.97 Yin et al.86 reported a PdCu/Cu2O hybrid with a mesoporous hollow sphere structure obtained by depositing Pd species on Cu2O (Fig. 3e). Benefitting from the highly dispersed PdCu alloy on the ultrathin Cu2O shell, the PdCu/Cu2O hybrid exhibits excellent NitRR activity. As shown in Fig. 3f, the maximum values of Faraday efficiency, selectivity, and conversion rate of PdCu/Cu2O hybrid reach 94.32%, 96.7%, and 99.82% respectively, which are superior to those of Cu2O and commercial Cu2O. Further theoretical evidence demonstrates that the existence of PdCu alloy is more conducive to the formation of intermediate *N to regulate the reaction path of ammonia synthesis. Given the advantages of alloys, fabricating commercial catalysts at an industrial scale should be explored extensively. Their composition should be controlled and studied.98
Doping engineering
Doping is considered to be an important approach to optimize the electronic structure of catalysts, thus improving catalytic performance.99–105 The different electronegativity among elements induces electron transfer and shifts the Fermi energy level of the catalysts, which is crucial for intrinsic activity. Besides, the crystal structure of the catalyst is destroyed once the intrinsic atoms are substituted by heteroatoms. Hence, defects arise along with the change in the crystal structure and give more sites for the reaction intermediates. Additionally, doping is a significant method to increase the number of active sites. Carbon materials with many advantages are employed as catalysts.25,76,106 Initial studies have reported that carbon-based catalysts doped with heteroatoms can modulate their physicochemical properties and enhance catalytic activity.75,107–110 Li et al.33 developed a group of carbon-based aerogel catalysts doping different N species for highly efficient nitrate-to-ammonia reduction. Fig. 4a depicts the constructions of four N moieties which are defined as pyridinic-N (N1), pyrrolic-N (N2), graphitic-N (N3), and pyridinic-N-oxide (N4). The adsorption energy of NO3− on each N–C site was determined as −3.10 eV, −3.25 eV, −5.66 eV, and −3.84 eV, respectively (Fig. 4b). The highest adsorption energy of NO3− on N3 suggests this site was favorable for the nitrate reduction reaction. The electrochemical experiments further discovered that N–C-1000 with a maximum of N3 absolute content (1.55 at%) exhibited the highest FE of 95% and YR of 1.3 mg h−1 cm−2 at −0.7 V vs. RHE.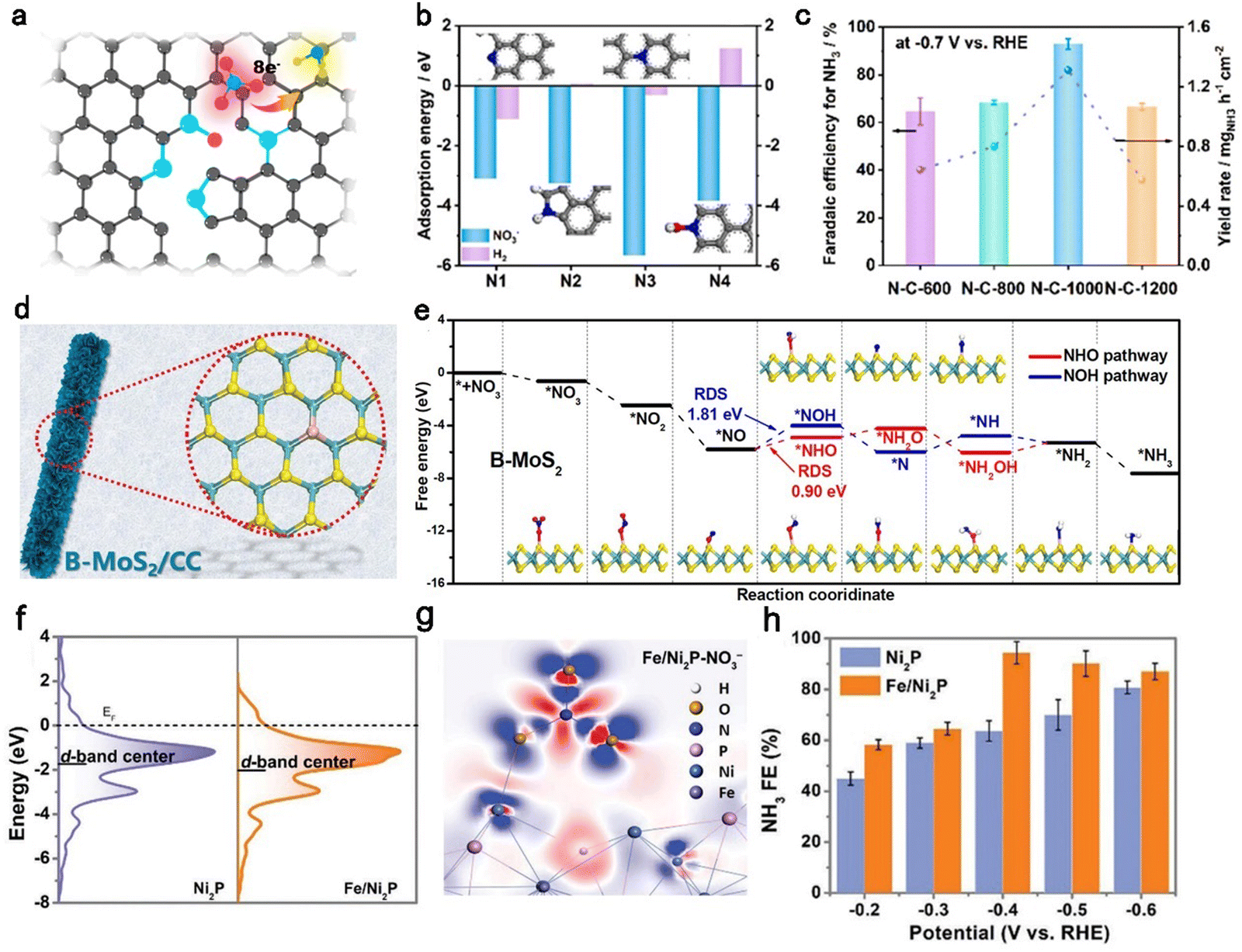 | ||
| Fig. 4 Doping strategy. (a) Schematic of the structure of the N–C-1000 catalyst. (b) The adsorption energy of NO3− and hydrogen on pyridinic-N, pyrrolic-N, graphitic-N, and pyridinic-N-oxide moieties, respectively. (c) Faradaic efficiency and yield rate of NH3 for N–C-1000.33 Copyright 2023, Elsevier. (d) Schematic of B–MoS2/CC. (e) Free energy diagram of the nitrate reduction to ammonia on B–MoS2.95 Copyright 2022, Elsevier. (f) The d-DOS of the Ni atom on the Fe/Ni2P and pristine Ni2P. (g) The charge density difference of Fe/Ni2P after absorbing NO3−. (h) FE of Ni2P and Fe/Ni2P.96 Copyright 2022, Wiley. | ||
This work emphasized the effect of the doping element species on catalytic behaviors in the NitRR.
Compared to the N element, the B element possesses a smaller atomic radius and moderately suitable electronegativity, providing it with a certain capacity to suppress the HER. Luo et al.95 reported a B-doped MoS2 nanosheet array on carbon cloth (B–MoS2/CC, Fig. 4d). The B–MoS2/CC reached a maximum FE of 92.3% and YR of 10.8 mg h−1 cm−2 at −0.7 V vs. RHE, while the MoS2/CC without doping B element only delivered a poor FE of 56.2% and YR of 5.1 mg h−1 cm−2, suggesting that the B-doping effectively improved the catalytic activity of the NitRR. DFT calculations further provided a theoretical understanding of the developed NitRR performance originating from B-doping. The NOH pathway (*NO–*NOH) and NHO pathway (*NO–*NH) were determined both on B–MoS2/CC (Fig. 4e) and MoS2/CC. Compared to the two pathways on MoS2/CC, the NOH pathway on B–MoS2/CC with the lowest rate-determining step energy barrier (0.90 eV) is more favorable for the conversion of NO3− to NH3.
Except for popular non-metal elements, metal dopants have attracted attention owing to their high activity. Zhang et al.96 synthesized an iron-doped nickel phosphides catalyst (Fe/Ni2P) for efficient NitRRs. As depicted in Fig. 4f, Fe doping regulated the electronic structure of Ni atoms with an obvious downshift of the d-band center from −1.73 eV (Ni2P) to 0.36 eV (Fe/Ni2P). Besides, the charge density differences of Fe/Ni2P and Ni2P after adsorption of NO3− illustrated different energy distributions around the active center Ni sites, implying the electronic structure of Ni2P was regulated (Fig. 4g). As a result, Fe/Ni2P reached a maximum FE of 94.3% with a corresponding YR of 4.17 mg h−1 cm−2 for NH3 at −0.4 V vs. RHE, which was far higher than that of Ni2P (Fig. 4h). Although all types of dopant atoms have their advantages, it is difficult to design and coordinate their respective advantages rationally. More studies should be conducted to achieve quantitative control of heteroatom doping.
