
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydrolytic depolymerisation of polyesters over heterogeneous ZnO catalyst†
Francesca
Liguori
 a,
Carmen
Moreno-Marrodán
a,
Werner
Oberhauser
a,
Elisa
Passaglia
b and
Pierluigi
Barbaro
*a
a,
Carmen
Moreno-Marrodán
a,
Werner
Oberhauser
a,
Elisa
Passaglia
b and
Pierluigi
Barbaro
*a
aConsiglio Nazionale delle Ricerche, Istituto di Chimica dei Composti Organo Metallici, Via Madonna del Piano 10, 50019 Sesto Fiorentino, Firenze, Italy. E-mail: pierluigi.barbaro@iccom.cnr.it
bConsiglio Nazionale delle Ricerche, Istituto di Chimica dei Composti Organo Metallici, Via Moruzzi 1, 56124 Pisa, Italy
First published on 3rd August 2023
Abstract
The hydrolytic depolymerisation of polylactic acid (PLA) and polyethylene terephthalate (PET) was accomplished in neat water using heterogenous ZnO catalysts. Up to 100% and 98% selectivity to the complete hydrolysis products was achieved at full PLA and PET conversion at 130 °C and 180 °C, respectively. The catalytic species could be partially (for PLA) recovered and reused, though with a gradual activity drop.
Sustainability spotlightPolyesters are among the most used plastics in everyday domestic and industrial applications, mainly in textiles and in food packaging. This involves their substantial contribution to the huge amount of plastic waste produced every year. Developing efficient depolymerisation processes may foster the transition from a linear to a circular economy model for plastics, where wastes are converted into reusable building blocks. However, most existing technologies for the chemical recycling of polyesters are unsustainable, requiring concentrated mineral acids or bases, soluble promoters, harsh conditions, organic reagents and the management of considerable amounts of by-products. Selectivity may also be an issue. All the above result in an underuse of waste plastic, for instance to produce fuel mixtures. In the present work, we show that the complete depolymerisation of PLA and PET can be achieved in high selectivity over easily separable heterogeneous ZnO catalysts, using neat water and mild reaction conditions. The advancements of the work are relevant to multiple targets defined by the SDG12 “Ensure sustainable consumption and production patterns”. |
Introduction
Plastic scraps accumulation in the environment is a major issue worldwide. Most plastics are oil-derived synthetic polymers recalcitrant to decomposition, and liable to release toxic components, thus representing severe pollutants for soils, oceans, crustaceans and rain.1 Presently, the majority of post-consumer plastics are landfilled (40%), leaked in the environment (20%) or incinerated (25%).2 Only a minor portion is mechanically recycled and reused.3 Besides being a threat to the ecosystem and health, plastic materials are a wastage of useful matter, if dumped. Indeed, waste plastics are valuable secondary raw materials for the production of reprocessable chemicals, which is the concept at the basis of the circular economy for plastics.4 This requires the implementation of selective depolymerisation processes,5 which can be accomplished by chemical recycling.‡ However, depolymerisation is not enough to achieve sustainability of plastic value chains. This must be performed at competitive economic and environmental costs.6 Unfortunately, the current technologies are poorly developed, mostly relying on organic solvents, considerable amounts of soluble promoters, toxic reagents or harsh reaction conditions, which ultimately result in the generation of large quantities of undesired products, costly downstream purifications and in high energy inputs.7 As a matter of fact, as low as 1% of waste plastics are chemically recycled to date.8 Therefore, new (catalytic) systems shall be developed, which enable the depolymerisation of plastics to occur selectively and effectively, under friendly conditions.9 Dedicated research policies address the topic both in the EU and in USA,10 whilst targets 12.4 and 12.5 of the UN's Sustainable Development Goals closely relate to the issue.11§Polyesters contribute with around 18% to the ca. 300 million tonnes plastic wastes globally generated every year,12 which accounts for their massive use in the packaging, beverage and textile industries (Fig. 1).13,14 Chemical recycling of polyesters may provide a variety of useful monomers, mainly depending on the cleaving agent used: acids in the case of water, esters for alcohols and glycols, amides in the case of amines. However, the existing solvolysis processes are flawed by the use of concentrated solutions of soluble metal salts (e.g. zinc acetate), strong mineral acids or bases, excess of (harmful) organic reagents (e.g. methanol), or high reaction temperatures, as well as in the management of considerable amounts salt by-products.15 Hydrogenolysis may be a cleaner option, though practically limited to bench, homogenous, and non-reusable metal complex catalysts.16 Aiming at a method for the selective depolymerisation of polyesters to reusable, added-value chemicals under sustainable conditions, in the present work we investigated the chemical recycling of market polyesters (PLA, PET), using water as lytic agent and solid catalysts.
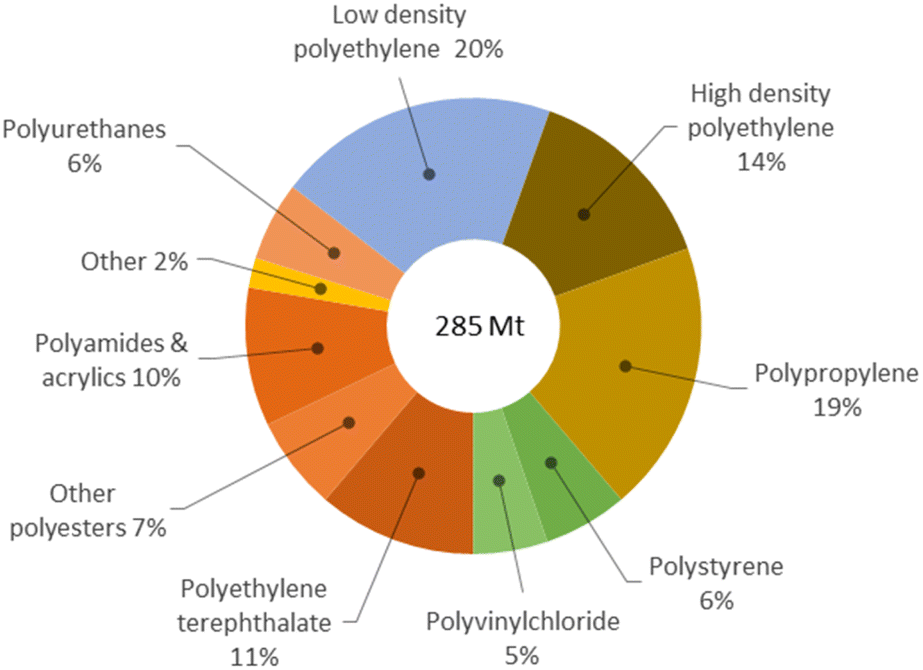 | ||
| Fig. 1 Global primary plastic waste generation in 2015 (million tons), according to type. Data from ref. 14. | ||
Results and discussion
Polylactic acid (PLA) is a bio-derived thermoplastic broadly used in microelectronics, textiles, biomedical and packaging applications.17 In 2019, the production of PLA was around 290 kt, mostly based on the processing of starch from corn, cassava, potato and sugar cane feeds.18 Due to the properties of being renewable sourced, soluble in a number of organic solvents and compostable, PLA is assuming a prevalent role as disposable commodity in the food sector (dishes, cutlery, containers) and in a variety of single-use products.19 As a consequence, manufacture of PLA is foreseen to increase to 560 kt in 2025.20 However, biodegradability of PLA often leads to a misunderstanding that it easily degrades in the environment, hence to a mismanagement of its “End-of-Life”. Actually, the complete depolymerisation of PLA can take up around a year in soil or in domestic composter, and 100 days in a composter at elevated temperature.21 Types of microorganism, pH, temperature and humidity have an effect on the rate of decomposition. PLA degradation in marine environments depends on several variables, and no changes were observed within a year.22 On the other hand, lactic acid (LA) is considered among the top platform molecules from biomass, allowing the production of a wide range of chemicals, thus underpinning a bio-based economy.23 Current demand of LA is around 400 kt per year, which is expected to grow significantly.24 Thus, in the light of