
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceActivating lattice oxygen by a defect-engineered Fe2O3–CeO2 nano-heterojunction for efficient electrochemical water oxidation†
Qiuping
Huang‡
a,
Guang-Jie
Xia‡
 b,
Bo
Huang
a,
Dongling
Xie
a,
Jianan
Wang
a,
Dan
Wen
a,
Dunmin
Lin
a,
Chenggang
Xu
a,
Lei
Gao
c,
Zhenduo
Wu
d,
Jinqi
Wu
e,
Fengyu
Xie
*a,
Wenhan
Guo
*b and
Ruqiang
Zou
*c
b,
Bo
Huang
a,
Dongling
Xie
a,
Jianan
Wang
a,
Dan
Wen
a,
Dunmin
Lin
a,
Chenggang
Xu
a,
Lei
Gao
c,
Zhenduo
Wu
d,
Jinqi
Wu
e,
Fengyu
Xie
*a,
Wenhan
Guo
*b and
Ruqiang
Zou
*c
aCollege of Chemistry and Materials Science, Sichuan Normal University, Chengdu 610066, P. R. China. E-mail: xiefengyu@sicnu.edu.cn
bDongguan Key Laboratory of Interdisciplinary Science for Advanced Materials and Large-Scale Scientific Facilities, School of Physical Sciences, Great Bay University, Dongguan 523000, China. E-mail: whguo@gbu.edu.cn
cSchool of Materials Science and Engineering, Peking University, Beijing 100871, China. E-mail: rzou@pku.edu.cn
dCity University of Hong Kong (Dongguan), Dongguan 523000, China
eShimadzu China Co. Ltd, Guangzhou, 510656, China
First published on 14th June 2024
Abstract
The sluggish anodic oxygen evolution reaction (OER) is currently the major hinderance for hydrogen production from water splitting. Iron-based materials are promising cost-effective candidates for OER electrocatalysts, however their low intrinsic activity limits their performance. Here we report a defect-engineered Fe2O3–CeO2 heterojunction with rich oxygen vacancies (Fe2O3@CeO2–OV), exhibiting ultralow overpotential of 172 mV at 10 mA cm−2 and 317 mV at 1000 mA cm−2, respectively, alongside superior stability and durability. Advanced characterization and density functional theory calculations demonstrate that defect engineering combined with heterojunction formation activates the lattice oxygen, switching the reaction pathway from the conventional adsorbate evolution mechanism (AEM) to the lattice oxygen mechanism (LOM). The oxygen vacancies are revealed to form preferably on CeO2 and induce not only electronic but also geometric modulation, contributing to strong Fe2O3–CeO2 interfacial interaction and charge transfer from CeO2 to Fe2O3, facilitating the O2 desorption and boosting the intrinsic activity.
Broader contextWater splitting powered by renewable power sources (like solar, wind or tide energy) is the most promising approach to produce green hydrogen for large-scale commercialization applications. The sluggish anodic oxygen evolution reaction (OER) is regarded as a major challenge for current overall water electrolysis research. Activating the lattice oxygen of oxide-based catalysts presents a fascinating new approach to switch the reaction pathway from a traditional adsorbate evolution mechanism (AEM) to the novel lattice oxygen mechanism (LOM), by passing the linear scaling limitations and thus reducing the overpotentials and thereby total energy consumption. In this work, we report a combination of defect engineering and heterojunction interfacial engineering to successfully construct a defect-rich Fe2O3–CeO2 nano-heterojunction material (denoted as Fe2O3@CeO2-OV) loaded onto porous nickel foam (NF) as self-supporting electrode for efficient OER catalysis. Our approach successfully activates the lattice oxygen, and this heterojunction electrode with rich oxygen vacancies exhibits excellent OER catalytic performance with a low potential of 172 mV at 10 mA cm−2 and 317 mV at 1000 mA cm−2, among the highest data for reported Fe-based catalysts, together with superior stability and durability. This work may provide new methodological and theoretical inspirations for researchers from broader research contexts. |
Introduction
Hydrogen as one of the cleanest energy carriers is essential for establishing a carbon-free sustainable eco-energy system.1,2 Electrocatalytic overall water splitting (OWS) combined with renewable power sources is a promising approach for mass production of green hydrogen from earth-abundant and pollutant-free water.3–6 The anodic oxygen evolution reaction (OER) is currently believed to be the major bottleneck of water electrolysis due to the sluggish reaction kinetics and limited performance of electrocatalysts.7,8 Catalysts following the traditional adsorbate evolution mechanism (AEM) could not overcome the intrinsic overpotential threshold of around 370 mV limited by the Sabatier's principle and linear-scaling relationship of key intermediates (*O, *OH, *OOH, etc.), impeding the improvement of catalytic performance.9,10 Recent studies have however revealed the critical role of lattice oxygen involved in the water oxidation process via formation and healing of surficial oxygen vacancies (OV) in certain oxide and (oxy)hydroxide materials, known now as the lattice oxygen mechanism (LOM).11 The LOM-based OER proceeds through decoupled proton and electron transfer, bypassing the limitation of the linear scaling AEM, thus reducing overpotential and thereby energy consumption. Therefore, developing novel OER electrocatalysts based on the LOM has become one of the research frontiers of water splitting.Yang Shao-Horn's group reported the early experimental verification of lattice oxygen participation in the OER for (La, Sr) CoO3−δ perovskite oxides by combining on-line electrochemical mass spectrometry (OLEMS) 18O-isotopic-labeled measurements and density functional theory (DFT) calculations,12 followed by numerous reports on other materials including perovskites,13 spinels,14 bimetallic oxyhydroxides15 and layered double hydroxides (LDHs).16 In addition to the above-mentioned materials, transition-metal oxides (TMOs) have been extensively studied as potential LOM-OER catalysts owing to their high stability, low cost and easy synthesis. Experimental and theoretical studies have shown that increasing the covalency of metal–oxygen bonds is key to triggering lattice-oxygen oxidation of TMOs.17 However, activation of lattice oxygen could conversely deteriorate the durability of materials due to the formation of coordination-unsaturated metal sites during the reaction, leading to metal leaching and thus collapse or phase transition of the base material.18 Consequently, achieving both high activity and stability, especially under industrially meaningful high current densities, remains a great challenge for most of the LOM-based OER electrocatalysts.
Iron (Fe) is the most abundant and inexpensive metal element widely applied, and Fe-based materials are promising candidates for large-scale water splitting, despite their unsatisfactory low catalytic performance.19 The presence of Fe could induce the downshift of the metal d-band to penetrate through the 2p band of oxygen ligands and increase the covalency of M–O, leading to trigger-on of the LOM route for the OER, as well as inhibit the dissolution of metal species.20 Unfortunately, Fe-based metal oxides following the LOM for efficient water splitting are rarely reported, mostly due to the low intrinsic activity of Fe-oxides.21 Constructing heterojunction nanostructures is a feasible way to modulate both the electronic structure and metal–O bonding properties of oxide materials. The different working functions and band structures of the two sides of the heterojunction would induce interfacial charge transfer and redistribution, contributing to the intriguing physiochemical properties,22 like the shifting of the d-band center,23 modulation of surficial electron density24 and largely increased concentration of local disorders and defects.25 Our group has recently reported the self-assembled bimetallic NiFe-MOF-74@NiFe-LDH nano-heterojunction self-supporting electrode as an electrocatalyst for efficient alkaline OER. DFT calculations revealed that the OER reaction took place at the interfacial oxygen defect sites, and the catalyst presented excellent OER performance aligning with the LOM pathway.26 These results demonstrate the promising potential of heterojunction materials in the LOM-based OER. In parallel to the above tactics, defect engineering is another promising strategy to modulate the intrinsic reactivity of surficial metal sites for TMOs. Defects play an essential role in the LOM-mediated OER, and the rich OV sites introduced by defect engineering could change the coordination environment of metal centers, so as to modulate both the M–O bonding strength and charge distribution, exposing more active sites and promoting charge transfer, ultimately enhancing the reactivity of lattice oxygens. Inspired by the aforementioned design rationales, defect engineering across heterojunction interfaces provides an intriguing approach to transform the OER mechanism of Fe-based metal oxides from the AEM to the LOM, breaking the linear limitation of the traditional AEM pathway and enhancing the intrinsic activity.
Herein, we report a defect-rich Fe2O3/CeO2 nano-heterojunction material (denoted as Fe2O3@CeO2–OV) loaded onto porous nickel foam (NF) as a self-supporting electrode for efficient OER catalysis. A two-step solvothermal reaction was applied to synthesize the heterojunction material, followed by an additional calcination process to create abundant oxygen vacancies on the catalyst surface. Diffractional and microscopic analyses revealed that the hierarchical hydrangea-like microstructures consisted of Fe2O3 nanoflowers covered with CeO2 nanoparticles, while X-ray photoelectron spectroscopy (XPS), X-ray absorption fine structure (XAFS) and electron paramagnetic resonance (EPR) confirmed the formation of additional oxygen vacancies. The combination of XPS, 18O-isotopic labelling mass spectroscopy and density functional theory (DFT) calculations validated the participation of lattice oxygen induced by the defect-rich initial structure, enabling the switching from the AEM to the LOM. Specifically, the oxygen vacancies significantly strengthened the interaction between Fe2O3 substrates and above CeO2 nanoparticles, enhancing their charge transfer at the heterojunction interface, facilitating the O2 desorption, thus promoting the intrinsic OER activity. The resultant defect-rich Fe2O3@CeO2–OV exhibited superior OER performance with a low overpotential of 172 mV at 10 mA cm−2, among the highest data reported for Fe-based catalysts. More intriguingly, Fe2O3@CeO2–OV could deliver a large current density of 1000 mA cm−2 at an ultralow overpotential of 317 mV. Excellent activity and stability were also verified in a membrane-based flow cell to simulate practical industrial scenarios, confirming the great potential of Fe2O3@CeO2–OV for commercial applications.
