
Ni2FeS4 as a highly efficient earth-abundant co-catalyst in photocatalytic hydrogen evolution†
Judith
Zander
and
Roland
Marschall
 *
*
Department of Chemistry, University of Bayreuth, Universitätsstraße 30, 95447 Bayreuth, Germany. E-mail: roland.marschall@uni-bayreuth.de
First published on 20th July 2023
Abstract
The earth abundant sulphide Ni2FeS4 was used as a highly efficient co-catalyst for the hydrogen evolution reaction (HER) over TiO2, boosting the activity of pure TiO2 (P25) by a factor of more than 8 under AM 1.5G simulated sunlight, thus presenting a promising alternative to platinum or rhodium co-catalysts. Low metal-sulphide loadings of only 0.5 wt% (0.29 wt% of metals) could be realised, thereby rivalling common loadings of noble metal co-catalysts at significantly lower material costs. The performance was stable under 1 sun irradiation and no decrease in the activity was observed over 20 h of irradiation. The synthesis of Ni2FeS4 is based on low-cost precursors and can be completed after only 1 min at 200 °C in the microwave, without need for toxic H2S, presenting an energy efficient and cost-effective possibility to obtain large amounts of such co-catalysts in a short time. This combination of a fast and cheap synthesis together with a high efficiency and stability makes Ni2FeS4 an outstanding candidate for the use as co-catalyst for the HER with sunlight.
Introduction
The development of a sustainable energy economy has increasingly come into the focus of current research. Apart from the generation of electric power from renewable sources, the synthesis of carbon-free fuels, such as H2 is required. Currently, H2 is to a large extent obtained via steam-reforming, however, and thus still contributing to the carbon footprint.1 Alternative approaches, such as water electrolysis (using electricity from sustainable energy sources) or photocatalysis, suffer from high costs and low efficiencies.2–5To improve the efficiency of photocatalysts, co-catalysts are added, that improve charge separation, reduce recombination rates, lower the overpotential, and provide active sites.6 For the hydrogen evolution half-reaction (HER), the most efficient co-catalysts are platinum, gold, or rhodium-based, hence an increased activity usually goes in hand with a higher system cost. Earth abundant co-catalysts are therefore desirable. Molecular co-catalysts have been designed and a good performance has been observed for transition metal complexes.7,8 However, many suffer from low long-term stabilities, hence, efforts have been made to design new heterogeneous transition metal co-catalysts.
While transition metal oxides, hydroxides and oxyhydroxides of Co, Fe and Ni have shown promise as electrocatalysts for the oxygen evolution reaction (OER), mainly alloys with molybdenum, metal phosphides and sulphides have been used to replace noble metals for the hydrogen evolution.9–13 Compared to oxides they exhibit a far higher conductivity. The active centres of major hydrogenase enzymes, contain either solely Fe, or Fe and Ni atoms together. Therefore, biomimetic approaches exploit compositional similarities.14,15
Especially sulphides of the earth abundant metals nickel and iron have emerged as highly efficient electrocatalysts for the HER.16,17 Thus, Faber et al. investigated FeS2, CoS2 and NiS2 for their activity in electrochemical HER, with CoS2 performing best.18 Even better activities were observed for ternary Fe–Co sulphides and ternary Fe–Ni sulphides and selenides.19–21 The activity of Ni and iron hydroxides, as well as sulphides in electrochemical water splitting could further be improved by a combination with MXene nanosheets, elucidating the advantages of composite formations.22,23
Good catalysts in the electrochemical HER are oftentimes also good co-catalysts in photocatalysis, since they fulfil a similar role. Thus, MoS2 and CoP are among both the most important earth-abundant electrocatalysts and co-catalysts.24–32 Additionally, nickel-based materials have shown promise as co-catalysts.33 Nickel nanoparticles themselves can already serve as co-catalysts, which has e.g. been shown on CdS.34–36 Ran et al. tested different Ni co-catalysts on ZnxCd1−xS, observing an enhancement effect for metallic Ni, as well as for NiS and Ni(OH)2.37 Similarly, Pareek et al. employed Ni and Co oxide and hydroxide co-catalysts on CdS.38 Other examples include Ni nitrides, phosphides, or Ni–Fe layered double hydroxides (LDH).39–42 NiSx can also be used as a co-catalyst, which has been shown again mainly on g-C3N4 and CdS.43–46 Interestingly, NiS can also be photochemically deposited on g-C3N4, as demonstrated by Zhao et al.47
Apart from CdS and g-C3N4 as photocatalyst materials, metallic Ni, as well as binary Ni and Fe sulphides could also improve the HER activity of TiO2. For example, Tran et al. employed Co and Ni nanoclusters on TiO2 to improve the HER activity under UV irradiation.48 Xiao et al. prepared atomically dispersed Ni on TiO2via a molten salt synthesis route.49 Moreover, Ni–Fe alloys were used as a co-catalyst on P25.50 When binary Ni or Fe sulphides were employed as co-catalysts, usually rather high sulphide loadings were required. Thus, Wang et al. synthesised composites of NiS and CuS on TiO2, with 5 wt% of each resulting in the highest activity enhancement.51 This is in agreement with an optimum loading of 7 at% of NiS on TiO2 found by Zhang et al.52 FeS2 was also used as a co-catalyst on TiO2, exploiting the good light harvesting abilities of the sulphide.53 The co-catalyst performance of NiSx could be further increased by the incorporation of Cu, which resulted in a decreased adsorption energy for S–Hads bonds, indicating, that additional metal centres can be beneficial.54
Compared to NiSx and FeSx, ternary nickel iron sulphides allow for more parameters to adjust the properties and in turn the catalytic activity. They possess an almost metallic conductivity and have already shown promise as electrocatalysts in multiple fields.55 Our group used Ni2FeS4 nanosheets for the electrochemical production of syn-gas.56 Additionally, the nickel- and iron-based thiospinel shows a low overpotential for the OER in alkaline electrolytes.55,57,58 The group of Apfel has successfully employed pentlandites, Fe4.5Ni4.5S8, for efficient electrocatalytic hydrogen evolution, with the partial replacement of sulphur by selenium further increasing the activity.59–61
Since co-catalysts are essentially electrocatalysts that additionally undergo efficient charge carrier exchange with the photocatalyst, we herein employed Ni2FeS4 as a co-catalyst for the first time to boost the hydrogen evolution activity of TiO2 under sunlight irradiation. Upon decoration with only 0.5 wt% (0.29 wt% of metals) of Ni2FeS4, the activity of pure P25 could be increased by a factor of more than 8 under AM 1.5G simulated sunlight, thus showing promise as an alternative to platinum or rhodium co-catalysts. This loading rivals common loadings of noble metal co-catalysts at significantly lower material costs. The performance was stable under 1 sun irradiation and no decrease in the activity was observed over 20 h hour of irradiation. The combination of a fast and low-cost synthesis together with a high efficiency and stability makes Ni2FeS4 an outstanding candidate for the use as co-catalyst for the HER with sunlight.
Experimental
Synthesis of Ni2FeS4
In a typical synthesis procedure 128.5 mg (2 eq., 0.5 mmol) of Ni(acac)2 (Sigma-Aldrich) and 88.3 mg (1 eqn (0.25) mmol) of Fe(acac)3 (Acros Organics) were dissolved in 5 mL of 1-phenylethanol (SigmaAldrich). 5 mL of benzyl mercaptan (Sigma-Aldrich) were added directly before the synthesis under stirring in a 30 mL borosilicate microwave vial. The solution was slowly heated under stirring in a microwave reactor (Anton Paar Monowave 400) up to 200 °C. The temperature was held for 1 to 30 min and subsequently the solution was cooled by compressed air. The product was precipitated with n-pentane, washed thrice with acetone/water mixtures and once with diethyl ether and then dried at 80 °C overnight in air. For storage, Ni2FeS4 was transferred to a glovebox.Decoration of P25 with Ni2FeS4
The respective wt% ratios of Ni2FeS4 and P25 were ground together in a mortar for 10 min under addition of low volumes of i-propanol, followed by annealing either in air in a muffle furnace for 2 h at 200 °C or in a tube furnace under argon.Photocatalysis
50 mg of the photocatalyst were ultrasonicated in approx. 20 mL of ultrapure water for 10 min. The dispersion was transferred to a home-made glass reactor. Water and methanol were added to a total volume of 150 mL containing 10 vol% of methanol. The dispersion was stirred and degassed with argon (25 mL min−1) prior to the measurement. The gas composition was analysed by gas chromatography (Shimadzu GC-2014) continuously throughout the experiment starting 30 min before switching on the lamp. After 30 min of gas monitoring, the irradiation by a solar simulator (150 W, Xe) equipped with an AM 1.5G filter (Newport) was started. After 5 hours of continued illumination, the lamp was turned off and the gas monitoring continued until the hydrogen concentration reached approx. 0. For measurements under UV irradiation, 150 mg were dispersed in 600 mL of a 10 vol% aqueous methanol solution. The dispersion was degassed with argon (100 mL min−1) prior to the experiment and subsequently irradiated for 5 h by a 700 W Hg lamp operated at 500 W (Peschl Ultraviolet). The gas composition was analysed by gas chromatography before, during and after the irradiation period.Material characterisation
Ni2FeS4 and P25 decorated with Ni2FeS4 were characterised with powder X-ray diffraction using either Cu Kα irradiation, or Ag Kα irradiation. For measurements with a Cu anode, a Malvern PANalytical Empyrean device was used, with an acceleration voltage of 40 kV, an emission current of 40 mA and Bragg–Brentano geometry. Measurements with an Ag anode were performed on a STOE STADI P Mythen2 4 K diffractometer, equipped with a Ge(111) monochromator and four Dectris MYTHEN2 R 1 K strip detectors.62 Hilgenberg capillaries (0.5 mm) were used and measurements were repeated and accumulated. X'Pert high score plus was used for the identification of crystal phases. Crystallite sizes were determined using the integral breadth method, which is the reciprocal crystallite size and calculated as the area of a reflection divided by its height, if the diffraction intensity is plotted versus the scattering vector.63 Raman measurements were performed on a Horiba Yvon Raman microscope, using a He–Ne-laser with a wavelength of 633 nm and a power of 11.5 W. The laser intensity was reduced down to 10%, or 25%. To observe oxidative changes, the power was increased to 50 or 100% for short periods. Spectra were despiked manually. For diffuse reflectance infrared Fourier transformed (DRIFT) spectroscopy, a Bruker Alpha II spectrometer was used. UV/vis/NIR measurements were conducted on a PerkinElmer Lambda 750 spectrometer, using a Praying Mantis (Harrick) and spectralon as white standard. The Kubelka–Munk function was used for the calculation of pseudo-absorption, F(R), according to:64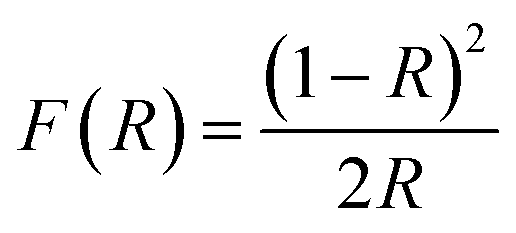
The band gap was calculate from a fit of the corresponding Tauc plots.65 (F(R)hν)1/n with n = 0.5 for direct band gaps and n = 2 for indirect ones. Physisorption measurements were performed on a Quadrasorb Evo device from Anton Paar QuantaTec at 77 K. Surface areas were determined using the Brunauer–Emmet–Teller (BET) model, using the software ASiQwin for data evaluation. Samples were degassed for 12 h at 120 °C prior to measurements. X-ray photoelectron spectroscopy (XPS) was performed on a Physical Electronics PHI VersaProbe III Scanning XPS Microprobe instrument. Al Kα irradiation, a beam voltage of 15 kV, a power of 50 W and a beam diameter of 200 μm were used. Step size was 0.8 eV with a time per step of 50 ms and a pass energy of 224 eV for survey spectra and 0.1 eV, 50/20 ms and 26 eV for high resolution spectra. Samples were continuously flooded with electrons and Ar+ at low energy. Data evaluation was done with CASA XPS,66 using a Shirley Background and Gaussian–Lorentzian profile functions (GL30). For charge correction C 1s was set to 248.8 eV. Morphology imaging by scanning electron microscopy (SEM) was performed on a Zeiss Leo 1530 device at an acceleration voltage of 3 kV, using Pt sputtering (Cressington Sputter Coater 208 HR). The same instrument was employed for energy-dispersive X-ray spectroscopy (EDX) at an acceleration voltage of 20 kV and using an ultra-dry EDX detector by Thermo Fisher Scientific NS7. For thermogravimetric analysis (TGA) coupled with mass spectrometry (MS) a Netzsch Jupiter STA 449C thermobalance and a Netzsch Aeolos QMS 402C quadrupole MS were used. A heating ramp of 2 K min−1 in synthetic air was employed. Selected solutions were examined after the photocatalytic experiments with ion chromatography. The solution was filtered through a 0.2 μm syringe filter and subsequently analysed by a Dionex Aquion system from Thermo Fisher, equipped with a Dionex IonPac AS9-HC column and IonPac AG9-HC guard column and a UV detector (λdet = 207 nm). 1 mM NaHCO3/80 mM Na2CO3 was used as eluent. For the electrochemical impedance spectroscopy (EIS), 10 mg of Ni2FeS4 were dispersed in 300 μL of i-propanol (p.a.) and 20 μL of a 5 wt% Nafion solution (Alfa Aesar) and dropcast onto carbon paper (Freudenberg H2315-C2) with the coated area restricted to 1 cm2 with Kapton tape. A three electrode H-cell setup was used, with 1 M KOH as the electrolyte, a platinum counter electrode and a RHE reference electrode (Gaskatel). A Parstat 3000A-DX potentiostat (Princeton Applied Research) and VersaStudios were employed.
Results and discussion
We have previously shown that a fast one-pot microwave-assisted synthesis of Ni2FeS4 is possible, using the metal acetlyacetonates and benzyl mercaptan as a sulphur source.56 The benzyl mercaptan is therein partially replacing the solvent 1-phenylethanol, that is known to directly take part in the reaction and condensation of organic metal precursors to oxides.67,68 Redox reactions have to occur during the reaction, as a change in the oxidation state of both nickel and iron from an initial Ni2+ and Fe3+ in the acetylacetonates to Ni3+ and Fe2+ in the sulphide is required.69 A possible reaction sequence based on the mechanism for oxide formation is depicted in Fig. 1.70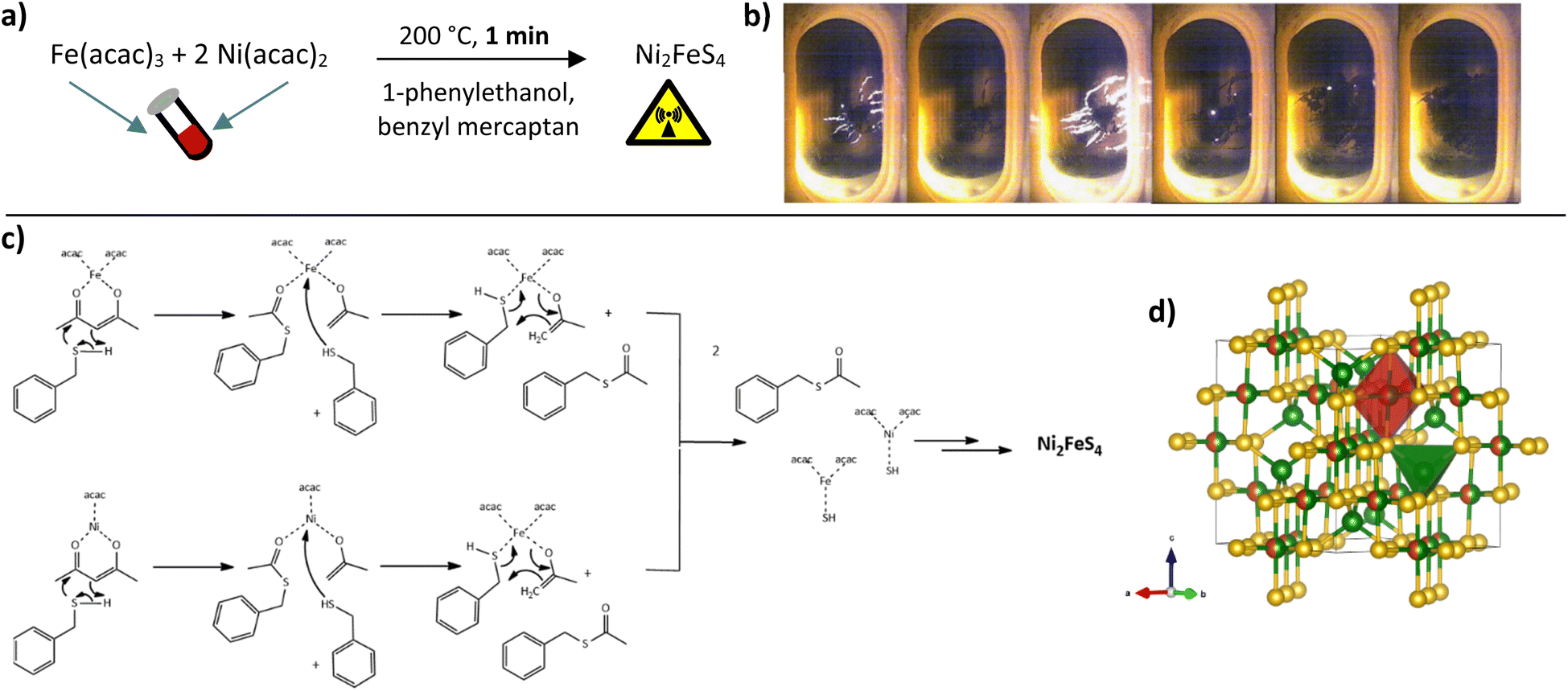 | ||
| Fig. 1 General reaction equation for the formation of Ni2FeS4 (a), photographs of observed light flashes and nanosheet formation (b), reaction sequence based on a nucleophilic attack of benzyl mercaptan similar to what was reported for alcohols (c) and crystal structure (d). | ||
The benzyl mercaptan to 1-phenylethanol ratio was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. A preferential/faster reaction with the sulphur precursor is assumed, hence a dilution with the alcohol is possible. The very fast reaction can be observed directly with an integrated camera (Fig. 1). Light flashes are visible during the first minutes of the synthesis, possibly either as a result of occurring redox processes, or as a consequence of strong microwave absorption by Ni-SH species, since the nanosheets can be observed to directly grow out of these spots along the wall of the microwave vessel, underlining that condensation and hotspots are directly connected.
:1. A preferential/faster reaction with the sulphur precursor is assumed, hence a dilution with the alcohol is possible. The very fast reaction can be observed directly with an integrated camera (Fig. 1). Light flashes are visible during the first minutes of the synthesis, possibly either as a result of occurring redox processes, or as a consequence of strong microwave absorption by Ni-SH species, since the nanosheets can be observed to directly grow out of these spots along the wall of the microwave vessel, underlining that condensation and hotspots are directly connected.
The appearance of light flashes is most pronounced during the first minute of the synthesis. Going in hand with this observation, we found that the reaction time can likewise be decreased down to one minute, without significant differences in the crystallinity of the obtained nanosheets and a complete retention of phase-purity, as confirmed by SEM and TEM images (Fig. 2, S1 and S2†) and high-resolution Ag XRD (Fig. 2). The measured separation of lattice planes in the sections are 2.2 and 2.6 Å, which corresponds to the (400) and (222) planes of a cubic thiospinel (compare ICDD reference 00-047-1740). The crystallite size is around 7 nm after a reaction time of 5 min, without observed growth upon prolonged heating, which is in good agreement with the visual monitoring of sheet growth. The same trend is reflected in the BET area that is approx. 80 m2 g−1 independent of the synthesis time (Fig. 2). EDX confirms an almost ideal nickel to iron ratio of 2, with possibly a slight decrease in the relative nickel ratio after prolonged heating (Table S1†). The metal to sulphur ratio (M:S) is with 0.8 to 0.9 slightly higher than the expected 0.75, likely due to partial surface oxidation and possibly lower sensitivity for sulphur in the EDX.
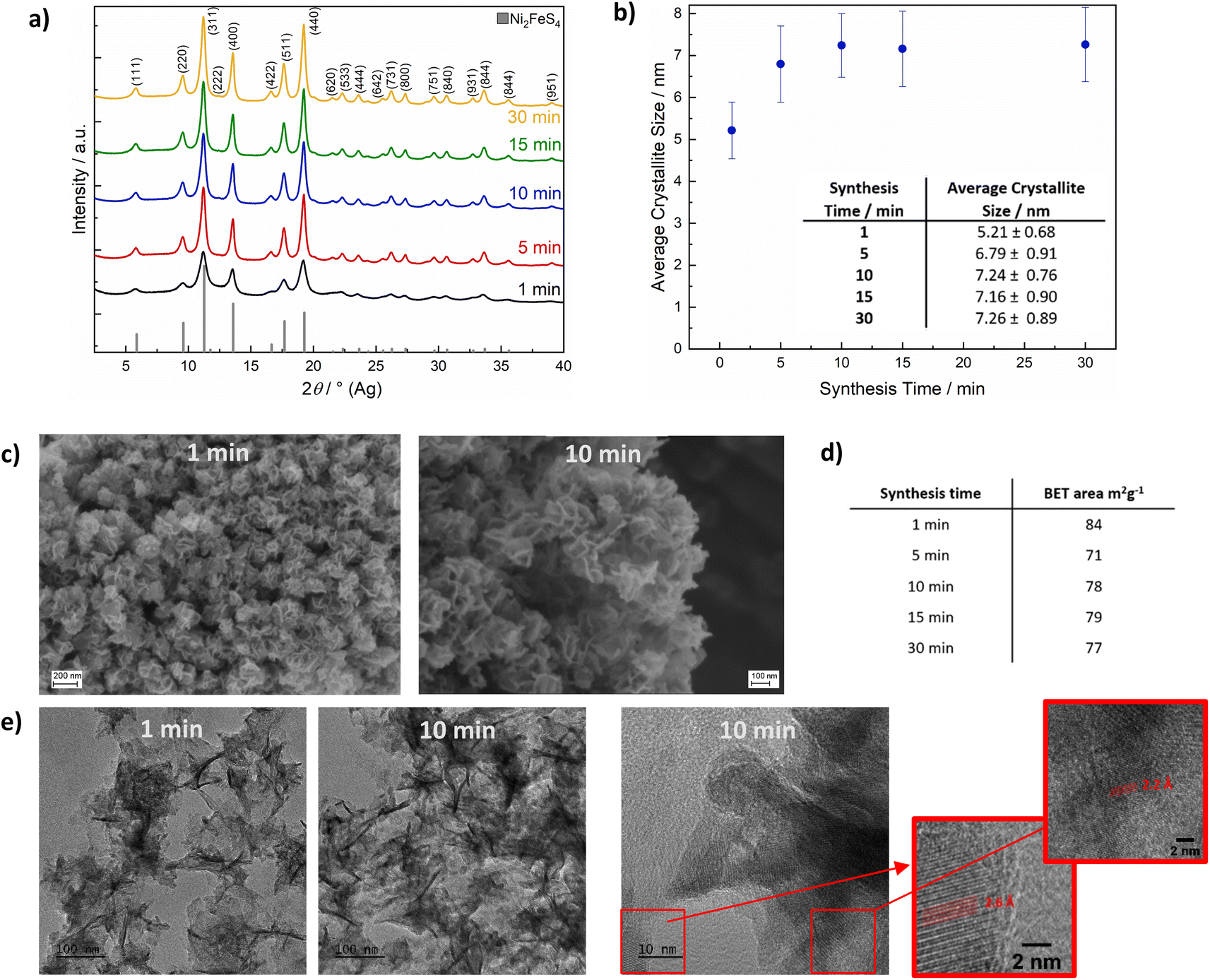 | ||
| Fig. 2 XRD patterns for Ni2FeS4 obtained after different synthesis times (a), corresponding crystallite sizes as determined by the integral breadth method (b), SEM images of Ni2FeS4 nanosheets obtained after 1 and 10 min (c) and BET surface areas (d). For the identification of reflections, the ICDD reference card 00-047-1740. Additionally, TEM images of Ni2FeS4 nanosheets obtained after 1 and 10 min are shown in (e). | ||
X-ray photoelectron spectroscopy (XPS) was conducted on Ni2FeS4 synthesised after short (1 min), medium (15 min) and long (30 min) reaction times in the microwave, to gain insights into the surface composition and to elucidate changes occurring during prolonged heating (Fig. 3 and S3†). Survey scans reveal a significantly lower M/S ratio of approx. 0.5 compared to EDX results, indicating an excess of sulphur on the surface (Table S1†). This could be an effect of organic residues from the synthesis, as also evidenced by the high carbon content. Partial surface oxidation is also confirmed by the presence of oxygen. The Ni to Fe ratio decreases from 2.35 to 1.88 upon an increase of the synthesis time from 1 to 30 min. A similar effect is also observed with EDX. This indicates a loss of Ni upon prolonged heating, which is mirrored in a decreasing M/S ratio. A similar loss of the more redox active and better microwave absorbing cation at the surface was observed in the synthesis of CuFe2O4, alongside cation migration of Cu2+.71 Possibly, an extended irradiation with microwaves results in changes in the Ni coordination as well. Santos-Carballal et al. suggested a normal spinel structure to be the thermodynamically more stable one, that is not formed in synthetic approaches due to kinetic reasons.72 Prolonged heating might thus lead to cation migration towards a thermodynamically more stable structure. The O 1s spectra additionally show a significant decrease in the amount of metal oxygen bonds with increasing synthesis duration, and an increase in the relative ratio of sulphates to hydroxides. To differentiate between sulphates and hydroxides/organics, the O 1s spectra was constrained to FWHM and binding energies reported by Legrand et al. for pentlandite.73 There is still some uncertainty in the relative ratio, as the constraints rely on accurate binding energy reference and the carbon species might change during continued heating as well – although no significant differences in the C 1s spectra were observed (Fig. S3†). The evidence for oxidic species would in turn suggest portions of Ni to exist in the oxidation state II+ instead of the further oxidised III+, indicating that oxidation is not yet fully completed after 1 min, but requires further microwave radiation.
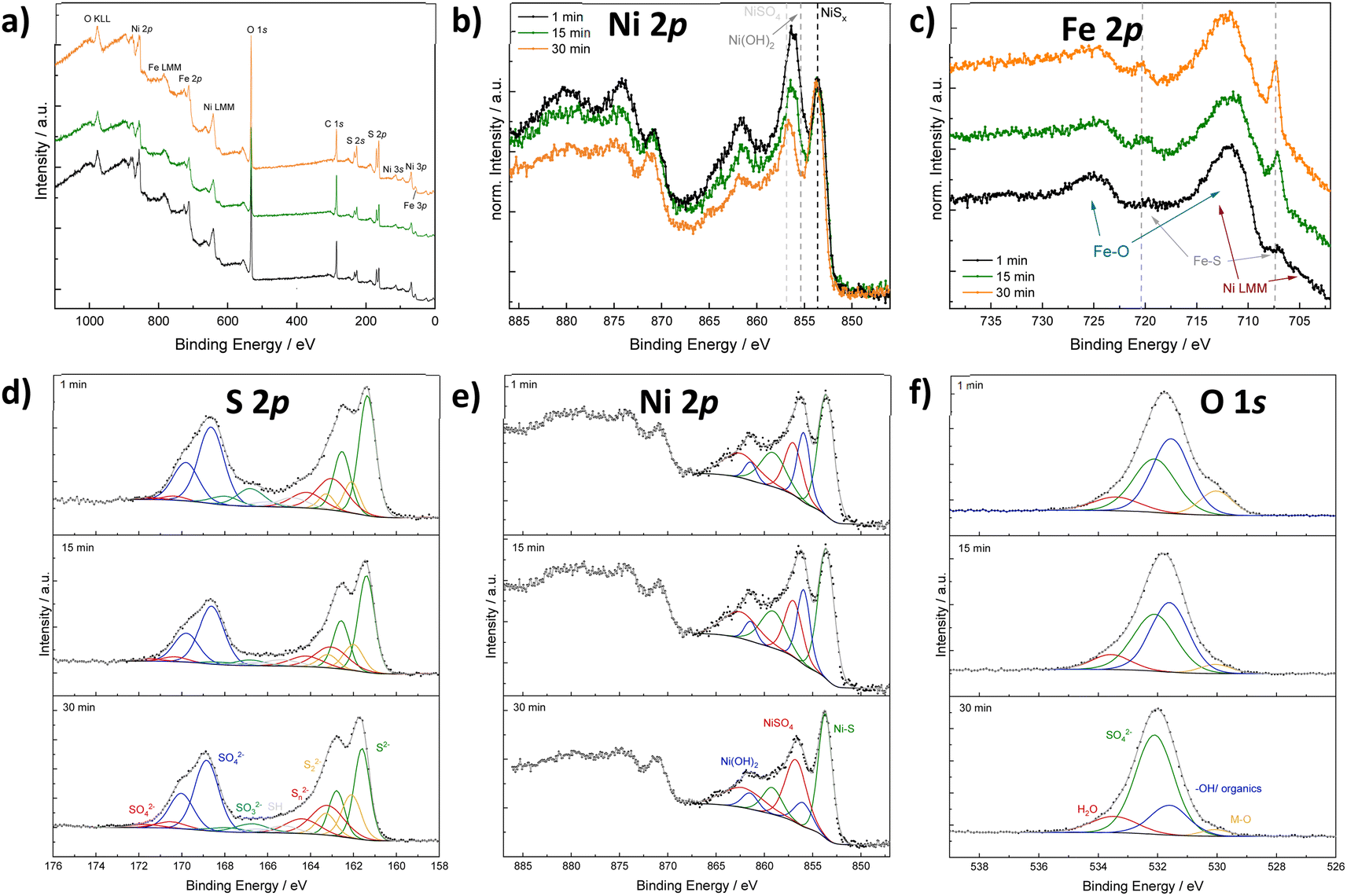 | ||
| Fig. 3 XP spectra for Ni2FeS4 obtained after different synthesis durations. Survey scan (a), Ni 2p spectra normalised to the Ni–S peak for qualitative comparison (b), normalised Fe 2p spectra (c), as well as fitted S 2p (d), Ni 2p (e) and O 1s spectra (f). | ||
The sulphur S 2p spectra mainly show the signals for S2− and SO42−, revealing a partial surface oxidation – something that is commonly recognised for sulphides that were handled in air, even if only shortly (Fig. 3).69 Moreover, disulphides and polysulphides are present in addition to at least one more species at slightly higher binding energies than those expected for polysulphides. These can probably be attributed to thiols together with sulphite species, that are especially pronounced after 1 min.74 The presence of SO32− species together with Ni2+ might be an indication that the oxidation of nickel is coupled to an oxidation of sulphur. Both sulphate and sulphide signal are shifted towards higher binding energies for Ni2FeS4 synthesised for 30 min (Fig. S3†), which might be attributed to a higher average oxidation states of the metal ions the sulphide ions are coordinating to. However, an effect of differences in the charge correction cannot be completely excluded. Fe 2p spectra are dominated by Ni L3M23M45 Auger peaks at 712 and 706 eV that prevent an accurate fit (Fig. 3). Still, an increase in the relative intensity of an iron sulphide species at approx. 707.5 eV and a decrease in the amount of oxidic species with time is observed in the normalised spectra, in good agreement with the O 1s spectra. Since the most intensive signal is the Ni Auger, spectra normalisation proceeds on this peak. Thus, a relative increase in the Fe–S peak can also be related to an actual decrease in the Ni LMM peak, which is in agreement with the loss of Ni with extended synthesis durations. An accurate fitting of the Ni 2p spectra is difficult, since at least three species (a sulphidic one, an oxidic one and a sulphate) are expected, all of which exhibit multiplet splitting.73,75 Additionally, the nickel sulphide peak might exhibit an asymmetric peak shape.76 Therefore, the spectra were approximated with one main peak for NiSx, Ni(OH)2 and NiSO4, respectively, and a satellite for each at 5.5 eV above the main signal. A decrease in the relative amount of oxidic nickel species, e.g. Ni(OH)2, with prolonged synthesis time is observed, in good agreement with the observations stated above. The binding energy for the main Ni sulphide peak is with 853.7 eV more in the range for what was observed for Ni–S in octahedral coordination,73 although half of the nickel species are expected to be in tetrahedral environment assuming a completely inverted structure.69,77,78 The binding energy for Fe(II) sulphide is with 707.4 eV in good agreement with what has been reported for an octahedral coordination.73,74 Hence, an inverted spinel structure is likely present, in agreement with literature.69
For practical application of a material for catalysis, stability is of crucial importance. Since sulphides are especially prone to oxidation under air, we performed TG-MS measurements to evaluate the stability of Ni2FeS4 at elevated temperatures in air (Fig. 4 and S4†). For all samples, an initial weight loss of 5 to 8% was observed, that corresponds to the desorption of water, but otherwise the sulphide is stable up to approx. 200 °C. A further temperature increase to 400 °C initially leads to an increase in the mass that can be attributed to surface oxidation and the formation of sulphates/oxides followed by a mass loss between 350 and 430 °C.69 The mass loss correlates with a peak for SO2 at ∼420 °C, together with an exothermic peak in the DSC, which can thus be attributed to a decomposition of Ni2FeS4 under formation of NiSO4. XRD patterns for Ni2FeS4 calcined for 2 h at 400 °C, i.e. after the mass loss, mainly show the reflections for NiSO4 (Fig. S5†). Additionally, CO2 is observed in low amounts, due to the decomposition of organic residues. A second major mass loss at temperatures above 600 °C is similar for all samples and corresponds to about 35%. It is due to the decomposition of NiSO4, which goes in hand with a second evolution of SO2 above 700 °C.79 Above 800 °C the mass is decreased to approx. 60%, in correlation to complete loss of sulphur and the formation of oxides (NiO and NiFe2O4) as observed in previous studies.56
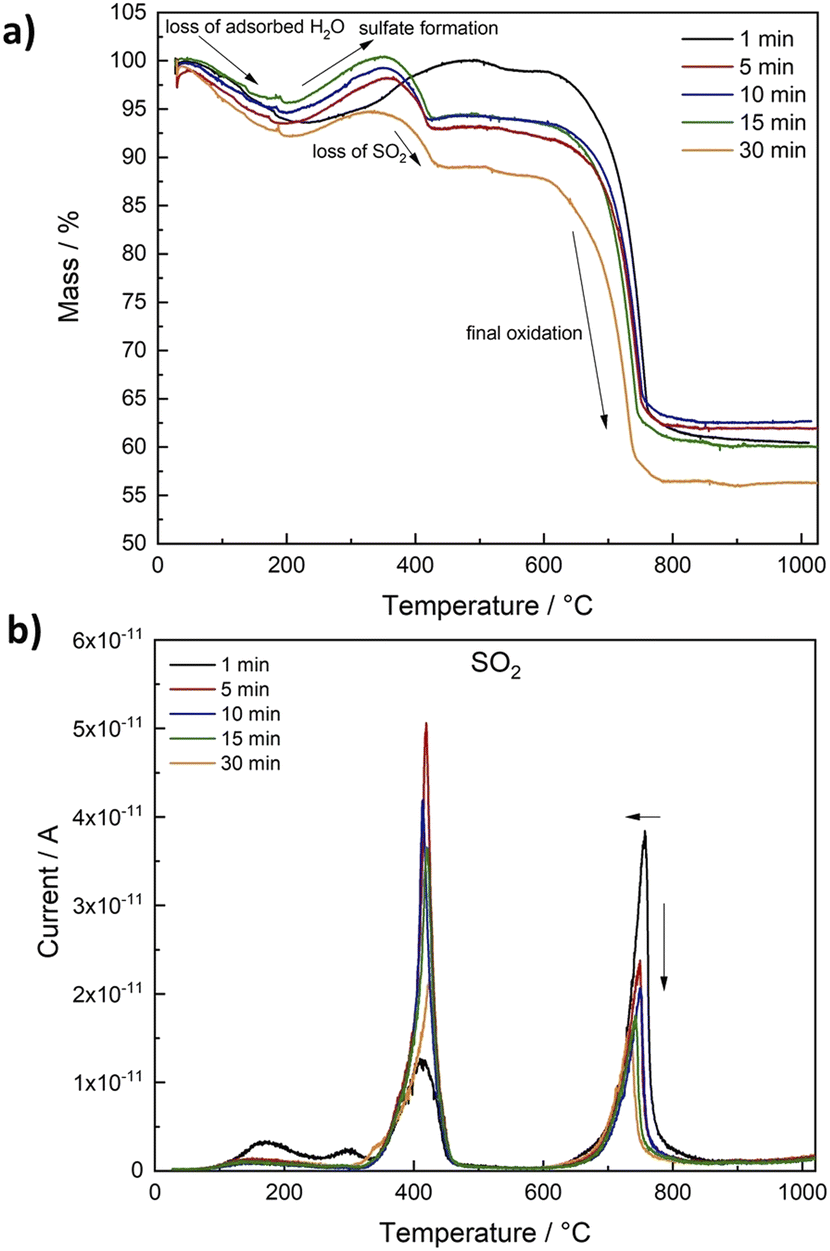 | ||
| Fig. 4 Relative mass loss during TG-MS measurements in air for Ni2FeS4 obtained after different synthesis durations (a) and SO2 evolution detected by MS depending on the temperature (b). | ||
The most striking difference between samples synthesised at different temperatures is the ratio of SO2 evolved at 400 °C (sulphate formation) and at 740 to 750 °C (decomposition of the metal sulphates). In the case of solely complete conversion of Ni2FeS4 to NiSO4 the first and the second peak for SO2 would be equal, which is roughly the case for the sample obtained after 30 min (Fig. S3†). Slightly more SO2 is evolved in the first step, possibly due to the presence of surface SO4-groups and incomplete conversion to NiSO4. The formation of NiSO4 at approx. 400 °C additionally requires the reduction of Ni3+. For samples obtained after synthesis times between 5 and 15 min, the peak in the DSC curves and the relation of both SO2 peaks is essentially the same. For the Ni2FeS4 synthesised for one minute, the first SO2 peak is significantly less intense than the second, and both are significantly smaller and larger, respectively, compared to all other samples. Notably, the initial oxidation of the sample obtained after 1 min also proceeds over a longer time range and the subsequent mass loss above 400 °C is lower. This might indicate that NiSO4 formation is favoured. Since the evolution of hot spots was not yet finished after 1 min, it can be assumed that the changes in the Ni and Fe valence were not yet complete/had not yet reached equilibrium conditions and more Ni2+ is still present in the structure after 1 min, partly bonded to oxygen as observed in the XP spectra. For Ni2FeS4 synthesised for 30 min, the initial SO2 evolution and the peak in the DSC are smaller again. The relative ratios of both peaks are listed in Table S2.† These changes in a sample that had experienced prolonged heating times might be due to changes in the relative ratio of nickel, possibly together with changes in the degree of inversion, as was observed in other microwave assisted synthesis routes of spinel oxides.71
Ni2FeS4 is absorbing light strongly over the entire visible light range and into the near infrared (NIR) region, as also apparent by its black colour (Fig. S6 and S7†). A band gap of around 2 eV is tentatively estimated based on the direct Tauc plot. By itself it is inactive in photocatalytic HER (see below), but the good light absorption characteristics together with high conductivity, as evidenced by a small charge transfer resistance in Nyquist plots (Fig. S8†), make it a suitable candidate for a co-catalyst in combination with an active photocatalyst. In this study we chose TiO2 (P25) as a model photocatalyst system, onto which we loaded Ni2FeS4 in different amounts between 0.1 and 10 wt% by grinding both constituents, followed by subsequent calcination for 2 h at 200 °C in argon, to improve the interparticular contact.
To evaluate the optimal amount of Ni2FeS4, we used a synthesis time of 30 min, i.e. the conditions employed in previous electrocatalytic experiments.56 XRD patterns (Cu anode) of the composites mainly show the reflections of TiO2 – only for higher loadings with Ni2FeS4 (5 and 10 wt%) the main reflection of the sulphide is visible (Fig. 5). Other bulk characterisation methods, like Raman (Fig. 5) or IR spectra (Fig. S7†) also mainly show the characteristic bands for TiO2 – which is metal–oxygen bonding vibrations at 920 cm−1 and additional bands for –OH vibrations of dangling bonds and adsorbed water – although the good light absorption properties become visible in an increased background noise in both techniques.80 Very weak bands for adsorbed organics, likely residues from the synthesis of Ni2FeS4, are also present. Small differences are observed in the Raman spectra of P25 decorated with Ni2FeS4, especially for higher loadings. Bands for Ni2FeS4 at 290 and 350 cm−1 are apparent even under relatively high laser intensity. When pure Ni2FeS4 was measured under the same conditions, a transformation to a pure inverse spinel, possibly NiFe2O4, was observed, likely due to the strong light absorption and thus sample heating under laser irradiation (Fig. S9†).81,82 Very weak bands were observed under reduced laser power. SEM images of a composite containing 5 wt% of Ni2FeS4 show homogeneous agglomeration of nanoparticles – mostly TiO2. EDX analysis over such an agglomerate mainly shows the expected signals for oxygen and titanium, evenly distributed over the entire agglomerate (Fig. 5). Signals for Ni and Fe appear in the same areas as those for TiO2 indicating homogeneous distribution of the sulphide. The changes during the annealing of ground Ni2FeS4 and P25 were also followed directly with TG-MS (Fig. S10†). A small signal for SO2 is observed during the initial heating. However, since only an m/z of 64 was monitored, this signal might also arise from adsorbed organic that was removed during heating. No SO2 signal is observed during the 2 h at 200 °C, proving the stability of the Ni2FeS4 during loading onto P25. Two peaks for SO2 are observed at 290 °C and at 433 °C, as a consequence of sulphate formation.
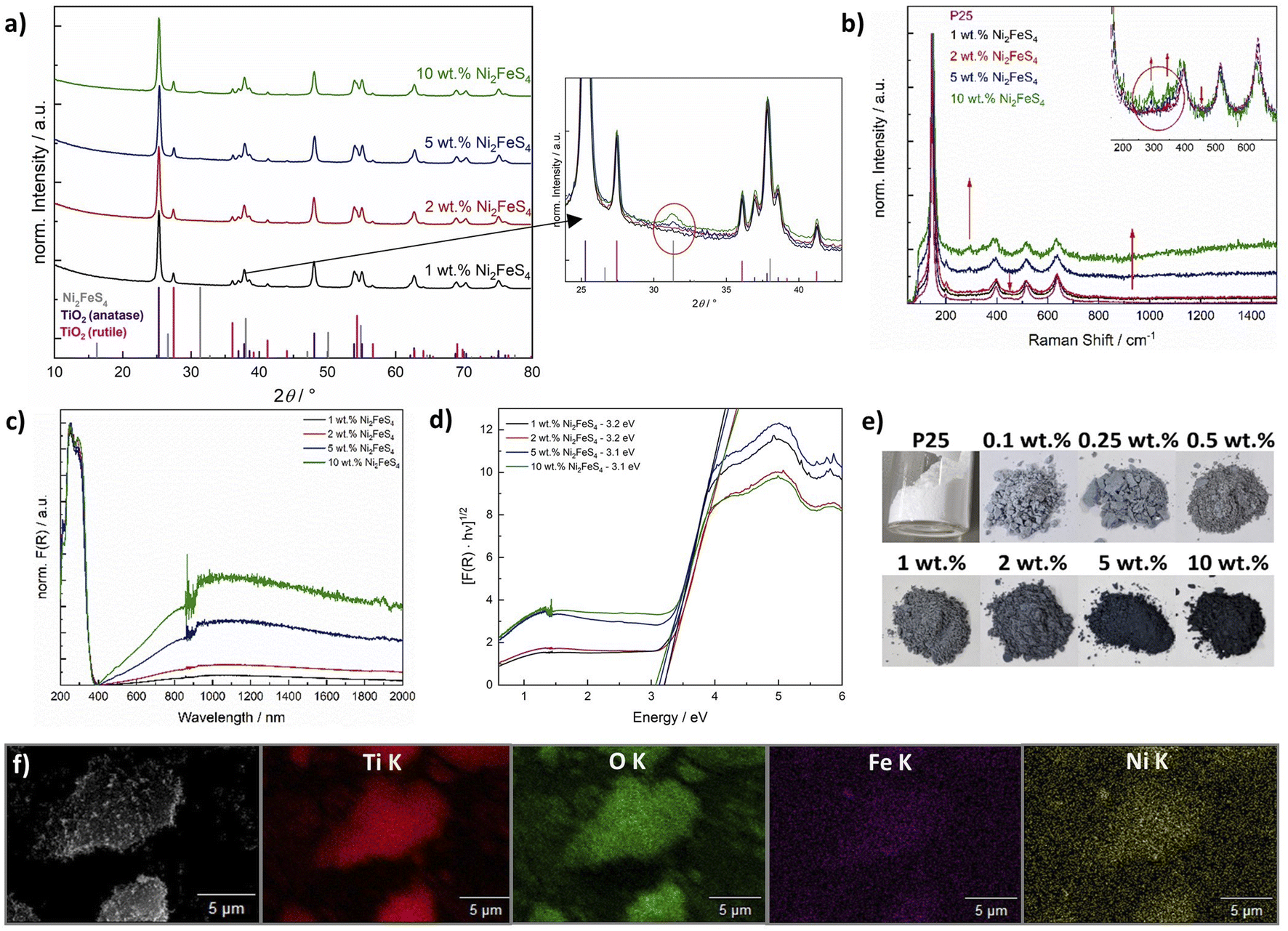 | ||
| Fig. 5 Cu-XRD patterns of P25 loaded with Ni2FeS4 with corresponding excerpt showing the main reflection of Ni2FeS4 (a), Raman spectra (b), UV/vis/NIR spectra (c and d) and photographs of P25 loaded with Ni2FeS4 (e), as well as EDX mapping of a P25 loaded with 5 wt% of Ni2FeS4 (f). (Due to all bulk methods being dominated by characteristics of TiO2, the characterisation after co-catalyst loadings of 0.1, 0.25 and 0.5 wt% is omitted here and shown in Fig. S11†). | ||
TiO2 loaded with Ni2FeS4 gains a greyish colouring, even at low loadings, which is also apparent in the UV/vis/NIR spectra (Fig. 5). For all composites a band gap of 3.1–3.2 eV is obtained from the indirect Tauc plots, in agreement with the expected band gap of 3.2 eV for anatase and that measured for P25 before and after Pt photo-deposition (Fig. S11†).83,84 The slightly higher value is due to the NIR absorption of Ni2FeS4 and especially problematic at higher co-catalyst loadings. The influence of absorption in the sulphide is well visible in an increased pseudo-absorption starting from 400 nm and reaching into the NIR region (Fig. 5d). As expected, the portion of absorbed visible and NIR light increases with an increasing content of Ni2FeS4.
The decorated TiO2 was then tested for photocatalytic hydrogen evolution under 1 sun simulated sunlight, using 10 vol% of methanol as hole scavenger (Fig. 6). While pure Ni2FeS4 is inactive under the employed conditions, calcined P25 (200 °C, 2 h) shows a hydrogen evolution rate of 3.4 μmol h−1 by itself. The activity of as received P25 is slightly lower, likely due to the desorption of water and organics during the annealing at 200 °C (Fig. S15†). The loading with Ni2FeS4 can boost the H2 production rate to 24 – 28 μmol h−1, largely independent of the loading over a wide range, demonstrating the effect of Ni2FeS4 as a co-catalyst as opposed to the function as a component in a heterojunction. Only at high loadings of 10 wt% of the sulphide, a diminished mass of active photocatalyst material and shadowing effects due to strong light absorption of Ni2FeS4 lead to a decrease in the observed activity. The amount of Ni2FeS4 can be decreased, without a loss of activity suggesting efficient charge transfer between Ni2FeS4 and TiO2. Even at low amounts of Ni2FeS4 of 0.5 wt%, which equals to 0.287 wt% of metals, an activity of 25 μmol h−1 is reached. Such a high activity enhancement at low loading makes Ni2FeS4 a very interesting noble metal free co-catalyst. The steady state activity for P25 with Ni2FeS4 is reached faster for lower co-catalyst loadings, possibly due to an initial activation phase. Compared to 0.5 wt% platinum on either as-obtained P25 or annealed P25, still more than 11% of the activity with Pt is reached (Fig. S15†) even at a lower metal loading. The relative activity compared to 0.1 wt% of Pt is with 16% even higher.
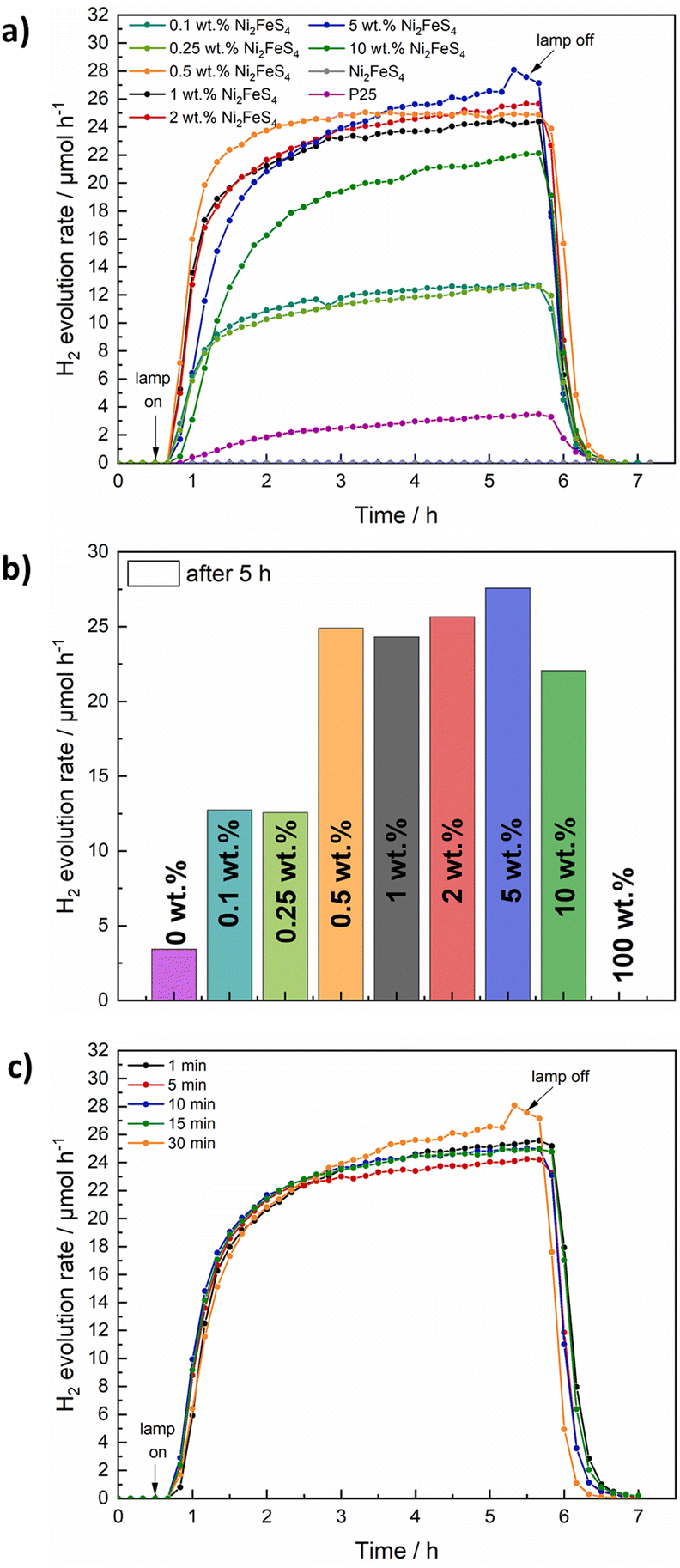 | ||
| Fig. 6 H2 evolution over P25 loaded with different amounts of Ni2FeS4 under 1 sun simulated sunlight (a and b). Additionally, the H2 evolution for 5 wt% Ni2FeS4@P25, with Ni2FeS4 obtained after different synthesis durations in the microwave is shown (c). The lamp was switched on after 30 min and turned off after 5 h of irradiation. | ||
In a next step the influence of the synthesis conditions of Ni2FeS4 on the performance as a co-catalyst on TiO2 was evaluated. A loading of 5 wt% was chosen, since the observed HER activity was highest (although similar to lower loadings) and the modification of TiO2 can be done more accurately for higher mass loadings/relative ratios of the sulphide. A similar relative loading is apparent in almost identical XRD patterns, Raman, DRIFT and UV/vis/NIR characteristics (Fig. S13†). Essentially the same activity is observed for all measured samples, independent of the synthesis time of Ni2FeS4, which is in very good agreement with the lack of differences observed in the characterisation, further confirming the complete reaction after only a couple of minutes (Fig. 6c). Interestingly, the differences in the amount of metal oxygen bonds observed by XPS and the loss of Ni upon extended time in the microwave did not appear to have an influence. Since the photocatalysis is performed in an aqueous environment, surface oxidation and formation of oxidic species, especially FeOOH and Ni(OH)2 that were observed for pentlandite likely occur anyway.73
To improve the sustainability, a calcination in an argon atmosphere is impedimental. Bulk Ni2FeS4 was therefore calcined for 2 h at 200 °C in air and the obtained XRD pattern only showed the expected reflections for the sulphide, without any additional by-phases (Fig. S14†). This is in good agreement with previous observations.56 Some surface oxidation likely occurs and has been observed for iron in Ni2FeS4,69 but since the M–O ratio did not influence the photocatalytic performance before, it might not have an impedimental effect here either. To validate the thermal stability even in a dilution with TiO2 and the influence of air calcination on the photocatalytic activity, 1 to 10 wt% of Ni2FeS4 (synthesised for 30 min) were loaded onto P25, followed by calcination in air. XRD measurements show a slightly lower, but still comparable intensity of the main reflection for Ni2FeS4 and no oxidation products thereof (Fig. S12†). A highly similar hydrogen evolution activity of around 25 μmol h−1 was observed for higher sulphide loadings, when compared to the calcination in argon (Fig. S15†). This is in good agreement with the observed bulk stability. When the concentration of Ni2FeS4 was decreased below 5 wt%, however, a decrease in the hydrogen evolution activity down to 14.5 μmol h−1 for 1 wt% of Ni2FeS4 was observed, indicating, that for higher dilutions of the sulphide in the oxide matrix, oxidation and thus partial decomposition might become problematic. A loss in the actual sulphide loading is also apparent in the UV/vis/NIR spectra of the air-calcined samples, which show a less intense absorption in the NIR region compared to the samples calcined in Ar (Fig. S14†) – thus suggesting partial oxidation. These experiments elucidate that a more expensive calcination in Ar can be avoided at the trade-off of higher required co-catalyst loadings.
To see if samples obtained after short reaction times are more prone to oxidation, the co-catalyst loading procedure in air was repeated for a 5 wt% loading with Ni2FeS4 synthesised for varied reaction times. All show a similar activity, with a synthesis time of only 1 min yielding the best result (Fig. S15†). At first glance this finding might seem counter-intuitive, but it is in good agreement with the slightly improved thermal stability for the 1 min sample observed in TG-MS-measurements (Fig. 4 and S4†).
The successful application of Ni2FeS4 as a co-catalyst on TiO2 requires a suitable band alignment, a large work function and a suitable conduction band potential for proton reduction. To get a rough idea about the respective band positions, they were calculated from the ionisation energies (Ei) and electron affinities (EEA) of the constituting atoms according to:85,86
| EVB = X − 4.44 eV + 0.5EBG |
and
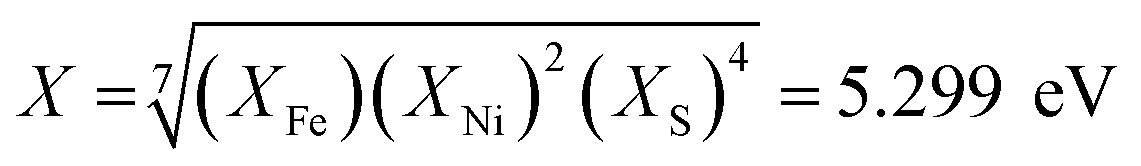
with an estimated band gap energy (EBG) of 2 eV, a valence band energy of 1.86 V and correspondingly a conduction band energy of −0.14 V vs. NHE is obtained. To estimate the Fermi level and thus the work function, Mott–Schottky analysis was performed, that yields a flat band potential of 0.49 V vs. RHE (Fig. 7). If a band diagram is drawn based on these values, and electron transfer in P25 to the conduction band of anatase is assumed83,87 excited electrons in TiO2 would be transferred to the lower energetic conduction band of Ni2FeS4 (Fig. 7). With the taken values, a small Schottky barrier would have to be overcome, however there exist multiple – partially contradicting – reports of work function and band positions in TiO2, depending on the conditions.84,87–89 The formed barrier would furthermore impede back-transfer and charge recombination. We decided on using the work function for TiO2 with surface adsorbed H2O/in water, since the flat band potential of Ni2FeS4 was likewise determined in aqueous electrolytes. In spite of uncertainties of exact band positions and of the direction of band bending the relative band positions of TiO2 and Ni2FeS4 should still be valid. If the conduction and valence band for TiO2 are calculated following the same approach as stated above, potentials of EVB = 2.97 eV and ECB = −0.23 eV are obtained. The deviations from experimentally observed values are due to the neglecting of crystal phase, interatomic interactions or environmental conditions, but the relative band positions to Ni2FeS4 and thus direction of charge carrier diffusion remain the same.
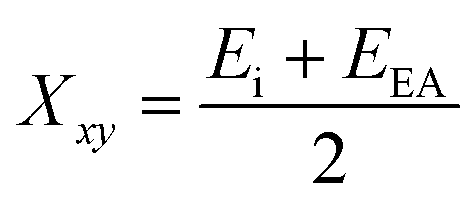
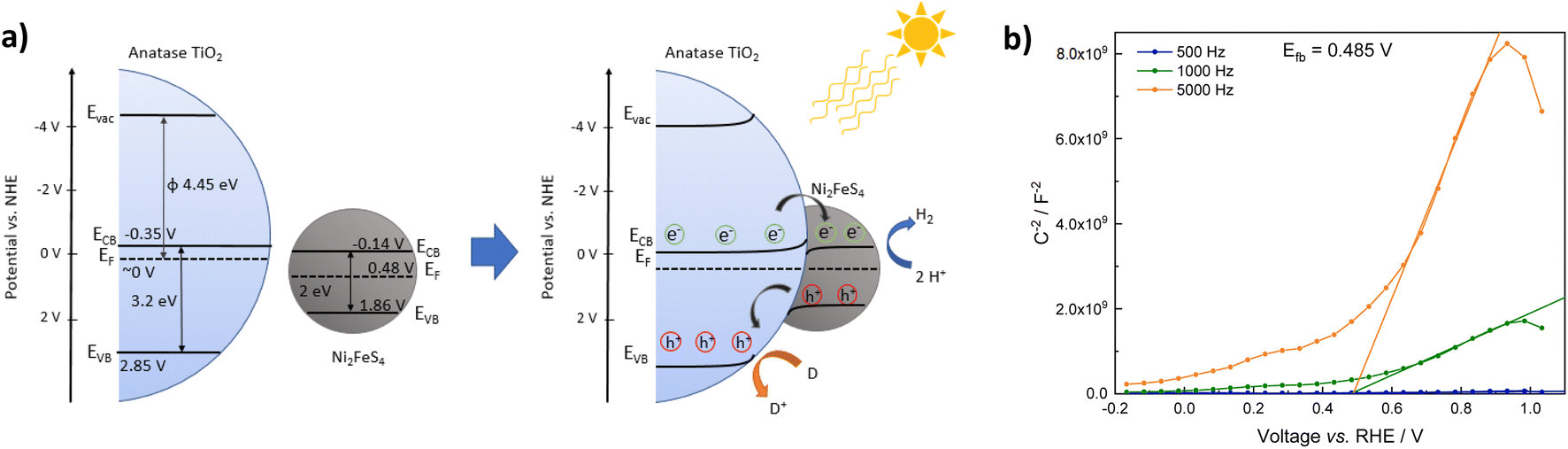 | ||
| Fig. 7 Band positions of TiO2 and Ni2FeS4 before and after contact (a) and Mott–Schottky plots for Ni2FeS4 in 0.5 M Na2SO4 (values are already converted to pH 0) (b). | ||
To verify the role of P25 as the photocatalyst and that of Ni2FeS4 as a co-catalyst, the photocatalytic experiment was repeated for a loading of 5 wt%, using 420 nm cut-off filter. No activity was observed, although light absorption and excitation in Ni2FeS4 should still occur. Additionally, we wanted to make sure that the observed activity increase is not in parts due to the use of methanol as a scavenger and thus the possibility of hydrogen generated via methanol oxidation in the dark, as observed for platinum.90 Therefore, the photocatalytic experiment was repeated for P25 and P25 decorated with 5 wt% of Ni2FeS4 using ethanol as a hole scavenger. The observed activity is about half that obtained in methanol, with a slower activation, due to the kinetically more impeded oxidation of ethanol and different oxidation mechanisms and products.91,92 The relative activity increase with and without co-catalyst loading is however essentially the same, proving that Ni2FeS4 is indeed acting as a co-catalyst (Fig. 8). Another question that arises when sulphides are used, is the stability under operating conditions. No decline in the hydrogen evolution rate was observed even when the measurement time was extended to 20 h (Fig. 8), demonstrating the extraordinary stable activity. XRD patterns of P25 loaded with 5 and 10 wt% of Ni2FeS4 reveal a slight decrease in the relative intensity of the sulphide reflection after photocatalysis (Fig. S16†). Some amorphisation is also observed for the photocatalytic experiment with pure Ni2FeS4, although no crystalline oxidation products were formed, highlighting the generally extraordinary photostability of the sulphide. At the same time the light absorption in the visible and NIR range is decreased (Fig. S17†), as is the additional band at 290 cm−1 in the Raman spectra. All these observations indicate, that Ni2FeS4 is partially oxidised/dissolved at the surface and thus perhaps not exclusively the active species under operating conditions, similar to what is known for sulphides, especially iron sulphides, in electrocatalysis.16,22 This finding is further underlined by the observation of SO32− and SO42− species with ion chromatography (Fig. S18†). Interestingly, SO42− was also found in the solution after the irradiation of pure Ni2FeS4 with 1 sun simulated sunlight, even though no hydrogen evolution was observed. Some dissolution of surface metal sulphate species is always expected in an aqueous environment, though.93 Thus, either the sulphide itself might undergo oxidation during the photocatalysis, or the SO42− stems from the sulphate species observed in XPS measurements. In any case, the evolving active species exhibits extraordinary stability and activity, as observed by the longtime photocatalytic testing. Ni2FeS4 also undergoes amorphisation to some extent in the absence of light. When it is dispersed for 24 hours in H2O or 10% aqueous methanol solutions an amorphisation is observed, although notably no reflections of hydroxides or oxyhydroxides are present (Fig. S5†). This would again support a partial dissolution of surface species. As a further verification of stability, we performed EDX mapping, XPS and Ag-XRD on P25 loaded with 5 wt% of Ni2FeS4 after the photocatalytic experiment for 20 h (Fig. S19†). The EDX mapping shows that Ni and Fe are still distributed homogeneously over the TiO2 surface. The Ag-XRD results elucidate partial amorphisation as observed for the HER experiments run for 5 h (Fig. S16†). The XPS survey spectra of 5 wt% of Ni2FeS4@P25 mainly show signals for Ti and O, even before the HER. However, after the 20 h experiment, Ni, Fe and S are still present. Sulphur mainly occurs in the form of sulphates. The intensity of Ni and Fe does not allow for a meaningful fit. Therefore, we additionally performed XPS analysis of Ni2FeS4 stirred for 48 h in water. Although the XRD results showed an amorphisation already after 24 h in water (Fig. S5†), the spectra are highly comparable to the as-synthesised material. The Ni 2p spectra still show signals for nickel sulphide, sulphate and hydroxide and the S 2p spectra show contributions from sulphide and sulphate species.
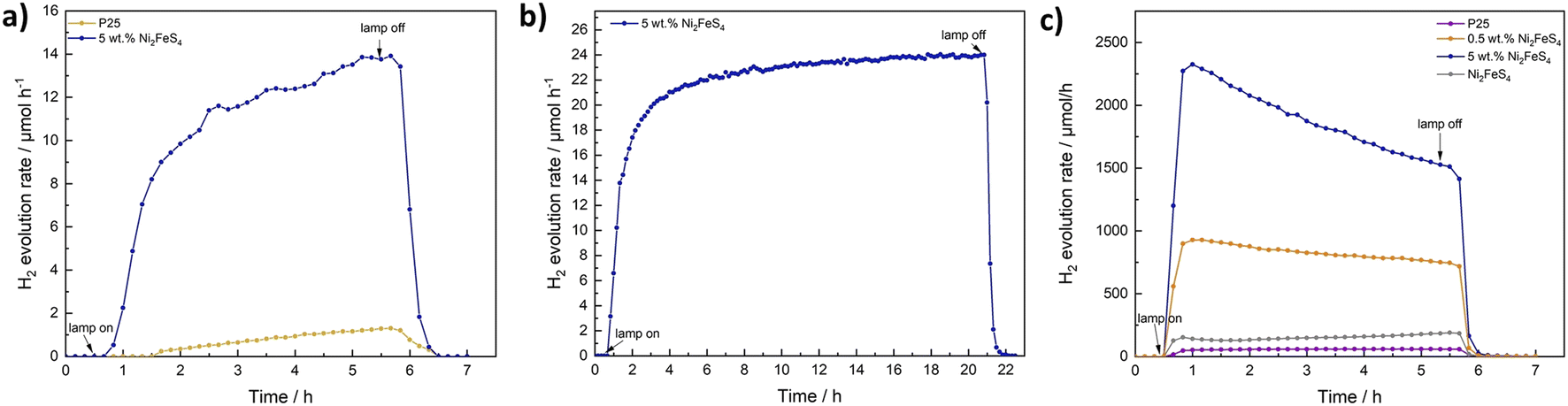 | ||
| Fig. 8 Photocatalytic HER over P25 and 5 wt% Ni2FeS4@P25 using ethanol as a scavenger (a), for longtime irradiation (1 sun) of 5 wt% Ni2FeS4@P25 in aqueous methanol solutions (b), as well as HER rates under UV irradiation (c). | ||
As a final test we wanted to examine the co-catalyst effect of the sulphide also under UV irradiation, i.e. the kind of irradiation still commonly employed for large band gap semiconductors such as TiO2, since these harsh conditions might put more stress on the sulphide co-catalyst. We therefore tested P25 and P25 loaded with 5 wt% of Ni2FeS4 under irradiation from a 500 W Hg lamp (Fig. 8). The factor of the activity enhancement is highly similar, but the performance is decreasing over the course of the experiment. Initially, the HER rate is boosted by a factor of 48 to over 2300 μmol h−1 for 150 mg of photocatalyst, while towards the end, the activity enhancement goes down to 1520 μmol h−1, which is approx. 26 times the HER rate over pure P25.
Interestingly, pure Ni2FeS4 is also active for the HER under UV light (Fig. 8). The curve for the HER rate shows a small hump in the first half hour after turning on the lamp. After that, the activity is continuously increasing. This might be explained by an in situ activation, that has been suggested for other transition metal chalcogenides and involves the formation of metal (in this case probably Ni) nanoparticles, that act as a co-catalyst.94 Such a process under high UV light irradiation might be the reason for the decrease in the activity as observed for Ni2FeS4 on P25, since it would change the electronic and chemical structure of the sulphide co-catalyst. Additionally, the UV light activity of Ni2FeS4 itself might influence the entire charge separation mechanism, since electrons are also excited in the sulphide. This could result in a heterojunction system and explain the larger factor of activity enhancement compared to the comparison under simulated sunlight. This assumption is supported by the far lower activity now observed for a loading of 0.5 wt% of Ni2FeS4, compared to the 5 wt% loading. Here, the activity is increased by a factor of only approx. 13, which is closer to the observations under simulated sunlight and can be attributed to the lower ratio of Ni2FeS4 and thus the lower number of photoexcited charges in the sulphide, preventing the formation of an efficient heterojunction. After the photocatalysis under intense UV light, some amorphisation is again observed in the XRD patterns, albeit without additional indications of by-phases. Still only the reflections for Ni2FeS4 are present, underlining the good stability against photocorrosion even under these harsh irradiation conditions (Fig. S16†).
The role of Ni2FeS4 as a co-catalyst was then finally underlined by testing it on Al-doped SrTiO3 (3%) for HER under 1 sun illumination (Fig. S20†). By itself, SrTiO3 is basically inactive for the HER under these conditions. The addition of 5 wt% of Ni2FeS4 could significantly boost the activity, although the observed H2 evolution rates were still far lower than those obtained for P25 in agreement with the generally lower HER activity reported in literature.95
Conclusions
Ni2FeS4 containing only earth-abundant elements can be synthesised via an energy efficient, high throughput microwave-assisted approach at 200 °C, requiring only 1 min of reaction time. Bulk characterisation methods support a complete reaction after such a short time. Ni2FeS4 was successfully used as a co-catalyst to improve the sunlight activity of large band gap semiconductors, such as TiO2 and SrTiO3, achieving an increase in the hydrogen evolution rate by a factor of 8 under 1 sun and by a factor of up to 48 under UV light. Very low co-catalyst loadings of 0.5 wt% can be used to achieve this outstanding HER rate enhancement. Additionally, an extraordinary stability was observed with no decrease in the hydrogen evolution being observed during 20 hours of photocatalysis, and no oxidation products of the sulphide co-catalyst were found afterwards. These findings underline the promise of Ni2FeS4 as a low-cost, earth abundant co-catalyst for photocatalysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Mirco Ade for SEM/EDX analysis and Jonas Jungmann for TEM analysis. Additionally, we are grateful to Lena Geiling for TGA-MS measurements, as well as Dr Morten Weiss and Lion Schumacher for XPS measurements (all University of Bayreuth). We are grateful to Dr Jana Timm and Jonas Jungmann for BET measurements and to Işil Merve Songür Sozak for assistance in the synthesis of Al-doped SrTiO3. We further thank the Bavarian Polymer institute (BPI) for usage of the XPS and SEM devices (KeyLabs Device Engineering and Electron and Optical Microscopy). J. Z. and R. M. gratefully acknowledge funding in the graduate school of the Bavarian Center for Battery Technology (BayBatt), University of Bayreuth, and by the Bavarian State Ministry of Science, Research and Arts within the scope of Solar Technologies Go Hybrid.References
- A. Pareek, R. Dom, J. Gupta, J. Chandran, V. Adepu and P. H. Borse, Mater. Sci. Energy Technol., 2020, 3, 319–327 CAS.
- L. Lin, T. Hisatomi, S. Chen, T. Takata and K. Domen, Trends Chem., 2020, 2, 813–824 CrossRef CAS.
- K. Takanabe, ACS Catal., 2017, 7, 8006–8022 CrossRef CAS.
- S. A. Grigoriev, V. N. Fateev, D. G. Bessarabov and P. Millet, Int. J. Hydrogen Energy, 2020, 45, 26036–26058 CrossRef CAS.
- M. Yu, E. Budiyanto and H. Tüysüz, Angew. Chem., Int. Ed., 2022, 61, e202103824 CAS.
- J. Yang, D. Wang, H. Han and C. A. N. Li, Acc. Chem. Res., 2013, 46, 1900–1909 CrossRef CAS PubMed.
- H. Chen, Z. Sun, S. Ye, D. Lu and P. Du, J. Mater. Chem. A, 2015, 3, 15729–15737 RSC.
- Y. Peng, L. Shang, Y. Cao, G. I. N. Waterhouse, C. Zhou, T. Bian, L. Z. Wu, C. H. Tung and T. Zhang, Chem. Commun., 2015, 51, 12556–12559 RSC.
- D. Li, J. Shi and C. Li, Small, 2018, 14, 1–22 Search PubMed.
- S. Li, E. Li, X. An, X. Hao, Z. Jiang and G. Guan, Nanoscale, 2021, 13, 12788–12817 RSC.
- M. S. Faber and S. Jin, Energy Environ. Sci., 2014, 7, 3519–3542 RSC.
- A. Li, Y. Sun, T. Yao and H. Han, Chem.–Eur. J., 2018, 24, 18334–18355 CrossRef CAS PubMed.
- Y. Yang, C. Zhou, W. Wang, W. Xiong, G. Zeng, D. Huang, C. Zhang, B. Song, W. Xue, X. Li, Z. Wang, D. He, H. Luo and Z. Ouyang, Chem. Eng. J., 2021, 405, 126547 CrossRef CAS.
- M. Isegawa, T. Matsumoto and S. Ogo, Dalton Trans., 2022, 51, 312–323 RSC.
- T. R. Simmons, G. Berggren, M. Bacchi, M. Fontecave and V. Artero, Coord. Chem. Rev., 2014, 270–271, 127–150 CrossRef CAS.
- S. Anantharaj, H. Sugime and S. Noda, Chem. Eng. J., 2021, 408, 127275 CrossRef CAS.
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- M. S. Faber, M. A. Lukowski, Q. Ding, N. S. Kaiser and S. Jin, J. Phys. Chem. C, 2014, 118, 21347–21356 CrossRef CAS PubMed.
- D. Kong, J. J. Cha, H. Wang, H. R. Lee and Y. Cui, Energy Environ. Sci., 2013, 6, 3553–3558 RSC.
- J. Yu, G. Cheng and W. Luo, J. Mater. Chem. A, 2017, 5, 15838–15844 RSC.
- H. Yu, Y. Xie, L. Deng, H. Huang, J. Song, D. Yu, L. Li and S. Peng, Inorg. Chem. Front., 2022, 9, 146–154 RSC.
- Y. Xie, H. Yu, L. Deng, R. S. Amin, D. Yu, A. E. Fetohi, M. Y. Maximov, L. Li, K. M. El-Khatib and S. Peng, Inorg. Chem. Front., 2022, 9, 662–669 RSC.
- L. Li, D. Yu, P. Li, H. Huang, D. Xie, C. C. Lin, F. Hu, H. Y. Chen and S. Peng, Energy Environ. Sci., 2021, 14, 6419–6427 RSC.
- S. Hong, D. P. Kumar, E. H. Kim, H. Park, M. Gopannagari, D. A. Reddy and T. K. Kim, J. Mater. Chem. A, 2017, 5, 20851–20859 RSC.
- K. Q. Lu, M. Y. Qi, Z. R. Tang and Y. J. Xu, Langmuir, 2019, 35, 11056–11065 CrossRef CAS PubMed.
- X. Shi, M. Fujitsuka, S. Kim and T. Majima, Small, 2018, 14, 1–9 Search PubMed.
- Z. Zhang, G. Liu, X. Cui, Y. Gong, D. Yi, Q. Zhang, C. Zhu, F. Saleem, B. Chen, Z. Lai, Q. Yun, H. Cheng, Z. Huang, Y. Peng, Z. Fan, B. Li, W. Dai, W. Chen, Y. Du, L. Ma, C. J. Sun, I. Hwang, S. Chen, L. Song, F. Ding, L. Gu, Y. Zhu and H. Zhang, Sci. Adv., 2021, 7, 1–10 Search PubMed.
- H. Xu, J. Yi, X. She, Q. Liu, L. Song, S. Chen, Y. Yang, Y. Song, R. Vajtai, J. Lou, H. Li, S. Yuan, J. Wu and P. M. Ajayan, Appl. Catal., B, 2018, 220, 379–385 CrossRef CAS.
- Z. Liang, Y. Xue, X. Wang, Y. Zhou, X. Zhang, H. Cui, G. Cheng and J. Tian, Chem. Eng. J., 2021, 421, 130016 CrossRef CAS.
- D. Zhao, B. Sun, X. Li, L. Qin, S. Kang and D. Wang, RSC Adv., 2016, 6, 33120–33125 RSC.
- S. Cao, Y. Chen, C. J. Wang, X. J. Lv and W. F. Fu, Chem. Commun., 2015, 51, 8708–8711 RSC.
- T. F. Jaramillo, K. P. Jørgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100–102 CrossRef CAS.
- Z. Wang, J. Fan, B. Cheng, J. Yu and J. Xu, Mater. Today Phys., 2020, 15, 100279 CrossRef.
- H. Wang, W. Chen, J. Zhang, C. Huang and L. Mao, Int. J. Hydrogen Energy, 2015, 40, 340–345 CrossRef CAS.
- T. Simon, N. Bouchonville, M. J. Berr, A. Vaneski, A. Adrović, D. Volbers, R. Wyrwich, M. Döblinger, A. S. Susha, A. L. Rogach, F. Jäckel, J. K. Stolarczyk and J. Feldmann, Nat. Mater., 2014, 13, 1013–1018 CrossRef CAS PubMed.
- Q. Zhao, J. Sun, S. Li, C. Huang, W. Yao, W. Chen, T. Zeng, Q. Wu and Q. Xu, ACS Catal., 2018, 8, 11863–11874 CrossRef CAS.
- J. Ran, J. Zhang, J. Yu and S. Z. Qiao, ChemSusChem, 2014, 7, 3426–3434 CrossRef CAS PubMed.
- A. Pareek, P. Paik and P. H. Borse, Dalton Trans., 2016, 45, 11120–11128 RSC.
- H. Qi, J. Wolfe, D. Fichou and Z. Chen, Nat. Publ. Gr., 2016, 30882 CAS.
- L. Li, J. Yi, X. Zhu, M. Zhou, S. Zhang, X. She, Z. Chen, H. M. Li and H. Xu, ACS Sustainable Chem. Eng., 2020, 8, 884–892 CrossRef CAS.
- S. Cao, Y. Chen, C. Wang, P. He and W. Fu, Chem. Commun., 2014, 50, 10427 RSC.
- Z. Sun, H. Chen, L. Zhang, D. Lu and P. Du, J. Mater. Chem. A, 2016, 4, 13289–13295 RSC.
- Y. Zhong, J. Yuan, J. Wen, X. Li, Y. Xu, W. Liu, S. Zhang and Y. Fang, Dalton Trans., 2015, 44, 18260–18269 RSC.
- B. Luo, R. Song, Z. Zeng and D. Jing, Appl. Surf. Sci., 2020, 511, 145646 CrossRef CAS.
- J. Sakizadeh, J. P. Cline, E. Wolfe, R. Thorpe, M. A. Snyder, C. J. Kiely and S. McIntosh, Green Chem., 2023, 25, 566–578 RSC.
- Y. Peng, Y. Zheng, Y. Yang, R. Jiang, G. Wang, Y. Zhang, E. Zhang, L. Zhao and C. Duan, J. Colloid Interface Sci., 2018, 514, 634–641 CrossRef.
- H. Zhao, H. Zhang, G. Cui, Y. Dong, G. Wang, P. Jiang, X. Wu and N. Zhao, Appl. Catal., B, 2018, 225, 284–290 CrossRef CAS.
- P. D. Tran, L. Xi, S. K. Batabyal, L. H. Wong, J. Barber and J. S. Chye Loo, Phys. Chem. Chem. Phys., 2012, 14, 11596–11599 RSC.
- M. Xiao, L. Zhang, B. Luo, M. Lyu, Z. Wang, H. Huang, S. Wang, A. Du and L. Wang, Angew. Chem., 2020, 132, 7297–7301 CrossRef.
- B. Tudu, N. Nalajala, K. Prabhakar Reddy, P. Saikia and C. S. Gopinath, ACS Sustainable Chem. Eng., 2021, 9, 13915–13925 CrossRef CAS.
- Q. Wang, G. Yun, Y. Bai, N. An, Y. Chen, R. Wang, Z. Lei and W. Shangguan, Int. J. Hydrogen Energy, 2014, 39, 13421–13428 CrossRef CAS.
- L. Zhang, B. Tian, F. Chen and J. Zhang, Int. J. Hydrogen Energy, 2012, 37, 17060–17067 CrossRef CAS.
- T. R. Kuo, H. J. Liao, Y. T. Chen, C. Y. Wei, C. C. Chang, Y. C. Chen, Y. H. Chang, J. C. Lin, Y. C. Lee, C. Y. Wen, S. S. Li, K. H. Lin and D. Y. Wang, Green Chem., 2018, 20, 1640–1647 RSC.
- W. Zhong, D. Gao, H. Yu, J. Fan and J. Yu, Chem. Eng. J., 2021, 419, 129652 CrossRef CAS.
- J. Jiang, Y. J. Zhang, X. J. Zhu, S. Lu, L. L. Long and J. J. Chen, Nano Energy, 2021, 81, 105619 CrossRef CAS.
- C. Simon, J. Zander, T. Kottakkat, M. Weiss, J. Timm, C. Roth and R. Marschall, ACS Appl. Energy Mater., 2021, 4, 8702–8708 CrossRef CAS.
- L. Wu, X. Shen, Z. Ji, J. Yuan, S. Yang, G. Zhu, L. Chen, L. Kong and H. Zhou, Adv. Funct. Mater., 2022, 2208170 Search PubMed.
- M. Zhou, Q. Weng, X. Zhang, X. Wang, Y. Xue, X. Zeng, Y. Bando and D. Golberg, J. Mater. Chem. A, 2017, 5, 4335–4342 RSC.
- B. Konkena, K. J. Puring, I. Sinev, S. Piontek, O. Khavryuchenko, J. P. Dürholt, R. Schmid, H. Tüysüz, M. Muhler, W. Schuhmann and U. P. Apfel, Nat. Commun., 2016, 7, 12269 CrossRef CAS PubMed.
- M. B. Z. Hegazy, K. Harrath, D. Tetzlaff, M. Smialkowski, D. Siegmund, J. Li, R. Cao and U.-P. Apfel, iScience, 2022, 25, 105148 CrossRef CAS.
- M. Smialkowski, D. Siegmund, K. Pellumbi, L. Hensgen, H. Antoni, M. Muhler and U. P. Apfel, Chem. Commun., 2019, 55, 8792–8795 RSC.
- S. L. J. Thomae, N. Prinz, T. Hartmann, M. Teck, S. Correll and M. Zobel, Rev. Sci. Instrum., 2019, 90, 043905 CrossRef PubMed.
- J. I. Langford and A. J. C. Wilson, J. Appl. Crystallogr., 1978, 11, 102–113 CrossRef CAS.
- P. Kubelka and F. Munk, Z. Tech. Phys., 1931, 265, 593–601 Search PubMed.
- J. Tauc, Mater. Res. Bull., 1968, 3, 37–46 CrossRef CAS.
- N. Fairley, V. Fernandez, M. Richard-Plouet, C. Guillot-Deudon, J. Walton, E. Smith, D. Flahaut, M. Greiner, M. Biesinger, S. Tougaard, D. Morgan and J. Baltrusaitis, Appl. Surf. Sci. Adv., 2021, 5, 100112 CrossRef.
- I. Bilecka, I. Djerdj and M. Niederberger, Chem. Commun., 2008, 886–888 RSC.
- R. Deshmukh and M. Niederberger, Chem.–Eur. J., 2017, 23, 8542–8570 CrossRef CAS PubMed.
- C. E. Mitchell, D. Santos-Carballal, A. M. Beale, W. Jones, D. J. Morgan, M. Sankar and N. H. De Leeuw, Faraday Discuss., 2021, 230, 30–51 RSC.
- M. Niederberger and G. Garnweitner, Chem.–Eur. J., 2006, 12, 7282–7302 CrossRef CAS PubMed.
- J. Zander, M. Weiss and R. Marschall, Adv. Energy Sustainability Res., 2023, 4, 2200184 CrossRef CAS.
- D. Santos-Carballal, A. Roldan, R. Grau-Crespo and N. H. De Leeuw, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 195106 CrossRef.
- D. L. Legrand, G. M. Bancroft and H. W. Nesbitt, Am. Mineral., 2005, 90, 1042–1054 CrossRef CAS.
- A. R. Pratt, I. J. Muir and H. W. Nesbitt, Geochim. Cosmochim. Acta, 1994, 58, 827–841 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- H. W. Nesbitt, D. Legrand and G. M. Bancroft, Phys. Chem. Miner., 2000, 27, 357–366 CrossRef CAS.
- C. Tenailleau, Am. Mineral., 2006, 91, 1442–1447 CrossRef CAS.
- M. G. Townsend, J. R. Gosselin, J. L. Horwood, L. G. Ripley and R. J. Tremblay, Phys. Status Solidi, 1977, 40, K25–K29 CrossRef CAS.
- A. G. Ostroff and R. T. Sanderson, J. Inorg. Nucl. Chem., 1959, 9, 45–50 CrossRef.
- G. Jeantelot, S. Ould-Chikh, J. Sofack-Kreutzer, E. Abou-Hamad, D. H. Anjum, S. Lopatin, M. Harb, L. Cavallo and J. M. Basset, Phys. Chem. Chem. Phys., 2018, 20, 14362–14373 RSC.
- K. R. Sanchez-Lievanos, J. L. Stair and K. E. Knowles, Inorg. Chem., 2021, 60, 4291–4305 CrossRef CAS PubMed.
- C. Simon, M. B. Zakaria, H. Kurz, D. Tetzlaff, A. Blösser, M. Weiss, J. Timm, B. Weber, U. P. Apfel and R. Marschall, Chem.–Eur. J., 2021, 27, 16990–17001 CrossRef CAS.
- X. Ruan, X. Cui, Y. Cui, X. Fan, Z. Li, T. Xie, K. Ba, G. Jia, H. Zhang, L. Zhang, W. Zhang, X. Zhao, J. Leng, S. Jin, D. J. Singh and W. Zheng, Adv. Energy Mater., 2022, 12, 2200298 CrossRef CAS.
- D. O. Scanlon, C. W. Dunnill, J. Buckeridge, S. A. Shevlin, A. J. Logsdail, S. M. Woodley, C. R. A. Catlow, M. J. Powell, R. G. Palgrave, I. P. Parkin, G. W. Watson, T. W. Keal, P. Sherwood, A. Walsh and A. A. Sokol, Nat. Mater., 2013, 12, 798–801 CrossRef CAS PubMed.
- R. G. Pearson, Inorg. Chem., 1988, 27, 734–740 CrossRef CAS.
- M. A. Butler, J. Electrochem. Soc., 1978, 125, 228 CrossRef CAS.
- P. Deák, J. Kullgren, B. Aradi, T. Frauenheim and L. Kavan, Electrochim. Acta, 2016, 199, 27–34 CrossRef.
- S. Kashiwaya, J. Morasch, V. Streibel, T. Toupance, W. Jaegermann and A. Klein, Surfaces, 2018, 1, 73–89 CrossRef.
- V. Mansfeldova, M. Zlamalova, H. Tarabkova, P. Janda, M. Vorokhta, L. Piliai and L. Kavan, J. Phys. Chem. C, 2021, 125, 1902–1912 CrossRef CAS.
- E. Antolini, Appl. Catal., B, 2018, 237, 491–503 CrossRef CAS.
- K. Sathiyan, R. Bar-Ziv, V. Marks, D. Meyerstein and T. Zidki, Chem.–Eur. J., 2021, 27, 15936–15943 CrossRef CAS.
- C. R. López, E. P. Melián, J. A. Ortega Méndez, D. E. Santiago, J. M. Doña Rodríguez and O. González Díaz, J. Photochem. Photobiol., A, 2015, 312, 45–54 CrossRef.
- S. Richardson and D. J. Vaughan, Mineral. Mag., 1989, 53, 213–222 CrossRef CAS.
- M. Pilarski, R. Marschall, S. Gross and M. Wark, Appl. Catal., B, 2018, 227, 349–355 CrossRef CAS.
- T. Alammar, V. Smetana, H. Pei, I. Hamm, M. Wark and A. V. Mudring, Adv. Sustainable Syst., 2021, 5, 2000180 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta02439c |
| This journal is © The Royal Society of Chemistry 2023 |