the above mentioned circular economy policies for plastics, the development of effective methods for the hydrolytic depolymerisation of post-consumer PLA to LA can be significant. Unfortunately, the applicability of biocatalysis for the production of LA from PLA at large is still uncertain.25 Despite the mild operative conditions, the low energy consumption and the possibility of stereo-specific synthesis of LA,26 this technology requires considerable amounts of specialized enzymes, a long time for the complete conversion of highly crystalline polymers and additional purifications to separate the monomer/oligomers mixtures usually obtained, which often results in poor LA yields.27 In addition, reaction temperature cannot be high when protein catalysts are used. As a matter of fact, enzymatic biodegradation of PLA often affects the morphology and the bulk properties of the material, rather than its molecular weight.28 All the above justify for the search of alternative hydrolytic depolymerisation routes based on chemical catalysts.29Thus, in a first set of experiments, the hydrolysis reaction of PLA (Mn 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100, GPC; 5630, 1H NMR) was carried out in neat water during 24 h reaction time, over a variety of commercial (TiO2, Nb2O5, Al2O3, Dowex) and home-made (ZnO, CeO2, boehmite) solid catalysts (Scheme 1). In line with previous observations,30 blank experiments in the absence of catalyst showed the reaction to occur in 100% and in 12% LA yield at 160 and 130 °C, respectively. Within the same timeframe, use of zinc oxide catalyst resulted in significantly higher LA yields at low temperatures (Fig. 2): 100% yield at 130 °C and 54% yield at 110 °C (Table 1). Compared to catalyst-free runs, use of the other catalysts did not improve PLA hydrolysis, with lower LA yields observed over purely acidic solids (Nb2O5, Dowex) (Table S1, Fig. S6†). Irrespective of the catalyst, no oligomers or products other than LA were detected by 1H NMR, HPLC, GC-Ms and GPC in the aqueous phase fraction of the reaction mixture recovered at the end of the catalytic runs, unless for very ineffective depolymerisations (i.e. ≤12% PLA conversion), for which small amounts of lactate dimer were observed (see ESI†).31 This finding can be explained in terms of high hydrolysis rate of water-soluble LA oligomers.32 LA oligomers were reported to be water-soluble for molar mass below 1000 g mol−1.33 Whenever PLA conversion was incomplete, a solid residue could be recovered showing to consist in a distribution of lower molecular weight PLA polymers (typically Mn ∼ 7000 by GPC and ∼3000 by 1H NMR, Table S1†). We then decided to focus on the hydrolysis of PLA over ZnO catalyst. The reaction was examined in detail at 130 °C, showing the process to be viable even upon decreasing the amount of catalyst used or the reaction time (Fig. 3, Table S2†). A 93% LA yield was obtained after 24 h upon lowering by 60% the catalyst amount, and a 98% LA yield was obtained after 18 h, using a fixed catalyst weight. The LA yield could be invariably brought to 100% by appropriate selection of the reaction conditions. After use in the hydrolysis process, the solid ZnO catalyst could be easily recovered by centrifugation and reused in further catalytic experiments under identical conditions, showing a slight drop (ca. 10%), but a pretty constant activity, after the first cycle (Fig. S7†). The aqueous solutions recovered after each cycle were analysed by ICP-OES, indicating a mean 23% wt Zn leaching in solution over three cycles. The amount of soluble Zn species leached in the catalytic solution was easily reduced by more than 99% via filtration over ion-exchange resin (see ESI†). The powder X-ray diffraction pattern (PXRD) of the solid catalyst recovered after use was identical to that of the original ZnO material (Fig. S9†). No catalytic activity was shown by the aqueous solution recovered after catalysis, within the experimental errors (ESI†): this rules out the contribution of homogeneous-phase active species leached in solution to the catalytic performance of the system.34 The above results indicate the ZnO catalyst to be heterogeneous under the hydrolytic depolymerisation conditions adopted. Activity decrease upon recycling can be tentatively attributed to both catalyst loss due to leaching and to sample manipulation. Additional experiments were performed to test the catalyst stability under operating conditions at full substrate conversion, i.e. in the presence of an equivalent amount of LA, confirming that ca. 80% wt solid ZnO could be recovered intact, whereas ICP data of the aqueous phase were compatible with the portion of Zn leached in solution (ESI†). On the basis of the experimental data and the known literature, we can hypothesize that the interaction between amphoteric ZnO and the incipient LA formed upon hydrolysis is responsible for the formation of soluble, yet catalytically inactive, zinc species.35 Experiments performed using PLA samples with higher molecular weights showed the hydrolysis reaction not to be significantly affected by the PLA chain length, which still resulted in 100% LA yield under the usual reaction conditions (Table 1). This suggests the hydrolytic depolymerisation of PLA to occur via an ester linkage random-scission process, as previously reported for homogenous catalysts.36
100, GPC; 5630, 1H NMR) was carried out in neat water during 24 h reaction time, over a variety of commercial (TiO2, Nb2O5, Al2O3, Dowex) and home-made (ZnO, CeO2, boehmite) solid catalysts (Scheme 1). In line with previous observations,30 blank experiments in the absence of catalyst showed the reaction to occur in 100% and in 12% LA yield at 160 and 130 °C, respectively. Within the same timeframe, use of zinc oxide catalyst resulted in significantly higher LA yields at low temperatures (Fig. 2): 100% yield at 130 °C and 54% yield at 110 °C (Table 1). Compared to catalyst-free runs, use of the other catalysts did not improve PLA hydrolysis, with lower LA yields observed over purely acidic solids (Nb2O5, Dowex) (Table S1, Fig. S6†). Irrespective of the catalyst, no oligomers or products other than LA were detected by 1H NMR, HPLC, GC-Ms and GPC in the aqueous phase fraction of the reaction mixture recovered at the end of the catalytic runs, unless for very ineffective depolymerisations (i.e. ≤12% PLA conversion), for which small amounts of lactate dimer were observed (see ESI†).31 This finding can be explained in terms of high hydrolysis rate of water-soluble LA oligomers.32 LA oligomers were reported to be water-soluble for molar mass below 1000 g mol−1.33 Whenever PLA conversion was incomplete, a solid residue could be recovered showing to consist in a distribution of lower molecular weight PLA polymers (typically Mn ∼ 7000 by GPC and ∼3000 by 1H NMR, Table S1†). We then decided to focus on the hydrolysis of PLA over ZnO catalyst. The reaction was examined in detail at 130 °C, showing the process to be viable even upon decreasing the amount of catalyst used or the reaction time (Fig. 3, Table S2†). A 93% LA yield was obtained after 24 h upon lowering by 60% the catalyst amount, and a 98% LA yield was obtained after 18 h, using a fixed catalyst weight. The LA yield could be invariably brought to 100% by appropriate selection of the reaction conditions. After use in the hydrolysis process, the solid ZnO catalyst could be easily recovered by centrifugation and reused in further catalytic experiments under identical conditions, showing a slight drop (ca. 10%), but a pretty constant activity, after the first cycle (Fig. S7†). The aqueous solutions recovered after each cycle were analysed by ICP-OES, indicating a mean 23% wt Zn leaching in solution over three cycles. The amount of soluble Zn species leached in the catalytic solution was easily reduced by more than 99% via filtration over ion-exchange resin (see ESI†). The powder X-ray diffraction pattern (PXRD) of the solid catalyst recovered after use was identical to that of the original ZnO material (Fig. S9†). No catalytic activity was shown by the aqueous solution recovered after catalysis, within the experimental errors (ESI†): this rules out the contribution of homogeneous-phase active species leached in solution to the catalytic performance of the system.34 The above results indicate the ZnO catalyst to be heterogeneous under the hydrolytic depolymerisation conditions adopted. Activity decrease upon recycling can be tentatively attributed to both catalyst loss due to leaching and to sample manipulation. Additional experiments were performed to test the catalyst stability under operating conditions at full substrate conversion, i.e. in the presence of an equivalent amount of LA, confirming that ca. 80% wt solid ZnO could be recovered intact, whereas ICP data of the aqueous phase were compatible with the portion of Zn leached in solution (ESI†). On the basis of the experimental data and the known literature, we can hypothesize that the interaction between amphoteric ZnO and the incipient LA formed upon hydrolysis is responsible for the formation of soluble, yet catalytically inactive, zinc species.35 Experiments performed using PLA samples with higher molecular weights showed the hydrolysis reaction not to be significantly affected by the PLA chain length, which still resulted in 100% LA yield under the usual reaction conditions (Table 1). This suggests the hydrolytic depolymerisation of PLA to occur via an ester linkage random-scission process, as previously reported for homogenous catalysts.36
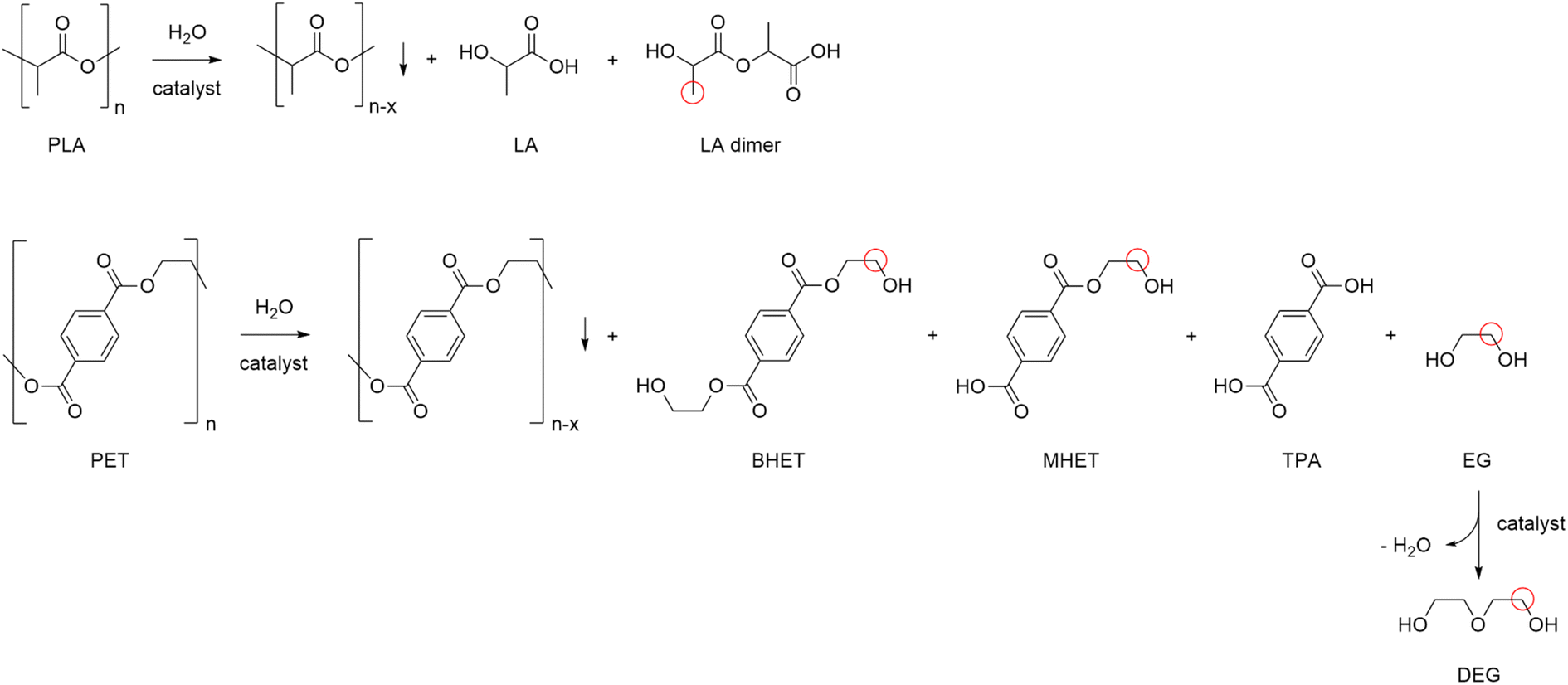 | ||
| Scheme 1 Schematic representation of the hydrolysis reaction of PLA (top) and PET (bottom) with products detected. Red circles indicate methyl and methylene proton groups used for evaluation of process selectivity in the aqueous phase via1H NMR. | ||
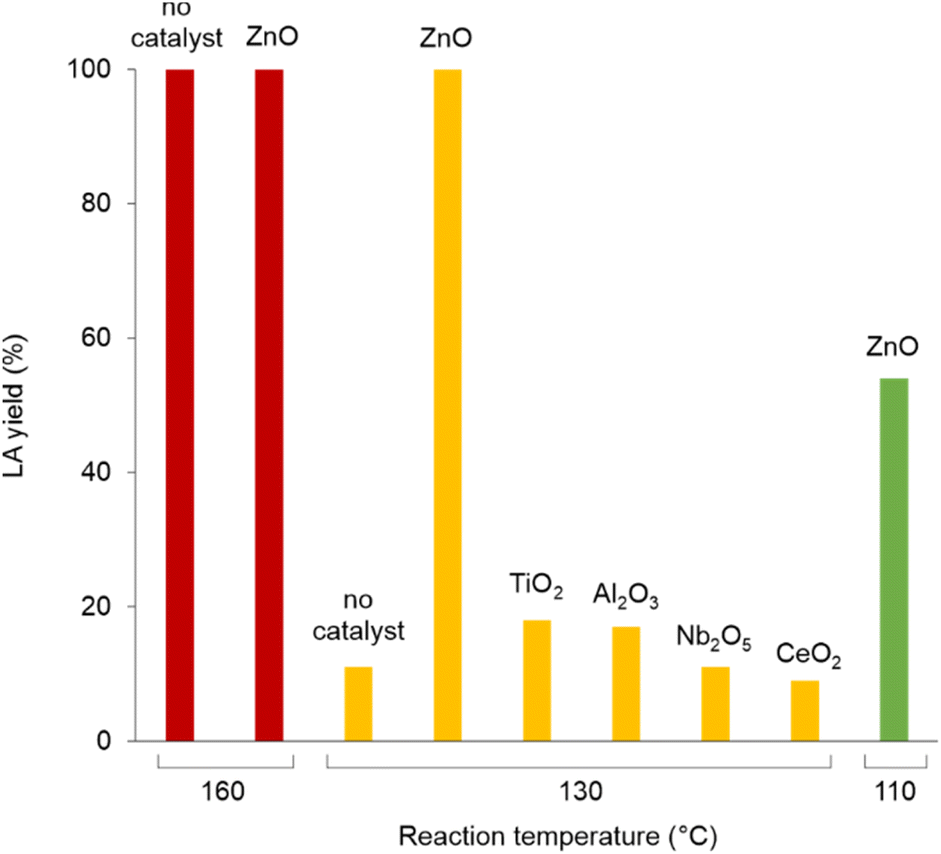 | ||
| Fig. 2 Selected data for the hydrolysis reaction of PLA over solid catalysts. Reaction conditions: 20 mg PLA, 10 mL water, 52 mg catalyst, 24 h reaction time. Data from HPLC analysis, based on number of PLA repetition units. | ||
| Catalyst | PLA Mnb | Temperaturec (°C) | PLA conversiond (%) | LA yielde (%) |
|---|---|---|---|---|
| a Reaction conditions: 10 mL H2O, 20 mg PLA (0.028 M based on PLA repetition units), 52 mg catalyst, reaction time 24 h. b From 1H NMR. c Reaction temperature. d PLA conversion, based on PLA mass loss. e Data from HPLC analysis, calculated on the basis of repetition units in PLA. Selectivity 100%. | ||||
| None | 5630 | 130 | 12 | 12 |
| ZnO | 5630 | 130 | 100 | 100 |
| ZnO | 5630 | 110 | 54 | 54 |
| ZnO | 10761 |
130 | 100 | 100 |
| ZnO | 30963 |
130 | 100 | 100 |
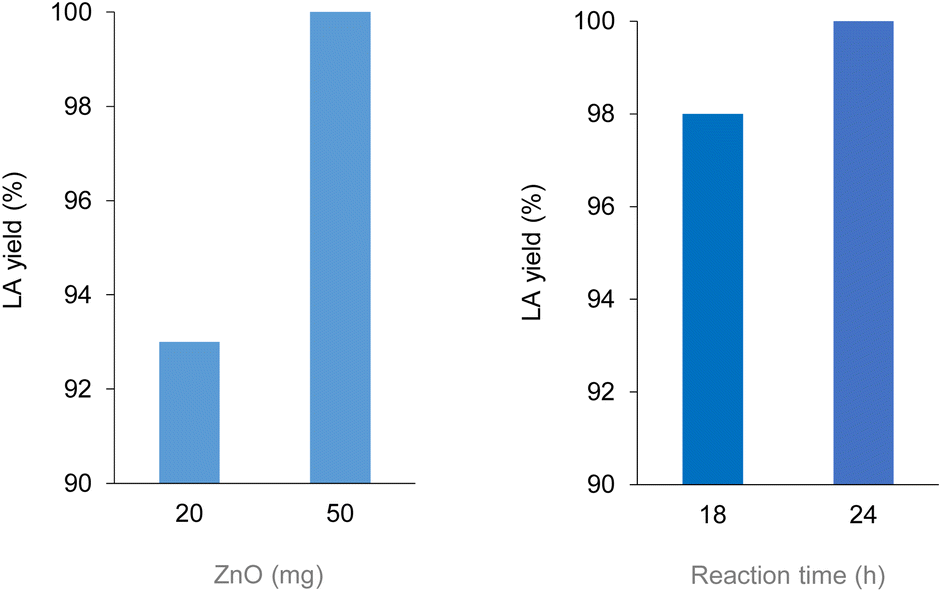 | ||
| Fig. 3 LA yield for the hydrolysis reaction of PLA over ZnO catalyst at 130 °C (20 mg PLA, 10 mL water). (Left) Effect of catalyst amount at fixed reaction time 24 h. (Right) Effect of reaction time at fixed catalyst amount 52 mg. Data from HPLC analysis, based on number of PLA repetition units. | ||
The above findings demonstrate that, despite PLA being insoluble in water under our experimental conditions,37 the use of a heterogeneous ZnO catalyst may effectively increase the hydrolysis reaction rate of PLA, so as to result in the complete chain cleavage to LA within acceptable reaction times, and even at temperatures significantly below the melting point of PLA (149–154 °C for Mn 5630).38 Compared to the catalyst-free system, use of ZnO provides an eight times higher PLA conversion within the same time frame at 130 °C, while it allows to achieve full PLA conversion at 30 °C lower operating temperature over the same reaction time (Table S1†), i.e. with significant energy consumption saving. Reusability of the catalyst provides additional benefits in terms of productivity/cost ratio of the overall process, where a fixed catalyst cost of 0.8 € per g is estimated.¶ The ZnO catalyst, which was obtained by calcination of the hydroxy-carbonate precursor at 400 °C, was characterised by a combination of solid-state techniques. The PXRD of the catalyst (Fig. S1 and S2†) indicated a hexagonal wurtzite phase (JCPDS card no. 36-1451).39 The crystallite size of the phase was of 28.4 nm, estimated by means of the Scherrer method, and based on the (101) Bragg reflex centred at 36.28° (2Θ). A combined SEM/HRTEM analysis showed the catalyst to consist in 100 ÷ 200 μm aggregates of a narrow size distribution of ca. 25 nm ZnO particles (Fig. 4 and S3†). The d-spacing of the lattice fringes is 0.26 nm (Fig. S4†), which agrees with the (002) crystallographic plane of the hexagonal structure observed by PXRD. The nitrogen isotherms (type IV, H3 hysteresis loop) were associated to mesopores with an average pore diameters 11.7 nm and a 7.1 m2 per g BET surface area (BJH desorption cumulative pore volume 0.024 cm3 g−1) (Fig. S5†). The role of the various solid catalysts herein investigated is difficult to ascertain, due to their very different morphologies, structures and acidic properties (type, strength, density). However, a detrimental effect of Brønsted acid-rich catalyst can be speculated.40
 | ||
| Fig. 4 Typical HRTEM image of ZnO catalyst after sonication. | ||
Catalytic methods for the molecular recycling of PLA to reusable monomers were described in the past.41 Several reports exist on the hydrolysis of PLA to LA by chemical catalysts.42 As for other polyesters, the inherent insolubility of PLA in water necessitates high temperatures to increase the water diffusion rate, and it is usually achieved using soluble catalysts to access the ester linkages in the bulk polymer, which makes the process economically unattractive.43 To the best of our knowledge, no heterogeneous catalysts have been previously reported for the complete hydrolysis of PLA to LA in neat water. On the industrial scale, the LOOPLA process provides lactate esters, or the LA mixtures thereof, via PLA depolymerisation with ethyl lactate at 140 °C, or with an excess of NaOH at 90 °C.44 NatureWorks recycles PLA wastes into reusable LA feeds through PLA hydrolysis mediated by nitric acid.45 Other patents describe the complete hydrolysis or alcoholysis of PLA, however using strong acids or bases (e.g. H2SO4, Ca(OH)2), soluble metal catalysts (e.g. FeCl3), or harsh reaction conditions.46 In a recent paper, the depolymerisation of PLA into reusable lactide and acrylic acid was reported using ionic liquids and an acidic co-catalyst.47
Having established ZnO to be an effective heterogeneous catalyst for the hydrolysis reaction of PLA, we then investigated the hydrolysis of a more complex co-polymer, namely polyethylene terephthalate (PET). PET is one of the most common synthetic organic polymers. The world production of PET is around 72 Mt per year (2019), which represents about 20% of all manufactured polymers.48 PET is a thermoplastic widely used in the food packaging (bottles, containers, ca. 24 Mt per year) and textiles sectors (clothing, furniture, ca. 48 Mt per year), with other minor technological applications (e.g. photovoltaic, electronic, adhesives, 3D printing).49 PET is not practically biodegradable.50 In 2019, 56 Mt of PET waste were generated, of which around 10 Mt were mechanically recycled, 14 Mt were incinerated and 30 Mt were landfilled or dispersed.51 Processes for the chemical recycling of PET have not yet reached commercial maturity,52 which justifies for methods of PET depolymerisation being intensively studied.53
Analogously to PLA, we accomplished the hydrolysis of PET in neat water over solid catalysts at significantly lower temperatures compared to catalyst-free experiment, with best performance observed for ZnO (Fig. 5 and Table S4†). The complete hydrolytic depolymerisation of PET provides equimolecular amounts of ethylene glycol (EG) and terephthalic acid (TPA), the latter being scarcely soluble in water at room temperature.54 For incomplete hydrolyses, besides EG and TPA, in the aqueous phase fraction of the mixture recovered at the end of the catalytic reactions, we detected variable quantities of sparingly water-soluble, partially hydrolysed monomers, namely bis(2-hydroxyethyl)terephthalate (BHET) and mono(2-hydroxyethyl)terephthalate (MHET), as well as the self-condensation product diethylene glycol (DEG) (Scheme 1). The amounts of the products obtained affects the selectivity of the hydrolysis process. Despite the depolymerisation of PET over ZnO was effective even at 150 °C, we explored in detail the reaction at 180 °C, because of the higher selectivity. After 24 h reaction time, the selectivity observed to the complete hydrolysis products was 98% and 94% at 180 °C and 165 °C, respectively, where a 4% molar amount of MHET was detected in the latter case (Table S5†). Formation of the partially hydrolysed compounds decreases with increasing reaction temperature, whereas a slight increase in DEG amount is observed. A conversion–selectivity diagram is reported in Fig. 6. A 98% EG yield was obtained at full PET conversion at 180 °C (Table 2), which is well below the melting point of PET. Compared to the catalyst-free experiment, a seven-fold higher EG yield was observed under identical reaction conditions (14%, Table 2, entry 1), whereas complete depolymerisation was achieved at ca. 60 °C lower operating temperature.55 No significant amounts of other products were detected in the aqueous phase by 1H NMR, HPLC, GC-Ms and GPC analysis. A section of the 1H COSY NMR spectrum recorded on the aqueous fraction recovered after catalysis at 165 °C is reported in Fig. 7. The effect of the reaction time and the catalyst amount used was investigated at fixed reaction temperature, showing no significant change of selectivity in both instances (Fig. 8 and Table S5†), where comparable depolymerisation yields were obtained even upon 50% reduction of the catalyst weight. As in the case of PLA hydrolysis, no catalytic activity was shown by the aqueous solution recovered after catalysis, which indicates the catalyst to be heterogeneous (ESI, section 3.2.2†), whereas, irrespective of the conditions, the amount of zinc leached in solution was below 1% of the amount used. At the end of each catalytic run, a white solid could be recovered by centrifugation of the reaction mixture, which contains the zinc species, the undissolved TPA formed and the residual insoluble polymer, if any. In this latter case, GPC data indicated the presence of CHCl3-soluble lower molecular weight PET oligomers. The solid recovered after PET hydrolysis was analysed, tested for catalyst reuse and for TPA yield. Recycling experiments without intermediate TPA separation resulted in a catalytic activity decrease of about 10% in each cycle (Fig. S11†). PXRD analysis of the solid revealed the incremental formation of a Znx(OH)y(TPA)z MOF-like heterostructure,56 which was confirmed by ZnO stability tests under PET catalytic hydrolysis conditions in the presence of an equivalent amount of TPA (ESI, Fig. S12†). Hydrolytic depolymerisation experiments using preformed ZnO–TPA MOF showed this species to have a lower catalytic activity than the original ZnO, and in line with that observed in recycling experiments (ESI†). On the above basis, we can assume that, in the case of PET hydrolysis and under our reaction conditions, the interaction between ZnO and the incipient TPA produced by depolymerisation leads to an insoluble, less active, MOF-like catalyst, which is responsible for the catalytic activity drop observed upon recycling. Noticeably, a mild basic treatment of the insoluble ZnO–MOF/TPA mixture obtained upon PET hydrolysis allowed to restore the catalytic activity, with concurrent TPA separation. Thus, addition of a stoichiometric amount of diluted NaOH to the solid recovered after the first catalytic cycle, followed by filtration, gave a solid Zn catalyst with almost the original catalytic activity (Fig. S11†), and a solution from which 99% purity TPA (by PXRD, NMR, IR and ICP analysis, Fig. S15–S17†) could be isolated after neutralisation in a yield comparable to that of EG (by gravimetry, ESI, Table S5†). Alternatively, to avoid any acid/base treatment, the excess of TPA accumulated after several catalytic cycles could be separated by sublimation of the overall solid recovered. After three catalytic cycles, this procedure gave 70 mg of 100% purity TPA on one hand (i.e. 27% yield based on the overall amount of TPA formed, including that part of the MOF generated), and the reusable ZnO–MOF catalyst on the other hand (ESI†). Some solid TPA was lost upon manipulation of the lab-scale quantities in this case.
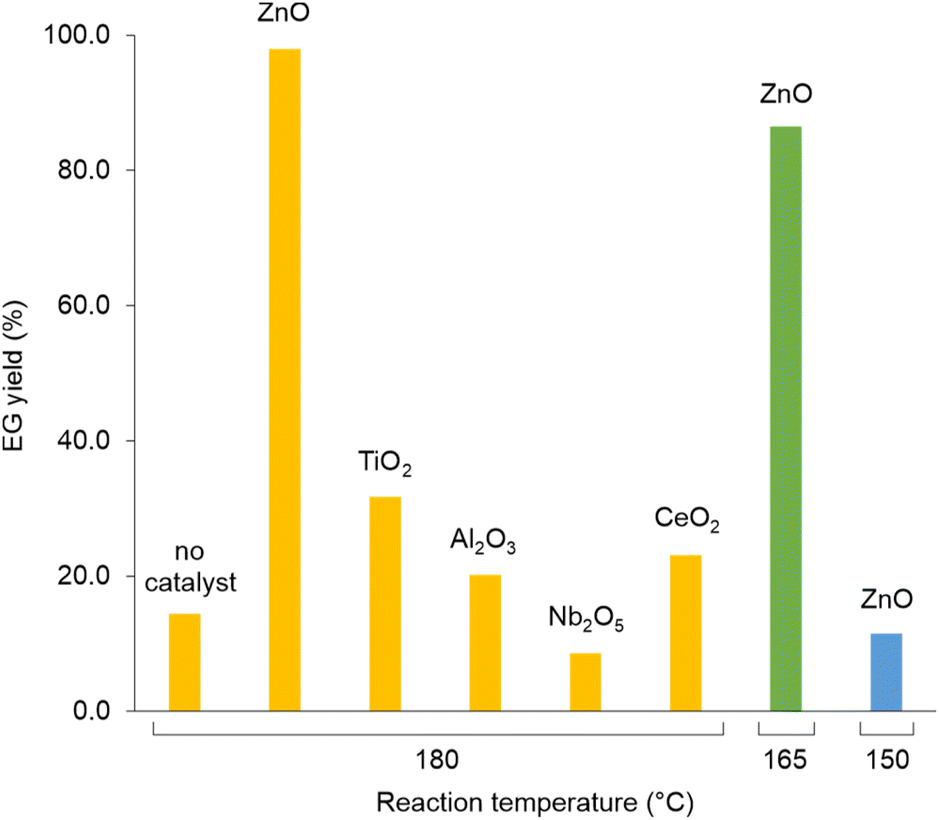 | ||
| Fig. 5 Selected data for the hydrolysis reaction of PET over heterogeneous catalysts. Reaction conditions: 100 mg PET, 15 mL water, 100 mg catalyst, 24 h reaction time. Data from HPLC analysis, based on number of PET repetition units. | ||
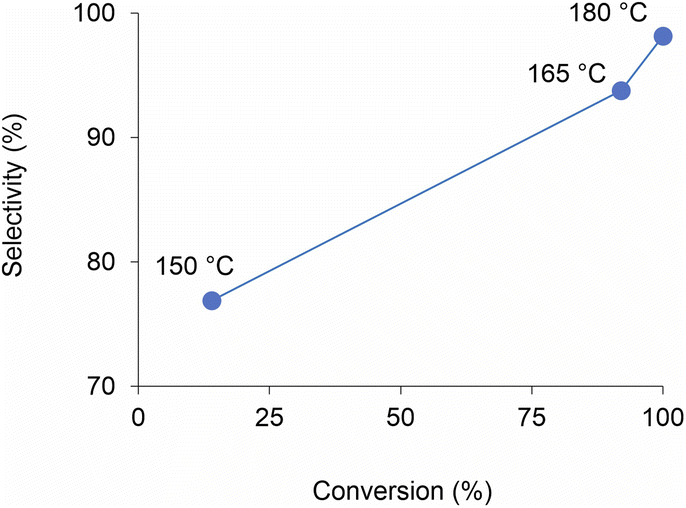 | ||
| Fig. 6 Diagram of PET conversion/selectivity to the complete hydrolysis products, as a function of reaction temperature, over ZnO catalyst. Aqueous phase. 24 h reaction time. | ||
| Catalyst | Temperatureb (°C) | PET conversionc (%) | EG yieldd (%) | Selectivitye (%) |
|---|---|---|---|---|
| a Reaction conditions: 15 mL H2O, 100 mg PET (0.035 M based on PET repetition units), 100 mg catalyst, reaction time 24 h. b Reaction temperature. c PET conversion, based on PET mass loss. d Aqueous phase. Data from HPLC analysis, calculated on the basis of repetition units in PET. e Aqueous phase. Selectivity to the complete hydrolysis products based on 1H NMR analysis. | ||||
| None | 180 | 15 | 14 | 96 |
| ZnO | 180 | 100 | 98 | 98 |
| ZnO | 165 | 92 | 86 | 94 |
| ZnO | 150 | 14 | 11 | 77 |
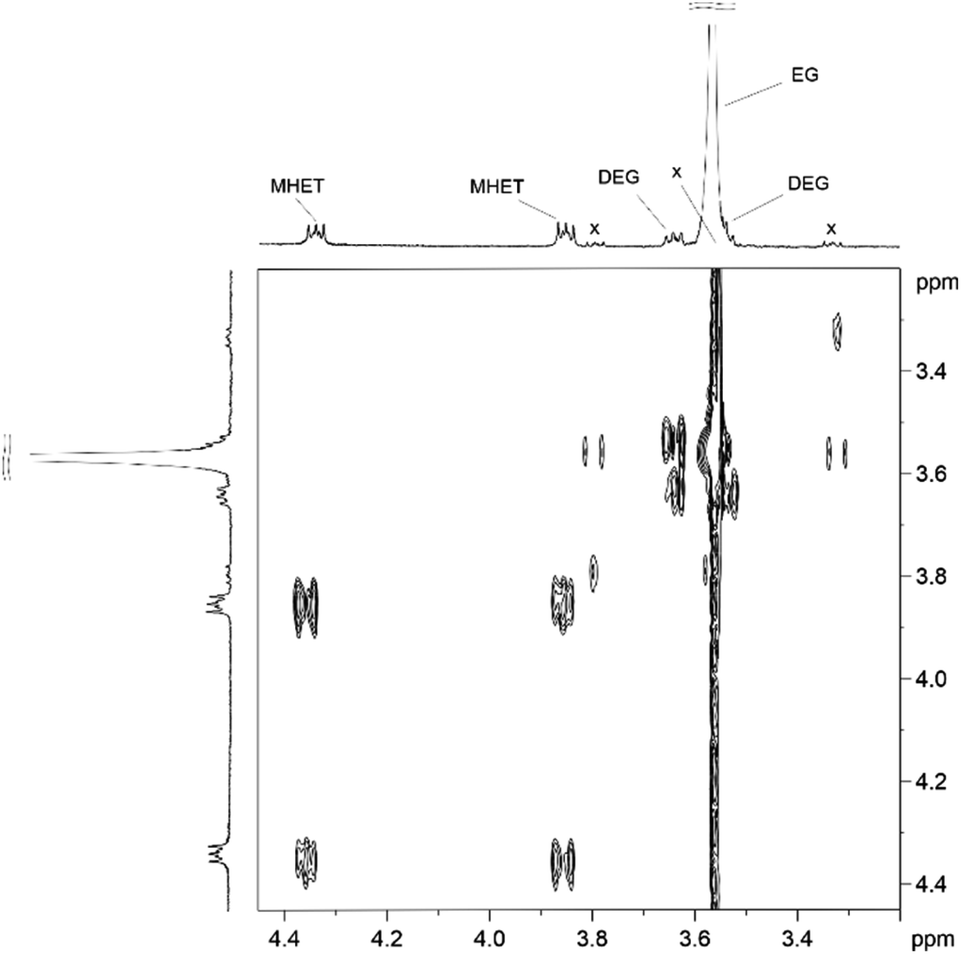 | ||
| Fig. 7 Section of the 1H COSY NMR spectrum in the aliphatic region (300.13 MHz, 296 K, D2O coaxial insert) of the aqueous phase mixture recovered at the end of catalytic hydrolysis reaction of PET over ZnO (165 °C, 24 h). x = unidentified –CH2CH2– containing product. | ||
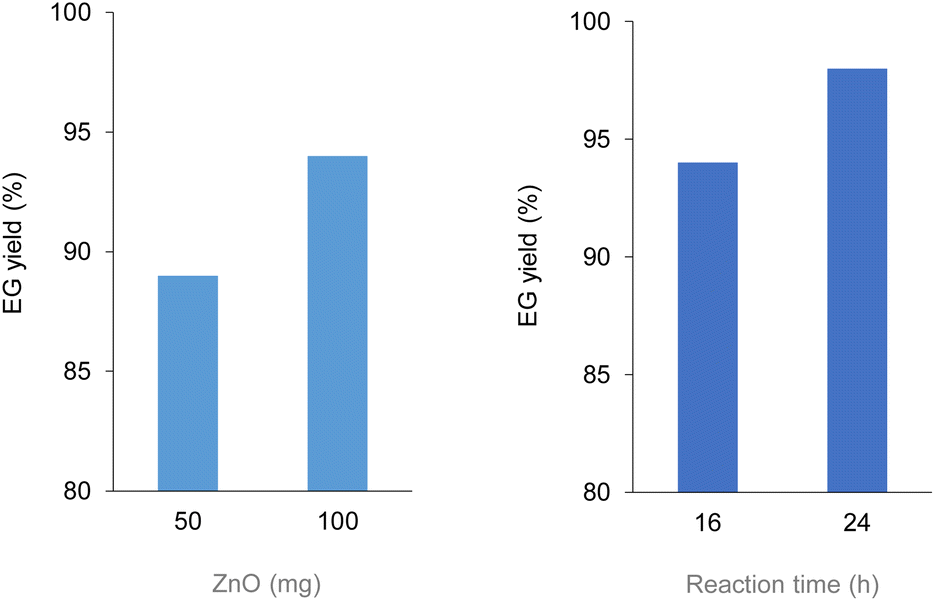 | ||
| Fig. 8 EG yield for the hydrolysis reaction of PET over ZnO catalyst at 180 °C (100 mg PET, 15 mL water). (Left) Effect of catalyst amount at fixed reaction time 16 h. (Right) Effect of reaction time at fixed catalyst amount 100 mg. Data from HPLC analysis, based on number of PET repetition units. | ||
TPA is a valuable chemical produced in a 40 Mt per year scale, and almost entirely employed in the manufacturing of PET and polybutylene terephthalate.57 The industrial synthesis of TPA is based on the Amoco process, which involves the use of petroleum-derived p-xylene as substrate, cobalt-manganese catalysts, bromide additives, acetic acid as solvent and harsh, corrosive reaction conditions.58 Purification, separation and quantification is a common issue for TPA obtained by chemical recycling of PET, mainly due to its poor solubility in water and in most common organic solvents, and it is usually achieved by an intensive, strong basic/acidic treatment.59
Several reviews describe the methods reported for the depolymerisation of PET, particularly via hydrolysis.60,61 Most of these require temperatures above 200 °C, concentrated alkali (e.g. NaOH) or acidic solutions (e.g. H2SO4), which results in corrosive reaction mixtures, considerable amounts of liquid and solid wastes and demanding purification procedures (particularly for EG), thus ending up in very expensive processes.62,63 Use of soluble transition metal salts was described for the catalytic hydrolysis of PET. Good depolymerisation yields were obtained using a 35:1 (w/w) excess of 70% wt aqueous ZnCl2 at 180 °C,64 and 1.5% wt ZnAc2 at 240 °C,65 respectively. More recently, a process for the hydrolysis of PET was claimed over solid acid zeolite catalysts under microwave heating.66 The NextChem company operates a demonstration plant for the depolymerisation of PET based on microwave-assisted alkaline hydrolysis (NaOH, KOH, or LiOH), using a technology co-licensed with the Swiss start-up Gr3n.67,68 A demonstration facility for the bio-catalytic hydrolysis of PET, showing good depolymerisation rate using an engineered enzyme, was started up by Carbios.69
Other depolymerisation processes of post-consumer PET, other than hydrolysis, are being commercialised. Although usually faster than hydrolysis, they necessitate (soluble) catalysts and organic solvents to give re-polymerisable phthalate esters. Scale-up of pilot plants have been announced either for methanolysis (by Eastman Chemical, Loop Industries, Renu) and glycolysis (Jeplan, Ioniqa, Garbo, Cure Technology, PerPETual, IBM) reactions. Comprehensive surveys can be found in the literature.7b,52a,70
The properties and the catalytic applications of zinc oxides were reviewed in the literature.71 Zinc oxide is categorised as non-toxic, although nano powders can be hazardous by inhalation. Soluble zinc salts are instead considered very toxic for aquatic life.72 ZnO was previously used in the catalytic glycolysis and methanolysis reaction of PET, to give BHET and dimethylterephthalate (DMT), respectively. In the former case, using ZnO supported onto 60 nm silica nanoparticles at 300 °C,73 and similarly for methanolysis, where a dispersion of 4 nm ZnO nanoparticles was used as pseudo-homogeneous catalyst at 170 °C.74 In the latter case, the catalyst could be reused with ca. 30% activity decrease after five runs, which was attributed to catalyst manipulation. A mechanism for the hydrolytic degradation of antimicrobial PLA/ZnO nanocomposites, in phosphate buffered or in NaOH solutions, has been proposed.75 Analogously to the above literature, the Zn centres were hypothesized to act as Lewis acid activators of the carbonyl ester bond, which then undergoes a nucleophile attack by water. In another report, it was suggested that the hydrolysis of PLA/ZnO nanocomposites occurs via anchoring of the acid end groups of PLA on a hydroxylated ZnO surface, followed by attack of the ester bond by hydroxyl anions.76 ZnO was previously used, in conjunction with ionic liquids, in the glycolysis reaction of polycarbonates in THF solution.77
Conclusions
In conclusion, we carried out a detailed investigation on the use of zinc oxide as catalyst for the hydrolysis reaction of polyesters. It was shown that ZnO is effective in achieving the complete depolymerisation of PLA and PET smoothly by a straightforward procedure. The system provides multiple benefits compared to conventional chemical recycling techniques, including:(1) Use of neat water as the only reagent.
(2) No need for additives, soluble promoters, co-solvents, strong acids/bases or homogenous catalysts.
(3) Mild reaction conditions (particularly, temperatures below the melting point of the polymer).
(4) >98% selectivity to the complete hydrolysis products at full polyester conversion.
(5) Recoverable and reusable heterogeneous catalytic species.
(6) Cheap and readily available catalyst.
(7) No management of excess of salt by-products.
(8) Obtainment of high added-value, reusable chemicals (LA, TPA), with no deed of organic solvents at any stage.
(9) Negligible leaching of Zn species in solution in the case of PET hydrolysis.
(10) Flexibility in terms of processable polyesters.
One limitation of the method is the decrease of catalytic activity upon catalyst recycling, which is attributable to the formation of Zn species having lower or no catalytic activity, and resulting from the interaction of ZnO with the nascent carboxylic acids. In the case of PLA hydrolysis, these species are water-soluble, thus leading to Zn loss in solution and to partial ZnO recovery. In the case of PET hydrolysis, insoluble MOF-like catalytically active compounds are formed, which are stable to high temperatures, but not to alkaline hydrolysis, which enables TPA recovery and catalytic activity regeneration.
The implementation of sustainable depolymerisation routes may promote the competitiveness of chemical recycling of plastics, particularly with respect to mechanical recycling, which is usually favoured.78,79 Advancements in the field should address both technological viability and low environmental impact.80 A solution to this may be provided by heterogeneous (metal) catalysis,81 which are still hampered by the lack of appropriate strategies.82,83 Overall, compared to conventional (non-catalysed) processes, the system herein examined contributes to sustainability mostly in terms of higher productivity, reduced energy inputs, minimisation of waste, no need of soluble promoters and, in the case of PET, enhanced selectivity. Elucidation of catalyst deactivation mechanism also contributes to the identification of factors affecting catalyst stability, hence to the design of improved catalysts. We believe that the results herein reported may be helpful in the development of effective heterogeneous catalysts for the chemical recycling of plastics. The approach may support the ambitious circular economy targets for plastics established in Europe,84 by producing key building blocks from secondary raw materials, while reducing the emissions of greenhouse gas and dependence on fossil sources.
Author contributions
F. Liguori and C. Moreno-Marrodán: investigation, formal analysis and validation. W. Oberhauser and E. Passaglia: investigation and formal analysis. P. Barbaro: supervision, and writing – original draft. All authors assisted in the conceptualization and in the critical review of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Thanks are due to Carlo Bartoli (ICCOM-CNR) for reactor design, Fabio Migliacci (ICCOM-CNR) for technical assistance, Dr Fulvio Ratto (IFAC-CNR) for samples lyophilisation, Centro Microscopie Elettroniche – CNR, Firenze, for HRTEM measurements and the Made in Italy – Circular and Sustainable (MICS) Extended Partnership funded by the European Union Next-Generation EU (Piano Nazionale di Ripresa e Resilienza (PNRR) – Missione 4, Componente 2, Investimento 1.3 – D.D. 1551.11-10-2022, PE00000004) for financial support.Notes and references
-
(a) R. Lehner, C. Weder, A. Petri-Fink and B. Rothen-Rutishauser, Environ. Sci. Technol., 2019, 53, 1748–1765 CrossRef CAS PubMed
; (b) L. Peng, D. Fu, H. Qi, C. Q. Lan, H. Yu and C. Ge, Sci. Total Environ., 2020, 698, 134254 CrossRef CAS PubMed
-
Ellen MacArthur Foundation, The New Plastics Economy: Rethinking the Future of Plastics & Catalysing Action, 2017 Search PubMed
-
(a)
Ellen MacArthur Foundation, Reuse – Rethinking Packaging, 2019 Search PubMed
-
(a)
Scientific Foresight Unit, Towards a Circular Economy – Waste Management in the EU, European Parliamentary Research Service, Brussels, PE 581.913, 2017 Search PubMed
-
(a) T. Thiounn and R. C. Smith, J. Polym. Sci., 2020, 58, 1347–1364 CrossRef CAS
-
The European Chemicals Agency, Chemical Recycling of Polymeric Materials from Waste in the Circular Economy, Final report, 2021 Search PubMed
-
(a)
Zero Waste Europe, Understanding the Environmental Impacts of Chemical Recycling – Ten Concerns with Existing Life Cycle Assessments, 2020 Search PubMed
-
McKinsey & Company, How Plastics Waste Could Transform the Chemical Industry, 2018 Search PubMed
-
(a) T. Keijer, V. Bakker and J. C. Slootweg, Nat. Chem., 2019, 11, 190–195 CrossRef CAS PubMed
-
(a)
European Commission, A European Strategy for Plastics in a Circular Economy, Brussels, 2018 Search PubMed
- D. Cole-Hamilton, Chem.–Eur. J., 2020, 26, 1894–1899 CrossRef CAS PubMed
-
M. W. Ryberg, A. Laurent and M. Hauschild, Mapping of Global Plastics Value Chain and Plastics Losses to the Environment, United Nations Environment Programme, 2018 Search PubMed
-
Plastics Europe, Plastics – The Facts 2021, Brussels, 2021 Search PubMed
- R. Geyer, J. R. Jambeck and K. Lavender Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed
- J. Payne and M. D. Jones, ChemSusChem, 2021, 14, 4041–4070 CrossRef CAS PubMed
-
(a) A. C. Fernandes, Green Chem., 2021, 23, 7330–7360 RSC
-
(a)
K. Masutani and Y. Kimura, in Poly(Lactic Acid) Science and Technology: Processing, Properties, Additives and Applications, ed. A. Jiménez, M. Peltzer and R. Ruseckaite, RSC, Cambridge, 2015, ch. 1 Search PubMed
- K. J. Jem and B. Tan, Adv. Ind. Eng. Polym. Res., 2020, 3, 60–70 Search PubMed
- B. Laycock, M. Nikolić, J. M. Colwell, E. Gauthier, P. Halley, S. Bottle and G. George, Prog. Polym. Sci., 2017, 71, 144–189 CrossRef CAS
- M. S. Singhvi, S. S. Zinjarde and D. V. Gokhale, J. Appl. Microbiol., 2019, 127, 1612–1626 CrossRef CAS PubMed
- P. Sangwan and D. Y. Wu, Macromol. Biosci., 2008, 8, 304–315 CrossRef CAS PubMed
- H. Tsuji and K. Suzuyoshi, Polym. Degrad. Stab., 2002, 75, 347–355 CrossRef CAS
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC
- M. Dusselier, P. Van Wouwe, A. Dewaele, E. Makshina and B. F. Sels, Energy Environ. Sci., 2013, 6, 1415–1442 RSC
-
(a) A. Folino, A. Karageorgiou, P. S. Calabrò and D. Komilis, Sustainability, 2020, 12, 6030 CrossRef CAS
- E. Feghali, L. Tauk, P. Ortiz, K. Vanbroekhoven and W. Eevers, Polym. Degrad. Stab., 2020, 179, 109241 CrossRef CAS
-
(a) M. Hajighasemi, B. P. Nocek, A. Tchigvintsev, G. Brown, R. Flick, X. Xu, H. Cui, T. Hai, A. Joachimiak, P. N. Golyshin, A. Savchenko, E. A. Edwards and A. F. Yakunin, Biomacromolecules, 2016, 17, 2027–2039 CrossRef CAS PubMed
-
(a) S. H. Lee, I. Y. Kim and W. S. Song, Macromol. Res., 2014, 22, 657–663 CrossRef CAS
- P. McKeown and M. D. Jones, Sustainable Chem., 2020, 1, 1–23 CrossRef
- H. Tsuji, T. Saeki, T. Tsukegi, H. Daimon and K. Fujie, Polym. Degrad. Stab., 2008, 10, 1956–1963 CrossRef
- K. Liu, X. Huang, E. A. Pidko and E. J. M. Hensen, ChemCatChem, 2018, 10, 810–817 CrossRef CAS PubMed
-
(a) S. K. Saha and H. Tsuji, Polym. Degrad. Stab., 2006, 91, 1665–1673 CrossRef CAS
- A. Höglund, K. Odelius and A. C. Albertsson, ACS Appl. Mater. Interfaces, 2012, 5, 2788–2793 CrossRef PubMed
- Maitlis’s
catalyst leaching test, see: J. P. Collman, K. M. Kosydar, M. Bressan, W. Lamanna and T. Garrett, J. Am. Chem. Soc., 1984, 106, 2569–2579 CrossRef CAS
-
(a) Y. Zhang, Y. Qi, Y. Yin, P. Sun, A. Li, Q. Zhang and W. Jiang, ACS Sustainable Chem. Eng., 2020, 8, 2865–2873 CrossRef CAS
- C. Shih, Pharm. Res., 1995, 12, 2036–2040 CrossRef CAS PubMed
- S. Sato, D. Gondo, T. Wada, S. Kanehashi and K. Nagai, J. Appl. Polym. Sci., 2013, 129, 1607–1617 CrossRef CAS
- H. Tsuji, H. Daimon and K. Fujie, Biomacromolecules, 2003, 4, 835–840 CrossRef CAS PubMed
- V. Srivastava, D. Gusain and Y. C. Sharma, Ceram. Int., 2013, 39, 9803–9808 CrossRef CAS
- Studies are ongoing in our labs.
- For a review on chemical recycling and depolymerisation of PLA, see:
(a) E. Gabirondo, A. Sangroniz, A. Etxeberria, S. Torres-Giner and H. Sardon, Polym. Chem., 2020, 11, 4861–4874 RSC
- For a review on hydrolysis reaction of PLA, see:
(a)
H. Tsuji, in Poly(Lactic Acid): Synthesis, Structures, Properties, Processing, and Applications, ed. R. A. Auras, L. T. Lim, S. E. M. Selke and H. Tsuji, Wiley, Hoboken, 2010, ch. 21 Search PubMed
-
(a) E. Castro-Aguirre, F. Iñiguez-Franco, H. Samsudin, X. Fang and R. Auras, Adv. Drug Delivery Rev., 2016, 107, 333–366 CrossRef CAS PubMed
-
(a)
https://www.futerro.com/what-renewtm/end-life, accessed July 2022;
(b)
P. Coszach, J. C. Bogaert and J. Willocq, US Pat., 0142958A1, Galactic S.A., 2012 Search PubMed
-
(a) E. T. H. Vink, K. R. Rábago, D. A. Glassner, B. Springs, R. P. O'Connor, J. Kolstad and P. R. Gruber, Macromol. Biosci., 2004, 4, 551–564 CrossRef CAS PubMed
- M. Niaounakis, Eur. Polym. J., 2019, 114, 464–475 CrossRef CAS
- K. Janssens, W. Stuyck, K. Stiers, J. Wery, M. Smet and D. E. De Vos, RSC Sustainability, 2023, 1, 83–89 RSC
- M. Rabnawaz, I. Wyman, R. Auras and S. Cheng, Green Chem., 2017, 19, 4737–4753 RSC
-
Eunomia, PET Market in Europe State of Play, 2022 Search PubMed
- X. Qi, W. Yan, Z. Cao, M. Ding and Y. Yuan, Microorganisms, 2022, 10, 39–64 CrossRef CAS PubMed
- Data from DEMETO project, https://www.demeto.eu, accessed August 2022.
-
(a)
Textile Exchange, Preferred Fiber & Materials Market Report, 2021 Search PubMed
- V. Beghetto, R. Sole, C. Buranello, M. Al-Abkal and M. Facchin, Materials, 2021, 14, 4782–4806 CrossRef CAS PubMed
- Y. Takebayashi, K. Sue, S. Yoda, Y. Hakuta and T. Furuya, J. Chem. Eng. Data, 2012, 57, 1810–1816 CrossRef CAS
- V. Sinha, M. R. Patel and J. V. Patel, J. Polym. Environ., 2010, 18, 8–25 CrossRef CAS
-
(a) Y. Hirai, K. Furukawa, H. Sun, Y. Matsushima, K. Shito, A. Masuhara, R. Ono, Y. Shimbori, H. Shiroishi, M. Schuette White and T. Yoshida, Microsyst. Technol., 2018, 24, 699–708 CrossRef CAS
-
A. Fuessl, M. Yamamoto and A. Schneller, Opportunities in Bio-Based Building Blocks for Thermoplastic Polymers, in Reference Module in Materials Science and Materials Engineering, Elsevier, 2016 Search PubMed
- R. A. F. Tomás, J. C. M. Bordado and J. F. P. Gomes, Chem. Rev., 2013, 113, 7421–7469 CrossRef PubMed
-
(a) L. Liu, D. Zhang, L. An, H. Zhang and Y. Tian, J. Appl. Polym. Sci., 2005, 95, 719–723 CrossRef CAS
-
M. Han, in Recycling of Polyethylene Terephthalate Bottles, ed. T. Sabu, R. Ajay, K., Krishnan, V. K. Abitha and G. T. Martin, Elsevier, 2018, ch. 5 Search PubMed
- E. Barnard, J. Jonathan, R. Arias and W. Thielemans, Green Chem., 2021, 23, 3765–3789 RSC
- S. K. Das, S. K. Eshkalak, A. Chinnappan, R. Ghosh, W. A. D. M. Jayathilaka, C. Baskar and S. Ramakrishna, Mater. Circ. Econ., 2021, 3, 9–31 CrossRef
-
(a)
J. Pitat, V. Holcik and M. A. Bacak, GB822834, 1959
- Y. Wang, Y. Zhang, H. Song, Y. Wang, T. Deng and X. Hou, J. Cleaner Prod., 2019, 208, 1469–1475 CrossRef CAS
- Y. Liu, M. Wang and Z. Pan, J. Supercrit. Fluids, 2012, 62, 226–231 CrossRef CAS
- M. J. Kang, H. J. Yu, J. Jegal, H. S. Kim and H. G. Cha, Chem. Eng. J., 2020, 398, 125655 CrossRef CAS
- https://nextchem.it/news/nextchem-completes-italys-1deg-demonstration-plant-pet-and-textiles-polyester-chemical, accessed December 2022.
-
M. Parravicini, M. Crippa and M. V. Bertele, WO014650A1, 2013
- V. Tournier,
et al.
, Nature, 2020, 580, 216–219 CrossRef CAS PubMed
- H. Li,
et al.
, Green Chem., 2022, 24, 8899–9002 RSC
-
(a) A. Moezzi, A. M. McDonagh and M. B. Cortie, Chem. Eng. J., 2012, 185–186, 1–22 CrossRef CAS
- ECHA Substance information, Zinc oxide, https://echa.europa.eu/substance-information/-/substanceinfo/100.013.839, accessed December 2022 Search PubMed.
-
(a) M. Imran, K. G. Lee, Q. Imtiaz, B. K. Kim, M. Han, B. G. Cho and D. H. Kim, J. Nanosci. Nanotechnol., 2011, 11, 824–828 CrossRef CAS PubMed
- J. T. Du, Q. Sun, X. F. Zeng, D. Wang, J. X. Wang and J. F. Chen, Chem. Eng. Sci., 2020, 220, 115642 CrossRef CAS
-
(a) E. Lizundia, P. Mateos and J. L. Vilas, Mater. Sci. Eng., C, 2017, 75, 714–720 CrossRef CAS PubMed
- M. Qu, H. Tu, M. Amarante, Y. Q. Song and S. S. Zhu, J. Appl. Polym. Sci., 2014, 40287 Search PubMed
- F. Iannone, M. Casiello, A. Monopoli, P. Cotugno, M. C. Sportelli, R. A. Picca, N. Cioffi, M. M. Dell'Anna and A. Nacci, J. Mol. Catal. A: Chem., 2017, 426, 107–116 CrossRef CAS
- C. Chariyachotilert, S. E. Selke, R. A. Auras and S. Joshi, J. Plast. Film Sheeting, 2012, 28, 314–335 CrossRef
- V. Piemonte, S. Sabatini and F. Gironi, J. Polym. Environ., 2013, 21, 640–647 CrossRef CAS
-
Closed Loop Partners, Transitioning to a Circular System for Plastics, 2021 Search PubMed
- K. V. Khopade, S. H. Chikkali and N. Barsu, Cell Rep. Phys. Sci., 2023, 4, 101341 CrossRef CAS
- M. Chu, Y. Liu, X. Lou, Q. Zhang and J. Chen, ACS Catal., 2022, 12, 4659–4679 CrossRef CAS
- A. J. Martín, C. Mondelli, S. D. Jaydev and J. Pérez-Ramírez, Chem, 2021, 7, 1–47 Search PubMed
-
M. Hestin, T. Faninger and L. Milios, Increased EU Plastics Recycling Targets: Environmental, Economic and Social Impact Assessment, Plastic Recyclers Europe, 2015 Search PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, characterisation and catalysis data, products analysis, additional tables and figures. See DOI: https://doi.org/10.1039/d3su00089c |
| ‡ Chemical recycling can be defined as “A process in which the molecular structure of a synthetic polymer is modified by selective breakage of the chain linkages to achieve depolymerisation, through the action of chemical agents and, preferably, a catalyst, to give the original monomer units or other monomeric entities reusable in a synthetic process”. See: G. W. Coates and Y. D. Y. L. Getzler, Nat. Rev. Mater., 2020, 5, 501–516. |
| § Target 12.4: “By 2020, achieve the environmentally sound management of chemicals and all wastes throughout their life cycle, in accordance with agreed international frameworks, and significantly reduce their release to air, water and soil in order to minimize their adverse impacts on human health and the environment”. Target 12.5: “By 2030, substantially reduce waste generation through prevention, reduction, recycling and reuse”. https://sdgs.un.org/goals/goal12 |
| ¶ The estimated cost of the ZnO catalyst is 0.8 € per g, based on the synthetic method herein adopted (see ESI,† page 4 for commercial products specifications, amounts and procedure). |
| This journal is © The Royal Society of Chemistry 2023 |