
Tuning the mechanoresponsive luminescence of rotaxane mechanophores by varying the stopper size†

Abstract
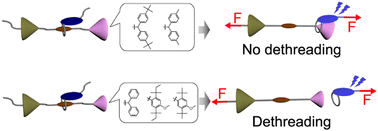
Mechanochromic mechanophores are useful molecular mechano-probes that can visualize forces in polymeric materials. Here, we report detailed studies on the correlation between the mechanoresponsive luminescent behaviour of rotaxane mechanophores and the size of the stopper used in the mechanophores. Five different stoppers were introduced at one end of the axle molecule, which featured an electron-deficient quencher group at its center. The same ring molecule carrying an electron-rich fluorophore was used in all rotaxanes. In solution, the blue fluorescence of the emitter is almost completely quenched for all rotaxanes, on account of charge-transfer interactions between the fluorophore and quencher. The mechanoresponsive character of these motifs was investigated by incorporating them into linear segmented polyurethanes (PUs). Sufficiently bulky stoppers prevent the ring of rotaxane from dethreading, and this leads to a fully reversible change of the emission intensity when films of the rotaxane-containing PUs are stretched and relaxed. This response is caused by the force-induced shuttling of the emitter-containing cycle away from and back to the quencher, without any breakage of the interlocked structure or dethreading. By contrast, small stoppers allow the rings to dethread from the axles of the rotaxanes, and the fluorescence turn-on triggered by deformation becomes fully or partially irreversible. In this case, the fraction of dethreaded rings and the residual fluorescence intensity gradually increases as samples are repeatedly strained. Molecular dynamics simulations show that the energy required for dethreading depends strongly on the size of the stopper, and confirm our experimental observations. Our data show that the response of rotaxane mechanophores can be easily varied between fully reversible and irreversible, and this allows one to create polymers with a broad range of mechanoresponsive luminescence behaviours.
- This article is part of the themed collection: 2023 Journal of Materials Chemistry C Most Popular Articles
 Please wait while we load your content...
Please wait while we load your content...