Single-atom engineering
Except for the above polyatomic doping, single-atom doping is a desirable design strategy to develop single-atom catalysts (SACs). SACs demonstrate high atomic utilization, distinct activity, and selectivity,112–116 which opens up a new avenue for designing efficient catalysts.117–123 Electronic structures of SACs are highly dependent on the electronegativity and ionic radius of metal centers, affecting the adsorption of reactive species. Additionally, the coordination environment, e.g., coordination atoms and numbers, greatly contributes to tailoring the electronic structure of SACs.28 Notably, carbon materials have the advantage of the support being sustainable and environmentally friendly,126 which are considered as important factors for SACs.127 Fe single-atom catalysts (Fe–PPy SACs) derived from ferric acetylacetonate-polypyrrole hydrogels were reported by Li et al.,78 possessing densely populated Fe active sites and demonstrated high activity for electroreduction of nitrate to ammonia. The Fe(II)–Nx sites as active centers in Fe–PPy SACs were embedded in carbon support, which were preferentially occupied by NO3− before being reduced to Fe(0)–Nx. Thus, the competitive hydrogen evolution reactions were inhibited for the lack of active sites to provide water molecules, while the Fe(0) sites in Fe nanoparticles (Fe NP) suffered from fierce competition. The single metal atoms were indicated by the HAADF-STEM image, which showed isolated bright spots with a diameter of ca. 0.1 nm. The electron energy loss spectrum (EELS) was collected at a local area of one atom marked in Fig. 5a. Iron, carbon and nitrogen elements were mainly distributed in the selected region in Fig. 5b. Additionally, SI-SECM was employed to time-dependently analyze the site density of the single-site Fe moiety with a dynamic oxidation state toward nitrate reduction and water dissociation at given potentials (Fig. 5c). Fe–PPy SACs reached a maximum FE of nearly 100% at −0.3 V vs. RHE with a corresponding YR of 2.75 mg NH3 h−1 cm−2. Besides, at least a twelve-fold turnover frequency difference was observed in Fe–PPy SACs and Fe NP, suggesting the higher producing-NH3 rate on isolated Fe atoms compared with bulk Fe. Single atomic Fe–N–C catalysts have attracted wide interest for different reactions.128–130 Particularly, Fe active sites are found in both Haber–Bosch catalysts and nitrogenase enzymes.131 Inspired by the single-site of iron, Wu et al.79 also designed a Fe single-atom catalyst (Fe SAC) with isolated Fe atoms uniformly embedded in the carbon matrix for reducing NO3− towards NH3. The observed results of EXAFS and STEM (Fig. 5e) both demonstrated the Fe–N4 construction in Fe SAC. In detail, a single Fe atom was coordinated by surrounding four N atoms (Fig. 5d), implying the Fe active sites were atomically dispersed in the N-doped carbon (NC) substrate.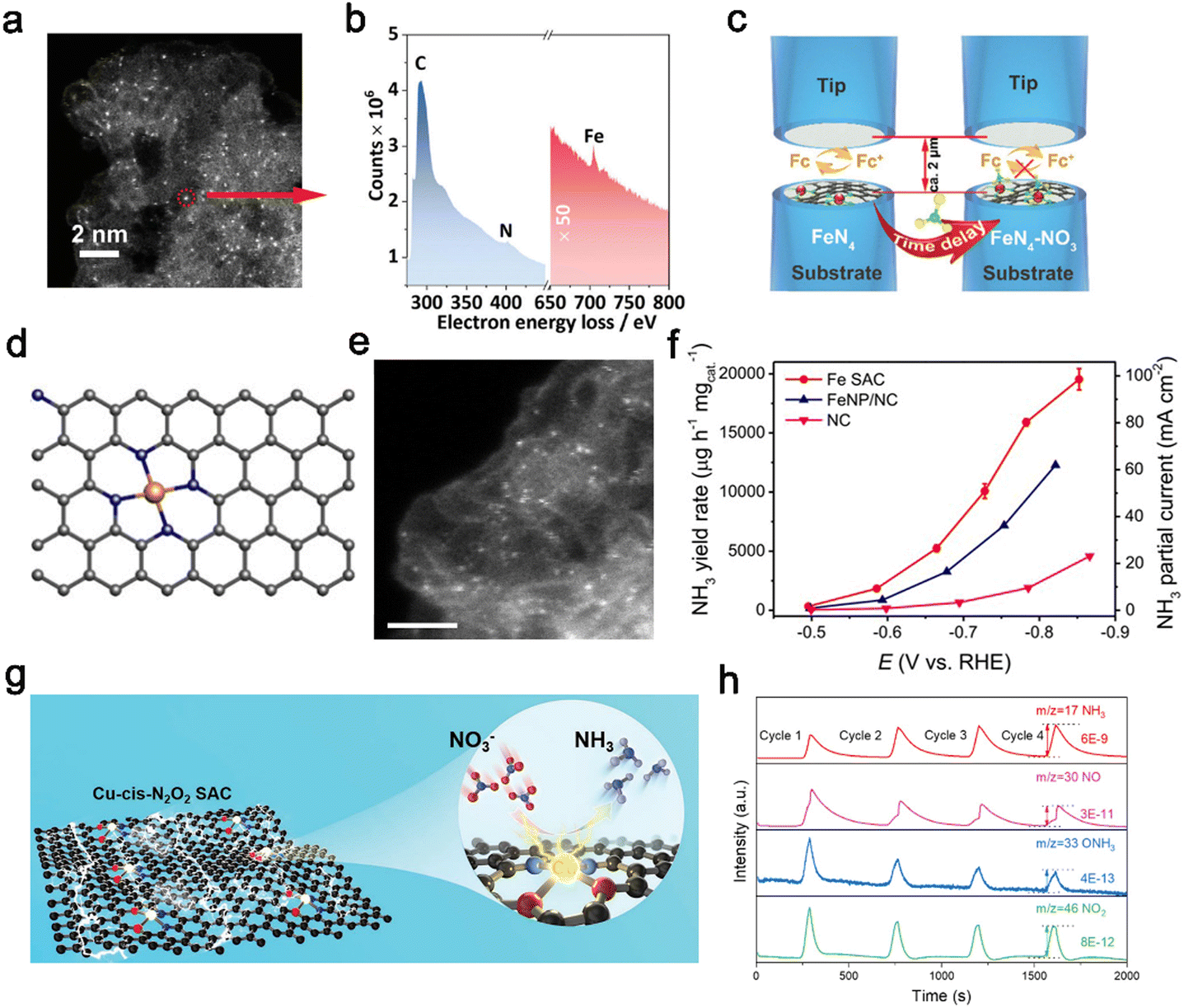 | ||
| Fig. 5 Single-atom engineering. (a) HADDF-STEM images of the Fe–PPy SACs. (b) The local EELS of the isolated Fe site. (c) Schematic of the SI-SECM setup for the titration of Fe sites.78 Copyright 2021, The Royal Society of Chemistry. (d) Schematic illusion of the Fe SAC. (e) AC MAADF-STEM. (f) NH3 yield rate and partial current density of Fe SAC, FeNP/NC, and NC.79 Copyright 2021, Springer. (g) Schematic illustration of the Cu-cis-N2O2 SAC. (h) DEMS measurements of nitrate reduction reaction over Cu-cis-N2O2 SAC.111 Copyright 2022, Wiley. | ||
Electrochemical tests showed that the Fe SAC delivered an impressive YR for NH3 of ca. 20000 μg h−1 mgcat−1, while Fe nanoparticles only exhibited poor catalytic behaviors (Fig. 5f).
Metallic Cu has relatively high activity for nitrate reduction, but it suffers from poor stability and nitrite accumulation.134,135 Fortunately, Cu incorporated on carbon support has been studied as a Cu SAC, which presents a superior catalytic performance in comparison to bulk Cu. Cu has been incorporated into PTCDA (3,4,9,10-perylenetetracarboxylic dianhydride) for the NO3−-to-NH3 reduction reaction with the properties of the proton/electron transfer regulation and HER suppression.136 More importantly, Feng et al. found that Cu single-atom anchored on nitrogenated carbon nanosheets (Cu–N–C) enabled significant alleviation of nitrite production. Nitrite is often observed for metallic Cu catalysts.137 Furthermore, Cheng et al.111 regulated the locally coordinated atoms of the center Cu single atom from four N atoms to two N and two O atoms, forming a cis-configuration Cu single-atom catalyst (Cu-cis-N2O2, Fig. 5g). Moreover, the control catalysts Cu-trans-N2O2 in the trans-configuration (two N atoms on opposite sides) were prepared to better investigate the coordination symmetry effect. In situ online differential electrochemical mass spectrometry (DEMS) was employed to investigate the proposed reaction pathways. The intermediate species NH3, NO, ONH3 and NO2 as shown in 5 h, confirmed the reaction pathways. Furthermore, a comparison of the partial density of states of the Cu 3d orbitals in Cu-trans-N2O2 and Cu-cis-N2O2 indicated that Cu-cis-N2O2 was more polar due to the asymmetric structure. The high polarity sites of Cu-cis-N2O2 could accumulate more NO3− around the surface and lower the energy barrier for the limiting step of *NO–*ONH by forming a π-complex. Therefore, Cu-cis-N2O2 exhibited outstanding NitRR activity with the highest YR of 27.84 mg h−1 cm−2 and impressive durability of continuous operation for more than 2000 h.
Due to strong interactions between a single-atom and substrates, the composition of the substrate is another factor influencing the electronic structure of SAC catalysts. Stable metals and metallic oxides have been investigated as promising candidates to replace the carbon support for anchoring single atoms. The metal oxides possess poor crystallinity with abundant dangling bonds and defects, providing hosts for the dispersion of single atoms.141 Lee et al.77 reported a highly electron deficient (electrophile) f-block GdSA stabilized on the surface of an oxygen-defect-rich NiO (denoted as GdSA-D-NiO400) support for efficient NO3−-to-NH3 reduction. The synergistic coupling between the highly electron-deficient GdSA and the defective NiO substrate can significantly enhance the NH3 selectivity and yield rate by facilitating stronger NO3− adsorption, effectively stabilizing the NitRR intermediates and improving the protonation kinetics by capturing H* from water dissociation with a suppressed HER (Fig. 6a). As compared to D-NiO400, the GdSA-D-NiO400 with higher adsorption of NO3− was revealed by in situ/operando Raman measurement (Fig. 6b). The GdSA-D-NiO400 achieved a maximum NH3 FE of ca. 97% at −0.1 V vs. RHE and yield rate of ca. 628 μg mgcat.−1 h−1 for the alkaline NitRR, significantly outperforming the Gd nanoparticles (Gd-NPs), D-NiO, and defect-free GdSA-P-NiO400 (Fig. 6c).
