
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceXanthate-supported photo-iniferter (XPI)-RAFT polymerization: facile and rapid access to complex macromolecules†
Anne-Catherine
Lehnen
 ab,
Johannes
Gurke
ab,
Alain M.
Bapolisi
a,
Martin
Reifarth
ab,
Marek
Bekir
c and
Matthias
Hartlieb
*ab
ab,
Johannes
Gurke
ab,
Alain M.
Bapolisi
a,
Martin
Reifarth
ab,
Marek
Bekir
c and
Matthias
Hartlieb
*ab
aUniversity of Potsdam, Institute of Chemistry, Karl-Liebknecht-Straße 24-25, D-14476, Potsdam, Germany. E-mail: mhartlieb@uni-potsdam.de
bFraunhofer Institute for Applied Polymer Research (IAP), Geiselbergstraße 69, D-14476, Potsdam, Germany
cUniversity of Potsdam, Institute of Physics and Astronomy, Karl-Liebknecht-Straße 24-25, D-14476, Potsdam, Germany
First published on 29th November 2022
Abstract
Xanthate-supported photo-iniferter (XPI)-reversible addition–fragmentation chain-transfer (RAFT) polymerization is introduced as a fast and versatile photo-polymerization strategy. Small amounts of xanthate are added to conventional RAFT polymerizations to act as a photo-iniferter under light irradiation. Radical exchange is facilitated by the main CTA ensuring control over the molecular weight distribution, while xanthate enables an efficient photo-(re)activation. The photo-active moiety is thus introduced into the polymer as an end group, which makes chain extension of the produced polymers possible directly by irradiation. This is in sharp contrast to conventional photo-initiators, or photo electron transfer (PET)-RAFT polymerizations, where radical generation depends on the added small molecules. In contrast to regular photo-iniferter-RAFT polymerization, photo-activation is decoupled from polymerization control, rendering XPI-RAFT an elegant tool for the fabrication of defined and complex macromolecules. The method is oxygen tolerant and robust and was used to perform screenings in a well-plate format, and it was even possible to produce multiblock copolymers in a coffee mug under open-to-air conditions. XPI-RAFT does not rely on highly specialized equipment and qualifies as a universal tool for the straightforward synthesis of complex macromolecules. The method is user-friendly and broadens the scope of what can be achieved with photo-polymerization techniques.
Introduction
The use of light to control polymerization reactions offers significant advantages compared to more conventional approaches (e.g. thermal activation).1 High spatial and temporal control as demonstrated in 3D-printing applications,2 or selective activation of certain initiators,3,4 as well as decoupling from the reaction temperature5 are among them. In particular, radical polymerization processes with their high tolerance for functional groups and reaction conditions benefit from photo-activation.1 Among reversible-deactivation radical polymerization (RDRP) methods, reversible addition–fragmentation chain-transfer (RAFT) polymerization is among the most frequently used techniques.6 RAFT polymerization is highly tolerant to various conditions and functionalities, and offers control over the polymerization of multiple monomer families.7 Among other techniques such as copper-mediated,8–11 or nitroxide-mediated polymerization,12,13 the RAFT process has been adapted to utilize light as an energy source in different ways.7,14,15 A common strategy is the use of photo-initiators as a radical source. However, general limitations of RAFT polymerization such as the restricted livingness and implementation of initiator derived end groups remain in place.16 To overcome these issues, Boyer and coworkers developed photo-electron/energy-transfer (PET)-RAFT polymerization.17 Here, a photo-catalyst is used, which harvests light and transfers energy or an electron to the chain transfer agent (CTA) controlling the RAFT polymerization.18,19 Subsequently, the CTA undergoes homolytic bond cleavage and produces a radical, which acts as an initiator for polymerization. However, it is also possible to induce photo-dissociation of CTAs directly by excitation of electronic transitions of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S bond, that is part of most CTAs.20 The following β-fragmentation results in a transient R-radical, which acts as an initiator for polymerization, as well as a persistent thiocarbonylthio radical. The latter species can deactivate growing chains in a reversible fashion, leading to an increased livingness of this method when compared to a conventional RAFT polymerization. This photo-iniferter (PI) process was first described by Otsu in 1982,21 and was later utilized in the framework of RAFT polymerization.20
S bond, that is part of most CTAs.20 The following β-fragmentation results in a transient R-radical, which acts as an initiator for polymerization, as well as a persistent thiocarbonylthio radical. The latter species can deactivate growing chains in a reversible fashion, leading to an increased livingness of this method when compared to a conventional RAFT polymerization. This photo-iniferter (PI) process was first described by Otsu in 1982,21 and was later utilized in the framework of RAFT polymerization.20
Depending on the photo-chemical properties of the CTA, PI-RAFT polymerization can be performed using UV22 or visible light.23,24 The method can be oxygen tolerant under certain conditions,25–27 and was used to produce ultra-high molecular weight polymers.28,29 Also, restrictions regarding the monomer order in block copolymers can be avoided.30,31 Despite these advantages, PI-RAFT polymerization has not yet gained significant traction in the polymer community.
Alongside side reactions of certain CTAs under irradiation,32 a major reason is likely a necessary trade-off between efficiency (in terms of conversion and reaction time) and control over the molecular weight distribution.
Xanthates are powerful photo-iniferters.28,33,34 They can be activated via excitation of the spin forbidden n–π* transition using UV-light, leading to β-fragmentation and rapid polymerization.28 However, chain transfer coefficients with more activated monomers (MAM)s are usually low, resulting in broad molecular weight distributions.34 And while Sumerlin and coworkers could show that for dimethylacrylamide,28,35 polymerizations are fast and controlled, for the control of MAMs in a broader sense, other classes of CTAs like dithioesters or trithiocarbonates are more suitable.36 However, they suffer from a comparably poor radical yield in PI-RAFT settings, rendering reaction times long and conversions not always high. In addition, degradation reactions of the thiocarbonylthio radical can diminish the livingness of the process during extended irradiation.37 And while certain combinations of monomer and CTA show impressive results,28 a general, easy-to-use methodology for efficient PI-RAFT polymerization is still lacking. We set out to overcome these restrictions and offer a user-friendly method with more universal access to well-defined polymers via light-induced RAFT polymerization within reasonable time frames, achieving high conversion, and without the use of photocatalysts. The solution is comparably simple: mixing xanthates with other CTAs that have a wider scope in terms of molecular weight control will combine the merits from both worlds. The group of Anastasaki has previously shown that a mixture of CTAs of different qualities enables control over the dispersity of the resulting macromolecules.38,39 Even with vastly different chain transfer coefficients, both CTAs are involved in chain transfer equilibria and the resulting molecular weight control is a product of both CTA's capabilities.40 However, the varying photo-activity of certain CTAs was to the best of our knowledge not yet considered in this context.
In our proposed method the xanthate is only used in relatively minor quantities, to not influence the overall dispersity in a significant manner, while still being able to boost radical production and enable fast and highly living polymerization reactions, producing polymers that are inherently photo-active and can be chain-extended without the necessity for additional catalysts, sensitizers or initiator molecules. In this scenario, the xanthate acts similarly to a photo-initiator, however with two important differences: (1) it will still take part in chain transfer equilibria40 and thus will be introduced as an end group in the polymer population; (2) the reversible deactivation provided by thiocarbonylthio radicals leads to an increased livingness,34 likely as a result of the persistent radical effect.41 Hence the xanthate can be added to an excess of a different CTA, and while the main CTA (CTA-2 from here on) maintains a narrow molecular weight distribution by efficient and fast chain transfer processes, the xanthate will fuel the polymerization, restrict termination by reversible deactivation, and create polymers that can be reactivated under irradiation. We refer to this process, in which a xanthate is mixed with a CTA of a different nature in a photo-initiated polymerization as xanthate-mediated photo-iniferter (XPI)-RAFT polymerization (Scheme 1). We will demonstrate that XPI-RAFT polymerization is a user-friendly and robust tool to broaden the scope of what can be achieved with photo-polymerization techniques.
 | ||
| Scheme 1 Illustration of the XPI-RAFT concept: a combination of two CTAs enables fast and controlled photopolymerization without additional additives. A coffee-theme is chosen as it will later be shown that the method is robust enough to produce multiblock copolymers in a coffee mug under open-to-air conditions. | ||
Results and discussion
To probe the proposed concept of XPI-RAFT polymerization, a closer look at the photo-activation of both CTAs is necessary. The direct photo-activation of CTAs is most efficient when their n–π* band is excited.42 Despite the spin forbidden nature of this transition, the quantum yield of the respective β-fragmentation seems to be significantly higher.23 The position of this band is strongly dependent on CTA substitution: while in trithiocarbonates, light absorption based on the n–π* transition is in the visible spectrum (∼450 nm), in xanthate-based CTAs, the n–π* band is located in the UV region (∼350 nm).On comparing the PI-RAFT polymerization of O-ethyl-S-(1-carboxy)methyl xanthate (Xan) with that of propanoic acid butyl trithiocarbonate (PABTC) (Fig. S1†) it becomes clear that the xanthate easily outperforms PABTC at all used wavelengths (Table S10†). This was even accomplished under visible light irradiation (450 nm) despite Xan possessing no noteworthy absorption capacity in this spectral region (Fig. S2†).
Hence Xan can be used to boost radical production when mixed with a second CTA (Fig. 1). During the reaction, mainly the xanthate is activated by light and fragments into an R-radical (transient), as well as a xanthate radical (persistent). The R-radical initiates the polymerization but also reacts with CTA-2 in a pre-RAFT equilibrium. CTA-2 distributes the radicals among chains (chain transfer between CTAs of equal nature is not depicted) and the persistent xanthate radical (or to an extent CTA-2 based radicals as well) eventually deactivates the growing chain. Thus, the final polymer possesses different combinations of end groups depending on the composition of the CTA mixture, as obvious form Fig. 1c (full spectra in Fig. S4†). Here, a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of PABTC and Xan, together with NAM as a monomer (DP 10) at a lamp power of 2 W (365 nm, 4.41 mW cm−2) was used as a model. The ratio of ω-end groups remains similar throughout the polymerization, and also after precipitation, as monitored by NMR spectroscopy. In electrospray ionization mass spectrometry (ESI-MS) three of the four possible combinations of end groups are visible (Fig. S5†). The fourth combination (R-group and Z-group from Xan) is likely not detectable due to its low prevalence (∼1% of chains in the case of ideal mixing based on statistical considerations).
:1 mixture of PABTC and Xan, together with NAM as a monomer (DP 10) at a lamp power of 2 W (365 nm, 4.41 mW cm−2) was used as a model. The ratio of ω-end groups remains similar throughout the polymerization, and also after precipitation, as monitored by NMR spectroscopy. In electrospray ionization mass spectrometry (ESI-MS) three of the four possible combinations of end groups are visible (Fig. S5†). The fourth combination (R-group and Z-group from Xan) is likely not detectable due to its low prevalence (∼1% of chains in the case of ideal mixing based on statistical considerations).
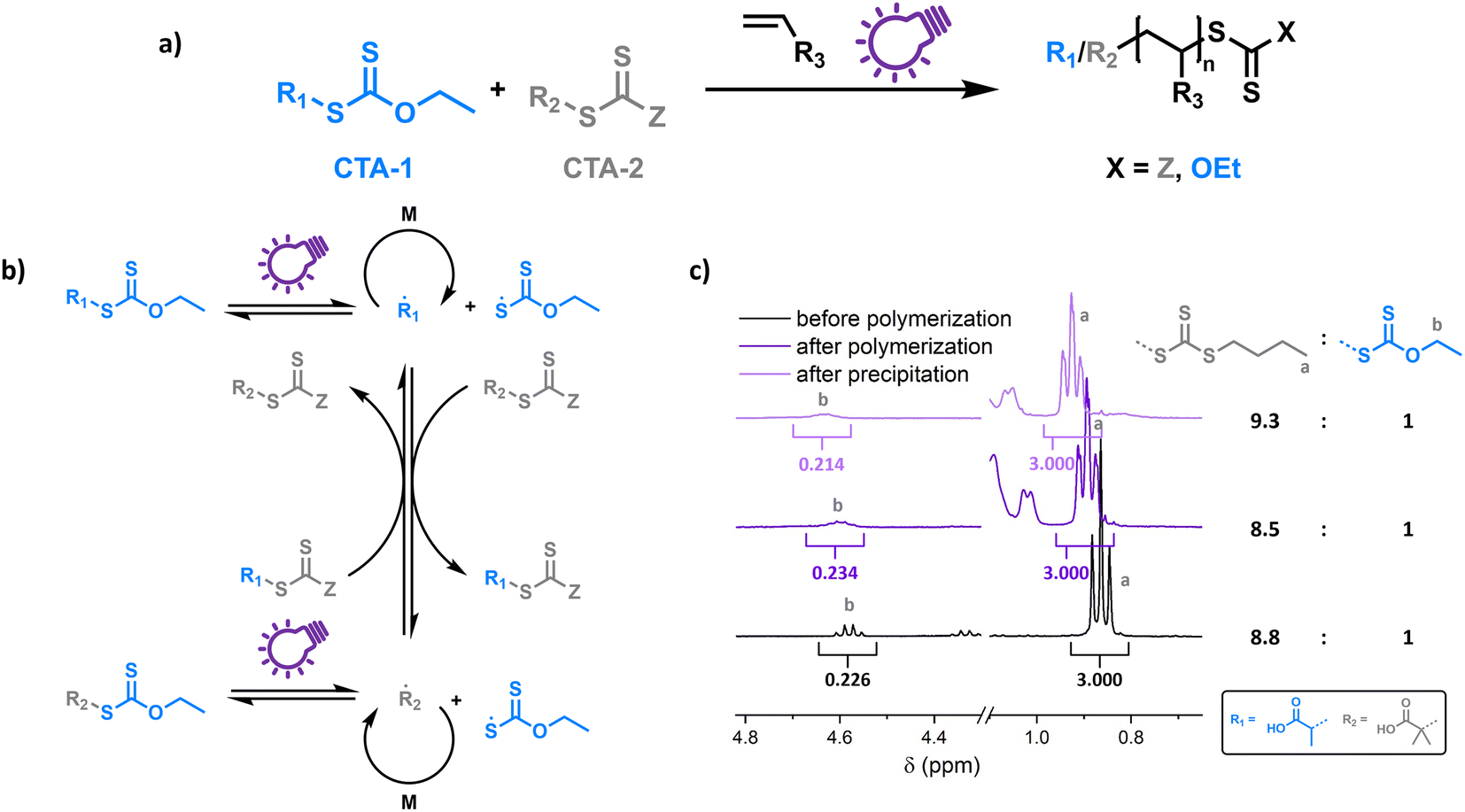 | ||
| Fig. 1 Schematic representation of XPI-RAFT polymerization (a), and the underlying transfer mechanism from xanthate to CTA-2 (chain transfer between CTAs of equal nature and photo-activation of CTA-2 is not pictured) (b), as well as end group distribution before and after polymerization of a PNAM10 using a 9:1 mixture of CTA-2 (BABTC) and Xan as monitored by 1H-NMR spectroscopy (c). | ||
This shows that both CTAs are involved in chain transfer equilibria and xanthate based thiocarbonylthio groups are introduced as terminal functionalities in the polymer population, which has an important consequence as the produced polymers are inherently photo-active and can be chain extended without the addition of catalysts, photo sensitizers or exogenous initiators (vide infra). This is in sharp contrast to PET-RAFT polymerization, where activation depends on the presence of a photocatalyst. While coupling of catalysts to polymer chains to create intrinsically photo-active polymers has been described,43–45 this strategy takes considerable effort in CTA or monomer design, whereas in XPI-RAFT polymerization a simple mix and match protocol is sufficient to achieve this result. Unlike in other PI-RAFT polymerization protocols, in the present study photo-activation (mediated by the xanthate) is decoupled from control over the molecular weight distribution (mediated by CTA-2), which is a significant improvement, as the CTA can be adjusted to the monomer without compromising photo-activation. The ratio between Xan and CTA-2 is essential for an optimal outcome of such an XPI-RAFT setting (Fig. S3†). While at low Xan contents, conversion is low, a prevalence of Xan will broaden molecular weight distributions due to the lack of efficient chain transfer. When screening with PABTC we found that 10–20% of Xan leads to optimal results: little to no observable broadening of the molecular weight distribution by using Xan paired with very high conversion within 2 h. For this process we used a standard laboratory UV-lamp (365 nm) with 2 W of power and a measured intensity of 4.41 mW cm−2 (from here on referred to as a 2 W lamp).
A closer look at the kinetics of the process using N-acryloyl morpholine (NAM) as a monomer and PABTC as CTA-2 (Fig. 2b) reveals the acceleration provided by xanthate. When only PABTC was used as a CTA, barely any conversion was achieved after 3 h (11%), while almost quantitative monomer consumption was accomplished with a 1:9 Xan/PABTC mixture in the same time frame and solvent. The rate of the polymerization depends on the type of solvent used, which might be linked to the increased polymerization rate of acrylamides in polar solvents and in particular water.46 It should be noted that the estimated optical penetration depth at this wavelength is still higher than the diameter of the vials (1 cm) used for these studies (Table S9†) and a relatively homogeneous activation can be assumed. Also, at higher concentrations up to bulk conditions, polymerization was possible where a slightly reduced control and conversion were observed without solvent (Fig. S6†).
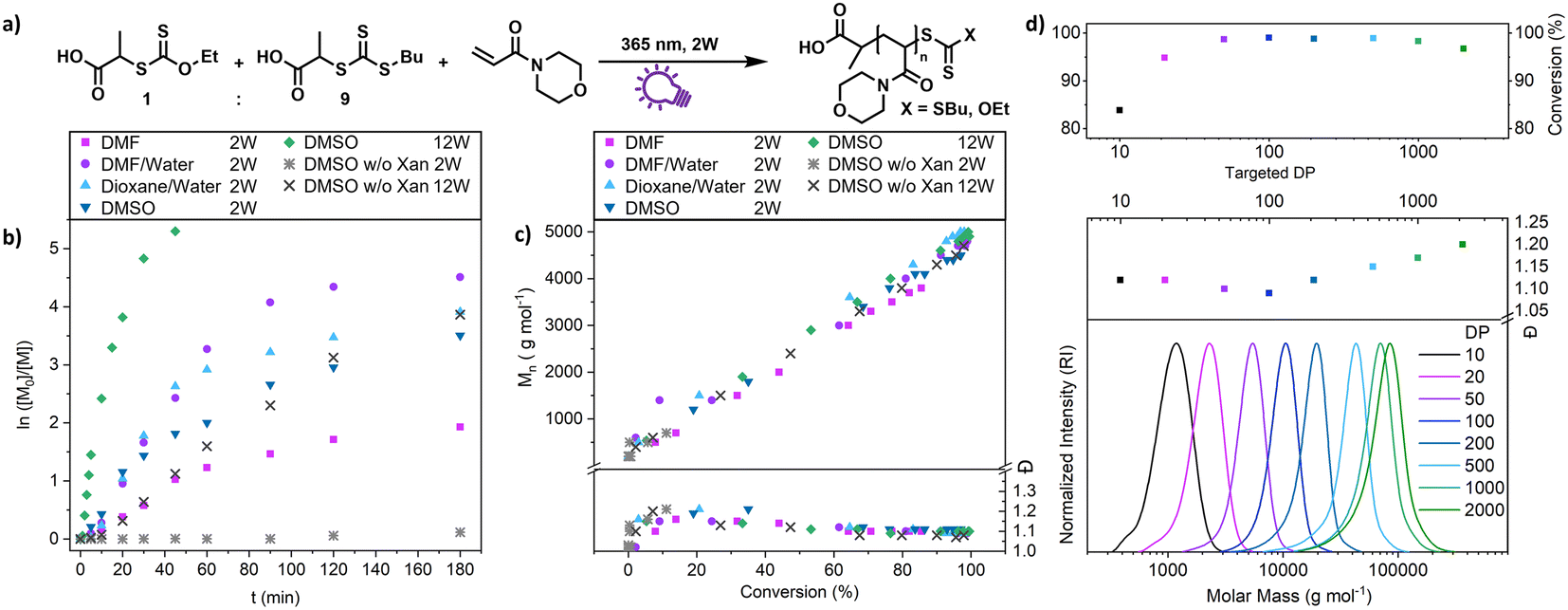 | ||
| Fig. 2 Kinetic investigation of XPI-RAFT polymerization of NAM using a 1:9 mixture of Xan and PABTC as CTAs (a). Polymerizations were performed using different solvents at a lamp power of 2 W (365 nm and 4.41 mW cm−2) or 12 W (365 nm and 27.39 mW cm−2). Control experiments without Xan were also performed. Conversion (b), as well as molecular weight distribution (c) was monitored under all conditions. (d) XPI-RAFT polymerization of NAM targeting various DPs at a constant concentration of monomer (2 mol L−1) and reaction time (3 h). Size-exclusion chromatography (SEC) traces were recorded in THF using poly(styrene) (PS) as calibration, and conversion was probed via1H-NMR spectroscopy. | ||
The declining nature of the curves in the log-plot (Fig. 2b) seems unintuitive at a first glance, as with a constant concentration of radicals (as expected for PI-RAFT) a pseudo first order kinetic and hence a linear evolution of logarithmic conversion is expected. In the first attempt to examine this behaviour a time-dependent monitoring of the process, as well as a kinetic modelling has been conducted (Fig. S7–S9,† reaction monitored via1H-NMR spectroscopy and modelled with Matlab). While CTA-2 (PABTC) is consumed quickly via chain transfer reactions, the signals associated with intact Xan are visible at much longer reaction times. Even though the simulation is fitted in decent agreement with the consumption of the CTAs, the modelled logarithmic conversion of the monomer deviates significantly from the experimental data towards the end of the reaction. The XPI-RAFT model predicts the conversion of the monomer with varying molar fractions of xanthate to a certain degree (r2 = 0.7841). However, the experimentally determined monomer consumption shows two separated linear regions in the logarithmic conversion plot (Fig. S7†). If the model is run on the assumption that photo-fragmentation of a polymer bound CTA is considerably slower compared to that of the small molecule CTA, the prediction of monomer conversion is improved significantly (r2 = 0.9706, Fig. S9†).
Taking both modelling and the experiment into account we hypothesize a less effective photo-induced fragmentation of Xan from the end of a polymer compared to that of free xanthate, causing a drop of the apparent kp. This observation fits well with the difference in radical stabilization energy of an acrylate (resembling the R-group) and an acrylamide (resembling a macro-CTA that is reactivated).47 As the radical formed by acrylamide is less stabilized, a β-fragmentation would be less favourable from the macro-CTA, leading to an overall lower radical concentration. In this case the reaction would slow down once free Xan is consumed. Exploring the nature of this process will be part of future studies.
Nevertheless, polymerization proceeds in a highly controlled manner in each case (Fig. S10†) and all reactions showed an excellent linear increase of molecular weight with conversion (Fig. 2c), while dispersity values are levelling out at ∼1.1. The reaction speed is scalable with light intensity as demonstrated by using a 12 W setup (combination of three 4 W UV lamps, each with a measured intensity of 9.13 mW cm−2 at the sample position) where the process reaches a conversion of >90% after 15 min (Fig. 2b, green diamonds). At these intensities (27.39 mW cm−2) PABTC can be activated in the absence of Xan to proceed to similar conversions that are achievable with Xan and a 2 W lamp. However, this strategy reduces the livingness of the chains drastically (vide infra).
We were also interested in how versatile the method is in terms of achievable polymer length and tested various targeted degrees of polymerizations (DPs) at a constant concentration of monomer (2 mol L−1 of NAM, Fig. 2d). The control over the molecular weight and its distribution is excellent at all targeted DPs between 10 and 2000. Impressively, XPI-RAFT polymerization is very efficient, even when the active component is diluted significantly, as demonstrated with high conversions at a very low Xan concentration (conversion > 95% after 3 h reaction time at a targeted DP of 2000, Table S3†).
It should be noted that in principle rapid and controlled polymerizations can also be achieved using PI-RAFT at very high light intensities (∼600 mW cm−2).26 However, this requires highly specialized equipment that also has to be tuned to the respective CTA. Using XPI-RAFT polymerization, where radical production is decoupled, readily available and low intensity lamps can be used.
While control and reaction rates are useful benchmarks, we were also interested in the general scope of XPI-RAFT polymerization. This was probed by using different combinations of monomer and CTA-2 while maintaining Xan as the main radical source (Table 1, Fig. S11†). Both main components are important as the quality of radical stabilization influences chain transfer processes as well as re-initiation of the end standing xanthate. Thus, a mismatch of quality in the R-group between the two CTAs or using a monomer, which results in radicals that are better stabilized than the R-group of Xan might result in complications. For each combination of monomer and CTA-2, the DP was varied to also test the scope regarding polymer length.
| Monomer | CTA-2 | Light source | Target DP | Reaction time (h) | Conversione (%) | M n (theo) g mol−1 | M n g mol−1 | Đ |
|---|---|---|---|---|---|---|---|---|
|
a Monomer concentration was 2 mol L−1, the solvent was DMSO, and Xan with a Xan/CTA-2 ratio of 9:1 was used as a photo-active component for all reactions.
b 2 W (4.41 mW cm−2), 365 nm.
c 24 W, 450 nm.
d 1 mL vial placed on the LED light source of an Apple iPad® Pro 1 (2018) 11.0′′.
e NMR.
f SEC.
g Dioxane/water was used as the solvent.
h Values in brackets without the addition of Xan.
|
||||||||
| NAM | BABTC | UVb | 50 | 4 | >99 | 7600 | 5100 | 1.1 |
| 200 | >99 | 29500 |
19400 |
1.2 | ||||
| 500 | >99 | 70800 |
42800 |
1.1 | ||||
| NIPAM | PABTC | UVb | 50 | 4 | 78 | 4700 | 1900 | 1.2 |
| 200 | 93 | 21300 |
8500 | 1.1 | ||||
| 500 | 91 | 51700 |
19900 |
1.3 | ||||
| MA | PABTC | UVb | 50 | 4 | 92 | 4200 | 5000 | 1.1 |
| 200 | 94 | 16400 |
19400 |
1.1 | ||||
| 500 | 89 | 38600 |
44500 |
1.1 | ||||
| MMA | CPCETC | UVb | 50 | 20 | 98 | 5200 | 4500 | 1.2 |
| 200 | 95 | 19300 |
12900 |
1.4 | ||||
| 500 | 88 | 44400 |
26000 |
1.6 | ||||
| DMAEMA | CPDTB | UVb,g | 50 | 20 | 80 | 6600 | 3600 | 1.2 |
| 200 | 75 | 23900 |
16700 |
1.3 | ||||
| 500 | 81 | 63900 |
40300 |
1.4 | ||||
| PEGA | PABTC | UVb,g | 10 | 4 | 95 | 4600 | 4900 | 1.1 |
| 20 | 93 | 8900 | 7700 | 1.1 | ||||
| 50 | 92 | 22100 |
13400 |
1.1 | ||||
| NIPAM | PABTC | Bluec | 50 | 4 | 75 (54)h | 4500 | 1900 | 1.1 |
| 200 | 78 (27)h | 17900 |
7100 | 1.2 | ||||
| 500 | 74 (36)h | 42100 |
19600 |
1.3 | ||||
| NAM | PABTC | iPad®d | 50 | 10 | 85 | 6200 | 4300 | 1.1 |
The first variation that was probed was the quality of the R-group of CTA-2. We used NAM and Xan in combination with tert-butanoic acid butyl trithiocarbonate (BABTC), a CTA with a tertiary R-group. The polymerization proceeded flawlessly with high conversions and excellent definition of the resulting polymers. Also, N-isopropyl acrylamide (NIPAM), as well as methyl acrylate (MA), both examples for fast propagating monomers showed satisfying results for all targeted polymer lengths. Slow propagating monomers forming tertiary radicals proved to be more challenging. First, we probed methyl methacrylate (MMA) using cyano pentanoic acid carboxyethyl trithiocarbonate (CPCETC) as the CTA. While reaction times had to be adjusted to 20 h to accommodate the monomer, the achieved conversions were still high. Here, the targeted DP of the polymer had an impact on the definition of the product. PMMA with a DP of 50 could be produced with a dispersity of 1.2; however, the molecular weight distribution broadened for longer polymers. It should be noted that curves were still unimodal (Fig. S11†) hinting toward a controlled process. A possible explanation would be that low chain transfer coefficients, as expected of a combination of Xan and MMA are the reason for these observations. When the same polymerization was performed using thermal initiation (Fig. S12†), control was increased supporting this hypothesis. If chains featuring a Xan end group would only be reactivated via photolysis, a low molecular weight tailing, as observed here would be the result.
As all previous combinations were between Xan and a trithiocarbonate, we also wanted to probe a different type of CTA in an XPI-RAFT protocol. Using cyano pentanoic acid dithiobenzoate (CPDTB) as CTA-2 in combination with dimethyl aminoethyl methacrylate (DMAEMA), it was possible to produce defined polymers in good conversions. We also included a poly(ethylene glycol) (PEG)-based monomer (480 g mol−1) featuring an acrylate group (PEGA) to demonstrate the capability of polymerizing more bulky repeating units. As we found Xan to be photo-active under visible light irradiation as well, the polymerization of NIPAM was also attempted using blue light (∼450 nm).
The obtained results are similar to the outcome of UV-mediated polymerization, while a higher light intensity (factor 6) was necessary due to limited spectral overlap. While here PABTC plays a more substantial role as a photo-iniferter (values in brackets in Table 1 are from polymerizations without Xan), the reactions are still much more efficient as an XPI-RAFT process. Even the white LED of an iPad® was able to drive the reaction when a vial containing the reaction solution was placed directly on the light source (Fig. S13†). The results in Table 1 indicate that the XPI-RAFT protocol can be used rather universally, without a major restriction regarding the nature of CTA-2 or the monomer, but with limitations regarding the achievable definition for tertiary monomers. Thus, the XPI-RAFT method essentially disconnects the activation wavelength from chain transfer, without requiring an exogenous photo-catalyst, as xanthate is still introduced as an end group in the produced polymers enabling efficient photo-activation (vide infra). The deviation from theoretical molecular weights can be explained with the mismatching SEC calibration. It should be noted that attempts to polymerize styrene yielded relatively low conversion (<50%, data not shown). Elucidating this will be part of future investigations.
As PI-RAFT has been shown to be tolerant toward the presence of limited amounts of oxygen,25,26 we were interested in knowing if this feature can also be achieved in XPI-RAFT polymerization. We found that the reaction can be performed open to air and without degassing (Fig. 3a and b). After oxygen was consumed during a short inhibition period polymerization proceeded without a decrease in the polymerization rate or control. Only when the polymerization mixture was stirred, the constant intake of O2 inhibited the process. The photostability of Xan was further probed using UV-vis spectroscopy of diluted solutions of Xan in DMSO. When illuminated at 365 nm using a similar intensity as that used for polymerizations, oxygen-free solutions showed stable absorption spectra, while the intensity of the peak associated with the π–π*-band of Xan was decreasing constantly during irradiation (Fig. 3c and d).
 | ||
| Fig. 3 Oxygen tolerance of XPI-RAFT polymerization. (a and b) Log-plots of the polymerization of NAM (2 mol L−1) in DMSO under different conditions using a mixture of PABTC and Xan (9:1) and targeting a DP of 50. Conversion (a) was measured via1H-NMR spectroscopy (CDCl3, 400 MHz) and molecular weight distribution (b) was determined via SEC in THF (PS-calibration); (c and d) photo-degradation of Xan in the presence and absence of oxygen. (c) Degradation kinetics under illumination with 365 nm light (I = 4.13 mW cm−2) under oxygen free conditions (0.02 mM of Xan in DMSO) and in the presence of oxygen (0.08 mM of Xan in DMSO); (d) time dependent UV-vis spectra of Xan under these conditions. The presence of oxygen seems to quench the UV-vis absorption of Xan. | ||
This indicates that the process that removes oxygen from solution during irradiation is also degrading the CTA. For polymerization reactions at a targeted DP of 50 this is not obvious as here the CTA concentration is three orders of magnitude higher while the oxygen concentration remains similar. Hence, XPI-RAFT polymerization can tolerate certain amounts of oxygen. However, for more sophisticated tasks, where a high livingness is essential or where Xan concentrations are very low, removal of oxygen prior to polymerization is still important.
Having shown that XPI-RAFT polymerization is a rather universal strategy which is also very robust, we wanted to demonstrate that the method can be utilized for the screening of polymeric materials in a simple well plate setup. Experiments using a well plate format have been described previously, but they relied on high reaction temperatures and the availability of a thermocycler,48 or on the presence of a photocatalyst using PET-RAFT polymerization.49
To demonstrate the ease of performing such a screening using the XPI-RAFT methodology, we set up a high throughput polymerization in a 96 well plate using NAM as a model monomer with a mixture of PABTC and Xan, and dioxane/water as the reaction medium (Fig. 4a). As it has been shown that the mole fraction of Xan in the CTA mixture influences the dispersity of the resulting polymer, the Xan content was chosen as one of the parameters to be varied in this experiment. Controlled dispersity38,40 can be beneficial for many aspects including interfacial properties,50 self-assembly,51 or biological activity.52 As such, the Xan content in the CTA mixture was varied between 10 and 90% in order to control Đ. Moreover, we wanted to probe whether this control can be exercised at different polymer lengths.
 | ||
| Fig. 4 High throughput screening of the polymerization of NAM using XPI-RAFT polymerization in a well plate format. (a) Reaction scheme including reaction conditions, and the ratio of CTAs was varied. (b) Picture of the well plate containing 63 reaction mixtures. The targeted DP was varied between 20 and 1280 by dilution of CTAs before monomer addition (factor 2), and the mole fraction of Xan (χ) was varied between 10 and 90%. (c) Conversion of each polymerization as determined by 1H-NMR spectroscopy. (d) SEC analysis (THF, PS calibration) of each sample. (e) SEC curves of individual polymers. | ||
Therefore, the targeted DP was varied as a second parameter. The respective well plate (Fig. 4b) contained 63 individual polymerization reactions and was prepared in about 1 h using stock solutions and serial dilution techniques. The plate was covered using a silicon mat to prevent spills and excessive intake of oxygen. Otherwise, no deoxygenation was performed (neither before nor after pipetting). The plate was subsequently illuminated from the bottom using 2 UV lamps (Fig. S14†). Impressively, the conversion as determined by NMR spectroscopy was almost quantitative for most of the wells (Fig. 4c).
Only for the three samples with high targeted DPs and low content of Xan, polymerization did not occur as here the active species was too diluted to consume the oxygen in solution and start the polymerization. From the sample with the lowest xanthate amount that is able to produce a polymer, the minimal concentration for successful deoxygenation and polymerization in a closed vessel can be approximated (0.625 mmol L−1). SEC measurements of the residual 60 polymers revealed excellent control over the molecular weight. Regardless of the CTA mixture composition, the measured Mn values correlate nicely with the targeted DP. The dispersity of individual polymers was dependent on the mole fraction of Xan used. Due to limited chain transfer capabilities of xanthate, molecular weight distribution broadened along the series. Unimodality of the curves and molecular weight control strongly suggest a controlled polymerization (Fig. 4e). This observation was true for all targeted DPs even though at high molecular weights Đ was already elevated at low Xan contents, which is potentially a result of the presence of oxygen in the polymerization mixture in combination with low Xan concentrations.
A correlation of the Đ value with the composition of the CTA mixture can be observed for each DP value (Fig. S15†)
Overall, this experiment shows that XPI-RAFT polymerization is a suitable protocol for screening reactions in a straightforward manner, using a well plate format without the necessity of degassing. In addition, as the method relies on a mixture of CTAs with different chain transfer coefficients, the dispersity of the resulting polymers can inherently be controlled over a wide range of targeted molecular weights by simply changing the ratio of CTAs.
An important feature of PI-RAFT polymerization is that polymers possessing a CTA end group can be reactivated via light and chain transfer without the addition of an additional CTA or initiator. As in XPI-RAFT polymerization, xanthate is incorporated into the polymer population (Fig. 1c), and it is expected that with this method also a simple reactivation is possible. To prove this, and also to demonstrate the straightforward nature of the XPI-RAFT process, various block copolymers were produced in a one-pot procedure (Fig. 5). After the first segment reached a high conversion (>95%) a second monomer was added, and polymerization was continued under UV-light irradiation. Monomer consumption was probed via1H-NMR spectroscopy and irradiation was continued until the desired conversion was achieved. A tailing, indicating incomplete chain extension is visible in SEC traces, but can be expected in the rapid synthesis of high molecular weight block copolymers. It should be emphasized that the synthesis of triblock copolymers with a total DP of 1000 (PNAM500-b-PNIPAM200-b-PMA300) represents a non-trivial task, which would at least require optimization of the used initiator amounts in conventional RAFT polymerization. Using XPI-RAFT this polymer could be produced in 13 h reaction time, without the necessary screening of parameters and with relative ease (standard laboratory UV-lamp, simple degassing strategy, no stirring, and no additional reagents).
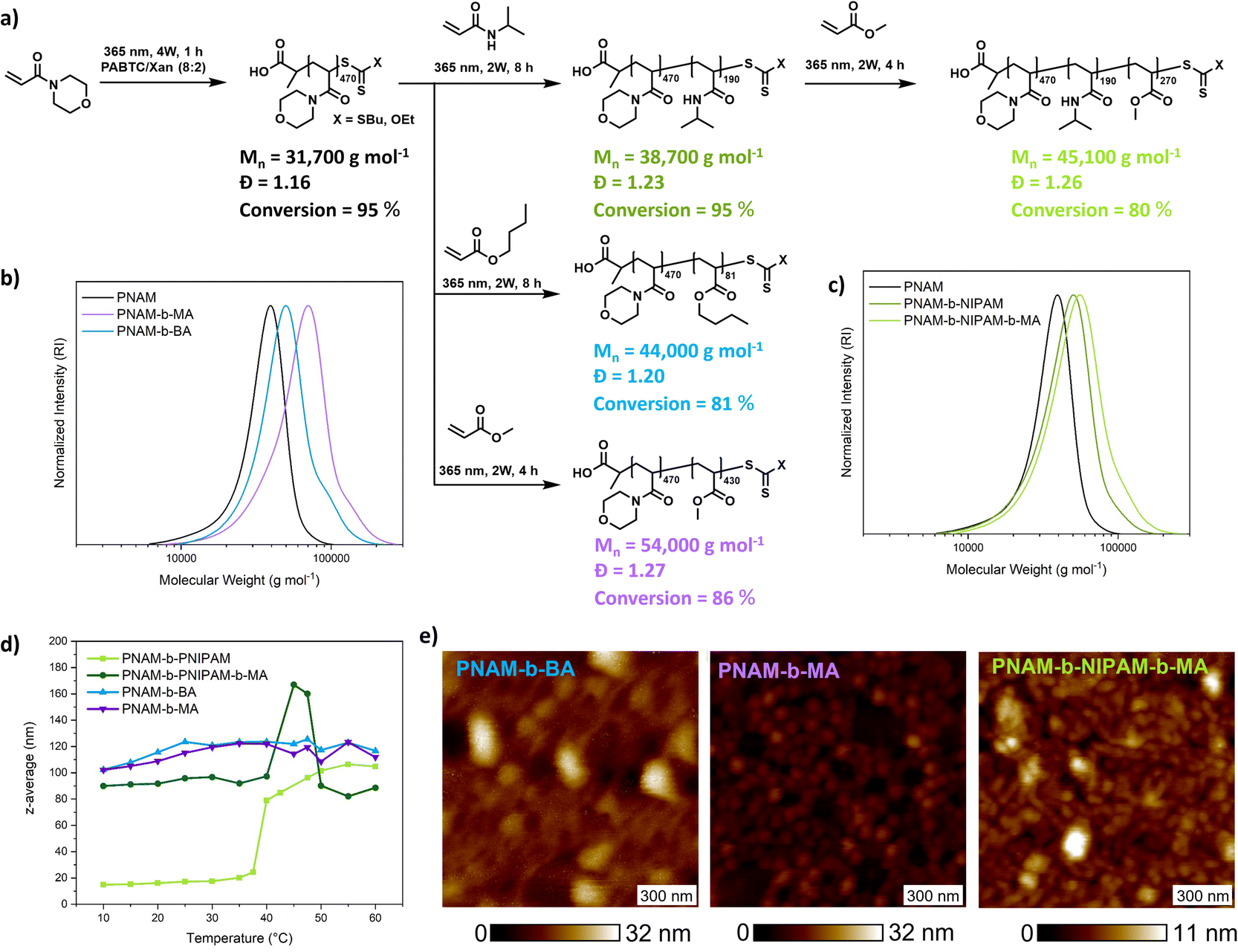 | ||
| Fig. 5 Synthesis of block copolymers using the XPI-RAFT methodology. (a) Schematic depiction of the block copolymer sequence and composition, (b) SEC traces of diblock copolymers based on BA and MA, (c) di and triblock copolymers based on NIPAM, (d) self-assembly of block copolymers in aqueous medium as a function of temperature via DLS, and (e) AFM height micrographs of self-assembled block copolymers. | ||
These findings also prove that xanthate is incorporated into the polymer and can be reactivated, although with a slower reaction rate, when compared to the first block, which is in accordance with the behavior observed in kinetic experiments. In addition, the method is highly adjustable, as conversion can be probed at any point of the reaction without disturbing the livingness, and polymerization can be continued until the desired monomer consumption is achieved. To illustrate the quality of the produced block copolymers, their self-assembly in aqueous solution was investigated. Based on their composition, diblock copolymers formed defined aggregates at room temperature or upon heating as indicated by DLS (Fig. 5d and S15†). Furthermore, atomic force microscopy (AFM) images reveal the morphology of surface-deposited assemblates (Fig. 5e).
All samples showed objects in a size range that is expected for micellar morphologies matching the values recorded in DLS. While PNAM-b-BA showed aggregation, PNAM-b-MA formed well defined particles.
Encouraged by these results we set out to probe the robustness and livingness of XPI-RAFT polymerization. The synthesis of a multiblock copolymer is an excellent benchmark in this context as a failure to re-initiate can be visualized in the size distribution via SEC. A further aim was to demonstrate that tedious parameter optimization or specialized conditions are unnecessary using an XPI-RAFT protocol, and that complex multiblock copolymer structures can be produced in an easy and rapid manner.
An alternating sequence of NAM and NIPAM was produced with a DP of 50 for each block, using an 8:2 mixture of PABTC and Xan (Fig. 6a). The conversion for each step was driven to above 90% to render an intermediate purification unnecessary. The polymerization proceeds with excellent definition of the resulting multiblocks (Fig. 6b and c). The molecular weight increases linearly over the block addition. An offset to the theoretical values is expected due to the mismatching calibration (PS). SEC curves display a significant shift upon each chain extension illustrating the livingness of XPI-RAFT polymerization. The evolution of a low molecular weight tailing, associated with dead chain formation is visible. This is expected in RAFT-based multiblock synthesis and is here likely associated with irreversible side reactions of CTA-2. Indeed, PABTC can also be activated by the used light source and similar CTAs were described to show irreversible degradation after longer irradiation times.53 Still the overall definition can be considered excellent for such a highly segmented macromolecule (12 blocks each with a DP of 50, Mn = 42400 g mol−1, and Đ = 1.33). While reaction times increase over the course of the procedure (Table S4†), the overall reaction time of 123 h is still moderate for a polymer containing 12 individual blocks. It should be emphasized that similar approaches using conventional RAFT polymerization were limited to a relatively short segment length (DP ∼ 10).54 While the synthesis of a polymer with the herein described length and definition would be highly difficult using conventional initiation, XPI-RAFT has the additional advantage that no tedious optimization of reaction parameters is necessary. The same multiblock synthesis was also performed at a higher light intensity (4 W instead of 2 W lamp power Fig. S20 and S21†), and while the final product was slightly less defined the overall outcome was similar, and only 91.5 h of reaction time were necessary. This demonstrates that the reaction times in XPI-RAFT can be scaled down using higher light intensities without a major compromise on the definition of the product. A similar multiblock copolymer with a targeted DP of 100 per block and 8 distinct segments could also be produced (Mn = 45900 g mol−1 and Đ = 1.28) (Fig. S22†).
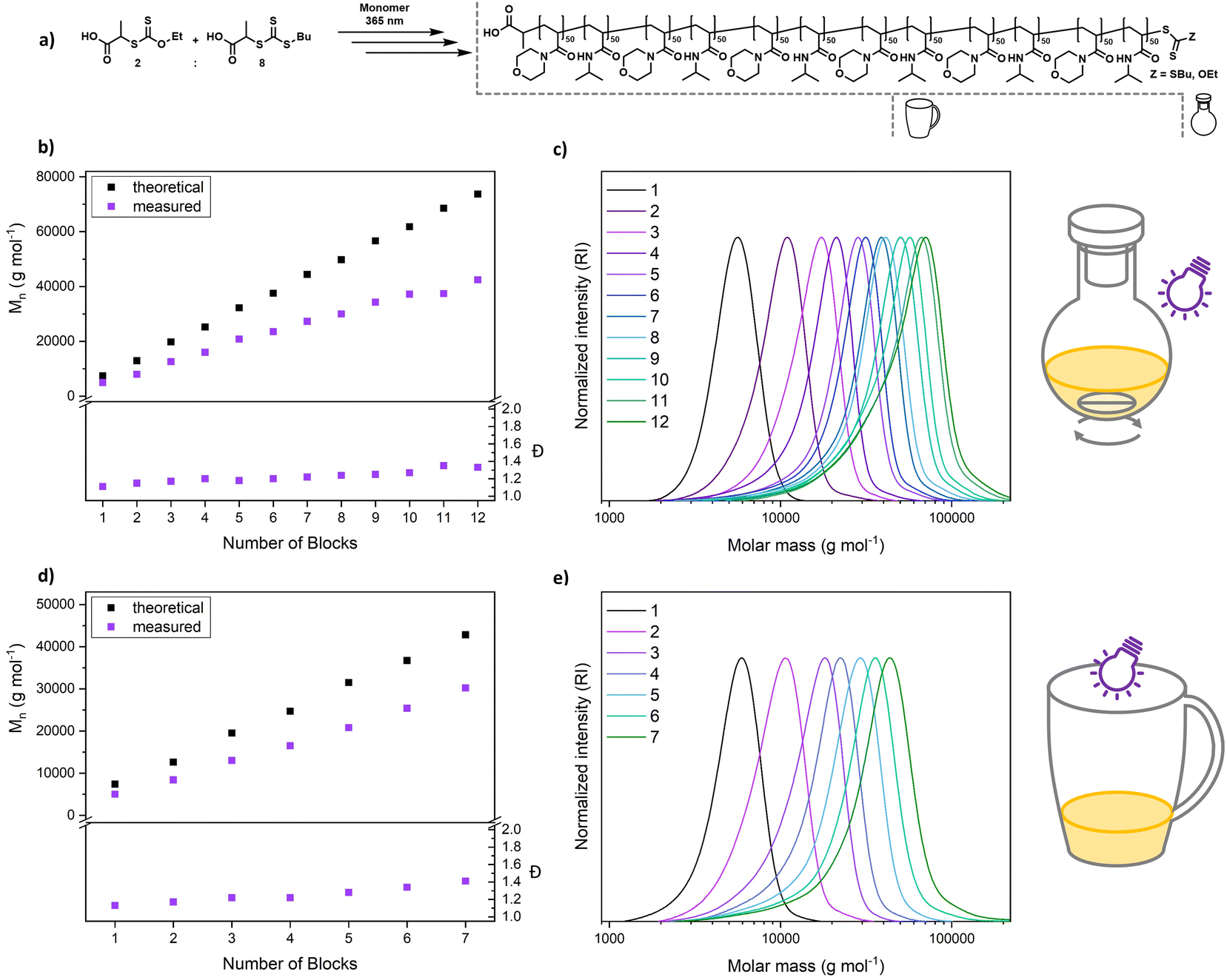 | ||
| Fig. 6 Synthesis of multiblock copolymers via XPI-RAFT polymerization by sequential monomer addition of NAM and NIPAM in an alternating fashion (a). The synthesis was performed in a flask under stirring using a 2 W lamp (b and c), as well as in a coffee mug without stirring or degassing, and under open-to-air conditions using a 4 W lamp (d and e). | ||
To demonstrate that the capabilities of XPI-RAFT polymerization go beyond what can be achieved without xanthate, a multiblock synthesis using only PABTC was attempted. Here the 12 W setup described in Fig. 2 was used to achieve reaction kinetics similar to that of XPI-RAFT polymerization. However, the degradation of the CTA under intense UV light53 becomes obvious rapidly by drastically increased reaction times after the first block (Table S8†), as well as multimodal SEC curves and fast increasing Đ values (Fig. S24†). This comparison impressively shows the advantages of XPI-RAFT polymerization over direct UV-light activation of trithiocarbonates.
To demonstrate the ease of preparation of complex macromolecular architectures using XPI-RAFT, we performed a multiblock synthesis in a coffee mug without degassing (Fig. 6d, e, S18 and S19†). As, in the absence of active mixing, the reaction is relatively oxygen tolerant, polymerization was performed open to air but without stirring. All components (monomer, CTA, xanthate, and solvent mixture (water/dioxane)) were simply added to the mug and the reaction was started directly by positioning a 4 W (365 nm and 9.13 mW cm−2) UV lamp on top of the cup (Fig. S18†). After polymerization reached high conversion as probed by 1H NMR, the next monomer (in solvent) was added directly to the reaction mixture and the synthesis was continued (without degassing). We were able to produce a heptablock copolymer with remarkably low dispersity (7 blocks, each with a DP of 50, Mn = 30200 g mol−1, and Đ = 1.41) under these simple conditions. After block 7 it was not possible to reach sufficient conversion within a reasonable time frame. It is likely that the Xan-end groups degraded over the reaction time in the presence of oxygen, as also indicated by the UV-vis experiments (Fig. 3c and d). Expectedly, livingness decreases faster under aerobic conditions and dispersity increases faster than in the closed vessel. Still, the ability to produce such a complex and defined macromolecule using breakfast tableware as a reaction vessel without any active measures to remove oxygen, underlines the potential and robustness of XPI-RAFT polymerization.
Conclusions
We developed a photo polymerization strategy based on the RAFT methodology, in which photo-activation is decoupled from molecular weight control, and all components are still introduced into the final material. This xanthate-supported photo-iniferter (XPI)-RAFT polymerization is performed by addition of small amounts of xanthate, acting as a photo-iniferter to a conventional RAFT polymerization. While polymerization is controlled by the main CTA, the xanthate enables highly efficient photo-activation and an excellent livingness. As both the CTA and the xanthate are introduced into the polymer as end groups, an efficient reactivation can be accomplished via light. We demonstrated the modularity of the process by combining xanthate with various combinations of CTA and monomer and using different solvents resulting in rapid and controlled photo polymerization. XPI-RAFT was also shown to be oxygen tolerant and could be used for high throughput screening of polymerizations in a well plate format, controlling the molecular weight and Đ over a wide range. Reactivation was illustrated by the synthesis of di- and triblock copolymers, as well as multiblock copolymers with high molecular weight and excellent definition (dodecablock copolymer, Mn = 42400 g mol−1, and Đ = 1.33). The practical and user-friendly nature of XPI-RAFT polymerization was illustrated by performing a multiblock synthesis under open-to-air conditions in a coffee mug.
In contrast to PET-RAFT polymerization or conventional RAFT-protocols, no external catalyst, sensitizer, or radical source is necessary and the produced polymers are inherently photo-active, enabling straightforward chain extension using just an additional monomer and light. Using the XPI-RAFT methodology, a broad range of conventional RAFT processes can be turned into rapid and robust photo polymerization with high livingness without a necessity for specialized equipment, tedious optimization or laborious reaction setups, enabling straightforward access to complex macromolecules.
Data availability
All data required to reproduce the results are presented in the main manuscript or the ESI.†Author contributions
M. H. conceived the idea and developed the concept. A.-C. L., A. M. B., M. R., M. B. and M. H. were involved in synthesis and characterization. J. G. performed the modelling. The manuscript was written by M. H. and A.-C. L. and reviewed and edited by all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge funding by the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG) (Emmy-Noether-Program, HA 7725/2-1 (AL, AB, MH), funding numbers 471323994 (MR) and BE 7745/1-1 (MB)). The authors also thank of Prof. Dr Heiko Möller, Dr Matthias Heydenreich, and Angela Krtitschka at the NMR core facility of the Institute of Chemistry (University of Potsdam). Prof. Dr Helmut Schlaad and Sascha Prentzel from the Institute of Chemistry (University of Potsdam) are gratefully acknowledged for providing the facility to perform SEC measurements. J. G. acknowledges the support from the German Research Foundation (DFG via Research Fellowships Gz. GU 2073/2-2). This DFT calculation was performed using the compute cluster for High Performance Computing operated by the Center for Information Technology and Media Management of the University of Potsdam (https://www.uni-potsdam.de/en/zim/angebote-loesungen/hpc). Johannes Martin (Fraunhofer IAP, Potsdam) is acknowledged for ESI-MS measurements.Notes and references
- M. Chen, M. Zhong and J. A. Johnson, Chem. Rev., 2016, 116, 10167–10211 CrossRef CAS.
- Z. Zhang, N. Corrigan, A. Bagheri, J. Jin and C. Boyer, Angew. Chem., 2019, 58, 17954–17963 CrossRef CAS PubMed.
- J. Xu, S. Shanmugam, C. Fu, K.-F. Aguey-Zinsou and C. Boyer, J. Am. Chem. Soc., 2016, 138, 3094–3106 CrossRef CAS PubMed.
- S. Shanmugam, J. Cuthbert, T. Kowalewski, C. Boyer and K. Matyjaszewski, Macromolecules, 2018, 51, 7776–7784 CrossRef CAS.
- P.-E. Millard, L. Barner, M. H. Stenzel, T. P. Davis, C. Barner-Kowollik and A. H. E. Müller, Macromol. Rapid Commun., 2006, 27, 821–828 CrossRef CAS.
- N. P. Truong, G. R. Jones, K. G. E. Bradford, D. Konkolewicz and A. Anastasaki, Nat. Rev. Chem., 2021, 5, 859–869 CrossRef CAS.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- X. Pan, N. Malhotra, A. Simakova, Z. Wang, D. Konkolewicz and K. Matyjaszewski, J. Am. Chem. Soc., 2015, 137, 15430–15433 CrossRef CAS.
- T. Zhang, T. Chen, I. Amin and R. Jordan, Polym. Chem., 2014, 5, 4790–4796 RSC.
- A. Anastasaki, V. Nikolaou, G. Nurumbetov, P. Wilson, K. Kempe, J. F. Quinn, T. P. Davis, M. R. Whittaker and D. M. Haddleton, Chem. Rev., 2016, 116, 835–877 CrossRef CAS.
- M. Vorobii, O. Pop-Georgievski, A. de los Santos Pereira, N. Y. Kostina, R. Jezorek, Z. Sedláková, V. Percec and C. Rodriguez-Emmenegger, Polym. Chem., 2016, 7, 6934–6945 RSC.
- Y. Guillaneuf, D. Bertin, D. Gigmes, D.-L. Versace, J. Lalevée and J.-P. Fouassier, Macromolecules, 2010, 43, 2204–2212 CrossRef CAS.
- D.-L. Versace, J. Lalevée, J.-P. Fouassier, D. Gigmes, Y. Guillaneuf and D. Bertin, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2910–2915 CrossRef CAS.
- T. G. McKenzie, Q. Fu, M. Uchiyama, K. Satoh, J. Xu, C. Boyer, M. Kamigaito and G. G. Qiao, Adv. Sci., 2016, 3, 1500394 CrossRef.
- R. Chapman, K. Jung and C. Boyer, in RAFT Polymerization, Wiley VCH, 2021, pp. 611–645, DOI:10.1002/9783527821358.ch12.
- M. H. Stenzel and C. Barner-Kowollik, Mater. Horiz., 2016, 3, 471–477 RSC.
- J. Xu, K. Jung, A. Atme, S. Shanmugam and C. Boyer, J. Am. Chem. Soc., 2014, 136, 5508–5519 CrossRef CAS.
- M. L. Allegrezza and D. Konkolewicz, ACS Macro Lett., 2021, 10, 433–446 CrossRef CAS.
- J. Phommalysack-Lovan, Y. Chu, C. Boyer and J. Xu, Chem. Commun., 2018, 54, 6591–6606 RSC.
- M. Hartlieb, Macromol. Rapid Commun., 2022, 43, 2100514 CrossRef CAS PubMed.
- T. Otsu, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2121–2136 CrossRef CAS.
- M. Chen and J. A. Johnson, Chem. Commun., 2015, 51, 6742–6745 RSC.
- T. G. McKenzie, Q. Fu, E. H. H. Wong, D. E. Dunstan and G. G. Qiao, Macromolecules, 2015, 48, 3864–3872 CrossRef CAS.
- J. Xu, S. Shanmugam, N. A. Corrigan and C. Boyer, in Controlled Radical Polymerization: Mechanisms, American Chemical Society, 2015, ch. 13, vol. 1187, pp. 247–267 Search PubMed.
- Q. Fu, K. Xie, T. G. McKenzie and G. G. Qiao, Polym. Chem., 2017, 8, 1519–1526 RSC.
- J. R. Lamb, K. P. Qin and J. A. Johnson, Polym. Chem., 2019, 10, 1585–1590 RSC.
- K. M. Burridge, N. De Alwis Watuthanthrige, C. Payne, R. C. Page and D. Konkolewicz, J. Polym. Sci., 2021, 59, 2530–2536 CrossRef CAS.
- R. N. Carmean, T. E. Becker, M. B. Sims and B. S. Sumerlin, Chem, 2017, 2, 93–101 CAS.
- R. N. Carmean, M. B. Sims, C. A. Figg, P. J. Hurst, J. P. Patterson and B. S. Sumerlin, ACS Macro Lett., 2020, 9, 613–618 CrossRef CAS.
- C. P. Easterling, Y. Xia, J. Zhao, G. E. Fanucci and B. S. Sumerlin, ACS Macro Lett., 2019, 8, 1461–1466 CrossRef CAS PubMed.
- J. Li, X. Pan, N. Li, J. Zhu and X. Zhu, Polym. Chem., 2018, 9, 2897–2904 RSC.
- P. Lambrinos, M. Tardi, A. Polton and P. Sigwalt, Eur. Polym. J., 1990, 26, 1125–1135 CrossRef CAS.
- B. Zhao, J. Li, Y. Xiu, X. Pan, Z. Zhang and J. Zhu, Macromolecules, 2022, 55, 1620–1628 CrossRef CAS.
- A.-C. Lehnen, J. A. M. Kurki and M. Hartlieb, Polym. Chem., 2022, 13, 1537–1546 RSC.
- R. A. Olson, M. E. Lott, J. B. Garrison, C. L. G. I. V. Davidson, L. Trachsel, D. I. Pedro, W. G. Sawyer and B. S. Sumerlin, Macromolecules, 2022, 55, 8451–8460 CrossRef CAS.
- D. J. Keddie, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 2012, 45, 5321–5342 CrossRef CAS.
- J. F. Quinn, L. Barner, C. Barner-Kowollik, E. Rizzardo and T. P. Davis, Macromolecules, 2002, 35, 7620–7627 CrossRef CAS.
- M.-N. Antonopoulou, R. Whitfield, N. P. Truong, D. Wyers, S. Harrisson, T. Junkers and A. Anastasaki, Nat. Chem., 2022, 14, 304–312 CrossRef CAS PubMed.
- T. Nwoko, N. De Alwis Watuthanthrige, B. Parnitzke, K. Yehl and D. Konkolewicz, Polym. Chem., 2021, 12, 6761–6770 RSC.
- R. Whitfield, K. Parkatzidis, N. P. Truong, T. Junkers and A. Anastasaki, Chem, 2020, 6, 1340–1352 CAS.
- H. Fischer, Chem. Rev., 2001, 101, 3581–3610 CrossRef CAS PubMed.
- J. D. Coyle and H. A. J. Carless, Chem. Soc. Rev., 1972, 1, 465–480 RSC.
- P. R. Judzewitsch, N. Corrigan, E. H. H. Wong and C. Boyer, Angew. Chem., Int. Ed., 2021, 60, 24248–24256 CrossRef CAS.
- J. Xu, S. Shanmugam and C. Boyer, ACS Macro Lett., 2015, 4, 926–932 CrossRef CAS.
- K. Bell, S. Freeburne, A. Wolford and C. W. Pester, Polym. Chem., 2022, 13, 6120–6126 RSC.
- A. Valdebenito and M. V. Encinas, Polym. Int., 2010, 59, 1246–1251 CrossRef CAS.
- N. M. Bingham, Z. Abousalman-Rezvani, K. Collins and P. J. Roth, Polym. Chem., 2022, 13, 2880–2901 RSC.
- P. Gurnani, T. Floyd, J. Tanaka, C. Stubbs, D. Lester, C. Sanchez-Cano and S. Perrier, Polym. Chem., 2020, 11, 1230–1236 RSC.
- G. Ng, J. Yeow, R. Chapman, N. Isahak, E. Wolvetang, J. J. Cooper-White and C. Boyer, Macromolecules, 2018, 51, 7600–7607 CrossRef CAS.
- M. Romio, B. Grob, L. Trachsel, A. Mattarei, G. Morgese, S. N. Ramakrishna, F. Niccolai, E. Guazzelli, C. Paradisi, E. Martinelli, N. D. Spencer and E. M. Benetti, J. Am. Chem. Soc., 2021, 143, 19067–19077 CrossRef CAS.
- A.-L. Buckinx, M. Rubens, N. R. Cameron, C. Bakkali-Hassani, A. Sokolova and T. Junkers, Polym. Chem., 2022, 13, 3444–3450 RSC.
- B. P. Mowery, A. H. Lindner, B. Weisblum, S. S. Stahl and S. H. Gellman, J. Am. Chem. Soc., 2009, 131, 9735–9745 CrossRef CAS.
- H. Wang, Q. Li, J. Dai, F. Du, H. Zheng and R. Bai, Macromolecules, 2013, 46, 2576–2582 CrossRef CAS.
- G. Gody, T. Maschmeyer, P. B. Zetterlund and S. Perrier, Nat. Commun., 2013, 4, 2505 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, additional figures, scheme, tables, and scripts. See DOI: https://doi.org/10.1039/d2sc05197d |
| This journal is © The Royal Society of Chemistry 2023 |