Results and discussion
The synthetic process of the self-supporting Fe2O3@CeO2–OV nano-heterojunction electrode on NF is illustrated in Fig. 1a and detailed experimental protocols are given in the ESI.† Initially, self-supported Fe-based metal–organic framework (MOF) MIL-53 nanosheet arrays were grown onto pre-cleansed NF sheets via solvothermal reactions in the mixed solution of Fe3+ and terephthalic acid (TPA) in N,N-dimethylformamide (DMF). Successful formation of MIL-53 on the NF surface was confirmed by powder X-ray diffraction (PXRD), Fourier transform infrared spectroscopy (FT-IR) and Raman spectroscopy, in accordance with previous reports27 (Fig. S1, ESI†). The MOF precursors were subjected to a secondary hydrothermal treatment in Ce3+-containing solution to obtain Fe2O3@CeO2 nano-heterojunction electrodes with nanoflower-like morphology, which were further annealed in an argon atmosphere to acquire defect-engineered Fe2O3@CeO2–OV electrodes. For comparison, two comparative samples, named Fe2O3 (synthesized directly from Fe3+ instead of MOF precursors) and CeO2 (without the addition of Fe3+ species) were also synthesized. PXRD patterns were collected for all samples to determine the phase information (Fig. 1b). After the secondary hydrothermal treatment, the peaks of MIL-53 disappeared, replaced by the peaks of Fe2O3 (JCPDS: 33-0644) and CeO2 (JCPDS: 34-0394), indicating the complete transformation of MOF precursors to oxide materials. The weak diffraction intensities of Fe2O3 peaks might be due to the low crystallinity and amorphization during hydrolysis of MOF precursors, while the signals from Fe2O3 further weakened after annealing, due to the introduction of additional oxygen vacancies and deterioration of local coordination structures. Rietveld refinement further verified that Fe2O3 and CeO2 phases coexisted with no other detectable impurities in Fe2O3@CeO2 and Fe2O3@CeO2–OV samples (Fig. S2, ESI†).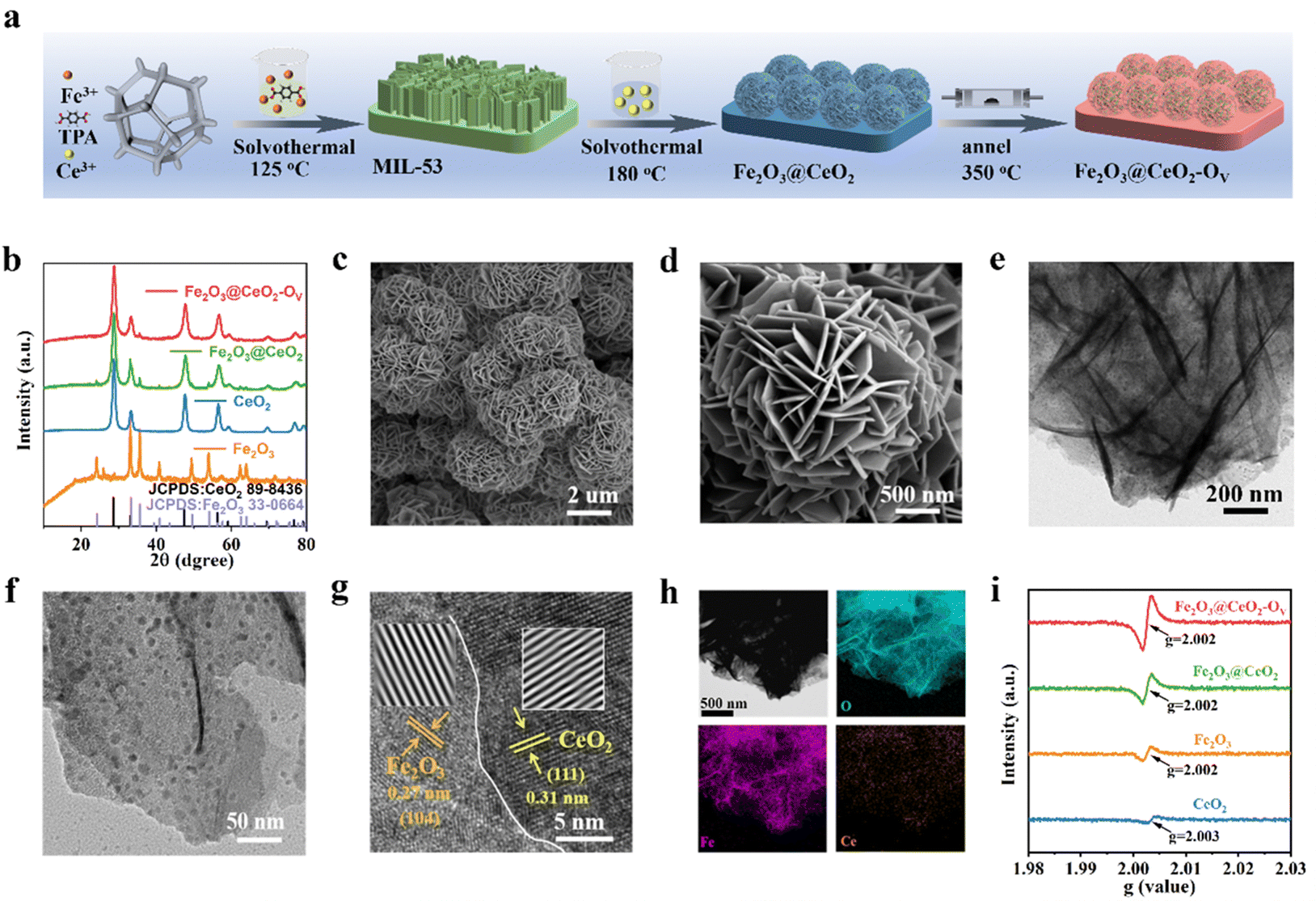 | ||
| Fig. 1 Characterization of Fe2O3@CeO2–OV. (a) Schematic illustration of the synthetic process for Fe2O3@CeO2–OV. (b) XRD patterns of Fe2O3@CeO2–OV, Fe2O3@CeO2, Fe2O3 and CeO2. (c) and (d) SEM images of Fe2O3@CeO2–OV. (e) and (f) TEM images of Fe2O3@CeO2–OV. (g) HRTEM image of Fe2O3@CeO2–OV. (h) EDX elemental mapping of Fe2O3@CeO2–OV. (i) EPR signals for Fe2O3@CeO2–OV, Fe2O3@CeO2, Fe2O3 and CeO2. | ||
Fourier-transform infrared (FT-IR) spectroscopy was applied to detect the surface functional groups, and the infrared spectra of Fe2O3@CeO2–OV, Fe2O3@CeO2, Fe2O3, and CeO2 (Fig. S3a, ESI†) samples were collected in the wavenumber range of 4000–500 cm−1. Characteristic peaks are observed at 545 and 557 cm−1, corresponding to the asymmetric tensile vibrations of Ce–O bonds and Fe–O, respectively. The vibrations at ∼1643 and ∼3440 cm−1 are attributed to OH− groups in water, and the two peaks detected at ∼1392 cm−1 and ∼1564 cm−1 arising from the –COO− groups are attributed to dissolved atmospheric CO2 and carboxylate in the ligand.28 The formation of the Fe2O3–CeO2 heterojunction structure was also confirmed via Raman spectroscopy (Fig. S3b, ESI†). For the CeO2 sample, a distinct Raman band is observed at ∼464 cm−1, corresponding to the F2g vibration of fluorite CeO2. Characteristic Raman bands of Fe2O3 are present at 221, 289, 406 and 653 cm−1. For the heterojunction samples, both bands from CeO2 and Fe2O3 are observable, in accordance with the coexistence of the two phases from PXRD patterns. Broadening of the F2g peak of CeO2 after heterojunction formation is observed, indicating the increased structural distortion at the Fe2O3–CeO2 heterojunction interface.29 Notably, after the annealing process, the Raman peaks show a significant blueshift for the defect-engineered Fe2O3@CeO2–OV sample, indicating the expansion of the lattice and decrease of crystallinity due to the introduction of abundant oxygen vacancies.
The morphological evolution of the catalysts was analyzed by field emission scanning electron microscopy (FESEM). After the initial solvothermal reaction, ultrathin MIL-53 nanosheet arrays were covered on the NF substrate surface (Fig. S4, ESI†). Through the Ce-assisted templated hydrolysis of the MOF precursor during the secondary hydrothermal reaction, the nanosheet arrays of MIL-53 transformed into self-assembled nanoflower structures for the Fe2O3@CeO2 heterojunction material (Fig. S5, ESI†). The hydrangea-like nanoflower microstructures with the scale around 1.5–3 μm comprised thin nanosheets with a thickness around 13 nm, endowing the material with hierarchical structures with abundant mass diffusion channels, integrated charge transport pathways and highly exposed active planes. Compared with the biphasic oxide materials derived from MOF precursor, the Fe2O3 sample directly from solution deposition (Fig. S6, ESI†) shows a similar vertically intergrown nanosheet array morphology with the MIL-53 precursor, while the CeO2 sample without the addition of Fe sources presents thick and smooth nanoplate morphology (Fig. S7, ESI†). Both samples show the absence of hydrangea-like morphology, proving that both the MOF precursor and Ce4+ ions play essential roles in the formation of such delicate micro-nanostructures. After the final annealing process, defect-rich Fe2O3@CeO2–OV retained the nanoflower structure of Fe2O3@CeO2 without obvious morphology change, confirming the integrity of the heterojunction backbone during defect engineering processing (Fig. 1c, d and Fig. S8, ESI†). To further investigate the detailed interfacial structure of the nano-heterojunction, transmission electron microscopy (TEM) was applied to analyze the microstructures of Fe2O3@CeO2–OV (Fig. 1e, f and Fig. S9, ESI†), in comparison with other samples (Fig. S10–S12, ESI†). As shown in the TEM images of various magnifications of Fe2O3@CeO2–OV, numerous ultrafine nanocrystals with sizes below 10 nm are randomly distributed on the intergrown nanosheets. A high-resolution TEM (HRTEM) image in Fig. 1g focusing on the boundary between the nanosheet and nanoparticle shows obvious lattice fringes of distinct orientations and spacings on the two sides of the boundary. The lattice fringes on the nanosheet side with 0.27 nm spacing are ascribed to the (104) plane of Fe2O3, and the (111) plane of CeO2 with an interplanar distance of 0.31 nm is attributed to the lattice fringes on the nanoparticle side, in accordance with the selective area electron diffraction (SAED) patterns in Fig. S9f (ESI†), where diffraction rings corresponding to Fe2O3 (104) and CeO2 (111) planes are observed. Formation of the Fe2O3–CeO2 heterojunction was also evidenced by elemental mapping of the energy dispersive spectrometry (EDS) results (Fig. 1h and Fig. S13–S15, ESI†). Both metallic elements (Fe or Ce) and O show homogeneous distribution in CeO2 (Fig. S13, ESI†) and Fe2O3 (Fig. S14, ESI†) samples, confirming their single-phase structures. In contrast, for the heterogeneous Fe2O3@CeO2 (Fig. S15, ESI†) and Fe2O3@CeO2–OV (Fig. 1h), Fe and O are also homogeneously distributed throughout the materials, while Ce is located as randomly distributed high-concentration discrete islands, in agreement with the nanoparticle feature of the secondary CeO2 phase on continuous Fe2O3 nanosheet substrates. Consequently, combined with diffraction, spectral and microscopic analyses, the successful construction of the Fe2O3–CeO2 nano-heterojunction in the Fe2O3@CeO2–OV micro-nano-hydrangeas was confirmed in the form of highly-dispersed ultrafine CeO2 nanoparticles imbedded onto Fe2O3 nanosheets.
XPS measurements were applied to investigate the surficial chemical states and charge transfer behaviors of the samples. From the survey spectra in Fig. S16 (ESI†), the surface of Fe2O3@CeO2–OV comprises elements such as Ce, Fe, O and C, consistent with the findings from EDS analysis. Signals of Ni are also detected from the NF substrates. The high-resolution O 1s spectrum of Fe2O3@CeO2–OV shows the presence of three types of oxides, M–O (529.6 eV), M–OH (531.7 eV) and adsorbed H2O (533.5 eV). The surfaces of the heterojunction samples (Fe2O3@CeO2–OV and Fe2O3@CeO2) resemble that of Fe2O3 (M–O at 529.8 eV) more than that of CeO2 (M–O at 529.3 eV). The asymmetric nature of the M–OH peak indicates the presence of oxygen vacancies, and thus the peak ratio of M–OH/M–O could be applied for rough assessment of oxygen vacancy concentration. Noteworthily, for Fe2O3@CeO2–OV, the M–O signals are seriously weakened, while M–OH signals significantly strengthen together with adsorbed H2O signals as compared with Fe2O3@CeO2, suggesting abundant surficial oxygen vacancies. Also, all the O 1s peaks shift to higher bonding energy in the defect-rich sample, suggesting alleviation of M–O bonding strength. The Ce 3d spectrum of Fe2O3@CeO2–OV shows two sets of doublet peaks corresponding to Ce3+ (green peaks at 882.3, 886.1, 900.5, 907.4 and 904.9 eV) and Ce4+ (blue peaks at 883, 885.5, 889.2, 898.6, 901.1 and 917.4 eV),30 while peaks corresponding to Ce3+ are not observed for CeO2 and Fe2O3@CeO2, which is ascribed to the spontaneous deoxygenation of Ce at high-temperature following the formula: 2Ce4+ + OL2− ↔ 2Ce3+ + OV + 1/2O2, where the lattice oxygen OL is released as oxygen gas to leave an oxygen vacancy. For the Fe 2p spectra of Fe2O3@CeO2–OV and Fe2O3@CeO2, the case is much more complicated due to the overlap of Fe 2p signals with Ni and Ce Auger peaks (cyan Ni LMM and blue Ce MNN peaks in Fig. S16, ESI†) in binding energy.31–33 After careful peak deconvolution, the Fe 2p spectrum of Fe2O3@CeO2–OV can be deconvoluted into a series of peaks: Fe2+ (green peaks, 2p3/2 at 711.2 eV, 2p1/2 at 724.3 eV, and satellite peak at 724 eV), and Fe0 2p3/2 (707 eV). Interestingly, the Fe3+ peaks observed in Fe2O3 and Fe2O3@CeO2 are not observed for Fe2O3@CeO2–OV, while low-valence Fe states including Fe2+ and even metallic Fe(0) are present. These results confirm that the formation of low-valence Ce3+ and rich oxygen vacancies induces significant charge transfer and cross-interface electron density redistribution from the CeO2 side to the Fe2O3 side of the heterojunction, leading to reduction of Fe centers. Formation of abundant oxygen vacancies was more directly confirmed by EPR as shown in Fig. 1i, where the strongest magnetic signals at g = 2.003 for Fe2O3@CeO2–OV indicated the highest concentration of unpaired electrons as compared with other samples, which were usually induced by oxygen vacancies.34
The chemical states and the localized coordination environment of the samples were further investigated by XAFS spectroscopy. Fig. 2a shows the Fe K-edge X-ray absorption near-edge structures (XANES) profiles of Fe2O3@CeO2, Fe2O3@CeO2–OV, Fe2O3 samples and Fe foil as references. The heterojunction Fe2O3@CeO2 sample shows similar absorption curve shape and peak positions with the single-phase Fe2O3 sample, indicating that Fe in Fe2O3@CeO2 mainly exists in the form of Fe3+ in FeO6 octahedra.35 After calcination, the defect-engineered Fe2O3@CeO2–OV shows an obvious shift of the absorption edge to lower energy, approaching that of Fe foil, indicating the decreased Fe valence state between Fe(0) and Fe(III), in accordance with the Ce-reduction-induced charge transfer from the CeO2 side to the Fe2O3 side of the heterojunction from XPS results.36 Also, Fe2O3@CeO2–OV shows a flattened white line peak at 7126.7 eV, with reduced white-line peak intensity compared with Fe2O3@CeO2, corresponding to less electron transfer from Fe centers to nearby coordination atoms (O in this case), further probing the increase of localized structural disorder after defect engineering. Fig. 2b shows the Fourier-transformed Fe K-edge extended X-ray absorption fine structure (EXAFS) curves of all the samples. Compared with Fe foil, no Fe–Fe signals around 2.21Å are observed for the three samples, proving the absence of metallic Fe species. Two prominent peaks at 1.51 Å and 2.62 Å are observed for Fe2O3@CeO2 and Fe2O3, which could be assigned to first-shell Fe–O and second-shell Fe–Fe/Ce, respectively. The increased intensity of the 2.62 Å peak for Fe2O3@CeO2 could be attributed to the additional second-shell Fe–Ce interactions from heterojunction formation. The defect-rich Fe2O3@CeO2–OV however shows distinct EXAFS features. The first peak shifts to 1.75 Å with dramatic broadening, while the second peak almost disappears. Such obvious change is evident for the breaking of long-range ordered structures due to defect-induced lattice distortion and amorphization. Quantitative EXAFS fitting analyses were performed for all the samples to investigate the coordination configuration, where the structural parameters are listed in Table S1 (ESI†) (original and fitting curves of separated samples in both k and R spaces are given in Fig. S17, ESI†). Both Fe2O3 and Fe2O3@CeO2 adopt a typical Fe2O3-typed coordination structure with 6 first-shell Fe–O and 12 second-shell Fe-Fe coordination numbers, while the Fe–O coordination number of Fe2O3@CeO2–OV decreases to ∼5.0, with expanded Fe–O bond length from 1.99 Å to 2.07 Å, and signals from second-shell almost disappear, confirming the formation of abundant oxygen vacancies and increased structural disorder. A wavelet transform (WT) of Fe K-edge EXAFS signals for Fe2O3@CeO2–OV is displayed in Fig. 2c, showing only one intensity maximum at ∼5.26 Å−1 in k space, which expands to a higher R region as compared with that of Fe2O3 and Fe2O3@CeO2 (Fig. S18, ESI†), attributed to extended Fe–O backscattering contribution.37
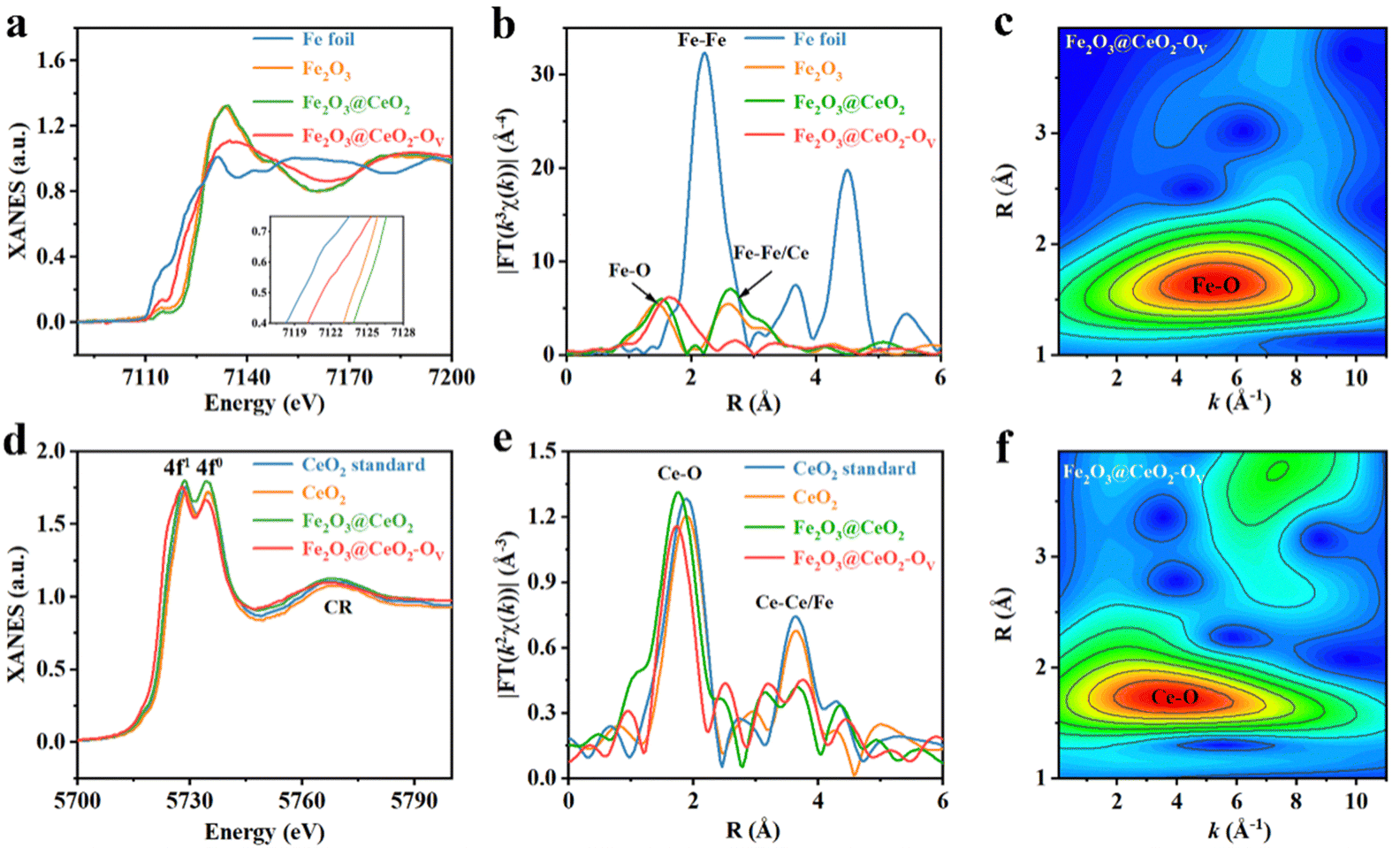 | ||
| Fig. 2 XAFS characterization. (a) Fe K-edge XANES spectra and (b) Fourier-transformed k3-weighted EXAFS spectra of Fe2O3@CeO2–OV, Fe2O3@CeO2, Fe2O3 and Fe foil as reference. (c) WT-EXAFS contour plot of Fe K-edge signals of Fe2O3@CeO2–OV. (d) Ce L3-edge XANES spectra and (e) Fourier-transformed k2-weighted EXAFS spectra of Fe2O3@CeO2–OV, Fe2O3@CeO2, CeO2 and commercial CeO2 as reference. (f) WT-EXAFS contour plot of Ce L3-edge signals of Fe2O3@CeO2–OV. | ||
Fig. 2d shows the Ce L3-edge XANES profiles of Fe2O3@CeO2, Fe2O3@CeO2–OV and CeO2, together with a commercial CeO2 sample as the Ce standard. Two white-line edge absorption peaks at 5728.7 eV and 5734.2 eV are identified, corresponding to a transition from the Ce 2p3/2 state to unoccupied Ce 5d states mixed with 4f1 or 4f0 final states, confirming the coexistence of Ce3+/Ce4+ mixed oxidation states. The white-line edge of Fe2O3@CeO2–OV is located at the lowest energy, accompanied by decreased intensity of the 4f0 edge, indicating the lowered Ce valence and increased Ce3+/Ce4+ ratio due to the formation of increased oxygen vacancies. The continuum resonance (CR) bulk peak is attributed to the combination of Ce–Fe/Ce scattering. The Ce L3-edge EXAFS spectra of all the Ce-containing samples are shown in Fig. 2e, which show two main features. The major peak around 1.75 Å is related to first-shell Ce–O coordination, as confirmed by quantitative EXAFS curve fitting (Fig. S19 and Table S2, ESI†). Notably, additional Ce–O scattering paths are fitted, with longer bond lengths than the typical Ce–O bonds in CeO2 with lengths between 2.2 and 2.3 Å for both Fe2O3@CeO2 and Fe2O3@CeO2–OV, hinting the contributions from interfacial Fe–O–Ce signals. Interestingly, the defect-rich Fe2O3@CeO2–OV shows decreased first-shell Ce–O coordination number of ∼6.2; however with increased coordination from the extended 3.16 Å Ce–O bond, arising from the stronger adhesion of the CeO2 nanoparticles to the Fe2O3 nanosheet surface due to oxygen-vacancy-induced evolution of atomic configuration, as later discussed in the calculation part. The second peak should be related to the second-nearest Ce–Ce or Ce–Fe scattering, and the slight change of shape in this peak contains fingerprint information indicative of redistributed middle-range lattice structures due to heterojunction and defect formation, respectively. The WT-EXAFS contour plot of Fe2O3@CeO2–OV in Fig. 2f and Fig. S20 (ESI†) shows an intensity maximum expanded between 2 and 6 Å−1 in k space, attributed to contributions from various Ce–O backscattering of varying bond lengths. A weaker peak from Ce–Ce/Fe backscattering signals is also identified in the high R region. Defect-induced structural variation was also validated by X-ray pair-distribution function (PDF) analyses. As shown in Fig. S21 (ESI†), the PDF profiles of Fe2O3@CeO2–OV and Fe2O3@CeO2 mostly resemble that of CeO2 instead of Fe2O3, possibly owing to the high surface coverage of CeO2 nanoclusters and the low crystallinity of the Fe2O3 substrates. Simulation reveals that the first three peaks of both Fe2O3@CeO2–OV and Fe2O3@CeO2 could be attributed to Ce–O, O–O and Ce–Ce of CeO2, respectively (Fig. S22, ESI†). For Fe2O3@CeO2–OV, the Ce–Ce and Ce–O peaks are enhanced, while the O–O peak intensity decreases as compared with Fe2O3@CeO2, in accordance with the lower Ce–O coordination numbers from EXAFS results, indicating the loss of lattice oxygen and formation of oxygen vacancies upon the defect engineering process.30,38
The electrochemical water oxidation performance of all the samples was further evaluated in a three-electrode system in 1.0 M KOH alkaline aqueous media. Fig. 3a shows the back-scan linear sweep voltammetry (LSV) OER polarization curves with 90% iR-correction manually compensated. Among all the samples, the defect-engineered heterojunction electrode Fe2O3@CeO2–OV exhibits the lowest overpotential of 172 mV and 201 mV to attain current densities of 10 and 100 mA cm−2, respectively, dramatically outperforming that of Fe2O3@CeO2 (217 and 261 mV), Fe2O3 (252 and 297 mV), CeO2 (242 and 306 mV), MIL-53 (268 and 326 mV) and commercial RuO2 (187 and 296 mV). Fe2O3@CeO2–OV also performs excellently under large current conditions, requiring only 317 mV to reach 1000 mA cm−2 current density, a typical condition for industrial electrolyzers. Such pronounced performance outperforms those of most of the reported Fe-based oxide/hydroxide/oxyhydroxide materials and those of even many noble-metal-based catalysts, as shown in Fig. 3b and Table S3 (ESI†). Tafel slope values were calculated to understand the intrinsic kinetics of catalysts. As shown in Fig. 3c, Fe2O3@CeO2–OV shows the lowest Tafel slope of 58 mV dec−1 among all the samples, lower than Fe2O3@CeO2 (62 mV dec−1), Fe2O3 (73 mV dec−1) and CeO2 (77 mV dec−1), and notably also lower than RuO2 (103 mV dec−1), proving the promoting synergistic effects of abundant heterojunction interfaces and oxygen defects. Electrochemical impedance spectroscopy (EIS) measurements were applied to investigate the charge transfer behaviors of samples. From the Nyquist plots in Fig. 3d, the heterojunction Fe2O3@CeO2 shows a charge-transfer resistance (Rct) of 13.09 Ω, much lower than that of single-component Fe2O3 (26.71 Ω) and CeO2 (49.72 Ω), confirming that the formation of heterojunction significantly increases the charge transfer owing to interfacial charge redistribution. Defect engineering further modulated the electronic structure of the heterojunction and improved the conductivity, where defect-rich Fe2O3@CeO2–OV presented the lowest Rct of 10.66 Ω, indicating the fast electron transfer between the electrode–electrolyte interfaces. It is also worth mentioning that the in situ grown electrodes exhibit much lower Rct than commercial RuO2 simply loaded onto the NF substrate (79.44 Ω), further proving the advantage of our self-supporting electrodes with hierarchical structures and interconnected conductive networks.
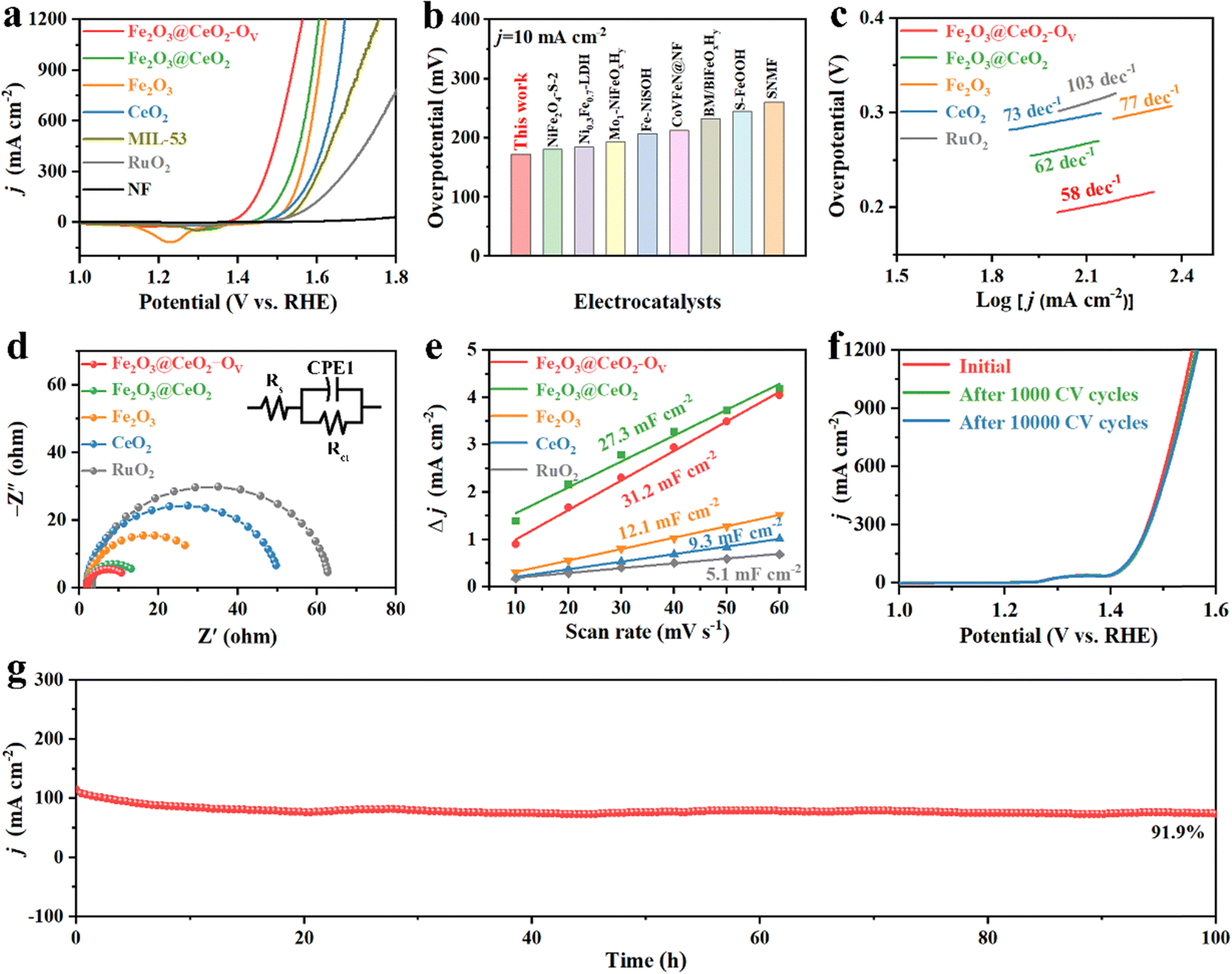 | ||
Fig. 3 OER performance of the prepared electrodes. (a) LSV negative scan curves in 1.0 M KOH with 90%-iR compensation. (b) Comparison of overpotentials at a current density of 10 mA cm−2 with reported Fe-based electrocatalysts. (c) Corresponding Tafel plots. (d) EIS plots of samples, and the insert shows the equivalent circuit. (e) Cdl calculations of the Fe2O3@CeO2–OV, Fe2O3@CeO2, Fe2O3, CeO2 and RuO2 samples. (f) LSV curves of Fe2O3@CeO2–OV, before and after 1000 and 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 CV cycles in 1.0 M KOH for the OER. (g) Chronoamperometric stability tests of Fe2O3@CeO2–OV. 000 CV cycles in 1.0 M KOH for the OER. (g) Chronoamperometric stability tests of Fe2O3@CeO2–OV. | ||
Electrochemical surface area (ECSA) is an important criterion to represent the density of intrinsic surface active sites of catalysts. The ECSA values were calculated accordingly as proportional to the electrochemical double-layer capacitance (Cdl) in Fig. 3e. Cdl values were calculated from cyclic voltammetry (CV) scans of different scan rates (10, 20, 30, 40, 50 and 60 mV s−1) in the non-redox region, as shown in Fig. S23 (ESI†). Fe2O3@CeO2–OV has the highest Cdl value of 31.2 mF cm−2, followed by Fe2O3@CeO2 (27.3 mF cm−2), Fe2O3 (12.1 mF cm−2), and CeO2 (9.3 mF cm−2), confirming that the formation of the heterojunction and following defect engineering effectively increase the number of surficial active sites. These results are also supported by the geometric surface areas calculated using the Brunauer–Emmett–Teller (BET) method from 77 K N2 sorption isotherms, as presented in Fig. S24 (ESI†), where Fe2O3@CeO2–OV shows the highest BET area of 75.44 m2 g−1. The OER LSV polarization curves were further normalized by using calculated ECSAs to exclude the influence of structural and geometrical variations of electrodes and present the intrinsic catalytic activities of samples (Fig. S25, ESI†). After ECSA-normalization, the superiority of Fe2O3@CeO2–OV over other samples becomes even more pronounced with a much lower OER onset, requiring only 226 mV to reach a current density of 10 mA cm−2 ECSA, which is significantly higher those of than commercial RuO2 (0.1136 mA cm−2 ECSA) at the same overpotential. The high intrinsic activity of Fe2O3@CeO2–OV for OER catalysis was further determined by calculating the turn-over frequency (TOF) values of singular active sites under certain given potentials. TOF values were estimated from the redox peaks of transition metals in CV scans of different scan rates, assuming a one-electron process for oxidation of metal centers (Fig. S26, ESI†). Fe2O3@CeO2–OV shows the highest TOF values of 0.05, 0.18 and 0.431 s−1 at overpotentials of 200, 250 and 300 mV, predominantly higher than those of other samples. These combined analyses provide solid evidence that construction of heterojunction nanostructures and the following defect engineering process could enhance interfacial charge transfer and increase both the catalytic active surface areas and intrinsic catalytic activities of singular active sites by effective modulation of interfacial geometric and electronic structures.