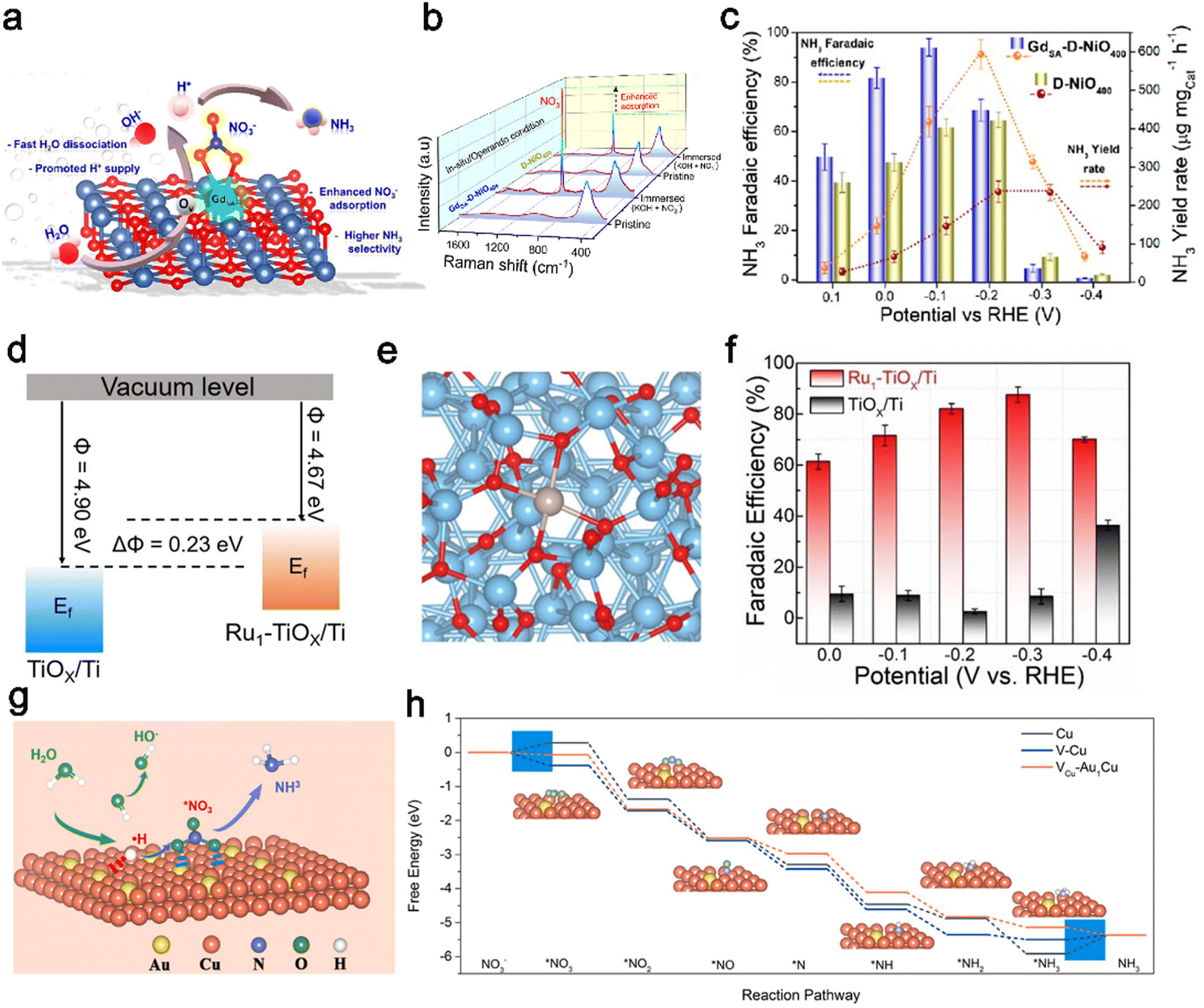 | ||
| Fig. 6 Single-atom engineering. (a) The proposed electrocatalytic nitrate reduction mechanism for the GdSA-D-NiO surface assisted via fast proton capture from H2O. (b) In situ Raman spectra for GdSA-D-NiO400 and D-Ni400 recorded under dry conditions and after immersion in electrolyte (KOH + NO3−). (c) FE and mass-normalized YR of GdSA-D-NiO400 and D-NiO400.77 Copyright 2022, American Chemical Society. (d) Schematic illustration of the work functions of Ru1–TiOx/Ti and TiOx/Ti. Ef refers to the Fermi level. (e) Structural model of Ru1–TiOx/Ti. (f) FE of NH3 at various potentials.124 Copyright 2022, Wiley. (g) Schematic illustration of the reaction mechanism for the NitRR on the Vcu-Au1CuSAAs surface. (h) Free energy diagrams for the NitRR to NH3 on Cu NSs(111), V-Cu NSs(111) and Vcu-Au1CuSAAs(111) surfaces.125 Copyright 2023, Wiley. | ||
Similarly, Yao et al.124 proposed an inherent oxide anchoring strategy to synthesize a Ru single-atom monolithic electrode by fastening isolated Ru atoms on the amorphous layer of the Ti support (Ru1–TiOx/Ti). This method enhanced the metal–support interactions (MSI) effect between Ru single atoms and the Ti support, which played a key role in catalytic performance. Compared with TiOx/Ti (4.9 eV), the work function of Ru1–TiOx/Ti was reduced by 0.23 eV and accompanied by the upward shift of its Fermi level, indicating that metal Ru populated with more electrons into TiOx/Ti (Fig. 6d). The charge density difference of Ru1–TiOx/Ti in Fig. 6e further visualized the enhanced electronic MSI owing to a Bader charge transfer of 0.66 e from the Ru atom to the Ti support. Meanwhile, Ru1–TiOx/Ti exhibited impressive improvement in electrocatalytic behavior for the NitRR. As shown in Fig. 6f, Ru1–TiOx/Ti reached a maximum FE of 87.6% at −0.3 V with a superior YR of 22.2 mol g−1 h−1, far higher than that of TiOx/Ti.
Additionally, Zhang et al.125 synthesized a highly active Au single-atom catalyst for the NitRR by dispersing Au atoms on a Cu(111) surface with further construction of Cu vacancies (VCu-Au1Cu SAAs). The Cu vacancies and Au atoms coordinate to regulate the local electronic structure so that VCu-Au1Cu SAAs has a stronger ability to capture and crack H2O molecules into *H. As shown in Fig. 6g, the obtained *H can be devoted to the hydrogenation processes of NO3− and intermediates to ammonia. The free energy on VCu-Au1Cu SAAs surfaces is decreased gradually without any uphill step (Fig. 6h), demonstrating that the formation of NH3 from NO3− on the VCu-Au1Cu SAAs(111) was thermodynamically favorable. However, there were energy barriers on the reaction pathways of pure Cu nanosheets (Cu NSs) and Cu NSs with vacancies (V-Cu NSs), resulting in poor NitRR activity and selectivity. Moreover, VCu-Au1Cu SAAs(111) exhibited a maximum FE of 98.7% and the highest NH3 selectivity of 94.5%, much higher than that of Cu NSs (72.9%, 64.6%) and V-Cu NSs (81.1%, 81.4%). Overall, the rapid development of single-atom catalysis in the NitRR field suggests that it is reasonable to obtain robust SACs with high stability, selectivity and activity for industrial important reactions. The development of SACs as tunable catalysts is interesting and promising.
Size regulation
The catalytic behaviour of ammonia electrosynthesis is dependent upon the electronic structure of the catalyst, determining the adsorption of reactants and other reaction intermediates.145 Previous literature showed that the N2 conversion into NH3 catalyzed by metal nanoparticles (NPs) and nanoclusters (NCs) was relevant to the size of the catalyst.145–148 With the size of active sites decreasing, more unsaturated coordination sites are exposed and a more efficient atom-utilization rate is achieved. Metal clusters with sizes below about 2 nm show a discrete electronic structure and molecule-like properties. These features are crucial for the performance of catalysts. From the perspective of nitrate electroreduction, electrocatalysts should be designed for the simultaneous enhancement of the M–N bond and activation of nitrate. The regulation in the size of the catalyst would cause a dramatic tuning in the electronic environment of active centers, which could affect the intrinsic activity of the catalyst for the NitRR. Recently, Li et al.132 reported that Ru-based catalysts with sizes ranging from nanoparticles to nanoclusters demonstrated different performances. The photo-deposition of size-defined Ru nanoclusters (NCs, average size: ca. 1.66 nm) on TiO2 nanotubes (NTs) showed enhanced activity for NO3−-to-NH3 conversion with a maximum yield of ca. 600 μg h−1 cm−2 and a Faradaic efficiency (FE) of >90% across a broad range of potential in comparison with electrodeposited Ru nanoparticles (NPs, average size: ca. 23.78 nm) on TiO2 NTs (Fig. 7b and c). In particular, the theoretical calculations revealed that the Ru nanoclusters catalyst with the strong metal/substrate interaction and unsaturated coordination state enabled stronger NO3− affinity, the lower energy barrier of *NHO intermediate, and the suppressed HER compared with Ru nanoparticles, facilitating the NO3−-to-NH3 conversion.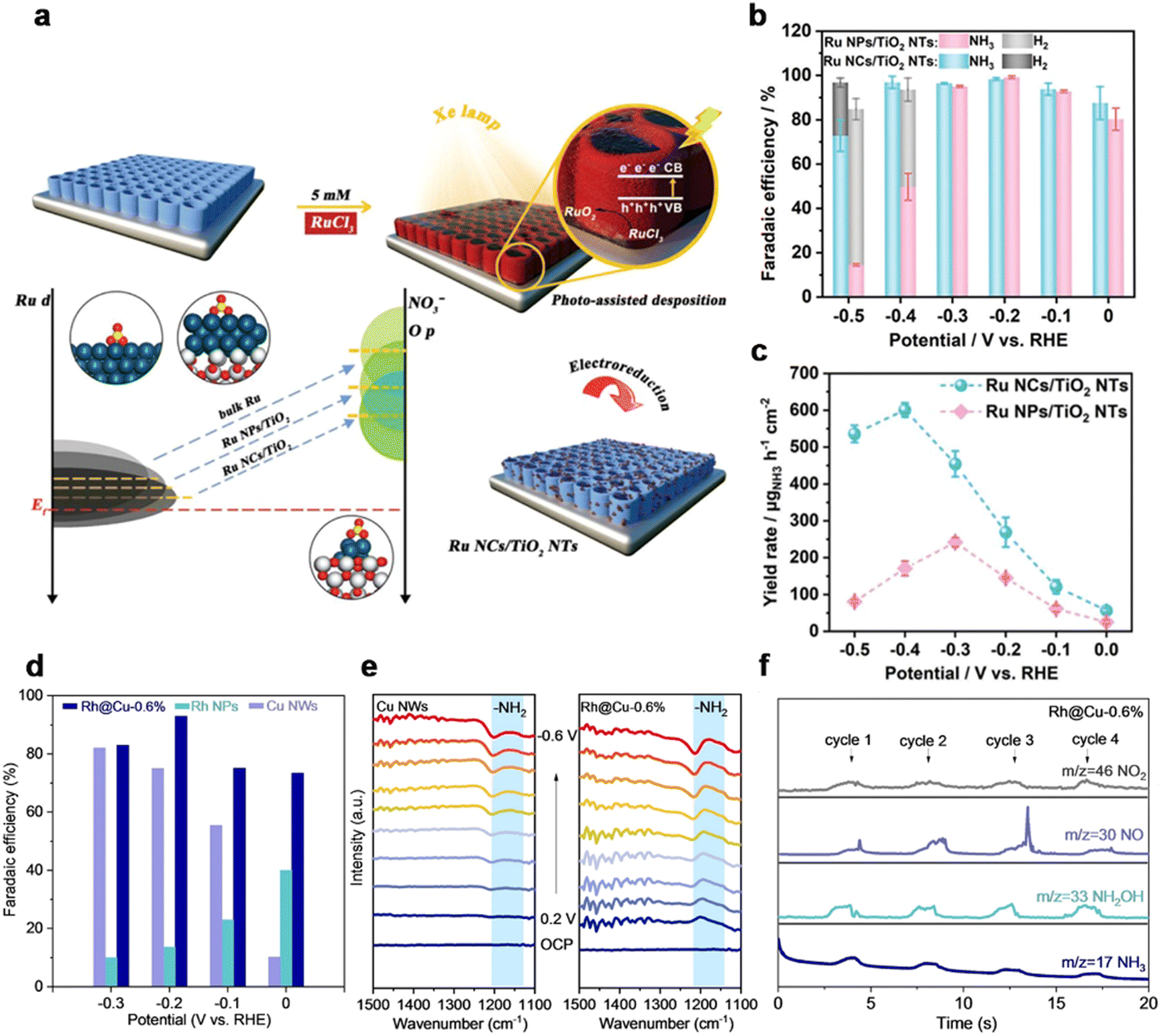 | ||
| Fig. 7 Size regulation. (a) Schematic of Ru nanoclusters loaded on TiO2 NTs via irradiation. (b) FE of NH3 and H2 for Ru NCs/TiO2 NTs and Ru NPs/TiO2 NTs at varying potentials. (c) The YR of NH3 for Ru NCs/TiO2 NTs and Ru NPs/TiO2 NTs.132 Copyright 2023, Wiley. (d) FE of NH3 over Cu NWs, Rh@Cu-0.6% and carbon cloth loaded with Rh NPs. (e) Electrochemical in situ infrared spectroscopy (IR) of Rh@Cu-0.6% and Cu NWs with different potentials at 0.1 M Na2SO4 electrolyte (pH 11.5) with 0.1 M KNO3. (f) Differential electrochemical mass spectrometry (DEMS) measurements of the NITRR over Rh@Cu-0.6%.133 Copyright 2022, Wiley. | ||
Additionally, Lou et al. prepared Rh clusters and Rh single-atoms dispersed onto Cu nanowires (NWs) for the NitRR. Rh@Ru-0.6% delivered the highest performance with an FE of 93% at −0.2 V vs. RHE and the highest ammonia yield rate of 1.27 mmol h−1 cm−2 compared with other catalysts with both lower and higher Rh loadings (Fig. 7d).133 Higher loading of Rh led to nanoparticles and lower loading resulted in too little Rh doping. In situ infrared spectroscopy (IR) results (Fig. 7e) showed an obvious –NH2 peak when Rh@Cu-0.6% catalyzed the NITRR. On-line differential electrochemical mass spectrometry (DEMS) was used to detect the intermediates during the NITRR (Fig. 7h). DEMS, in situ Raman and theoretical calculations proved that the high activity was attributed to the synergistic catalytic cooperation between Rh and Cu sites. Rh single atoms and Rh clusters provided H to *NO intermediates absorbed on Cu, thus promoting the hydrogenation step and ammonia formation. Ammonia synthesis is sensitive to the electronic states. Regulation of the catalyst's size would cause a dramatic change in electronic structure, which would promote the activity. However, the small size of catalysts might degrade into larger-sized aggregates during the catalytic process, leading to a decline in activity. So, it is crucial to explore a suitable support to immobilize the clusters and keep them stable.
