
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic photochemical enantioselective α-alkylation with pyridinium salts†
Santhivardhana Reddy
Yetra
 ,
Nathan
Schmitt
and
Uttam K.
Tambar
*
,
Nathan
Schmitt
and
Uttam K.
Tambar
*
Department of Biochemistry, The University of Texas Southwestern Medical Center, 5323 Harry Hines Boulevard, Dallas, Texas 75390-9038, USA. E-mail: Uttam.Tambar@utsouthwestern.edu
First published on 8th December 2022
Abstract
We have developed a chiral amine catalyzed enantioselective α-alkylation of aldehydes with amino acid derived pyridinium salts as alkylating reagents. The reaction proceeds in the presence of visible light and in the absence of a photocatalyst via a light activated charge-transfer complex. We apply this photochemical stereoconvergent process to the total synthesis of the lignan natural products (−)-enterolactone and (−)-enterodiol. Mechanistic studies support the ground-state complexation of the reactive components followed by divergent charge-transfer processes involving catalyst-controlled radical chain and in-cage radical combination steps.
Introduction
Carbonyl compounds with α-stereocenters are key components of many biologically active molecules, including pharmaceutical drugs and secondary metabolites.1 To access this important class of compounds, chemists have developed numerous transformative concepts in asymmetric catalysis for the enantioselective α-alkylation of enolates.2 While these reactions are often categorized by the mode of catalysis and the enolate precursor, an underappreciated component of these processes is the identity of the alkylating reagents that are coupled with enolates. Commonly employed alkylating reagents include alkyl halides and sulfonates (2, Scheme 1A).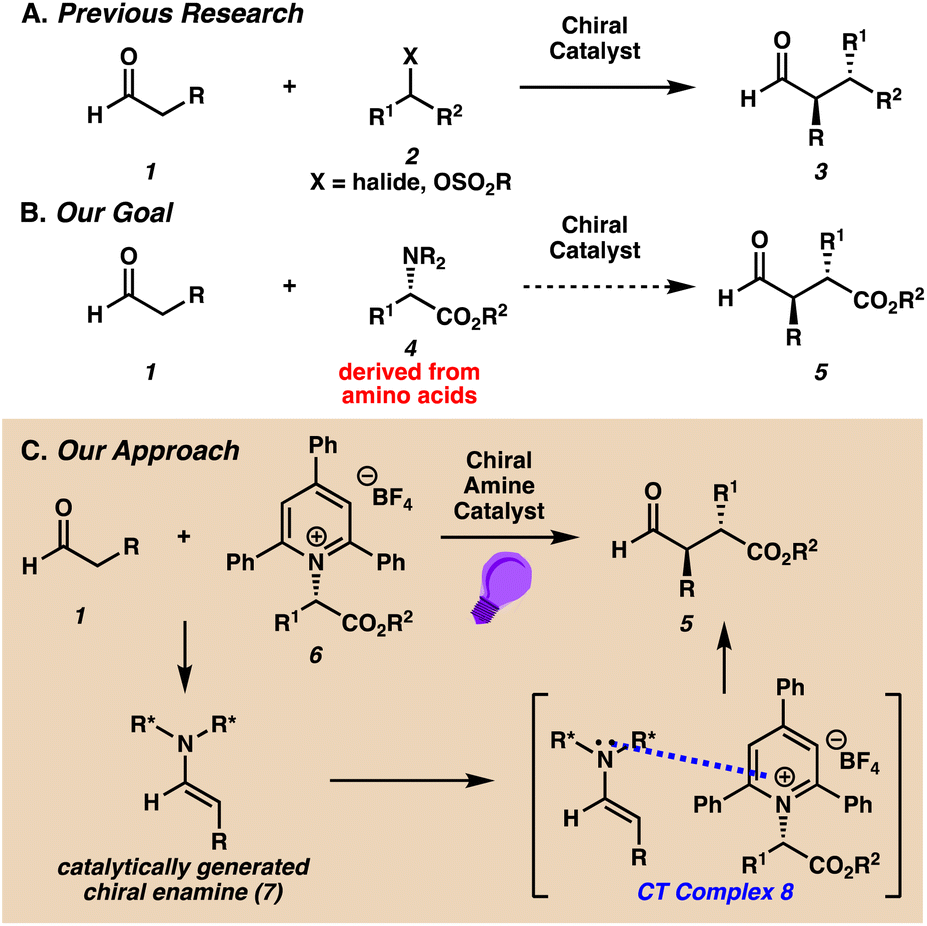 | ||
| Scheme 1 Strategy for catalytic enantioselective α-alkylation with amino acid derivatives. | ||
Recent elegant examples of photochemical enantioselective α-functionalizations of carbonyl compounds have been developed with alkyl halides.3–6 Notably, Melchiorre and co-workers pioneered the use of α-bromoketones and benzylic bromides as alkylating agents via light activated charge-transfer complexes.5 We were interested in developing a complementary approach for the catalytic enantioselective α-alkylation of aldehydes 1 based on renewable and sustainable sources of alkylating reagents. We identified amino acid derived substrates 4 as ideal reagents for enantioselective alkylations (Scheme 1B), as they possess several inherent advantages over traditionally used alkyl halides with respect to abundance, stability, versatility, and ease of preparation.7 In light of the poor electrophilicity of amino acid derivatives in enolate alkylations, we were motivated to devise a strategy for the activation of this class of substrates.
We report a catalytic photochemical enantioselective α-alkylation of aldehydes with amino acid derived pyridinium salts as alkylating reagents (Scheme 1C). These compounds are air and moisture stable crystalline solids that can be easily purified and stored for extended periods of time. Moreover, pyridinium salts can be generated on preparative scale from the facile condensation of amino acid derivatives and pyrylium salts.8
We hypothesized that pyridinium salts could form ground-state encounter complexes with catalytically generated electron rich chiral enolate equivalents.9,10 Our approach was supported by an early report from Katritzky on the α-benzylation of diethylmalonate with pyridinium salts of benzylamine, which he postulated proceeds through the formation of light activated charge-transfer (CT) complexes.11 More recently, Melchiorre has demonstrated the formation of CT complexes between enamines and alkyl halides.5 In our case, the generation of chiral enamine 7 from the condensation of aldehyde substrate 1 and a chiral amine catalyst could form CT complex 8 with pyridinium salt 6, which would then undergo stereoselective C–C bond formation in the presence of visible light.
Our proposal to utilize pyridinium salts in enantioselective α-alkylations is motivated by their storied history as radical precursors.12–16 More recently, pyridinium salts have been utilized in deaminative transformations through activation by photoredox catalysis17 or the formation of CT complexes with electron rich molecules.18 Despite the widespread application of pyridinium salts as radical precursors, the use of these substrates in catalytic enantioselective transformations is rare,19 and enantioselective reactions with prochiral enolate equivalents is unprecedented.
Results
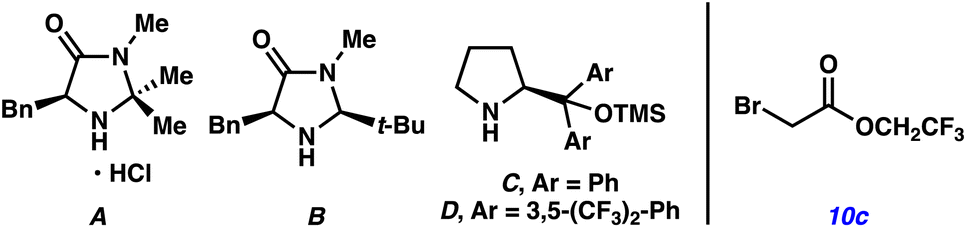
We initiated our studies by coupling hydrocinnamaldehyde 9 with Katritzky salt 10a derived from the ethyl ester of glycine as the alkylating agent (Table 1). In the presence of MacMillan's amine catalyst A, 2,6-lutidine, and purple light (390 nm) in CH2Cl2, we observed trace amounts of product 11a in 58% ee (entry 1). Interestingly, the deaminated byproduct of Katritzky salt 10a and the corresponding 2,4,6-triphenylpyridine were formed, suggesting the formation of a light activated CT complex. We reasoned that the radical philicity of intermediates generated upon charge transfer between the enamine of aldehyde 9 and pyridinium 10a may not be matched for the desired C–C bond forming event.20 To test this hypothesis, we subjected electron-deficient Katritzky salt 10b derived from the 2,2,2-trifluoroethyl ester of glycine to the reaction conditions (entry 2). Gratifyingly, we obtained α-alkylation product 11b in 36% yield and 60% ee, presumably via a more electron-deficient α-carboxy radical. In a Lewis basic medium such as DMA, the desired product was formed in 40% yield and 92% ee (entry 3). Although other amine catalysts B–D also furnished product 11b (entries 4–6), catalyst A was still best for enantioselectivity.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Alkylating agent | Catalyst | Solvent | Additive | Yieldb (%) | eec (%) |
| a Reaction conditions: 9 (0.30 mmol), 10 (0.1 mmol), catalyst (20 mol%), 2,6-lutidine (0.1 mmol) NaI (0.1 mmol), H2O (1.0 mmol), 4 °C, 24 h. b Isolated yield. c Enantiomeric excess determined by chiral HPLC analysis of a lactone derivative (see ESI). d Reaction conducted at 23 °C. e Irradiation with 370 nm Kessil lamp. f Irradiation with 427 nm Kessil lamp. g No light. | ||||||
| 1 | 10a | A | CH2Cl2 | — | 5 | 58 |
| 2 | 10b | A | CH2Cl2 | — | 36 | 60 |
| 3 | 10b | A | DMA | — | 40 | 92 |
| 4 | 10b | B | DMA | — | 22 | 5 |
| 5 | 10b | C | DMA | — | 55 | 15 |
| 6 | 10b | D | DMA | — | 52 | 23 |
| 7 | 10b | A | DMA | NaI | 65 | 92 |

|

|

|

|

|

|

|
| 9 |

|
A | DMA | NaI, H2O | <5 | — |
| 10d | 10b | A | DMA | NaI, H2O | 80 | 46 |
| 11e | 10b | A | DMA | NaI, H2O | 62 | 92 |
| 12f | 10b | A | DMA | NaI, H2O | 18 | 92 |
| 13g | 10b | A | DMA | NaI, H2O | <5 | — |
|
||||||
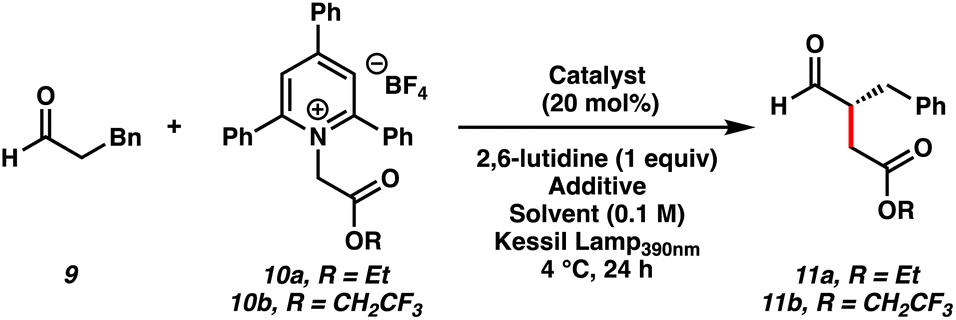
With enantioselectivity optimized, we focused on improving the yield of the reaction by employing additives that could enhance the ground-state complexation of the reaction components. Ultimately, the addition of stoichiometric NaI resulted in an increase in yield to 65% (entry 7), presumably through the formation of a multicomponent CT complex.21 Inclusion of water to help solubilize NaI further improved the yield to 75% while maintaining the enantioselectivity at 92% ee, which represented the optimal conditions for the reaction (entry 8). Interestingly, while α-bromoketones have been demonstrated to be competent acceptors in CT complexes with catalytically generated enamines,5 the corresponding 2,2,2-trifluoroethyl ester of α-bromoacetic acid (10c) was not a suitable alkylating agent under our optimized conditions (entry 9). This result highlights a unique advantage of pyridinium derived alkylating agents over the more traditionally used alkyl bromides in enantioselective α-alkylations.
We performed a series of control experiments to gain insight into the reaction (entries 10–13). Conducting the reaction at room temperature instead of 4 °C resulted in a loss of enantioselectivity (entry 10). Irradiation with various wavelengths of light resulted in diminished yields (entries 11–12), which confirmed the importance of activating the ground-state encounter complex at the appropriate wavelength. Furthermore, no product was observed in the absence of light (entry 13).
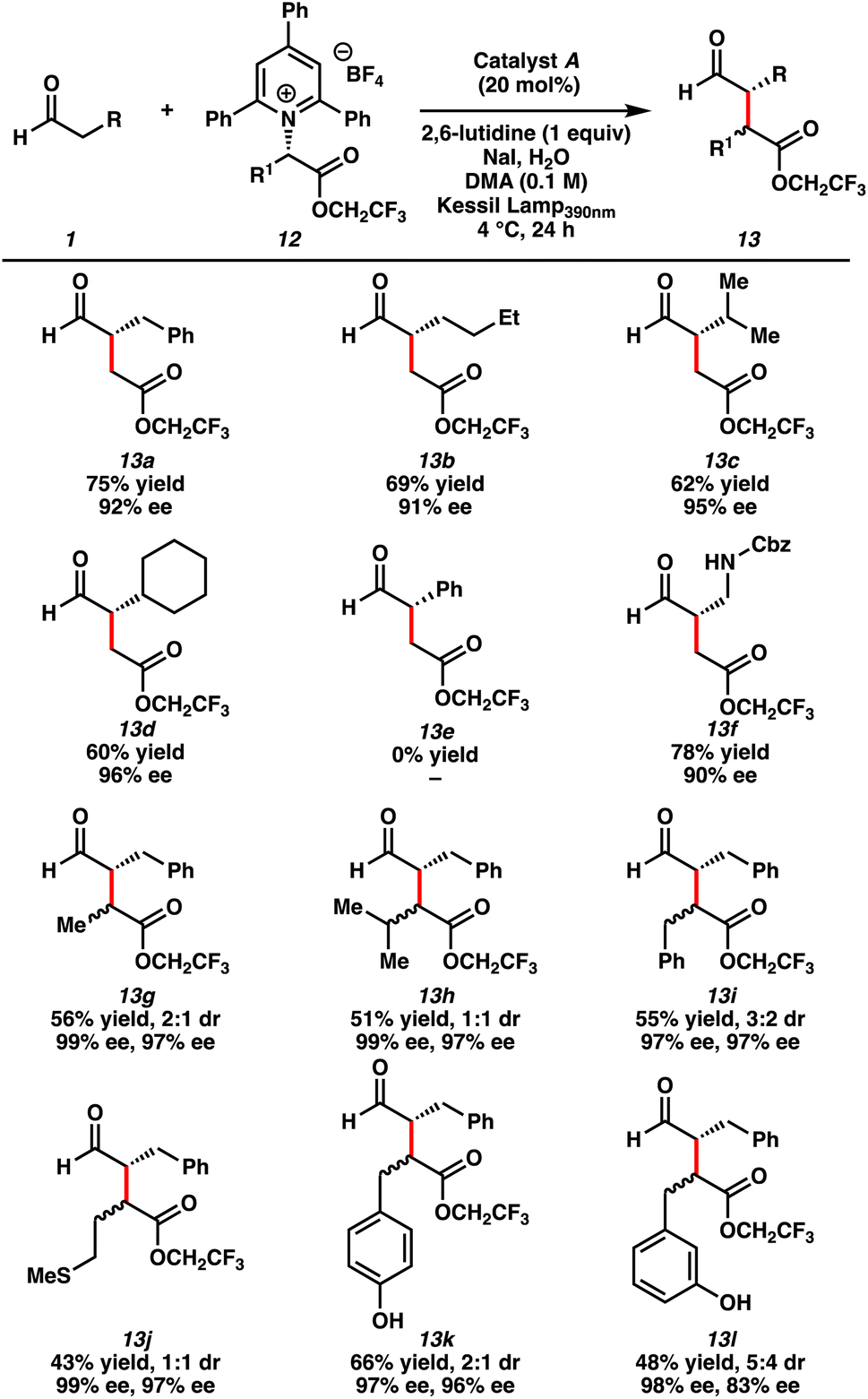
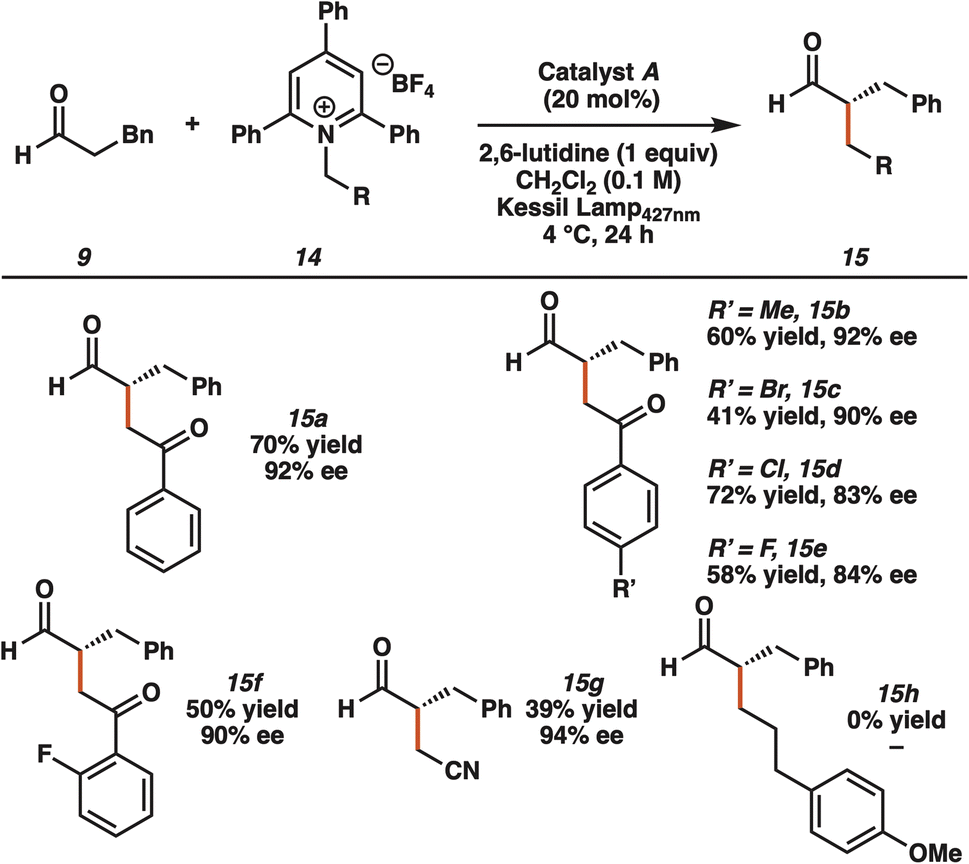
With optimal reaction conditions identified, we examined the substrate scope of the transformation (Table 2). The Katritzky salt of the trifluoroethyl ester of glycine was coupled with various aldehydes to furnish the desired products in synthetically useful yields and greater than 90% ee (13a–d). We obtained products from linear aldehydes (13a–b) and branched aldehydes (13c–d). Although we did not observe any product with phenylacetaldehyde (13e), a carbamate functionalized aldehyde yielded the desired alkylation product (13f). Enantioenriched alkylation products were also formed from the coupling of hydrocinnamaldehyde and Katritzky salts derived from various natural amino acids, such as alanine (13g), valine (13h), phenylalanine (13i), methionine (13j), and tyrosine (13k). In addition, we generated the alkylated product derived from an unnatural amino acid (13l). The products were generated with high enantioselectivity but poor diastereoselectivity, presumably because of the formation of open-shell intermediates (vide infra).
| a Reaction conditions: aldehyde (0.30 mmol), pyridinium salt (0.1 mmol), catalyst A (20 mol%), 2,6-lutidine (0.1 mmol), NaI (0.1 mmol), H2O (1.0 mmol), DMA (0.1 M), Kessil lamp390 nm, 4 °C, 24 h. |
|---|
|
To demonstrate the utility of this new mode of activation with other classes of substrates, we reacted hydrocinnamaldehyde 9 with Katritzky salts derived from various amines (Table 3). Notably, these reactions were performed in CH2Cl2 in the absence of NaI and water. An electron-withdrawing group adjacent to the amine functionality was necessary for reactivity. For example, several 2-aminoacetophenone pyridinium salts were compatible substrates for the enantioselective transformation (15a–f). Through optimization, 427 nm irradiation performed as well as 390 nm for the aminoketone derived pyridinium salts, presumably because of different photophysical properties of the charge transfer complexes. With 427 nm being lower in energy, we chose to move forward with this wavelength. We also observed the desired product derived from aminoacetonitrile (15g). However, the Katritzky salt derived from 3-phenyl-1-propylamine did not yield alkylation product 15h under the reaction conditions. The requirement for an electron-withdrawing group in the pyridinium salt further demonstrates the importance of matching the radical philicities of intermediates generated upon light activated charge transfer.
| a Reaction conditions: aldehyde (0.30 mmol), pyridinium salt (0.1 mmol), catalyst A (20 mol%), 2,6-lutidine (0.1 mmol), CH2Cl2 (0.1 M), Kessil lamp427 nm, 4 °C, 24 h. |
|---|
|
The synthetic utility of our new catalytic method is highlighted by the enantioselective total synthesis of the lignan natural products (−)-enterolactone 17 and (−)-enterodiol 18 (Scheme 2).22 Under optimal conditions, 3-(3-hydroxyphenyl)propanal 16 and the pyridinium salt of racemic m-tyrosine (12l) reacted in a stereoconvergent process to form the α-alkylated product, which was subjected to reductive conditions without purification to furnish (−)-enterolactone 17 and its epimer (epi-17) in 46% yield over 2 steps. Although the diastereomeric lactones were obtained in high ee but poor dr, we recognized the opportunity to epimerize the mixture for the synthesis of more complex structures with high diastereoselectivity. Therefore, the diastereomers were subjected to LHMDS and TMSCl to yield (−)-enterolactone 17 in 81% yield, 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr, and 97% ee upon diastereoselective protonation of the enolate intermediate. Reduction with LiAlH4 resulted in the formation of (−)-enterodiol 18 in 70% yield and 97% ee as a single diastereomer. Optical rotations of the synthetic samples of the two natural products also confirmed the absolute stereochemistry of the alkylation products obtained in our enantioselective reaction.
:1 dr, and 97% ee upon diastereoselective protonation of the enolate intermediate. Reduction with LiAlH4 resulted in the formation of (−)-enterodiol 18 in 70% yield and 97% ee as a single diastereomer. Optical rotations of the synthetic samples of the two natural products also confirmed the absolute stereochemistry of the alkylation products obtained in our enantioselective reaction.
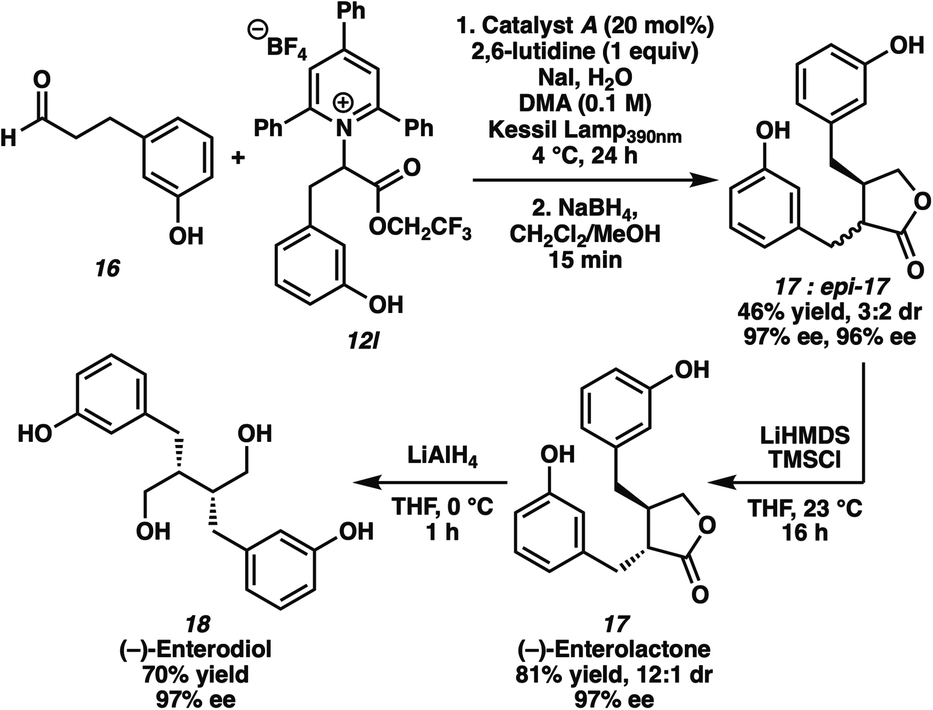 | ||
| Scheme 2 Synthesis of lignan natural products via enantioselective α-alkylation with pyridinium salts. | ||
Based on our initial proposal of a light activated CT complex between the pyridinium substrate and a catalytically generated enamine (Scheme 1C), we performed a series of experiments to gain insight into the mechanism of the photochemical process. The subjection of either enantiomer of Katritzky salt 12g to the optimized reaction conditions with aldehyde 9 resulted in the stereoconvergent formation of the same major enantiomer of both diastereomers of product 13g (Fig. 1A). These experiments with both enantiomers of Katritzky salt 12g, in combination with the absence of product formation in the presence of 1 equivalent of TEMPO,23 are consistent with the formation of α-carboxy radical 19 as a common intermediate, which also accounts for the low diastereoselectivity in the transformation.
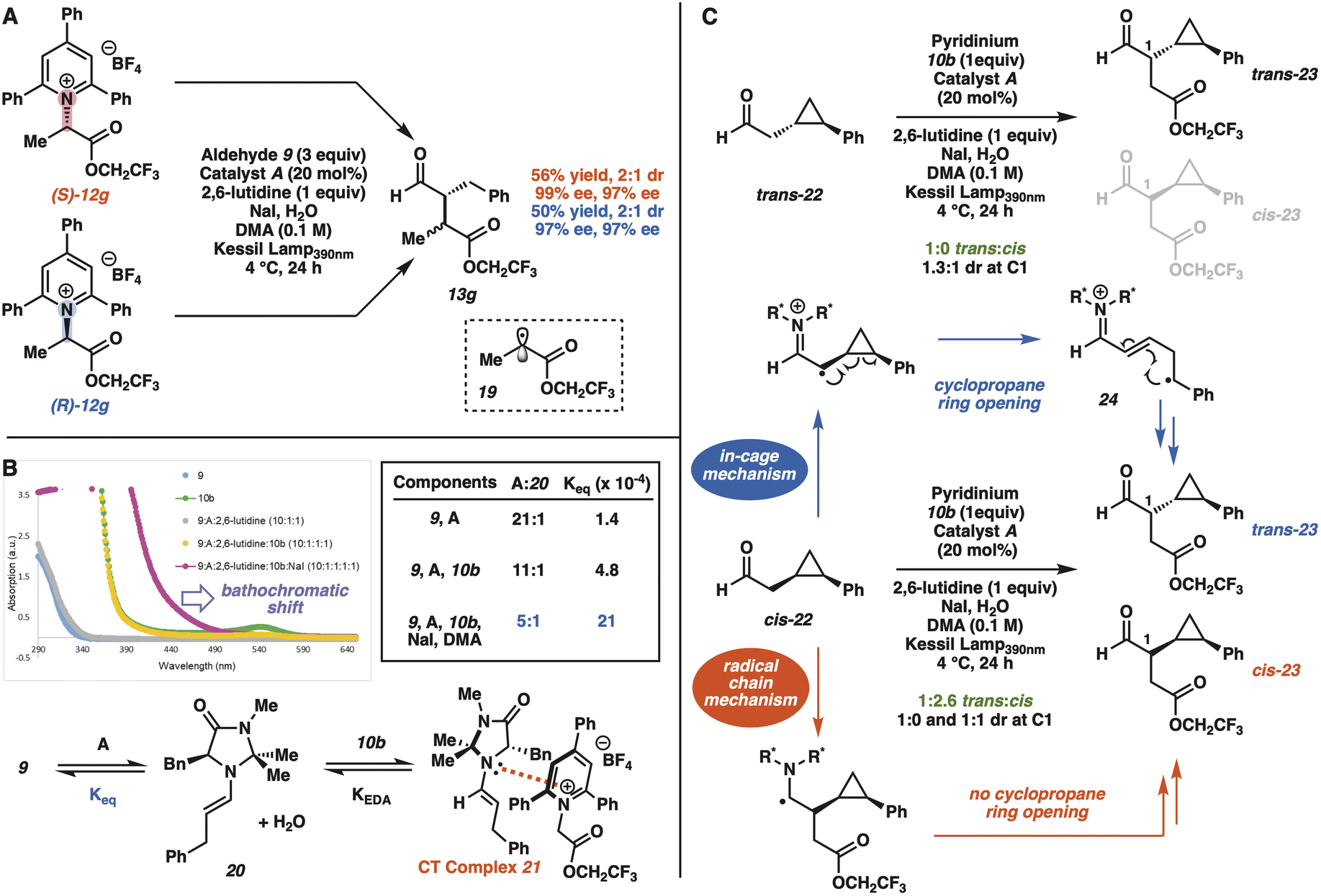 | ||
| Fig. 1 Mechanistic experiments. | ||
We were also interested in gaining insight into the role of NaI in the catalytic process. The use of 50 mol% NaI led to 58% isolated yield of the coupled product 11b. In addition, the pyridinium substrate 10b was not converted to the α-iodoester in the presence of NaI and absence of aldehyde.23 However, NaI caused a bathochromatic shift into the purple region of the absorption spectrum (Fig. 1B). Moreover, the inclusion of NaI had a profound impact on the equilibrium of enamine formation, presumably by affecting the equilibrium of CT complex 21 (Fig. 1B). Therefore, we believe NaI may affect the identity of the CT complex by forming a ternary complex with the catalytically generated enamine and pyridinium substrate.24
Next, we used the trans and cis isomers of the radical probe 22 as the aldehyde component to determine whether the enantioselective reaction proceeds through a radical chain or in-cage radical combination process (Fig. 1C and S7†).4g,5a Radical probe trans-22 exclusively formed the alkylation product trans-23 as a mixture of diastereomers at C1, which is consistent with either a radical chain or in-cage radical combination. With radical probe cis-22, the expectation was that an in-cage radical process would exclusively furnish the thermodynamically stable alkylation product trans-23via acyclic intermediate 24.5a Alternatively, the alkylation product cis-23 would form exclusively if the reaction proceeded through a radical chain.4g Surprisingly, starting from radical probe cis-22, we isolated both trans and cis isomers of alkylation product 23 in a 1:2.6 ratio.
These observations suggest that the catalytic enantioselective reaction may proceed simultaneously through two highly enantioselective processes (Scheme 3 and Fig. S7†): an in-cage radical combination mechanism (path a) and a radical chain mechanism (path b). Although we cannot rule out the possibility of a radical chain mechanism with cyclopropane ring opening as an off-cycle process, we believe the measured quantum yield of 4 may be more consistent with the co-existence of two distinct mechanisms.23
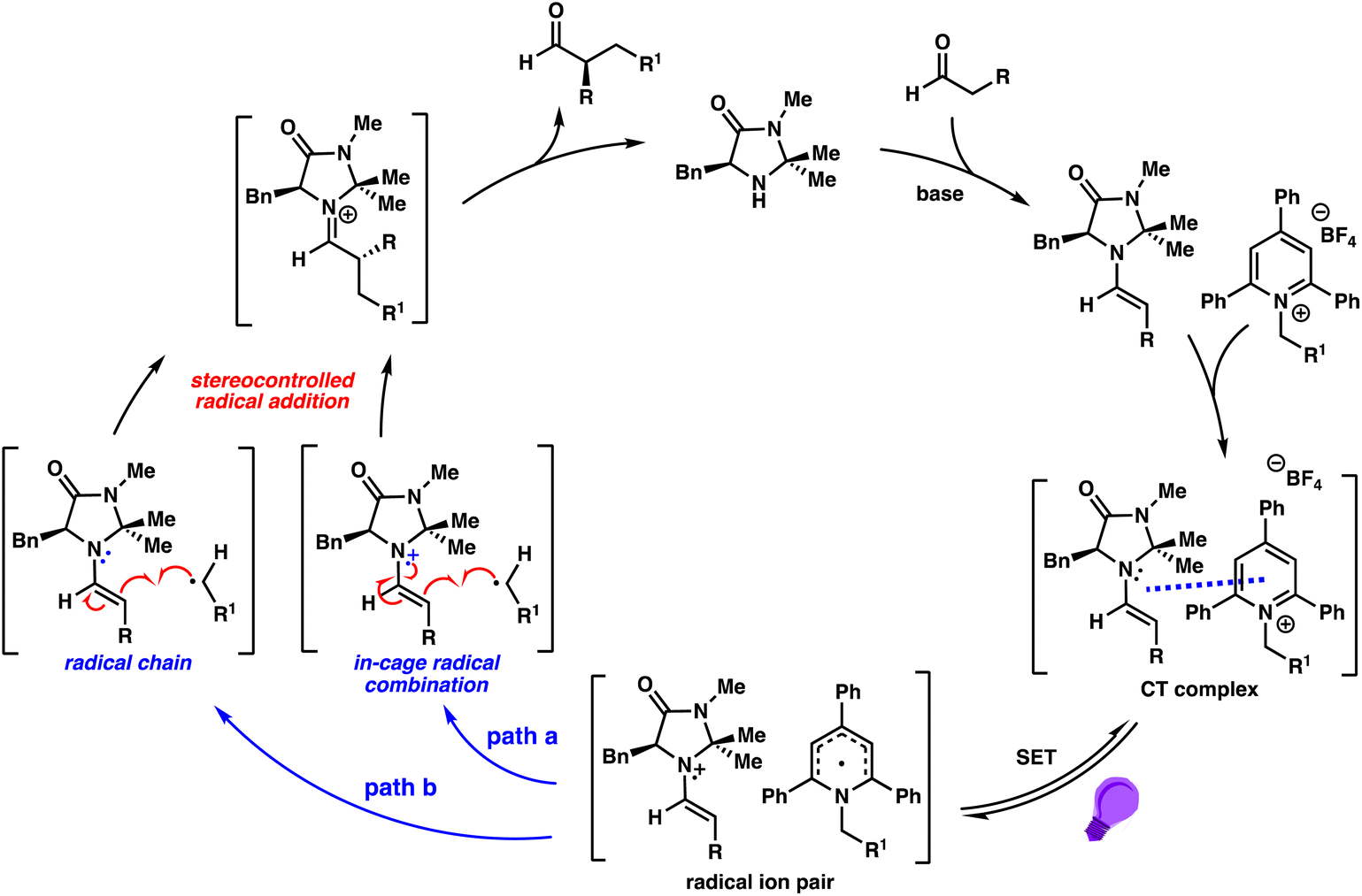 | ||
| Scheme 3 Proposed mechanism. | ||
Conclusions
In summary, we have developed a catalytic enantioselective alkylation of aldehydes with pyridinium salts derived from amino acids and other α-stabilized amines. The reaction is enabled by a visible light activated CT complex between electron-deficient pyridinium salts and electron-rich components of the reaction. The mild conditions are compatible with several functional groups, enabling the enantioselective synthesis of lignan natural products. Future studies will examine the mechanism of this process in more detail. We anticipate this approach may be extended to the photochemical catalytic enantioselective alkylation of several classes of carbonyl compounds with pyridinium salts based on other modes of catalysis.Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by W. W. Caruth Jr Endowed Scholarship, Bonnie Bell Harding Professorship in Biochemistry, Welch Foundation (I-1748), National Institutes of Health (R01GM102604), American Chemical Society Petroleum Research Fund (59177-ND1), Teva Pharmaceuticals Marc A. Goshko Memorial Grant (60011-TEV), and Sloan Research Fellowship. We also thank our diverse collection of lab members for creating a supportive environment for research.Notes and references
- (a) V. Farina, J. T. Reeves, C. H. Senanayake and J. J. Song, Chem. Rev., 2006, 106, 2734 CrossRef CAS PubMed; (b) N. A. McGrath, M. Brichacek and J. T. Njardarson, J. Chem. Educ., 2010, 87, 1348 CrossRef CAS; (c) R. M. Wilson and S. J. Danishefsky, Angew. Chem., Int. Ed., 2010, 49, 6032 CrossRef CAS; (d) W. H. Brooks, W. C. Guida and K. G. Daniel, Curr. Top. Med. Chem., 2011, 11, 760 CrossRef CAS PubMed; (e) D. J. Newman and G. M. Cragg, J. Nat. Prod., 2020, 83, 770 CrossRef CAS.
- (a) D. L. Hughes, in Alkylation of enolates, Springer-Verlag, 1999, pp. 1273–1294 Search PubMed; (b) T. Wirth, New strategies to α-alkylated α-amino acids, Wiley-VCH Verlag GmbH, 2000, pp. 26–33 Search PubMed; (c) K. Koga, Organomet. News, 2003, 3, 100 Search PubMed; (d) K. Maruoka and T. Ooi, Chem. Rev., 2003, 103, 3013 CrossRef CAS; (e) J. Eames and M. J. Suggate, Angew. Chem., Int. Ed., 2005, 44, 186 CrossRef CAS; (f) M. Braun and T. Meier, Synlett, 2006, 5, 661 CrossRef; (g) M. Ikunaka and K. Maruoka, in Asymmetric phase-transfer catalysis for the production of non-proteinogenic α-amino acids, Wiley-VCH Verlag GmbH & Co. KGaA, 2010, pp. 151–169 Search PubMed; (h) C. M. Reeves and B. M. Stoltz, in Catalytic enantioselective alkylation of prochiral ketone enolates, Wiley-VCH Verlag GmbH & Co. KGaA, 2012, pp. 1–10 Search PubMed; (i) S. Shirakawa and K. Maruoka, in Asymmetric α-functionalization of carbonyl compounds and alkylation of enolates, Georg Thieme Verlag, 2012, pp. 601–615 Search PubMed; (j) M. Remes and J. Vesely, In α-Alkylation of carbonyl compounds, John Wiley & Sons, Inc., 2013, pp. 267–312 Search PubMed; (k) J. C. Hethcox, S. E. Shockley and B. M. Stoltz, ACS Catal., 2016, 6, 6207 CrossRef CAS; (l) U. Kazmaier, Org. Chem. Front., 2016, 3, 1541 RSC; (m) S. G. Davies, A. M. Fletcher, P. M. Roberts and J. E. Thomson, Tetrahedron: Asymmetry, 2017, 28, 1842 CrossRef CAS; (n) A. Cruz, I. I. Padilla-Martinez and M. E. Bautista-Ramirez, Curr. Org. Synth., 2018, 15, 38 CrossRef CAS; (o) S. E. Denmark and G. L. Beutner, Angew. Chem., Int. Ed., 2008, 47, 1560 CrossRef CAS; (p) S. Mondal, F. Dumur, D. Gigmes, M. P. Sibi, M. P. Bertrand and M. Nechab, Chem. Rev., 2022, 122, 5842 CrossRef CAS; (q) J. Guo, X. Liu, C. He, F. Tan, S. Dong and X. Feng, Chem. Commun., 2018, 54, 12254 RSC; (r) H. Hu, J. Xu, W. Liu, S. Dong, L. Lin and X. Feng, Org. Lett., 2018, 20, 5601 CrossRef CAS; (s) Y. Luo, Q. Wei, L. Yang, Y. Zhou, W. Cao, Z. Su, X. Liu and X. Feng, ACS Catal., 2022, 12, 12984 CrossRef CAS; (t) J. Xu, Z. Zhong, M. Jiang, Y. Zhou, X. Liu and X. Feng, CCS Chem., 2020, 3, 1894 CrossRef.
- (a) D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77 CrossRef CAS PubMed; (b) D. A. Nagib, M. E. Scott and D. W. C. MacMillan, J. Am. Chem. Soc., 2009, 131, 10875 CrossRef CAS; (c) H.-W. Shih, M. N. Vander Wal, R. L. Grange and D. W. C. MacMillan, J. Am. Chem. Soc., 2010, 132, 13600 CrossRef CAS PubMed; (d) E. R. Welin, A. A. Warkentin, J. C. Conrad and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2015, 54, 9668 CrossRef CAS.
- (a) M. Neumann, S. Füldner, B. König and K. Zeitler, Angew. Chem., Int. Ed., 2011, 50, 951 CrossRef CAS PubMed; (b) M. Cherevatskaya, M. Neumann, S. Füldner, H. Christoph, K. Susanne, D. Stephan, P. Arno, Z. Kirsten and K. Burkhard, Angew. Chem., Int. Ed., 2012, 51, 4062 CrossRef CAS; (c) K. Fidaly, C. Ceballos, A. Falguières, M. S.-I. Veitia, A. Guy and C. Ferroud, Green Chem., 2012, 14, 1293 RSC; (d) P. Wu, P. C. He, J. Wang, X. Peng, X. Li, Y. An and C. Duan, J. Am. Chem. Soc., 2012, 134, 14991 CrossRef CAS PubMed; (e) P. Riente, A. M. Adams, J. Albero, E. Palomares and M. A. Pericàs, Angew. Chem., Int. Ed., 2014, 53, 9613 CrossRef CAS; (f) Y. Zhu, L. Zhang and S. Luo, J. Am. Chem. Soc., 2014, 136, 14642 CrossRef CAS; (g) A. Gualandi, M. Marchini, L. Mengozzi, M. Natali, M. Lucarini, P. Ceroni and P. G. Cozzi, ACS Catal., 2015, 5, 5927 CrossRef CAS; (h) X. Li, J. Wang, D. Xu, Z. Sun, Q. Zhao, W. Peng, Y. Li, G. Zhang, F. Zhang and X. Fan, ACS Sustainable Chem. Eng., 2015, 3, 1017 CrossRef CAS; (i) T. Rigotti, A. Casado-Sánchez, S. Cabrera, R. Pérez-Ruiz, M. Liras, V. A. de la Peña O'Shea and J. A. Alemán, ACS Catal., 2018, 8, 5928 CrossRef CAS; (j) A. Gualandi, M. Marchini, L. Mengozzi, H. T. Kidanu, A. Franc, P. Ceroni and P. G. Cozzi, Eur. J. Org. Chem., 2020, 1486 CrossRef CAS.
- (a) E. Arceo, I. D. Jurberg, A. Álvarez-Fernández and P. Melchiorre, Nat. Chem., 2013, 5, 750 CrossRef CAS PubMed; (b) M. Silvi, E. Arceo, I. D. Jurberg, C. Cassani and P. Melchiorre, J. Am. Chem. Soc., 2015, 137, 6120 CrossRef CAS PubMed; (c) A. Bahamonde and P. Melchiorre, J. Am. Chem. Soc., 2016, 138, 8019 CrossRef CAS PubMed; (d) G. Filippini, M. Silvi and P. Melchiorre, Angew. Chem., Int. Ed., 2017, 56, 4447 CrossRef CAS PubMed.
- (a) Y.-Q. Zou, F. M. Hörmann and T. Bach, Chem. Soc. Rev., 2018, 47, 278 RSC; (b) M. Silvi and P. Melchiorre, Nature, 2018, 554, 41 CrossRef CAS; (c) S. Roy, H. Paul and I. Chatterjee, Eur. J. Org. Chem., 2022, 25, e202200446 Search PubMed.
- (a) C. O. Tuck, E. Pérez, I. T. Horváth, R. A. Sheldon and M. Poliakoff, Science, 2012, 337, 695 CrossRef CAS PubMed; (b) T. M. Lammens, M. C. R. Franssen, E. L. Scott and J. P. M. Sanders, Biomass Bioenergy, 2012, 44, 168 CrossRef; (c) T. Yan, B. L. Feringa and K. Barta, Sci. Adv., 2017, 3, eaao6494 CrossRef PubMed.
- (a) A. R. Katritzky, R. H. Manzo, J. M. Lloyd and R. C. Patel, Angew. Chem., Int. Ed., 1980, 19, 306 CrossRef; (b) S. Sowmiah, J. M. S. S. Esperança, L. P. N. Rebelo and C. A. M. Afonso, Org. Chem. Front., 2018, 5, 453 RSC.
- (a) R. S. Mulliken, J. Am. Chem. Soc., 1950, 72, 600 CrossRef CAS; (b) R. S. Mulliken, J. Phys. Chem., 1952, 56, 801 CrossRef CAS; (c) J. K. Kochi, Angew. Chem., Int. Ed., 1988, 27, 1227 CrossRef; (d) R. A. Marcus, Angew. Chem., Int. Ed., 1993, 32, 1111 CrossRef; (e) S. V. Rosokha and J. K. Kochi, Acc. Chem. Res., 2008, 41, 641 CrossRef CAS.
- (a) C. G. S. Lima, T. M. Lima, M. Duarte, I. D. Jurberg and M. W. Paixão, ACS Catal., 2016, 6, 1389 CrossRef CAS; (b) G. E. M. Crisenza, D. Mazzarella and P. Melchiorre, J. Am. Chem. Soc., 2020, 142, 5461 CrossRef CAS; (c) Y.-Q. Yuan, S. Majumder, M.-H. Yang and S.-R. Guo, Tetrahedron Lett., 2020, 61, 151506 CrossRef CAS; (d) X. Cheng, A. Lei, T.-S. Mei, H.-C. Xu, K. Xu and C. Zeng, CCS Chem., 2022, 4, 1120 CrossRef CAS.
- A. R. Katritzky, G. Z. de Ville and R. C. Patel, Tetrahedron Lett., 1980, 21, 1723 CrossRef CAS.
- (a) N. F. Eweiss, A. R. Katritzky, P.-L. Nie and C. A. Ramsden, Synthesis, 1977, 1977, 634 CrossRef; (b) A. R. Katritzky, Tetrahedron, 1980, 36, 679 CrossRef CAS; (c) A. R. Katritzky and C. M. Marson, Angew. Chem., Int. Ed., 1984, 23, 420 CrossRef.
- (a) F.-S. He, S. Ye and J. Wu, ACS Catal., 2019, 9, 8943 CrossRef CAS; (b) Y. Pang, D. Moser and J. Cornella, Synthesis, 2020, 52, 489 CrossRef CAS; (c) S. L. Rössler, B. J. Jelier, E. Magnier, G. Dagousset, E. M. Carreira and A. Togni, Angew. Chem., Int. Ed., 2020, 59, 9264 CrossRef PubMed.
- (a) C. H. Basch, J. Liao, J. Xu, J. J. Piane and M. P. Watson, J. Am. Chem. Soc., 2017, 139, 5313 CrossRef CAS PubMed; (b) W. Guan, J. Liao and M. P. Watson, Synthesis, 2018, 50, 3231 CrossRef CAS; (c) J. Liao, W. Guan, B. P. Boscoe, J. W. Tucker, J. W. Tomlin, M. R. Garnsey and M. P. Watson, Org. Lett., 2018, 20, 3030 CrossRef CAS; (d) M. E. Hoerrner, K. M. Baker, C. H. Basch, E. M. Bampo and M. P. Watson, Org. Lett., 2019, 21, 7356 CrossRef CAS PubMed.
- (a) J. Liao, C. H. Basch, M. E. Hoerrner, M. R. Talley, B. P. Boscoe, J. W. Tucker, M. R. Garnsey and M. P. Watson, Org. Lett., 2019, 21, 2941 CrossRef CAS PubMed; (b) R. Martin-Montero, V. R. Yatham, H. Yin, J. Davies and R. Martin, Org. Lett., 2019, 21, 2947 CrossRef CAS PubMed; (c) S. Ni, C.-X. Li, Y. Mao, J. Han, Y. Wang, H. Yan and Y. Pan, Sci. Adv., 2019, 5, eaaw9516 CrossRef CAS PubMed; (d) H. Yue, C. Zhu, L. Shen, Q. Geng, K. J. Hock, T. Yuan, L. Cavallo and M. Rueping, Chem. Sci., 2019, 10, 4430–4435 RSC.
- S.-Z. Sun, C. Romano and R. Martin, J. Am. Chem. Soc., 2019, 141, 16197 CrossRef CAS PubMed.
- (a) F. J. R. Klauck, M. J. James and F. Glorius, Angew. Chem., Int. Ed., 2017, 56, 12336 CrossRef CAS; (b) M. Ociepa, J. Turkowska and D. Gryko, ACS Catal., 2018, 8, 11362 CrossRef CAS; (c) M.-M. Zhang and F. Liu, Org. Chem. Front., 2018, 5(23), 3443 RSC; (d) F. J. R. Klauck, H. Yoon, M. J. James, M. Lautens and F. Glorius, ACS Catal., 2019, 9, 236 CrossRef CAS; (e) J. Yi, S. O. Badir, L. M. Kammer, M. Ribagorda and G. A. Molander, Org. Lett., 2019, 21, 3346 CrossRef CAS PubMed; (f) Z.-F. Zhu, M.-M. Zhang and F. Liu, Org. Biomol. Chem., 2019, 17, 1531 RSC; (g) G. R. Mathi, Y. Jeong, Y. Moon and S. Hong, Angew. Chem., Int. Ed., 2020, 59, 2049 CrossRef CAS PubMed.
- (a) J. Hu, G. Wang, S. Li and Z. Shi, Angew. Chem., Int. Ed., 2018, 57, 15227 CrossRef CAS PubMed; (b) F. Sandfort, F. Strieth-Kalthoff, F. J. R. Klauck, M. J. James and F. Glorius, Chem.–Eur. J., 2018, 24, 17210 CrossRef CAS PubMed; (c) J. Wu, L. He, A. Noble and V. K. Aggarwal, J. Am. Chem. Soc., 2018, 140, 10700 CrossRef CAS PubMed; (d) J. Hu, B. Cheng, X. Yang and T.-P. Loh, Adv. Synth. Catal., 2019, 361, 4902 CrossRef CAS; (e) M. J. James, F. Strieth-Kalthoff, F. Sandfort, F. J. R. Klauck, F. Wagener and F. Glorius, Chem.–Eur. J., 2019, 25, 8240 CrossRef CAS; (f) J. Wu, P. S. Grant, X. Li, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2019, 58, 5697 CrossRef CAS PubMed; (g) M. Lübbesmeyer, E. G. Mackay, M. A. R. Raycroft, J. Elfert, D. A. Pratt and A. Studer, J. Am. Chem. Soc., 2020, 142, 2609 CrossRef PubMed.
- A conceptually distinct study was recently reported for the Ni-catalyzed enantioselective hydroamination of amino alkenes with pyridinium salts: S.-Z. Sun, Y.-M. Cai, D.-L. Zhang, J.-B. Wang, H.-Q. Yao, X.-Y. Rui, R. Martin and M. Shang, J. Am. Chem. Soc., 2022, 144, 1130 CrossRef CAS PubMed.
- F. Parsaee, M. C. Senarathna, P. B. Kannangara, S. N. Alexander, P. D. E. Arche and E. R. Welin, Nat. Rev. Chem., 2021, 5, 486 CrossRef CAS.
- (a) C.-S. Zhang, L. Bao, K.-Q. Chen, Z.-X. Wang and X.-Y. Chen, Org. Lett., 2021, 23, 1577 CrossRef CAS PubMed; (b) M.-C. Fu, R. Shang, B. Zhao, B. Wang and Y. Fu, Science, 2019, 363, 1429 CrossRef CAS PubMed.
- For a recent review of photocatalysis in natural product synthesis, see: M. S. Galliher, B. J. Roldan and C. R. J. Stephenson, Chem. Soc. Rev., 2021, 50, 10044 RSC.
- See ESI† for details.
- We cannot rule out the possibility that DMA and other reaction components, such as 2,6-lutidine, also impact the nature of the CT complex.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization data, and spectral data (PDF). See DOI: https://doi.org/10.1039/d2sc05654b |
| This journal is © The Royal Society of Chemistry 2023 |