
Tuning the mechanoresponsive luminescence of rotaxane mechanophores by varying the stopper size†
Keiko
Hiratsuka
a,
Tatsuya
Muramatsu
a,
Takuya
Seki
b,
Christoph
Weder
 c,
Go
Watanabe
*bd and
Yoshimitsu
Sagara
*ae
c,
Go
Watanabe
*bd and
Yoshimitsu
Sagara
*ae
aDepartment of Materials Science and Engineering, Tokyo Institute of Technology, 2-12-1 Ookayama, Meguro-ku, Tokyo 152-8552, Japan. E-mail: sagara.y.aa@m.titech.ac.jp
bDepartment of Physics, School of Science, Kitasato University, 1-15-1 Kitasato, Minami-ku, Sagamihara, Kanagawa 252-0373, Japan. E-mail: go0325@kitasato-u.ac.jp
cAdolphe Merkle Institute, University of Fribourg, Chemin des Verdiers 4, CH-1700, Fribourg, Switzerland
dKanagawa Institute of Industrial Science and Technology (KISTEC), 705–1 Shimoizumi, Ebina, Kanagawa 243–0435, Japan
eLiving Systems Materialogy (LiSM) Research Group, International Research Frontiers Initiative (IRFI), Tokyo Institute of Technology 4259 Nagatsuta-cho, Midori-ku, Yokohama 226-8501, Japan
First published on 17th February 2023
Abstract
Mechanochromic mechanophores are useful molecular mechano-probes that can visualize forces in polymeric materials. Here, we report detailed studies on the correlation between the mechanoresponsive luminescent behaviour of rotaxane mechanophores and the size of the stopper used in the mechanophores. Five different stoppers were introduced at one end of the axle molecule, which featured an electron-deficient quencher group at its center. The same ring molecule carrying an electron-rich fluorophore was used in all rotaxanes. In solution, the blue fluorescence of the emitter is almost completely quenched for all rotaxanes, on account of charge-transfer interactions between the fluorophore and quencher. The mechanoresponsive character of these motifs was investigated by incorporating them into linear segmented polyurethanes (PUs). Sufficiently bulky stoppers prevent the ring of rotaxane from dethreading, and this leads to a fully reversible change of the emission intensity when films of the rotaxane-containing PUs are stretched and relaxed. This response is caused by the force-induced shuttling of the emitter-containing cycle away from and back to the quencher, without any breakage of the interlocked structure or dethreading. By contrast, small stoppers allow the rings to dethread from the axles of the rotaxanes, and the fluorescence turn-on triggered by deformation becomes fully or partially irreversible. In this case, the fraction of dethreaded rings and the residual fluorescence intensity gradually increases as samples are repeatedly strained. Molecular dynamics simulations show that the energy required for dethreading depends strongly on the size of the stopper, and confirm our experimental observations. Our data show that the response of rotaxane mechanophores can be easily varied between fully reversible and irreversible, and this allows one to create polymers with a broad range of mechanoresponsive luminescence behaviours.
Introduction
Mechanochromic mechanophores, which change their optical absorption and/or photoluminescence (PL) when a mechanical force is applied, are receiving attention because of their potential use as reporters that can visualize stresses and strains in polymers.1–3 Since spiropyran was reported as the first mechanochromic mechanophore in 2009,4 a variety of mechanophores have been reported to show mechanochromic effects that are rooted in the scission of covalent bonds.5–33 Some mechanochromic mechanophores which need scission of covalent bonds for activation require time or additional energy such as light irradiation or thermal treatment to recover initial molecular structures,6–9,11–16 except for a few mechanophores.33 The other mechanochromic mechanophores that rely on breakage of covalent bonds are inherently irreversible.21–31 Therefore, such mechanophores would be used as mechanoprobes to assess past (cumulative) force effects. The investigation of instantaneous effects requires mechanophores that change their photophysical properties instantly and reversibly. For such purposes, mechanophores that function without covalent bond scission and instantly revert back to their original state when the force is removed are preferable.34–56 Supramolecular mechanophores in which the assembly of multiple chromophores, luminophores, and/or quenchers and therewith the optical properties can be mechanically altered have been shown useful in this context.2 Rotaxanes,34–38 cyclophanes,39,40 and loop-forming motifs41–43 have been demonstrated to reversibly change their PL properties in response to mechanical force. While the De Bo group reported force-induced breakages of covalent bonds in several rotaxanes,57,58 we demonstrated that the molecular shuttling effect in rotaxanes can be exploited to achieve reversible mechanoresponsive luminescence without any covalent bond scission.34–37 Our previously reported rotaxane mechanophores are composed of a cyclic molecule with an appended luminophore and a rod-like axle molecule containing a matching quencher in the middle and two stoppers at both ends. The quencher and the cycle show weak charge transfer interactions, which in the force-free state causes the formation of an inclusion complex and quenching of the luminophore. Photo-induced electron transfer (PET) would also contribute to the quenching. When a sufficiently large mechanical force is applied through polymers attached to the cycle and the axle, the cycle slides along the axle, and the ensuing spatial separation between the quencher and the luminophore limits their electronic interactions or PET. Therefore, strong PL was observed when films of elastic polymers in which the mechanophores were covalently embedded were uniaxially deformed.35–37 Upon removal of the force, the films contract and the emission is instantly quenched again. The response is reversible over many stretch and release cycles when rotaxanes are used that contain tetraphenylmethane units equipped with three tert-butyl groups as stoppers, suggesting that mechanically induced dethreading does not occur.35–37 However, when a rotaxane mechanophore with a 1,4-di-tert-butylbenzene stopper was employed, reversible as well as irreversible changes of the PL intensity were observed. The latter irreversible behaviour is caused by force-induced dethreading.34Here we demonstrate that the efficiency of force-induced dethreading of rotaxane mechanophores can be controlled by the stopper size and that this allows one to create polymers with a broad range of mechanoresponsive luminescence behaviours. Although force-induced dethreading of rotaxanes has been already reported by several groups,59–61 this effect was primarily studied by molecular force spectroscopy and no systematic investigations of how the molecular structures of the stopper groups affect the dethreading have been carried out. Here we combine molecular dynamics simulations, which represent a useful tool for revealing the dynamics of the rotaxanes at the atomic level,62,63 with in situ PL measurements of rotaxane-containing PUs to show that the dethreading efficiency scale with the size of the stoppers. We demonstrate that judicious choice of stoppers can be used to tailor the response of rotaxane mechanophores between fully reversible and irreversible (Fig. 1).
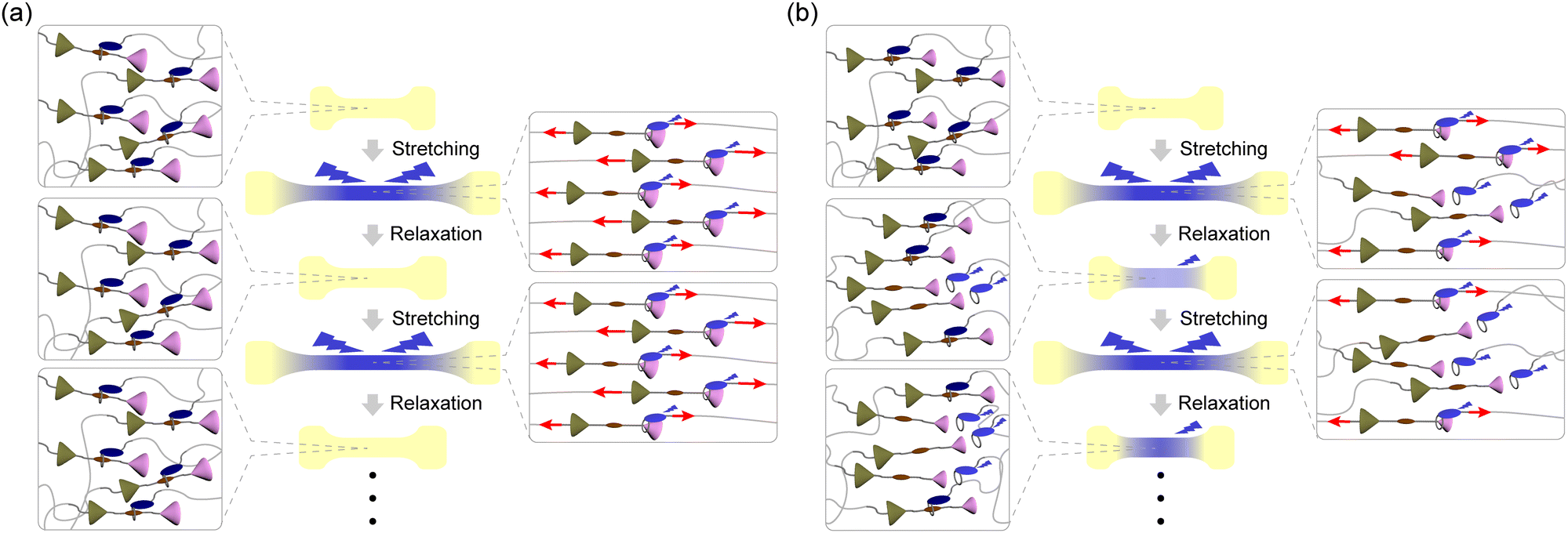 | ||
| Fig. 1 Schematic illustrations of mechanoresponsive luminescence exhibited by rotaxane mechanophore equipped with (a) sufficient bulky stoppers and (b) small stoppers in polyurethane films. In the initial state, the quencher (brown) is intercalated by the ring featuring luminophore (blue). When the films in panel (a) are stretched, bulky stoppers (pink) fully prevent the rings from dethreading. In the case of panel (b), some rings can pass the smaller stopper (pink). The number of dethreaded rings increases with the number of stretch and release cycles. | ||
Results and discussion
We designed and synthesized a series of rotaxanes (Rot1-5) in which the chemical structure of the stopper facing the end of the rod molecule towards which the cyclic molecule is drawn upon application of force changes (Fig. 2). Bis(4-tert-butylphenyl)methane, bis(4-methylphenyl)methane, diphenylmethane, 1,4-di-tert-pentylbenzene, and 1,4-di-tert-butylbenzene groups were incorporated in Rot1, Rot2, Rot3, Rot4, and Rot5, respectively. In all rotaxanes, the same diphenylmethane-based stopper featuring one tert-butyl and one 1-hydroxy-2-methylpropyl group was used to cap the second end of the axle molecule. Hydroxy groups connected to the common stopper and to the cycle-appended luminophore were used to covalently incorporate the mechanophores into polymer backbones, which can transfer mechanical forces to these sites (Fig. 2). A pyromellitic diimide (PMDI) was embedded as a quencher in the center of the axle molecule and a blue-light emitting 1-phenyl-6-(phenylethynyl)pyrene, which differs from the 9,10-(phenylethynyl)anthracene used in previous experiments,34 formed part of the cycle. The current emitter was chosen because 1,6-disubstituted pyrene derivatives show high quantum efficiency.64–66 Besides, the PMDI and pyrene groups have been reported to form charge transfer (CT) complexes.67,68 The luminophore was appended with a tetraethyleneglycol chain in order to improve the solubility of the cyclic building block. This aspect is important for the yield of the Cu(I)-catalyzed Huisgen click reactions69,70 between the axle molecule featuring a terminal alkyne and the azide group-containing stoppers, which were used for the rotaxane synthesis. This reaction requires a high concentration of all components for the pseudo rotaxane formation to be efficient (ESI†). Yields of 3–17% were attained for this step, and all rotaxanes were fully characterized by 1H and 13C NMR spectroscopy and high-resolution electrospray ionization mass spectrometry (ESI†). In each case, the 1H NMR spectra display diagnostic shifts of the peaks corresponding to the aromatic protons that confirm the rotaxane formation (Fig. S1, ESI†). Ring molecule Py was synthesized as a reference compound.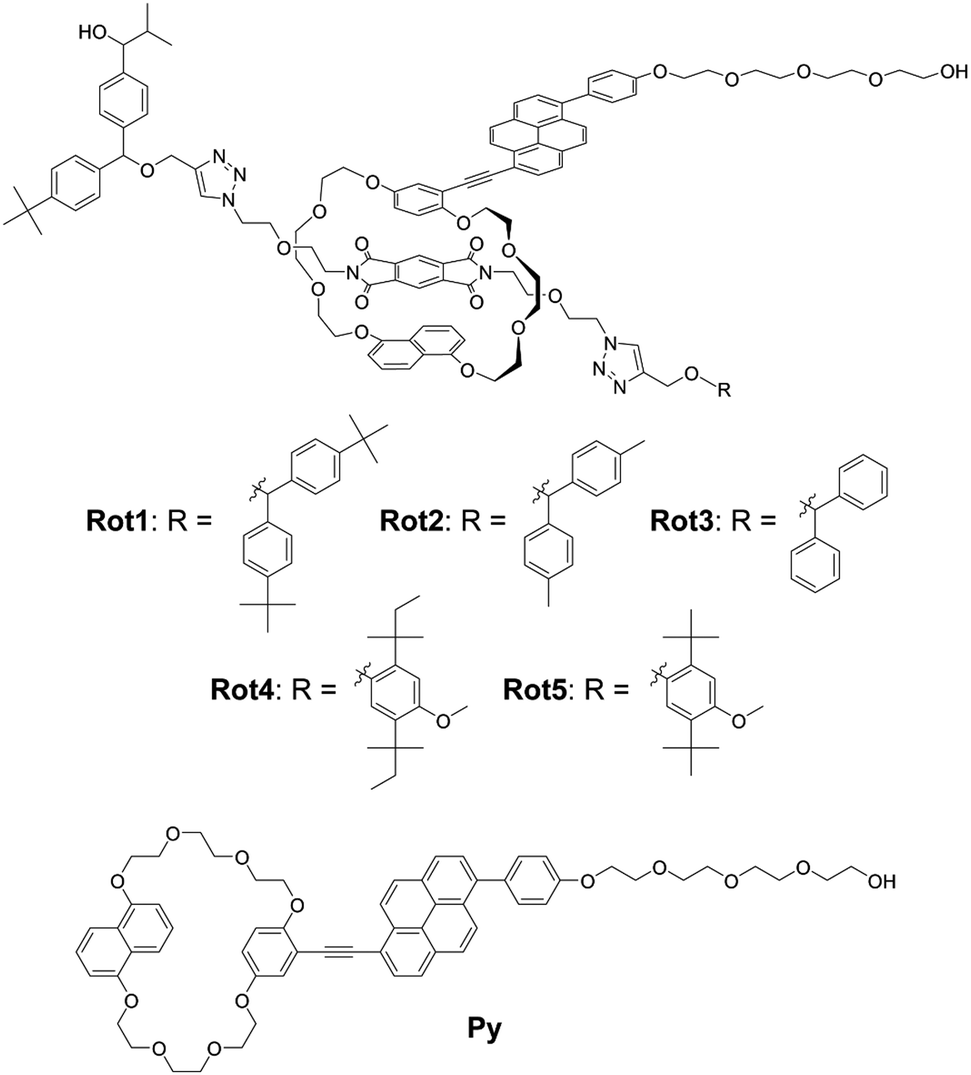 | ||
| Fig. 2 Chemical structures of rotaxane-based supramolecular mechanophores Rot1-5 and ring reference Py. | ||
The UV-vis absorption and PL spectra of all rotaxanes and reference Py were first measured in chloroform (c = 1.0 × 10−5 M) (Fig. 3 and Fig. S2, ESI†). The absorption spectrum of Py shows an absorption band with peaks at 388 and 407 nm and another sharp peak at 296 nm (Fig. 3a). The PL spectrum has peaks at 430 and 449 nm (Fig. 3b) and the PL quantum yield Φ = 0.85 is high. These spectral features are comparable to those of a reported 1-phenyl-6-(phenylethynyl)pyrene derivative.35 The absorption spectra of rotaxanes Rot1-5 display similar peaks as Py, but in each case the absorption spectrum shows a tail towards longer wavelengths, which is ascribed to CT complex formation between the luminophore and PMDI. In all rotaxanes, the fluorescence of the emitter is completely quenched. As expected, the absorption and emission spectra of all rotaxanes are practically identical, i.e., the nature of the stopper does not affect the photophysical properties in the force-free state in the solution.
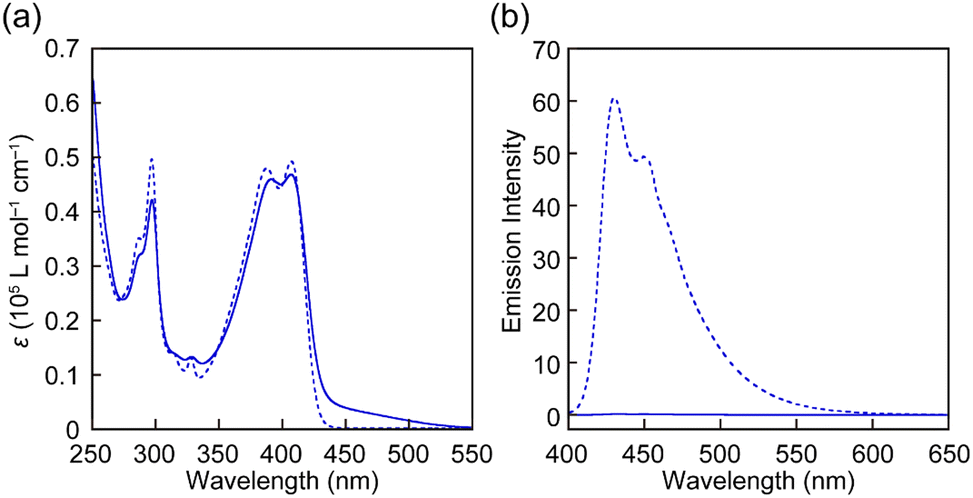 | ||
| Fig. 3 (a) UV-vis absorption and (b) PL spectra of Rot-1 (solid line) and Py (dotted line) in chloroform (c = 1.0 × 10−5 M). The excitation wavelength was 380 nm. | ||
Rot1-5 were separately incorporated into linear segmented polyurethanes (PUs) to investigate the mechanoresponsive luminescence. The specific composition was selected as it shows high elasticity and extensibility and is thus suitable to probe the response of supramolecular mechanophores.34–37,39–42 Moreover, the same PU platform was used in previous studies,34–37,39–41 which makes it possible to evaluate and compare the mechanoresponsive behaviour of the motifs investigated here with other mechanophores. To enable a direct comparison of the mechanoresponses, polymers of otherwise identical composition and similar number-averaged molecular weights (Mn) were made and processed under identical conditions (see below and ESI†). The PUs were synthesized through dibutyltin dilaurate catalyzed polyaddition reactions between the respective rotaxane (≈ 0.002 wt%), telechelic poly(tetrahydrofuran)diol (Mn ≈ 2000 g mol−1), 1,4-butanediol, and 4,4′-methylenebis(phenylisocyanate), which were carried out in THF and at room temperature (r.t.). The Mn of the resulting polymers Rot1-PU, Rot2-PU, Rot3-PU, Rot4-PU, and Rot5-PU ranges from 116 kg mol−1 to 139 kg mol−1. The 1H NMR spectra of these polymers display peaks that are characteristic of the polyurethanes, but no signals corresponding to rotaxane are discernible due to their low concentration (Fig. S3, ESI†). However, their incorporation was confirmed by UV-vis absorption spectra in DMF solutions, which display similar absorption bands as the rotaxanes in chloroform (Fig. S4, ESI†). The PL of the blue emitter remains quenched in DMF solutions of Rot1-PU, Rot2-PU, Rot4-PU, and Rot5-PU (Fig. S4, ESI†). By contrast, the solution of Rot3-PU exhibits blue PL that is derived from the emitter. We ascribe this to the fact that the Py ring gradually passes the diphenylmethane stopper. To probe under which conditions Py can dethread from the different rotaxanes in solution, the fluorescence intensities of Rot1 and Rot3 solutions in toluene were examined before and after heating the solutions for 3 h at 90 °C (Fig. 4). In the case of Rot1 both solutions displayed almost no fluorescence and heating had practically no effect. By contrast, the fluorescence intensity of the Rot3 solution significantly increased upon heating, due to heat-induced dethreading. HPLC analysis confirmed this effect, revealing the formation of free Py (Fig. S5, ESI†). Experiments with Rot3 solutions in THF show that dethreading also occurs at r.t. for this rotaxane, although the process is much slower than at 90 °C (Fig. S6, ESI†).
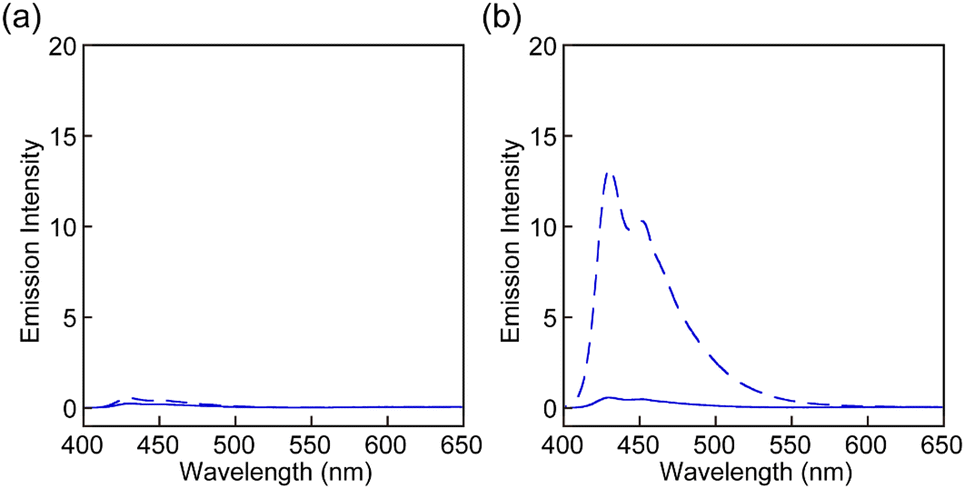 | ||
| Fig. 4 PL spectra of the toluene solution of (a) Rot1 and (b) Rot3 before (solid lines) and after (dashed lines) heating for 3 h at 90 °C. The concentration of the solutions did not change upon heating. The PL spectra were recorded with the excitation light of 380 nm at r.t. | ||
Thin films of Rot1-PU, Rot2-PU, Rot3-PU, Rot4-PU, and Rot5-PU were prepared by solution-casting from THF (ESI†). As expected, the different polymers display almost identical mechanical and thermal properties (Fig. S7–S10 and Table S1, ESI†). The tensile tests (Fig. S7, ESI†) reveal Young's moduli of 10–13 MPa, a strain at break of ca. 700%, and a stress at break of 44–70 MPa. Dynamic mechanical analysis shows that all polymers display a rubbery regime from –10 to 150 °C (Fig. S8, ESI†). Furthermore, the dynamic scanning calorimetry and thermogravimetric analysis traces are also similar to each other (Fig. S9 and S10, ESI†). The differences shown between experiments are statistically insignificant (Table S1, ESI†) and appear to be caused by sample-to-sample variations, and not the mechanophore type. The properties are also similar to previously reported linear segmented polyurethanes,34–37,39,40 suggesting that the incorporation of the rotaxanes with very low concentrations does not lead to specific “sliding ring” effects.
The mechanoresponsive PL of films of all polymers was investigated by in situ PL spectroscopy measurements that were combined with multi-cycle tensile tests. Before stretching, the Rot1-PU, Rot2-PU, Rot4-PU, and Rot5-PU films show very weak blue emission from the luminophore (Fig. 5). The absence of any correlation between stopper size and intensity suggests that the emission is caused by mechanophores that were trapped in the activated state during processing.39,40 The PL spectra of these films also display a faint emission band between 520 and 700 nm, which is ascribed to emission from a CT complex between the luminophore and the quencher. This is supported by the fact that a cyclophane-based mechanophore containing a pair of 1,6-bis(phenylethynyl)pyrene and PMDI exhibits CT complex emission at a similar wavelength.40 Unlike the other polymers, the pristine Rot3-PU films show strong PL that is clearly associated with the luminophore, reflecting considerable dethreading of Py during the solvent casting process.
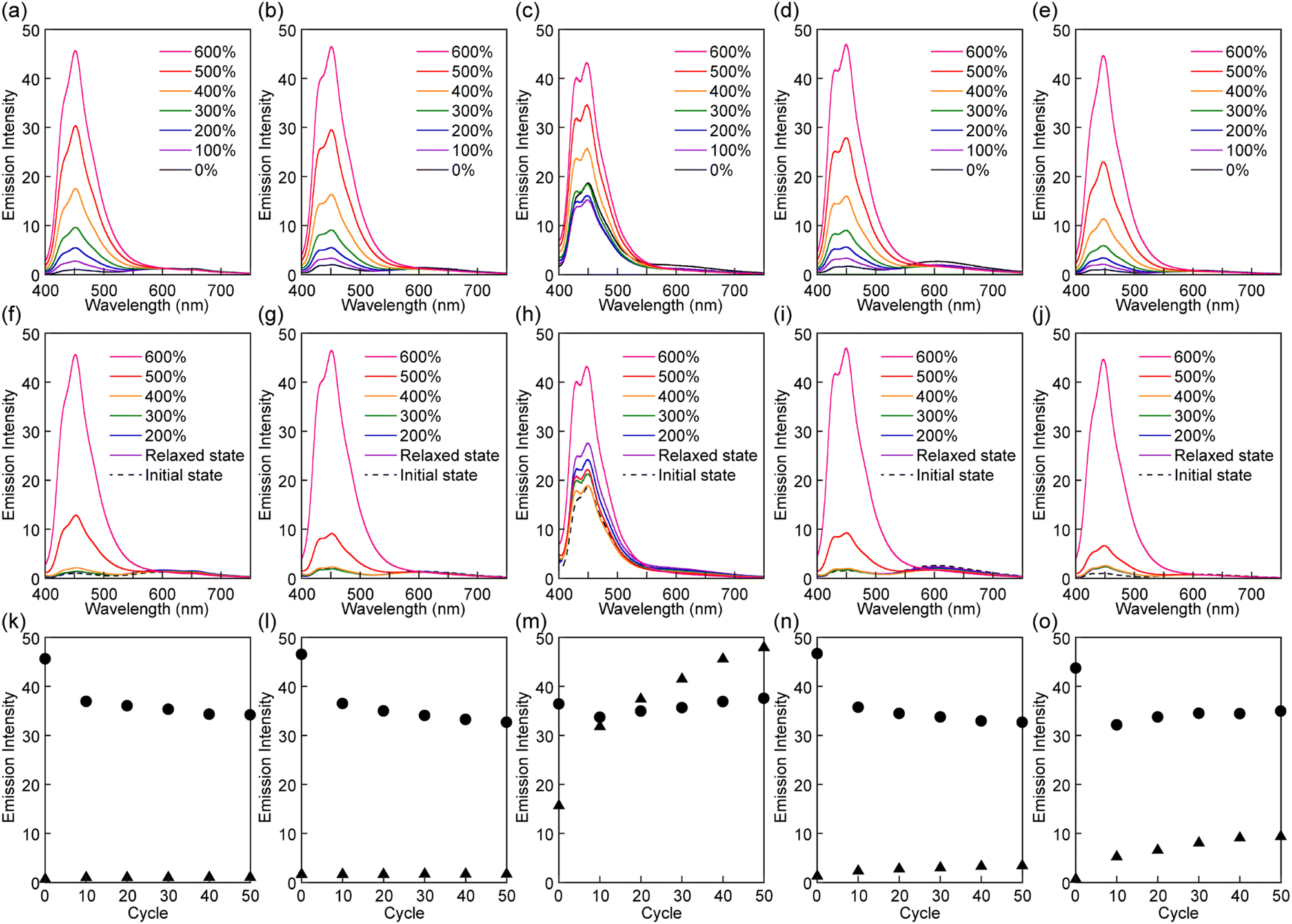 | ||
| Fig. 5 Mechanoresponsive PL of films of Rot1-PU (a, f and k), Rot2-PU (b, g and l), Rot3-PU (c, h and m), Rot4-PU (d, i and n), and Rot5-PU (e, j and o). The top and middle panels show change in PL spectra upon the first stretching (top) and relaxation (middle) cycle. All spectra were recorded at the indicated strain. The bottom panels represent plots of the emission intensity at 450 nm recorded in cyclic tensile tests at strains of 600% (circles) and in the force-free state (triangles). All PL data were recorded with excitation at 380 nm. | ||
When the Rot1-PU, Rot2-PU, Rot4-PU, and Rot5-PU films were uniaxially stretched, the intensity of the emission from the blue emitter gradually increased with the applied stress (Fig. 5a, b, d, and e). For example, the emission intensity of the Rot1-PU film at 449 nm at a strain of 600% is 45 times higher than that before deformation, and similar responses were recorded for the other materials. Upon releasing the applied force, the original, little-emissive states are recovered (Fig. 5f, g, i, and j). The significant, reversible change in the emission intensity observed in the first stretch and release cycle appears to be largely driven by the force-induced shuttling function of the rotaxanes, whereas dethreading events that would cause irreversible changes appear to be rare. The Rot3-PU films also show an incremental increase of the monomer emission upon elongation after an initial decrease up to an elongation of 100% (Fig. 5c). The initial decremental effect is ascribed to decrease of excited luminophores in the irradiated area due to film-thinning upon stretching. Since the initial emission intensity of the Rot3-PU films is already quite pronounced, the emission contrast observed upon deformation is much lower than in the case of the other films. Furthermore, the emission intensity of the Rot3-PU films after the first stretch and relax cycle is higher than that before stretching (Fig. 5h). Thus, only one single elongation is sufficient to induce dethreading of a significant fraction of the rotaxane mechanophores, reflecting that the Py cycle can easily slip past the diphenylmethane stopper used in Rot3, as seen in the thermal experiments.
Further cyclic tensile tests reveal that dethreading is also possible in some of the other rotaxanes and revealed differences between rotaxanes Rot1, Rot2, Rot4 and Rot5. Plots showing the fluorescence intensity at 450 nm of all films in the stretched state at a strain of 600% and in the relaxed state over 50 stretch and release cycles are shown in Fig. 5k, l, n and o. In all films, the intensity observed in the stretched state drops in the first few cycles, due to irreversible changes in the domain structure of segmented PUs upon stretching. No increase in emission intensity was measured under relaxed conditions for the Rot1-PU and Rot2-PU films over 50 stretching cycles, which indicates that under the mechanical conditions applied, the Py ring cannot slide past the bis(4-tert-butylphenyl)methane and bis(4-methylphenyl)methane stoppers. Thus, rotaxanes Rot1 and Rot2 can be considered as fully reversible supramolecular mechanophores that show only reversible PL changes. By contrast, the Rot4-PU and Rot5-PU films exhibit a gradual increase in monomer emission intensity as the number of deformation cycles increases, reflecting that the fraction of dethreaded Py rings increases with each cycle stretching. A comparison of the data (Fig. 5n and o) reflects that the ring can more easily pass the 1,4-di-tert-butylbenzene than the 1,4-di-tert-pentylbenzene stopper, as the relative residual PL intensity of the Rot5-PU films after 50 cycles is about 2.4 times as high as that of Rot4-PU films. Thus, the two additional methyl groups in 1,4-di-tert-pentylbenzene considerably reduce the dethreading efficiency. The data collected in the cyclic tensile tests clarified that the mechanoresponsive PL responses of Rot4-PU and Rot5-PU films also show a large reversible component, and therefore rotaxanes Rot4 and Rot5 can be considered as dual-type supramolecular mechanophores.34 A much more dramatic increase of the PL emission intensity in the relaxed state was observed for the Rot3-PU films (Fig. 5m), in which case differences between the relaxed and the stretched state disappear after only a few cycles. Thus, Rot3 is an extremely sensitive mechanophore that shows irreversible PL changes at presumably extremely low forces.
To correlate the force-induced dethreading of the ring from the rotaxane with the size of the stopper were also confirmed by MD simulations performed for Rot1, Rot3, and Rot4. The MD simulations of the rotaxanes in diethyl ether (DEE) without any restraints and external forces show that Rot1, Rot3, and Rot4 are thermally stable so that the ring molecule cannot slide past these stoppers at 300 K (see Movies S1–S3, ESI†).
The steered MD simulations (Fig. 6) were also performed for exploring the possible pathway of the ring molecule being pulled away from the axle molecule by applying an external force (see Movies S4–S6, ESI†). Since the axle molecule was pulled at a constant velocity in the steered MD simulations of this study, the time dependence of the external pulling force can be investigated as shown in Fig. S11 (ESI†). The pulling forces increase linearly until the ring molecule is about to pass through the stopper of the rod molecule. Beyond this point, the values of the pulling force rapidly decrease to zero. The maximum values of the time-dependent pulling force are 2.71 × 103 kJ mol−1 nm−1 at 9.2 ns for Rot1, 9.61 × 102 kJ mol−1 nm−1 at 6.6 ns for Rot3, and 1.38 × 103 kJ mol−1 nm−1 at 7.3 ns for Rot4. The data indicates that the external force required to pull the ring molecule away from the rotaxane decreases in the order Rot1 > Rot4 >Rot3.
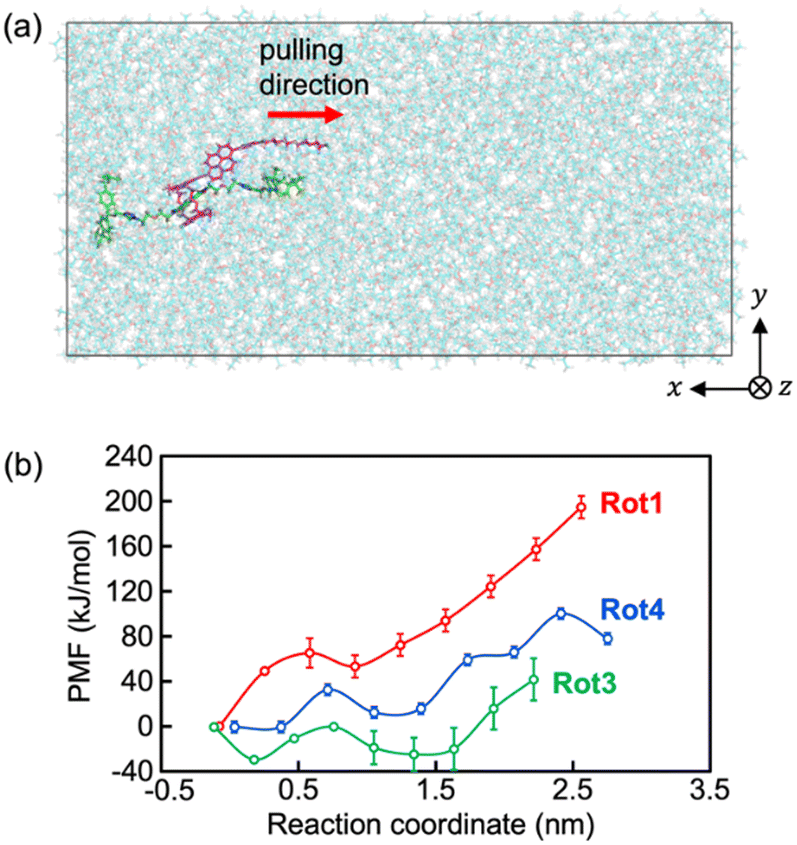 | ||
| Fig. 6 (a) Steered MD simulation snapshot of Rot1 in the initial state. The ring and axle molecules are shown as purple and green sticks, respectively. Surrounding thin sticks in cyan represent DEE. (b) Potential of mean force (PMF) profiles of Rot1, Rot3, and Rot4. The error bars depict only the statistical uncertainty from the connection of probability density by the weighted histogram method. | ||
For a more quantitative discussion, the free energy barriers for dethreading of the rotaxanes were estimated using the potential of mean force (PMF). As shown in Fig. 6b, the PMFs for pulling the ring molecule of the rotaxane past the stopper are 195 ± 10 kJ mol−1 for Rot1, 71 ± 19 kJ mol−1 for Rot3, and 101 ± 29 kJ mol−1 for Rot4. Since the thermal energy of 2 molecules at 300 K can be estimated about 5 kJ mol−1, all the rotaxanes simulated were energetically stable. While the PMF values for Rot1 and Rot4 correspond to the maximum, that for Rot3 was obtained from the difference between the minimum and maximum. This is because the initial structure of the first umbrella sampling simulation might be not the thermally most stable. The analysis of the PMF shows that it is required more energy and pulling force for dethreading of the rotaxanes of which the stopper of the rod molecule is larger or bulkier.
Conclusions
In conclusion, we demonstrated that force-induced dethreading of the ring of rotaxane from the rod group depends on the stopper bulkiness. Whether the dethreading occurs or not determines the mechanoresponsive luminescence of the rotaxane mechanophores. Our experimental and MD simulations results show clearly a correlation between the threading efficiency and the bulkiness of the stopper. The two large stoppers, bis(4-tert-butylphenyl)methane (Rot1) and bis(4-methylphenyl)methane (Rot2), completely prevent dethreading at the forces that the mechanophores experience upon deformation of polyurethane films. By contrast, the ring can easily slide past the diphenylmethane group (Rot3). The 1,4-di-substituted benzene-based stoppers show an intermediate behaviour. Intriguingly, the resistance to dethreading is extremely sensitive to the nature of auxiliary substituents attached to the benzene moieties. For example, changing the design from tert-pentyl (Rot4) to tert-butyl (Rot5) groups considerably affects the capability for dethreading. Our data show that the response of rotaxane mechanophores can be easily varied between fully reversible and irreversible, and this allows one to create polymers with a broad range of mechanoresponsive luminescence behaviours.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Mr Masato Koizumi at Open Facility Center, Tokyo Institute of Technology for HRMS measurements. This work was partially supported by Japan Science Technology Agency (JST), PRESTO (No. JPMJPR17P6) and FOREST (No. JPMJFR201N). This work was partially supported by JSPS KAKENHI (grant no. JP18H02024, JP19H02537, JP19H05718, JP20H05198, and JP 22H04531). Financial support through the National Center of Competence in Research (NCCR) Bio-Inspired Materials, a research instrument of the Swiss National Science Foundation (SNF) as well as funding from the Adolphe Merkle Foundation is gratefully acknowledged. The computations were partially performed at the Research Center for Computational Science, Okazaki, Japan (Project: 21-IMS-C043, 22-IMS-C043).Notes and references
- Y. Chen, G. Mellot, D. Van Luijk, C. Creton and R. P. Sijbesma, Chem. Soc. Rev., 2021, 50, 4100–4140 RSC.
- H. Traeger, D. J. Kiebala, C. Weder and S. Schrettl, Macromol. Rapid Commun., 2021, 2000573 CrossRef CAS PubMed.
- S. He, M. Stratigaki, S. P. Centeno, A. Dreuw and R. Göstl, Chem. - Eur. J., 2021, 27, 15889–15897 CrossRef CAS PubMed.
- D. A. Davis, A. Hamilton, J. Yang, L. D. Cremar, D. Van Gough, S. L. Potisek, M. T. Ong, P. V. Braun, T. J. Martinez, S. R. White, J. S. Moore and N. R. Sottos, Nature, 2009, 459, 68–72 CrossRef CAS PubMed.
- M. J. Robb, T. A. Kim, A. J. Halmes, S. R. White, N. R. Sottos and J. S. Moore, J. Am. Chem. Soc., 2016, 138, 12328–12331 CrossRef CAS PubMed.
- M. E. McFadden and M. J. Robb, J. Am. Chem. Soc., 2019, 141, 11388–11392 CrossRef CAS PubMed.
- B. A. Versaw, M. E. McFadden, C. C. Husic and M. J. Robb, Chem. Sci., 2020, 11, 4525–4530 RSC.
- M. E. McFadden and M. J. Robb, J. Am. Chem. Soc., 2021, 143, 7925–7929 CrossRef CAS PubMed.
- Z. Wang, Z. Ma, Y. Wang, Z. Xu, Y. Luo, Y. Wei and X. Jia, Adv. Mater., 2015, 27, 6469–6474 CrossRef CAS PubMed.
- Y. Lin, M. H. Barbee, C.-C. Chang and S. L. Craig, J. Am. Chem. Soc., 2018, 140, 15969–15975 CrossRef CAS PubMed.
- G. R. Gossweiler, G. B. Hewage, G. Soriano, Q. Wang, G. W. Welshofer, X. Zhao and S. L. Craig, ACS Macro Lett., 2014, 3, 216–219 CrossRef CAS PubMed.
- F. Verstraeten, R. Göstl and R. P. Sijbesma, Chem. Commun., 2016, 52, 8608–8611 RSC.
- T. Kosuge, X. Zhu, V. M. Lau, D. Aoki, T. J. Martinez, J. S. Moore and H. Otsuka, J. Am. Chem. Soc., 2019, 141, 1898–1902 CrossRef CAS PubMed.
- K. Imato, A. Irie, T. Kosuge, T. Ohishi, M. Nishihara, A. Takahara and H. Otsuka, Angew. Chem., Int. Ed., 2015, 54, 6168–6172 CrossRef CAS PubMed.
- T. Wang, N. Zhang, J. Dai, Z. Li, W. Bai and R. Bai, ACS Appl. Mater. Interfaces, 2017, 9, 11874–11881 CrossRef CAS PubMed.
- T. Sumi, R. Goseki and H. Otsuka, Chem. Commun., 2017, 53, 11885–11888 RSC.
- Y. Lu, H. Sugita, K. Mikami, D. Aoki and H. Otsuka, J. Am. Chem. Soc., 2021, 143, 17744–17750 CrossRef CAS PubMed.
- S. Kato, S. Furukawa, D. Aoki, R. Goseki, K. Oikawa, K. Tsuchiya, N. Shimada, A. Maruyama, K. Numata and H. Otsuka, Nat. Commun., 2021, 12, 126 CrossRef CAS PubMed.
- J. Kida, K. Imato, R. Goseki, D. Aoki, M. Morimoto and H. Otsuka, Nat. Commun., 2018, 9, 3504 CrossRef PubMed.
- S. Kato, K. Ishizuki, D. Aoki, R. Goseki and H. Otsuka, ACS Macro Lett., 2018, 7, 1087–1091 CrossRef CAS PubMed.
- Z. S. Kean, G. R. Gossweiler, T. B. Kouznetsova, G. B. Hewage and S. L. Craig, Chem. Commun., 2015, 51, 9157–9160 RSC.
- J. M. Clough, J. van der Gucht and R. P. Sijbesma, Macromolecules, 2017, 50, 2043–2053 CrossRef CAS PubMed.
- Z. Chen, J. A. M. Mercer, X. Zhu, J. A. H. Romaniuk, R. Pfattner, L. Cegelski, T. J. Martinez, N. Z. Burns and Y. Xia, Science, 2017, 357, 475–479 CrossRef CAS PubMed.
- Y. Chen, A. J. H. Spiering, S. Karthikeyan, G. W. M. Peters, E. W. Meijer and R. P. Sijbesma, Nat. Chem., 2012, 4, 559–562 CrossRef CAS PubMed.
- R. Göstl and R. P. Sijbesma, Chem. Sci., 2016, 7, 370–375 RSC.
- C. Baumann, M. Stratigaki, S. P. Centeno and R. Göstl, Angew. Chem., Int. Ed., 2021, 60, 13287–13293 CrossRef CAS PubMed.
- J. Yang, M. Horst, S. H. Werby, L. Cegelski, N. Z. Burns and Y. Xia, J. Am. Chem. Soc., 2020, 142, 14619–14626 CrossRef CAS PubMed.
- J. Yang, M. Horst, J. A. H. Romaniuk, Z. Jin, L. Cegelski and Y. Xia, J. Am. Chem. Soc., 2019, 141, 6479–6483 CrossRef CAS PubMed.
- J. R. Hemmer, C. Rader, B. D. Wilts, C. Weder and J. A. Berrocal, J. Am. Chem. Soc., 2021, 143, 18859–18863 CrossRef CAS PubMed.
- M. Karman, E. Verde-Sesto, C. Weder and Y. C. Simon, ACS Macro Lett., 2018, 7, 1099–1104 CrossRef CAS PubMed.
- M. Karman, E. Verde-Sesto and C. Weder, ACS Macro Lett., 2018, 7, 1028–1033 CrossRef CAS PubMed.
- Y. Pan, H. Zhang, P. Xu, Y. Tian, C. Wang, S. Xiang, R. Boulatov and W. Weng, Angew. Chem., Int. Ed., 2020, 59, 21980–21985 CrossRef CAS PubMed.
- H. Qian, N. S. Purwanto, D. G. Ivanoff, A. J. Halmes, N. R. Sottos and J. S. Moore, Chem, 2021, 7, 1080–1091 CAS.
- T. Muramatsu, Y. Okado, H. Traeger, S. Schrettl, N. Tamaoki, C. Weder and Y. Sagara, J. Am. Chem. Soc., 2021, 143, 9884–9892 CrossRef CAS PubMed.
- Y. Sagara, M. Karman, A. Seki, M. Pannipara, N. Tamaoki and C. Weder, ACS Cent. Sci., 2019, 5, 874–881 CrossRef CAS PubMed.
- T. Muramatsu, Y. Sagara, H. Traeger, N. Tamaoki and C. Weder, ACS Appl. Mater. Interfaces, 2019, 11, 24571–24576 CrossRef CAS PubMed.
- Y. Sagara, M. Karman, E. Verde-Sesto, K. Matsuo, Y. Kim, N. Tamaoki and C. Weder, J. Am. Chem. Soc., 2018, 140, 1584–1587 CrossRef CAS PubMed.
- R. Sandoval-Torrientes, T. Carr and G. De Bo, Macromol. Rapid Commun., 2021, 42, 2000447 CrossRef CAS PubMed.
- Y. Sagara, H. Traeger, J. Li, Y. Okado, S. Schrettl, N. Tamaoki and C. Weder, J. Am. Chem. Soc., 2021, 143, 5519–5525 CrossRef CAS PubMed.
- S. Thazhathethil, T. Muramatsu, N. Tamaoki, C. Weder and Y. Sagara, Angew. Chem., Int. Ed., 2022, 61, e202209225 CAS.
- H. Traeger, Y. Sagara, D. J. Kiebala, S. Schrettl and C. Weder, Angew. Chem., Int. Ed., 2021, 60, 16191–16199 CrossRef CAS PubMed.
- H. Traeger, Y. Sagara, J. A. Berrocal, S. Schrettl and C. Weder, Polym. Chem., 2022, 13, 2860–2869 RSC.
- K. Imato, R. Yamanaka, H. Nakajima and N. Takeda, Chem. Commun., 2020, 56, 7937–7940 RSC.
- Q. Verolet, A. Rosspeintner, S. Soleimanpour, N. Sakai, E. Vauthey and S. Matile, J. Am. Chem. Soc., 2015, 137, 15644–15647 CrossRef CAS PubMed.
- A. Colom, E. Derivery, S. Soleimanpour, C. Tomba, M. D. Molin, N. Sakai, M. González-Gaitán, S. Matile and A. Roux, Nat. Chem., 2018, 10, 1118–1125 CrossRef CAS PubMed.
- J. García-Calvo, J. Maillard, I. Fureraj, K. Strakova, A. Colom, V. Mercier, A. Roux, E. Vauthey, N. Sakai, A. Fürstenberg and S. Matile, J. Am. Chem. Soc., 2020, 142, 12034–12038 CrossRef PubMed.
- A. E. Früh, F. Artoni, R. Brighenti and E. Dalcanale, Chem. Mater., 2017, 29, 7450–7457 CrossRef.
- G. A. Filonenko and J. R. Khusnutdinova, Adv. Mater., 2017, 1700563 CrossRef PubMed.
- G. A. Filonenko, D. Sun, M. Weber, C. Müller and E. A. Pidko, J. Am. Chem. Soc., 2019, 141, 9687–9692 CrossRef CAS PubMed.
- G. A. Filonenko, J. A. M. Lugger, C. Liu, E. P. A. van Heeswijk, M. M. R. M. Hendrix, M. Weber, C. Müller, E. J. M. Hensen, R. P. Sijbesma and E. A. Pidko, Angew. Chem., Int. Ed., 2018, 57, 16385–16390 CrossRef CAS PubMed.
- T. Yamakado and S. Saito, J. Am. Chem. Soc., 2022, 144, 2804–2815 CrossRef CAS PubMed.
- R. Kotani, S. Yokoyama, S. Nobusue, S. Yamaguchi, A. Osuka, H. Yabu and S. Saito, Nat. Commun., 2022, 13, 303 CrossRef CAS PubMed.
- Y. Goto, S. Omagari, R. Sato, T. Yamakado, R. Achiwa, N. Dey, K. Suga, M. Vacha and S. Saito, J. Am. Chem. Soc., 2021, 143, 14306–14313 CrossRef CAS PubMed.
- M. Raisch, W. Maftuhin, M. Walter and M. Sommer, Nat. Commun., 2021, 12, 4243 CrossRef CAS PubMed.
- M. Taki, T. Yamashita, K. Yatabe and V. Vogel, Soft Matter, 2019, 15, 9388–9393 RSC.
- H. Hu, X. Cheng, Z. Ma, R. P. Sijbesma and Z. Ma, J. Am. Chem. Soc., 2022, 144, 9971–9979 CrossRef CAS PubMed.
- M. Zhang and G. De Bo, J. Am. Chem. Soc., 2019, 141, 15879–15883 CrossRef CAS PubMed.
- M. Zhang and G. De Bo, J. Am. Chem. Soc., 2018, 140, 12724–12727 CrossRef CAS PubMed.
- G. De Bo, Chem. Sci., 2018, 9, 15–21 RSC.
- B. Brough, B. H. Northrop, J. J. Schmidt, H.-R. Tseng, K. N. Houk, J. F. Stoddart and C.-M. Ho, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8583–8588 CrossRef CAS PubMed.
- A. Dunlop, J. Wattoom, E. A. Hasan, T. Cosgrove and A. N. Round, Nanotechnology, 2008, 19, 345706 CrossRef PubMed.
- S. Du, H. Fu, X. Shao, C. Chipot and W. Cai, J. Phys. Chem. C, 2018, 122, 9229–9234 CrossRef CAS.
- Z. Wu, S. Wang, Z. Zhang, Y. Zhang, Y. Yin, H. Shi and S. Jiao, RSC Adv., 2022, 12, 30495–30500 RSC.
- Y. Sagara, T. Komatsu, T. Ueno, K. Hanaoka, T. Kato and T. Nagano, J. Am. Chem. Soc., 2014, 136, 4273–4280 CrossRef CAS PubMed.
- A. Yoshizawa and M. Inouye, ChemPhotoChem, 2018, 2, 353–356 CrossRef CAS.
- H. Maeda, T. Maeda, K. Mizuno, K. Fujimoto, H. Shimizu and M. Inouye, Chem. – Eur. J., 2006, 12, 824–831 CrossRef CAS PubMed.
- S. Amemori, K. Kokado and K. Sada, Angew. Chem., Int. Ed., 2013, 52, 4174–4178 CrossRef CAS PubMed.
- S. B. Krishnan, R. Krishnan and K. R. Gopidas, Chem. – Eur. J., 2018, 24, 11451–11460 CrossRef CAS PubMed.
- Q. Wang, T. R. Chan, R. Hilgraf, V. V. Fokin, K. B. Sharpless and M. G. Finn, J. Am. Chem. Soc., 2003, 125, 3192–3193 CrossRef CAS PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tc00330b |
| This journal is © The Royal Society of Chemistry 2023 |