Nanoconfinement
The fabrication of various nanostructures brings significant opportunities for electrocatalyst designs. One should take notice of the fact that the nanoconfinement effect of an electrocatalyst stems from a unique spatial environment, which would confine the reactant and thus promote interaction between the reactant molecule and the catalyst surface.149–153 This section induces the nanoconfinement effects in electrocatalysts for confining intermediates during the NitRR and the recent progress in this field. Li et al.138 reported the incorporation of CuOx active species into a TiO2-nanotube reactor for the NitRR (Fig. 8a). With the help of the experiment and finite element simulations, the nanotubular electrode exhibited remarkedly diminished NO2− in contrast to planar geometry, confirming the confinement effect toward intermediates (Fig. 8c). Thus, the nanoreactor performed a FE of 92.23% and yield rate of 1241.81 μg h−1 cm−2 for ammonia production (Fig. 8b). Furthermore, theoretical calculations were used to give insight into the NitRR mechanism, where TiO2/Cu2O/Cu active species with a lower energy barrier of *NHO formation facilitated more favorable NO3−-to-NH3 conversion efficiency as well as inhibited the HER. Given the confinement effect in strengthening reaction kinetics, Li et al.139 prepared a Cu single-atom gel (Cu SAG) electrocatalyst with spatial confinement. The aberration-corrected high-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) analysis with the bright spots marked in red circles confirmed the carbon-supported isolated single Cu atoms (Fig. 8d). Cu SAAs revealed a higher NitRR performance in comparison with Cu foil and PPy–C (Fig. 8e). In addition, finite element simulations were performed to demonstrate how the nanoconfinement effect promoted the transformation of the intermediate NO2− within 3D channels of the SAA catalyst during the NitRR.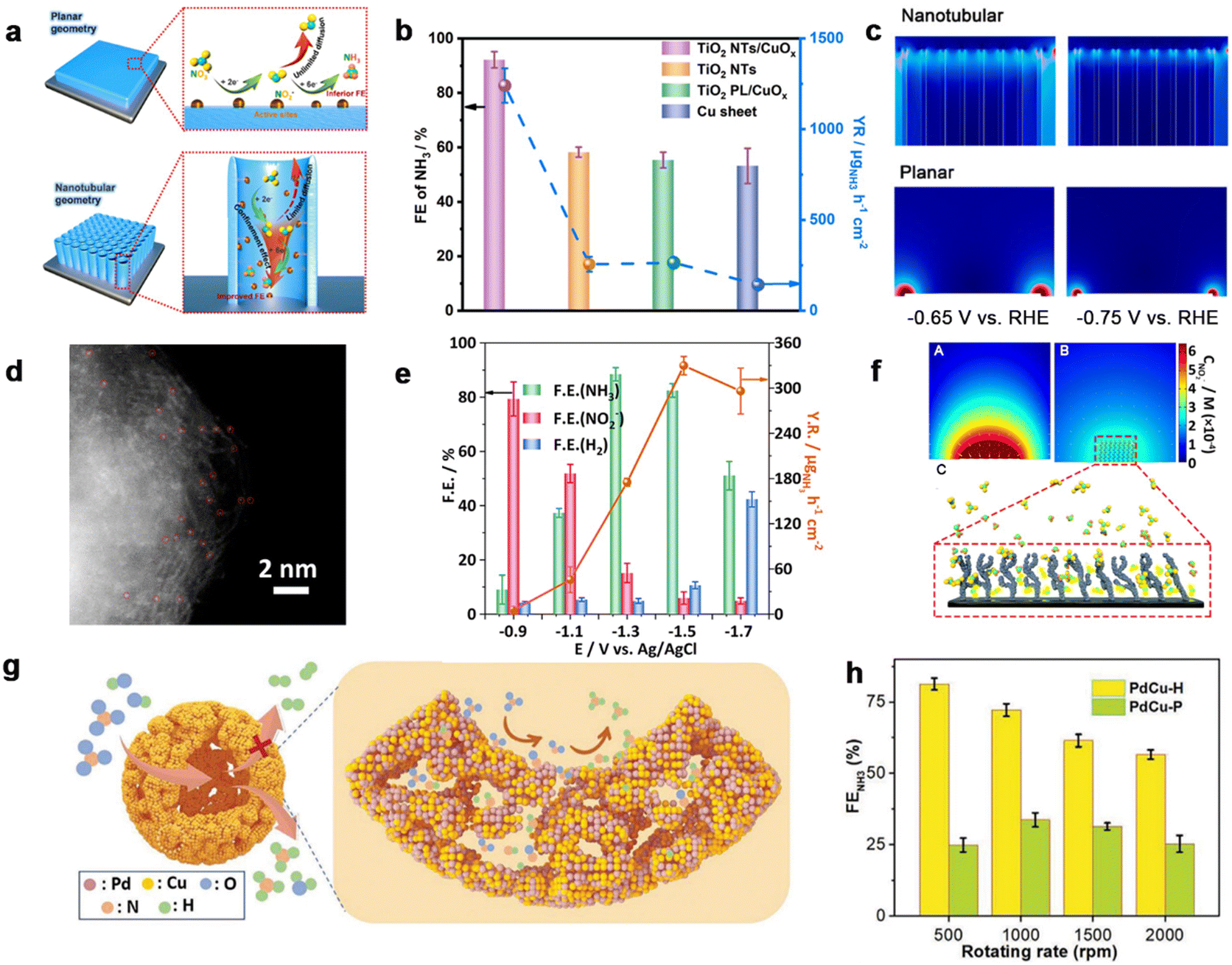 | ||
| Fig. 8 Confinement effect. (a) Schematic illustration of NO3−-to-NH3 reduction over an electrode with nanotubular geometries. (b) Comparison of FE of NH3 over TiO2 NTs/CuOx, TiO2 NTs, TiO2 PL/CuOx and Cu sheet at −0.75 V vs. RHE. (c) Simulated NO2− concentration distribution on nanotubular and planar electrodes.138 Copyright 2022, Elsevier. (d) Aberration-corrected HAADF-STEM image of Cu SAAs. (e) Faradaic efficiency of products and NH3 yield rate of Cu SAAs, Cu foil, and PPy–C at −0.9 V (vs. RHE). (f) A flat electrode (A) and a porous electrode (B) at the same potential, where NO3−/NO2− and NO2−/NH4+ transformation simultaneously occur. Schematic diffusion process of NO2− in interconnecting networks (C).139 Copyright 2023. (f) Schematic illustrations for NitRR over PdCu–P with high selectivity. (g) The relationships between FE of NH3 and rotating rates over PdCu–H, PdCu–P, and Pd–P.140 Copyright 2023, Wiley. | ||
Moreover, the PdCu hollow (PdCu–H) catalyst was prepared via in situ reduction and nucleation of PdCu nanocrystals along a self-assembled micelle of a well-designed surfactant,140 which aimed to confine the intermediates and promote selective NH3 electrosynthesis from nitrate (Fig. 8g). The PdCu–H catalyst disclosed a high NH3 FE of 87.3% and a remarkable NH3 yield rate of 0.551 mmol h−1 mg−1 at −0.3 V vs. RHE (Fig. 8h). Mechanism studies were investigated to provide details of the PdCu–H promoted selectivity of NitRR of PdCu–H for NitRR, suggesting that the confinement of a hollow structure not only activated NO3− but also facilitated the deeper electroreduction to NH3.
With the development of nanomaterials, nanoconfinement is becoming more important in electrochemistry. The above studies have investigated how the nanoconfinement tunes the NitRR reaction. There should be more studies on the influence of nanoconfinement on the kinetics and selectivities of electrochemical reactions.
Tandem catalysis
Electroreduction of nitrate to ammonia in microorganisms is a tandem process: combining nitrate reductase enzymes reducing NO3− to NO2− and nitrite reductase converting NO2−-to-NH3.154–156 The enzyme tandem system allows efficient NH3 generation based on coordination binding with NO3− and NO2−,157 meanwhile the concept of tandem reaction is implemented in electrocatalyst design for the NitRR. Schuhmann et al. coupled the potential-dependent intermediated phase of transition metals as cooperative catalytic sites for cascade NO3− to NH3 conversion, demonstrating the tandem catalyst design strategy (Fig. 9a).142 In this work, Cu/Co-base binary metal sulfides were electrochemically transformed into Cu/CuOx–Co/CoO hybrids. The rate constants k1 and k2 of each catalyst for NO3−-to-NO2− and NO2−-to-NH3 reactions were evaluated to validate that Cu and Co active phases synergistically catalyze the tandem NitRR (Fig. 9b).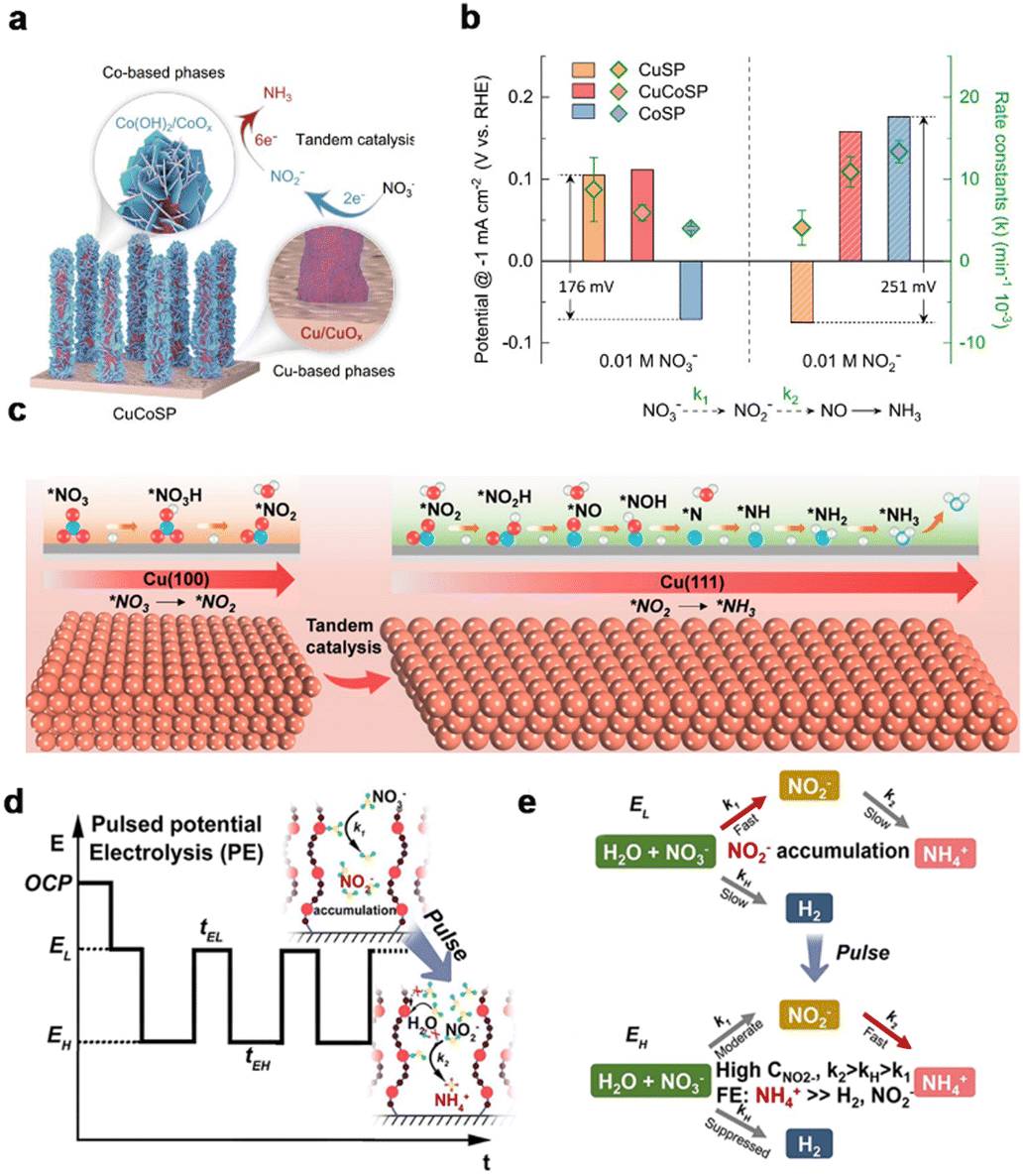 | ||
| Fig. 9 Tandem catalysis. (a) Schematic illustration of the structure of the Cu/Co-based binary “tandem catalyst”. (b) The LSV-derived potentials and the calculated reaction constants for NO3− and NO2− reduction on CuSP, CoSP and CuCoSP catalysts.142 Copyright 2022, Springer. (c) Tandem interaction of Cu(100) and Cu(111) facets.143 Copyright 2023, Wiley. (d) Profile of the applied potential for PE. (e) Reaction pathways of the NO3RR through the tandem NO2− accumulation–conversion process with PE.144 Copyright 2023, American Chemical Society. | ||
Recently, a tandem catalyst has been developed for enhancing NH3 selectivity. Inspired by Cu facets with different adsorption of reactants and intermediates, Wang et al.143 developed Cu nanosheets in situ derived from CuO nanosheets to catalyze the NitRR for NH3 production. The catalyst delivered an improved NH3 yield rate of 1.41 mmol h−1 cm−2 and FE of ca. 88% at 365 mA cm−2. Electrochemical studies and DFT calculations featured the tandem interaction of the Cu(100) and Cu(111) facets. The Cu(100) facets were responsible for the absorption of NO3− and conversion of NO3−-to-NO2−, while the Cu(111) facets contribute to the hydrogenation of NO2−, thereby promoting the tandem catalysis from NO3− to NH3 (Fig. 9c). As further proof of the advantages of tandem catalysts, Li et al. constructed a Cu single-atom gel electrocatalyst for highly efficient nitrate-to-ammonia conversion enabled by tandem catalysis of unfavorable intermediates.144 A pulse electrosynthesis strategy was proposed (Fig. 9d) to accumulate the localized nitrite. A locally concentrated NO2− enabled tandem accumulation–conversion process promoted nitrate-to-ammonia electro reduction (Fig. 9e). In summary, the development of materials for tandem electrocatalysis has attracted attention. Tandem catalysts with multifunctional active sites can promote the selectivity and efficiency of production, by eliminating the unnecessary separation steps and reducing the cost and energy consumption. Additionally, toxic or unstable intermediates can be decreased during tandem catalysis.
Summary and outlook
The imbalance in the global nitrogen cycle has attracted wide attention, as it causes considerable environmental and human health problems. The removal of nitrates from wastewater has been achieved using different processes including physical, chemical, and biological technologies. Among them, electrocatalytic nitrate reduction has attracted much attention because of various advantageous features, such as mild operation conditions, high efficiency, and no sludge generation. Besides, this process does not depend on fossil fuels but uses renewable electricity without carbon dioxide emission. Additionally, NH3 is a key commodity product, which can be used in fertilizers, promising energy carriers, and chemical precursors. From the perspective of addressing environmental pollution and converting waste into value-added products, electroreduction of nitrate to NH3 opens up the field for an alternative, sustainable route for ammonia synthesis. The storage of large amounts of ammonium nitrate poses a serious explosion risk. Hence, the electrocatalytic NO3−-to-NH3 conversion can facilitate the production of ammonia on demand, lowering the risk of storage of ammonium nitrate. However, the nitrate reduction reaction is a complicated process including multiple proton and electron transfer. It is hoped to construct catalysts with stability and specific geometric structures, facilitating activation of nitrate and enhancing the mass transfer of protons and electrons. Currently, it is necessary to explore design strategies for electrocatalysts for the practical application of electrocatalytic nitrate reduction technology. The composition, geometric structure, and size of the catalyst are all key factors that affect the activity of the catalyst. In this review, we have summarized catalyst design strategies for improving the activity and efficiency of the nitrate reduction reaction. First, we discussed the electrocatalytic nitrate reduction reaction mechanism. Then, recent catalyst design strategies for the NitRR were summarized including alloying, doping, single-atom engineering, size regulation, nanoconfinement effect, and tandem catalysis, which are highlighted in the development of efficient catalysts as shown in Table 1. In particular, doping and alloying engineering change the surface electronic structure of active sites, while single-atom engineering and size regulation not only increase the exposure sites but also regulate the electronic structure. Besides, confinement controls the mass transport and tandem catalysis involves cooperative sites that facilitate the proton–electron transfer. Although such strategies enable boosting catalytic efficiency and activity, challenges remain in the future development of this field.(1) Insight into the mechanism of the nitrate reduction reaction on catalysts provides meaningful guidance for choosing appropriate design strategies. More attention should be paid to tailoring the adsorption strength of NO3− and Had. Strong adsorption of NO3− on the catalyst would be favorable for the NitRR. When the proton affinity of catalyst active sites is high, the competing HER dominates the majority of active sites. The favorable adsorption of Had. not only supplies protons for deoxygenation and protonation steps but also prohibits the formation of H2. Thus, the coverage of Had. should be adjusted by regulating the electronic structure of catalysts. Theoretical calculation is one of the most important methods to describe the electronic structures of catalysts. Therefore, one would focus on the rational theoretical simulation, which could optimize the adsorption of active intermediates and thus suppress the competing side reactions.
(2) During the NitRR process, in situ and operando characterization methods are significant for monitoring the change of catalyst structure and intermediate generation at a molecular level. For example, attenuated total reflectance surface-enhance infrared absorption spectroscopy,159 surface-enhanced Raman spectroscopy,160 and differential electrochemical mass spectrometry26 would contribute to providing the adsorption states and conformation of intermediates on the catalyst surface. Additionally, scanning electrochemical microscopy and scanning electrochemical cell microscopy have been recognized as powerful tools for studying the local information at the catalyst surface during various reactions, including products, intermediates, active sites, kinetic rates, etc.161–165 Investigating real-time working state information of the catalyst is crucial for understanding the effects of structure–activity relationships for the nitrate reduction reaction and giving directions for the future electrocatalyst design strategies, thereby promoting the efficiency of this reaction.
(3) With regard to designing an electrocatalyst, stability is another important parameter. It is evaluated by consecutive recycling tests at the optimal potential, and a slight fluctuation in the yield rate and faradaic efficiency of the product ammonia represents the good stability of the catalyst. Good stability of catalysts is a prerequisite for industrial applications, while the reported catalysts hardly meet the durability standard. After long-time operation, catalysts may encounter related problems e.g., geometric structure collapse, active sites losing activity, corrosion, and others. Along with high efficiency and selectivity, enhancing stability should be considered as part of catalyst designing. We should choose the cathode material with high stability and excellent chemical and physical properties. Besides, we aim to achieve a controllable morphological structure to give good stability and high activity through tuning the edge structure, doping and some other methods.166–168
(4) Hydrogen from the competing HER at a large-scale nitrate electroreduction should not be ignored. From the perspective of safety, a mass of hydrogen is prominent. When the concentration of nitrate is low, the HER becomes serious. More research on the selectivity of catalysts for hindering H2 generation is required. The change in the electronic structure of catalysts could regulate the adsorption of hydrogen to favor ammonia production with suppressed hydrogen evolution.
(5) Moreover, upcycling of nitrate wastes into value-added ammonia is very interesting. It is necessary to extract and collect high-purity ammonia products. Designing catalysts with industrial-relevant current density is one of the key factors for the green and large-scale production of ammonia. Meanwhile, researchers should try to design superior reaction and production conditions, feasible solutions with low energy consumption and low carbon for ammonia collection due to the original intention of environmental protection.
(6) Nowadays, 90% of synthetic ammonia is produced using the Haber–Bosch process, which consumes about 500 kJ to produce 1 mol of NH3, equivalent to nearly 4 kW h per kilogram of ammonia produced. The application of reported electrocatalysts to the actual ammonia synthesis industry requires assessing their power consumption in terms of their cell voltage, partial current density, and productivity. For example, the strained ruthenium nanocluster catalyst requires about 22 kW h per kilogram of ammonia produced. However, the price of electricity will constitute a large part of the overall expenses, thus, the cost required for the electrochemical synthesis of marketable NH4NO3 can be directly assessed according to the local electricity price. As a result, the production cost of electrocatalytic ammonia synthesis (approximately $776 per metric ton ammonium nitrate) is very close to that of the current Haber–Bosch process ($560 per metric ton). On the other hand, the electricity supply can be replaced by renewable energy in the future, which promotes the reduction of production costs. Overall, the electrochemical synthesis of ammonia has potential economic benefits and is a promising candidate to replace the Haber–Bosch process.
By taking a forward outlook, further advancing the field of electroreduction of nitrate to ammonia may be decisive for an environment compatible energy economy. A well-designed electrocatalyst can boost the activity and selectivity of the NitRR.139,169 We hope this review helps researchers to develop new catalyst strategies to realize green ammonia electrosynthesis from nitrate under mild conditions. In addition, outstanding electrocatalysts are encouraged to promote the nitrate electroreduction technology from the laboratory to industry, thereby managing nitrate-rich wastewater.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. L. thanks the National Natural Science Foundation of China (52202372), the Sichuan Science and Technology Program (2023NSFSC0436, 2023NSFSC0089), and the Fundamental Research Funds for the Central Universities (YJ2021151). G. Y. acknowledges the funding support from the U.S. Department of Energy, Office of Science, Basic Energy Sciences under Award DE-SC0019019, Norman Hackerman Award in Chemical Research by the Welch Foundation, and Camille Dreyfus Teacher-Scholar Award.References
- J. N. Galloway, A. R. Townsend, J. W. Erisman, M. Bekunda, Z. Cai, J. R. Freney, L. A. Martinelli, S. P. Seitzinger and M. A. Sutton, Science, 2008, 320, 889–892 CrossRef CAS.
- D. E. Canfield, A. N. Glazer and P. G. Falkowski, Science, 2010, 330, 192–196 CrossRef CAS PubMed.
- J. G. Chen, R. M. Crooks, L. C. Seefeldt, K. L. Bren, R. M. Bullock, M. Y. Darensbourg, P. L. Holland, B. Hoffman, M. J. Janik, A. K. Jones, M. G. Kanatzidis, P. King, K. M. Lancaster, S. V. Lymar, P. Pfromm, W. F. Schneider and R. R. Schrock, Science, 2018, 360, eaar6611 CrossRef PubMed.
- M. Duca and M. T. M. Koper, Energy Environ. Sci., 2012, 5, 9726–9742 RSC.
- R. J. Diaz and R. Rosenberg, Science, 2008, 321, 926–929 CrossRef CAS.
- S. Garcia-Segura, M. Lanzarini-Lopes, K. Hristovski and P. Westerhoff, Appl. Catal., B, 2018, 236, 546–568 CrossRef CAS.
- P. J. Weyer, J. R. Kantamneni, X. Lu, M. H. Ward and J. R. Cerhan, Epidemiology, 2008, 12, 327–338 CrossRef.
- J. O. Lundberg, E. Weitzberg, J. A. Cole and N. Benjamin, Nat. Rev. Microbiol., 2004, 2, 593–602 CrossRef CAS.
- N. Gruber and J. N. Galloway, Nature, 2008, 451, 293–296 CrossRef CAS.
- C. Yu, X. Huang, H. Chen, H. C. J. Godfray, J. S. Wright, J. W. Hall, P. Gong, S. Ni, S. Qiao, G. Huang, Y. Xiao, J. Zhang, Z. Feng, X. Ju, P. Ciais, N. C. Stenseth, D. O. Hessen, Z. Sun, L. Yu, W. Cai, H. Fu, X. Huang, C. Zhang, H. Liu and J. Taylor, Nature, 2019, 567, 516–520 CrossRef CAS PubMed.
- R. Epsztein, O. Nir, O. Lahav and M. Green, Chem. Eng. J., 2015, 279, 372–378 CrossRef CAS.
- A. D. Fonseca, J. G. Crespo, J. S. Almeida and M. A. Reis, Energy Environ. Sci., 2000, 34, 1557–1562 CAS.
- F. Rezvani, M.-H. Sarrafzadeh, S. Ebrahimi and H.-M. Oh, Environ. Sci. Pollut. Res., 2019, 26, 1124–1141 CrossRef CAS.
- S. Xu, K. Lan, J. Li, T. He and C. Hu, Sep. Purif. Technol., 2018, 204, 281–289 CrossRef CAS.
- N. Singh and B. R. Goldsmith, ACS Catal., 2020, 10, 3365–3371 CrossRef CAS.
- B. P. Chaplin, Acc. Chem. Res., 2019, 52, 596–604 CrossRef CAS PubMed.
- Y. Liu, K. Liu, P. Wang, Z. Jin and P. Li, Carbon Neutrality, 2023, 2, 14 CrossRef.
- J. M. McEnaney, S. J. Blair, A. C. Nielander, J. A. Schwalbe, D. M. Koshy, M. Cargnello and T. F. Jaramillo, ACS Sustainable Chem. Eng., 2020, 8, 2672–2681 CrossRef CAS.
- J. Liang, Q. Liu, A. A. Alshehri and X. Sun, Nano Res. Energy, 2022, 1, e9120010 CrossRef.
- K. E. Lamb, M. D. Dolan and D. F. Kennedy, Int. J. Hydrogen Energy, 2019, 44, 3580–3593 CrossRef CAS.
- J. Cha, T. Lee, Y.-J. Lee, H. Jeong, Y. S. Jo, Y. Kim, S. W. Nam, J. Han, K. B. Lee, C. W. Yoon and H. Sohn, Appl. Catal., B, 2021, 283, 119627 CrossRef CAS.
- C. Smith, A. K. Hill and L. Torrente-Murciano, Energy Environ. Sci., 2020, 13, 331–344 RSC.
- G. Meng, T. Wei, W. Liu, W. Li, S. Zhang, W. Liu, Q. Liu, H. Bao, J. Luo and X. Liu, Chem. Commun., 2022, 58, 8097–8100 RSC.
- T. Wei, H. Bao, X. Wang, S. Zhang, Q. Liu, J. Luo and X. Liu, ChemCatChem, 2023, 15, e202201411 CrossRef CAS.
- D. Qi, F. Lv, T. Wei, M. Jin, G. Meng, S. Zhang, Q. Liu, W. Liu, D. Ma, M. S. Hamdy, J. Luo and X. Liu, Nano Res. Energy, 2022, 1, e9120022 CrossRef.
- X. Zou, C. Chen, C. Wang, Q. Zhang, Z. Yu, H. Wu, C. Zhuo and T. C. Zhang, Sci. Total Environ., 2021, 800, 149645 CrossRef CAS.
- J. Yang, P. Sebastian, M. Duca, T. Hoogenboom and M. T. M. Koper, Chem. Commun., 2014, 50, 2148–2151 RSC.
- P. Wang, Z. Jin, P. Li and G. Yu, Chem Catal., 2022, 2, 1277–1287 CrossRef CAS.
- J.-Y. Zhu, Q. Xue, Y.-Y. Xue, Y. Ding, F.-M. Li, P. Jin, P. Chen and Y. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 14064–14070 CrossRef CAS.
- X. Fu, X. Zhao, X. Hu, K. He, Y. Yu, T. Li, Q. Tu, X. Qian, Q. Yue, M. R. Wasielewski and Y. Kang, Appl. Mater. Today, 2020, 19, 100620 CrossRef.
- Y. Wang, W. Zhou, R. Jia, Y. Yu and B. Zhang, Angew. Chem., Int. Ed., 2020, 59, 5350–5354 CrossRef CAS PubMed.
- J. Ding, X. Hou, Y. Qiu, S. Zhang, Q. Liu, J. Luo and X. Liu, Inorg. Chem. Commun., 2023, 151, 110621 CrossRef CAS.
- R. Li, T. Gao, P. Wang, W. Qiu, K. Liu, Y. Liu, Z. Jin and P. Li, Appl. Catal., B, 2023, 331, 122677 CrossRef CAS.
- Y. Yao, S. Zhu, H. Wang, H. Li and M. Shao, Angew. Chem., Int. Ed., 2020, 59, 10479–10483 CrossRef CAS PubMed.
- Y. Wang, C. Wang, M. Li, Y. Yu and B. Zhang, Chem. Soc. Rev., 2021, 50, 6720–6733 RSC.
- R. Lange, E. Maisonhaute, R. Robin and V. Vivier, Electrochem. Commun., 2013, 29, 25–28 CrossRef CAS.
- D. Xu, Y. Li, L. Yin, Y. Ji, J. Niu and Y. Yu, Front. Environ. Sci. Eng., 2018, 12, 9 CrossRef.
- J. Gao, B. Jiang, C. Ni, Y. Qi and X. Bi, Chem. Eng. J., 2020, 382, 123034 CrossRef CAS.
- A. R. Cook, N. Dimitrijevic, B. W. Dreyfus, D. Meisel, L. A. Curtiss and D. M. Camaioni, J. Phys. Chem. A, 2001, 105, 3658–3666 CrossRef CAS.
- Y. Li, Y. K. Go, H. Ooka, D. He, F. Jin, S. H. Kim and R. Nakamura, Angew. Chem., Int. Ed., 2020, 59, 9744–9750 CrossRef CAS.
- R. Jia, Y. Wang, C. Wang, Y. Ling, Y. Yu and B. Zhang, ACS Catal., 2020, 10, 3533–3540 CrossRef CAS.
- Z. Chen, J. Chen, G. Barcaro, T. M. Budnyak, A. Rokicińska, R. Dronskowski, S. Budnyk, P. Kuśtrowski, S. Monti and A. Slabon, Catal. Sci. Technol., 2022, 12, 3582–3593 RSC.
- X. Lu, H. Song, J. Cai and S. Lu, Electrochem. Commun., 2021, 129, 107094 CrossRef CAS.
- G. E. Dima, A. C. A. de Vooys and M. T. M. Koper, J. Electroanal. Chem., 2003, 554–555, 15–23 CrossRef CAS.
- C. Zhou, J. Bai, Y. Zhang, J. Li, Z. Li, P. Jiang, F. Fang, M. Zhou, X. Mei and B. Zhou, J. Hazard. Mater., 2021, 401, 123232 CrossRef CAS PubMed.
- H. O. N. Tugaoen, S. Garcia-Segura, K. Hristovski and P. Westerhoff, Sci. Total Environ., 2017, 599–600, 1524–1551 CrossRef PubMed.
- M. Fedurco, P. Kedzierzawski and J. Augustynski, J. Electrochem. Soc., 1999, 146, 2569 CrossRef CAS.
- R. Tenne, K. Patel, K. Hashimoto and A. Fujishima, J. Electroanal. Chem., 1993, 347, 409–415 CrossRef CAS.
- M. C. P. M. da Cunha, M. Weber and F. C. Nart, J. Electroanal. Chem., 1996, 414, 163–170 Search PubMed.
- H. Xu, Y. Ma, J. Chen, W.-X. Zhang and J. Yang, Chem. Soc. Rev., 2022, 51, 2710–2758 RSC.
- A. C. A. de Vooys, M. T. M. Koper, R. A. van Santen and J. A. R. van Veen, Electrochim. Acta, 2001, 46, 923–930 CrossRef CAS.
- A. S. Dutton, J. M. Fukuto and K. N. Houk, Inorg. Chem., 2005, 44, 4024–4028 CrossRef CAS PubMed.
- T. Yoshioka, K. Iwase, S. Nakanishi, K. Hashimoto and K. Kamiya, J. Phys. Chem. C, 2016, 120, 15729–15734 CrossRef CAS.
- J. Zheng, T. Lu, T. M. Cotton and G. Chumanov, J. Phys. Chem. B, 1999, 103, 6567–6572 CrossRef CAS.
- J. Yang, M. Duca, K. J. P. Schouten and M. T. M. Koper, J. Electroanal. Chem., 2011, 662, 87–92 CrossRef CAS.
- Y. Zeng, C. Priest, G. Wang and G. Wu, Small Methods, 2020, 4, 2000672 CrossRef CAS.
- F. T. Bonner and M. N. Hughes, Comments Inorg. Chem., 1988, 7, 215–234 CrossRef CAS.
- J. F. E. Gootzen, P. G. J. M. Peeters, J. M. B. Dukers, L. Lefferts, W. Visscher and J. A. R. van Veen, J. Electroanal. Chem., 1997, 434, 171–183 CrossRef CAS.
- I. Katsounaros and G. Kyriacou, Electrochim. Acta, 2008, 53, 5477–5484 CrossRef CAS.
- P. M. Tucker, M. J. Waite and B. E. Hayden, J. Appl. Electrochem., 2004, 34, 781–796 CrossRef CAS.
- G. E. Dima, G. L. Beltramo and M. T. M. Koper, Electrochim. Acta, 2005, 50, 4318–4326 CrossRef CAS.
- T. Chen, H. Li, H. Ma and M. T. M. Koper, Langmuir, 2015, 31, 3277–3281 CrossRef CAS PubMed.
- M. A. Hasnat, N. Ahamad, S. M. Nizam Uddin and N. Mohamed, Appl. Surf. Sci., 2012, 258, 3309–3314 CrossRef CAS.
- J. Lim, C.-Y. Liu, J. Park, Y.-H. Liu, T. P. Senftle, S. W. Lee and M. C. Hatzell, ACS Catal., 2021, 11, 7568–7577 CrossRef CAS.
- J. Li, G. Zhan, J. Yang, F. Quan, C. Mao, Y. Liu, B. Wang, F. Lei, L. Li, A. W. M. Chan, L. Xu, Y. Shi, Y. Du, W. Hao, P. K. Wong, J. Wang, S.-X. Dou, L. Zhang and J. C. Yu, J. Am. Chem. Soc., 2020, 142, 7036–7046 CrossRef CAS PubMed.
- M. R. Ehrenburg, A. I. Danilov, I. G. Botryakova, E. B. Molodkina and A. V. Rudnev, J. Electroanal. Chem., 2017, 802, 109–117 CrossRef CAS.
- K.-W. Kim, S.-M. Kim, Y.-H. Kim, E.-H. Lee and K.-Y. Jee, J. Am. Chem. Soc., 2007, 154, E145 CAS.
- H. Liu, J. Park, Y. Chen, Y. Qiu, Y. Cheng, K. Srivastava, S. Gu, B. H. Shanks, L. T. Roling and W. Li, ACS Catal., 2021, 11, 8431–8442 CrossRef CAS.
- M. Dortsiou and G. Kyriacou, J. Electroanal. Chem., 2009, 630, 69–74 CrossRef CAS.
- S. Cattarin, J. Appl. Electrochem., 1992, 22, 1077–1081 CrossRef CAS.
- C. Wang, Z. Liu, T. Hu, J. Li, L. Dong, F. Du, C. Li and C. Guo, ChemSusChem, 2021, 14, 1825–1829 CrossRef CAS PubMed.
- V. R. Stamenkovic, B. Fowler, B. S. Mun, G. Wang, P. N. Ross, C. A. Lucas and N. M. Marković, Science, 2007, 315, 493–497 CrossRef CAS.
- D. Kim, J. Resasco, Y. Yu, A. M. Asiri and P. Yang, Nat. Commun., 2014, 5, 4948 CrossRef CAS PubMed.
- Y. Wang, A. Xu, Z. Wang, L. Huang, J. Li, F. Li, J. Wicks, M. Luo, D.-H. Nam, C.-S. Tan, Y. Ding, J. Wu, Y. Lum, C.-T. Dinh, D. Sinton, G. Zheng and E. H. Sargent, J. Am. Chem. Soc., 2020, 142, 5702–5708 CrossRef CAS PubMed.
- Z. Dong, H. Yang, Z. Liu, P. Chen, Y. Chen, X. Wang and H. Chen, Fuel, 2021, 303, 121237 CrossRef CAS.
- S. A. Shah, Z. Ji, X. Shen, X. Yue, G. Zhu, K. Xu, A. Yuan, N. Ullah, J. Zhu, P. Song and X. Li, ACS Appl. Energy Mater., 2019, 2, 4075–4083 CrossRef CAS.
- A. Kumar, J. Lee, M. G. Kim, B. Debnath, X. Liu, Y. Hwang, Y. Wang, X. Shao, A. R. Jadhav, Y. Liu, H. Tüysüz and H. Lee, ACS Nano, 2022, 16, 15297–15309 CrossRef CAS PubMed.
- P. Li, Z. Jin, Z. Fang and G. Yu, Energy Environ. Sci., 2021, 14, 3522–3531 RSC.
- Z.-Y. Wu, M. Karamad, X. Yong, Q. Huang, D. A. Cullen, P. Zhu, C. Xia, Q. Xiao, M. Shakouri, F.-Y. Chen, J. Y. Kim, Y. Xia, K. Heck, Y. Hu, M. S. Wong, Q. Li, I. Gates, S. Siahrostami and H. Wang, Nat. Commun., 2021, 12, 2870 CrossRef CAS.
- S. Wei, W. Liu, C. Yang, P. Bai, X. Kong, W. Sun and L. Xu, Mater. Chem. Front., 2023, 7, 4723–4743 RSC.
- D. E. Kim and D. Pak, Chemosphere, 2019, 228, 611–618 CrossRef CAS PubMed.
- A. P. Leontiev, O. A. Brylev and K. S. Napolskii, Electrochim. Acta, 2015, 155, 466–473 CrossRef CAS.
- L. Mattarozzi, S. Cattarin, N. Comisso, R. Gerbasi, P. Guerriero, M. Musiani, L. Vázquez-Gómez and E. Verlato, ECS Electrochem. Lett., 2013, 2, D58 CrossRef CAS.
- M. Luo and S. Guo, Nat. Rev. Mater., 2017, 2, 17059 CrossRef CAS.
- W. Yu, J. Yu, M. Huang, Y. Wang, Y. Wang, J. Li, H. Liu and W. Zhou, Energy Environ. Sci., 2023, 16, 2991–3001 RSC.
- H. Yin, Z. Chen, S. Xiong, J. Chen, C. Wang, R. Wang, Y. Kuwahara, J. Luo, H. Yamashita, Y. Peng and J. Li, Chem Catal., 2021, 1, 1088–1103 CrossRef CAS.
- H. Miura and T. Shishido, Chem. Lett., 2021, 50, 346–352 CrossRef CAS.
- A. Lima, C. Coutanceau, J. M. LÉger and C. Lamy, J. Appl. Electrochem., 2001, 31, 379–386 CrossRef CAS.
- G. Yang, Q. Zhang, H. Yu and F. Peng, Particuology, 2021, 58, 169–186 CrossRef CAS.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- H. Huang, H. Jia, Z. Liu, P. Gao, J. Zhao, Z. Luo, J. Yang and J. Zeng, Angew. Chem., Int. Ed., 2017, 56, 3594–3598 CrossRef CAS PubMed.
- S. Roy, P. Pachfule and Q. Xu, Eur. J. Inorg. Chem., 2016, 4353–4357 CrossRef CAS.
- L. Fang, S. Wang, C. Song, X. Yang, Y. Li and H. Liu, J. Hazard. Mater., 2022, 421, 126628 CrossRef CAS.
- Y. Liu, B. Deng, K. Li, H. Wang, Y. Sun and F. Dong, J. Colloid Interface Sci., 2022, 614, 405–414 CrossRef CAS.
- Y. Luo, K. Chen, P. Shen, X. Li, X. Li, Y. Li and K. Chu, J. Colloid Interface Sci., 2023, 629, 950–957 CrossRef CAS.
- R. Zhang, Y. Guo, S. Zhang, D. Chen, Y. Zhao, Z. Huang, L. Ma, P. Li, Q. Yang, G. Liang and C. Zhi, Adv. Energy Mater., 2022, 12, 2103872 CrossRef CAS.
- N. Comisso, S. Cattarin, S. Fiameni, R. Gerbasi, L. Mattarozzi, M. Musiani, L. Vázquez-Gómez and E. Verlato, Electrochem. Commun., 2012, 25, 91–93 CrossRef CAS.
- P. Zhou, P. Niu, J. Liu, N. Zhang, H. Bai, M. Chen, J. Feng, D. Liu, L. Wang, S. Chen, C. T. Kwok, Y. Tang, R. Li, S. Wang and H. Pan, Adv. Funct. Mater., 2022, 32, 2202068 CrossRef CAS.
- J. Duan, S. Chen, M. Jaroniec and S. Z. Qiao, ACS Catal., 2015, 5, 5207–5234 CrossRef CAS.
- R. Kumar, S. Sahoo, E. Joanni, R. K. Singh, K. Maegawa, W. K. Tan, G. Kawamura, K. K. Kar and A. Matsuda, Mater. Today, 2020, 39, 47–65 CrossRef CAS.
- P. Xu, D. Wu, L. Wan, P. Hu and R. Liu, J. Colloid Interface Sci., 2014, 421, 160–164 CrossRef CAS.
- W. Chen, C. Ge, J. T. Li, J. L. Beckham, Z. Yuan, K. M. Wyss, P. A. Advincula, L. Eddy, C. Kittrell, J. Chen, D. X. Luong, R. A. Carter and J. M. Tour, ACS Nano, 2022, 16, 6646–6656 CrossRef CAS.
- Q. Zhang, D. Zhang, Y. Zhou, J. Qian, X. Wen, P. Jiang, L. Ma, C. Lu, F. Feng and X. Li, ChemistrySelect, 2022, 7, e202102581 CrossRef CAS.
- P. Zhang, Z. Wang, P. Wang, X. Hui, D. Zhao, Z. Zhang and L. Yin, ACS Appl. Energy Mater., 2022, 5, 3359–3368 CrossRef CAS.
- Y. Liu, Y. Ying, L. Fei, Y. Liu, Q. Hu, G. Zhang, S. Y. Pang, W. Lu, C. L. Mak, X. Luo, L. Zhou, M. Wei and H. Huang, J. Am. Chem. Soc., 2019, 141, 8136–8145 CrossRef CAS.
- Z. Jin, P. Li and D. Xiao, ChemSusChem, 2017, 10, 483–488 CrossRef CAS.
- X. Wan, F. Shen, J. Hu, M. Huang, L. Zhao, Y. Zeng, D. Tian, G. Yang and Y. Zhang, Int. J. Biol. Macromol., 2021, 180, 51–60 CrossRef CAS.
- G. Nazir, A. Rehman and S.-J. Park, J. CO2 Util., 2020, 42, 101326 CrossRef CAS.
- M. Demir, A. A. Farghaly, M. J. Decuir, M. M. Collinson and R. B. Gupta, Mater. Chem. Phys., 2018, 216, 508–516 CrossRef CAS.
- J. P. Paraknowitsch and A. Thomas, Energy Environ. Sci., 2013, 6, 2839–2855 RSC.
- X.-F. Cheng, J.-H. He, H.-Q. Ji, H.-Y. Zhang, Q. Cao, W.-J. Sun, C.-L. Yan and J.-M. Lu, Adv. Mater., 2022, 34, 2205767 CrossRef CAS.
- C. Zhu, S. Fu, Q. Shi, D. Du and Y. Lin, Angew. Chem., Int. Ed., 2017, 56, 13944–13960 CrossRef CAS PubMed.
- L. Wang, J. Wang, X. Gao, C. Chen, Y. Da, S. Wang, J. Yang, Z. Wang, J. Song, T. Yao, W. Zhou, H. Zhou and Y. Wu, J. Am. Chem. Soc., 2022, 144, 15999–16005 CrossRef CAS PubMed.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS.
- W. Zhang, X. Qin, T. Wei, Q. Liu, J. Luo and X. Liu, J. Colloid Interface Sci., 2023, 638, 650–657 CrossRef CAS PubMed.
- T. Gao, L. Qiu, M. Xie, Z. Jin, P. Li and G. Yu, Mater. Horiz., 2023, 10, 4270–4277 RSC.
- H. Niu, Z. Zhang, X. Wang, X. Wan, C. Shao and Y. Guo, Adv. Funct. Mater., 2021, 31, 2008533 CrossRef CAS.
- Y. Pan, R. Lin, Y. Chen, S. Liu, W. Zhu, X. Cao, W. Chen, K. Wu, W.-C. Cheong, Y. Wang, L. Zheng, J. Luo, Y. Lin, Y. Liu, C. Liu, J. Li, Q. Lu, X. Chen, D. Wang, Q. Peng, C. Chen and Y. Li, J. Am. Chem. Soc., 2018, 140, 4218–4221 CrossRef CAS PubMed.
- H. T. Chung, D. A. Cullen, D. Higgins, B. T. Sneed, E. F. Holby, K. L. More and P. Zelenay, Science, 2017, 357, 479–484 CrossRef CAS PubMed.
- L. Wang, W. Zhang, S. Wang, Z. Gao, Z. Luo, X. Wang, R. Zeng, A. Li, H. Li, M. Wang, X. Zheng, J. Zhu, W. Zhang, C. Ma, R. Si and J. Zeng, Nat. Commun., 2016, 7, 14036 CrossRef CAS PubMed.
- H. He, H. H. Wang, J. Liu, X. Liu, W. Li and Y. Wang, Molecules, 2021, 26, 6501 CrossRef CAS PubMed.
- Y. Liu, W. Qiu, P. Wang, R. Li, K. Liu, K. M. Omer, Z. Jin and P. Li, Appl. Catal., B, 2024, 340, 123228 CrossRef CAS.
- M. Xie, S. Tang, Z. Li, M. Wang, Z. Jin, P. Li, X. Zhan, H. Zhou and G. Yu, J. Am. Chem. Soc., 2023, 145, 13957–13967 CrossRef CAS.
- Y. Yao, L. Zhao, J. Dai, J. Wang, C. Fang, G. Zhan, Q. Zheng, W. Hou and L. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202208215 CrossRef CAS PubMed.
- Y. Zhang, X. Chen, W. Wang, L. Yin and J. C. Crittenden, Appl. Catal., B, 2022, 310, 121346 CrossRef CAS.
- Z. Chen, S. Mitchell, F. Krumeich, R. Hauert, S. Yakunin, M. V. Kovalenko and J. Pérez-Ramírez, ACS Sustainable Chem. Eng., 2019, 7, 5223–5230 CrossRef CAS.
- S. Mitchell, E. Vorobyeva and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2018, 57, 15316–15329 CrossRef CAS PubMed.
- M. Liu, J. Lee, T.-C. Yang, F. Zheng, J. Zhao, C.-M. Yang and L. Y. S. Lee, Small Methods, 2021, 5, 2001165 CrossRef CAS.
- Y. Liu, Z. Zhao, W. Wei, X. Jin, G. Wang, K. Li and Y. Lin, ACS Appl. Nano Mater., 2021, 4, 13001–13009 CrossRef CAS.
- W.-J. Niu, Y.-Y. Yan, R.-J. Li, W.-W. Zhao, J.-L. Chen, M.-J. Liu, B. Gu, W.-W. Liu and Y.-L. Chueh, Chem. Eng. J., 2023, 456, 140858 CrossRef CAS.
- Y. Wan, J. Xu and R. Lv, Mater. Today, 2019, 27, 69–90 CrossRef CAS.
- W. Qiu, M. Xie, P. Wang, T. Gao, R. Li, D. Xiao, Z. Jin and P. Li, Small, 2023, 19, 2300437 CrossRef CAS PubMed.
- H. Liu, X. Lang, C. Zhu, J. Timoshenko, M. Rüscher, L. Bai, N. Guijarro, H. Yin, Y. Peng, J. Li, Z. Liu, W. Wang, B. R. Cuenya and J. Luo, Angew. Chem., Int. Ed., 2022, 61, e202202556 CrossRef CAS.
- C. Roy, J. Deschamps, M. H. Martin, E. Bertin, D. Reyter, S. Garbarino, L. Roué and D. Guay, Appl. Catal., B, 2016, 187, 399–407 CrossRef CAS.
- D. Çirmi, R. Aydın and F. Köleli, J. Electroanal. Chem., 2015, 736, 101–106 CrossRef.
- G.-F. Chen, Y. Yuan, H. Jiang, S.-Y. Ren, L.-X. Ding, L. Ma, T. Wu, J. Lu and H. Wang, Nat. Energy, 2020, 5, 605–613 CrossRef CAS.
- T. Zhu, Q. Chen, P. Liao, W. Duan, S. Liang, Z. Yan and C. Feng, Small, 2020, 16, 2004526 CrossRef CAS.
- W. Qiu, X. Chen, Y. Liu, D. Xiao, P. Wang, R. Li, K. Liu, Z. Jin and P. Li, Appl. Catal., B, 2022, 315, 121548 CrossRef CAS.
- P. Li, L. Liao, Z. Fang, G. Su, Z. Jin and G. Yu, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2305489120 CrossRef PubMed.
- X. Min and B. Liu, Small, 2023, 2300794 CrossRef CAS PubMed.
- N. Daelman, M. Capdevila-Cortada and N. López, Nat. Mater., 2019, 18, 1215–1221 CrossRef CAS PubMed.
- W. He, J. Zhang, S. Dieckhöfer, S. Varhade, A. C. Brix, A. Lielpetere, S. Seisel, J. R. C. Junqueira and W. Schuhmann, Nat. Commun., 2022, 13, 1129 CrossRef CAS.
- Y. Fu, S. Wang, Y. Wang, P. Wei, J. Shao, T. Liu, G. Wang and X. Bao, Angew. Chem., Int. Ed., 2023, 62, e202303327 CrossRef CAS.
- P. Li, R. Li, Y. Liu, M. Xie, Z. Jin and G. Yu, J. Am. Chem. Soc., 2023, 145, 6471–6479 CrossRef CAS PubMed.
- L. Li, Y.-F. Jiang, T. Zhang, H. Cai, Y. Zhou, B. Lin, X. Lin, Y. Zheng, L. Zheng, X. Wang, C.-Q. Xu, C.-T. Au, L. Jiang and J. Li, Chem, 2022, 8, 749–768 CAS.
- M. Zybert, A. Tarka, W. Patkowski, H. Ronduda, B. Mierzwa, L. Kępiński and W. Raróg-Pilecka, Catalysts, 2022, 12, 1285 CrossRef CAS.
- W. Raróg-Pilecka, E. Miśkiewicz, D. Szmigiel and Z. Kowalczyk, J. Catal., 2005, 231, 11–19 CrossRef.
- R. Li, T. Gao, W. Qiu, M. Xie, Z. Jin and P. Li, Nano Res., 2023 DOI:10.1007/s12274-023-6094-z.
- M. Seo and T. D. Chung, Curr. Opin. Electrochem., 2019, 13, 47–54 CrossRef CAS.
- J. Wordsworth, T. M. Benedetti, S. V. Somerville, W. Schuhmann, R. D. Tilley and J. J. Gooding, Angew. Chem., Int. Ed., 2022, 61, e202200755 CrossRef CAS PubMed.
- J. H. Bae, J.-H. Han and T. D. Chung, Phys. Chem. Chem. Phys., 2012, 14, 448–463 RSC.
- H. Li, J. Xiao, Q. Fu and X. Bao, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 5930–5934 CrossRef CAS.
- Y. Zhou, S. Chen, S. Xi, Z. Wang, P. Deng, F. Yang, Y. Han, Y. Pang and B. Y. Xia, Cell Rep. Phys. Sci., 2020, 1, 100182 CrossRef.
- M. Duca, J. R. Weeks, J. G. Fedor, J. H. Weiner and K. A. Vincent, ChemElectroChem, 2015, 2, 1086–1089 CrossRef CAS.
- R. D. Milton and S. D. Minteer, ChemPlusChem, 2017, 82, 513–521 CrossRef CAS PubMed.
- C. L. Ford, Y. J. Park, E. M. Matson, Z. Gordon and A. R. Fout, Science, 2016, 354, 741–743 CrossRef CAS PubMed.
- C. Coelho and M. J. Romão, Protein Sci., 2015, 24, 1901–1911 CrossRef CAS PubMed.
- G. Zhang, X. Li, K. Chen, Y. Guo, D. Ma and K. Chu, Angew. Chem., Int. Ed., 2023, 62, e202300054 CrossRef CAS PubMed.
- Y. Yao, S. Zhu, H. Wang, H. Li and M. Shao, J. Am. Chem. Soc., 2018, 140, 1496–1501 CrossRef CAS PubMed.
- M. J. Weaver, J. Raman Spectrosc., 2002, 33, 309–317 CrossRef CAS.
- Z. Jin, Anal. Chem., 2023, 95, 6477–6489 CrossRef CAS PubMed.
- Z. Jin and A. J. Bard, Angew. Chem., Int. Ed., 2021, 60, 794–799 CrossRef CAS PubMed.
- Z. Jin, P. Li, Z. Fang and G. Yu, Acc. Chem. Res., 2022, 55, 759–769 CrossRef CAS.
- Z. Jin, P. Li, Y. Meng, Z. Fang, D. Xiao and G. Yu, Nat. Catal., 2021, 4, 615–622 CrossRef CAS.
- H. Li, Y. Guo and Z. Jin, Carbon Neutrality, 2023, 2, 22 CrossRef.
- T. Kajana, A. Pirashanthan, D. Velauthapillai, A. Yuvapragasam, S. Yohi, P. Ravirajan and M. Senthilnanthanan, RSC Adv., 2022, 12, 18041–18062 RSC.
- Z. Zhang, S. Tang, X. Lin, C. Liu, S. Hu and Q. Huang, Appl. Surf. Sci., 2022, 584, 152546 CrossRef CAS.
- R. He, X. Huang and L. Feng, Energy Fuels, 2022, 36, 6675–6694 CrossRef CAS.
- Y. Zhang, S. Li, W. Zheng and X. Wang, Nano Res. Energy, 2023, 2, e9120068 CrossRef.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |