
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced nickel catalysts for producing electrolytic hydrogen
Rosaria
Ciriminna
 * and
Mario
Pagliaro
*
* and
Mario
Pagliaro
*
Istituto per lo Studio dei Materiali Nanostrutturati, CNR, via U. La Malfa 153, 90146 Palermo, Italy. E-mail: rosaria.ciriminna@cnr.it; mario.pagliaro@cnr.it
First published on 11th July 2023
Abstract
Low-cost and durable nickel-based catalysts are conventionally used in alkaline water electrolysers. Lowering the cost of electrolytic hydrogen produced via alkaline water electrolysis, particularly when using electricity sourced from intermittent renewable energy sources, is a sustainable development goal of global environmental and societal relevance. This, in practice, requires lowering the electrolytic cell overpotential while retaining high electrocatalyst durability typical of Ni-based catalytic materials. The most promising routes, we argue in this study, are the nanostructured and single-atom catalysts coupled to 3D printing for the deposition of the electrocatalytic layer on electrodes of large active area and optimal shape.
Sustainability spotlightAlong with the Li-ion battery, solar hydrogen, namely hydrogen produced by splitting water using renewable solar energy in any of its main three forms (sunlight, wind, and water fall), is the main energy storage technology en route from today's energy mix to a renewable energy only future. Electrolytic hydrogen produced splitting water using low cost renewable electricity at the surface of low cost, Ni-based electrodes in low cost alkaline water electrolyzers therefore is one of the key new energy technologies. Lowering the cost of electrolytic H2 requires to develop new Ni-based electrodes of ultralow overpotential and high stability in alkaline electrolyte. This study identifies the most promising routes to achieve these urgent objectives. |
1. Introduction
Alongside the Li-ion battery, hydrogen prepared via water electrolysis using electricity from today's abundant and low-cost electricity sourced from photovoltaic modules, waterfall, and wind turbines, is the main electrical energy storage technology.1 Contrary to what is often read in published literature,2 or in the introductory section of research articles dealing with advances in electrolytic H2 production, electrolytic hydrogen is not produced using “expensive noble metal components such as platinum and iridium”2 that would be “the preferred catalysts for producing hydrogen through electrolysis at scale”.2In nearly all commercial electrolysers, alkaline water electrolysis (AWE) is used to split water molecules at the surface of low-cost nickel-based electrodes in electrolysis cells operated at 80 °C in 30% KOH.3 The process is used worldwide to produce about 1% of the global amount of hydrogen manufactured yearly. Alongside AWE, more capital-intensive (but more efficient) proton exchange membrane (PEM) electrolysis is also used to prepare electrocatalytic hydrogen.4 Both have advantages and disadvantages working in alkaline and acid media. The reason why AWE over Ni-based catalysts is discussed in this study is similar to that identified by Ayers for PEM electrolysers, “while catalyst cost currently is not the major driver for PEM electrolysis cost, it will become significant as other costs come down and as demand for precious metals increases”.4
Nearly the whole H2 output (∼55 million t of hydrogen annually, used in a wide range of chemical productions)5 is obtained from the petrochemical industry via steam reforming of natural gas methane. The chemical industry needs hydrogen chiefly (but not only) to refine petroleum and make ammonia, methanol, and hydrogen peroxide. The hydrogen market, valued at $ 150 billion in 2017, is growing at a fast pace driven by the increasing demand for fertilizers and crude oil refinement (crude oil of decreasing quality requires more hydrogen for refining).5
Electrolytic hydrogen is sold to pharmaceutical and food companies, which need highly pure (99.999%) electrolytic H2 in place of hydrogen obtained from steam methane (and refinery/chemical off-gases) reforming.3 Besides the low cost of the catalyst, the key advantage of the AWE technology lies in its exceptional durability, with demonstrated MW-scale electrolyzer lifetimes of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h (>30 years).6
000 h (>30 years).6
An excellent historic account of industrial water electrolysis discovery and uptake has been recently published by German scholars.7 Remarkably, in the early 1900s industrial water electrolysis had already reached “a respectable level”,6 thanks to cells able to operate at several hundreds A of current load (running on the direct current supplied by large dynamos) in the bipolar arrangement based on the filter-press design patented by Schmidt in 1899,8 and commercialized in Switzerland as early as of 1902.3,7
The cost of H2 from electrolysis was indeed five times lower than hydrogen that was made via chemical processes such as the reduction of acidic protons in the solution with metallic powdered Zn.7 Accordingly, more than 400 industrial alkaline electrolyzers were already operating in the early 1900s3,7 chiefly installed at companies needing H2 and O2 for welding and cutting applications due to the very high temperature of the oxy-hydrogen flame.3
Originally, the cathode material for AWE electrolyzers was made of steel and the anode material of Ni or Ni-coated steel.9 In 1954 Justi and Winsel in Germany discovered that the highly porous nickel developed in 1926 by Raney,10 with its large surface area and high density of active sites, was a significantly better electrocatalyst for H2 evolution than unmodified Ni.11
Lowering the cost of electrolytic H2 requires the development of more efficient electrodes. As surprising as it may seem, indeed, the manufacturing cost of electrolytic H2 has barely declined compared to the early 1900s when Schmidt's bipolar electrolyzer was commercialized. Calculations of the hydrogen production costs using electrolyzers developed at the end of the 19th century compared to those state of the art in 2017 unveil that no “noticeable cost reduction so far”12 has been achieved. According to Bergmann, not only electricity cost had barely changed, amounting to about €0.10 per kW h (€100 per MW h), but also the investment cost to build an electrolyzer has remained practically the same.12 As a result, assuming an electrical energy demand of about 55 kWh kg−1 H2, the hydrogen production cost would be 5.50 € per kg, namely three times higher than the value targeted for 2030.7
From the new generation RANEY® Ni obtained via physical vapour deposition of Al onto Ni substrate,13 through porous Ni/Co alloys,14 several new Ni-based catalysts have been developed both in hydrogen and oxygen evolution reactions. However, nearly no chemistry or process engineering research paper has entered the stage of application yet.15 According to Segets and Apfel, most reports inadequately address the utilization of the new catalysts, as well as electrode and cell concepts, that might be suitable for the market.15 Similarly, reviewing in 2017, the use of Ni-based electrocatalysts for oxygen reduction, oxygen, and hydrogen evolution reactions, Kim and co-workers concluded that most new catalysts, despite very high initial catalytic efficiencies, show poor electrochemical stability, especially under highly concentrated alkaline conditions.16
This study suggests that, from a practical viewpoint, the most promising routes to lower the cost of electrolytic hydrogen made via AWE mediated by Ni-based electrocatalysts involve the development of (i) new generation catalysts either nanostructured or single-atom catalysts (SACs) of inherently lower overpotential, and (ii) of newly shaped catalytic electrodes obtained via 3D printing.
Generally consisting of catalytic materials in which metal atoms are bound to the support surface through heteroatoms such as oxygen and nitrogen or defective metal active sites,17 SACs are extremely active catalytic materials for which the major challenge, the lack of efficient synthetic routes affording SACs of high catalytic loading and the poor stability at high temperature, are eventually being addressed.18,19 Finally, we suggest three main reasons why new Ni-based electrodes of lower overpotential based on nanoscale or single-atom Ni structures are likely to replace soon the conventional Ni-based electrodes currently used on an industrial scale to produce hydrogen (and oxygen) via AWE.
2. New catalyst structures
2.1 Nanostructured catalysts
The polymorph of nickel hydroxide α-Ni(OH)2 consisting of layers of β-Ni(OH)2, oriented parallel to the crystallographic ab-plane, shows relatively good activity20 in the four electron–proton coupled oxygen evolution reaction (OER, eqn (1)):21| 2H2O(l) + 4 × 1.23 eV → O2 + 4H+ + 4e− | (1) |
With its four electron reaction paths (eqn (1)–(4)), the OER involves four electron transfers [eqn (2)–(5)]:21
| H2O(l) + * → OH* + (H+ + e−) | (2) |
| OH* → O* + (H+ + e−) | (3) |
| O* + H2O(l) → OOH* + (H+ + e−) | (4) |
| OOH* → * + O2(g) + (H+ + e−) | (5) |
The OOH* adsorption (eqn (5)) is the rate-determining step (* stands for an active site on the surface, O*, OH* and OOH* are the adsorbed intermediates, and (l) and (g) denote the liquid and the gas phases).
In 2020, Xue and co-workers in Singapore discovered that nickel hydroxide with a two-dimensional nanoribbon structure (NR-Ni(OH)2, 10–20 nm in length, 2–5 nm in width, and two or three layers in thickness), is significantly more active than β-Ni(OH)2.22 In detail, the nanostructured catalyst obtained via a straightforward electro-oxidation route has an 18 mV dec−1 lower Tafel slope and a ∼150 mV lower overpotential than those of β-Ni(OH)2. Furthermore, the catalyst showed good stability with the overpotential remaining below 200 mV at 10 mA cm−2 after 10 days of operation.
Calculations suggest that the reduction in the overpotential of NR-Ni(OH)2 can be ascribed to its easier OOH* adsorption by the active tetra-coordinated Ni edge sites when compared to hexa-coordinated Ni atoms of β-Ni(OH)2.22
The application potential of the nanostructured NR-Ni(OH)2 catalyst was widened by the subsequent discovery that by illuminating with visible light of 1 sun solar irradiance (1000 W m−2) an electrode made of the same nickel oxyhydroxide-based ribbon-like material electrodeposited on carbon, resulted in a dramatic improvement in the OER rate.23 In detail, after irradiating the electrode kept in a thermostatic bath wherein cooling water was used to keep the temperature of the system constant, the current density nearly doubled from 13.5 to 25 mA cm−2 within 4.3 h. It was enough to switch off the light to observe the current intensity returning to the original value of 13.5 mA cm−2 within 4.4 h.
The light-induced process is fully reversible having been observed during prolonged operation (100 h) of the cell. We briefly remind that chronoamperometry is used to determine the effect of an electrocatalyst on the electrolysis reaction rate because it measures a current whose value depends on the rate of reduction (or oxidation) reaction occurring at the electrode.24
Taking into consideration the entire resistance (Ru + Rs) for iR correction including the resistance Rs of the electrolyte, the resistance of the catalytic material NR-NiOOH (Ru) under dark and in the light were relatively similar.
In brief, new industrial processes to produce electrocatalytic hydrogen and oxygen over the low-cost NR-NiOOH catalyst would involve transparent cells with direct sunlight (or light emitted by energy-efficient LEDs) further accelerating the alkaline water electrolysis, thereby lowering the amount of electrical energy needed to split water.
2.2 Single-atom catalysts
In 2017 Park and co-workers in South Korea reported the first highly active single-atom nickel catalyst for the OER reaction.25 Consisting of Ni atoms bound to the structure of g-C3N4 simply obtained by the thermal treatment at 520 °C (under N2 flow) of a mixture of melamine and NiCl2·6H2O in ethanol, the catalyst consisting of a light brown powder was mixed with a small amount of a solution of the perfluorinated ion-exchange resin Nafion (5% w/w) in aqueous aliphatic alcohols, and deposited on a glassy carbon electrode by simple solution dropping followed by drying for 1.5 h. No nanoparticle was observed via TEM. The field emission TEM image displays good dispersion and lack of agglomeration of Ni-containing species (Fig. 1).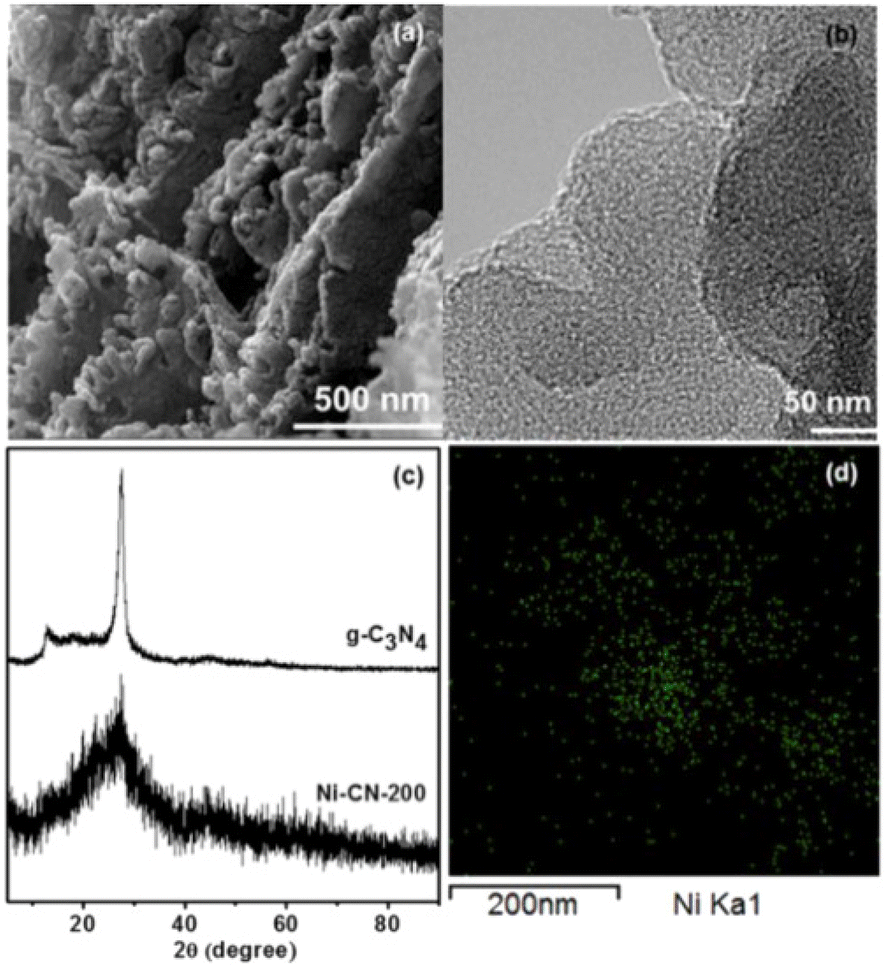 | ||
| Fig. 1 (a) SEM image of N-CN-200, (b) FE-TEM image of N-CN-200, (c) XRD patterns of g-C3N4 and N-CN-200 (d) elemental mapping images of Ni using FE-TEM of N-CN-200 (green dots indicate Ni atoms) [reproduced from ref. 25, with kind permission]. | ||
Evidence that Ni species are atomically dispersed in the samples via the coordination to the nitrogen atoms of g-C3N4 originates from the EXAFS analysis pointing to a 3.7 coordination number for Ni, with a Ni–N bond length of 2.1 Å. The SEM and TEM images showed a highly porous structure. The incorporation of Ni atoms into the g-C3N4 structure results in a surface area increase from 7.5 to 25.7 m2 g−1, whereas the XRD pattern of Ni-CN-200 showed the lack of typical in-plane crystallinity of g-C3N4 (the peak at 12.8° disappears and that at 27.6° lowers and broadens).
The electrocatalytic performance of the single-atom electrocatalytic material was investigated for the OER at a scan rate of 5 mV s−1 using a rotating disk electrode (RDE). The trend observed (not shown) showing less positive onset potentials of electrodes modified with SACs of increasing Ni content suggests that the tetra-coordinated Ni species on the g-C3N4 are the active species for the OER. The electrode modified with Ni-CN-200 exhibited the best electrocatalytic activity with an onset potential of 1.54 V (vs. RHE), 80 mV less positive compared to the glassy carbon electrode modified with g-C3N4. Among the five modified electrodes investigated, the Ni-CN-200-modified electrode had the lowest value, and only electrodes modified with expensive IrOx had better electrocatalytic activity than the former.
Finally, the Ni-CN-200-modified electrode showed excellent stability during the OER with only trace amounts (ppb) of Ni leaching from the stable molecular structure displayed in Fig. 3, which is reflected in the stable cyclic voltammograms of Ni-CN-200 modified electrode after 500 or 1000 cycles (Fig. 2).
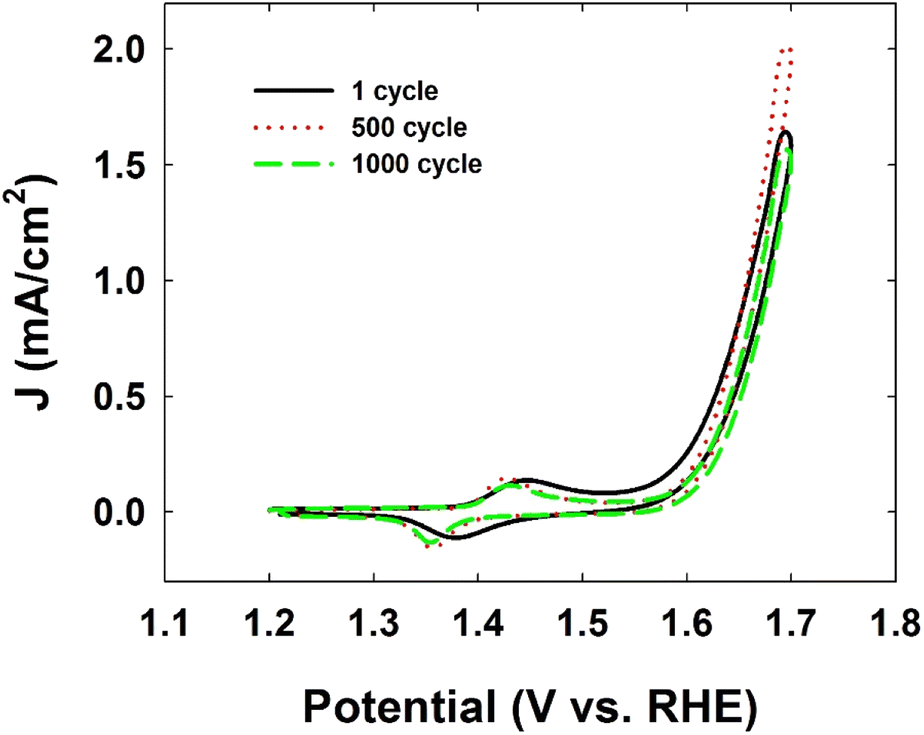 | ||
| Fig. 2 Cyclic voltammograms of Ni-CN-200 modified electrode for the durability test after 500 or 1000 cycles at a scan rate of 100 mV s−1. The electrolyte solution was Ar-saturated 1 M KOH [reproduced from ref. 25, with kind permission]. | ||
Similarly to what found for the oxyhydroxide-based ribbon-like NR-Ni(OH)2 electrodeposited on carbon,22 the team unveiled through EXAFS spectroscopy the presence in the SAC of two differently bound Ni atomic species denoted “EDGE-bonded” and BASAL-bonded” (Fig. 3).
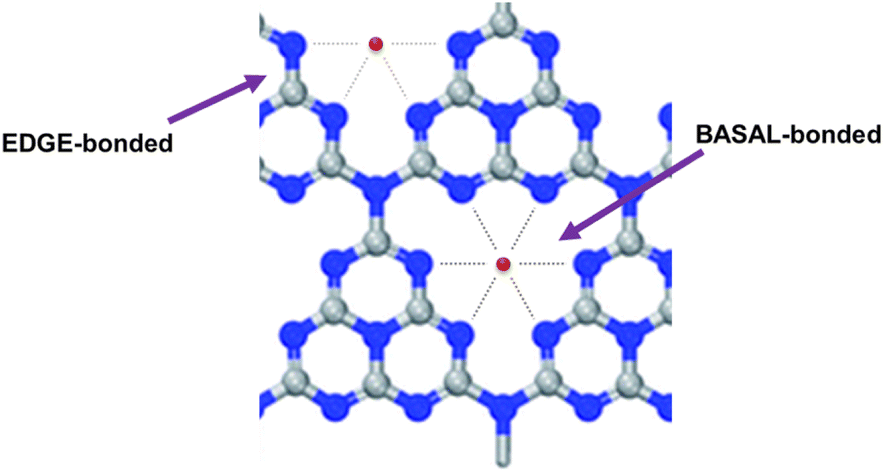 | ||
| Fig. 3 The expected local structure around Ni atoms (EDGE-bonded and BASAL-bonded). The EDGE-bonded structure is more fluctuant than the BASAL-bonded structure [reproduced from ref. 25, with kind permission]. | ||
The significantly higher OER activity of Ni-CN-200 was ascribed to the “more flexible bonding nature of Ni”.25 The researchers hypothesized that when a low amount of NiCl2 is used in the catalyst synthesis (such as in the case of Ni-CN-100 made using 100 mg of nickel chloride), energetically more favorable BASAL-bonded Ni atoms of higher coordination number with C3N4 are preferentially generated. Increasing to 200 mg, the amount of NiCl2 in the synthesis of the SAC (Ni-CN-200), Ni atoms get coordinated also to the edge positions of the catalyst molecular structure (Fig. 3).
In agreement with what will be reported for the nickel oxyhydroxide-based ribbon-like material,23 it is the dramatically enhanced OOH* adsorption (eqn (4), the rate-determining step in the OER)21 at the tetra-coordinated Ni edge sites (when compared to the Ni atoms fully coordinated with 6 nitrogen atoms), which is responsible for the enhanced activity of these Ni-based electrocatalysts.
The solution-dropping electrocatalyst deposition method used to functionalize the surface of the glassy carbon electrode with Ni-CN-200 naturally leads to the need, solved by 3D printing, to improve the efficiency and efficacy of electrode functionalization to develop enhanced electrodes for AWE.
2.3 3D-printed electrodes
In 2022, Tang and co-workers in China reported the first photo-curing 3D-printing method to directly manufacture structured Ni-based electrocatalysts with enhanced activity in the hydrogen evolution reaction (HER) in alkaline water electrolysis.26The 3D-printed electrode (denoted Ti–Ni NS, starting from a ceramic paste with ∼55 wt% solid loads) consists of a Ni substrate uniformly coated by the Ni–Ti non-crystalline catalyst, featuring a blocky porous structure causing closer connection and enhanced electron transfer between the catalyst and Ni substrate. The electrode was printed layer-by-layer using a digital light printing (DLP) printer (UV radiation wavelength of 405 nm, and residence time of 13 s). The thickness of each printing layer was 25 μm.
In closer detail, the printed ceramic paste consists of TMPTA (trimethylolpropane triacrylate), TPGDA (tripropylene glycol diacrylate), and PUA (polyurethane-acrylate) with a mass ratio of 3:4:3, to which NiO powder (40 wt%), YSZ (yttria-stabilized zirconia, 11 wt%) as a structural stabilizer, and TiC (4.5 wt%) as a doping agent were added alongside BYK-111, BYK-410, and (methylpropyl) trimethoxy silane (KH-570) sizing stabilizers, which make the printing paste stable even after 30 days.
The printed ceramic electrode (Fig. 4) was first ultrasonically cleaned using ethanol to remove the residues, followed by heat treatment in a tubular furnace and finally by reduction using a 5 vol% H2/Ar gas mixture.
 | ||
| Fig. 4 Printed ceramic electrode Ti–Ni NS obtained by digital light printing technology after degreasing, sintering, and reduction, respectively [reproduced from ref. 26, with kind permission]. | ||
The method allows printing electrodes with complex shapes with the unique degree of freedom typical of digital 3D printing (Fig. 4). Even without electrode shape optimization aimed at easing the release of H2 gas molecules (bubbles partly block the catalytically active surface area, lowering reactant diffusion and increasing the ohmic resistance), the 3D-printed Ti–Ni NS electrode (in column shape) for the alkaline HER had an overpotential of 34 mV to deliver a current density of 10 mA cm−2 and a Tafel slope of 74 mV dec−1, surpassing the commercial Pt/C catalyst and most of the state-of-the-art electrocatalysts.
Offering both low cost and printing accuracy, this electrode manufacturing method avoids deposition of the electrocatalyst by external means but rather produces a homogeneous catalytic electrode similar to what happens with sol–gel electrodeposited electrodes.27 The researchers further optimized the printing paste composition where Ti exists in the amorphous state with strong interaction with Ni, leading to increased active sites and improved electrolytic properties.
The HER process in alkaline solution consists of the Volmer (H2O + e− → Had + OH−), Heyrovsky (Had + H2O + e− → OH− + H2) and Tafel (Had + Had → H2) steps.
According to the volcanic shape of Trasatti's curve correlating activity and electrode material by the bond strength of intermediates to the electrode surface,28 Ni is on the left side of Pt. The weaker binding energy of Ni with Hads suggests lower adsorption of H atoms on the surface of the Ni electrode. This issue may be addressed by alloying Ni with the metals located on the right side of Pt in Trasatti's plot.
Quantum calculations aimed at identifying the catalytic effect of Ti incorporation into the Ni lattice show that Ti doping lowers both the water dissociation (ΔGH2O) and hydrogen adsorption (ΔGHad) energy barriers, thus enhancing HER (Ti doping enhances adsorption and activation of water molecules via significantly enhanced acid strength due to more vacant orbitals in the valence shell). The electrode demonstrated excellent stability during prolonged electrolysis. Tested for 100 h in the continuous electrolysis in 1 M KOH at a potential of −217 mV versus RHE, with an initial current density of 70 mA cm−2 the activity of the electrode remained unvaried (Fig. 5a). Negligible structural changes were observed in the XRD and XPS patterns of Ti–Ni NS before and after HER at 45 mA cm−2 for 40 h, with no formation of Ni oxides and negligible changes in the valence state (Fig. 5b–d).
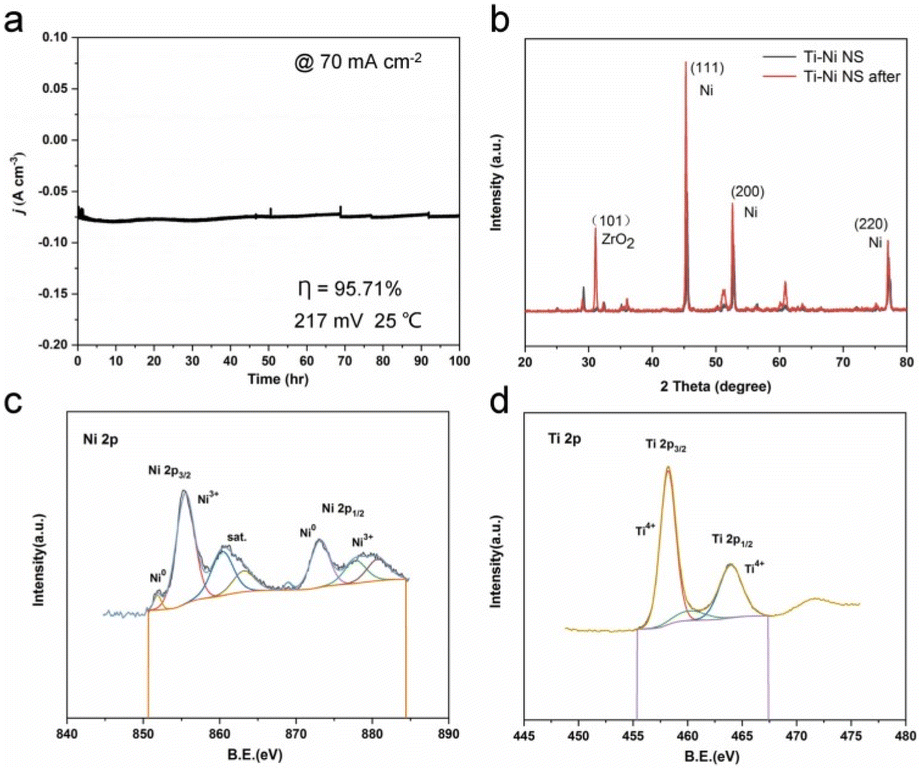 | ||
| Fig. 5 Long-term stability measurement of Ti–Ni NS. (a) The durability of Ti–Ni NS in constant potential testing for 100 h. (b) XRD patterns of Ti–Ni NS before and after the HER. (c and d) XPS spectra of Ti–Ni NS after the HER [reproduced from ref. 26, with kind permission]. | ||
The SEM images of the surface of the electrode before and after electrocatalysis (Fig. 6) showed retention of the gluten-like cubes after prolonged hydrogen evolution and no peel-off of the coated electrocatalytic layer from the substrate.
 | ||
| Fig. 6 SEM images of the Ti–Ni NS catalyst prior (left) and after 100 h electrocatalysis (right) [reproduced from ref. 26, with kind permission]. | ||
These findings are particularly promising also taking into account that similar 3D-printed hierarchically porous Ni-based electrocatalysts of uniform chemical composition with multiscale porosity are ideally suited to carry out the electrocatalytic reactions under flow, replacing the traditional planar (static) electrode configuration.
This was recently demonstrated by a team of researchers from China and the USA who used a 3D-printed nickel molybdenum (NiMo) electrocatalyst from a resol-based aerogel precursor for the HER in 1.0 M KOH in an H-cell (a divided electrochemical cell, named after its similarity with letter H) in which the electrolyte flow direction was orthogonal to the electrode surface.29 The flow-through configuration of the 3D-printed electrocatalyst increases the accessible surface area and roughness factor, reducing mass-transport limitations and promoting the efficient removal of H2 bubbling from the electrode surface. Without electrolyte flow, an overpotential of 60 mV was measured at 10 mA cm−2 current density (Fig. 7a), whereas under the electrolyte flow an average overpotential of 45 mV at 10 mA cm−2 over 24 h was observed.
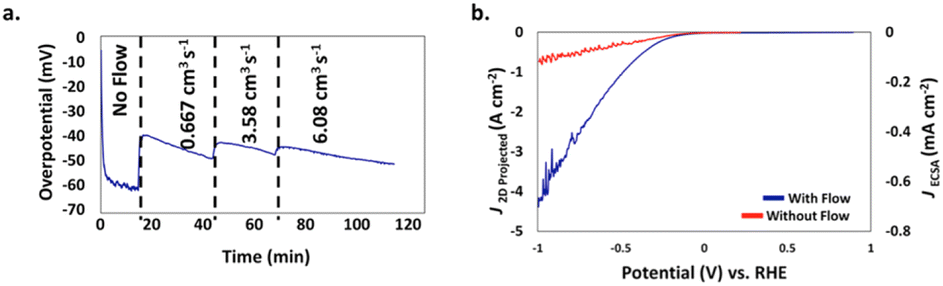 | ||
| Fig. 7 (a) Impact of flow rate on the overpotential of the 3D printed NiMo flow-through electrode for HER at J2D,projected = 10 mA cm−2 as a function of time. (b) J2D,projected (left axis) and J3D,ECSA (right axis) of the NiMo electrodes as a function of the potential vs. RHE in a traditional electrode configuration (red) and in a flow-through configuration at a flow rate of 3.58 cm3 s−1 (blue) [reproduced from ref. 29, with kind permission]. | ||
The measurement of the overpotential is difficult due to the formation of H2 gas bubbles on the electrode surface. As flow starts, bubbles free the surface and the overpotential decreases to ∼40 mV. Further increases in flow rate (in two steps from 0.667 to 6.08 cm3 s−1) barely impacted the overpotential. Fig. 7b shows evidence of significant overpotential differences with or without the electrolyte flow, particularly at high current densities (for example, at 500 mA cm−2 the overpotential was −0.36 V with flow and −0.77 V without flow).
3. Perspectives and conclusions
Three main reasons suggest that new Ni-based electrodes of lower overpotential are likely to soon replace the conventional Ni-based electrodes (RANEY® Ni perforated stainless steel as the cathode and stainless steel as the anode) widely used on an industrial scale to produce highly pure electrolytic hydrogen (and oxygen) via alkaline water electrolysis.Firstly, the advanced design of alkaline electrolyzers using zero-gap electrodes and thinner diaphragms to increase current density has already reduced their performance gap in comparison to PEM technology.30 Alkaline electrolyzers are the only ones in the industry to have shown exceptional durability, reaching lifetimes above 30 years. The key component of the electrolyzer that remains to be significantly advanced is the catalytic electrode (see below).
Secondly, the nanochemistry-based chemical methods to produce the said new electrodes are efficient, low-cost, and highly reproducible. In other words, they do not require expensive, special equipment to deposit the electrocatalytic layer on the electrode substrate, and can be reproducibly applied to the mass production of electrodes for new electrolyzers.
Thirdly, the high stability of the new generation Ni-based electrodes makes them ideally suited to be applied in continuous flow electrolyzers for producing H2 at lower capital and operational costs using highly intermittent wind and sunlight as renewable sources of electrical energy.
Pagliaro has suggested how, driven by technology, societal and environmental megatrends, chemical productions, including those of bulk chemicals produced in huge centralized chemical plants, will progressively switch to small flow reactors where production will take place continuously under mild reaction conditions sourcing the reactants from biological resources, rather than from hydrocarbons.31 Hydrogen is no exception.
Its current industrial production via steam reforming of methane and other hydrocarbons in large petrochemical plants will increasingly be replaced by multiple, new generation electrolyzers running under flow sourcing the fuel from the renewable material source par excellence, water, using low-cost renewable electricity.
Getting to the electrolyzer industry, the history of the industrial uptake of completely new electrodes in the chemical industry suggests useful lessons. Trasatti has recounted how dimensionally stable anodes comprised of conductive oxide “were essentially unknown in electrochemistry and they remained undisclosed for at least 7 years after their invention”,32 while industry was in fact first testing their performance. In a few years since their invention, in 1968 the new electrodes were successfully applied to industrial electrochemical productions, reducing operational and investment costs.
It is therefore instructive to learn that from the industrial perspective of the world's largest alkaline electrolyzer manufacturer, the key enablers to enhance the efficiency and capacity of industrial cell stacks are the increase of (i) electrode size and active area, and (ii) current density.33 As mentioned above, 3D printing allows for obtaining large-size electrodes coated with a nanostructured Ni-based electrocatalytic layer of significantly larger surface area and stability, capable to withstand large current densities without degrading. Furthermore, 3D printing allows easy obtainment of electrodes whose optimal shapes effectively promote the removal of H2 and O2 bubbles from the electrode surface, avoiding the rise in overpotential.
For these reasons, upon reviewing selected examples of new-generation Ni-based electrocatalysts for enhanced alkaline water electrolysis, this study suggests that the most promising routes to lower the cost of electrolytic hydrogen made via AWE involve the development of nanostructured or single-atom catalysts of inherently lower overpotential, subsequently deposited via 3D printing onto catalytic electrodes of optimal shapes.
In the course of the 2010s, the dramatic fall in renewable electricity generation costs following the global uptake of wind and photovoltaic34 renewable energy technologies, led to revamping alkaline water electrolysis research in most industrialized countries.35 The main outcomes of the first new wave of developmental research are the need to enhance the electrocatalytic activity of the cell anode for OER, and “appropriate management of the gas bubble phenomena”.35
Enhanced collaboration between academic researchers and industry is, once more, needed to accelerate practically useful innovation. For example, even in South Korea, which is hosting one of the world's most advanced industrial economies where the world's best-selling H2-fuelled fuel cell electric vehicle is successfully produced,36 the first Ni-based single-atom catalyst for the OER reaction introduced in 2017 (ref. 25) has not yet been tested on a large scale.37
The above-mentioned new collaborations will guide research chemists to develop practically useful new-generation catalysts suitable for use in compact electrolyzers made according to the advanced design and lean production principles. Using recent research outcomes to foster student creativity,38 this study, in conclusion, provides further examples of how to advance education in electrochemistry: a global need to provide industry with a higher number of highly qualified young researchers in this eminent cross-disciplinary field of today's chemistry research.39
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This article is dedicated to the memory of Professor Sergio Trasatti (1937–2021) for all he has done for the progress of electrochemistry, including his educational work. We thank Professor Sungjin Park, Inha University, for valuable correspondence on the topics of this study. Thanks to Piano Operativo della Ricerca “Ricerca e sviluppo sull'idrogeno” financed by the EU – NextGenerationEU – M2C2 Investment 3.5, for funding within the “D.1.1.6.3 Sintesi e caratterizzazione morfologico strutturale di catalizzatori per l'evoluzione di ossigeno (almeno 4 tipologie) e di idrogeno (almeno 4 tipologie) e membrane (almeno 4 tipologie) per operazioni in ambiente alcalino” research line. We thank Dr Salvatore Romeo, Istituto per lo Studio dei Materiali Nanostrutturati, CNR, for administrative assistance.References
- M. Pagliaro, A. G. Konstandopoulos, R. Ciriminna and G. Palmisano, Solar hydrogen: fuel of the near future, Energy Environ. Sci., 2010, 3, 279–287, 10.1039/b923793n.
- Georgia Institute of Technology, A catalyst for more efficient green hydrogen production, ScienceDaily, January 14, 2022, https://www.sciencedaily.com/releases/2022/01/220114153429.htm, accessed July 3, 2023 Search PubMed.
- N. Guillet and P. Millet, Alkaline water electrolysis, in Hydrogen Production, ed. A. Godula-Jopek, Wiley-VCH, Weinheim, 2015, pp. 117–166 Search PubMed.
- K. Ayers, The potential of proton exchange membrane-based electrolysis technology, Curr. Opin. Electrochem., 2019, 18, 9–15, DOI:10.1016/j.coelec.2019.08.008.
- Hydrogen Council, Hydrogen Scaling Up, Brussels, 2017, https://hydrogencouncil.com/wp-content/uploads/2017/11/Hydrogen-Scaling-up_Hydrogen-Council_2017.compressed.pdf, accessed July 3, 2023 Search PubMed.
- A. Buttler and H. Spliethoff, Current status of water electrolysis for energy storage, grid balancing and sector coupling via power-to-gas and power-to-liquids: A review, Renewable Sustainable Energy Rev., 2018, 82, 2440, DOI:10.1016/j.rser.2017.09.003.
- T. Smolinka, H. Bergmann, J. Garche and M. Kusnezoff, The history of water electrolysis from its beginnings to the present, in Electrochemical Power Sources: Fundamentals, Systems, and Applications: Hydrogen Production by Water Electrolysis, ed. T. Smolinka and J. Garche, Elsevier, Amsterdam, 2022, pp. 83–164, DOI:10.1016/B978-0-12-819424-9.00010-0.
- O. Schmidt, Apparat zur Elektrolyse von Wasser, German Pat., DE111131, 1899.
- P. A. Lessing, Materials for water electrolysis cells, in Materials for the Hydrogen Economy, ed. R. H. Jones and G. J. Thomas, CRC Press, Boca Raton (FL), 2007, pp. 38–41 Search PubMed.
- M. Raney, Method of producing finely-divided nickel, US Pat., US 1628190, 1927 Search PubMed.
- E. Justi and A. Winsel, Doppelskelett-Katalysator-Elektrode, German Pat., DE1019361B, 1954.
- H. Bergmann, Aus der Geschichte der Wasserelektrolyse - Wie es einst began. Mit einer Diskussion der Perspektiven, Galvanotechnik, 2017, 108 Search PubMed , Teil 1: 44–49, Teil 2: 260–267, Teil 3: 471–479..
- C. K. Kjartansdóttir, L. Pleth Nielsen and P. Møller, Development of durable and efficient electrodes for large-scale alkaline water electrolysis, Int. J. Hydrogen Energy, 2013, 38, 8221–8231, DOI:10.1016/j.ijhydene.2013.04.101.
- T. Veettil Vineesh, S. Mubarak, M. Gwan Hahm, V. Prabu, S. Alwarappan and T. N. Narayanan, Controllably alloyed, low density, free-standing Ni-Co and Ni-graphene sponges for electrocatalytic water splitting, Sci. Rep., 2016, 6, 31202, DOI:10.1038/srep31202.
- D. Siegmund, S. Metz, V. Peinecke, T. E. Warner, C. Cremers, A. Grevé, T. Smolinka, D. Segets and U.-P. Apfel, Crossing the valley of death: from fundamental to applied research in electrolysis, JACS Au, 2021, 1, 527–535, DOI:10.1021/jacsau.1c00092.
- V. Vij, S. Sultan, A. M. Harzandi, A. Meena, J. N. Tiwari, W.-G. Lee, T. Yoon and K. S. Kim, Nickel-based electrocatalysts for energy-related applications: oxygen reduction, oxygen evolution, and hydrogen evolution reactions, ACS Catal., 2017, 10, 7196–7225, DOI:10.1021/acscatal.7b01800.
- M. Pagliaro, Single-Atom Catalysis, Elsevier, Amsterdam, 2019 Search PubMed.
- R. Ciriminna, M. Ghahremani, B. Karimi, R. Luque and M. Pagliaro, Single-atom catalysis: a practically viable technology?, Curr. Opin. Green Sustainable Chem., 2020, 25, 100358, DOI:10.1016/j.cogsc.2020.100358.
- Y. Hu, B. Li, C. Yu, H. Fang and Z. Li, Mechanochemical preparation of single atom catalysts for versatile catalytic applications: A perspective review, Mater. Today, 2023, 63, 288–312, DOI:10.1016/j.mattod.2023.01.019.
- C. Luan, G. Liu, Y. Liu, L. Yu, Y. Wang, Y. Xiao, H. Qiao, X. Dai and X. Zhang, Structure effects of 2D materials on α-nickel hydroxide for oxygen evolution reaction, ACS Nano, 2018, 12, 3875–3885, DOI:10.1021/acsnano.8b01296.
- I. C. Man, H.-Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, Universality in oxygen evolution electrocatalysis on oxide surfaces, ChemCatChem, 2011, 3, 1159–1165, DOI:10.1002/cctc.201000397.
- X. Wang, H. J. Wu, S. B. Xi, W. S. V. Lee, J. Zhang, Z. H. Wu, J. O. Wang, T. D. Hu, L. M. Liu, Y. Han, S. W. Chee, S. C. Ning, U. Mirsaidov, Z. B. Wang, Y. W. Zhang, A. Borgna, J. Wang, Y. H. Du, Z. G. Yu, S. J. Pennycook and J. M. Xue, Strain stabilizing nickel hydroxide nanoribbons for efficient water splitting, Energy Environ. Sci., 2020, 13, 229, 10.1039/c9ee02565k.
- X. Wang, S. Xi, P. Huang, Y. Du, H. Zhong, Q. Wang, A. Borgna, Y.-W. Zhang, Z. Wang, H. Wang, Z. G. Yu, W. S. V. Lee and J. Xue, Pivotal role of reversible NiO6 geometric conversion in oxygen evolution, Nature, 2022, 611, 702–708, DOI:10.1038/s41586-022-05296-7.
- A quicker but similarly accurate method (sampled current voltammetry) at a constant steady-state applied voltage has lately been introduced: S. Anantharaj, S. Kundu and S. Noda, Worrisome exaggeration of activity of electrocatalysts destined for steady-state water electrolysis by polarization curves from transient techniques, J. Electrochem. Soc., 2022, 169, 014508, DOI:10.1149/1945-7111/ac47ec.
- S. Ohn, S. Y. Kim, S. K. Mun, J. Oh, Y. J. Sa, S. Park, S. H. Joo, S. J. Kwon and S. Park, Molecularly dispersed nickel-containing species on the carbon nitride network as electrocatalysts for the oxygen evolution reaction, Carbon, 2017, 124, 180–187, DOI:10.1016/j.carbon.2017.08.039.
- Z. Han, G. Wang, J. Zhang and Z. Tang, Direct photo-curing 3D printing of nickel-based electrocatalysts for highly-efficient hydrogen evolution, Nano Energy, 2022, 102, 107615, DOI:10.1016/j.nanoen.2022.107615.
- L. Liu and D. Mandler, Electrochemical deposition of sol-gel films, in Handbook of Sol-Gel Science and Technology, ed. L. Klein, M. Aparicio and A. Jitianu, Springer, Cham, Switzerland, 2018, pp. 531–568, DOI:10.1007/978-3-319-32101-1_113.
- P. Quaino, F. Juarez, E. Santos and W. Schmickler, Volcano plots in hydrogen electrocatalysis - uses and abuses, Beilstein J. Nanotechnol., 2014, 5, 846–854, DOI:10.3762/bjnano.5.96.
- I. Sullivan, H. Zhang, C. Zhu, M. Wood, A. J. Nelson, S. E. Baker, C. M. Spadaccini, T. Van Buuren, M. Lin, E. B. Duoss, S. Liang and C. Xiang, 3D printed nickel-molybdenum-based electrocatalysts for hydrogen evolution at low overpotentials in a flow-through configuration, ACS Appl. Mater. Interfaces, 2021, 13, 20260–20268, DOI:10.1021/acsami.1c05648.
- IRENA, Green Hydrogen Cost Reduction, Abu Dhabi, 2020 Search PubMed.
- M. Pagliaro, An industry in transition: The chemical industry and the megatrends driving its forthcoming transformation, Angew. Chem., Int. Ed., 2019, 58, 11154–11159, DOI:10.1002/anie.201905032.
- S. Trasatti, Electrocatalysis: understanding the success of DSA, Electrochim. Acta, 2000, 45, 2377–2385, DOI:10.1016/S0013-4686(00)00338-8.
- S. Szymanski, Nel Hydrogen: Electrolyser Solutions for Large Scale Hydrogen Production, California Energy Commission, July 2021, https://efiling.energy.ca.gov/getdocument.aspx?tn=239029, accessed July 3, 2023 Search PubMed.
- F. Meneguzzo, R. Ciriminna, L. Albanese and M. Pagliaro, The great solar boom: a global perspective into the far reaching impact of an unexpected energy revolution, Energy Sci. Eng., 2015, 3, 499–509, DOI:10.1002/ese3.98.
- A. N. Colli, H. H. Girault and A. Battistel, Non-precious electrodes for practical alkaline water electrolysis, Materials, 2019, 12, 1336, DOI:10.3390/ma12081336.
- Global sales of Hyundai Motor's hydrogen fuel cell Nexo surpass 10,000, FuelCellsWorks, January 4, 2023, https://fuelcellsworks.com/news/global-sales-of-hyundai-motors-hydrogen-fuel-cell-nexo-surpass-10000/, accessed July 3, 2023 Search PubMed.
- S. Park, personal communication to M. P., November 2022.
- M. Pagliaro, Chemistry education fostering creativity in the digital era, Isr. J. Chem., 2019, 59, 565–571, DOI:10.1002/ijch.201800179.
- R. Ciriminna, M. Ghahremani, F. Varmaghani, B. Karimi and M. Pagliaro, Improving education in electrochemistry via a modeling approach and focusing on green chemistry applications, Sustainable Chem. Pharm., 2023, 31, 100931, DOI:10.1016/j.scp.2022.100931.
| This journal is © The Royal Society of Chemistry 2023 |