Stability and durability are essential concerns for industrial applications. Industrial water splitting electrolyzers work under harsh conditions, thus requiring the electrodes to endure extreme chemical, electrochemical and mechanical corrosions from highly alkaline media, high current and vigorous shock of generated gas bubbles. Fig. S27 (ESI†) shows a multiple current step chronopotentiometry (MCSCP) curve of Fe2O3@CeO2–OV, where the current density step increment was set as 4 mA cm−2. As the current densities stepped, the potential immediately responded and stabilized, remaining mostly constant for 500 seconds. Such rapid response to current change and rapid equilibrium are indicative of the fast mass diffusion at the electrode surface, owing to the hierarchical three-dimensional nanoflower structures. These properties could enable the quick start and rapid response of the electrodes when utilized in industrial water electrolyzers, especially powered by a green-electricity power source (like solar panels or wind turbos) with unstable power supply. Accelerated degradation tests (ADT) were further conducted via CV cycling within the potential window of 1.20 to 1.50 V. As shown in Fig. 3f, the LSV curves remain mostly unchanged after ADT cycling, where the overpotential required to reach the current density of 100 mA cm−2 shows negligible change. Even up to a high current density of 1000 mA cm−2, the overpotential after 10000 CV cycles increases by only 9 mV, proving the structural and chemical stability of Fe2O3@CeO2–OV against varying current–potential conditions. SEM images of Fe2O3@CeO2 and Fe2O3@CeO2–OV after ADT tests (Fig. S28, ESI†) show that the hierarchical hydrangea-like nanoflower microstructures assembled from nanosheets were mostly well-retained, proving the excellent structural stability of the heterojunction samples against long-term electrochemical corrosion and mechanical wear. Chronoamperometric measurements under constant potentials were applied to access the steady-state long-term stability of the electrodes. The Fe2O3@CeO2–OV electrode could maintain the current density around 10 and 100 mA cm−2 for continuous 100 h at constant potentials of 1.424 and 1.509 V, respectively, with only minor current fluctuation and degradation (Fig. 3g and Fig. S29, ESI†). After 100 h of electrolysis at 1.509 V, the electrode could still retain 91.9% of the starting current density. XRD, Raman (Fig. S30, ESI†) and XPS (Fig. S31, ESI†) analyses after OER tests further confirm no significant change of the crystalline structure and elemental composition for Fe2O3@CeO2–OV. Specifically, an obvious blueshift of the CeO2 F2g peak is observed, corresponding to lattice expansion due to the formation of additional oxygen vacancies and surficial reconstruction during the reaction. The hierarchical micro–nano structure of the heterojunction is also well-retained without obvious morphology change, as confirmed by the SEM (Fig. S32, ESI†), TEM and HRTEM images (Fig. S33, ESI†) after OER testing. These results confirm that the Fe2O3@CeO2–OV electrocatalyst could operate steadily under both rapidly varying current–voltage conditions and long-term steady-state electrolysis, especially under industrial-level high current densities, thus presenting excellent stability and durability. Selectivity is another critical index for practical applications. Incomplete oxidation of water through the two-electron pathway into hydroperoxide (H2O2) may lead to corrosion of the polymeric components of the membrane electrode assembles (MEAs) through Fenton-like reaction, together with other undesirable side reactions. The Faraday efficiency (FE) of Fe2O3@CeO2–OV was thus evaluated via the rotating ring-disk electrode (RRDE) experiments in N2-saturated 1.0 M KOH solution. To confirm that the current was induced by water oxidation instead of side effects, a constant potential of 0.45 V was applied on the Pt ring electrode to reduce any oxygen generated at the disk electrode loaded with the Fe2O3@CeO2–OV electrocatalyst. As shown in Fig. 4a, both disk and ring currents are negligible until the oxygen evolution potential is reached at the disk electrode, and both increase concurrently as the potential further increases to a higher level. The FE at 1.55 V is calculated to be as high as 96.3%, indicating that water is completely oxidized into oxygen with negligible side reactions. The FE measurement was double-checked using a conventional water drainage method. At constant current densities of 100 mA cm−2 and 500 mA cm−2, the oxygen evolution rates were measured to be 0.09 and 0.18 mL min−1, respectively, corresponding to FEs of 99.99% and 99.92%, respectively (Fig. 4b). The actual oxygen production rates at different times are close to theoretical values, and the calculated FE values approach 100%, again validating the complete four-electron water oxidation of the defect-engineered heterojunction electrodes. Considering the excellent OER catalytic activity and stability of Fe2O3@CeO2–OV electrodes, we further applied the self-supporting electrodes in a two-electrode MEA-based flow cell to examine the overall water splitting performance under industrial conditions. The schematic diagram and photographic image of the testing cell are given in the insert in Fig. 4c and Fig. S34 (ESI†). The self-supporting Fe2O3@CeO2–OV electrode with the NF substrate was directly used as the anode, and a carbon paper loaded with a commercial Pt/C catalyst was used as the cathode, and both electrodes were pressed tightly against an anion exchange membrane. The 1.0 M KOH solution electrolyte flowed through the anode chamber. The MEA-based water splitting performance was validated using the potential-scan polarization curve, as shown in Fig. 4d. The cell shows promising performance, requiring a low cell voltage of 1.61 V and 1.80 V to reach current densities of 100 and 500 mA cm−2, respectively. The flow cell specifically demonstrates great potential in the high current density region, reaching 1000 mA cm−2 at a cell voltage of 1.93 V. When a current density of 1.0 A cm−2 was applied for 100 h, the cell operated steadily, with the cell voltage increasing from the initial 1.968 V to 2.019 V, exhibiting favorable stability. These results demonstrate the promising performance and stability of the defect-engineered Fe2O3@CeO2–OV heterojunction self-supporting electrodes in the industrial membrane-based electrolyzer under high current working conditions.
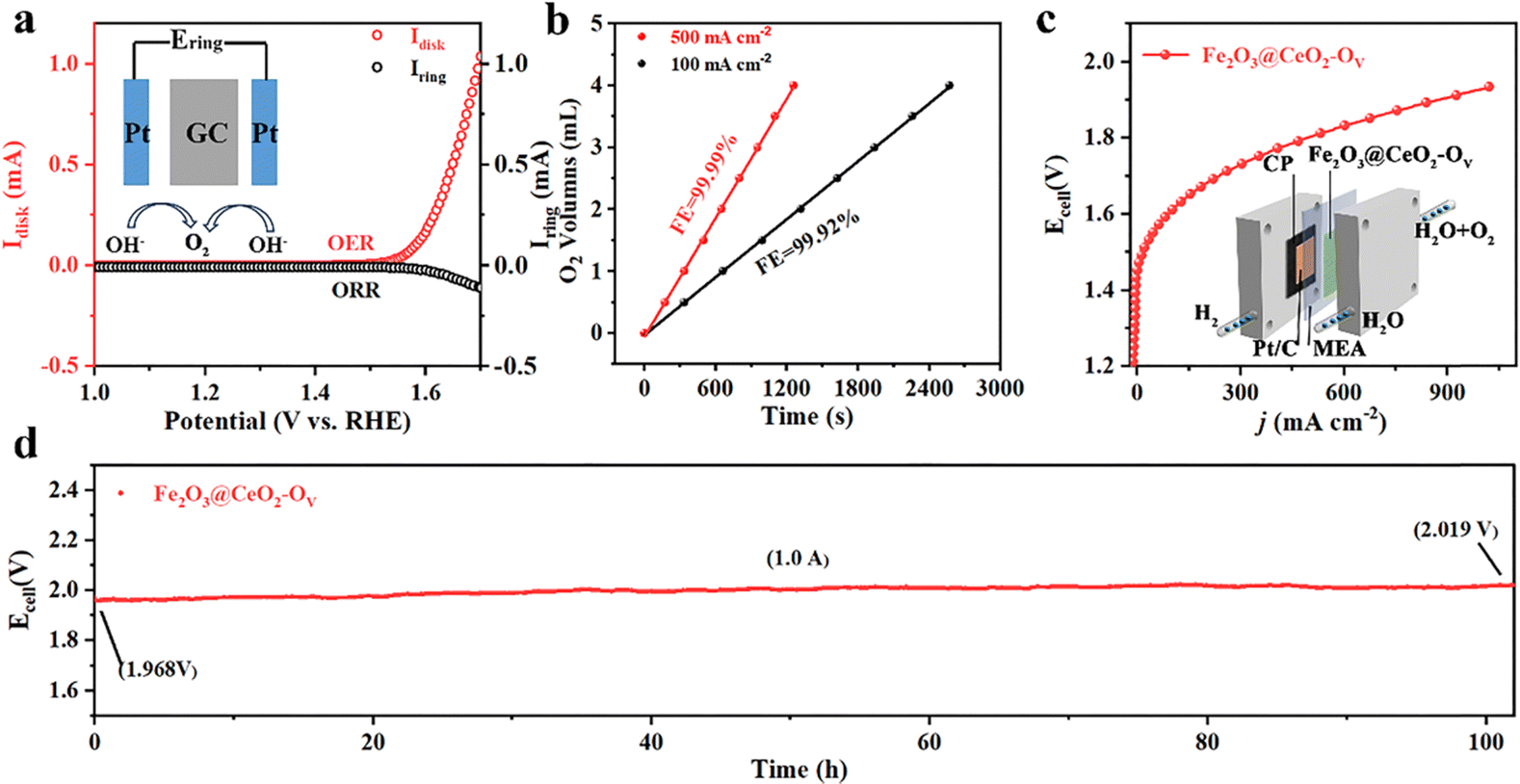 | ||
| Fig. 4 OER performance of the prepared electrodes. (a) RRDE testing results. The O2 generated at the disk electrode loaded with the Fe2O3@CeO2–OV catalyst was reduced at the Pt ring electrode at a constant potential of 0.45 V (vs. RHE) in N2-saturated 1.0 M KOH solution. (b) Oxygen evolution rates at constant current densities of 100 and 500 mA cm−2, respectively. (c) Overall water splitting performance of the Fe2O3@CeO2–OV anode coupled with the commercial Pt/C cathode in a membrane-based flow cell. (d) Chronopotentiometric curve of the Fe2O3@CeO2–OV‖Pt/C flow cell for overall water electrolysis for 100 h. Constant current densities of 1.0 A cm−2 were applied during the reaction. | ||
The large number of oxygen vacancies and outstanding OER performance of Fe2O3@CeO2–OV, especially the much lower OER onset potential and Tafel slop, indicate the substantial change of reaction kinetics and possible involvement of lattice oxygen. To identify the reaction pathway, the pH-dependence of the electrodes was evaluated. LSV curves were collected under different pH values ranging from 12.5 to 14 (Fig. 5a and Fig. S35, ESI†). Fe2O3@CeO2–OV shows much stronger pH dependence than Fe2O3@CeO2 and other comparison samples. To quantify the pH dependence of OER activity, the proton reaction orders on RHE scale (ρRHE, ρRHE = ∂log(j)/∂pH) were derived, which reflected the relationship between the OER reaction kinetics and proton activity. The ρRHE value (Fig. S36, ESI†) of Fe2O3@CeO2–OV is fitted to be 0.90, larger than that of Fe2O3@CeO2 (0.68), corresponding to a stronger pH-dependence, which implies non-cooperative proton-electron transfer and a LOM pathway rather than the conventional AEM during the OER.39 Generation of negatively charge oxygen-containing species (*O22− and *O2−) is another important feature of the LOM-based OER, thus detection of these intermediate species provides solid evidence of surficial oxygen involvement. Tetramethylammonium hydroxide (TMAOH) is a typical detector because the tetramethylammonium cation (TMA+) could bind strongly with the oxygen-containing species through electrostatic interaction and compete with the OER, thus hindering the OER kinetics. The KOH electrolyte was then replaced with the TMAOH solution for OER testing to verify the reaction mechanism. Fe2O3@CeO2–OV shows significant OER activity reduction, where the overpotential to reach 100 mA cm−2 current density increases by 192 mV, indicative of combination between TMA+ and negatively charged oxygen species, implying a LOM pathway. Comparatively, the reaction activity reduction of Fe2O3@CeO2 is not so pronounced, and the corresponding overpotential increment is only 128 mV, corresponding to an AEM mechanism.40 Variation of the pre-OER redox peaks and pseudocapacitive currents from CV plots under different scan rates (1 to 5 mV s−1) could be applied to study the OER kinetics (Fig. S37, ESI†). The relationship between the current (i) and sweep rate (ν) follows the equation: i = aνb, where the b value could be acquired as the slope from the linear fitting of log(i) against log(ν), and applied to elucidate the reaction mechanism. Specifically, the b value of 0.5 represents a completely diffusion-controlled process, whilst 1.0 indicates an ideal surficial capacitive process. The b value of Fe2O3@CeO2–OV is calculated to be 0.843, in contrast to the 0.506 for Fe2O3@CeO2, revealing that the OER reaction transforms from a typical diffusion-dominated process for Fe2O3@CeO2 into a majorly surface-controlled one. More specifically, the contribution from either surficial capacitive or diffusion component at a fixed potential V is quantified using the following formula: i(V) = k1V + k2V1/2, where k1V is attributed to the capacitance portion, and k2V1/2 is the diffusion fraction. As shown in Fig. 5b and c, the pseudocapacitive behavior contributes to most of the current densities for Fe2O3@CeO2–OV, especially at higher scan rates, contributing 70% of the redox current at a scan rate of 5 mV s−1, higher than that of Fe2O3@CeO2 (64%) under the same condition. The pseudocapacitive charging process of transition metal compounds is often related to the formation of abundant active centers at anodic potentials lower than the OER onset, thus boosting the following OER process.41,42
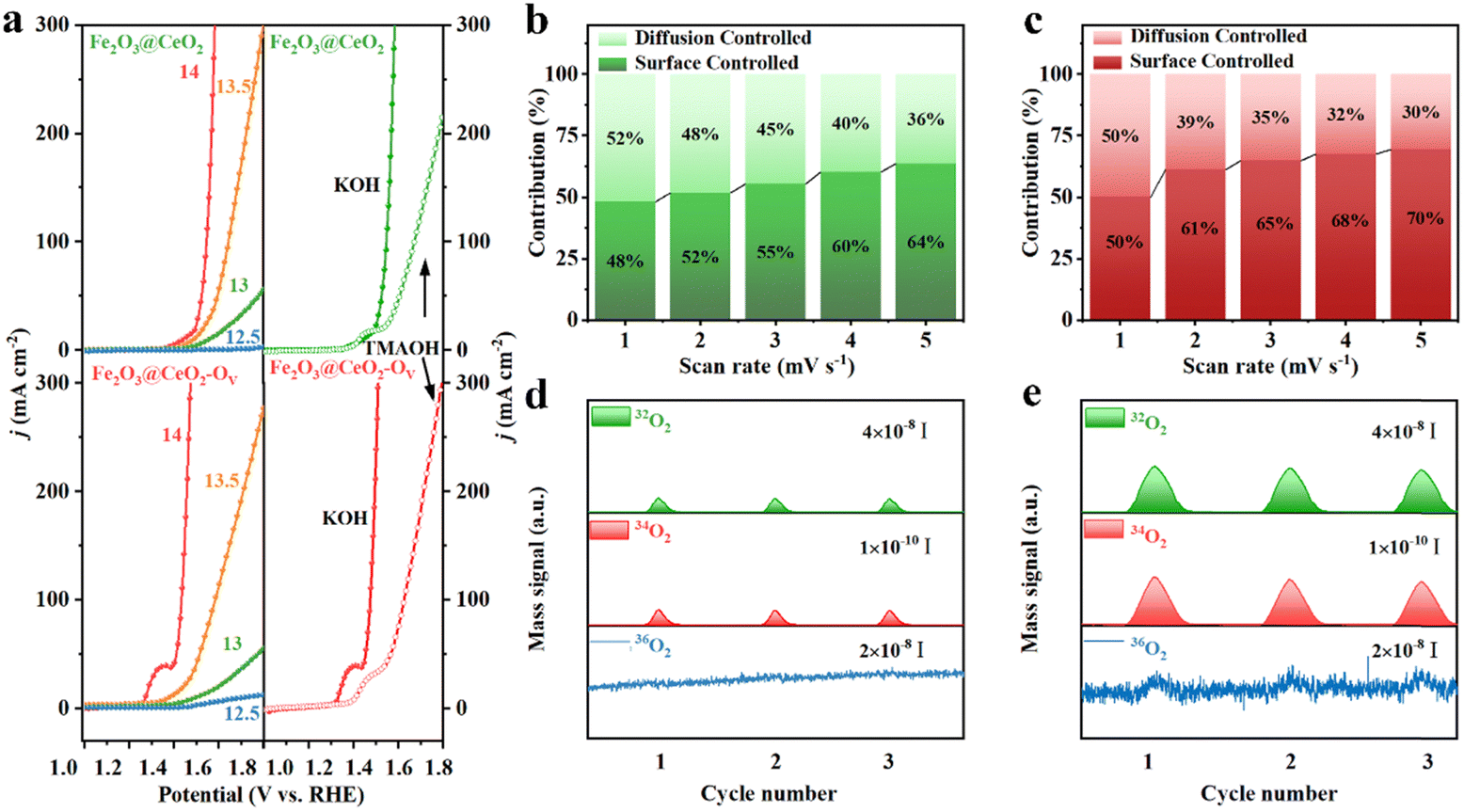 | ||
| Fig. 5 OER performance of the prepared electrodes. (a) LSV curves of Fe2O3@CeO2–OV and Fe2O3@CeO2 under different pH conditions (left) and in 1.0 M KOH or 1.0 M TMAOH, respectively (right). Contribution ratios of the capacitive and diffusion-controlled current densities vs. scan rates for (b) Fe2O3@CeO2 and (c) Fe2O3@CeO2–OV. DEMS results by 18O-isotopic labeling experiments for (d) Fe2O3@CeO2 and (e) Fe2O3@CeO2–OV. | ||
The above results provide abundant yet indirect evidence of the LOM pathway during the OER process for Fe2O3@CeO2–OV. To directly reveal the participation of lattice oxygen during the OER process, we performed operando isotopic 18O-labeled differential electrochemical mass spectrometry (DEMS) experiments for both Fe2O3@CeO2–OV and Fe2O3@CeO2. The working electrodes were firstly labeled by CV scanning in 18O-labeled 0.1 M KOH solution, where the surficial oxygen atoms would be exchanged by 18O if the LOM pathway was possible. The labeled electrodes were then tested for the OER by CV in 0.1 M KOH solution with regular H2O, and the gases generated were constantly monitored using mass spectra. Fig. 5d and e show the acquired DEMS spectra from three consecutive scans for Fe2O3@CeO2 and Fe2O3@CeO2–OV, respectively. Mass signals at m/z = 32, 34 and 36 were monitored, corresponding to 16O2, 16O18O, and 18O2 in the gas products during potential scans. As shown in Fig. S38 (ESI†), during the 18O-labeling process, obvious 18O2 signals were observed from the OER of H218O, and some 16O18O signals were also recorded due to surface oxygen exchange. Notably, for Fe2O3@CeO2–OV, some minor signals of 16O2 were also detected, corresponding to the release of two adjacent surface oxygen atoms through a dual-vacancy LOM pathway, which was absent for Fe2O3@CeO2. The participation of surface oxygen was further proven by measuring the labeled electrodes in H216O, where Fe2O3@CeO2–OV showed both signals from 16O18O and 18O2, confirming the exchange and release of surface oxygen atoms during the OER. Similarly, the signals from 18O2 were negligible from the background noise for Fe2O3@CeO2.
Conclusively, the DEMS results confirmed the production of LOM-related isotopic-labeled gas products, thus proving the involvement of surface oxygen atoms during the OER for the defect-engineered Fe2O3@CeO2–OV.12,19,43,44 To further understand the promoting effect of oxygen vacancies towards the OER for the defect-engineered Fe2O3@CeO2–OV, DFT+U calculations were implemented. The model of Fe2O3@CeO2–OV is established as a Ce10O20 cluster supported on the α-Fe2O3 (104) surface, as characterized by XRD in Fig. 1b. As shown in Fig. S39 (ESI†), the CeO2 favors to bind on the Fe-terminal surface of α-Fe2O3 instead of the O-terminal one. By comparing the oxygen vacancy (OV) formation energies of different situations, OV is found to preferably form on top of the CeO2 cluster, rather than at the heterojunction interface or on top of the Fe2O3 substrate, in accordance with the observed spontaneous deoxygenation of CeO2 upon heat treatment. After achieving the representative configurations of Fe2O3–CeO2 heterojunction catalysts without and with OV, the reaction pathway following the LOM was explored. Fig. S40 (ESI†) shows the corresponding geometric configurations of reaction intermediates for both catalysts. Interestingly, the already-existing OV (denoted by the blue dashed circle) does not serve directly as the reaction active site for the LOM, but instead plays the role of a modulator to activate another lattice oxygen (OL, the blue sphere), which combines with an adsorbed oxygen from solvent water (the purple sphere in Fig. 6a) to give out a molecule of O2, leaving an additional instantaneous oxygen vacancy (*□). The free energy diagram in Fig. 6a shows the superior activity of Fe2O3@CeO2–OV with the lower free energy increasing as compared with Fe2O3@CeO2 under standard basic conditions (pH = 14 and U = 0 V). The overpotential of the catalyst with OV, as determined in the step of OL–O formation (OH*(OL) + OH− → *(OL–O) + H2O + e−), is lowered by 0.32 V when OV was introduced. More importantly, without OV, the Fe2O3@CeO2 catalyst suffers from the extremely endergonic free energy of 1.43 eV during the O2 desorption (*(OL–O) → *□ + O2(g)), while the free energy increase dramatically lowers to 0.70 eV in the presence of OV. Considering that this O2 desorption is the key step in the LOM that can hardly be accelerated by the applied electro-potential, this result shows the counter-intuitive factor that the presence of a first OV could promote the O2 release to form another instantaneous oxygen vacancy *□ during the catalytic cycle, thus promoting OER activity. For comparison, we also evaluated the cases where LOM-active sites were located at the heterojunction interface with and without OV (Fig. S41, ESI†). The free energy diagram (Fig. S42, ESI†) confirms the promotion effect of a starting Ov at the interfacial site, but its activity is still inferior to that on top of CeO2 in Fig. 6a. These results imply the actual active sites to be on top of the CeO2 nanoparticles instead of on the Fe2O3 surface or at the CeO2/Fe2O3 interface.
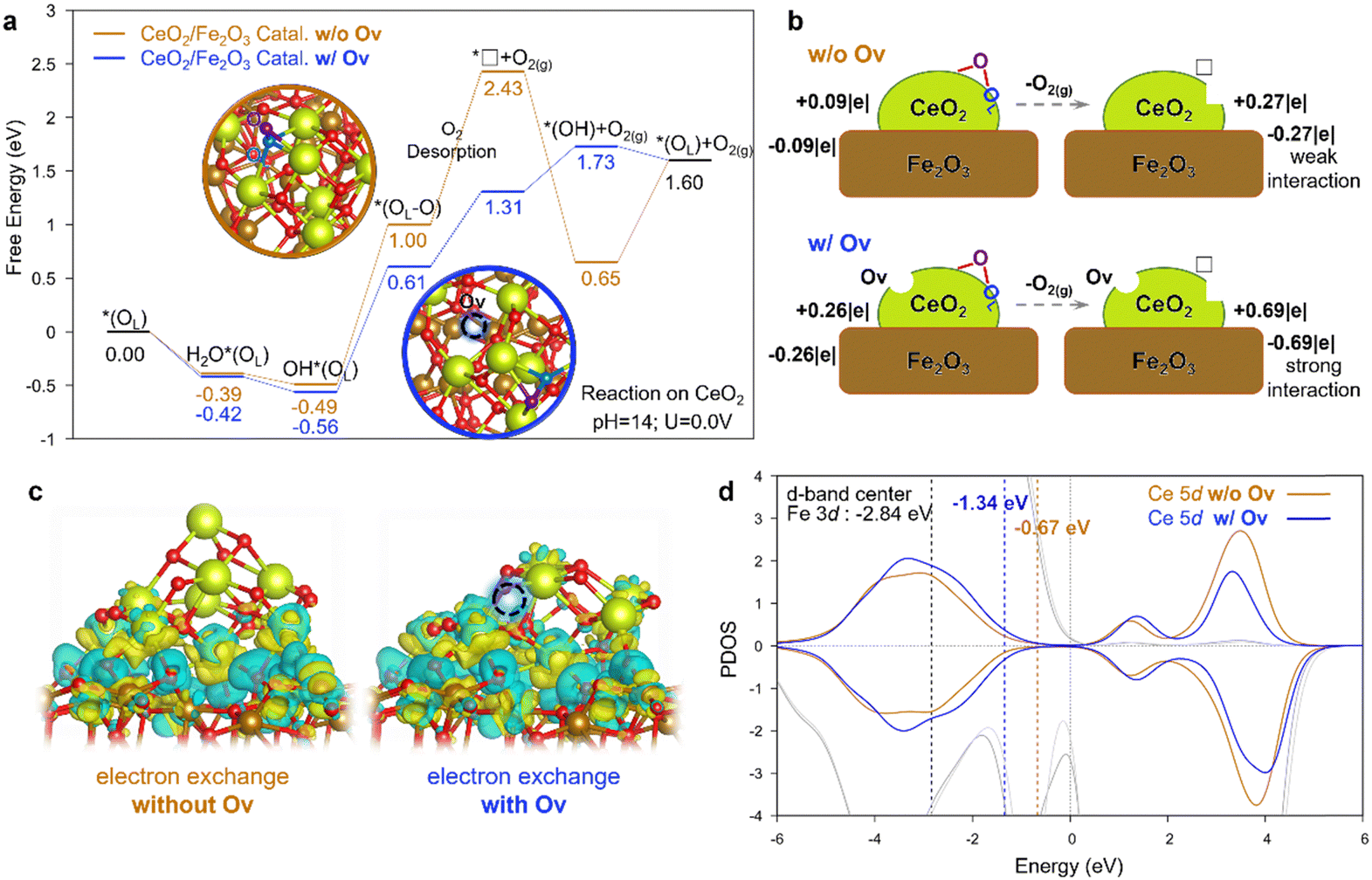 | ||
| Fig. 6 DFT calculations for LOM-based OER catalyzed by the Fe2O3@CeO2–OV and Fe2O3@CeO2 catalysts. (a) Free energy diagram for LOM-based OER steps. The detailed 3D configurations are shown in Fig. S40 (ESI†). Color code: Ce in yellow; Fe in brown; the O about to be removed in LOM in blue; the O from water in purple; the other O in red. The dotted lines represent the O2 desorption step. (b) Illustration of Bader charge changes during the O2 desorption step. (c) Charge density diagram showing the electron exchange between the CeO2 cluster and Fe2O3 support of the *□ intermediate after O2 desorption. Electron depletion and accumulation are depicted by yellow and blue areas, respectively. The isosurfaces are plotted at the value of ± 0.03 |e| Å−3. (d) Projected density of states (PDOS) of Ce 5d orbitals in the *□ intermediate of CeO2/Fe2O3 catalysts. The lighter and darker gray lines show the PDOS of surface Fe atoms with and without OV on CeO2, respectively. The yellow and blue numbers show the d-band-center of Ce without and with OV. | ||
To figure out the chemistry behind, the Bader charge analysis was performed (Fig. 6b). For Fe2O3@CeO2, when in contact with the Fe2O3 surface, the CeO2 cluster without OV takes a slightly positive charge (+0.09|e|), and the Fe2O3 surface is negative, indicating the electron transfer from CeO2 to Fe2O3, in consistent with experimental results. After O2 desorption, the positive charge of the CeO2 cluster is increased by 0.18|e| (i.e. a total of 0.27|e|). For Fe2O3@CeO2–OV with the presence of a starting OV, the CeO2−x cluster is more positive (+0.26|e|), and this charge dramatically increases by 0.43|e| after O2 desorption. The more significant charge polarization indicates the stronger interaction between the CeO2 cluster and Fe2O3 support. This trend could also be observed from the charge density distribution plot in Fig. 6c showing the electron exchange at the heterojunction interface. The cluster/support interaction is so strong for the *□ intermediate with OV that the CeO2−x cluster slightly changes its geometric configuration to take a closer contact with the support, like “butter soften on a hot pan”. The effect of strong cluster/support interaction can also be elucidated by the projected density of states (PDOS) and d-band center in Fig. 6d. Due to the presence of OV, the PDOS of Ce 5d shifts to a lower energy to better bind with Fe2O3. The d-band center of the Ce 5d orbital decreases from −0.67 eV to −1.34 eV when introducing the OV, which becomes more closer to the d-band center of Fe 3d orbitals (−2.84 eV). Upshift of the O 2p band center is generally believed to be a key parameter to index the reactivity of lattice oxygen.45,46 In our case, however, the situation is more complicated since two different metal d-bands coexist due to the formation of the oxide-heterojunction, and the strong cluster/support interaction would induce not only the modulation of electronic structures but also the geometric configuration, as was observed at the metal/oxide interface.47,48 The PDOS of 2p orbitals of O on the CeO2 cluster was also analyzed, as shown in Fig. S43 (ESI†). Without the starting OV for Fe2O3@CeO2, the O 2p moves closer to the Fermi level during the O2 dissociation of the LOM, indicating the reduction of the CeO2 cluster and the formation of Ce(III). In contrast, for Fe2O3@CeO2–OV with the presence of OV, the O 2p moves away from the Fermi level during the O2 dissociation of LOM due to the stronger electron interaction between the CeO2 cluster and the Fe2O3 support, which compensated the effect from O2 dissociation. (See detailed discussion after Fig. S43 in the ESI†). In general, the strong interaction between CeO2−x and Fe2O3 cancels out part of the free energy raise of the additional instantaneous oxygen vacancy (i.e. *□) during the O2 desorption in LOM by boosting electron transfer from CeO2−x to Fe2O3, thus contributing to the superior OER catalytic activity. One can postulate that such strong interaction at the heterojunction interface could promote the LOM in the OER over similar reducible oxide-heterojunction catalysts,49,50 the mechanism of which can be helpful for the design of new LOM electrocatalysts.
Conclusions
In summary, we demonstrated a defect-engineered Fe2O3@CeO2–OV nano-heterojunction material as highly efficient self-supporting alkaline OER electrocatalysts. The electrodes exhibited superior OER activity and durability, especially under industrial large-current density conditions and membrane-based flow cells. 18O-isotopic labelling experiments combined with a series of electrochemical measurements confirmed the participation of lattice oxygen, transforming the OER mechanism from the AEM to LOM. XPS, XAFS and DFT+U calculations revealed that oxygen vacancies predominantly existed on the surface of CeO2 nanoclusters, inducing strong charge transfer from CeO2 to Fe2O3 and electron density redistribution across the heterojunction interface. The presence of OV on CeO2 promoted strong cluster/support interaction and downshifted the Ce 5d band center closer to the Fe 3d bands, lowering the free energy barrier for both *(OL–O) formation and O2 desorption steps in the LOM, thus facilitating the OER. This work provides a novel strategy to construct high-performance iron-based OER catalysts, presenting a promising solution for cost-effective industrial water splitting applications.Author contributions
Q. H.: data curation, formal analysis, investigation, and writing – original draft. G. J. X.: methodology, data curation, formal analysis, and funding acquisition. B. H., D. X., and L. G.: formal analysis and analysis of supporting methodology. J. Wang, D. W., and L. G.: software and writing – original support draft. D. L., C. X., Z. W., and J. Wu: data curation and support visualization. F. X. and R. Z.: conceptualization. F. X., W. G. and R. Z.: funding acquisition, methodology, project administration, resources, supervision, and writing – review & editing. All the authors discussed the results and commented on the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 52203361, 22203041), the Basic and Applied Basic Research Foundation of Guangdong Province (No. 2023A1515012351, 2021A1515110406) and the Sichuan Department of Science and Technology Program of China (No. 2022YFG0299). We thank Materials Processing and Analysis Center, Peking University for assistance with TEM characterization. The XPS measurements and analyses were supported by Shimadzu Analytical Applications Center. The computational resources were supported from the Dongguan Key Laboratory of Artificial Intelligence Design for Advanced Materials, China.Notes and references
- Z. Y. Yu, Y. Duan, X. Y. Feng, X. Yu, M. R. Gao and S. H. Yu, Adv. Mater., 2021, 33, 2007100 CrossRef CAS PubMed.
- Y. Zhao, D. P. Adiyeri Saseendran, C. Huang, C. A. Triana, W. R. Marks, H. Chen, H. Zhao and G. R. Patzke, Chem. Rev., 2023, 123, 6257–6358 CrossRef CAS PubMed.
- S. Zhang, C. Tan, R. Yan, X. Zou, F. L. Hu, Y. Mi, C. Yan and S. Zhao, Angew. Chem., Int. Ed., 2023, 62, e202302795 CrossRef CAS PubMed.
- M. Vanags, G. Kulikovskis, J. Kostjukovs, L. Jekabsons, A. Sarakovskis, K. Smits, L. Bikse and A. Šutka, Energy Environ. Sci., 2022, 15, 2021–2028 RSC.
- L. Chong, G. Gao, J. Wen, H. Li, H. Xu, Z. Green, J. D. Sugar, A. J. Kropf, W. Xu and X.-M. Lin, Science, 2023, 380, 609 CrossRef CAS PubMed.
- X. Kang, F. Yang, Z. Zhang, H. Liu, S. Ge, S. Hu, S. Li, Y. Luo, Q. Yu, Z. Liu, Q. Wang, W. Ren, C. Sun, H.-M. Cheng and B. Liu, Nat. Commun., 2023, 14, 3607 CrossRef CAS PubMed.
- Y. Ma, G.-M. Mu, Y.-J. Miao, D.-M. Lin, C.-G. Xu, F.-Y. Xie and W. Zeng, Rare Met., 2022, 41, 844–850 CrossRef CAS.
- W. Cao, X.-H. Gao, J. Wu, A.-Q. Huang, H. Hu and Z.-W. Chen, ACS Catal., 2024, 14, 3640–3646 CrossRef CAS.
- N. Zhang and Y. Chai, Energy Environ. Sci., 2021, 14, 4647 RSC.
- J. Wang, Y. Gao, H. Kong, J. Kim, S. Choi, F. Ciucci, Y. Hao, S. Yang, Z. Shao and J. Lim, Chem. Soc. Rev., 2020, 49, 9154–9196 RSC.
- Y. Wang, R. Yang, Y. Ding, B. Zhang, H. Li, B. Bai, M. Li, Y. Cui, J. Xiao and Z.-S. Wu, Nat. Commun., 2023, 14, 1412 CrossRef CAS PubMed.
- A. Grimaud, O. Diaz-Morales, B. Han, W. T. Hong, Y.-L. Lee, L. Giordano, K. A. Stoerzinger, M. T. M. Koper and Y. Shao-Horn, Nat. Chem., 2017, 9, 457–465 CrossRef CAS PubMed.
- J. Jin, J. Yin, Y. Hu, Y. Zheng, H. Liu, X. Wang, P. Xi and C. H. Yan, Angew. Chem., Int. Ed., 2024, 63, e202313185 CrossRef CAS PubMed.
- K. Xiao, Y. Wang, P. Wu, L. Hou and Z. Q. Liu, Angew. Chem., Int. Ed., 2023, 135, e202301408 CrossRef.
- Y. Wu, Y. Zhao, P. Zhai, C. Wang, J. Gao, L. Sun and J. Hou, Adv. Mater., 2022, 34, 2202523 CrossRef CAS PubMed.
- Q. Zhang, W. Xiao, H. C. Fu, X. L. Li, J. L. Lei, H. Q. Luo and N. B. Li, ACS Catal., 2023, 13, 14975–14986 CrossRef CAS.
- X. Wang, H. Zhong, S. Xi, W. S. V. Lee and J. Xue, Adv. Mater., 2022, 34, 2107956 CrossRef CAS PubMed.
- Y. H. Wang, L. Li, J. Shi, M. Y. Xie, J. Nie, G. F. Huang, B. Li, W. Hu, A. Pan and W. Q. Huang, Adv. Sci., 2023, 10, 2303321 CrossRef CAS PubMed.
- Y. Ou, L. P. Twight, B. Samanta, L. Liu, S. Biswas, J. L. Fehrs, N. A. Sagui, J. Villalobos, J. Morales-Santelices, D. Antipin, M. Risch, M. C. Toroker and S. W. Boettcher, Nat. Commun., 2023, 14, 7688 CrossRef CAS PubMed.
- P. Ye, K. Fang, H. Wang, Y. Wang, H. Huang, C. Mo, J. Ning and Y. Hu, Nat. Commun., 2024, 15, 1012 CrossRef CAS PubMed.
- S. Xin, Y. Tang, B. Jia, Z. Zhang, C. Li, R. Bao, C. Li, J. Yi, J. Wang and T. Ma, Adv. Funct. Mater., 2023, 33, 2305243 CrossRef CAS.
- H. Y. Chen, L. Yang, R. X. Wang, W. J. Zhang, R. Liu, Y. Z. Yun, N. Wang, S. Ramakrishna, L. Jiao and Y. Z. Long, Small, 2023, 19, 2304086 CrossRef CAS PubMed.
- P. Kuang, Z. Ni, B. Zhu, Y. Lin and J. Yu, Adv. Mater., 2023, 35, 2303030 CrossRef CAS PubMed.
- X. Yang, C. Xing, B. Zhang, X. Liu, H. Liang, G. Luo, G. Zhang, Z. Li, S. Zhao, J. Zhang, G. Wang and Y. Qin, ACS Catal., 2022, 12, 10849–10856 CrossRef CAS.
- Y. Guo, J. Li, G. Yuan, J. Guo, Y. Zheng, Y. Huang, Q. Zhang, J. Li, J. Shen, C. Shu, J. Xu, Y. Tang, W. Lei and H. Shao, ACS Nano, 2023, 17, 18253–18265 CrossRef CAS PubMed.
- G. Mu, G. Wang, Q. Huang, Y. Miao, D. Wen, D. Lin, C. Xu, Y. Wan, F. Xie, W. Guo and R. Zou, Adv. Funct. Mater., 2023, 33, 2211260 CrossRef CAS.
- Y. Ma, Y. Miao, G. Mu, D. Lin, C. Xu, W. Zeng and F. Xie, Nanomaterials, 2021, 11, 1847 CrossRef CAS PubMed.
- D. Tyndall, M. J. Craig, L. Gannon, C. McGuinness, N. McEvoy, A. Roy, M. García-Melchor, M. P. Browne and V. Nicolosi, J. Mater. Chem. A, 2023, 11, 4067–4077 RSC.
- C. Li, Y. Nakagawa, M. Tamura, A. Nakayama and K. Tomishige, ACS Catal., 2020, 10, 14624–14639 CrossRef CAS.
- S. Liu, W. Xue, Y. Ji, W. Xu, W. Chen, L. Jia, T. Zhu, Z. Zhong, G. Xu, D. Mei and F. Su, Appl. Catal., B, 2023, 323, 122151 CrossRef CAS.
- B. Elsener, D. Atzei, A. Krolikowski, V. Rossi Albertini, C. Sadun, R. Caminiti and A. Rossi, Chem. Mater., 2004, 16, 4216–4225 CrossRef CAS.
- T. Liu, L. Zhao, D. Wang, J. Zhu, B. Wang and C. Guo, RSC Adv., 2013, 3, 25648–25651 RSC.
- A. Medvedeva, E. Makhonina, L. Pechen, Y. Politov, A. Rumyantsev, Y. Koshtyal, A. Goloveshkin, K. Maslakov and I. Eremenko, Materials, 2022, 15, 8225 CrossRef CAS PubMed.
- W. Li, J. Lv, D. Liu, W. Cai, X. Chen, Q. Huang, L. Wang and B. Wang, Chem. Mater., 2023, 35, 3892–3901 CrossRef CAS.
- N. Zhang, J. Wang, Q. Li, Y. Xin, L. Zheng, Y. Wang and Z. Zhang, Chem. Eng. J., 2021, 426, 131845 CrossRef CAS.
- X. Zhao, F. Wang, X. Kong, R. Fang and Y. Li, Nat. Commun., 2022, 13, 2591 CrossRef CAS PubMed.
- Z.-J. Chen, J. Dong, J. Wu, Q. Shao, N. Luo, M. Xu, Y. Sun, Y. Tang, J. Peng and H.-M. Cheng, Nat. Commun., 2023, 14, 4210 CrossRef CAS PubMed.
- M. Li, X. Wang, K. Liu, H. Sun, D. Sun, K. Huang, Y. Tang, W. Xing, H. Li and G. Fu, Adv. Mater., 2023, 35, 2302462 CrossRef CAS PubMed.
- Z. He, J. Zhang, Z. Gong, H. Lei, D. Zhou, N. Zhang, W. Mai, S. Zhao and Y. Chen, Nat. Commun., 2022, 13, 2191 CrossRef CAS PubMed.
- Y. Qin, Y. Liu, Y. Zhang, Y. Gu, Y. Lian, Y. Su, J. Hu, X. Zhao, Y. Peng, K. Feng, J. Zhong, M. H. Rummeli and Z. Deng, ACS Catal., 2023, 13, 256–266 CrossRef CAS.
- X. Chen, Q. Wang, Y. Cheng, H. Xing, J. Li, X. Zhu, L. Ma, Y. Li and D. Liu, Adv. Funct. Mater., 2022, 2112674 CrossRef CAS.
- Y. Zhai, X. Ren, Y. Sun, D. Li, B. Wang and S. Liu, Appl. Catal., B, 2023, 323, 122091 CrossRef CAS.
- C. Lin, J.-L. Li, X. Li, S. Yang, W. Luo, Y. Zhang, S.-H. Kim, D.-H. Kim, S. S. Shinde, Y.-F. Li, Z.-P. Liu, Z. Jiang and J.-H. Lee, Nat. Catal., 2021, 4, 1012–1023 CrossRef CAS.
- Y. Wen, P. Chen, L. Wang, S. Li, Z. Wang, J. Abed, X. Mao, Y. Min, C. T. Dinh, P. D. Luna, R. Huang, L. Zhang, L. Wang, L. Wang, R. J. Nielsen, H. Li, T. Zhuang, C. Ke, O. Voznyy, Y. Hu, Y. Li, W. A. Goddard Iii, B. Zhang, H. Peng and E. H. Sargent, J. Am. Chem. Soc., 2021, 143, 6482–6490 CrossRef CAS PubMed.
- F. Q. Wang, P. C. Zou, Y. Y. Zhang, W. L. Pan, Y. Li, L. M. Liang, C. Chen, H. Liu and S. J. Zheng, Nat. Commun., 2023, 14, 6019 CrossRef CAS PubMed.
- C. Rong, S. Wang, S. Xin, C. Jia, Q. Sun, Q. Zhang and C. Zhao, Energy Environ. Sci., 2024, 17, 4196–4204 RSC.
- T. W. van Deelen, C. Hernández Mejía and K. P. de Jong, Nat. Catal., 2019, 2, 955–970 CrossRef CAS.
- G.-J. Xia, Y. Fu, W. Cao, J. Li and Y.-G. Wang, Nano Res., 2024 DOI:10.1007/s12274-024-6625-2.
- B. Qiu, C. Wang, N. Zhang, L. Cai, Y. Xiong and Y. Chai, ACS Catal., 2019, 9, 6484–6490 CrossRef CAS.
- Y. Yang, M. Lin, Y. Song, G. Tuerhong, J. Dai, T. Zhang, D. Guo and L. Liu, J. Alloys Compd., 2022, 910, 164869 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee01588f |
| ‡ Qiuping Huang and Guang-Jie Xia contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |