
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTemplate-directed 2D nanopatterning of S = 1/2 molecular spins†
Kyungju
Noh
ab,
Luciano
Colazzo
*ac,
Corina
Urdaniz
ac,
Jaehyun
Lee
ab,
Denis
Krylov
ac,
Parul
Devi
ac,
Andrin
Doll
 d,
Andreas J.
Heinrich
ab,
Christoph
Wolf
*ab,
Fabio
Donati
*ab and
Yujeong
Bae
*ab
d,
Andreas J.
Heinrich
ab,
Christoph
Wolf
*ab,
Fabio
Donati
*ab and
Yujeong
Bae
*ab
aCenter for Quantum Nanoscience (QNS), Institute of Basic Science (IBS), 03760 Seoul, Republic of Korea. E-mail: colazzo.luciano@qns.science; wolf.christoph@qns.science; donati.fabio@qns.science; bae.yujeong@qns.science
bDepartment of Physics, Ewha Womans University, 03760 Seoul, Republic of Korea
cEwha Womans University, 03760 Seoul, Republic of Korea
dSwiss Light Source (SLS), Paul Scherrer Institut (PSI), 5232 Villigen, Switzerland
First published on 26th January 2023
Abstract
Molecular spins are emerging platforms for quantum information processing. By chemically tuning their molecular structure, it is possible to prepare a robust environment for electron spins and drive the assembly of a large number of qubits in atomically precise spin-architectures. The main challenges in the integration of molecular qubits into solid-state devices are (i) minimizing the interaction with the supporting substrate to suppress quantum decoherence and (ii) controlling the spatial distribution of the spins at the nanometer scale to tailor the coupling among qubits. Herein, we provide a nanofabrication method for the realization of a 2D patterned array of individually addressable Vanadyl Phthalocyanine (VOPc) spin qubits. The molecular nanoarchitecture is crafted on top of a diamagnetic monolayer of Titanyl Phthalocyanine (TiOPc) that electronically decouples the electronic spin of VOPc from the underlying Ag(100) substrate. The isostructural TiOPc interlayer also serves as a template to regulate the spacing between VOPc spin qubits on a scale of a few nanometers, as demonstrated using scanning tunneling microscopy, X-ray circular dichroism, and density functional theory. The long-range molecular ordering is due to a combination of charge transfer from the metallic substrate and strain in the TiOPc interlayer, which is attained without altering the pristine VOPc spin characteristics. Our results pave a viable route towards the future integration of molecular spin qubits into solid-state devices.
New conceptsWe demonstrate the fabrication of a surface-supported array of VOPc molecular spin qubit candidates with controlled spin–spin distance, electronic decoupling from the supporting substrate, and individual addressability. Achieving these three properties at once represents a major step forward with respect to previously investigated molecular spin qubit systems. In particular, realizing a minimal spacing of a few nanometers between spin centres strikes the balance between minimizing spin–spin decoherence whilst maintaining weak spin–spin interactions, which is a crucial trade-off in the design of interacting qubits systems. Throughout the templating process that is realized using an isostructural TiOPc monolayer, the VOPc molecular spins remain intact and well decoupled from the substrate, indicating the potential of these molecules to operate as spin qubits in such a molecular array. Using ab initio calculations, we identified the templating mechanism to stem from a thermally induced surface modification that enforces the minimal spacing between the spins through a combination of charge transfer from the surface and induced strain in the molecular layer. Our results establish an approach to realize molecular spin qubit platforms that are interfaceable with solid state devices. |
Introduction
Employing magnetic molecules as hosts for spin-based qubits offers an advantageous route towards quantum information processing. The rational design of molecular spin precursors can be used to fine-tune the spin environment and mitigate decoherence sources, e.g. spin–phonon coupling or magnetic noise coming from neighbouring nuclear and electron spins.1–4 Furthermore, molecules can be transferred onto a surface as individual units or self-assembled in 2D architectures, and individually addressed using high spatial resolution techniques such as scanning tunnelling microscopy (STM).5–7 A chemical bottom-up approach to produce extended arrays of molecular quantum units provides a route to tackling one of the main challenges for the implementation of molecular spins in the solid state, i.e. the scaling up to a large number of qubits with controllable interactions between them.8,9 However, as the magnetic noise increases with increasing spin density, balancing the coupling strength between the qubits by controlling the spin–spin distance at the atomic scale is a key factor towards multiqubit platforms.Numerous molecule-based spin qubits exhibit long spin coherence time when measured in magnetically diluted ensembles.10–12 As an example, spin-1/2 Vanadyl Phthalocyanine (VOPc) proved to be a good molecular qubit,13 with a spin coherence time of up to 1 μs at room temperature (RT) when diluted in the isostructural diamagnetic matrix of Titanyl Phthalocyanine (TiOPc). The long spin coherence time of VOPc stems from the limited overlap between the electron spin at the V dxy-orbital and the ligand's molecular orbitals, which provides decoupling from vibrations and neighbouring nuclear spins.14 However, the actual integration of VOPc molecular qubits into solid state quantum devices poses additional challenges. Firstly, the interaction with the substrate introduces an additional source of quantum decoherence that needs to be mitigated by suitable decoupling strategies. Secondly, an atomically precise spacing between the spin centres is required to propagate the spin–spin interactions among neighbouring molecular qubits.
The structural, electronic, and magnetic properties of VOPc on various surfaces have been studied using STM and X-ray magnetic circular dichroism (XMCD),15–19 showing various degrees of molecule–substrate interactions depending on the nature of the surface, ranging from charge transfer16 and chemical bonding17 on metallic substrates to the generation of Yu–Shiba–Rusinov states on superconductors.19 These investigations provide remarkable case studies to address the behaviour of a molecular spin qubit on various surfaces, which shows that such strong molecule–substrate interactions may potentially mask peculiar molecular spin attributes. Recently, weakly bound VOPc molecules on top of a graphene16,20 have shed light on the possibility of preserving the spin character of the molecular qubit by reducing the hybridization with the surface electrons. Besides these recent studies, however, the properties of surface-supported molecular spins in a decoupled environment and their use as building blocks in the realization of 2D nanopatterns remain largely unexplored.
In this work, we provide a 2D nanofabrication approach towards the engineering of a nanometer-spaced, regular array of molecular spin qubits. We combine STM, XMCD, and density functional theory (DFT) to demonstrate that the introduction of a diamagnetic TiOPc monolayer (ML) effectively decouples the VOPc molecular spins from the Ag(100) substrate electrons. Furthermore, a mild thermal treatment promotes the generation of uniformly spaced VOPc molecular patterns with a separation of ∼4.3 nm. Our calculations provide further insights into the concurrent variations of the molecular adsorption geometry that occurs within this regular arrangement and the underlying mechanism of thermally induced surface modification. Ultimately, we identify the related strain induced in the TiOPc layer as the driving mechanism for the long-range ordering arrangement, which induces minimal changes in the VOPc organic scaffold while preserving the V d-orbital structure and the relevant molecular spin feature.
Results and discussion
Our on-surface fabrication protocol involves organizing a VOPc molecular spin architecture on top of a diamagnetic TiOPc templating layer. The two molecules are isostructural (see the molecular model in Fig. 1a), with the only difference being the metal enclosed in the molecular scaffold. As shown in Fig. 1b, a closely packed TiOPc monolayer is readily obtained by depositing the molecules directly on Ag(100) at RT. Analogous closely packed molecular layers were also reported on other metallic surfaces like Au(111), Ag(111), and Cu(111).15,21 The dark and bright appearance of the molecules in the constant-current STM image can be attributed to the TiOPc in two different adsorption configurations, namely oxygen pointing to the vacuum side (O-up) and oxygen pointing to the surface (O-down), respectively.22 We find the as-deposited TiOPc layer to have a ratio of O-up and O-down molecules close to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, while annealing the sample at 570 K increases the abundance of O-up species (see Fig. S3, ESI†). These observations are in line with previous experiments on TiOPc/Ag(111)23 and indicate a lower adsorption energy for the O-up molecules. The TiOPc molecules show an intermolecular spacing of 1.44 nm as determined by low-energy electron diffraction (LEED) measurements (Fig. S1, ESI†), in agreement with previous reports,21 and are azimuthally rotated with respect to the molecular unit cell vector by about 26 ± 2 degrees (Fig. S2, ESI†).
:1, while annealing the sample at 570 K increases the abundance of O-up species (see Fig. S3, ESI†). These observations are in line with previous experiments on TiOPc/Ag(111)23 and indicate a lower adsorption energy for the O-up molecules. The TiOPc molecules show an intermolecular spacing of 1.44 nm as determined by low-energy electron diffraction (LEED) measurements (Fig. S1, ESI†), in agreement with previous reports,21 and are azimuthally rotated with respect to the molecular unit cell vector by about 26 ± 2 degrees (Fig. S2, ESI†).
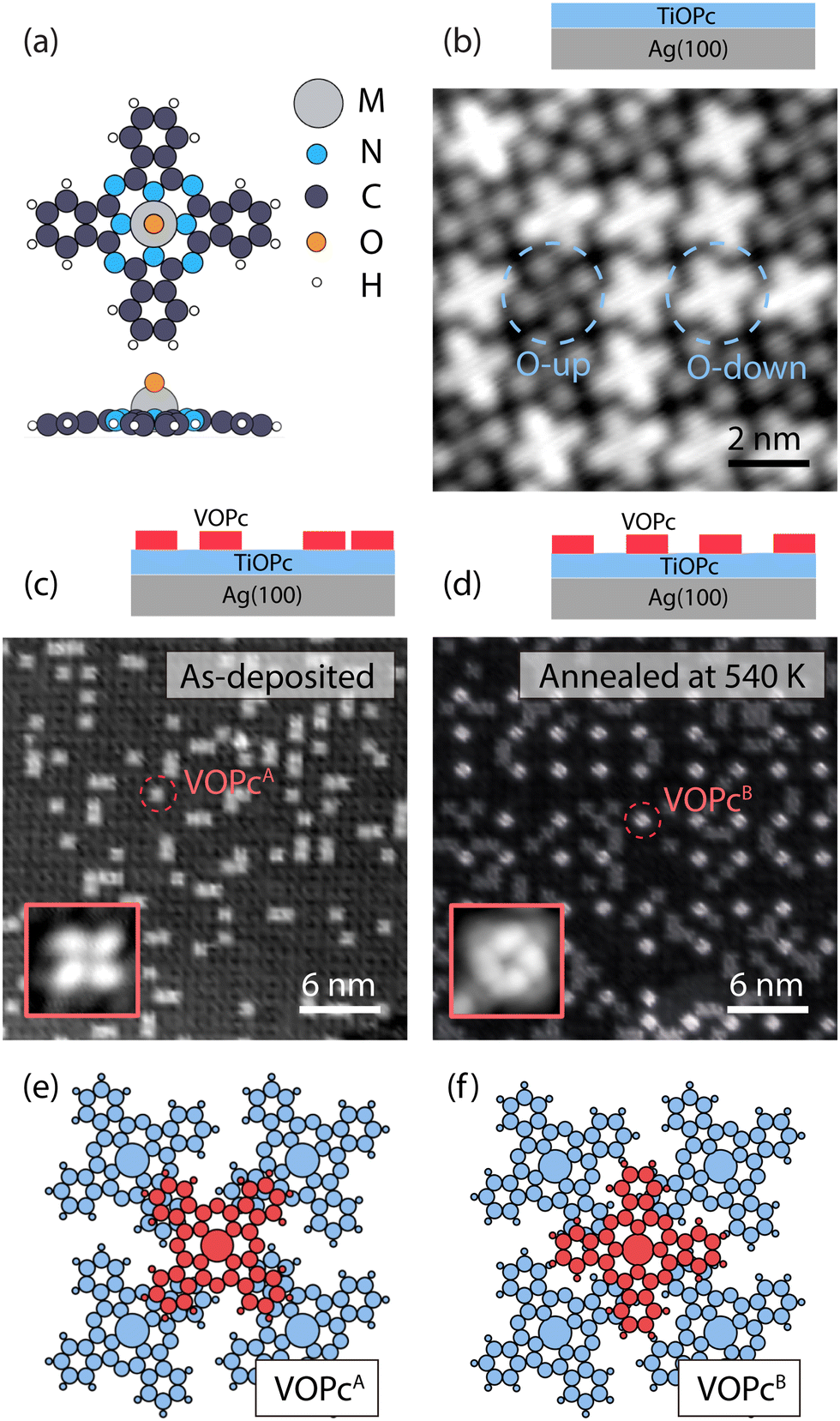 | ||
| Fig. 1 Metallo-phthalocyanine 2D architectures on Ag(100). (a) Molecular structure of a single non-planar MOPc molecule (M = Ti, V; upper: top view, lower: side view). (b) STM image of 1 ML TiOPc on Ag(100). The dashed circles show TiOPc molecules with two different adsorption configurations on Ag(100): Oxygen atom toward vacuum (O-up) and oxygen atom toward Ag(100) (O-down) (scan size: 10 × 10 nm2, VDC = 100 mV, Iset = 50 pA). (c) As-deposited and (d) post-annealed VOPc molecules with sub-ML coverage on 1 ML TiOPc on Ag(100). In addition to the VOPc molecules observed before the annealing (VOPcA), another type of VOPc molecule denoted as VOPcB appears after the thermal treatment (scan size: 30 × 30 nm2, VDC = 500 mV, Iset = 100 pA; inset: scan size of 2 × 2 nm2, VDC = 500 mV, Iset = 100 pA). (e) Schematic of the adsorption configurations of VOPcA and (f) VOPcB (red) on TiOPc (blue). | ||
Following the deposition of 1 ML TiOPc, with an entirely covered Ag(100) surface, the VOPc molecules were deposited at an approximately 0.3 ML coverage while maintaining the substrate at RT (Fig. 1c). The VOPc molecules on the TiOPc layer adsorb at the hollow sites of the TiOPc layer at an azimuthal angle of ∼46 ± 3 degrees with respect to the molecular unit cell vector of the TiOPc underneath (Fig. S2, ESI†) and show no recognizable short or long-range order. Hereafter, this configuration will be referred to as VOPcA.
After annealing the sample at 540 K, we observe the occurrence of an additional type of VOPc on the TiOPc layer (see Fig. 1d). Whilst some of VOPcA molecules remain unchanged, the newly appeared species (henceforth labeled as VOPcB) have a substantially different appearance in the constant-current STM image. VOPcB molecules display a larger apparent height compared to VOPcA when imaged at a sample bias of +0.5 V (Fig. 1d). They are organized in a homogeneously spaced square arrangement with a minimum distance of ∼4.3 nm among nearest neighbouring VOPcB molecules, which is 3 times of the unit cell length of the underlying TiOPc molecular layer. Similar to VOPcA, VOPcB adsorbs at the hollow sites between 4 underlying TiOPc molecules. The VOPcB molecules, however, have a 45-degrees azimuthal rotation when compared to the VOPcA counterpart (see insets in Fig. 1c and d). The existence of two different adsorption geometries implies two different stacking schemes for the phthalocyanine (Pc) scaffolds. The tentative molecular model of the adsorption configuration of VOPcA considers the overlap of the VOPcA isoindole units (the Pc arms) with the underlying TiOPc molecules in a π–π staggered stacking configuration (see Fig. 1e). On the other hand, the stacking of VOPcB molecules is interpreted as a quasi in-between stacking of the VOPc isoindoles with the underlying TiOPc molecules (Fig. 1f). The local C–H dipoles of the Pc periphery are responsible for the VOPc azimuthal rotations with respect to the underlying layer. A precise overlap between benzene rings would place the C–H dipoles in a condition of repulsive quadrupole interaction.24–26 However, such effect is attenuated in the rotated configuration of VOPcB.
The formation of VOPcB occurs under specific temperature and coverage. Following the aforementioned behaviour of TiOPc layer on the Ag(100) surface, by annealing at 570 K, we observe a correlation between the flip of TiOPc O-down into O-up in the 1st layer and the disappearance of VOPcB in favour of a significant regeneration of VOPcA species. This evidence indicates that TiOPc O-down molecules are needed to obtain VOPcB. In addition, while both VOPcA and VOPcB coexist at a VOPc coverage of 0.25 ML, only VOPcA is observed above 0.5 ML. The low coverage required for VOPcB formation suggests that the ordering mechanism may be driven by a modification of the TiOPc layer occurring over the scale of a few molecular units. To this extent, close-up images around VOPcB show a noticeable distortion of the surrounding TiOPc molecules (see Fig. S4, ESI†). We speculate that the accumulated strain in the TiOPc layer may not be energetically favourable when the VOPc coverage exceeds a critical VOPc coverage.
These results indicate that the pristine TiOPc layer acts as a template for the formation of the VOPcB phase by promoting the organization of the decoupled molecules. In addition, we find an analogous behaviour for the system composed of purely TiOPc molecules, with as-deposited TiOPcA-like species on a filled TiOPc layer transforming into TiOPcB-like species after thermal annealing (see Fig. S5, ESI†). Such observation suggests that the oxo ligand, which is a common feature in both VOPc and TiOPc molecules, plays a direct role in the formation of the long range ordered species. The underlying mechanism of VOPcB formation will be clarified later, with the support of DFT calculations.
In order to explore the magnetic properties of VOPc before and after the formation of the VOPcB phase, we performed X-ray absorption spectroscopy (XAS) and near edge X-ray absorption fine structure (NEXAFS) at the EPFL/PSI X-treme beamline of the Swiss Light Source.27 The spectra were measured over the L2,3 edge of V measured at T = 2.5 K and B = 6.8 T, with both photon beam and magnetic field perpendicular to the sample surface (see methods in the ESI†). The sample prepared with 0.25 ML of VOPc on top of 1 ML TiOPc (Fig. 2a, blue solid line) shows the distinctive VOPc total XAS.17–20,28 The three well-separated peaks at the L3 edge are generated by the excitation from the 2p3/2 core level to the crystal-field split 3d orbitals.28 Conversely, excitations from the 2p1/2 levels are merged into a single broad peak at the L2 edge as a result of the shorter lifetime of the excited states due to the L2–L3 Coster–Kronig decay.29 The XMCD, obtained from the difference of the two circular polarizations, indicates sizable magnetic moments localized at the 3d orbitals, as shown in Fig. 2b (blue solid line).19,20,28
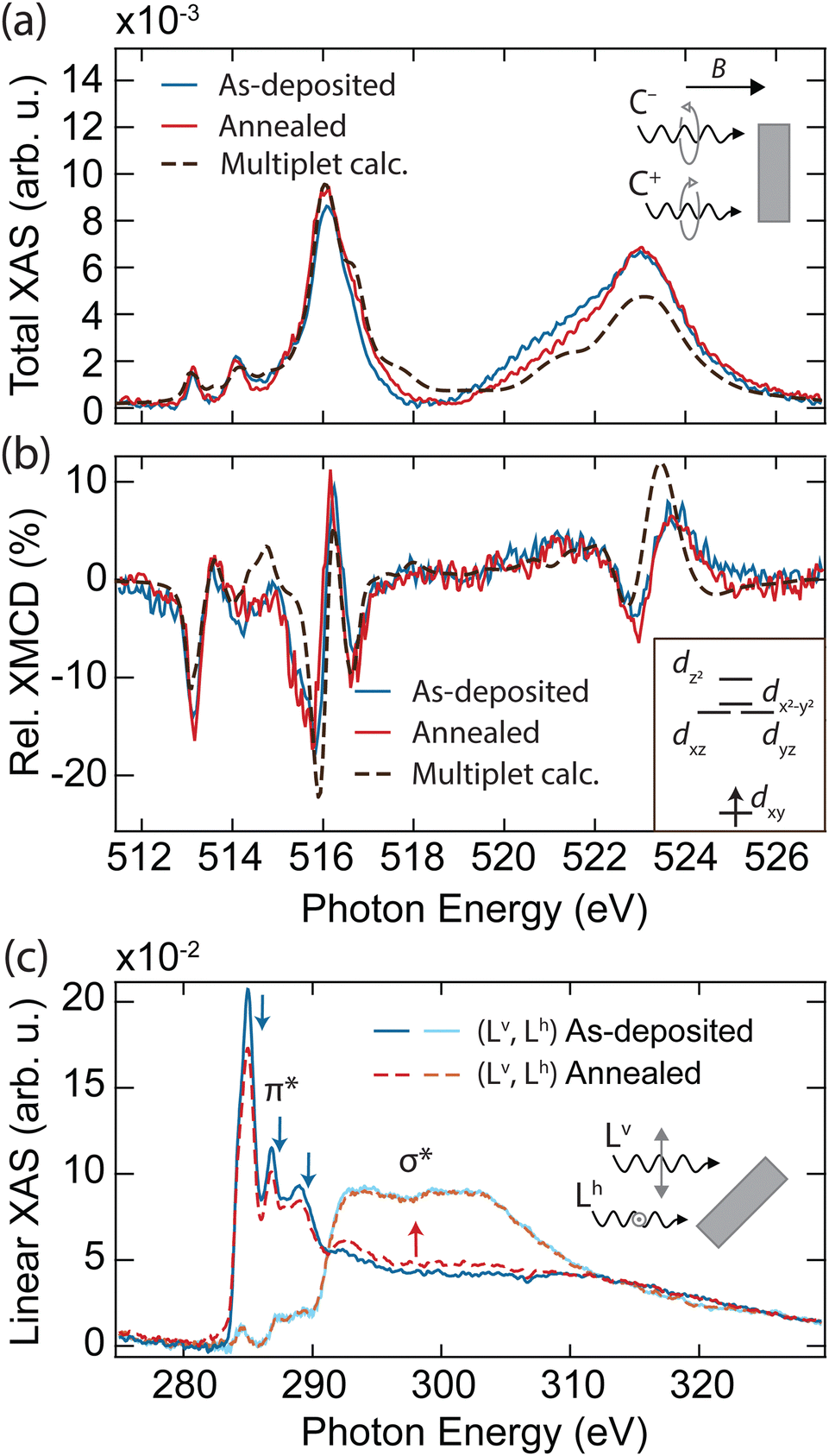 | ||
| Fig. 2 X-ray absorption spectroscopy of as-deposited and annealed VOPc/TiOPc layers on Ag(100). (a) Sum and (b) difference of the two circular polarization (right: C+, left: C−) giving the total XAS and XMCD at the V L2,3 edges (T = 2.5 K, B = 6.8 T, normal incidence). Dashed lines show the fit using a multiplet calculation. Inset in (b): electronic configuration and d orbital splitting obtained from the multiplet calculations. (c) Near-edge linear XAS at the C K edge showing individual vertical (Lv) and horizontal (Lh) polarization signals (T = 300 K, B = 0.05 T, grazing incidence). Spectral regions related to the π* and σ* resonances are indicated. Blue (red) arrows indicate the features whose intensity decreases (increases) upon annealing. In all panels, the absorption intensity is shown after subtraction of the absorption background from the substrate and indicated as the relative variation with respect to the pre-edge signal, see methods in the ESI.† The XMCD is expressed in percent over the total XAS signal (coverage of TiOPc = 1.15 ML, coverage of VOPc = 0.25 ML). | ||
To obtain further insight into the electronic and spin configuration of the V centre, we fit the experimental results with simulated spectra from multiplet calculations using the Quanty code.30 The fit was performed using a Bayesian optimization algorithm over a set of angular dependent circular and linear absorption spectra simultaneously (Fig. S7, ESI†), with the electron–electron interaction and on-site orbital energy as free parameters. This procedure allows us to infer the orbital occupation and splitting shown in the inset of Fig. 2b. In agreement with previous works,17–20,28 we find the V centre in a tetravalent configuration (V4+) with a single unpaired electron on the dxy orbital, well separated from the other d levels by more than 2.2 eV. Remarkably, both XAS and XMCD show no significant variations upon annealing and formation of the VOPcB molecules (Fig. 2a and b, red solid line, and Fig. S6, ESI†), indicating that the spin properties and crystal field at the V4+ centre remain essentially unchanged due to the extremely high localization of the unpaired electron into a well isolated atomic orbital. In addition, the marked difference with the XMCD spectra acquired on VOPc in direct contact with the Ag(100) substrate allows us to exclude the possible migration of the VOPc to the TiOPc layer (see Fig. S8, ESI†).
Linearly polarized absorption spectra at the C K-edge recorded with the sample surface at 60 degrees with the incident beam probe the ligand's orbital structure from both TiOPc and VOPc molecules and provide insights into the molecular planarity with respect to the surface. As shown in Fig. 2c, the spectra show characteristic features of excitations to π* and σ* orbitals, whose amplitude is strongly dependent on the direction of the X-ray polarization. The C K-edge spectrum displays π* resonances with maximum intensity in the vertical (Lv) polarization that are vastly attenuated in the horizontal (Lh) polarization, whereas the opposite occurs for σ* resonances. The strong linear dichroism of the π* and σ* resonances confirms that VOPc and TiOPc molecules lie with the molecule plane parallel to the surface,31 as was also observed in our STM measurements. According to previous reports, the peaks at 285.0 eV, 286.8 eV, and 289.0 eV (indicated by blue arrows in Fig. 2c) are generated by π* excitations located mainly at the phenyl subunits and pyrrolic molecular core, while the broad structures above 290 eV (red arrow) originate from the electronic transitions to the σ* of the isoindole subunits.32–34
Upon annealing, the amplitude of the signal at the π* orbitals detected in Lv polarization decreases, with part of the K edge intensity redistributing in the spectral region characteristic of the σ* bonds. A similar effect is also visible in the N K-edge recorded on the same sample (see Fig. S9, ESI†). Conversely, we observe no variation in the energy position and width of the NEXAFS peaks, and no emergence of new peaks, which would occur in the case of thermal degradation altering the molecular bonding scheme and orbital sequence.35,36 At the same time, no significant changes are observed in the linear absorption of Ti, V, and O edges, allowing us to ascertain the absence of demetallation and detachment of the apical oxygen from the molecule (see Fig. S10, ESI†). Hence, we interpret the changes in the C and N spectra as a consequence of the reduced molecular planarity of the TiOPc layer surrounding VOPcB molecules, as is also suggested by our STM images (see Fig. S4, ESI†).33,37,38 It is worth noting that the redistribution between π* and σ* features is only observed when the amount of VOPc molecules is close to 0.25 ML, which corresponds to the formation of the long-range ordered VOPcB molecules and the related strain into the TiOPc layer. For larger VOPc coverages, annealing at the same temperature does not induce any change in the C K edge, which correlates well with the absence of VOPcB molecules in such samples (Fig. S11, ESI†).
To gather further details about the electronic structure of VOPc molecules in the two different configurations (VOPcA and VOPcB), we compare STM images and differential conductance (dI/dV) measurements at the single molecular level with DFT. Both VOPc and TiOPc molecules adsorbed directly on the Ag(100) surface exhibit nearly featureless dI/dV curves (Fig. S12, ESI†). The lack of dI/dV features is not unusual for molecules in direct contact with a metallic surface, and is highly dependent on the degree of hybridization of the molecular scaffold with surface electrons.39–41 In contrast, as-deposited VOPc molecules adsorbed on top of the TiOPc decoupling layer show distinct peaks in dI/dV curves, which can be used to map their molecular orbital structure.40,42 The dI/dV measured at the centre of VOPcA shows resonance peaks at −1.65 V and 1.05 V (peaks A and B in Fig. 3a). On the other hand, when the tip is positioned at one of the peripheral carbon rings, two distinct peaks appear at −1.55 V and 0.95 V (peaks A′ and B′ in Fig. 3a). These two features can be attributed to the highest occupied (HOMO) and lowest unoccupied molecular orbitals (LUMO), respectively, resulting in a measured gap of 2.50 eV. Our DFT calculations indicate O-down as the energetically favoured adsorption configuration for VOPc on TiOPc, with the oxo ligand pointing towards the Ag surface. The resonances in the simulated dI/dV spectra (Fig. 3b) follow the same trend observed in the experiment, with a larger energy separation at the centre compared to the periphery of the molecules, albeit with a lower HOMO and LUMO gap owing to the well-known self-interaction problem of DFT.43,44
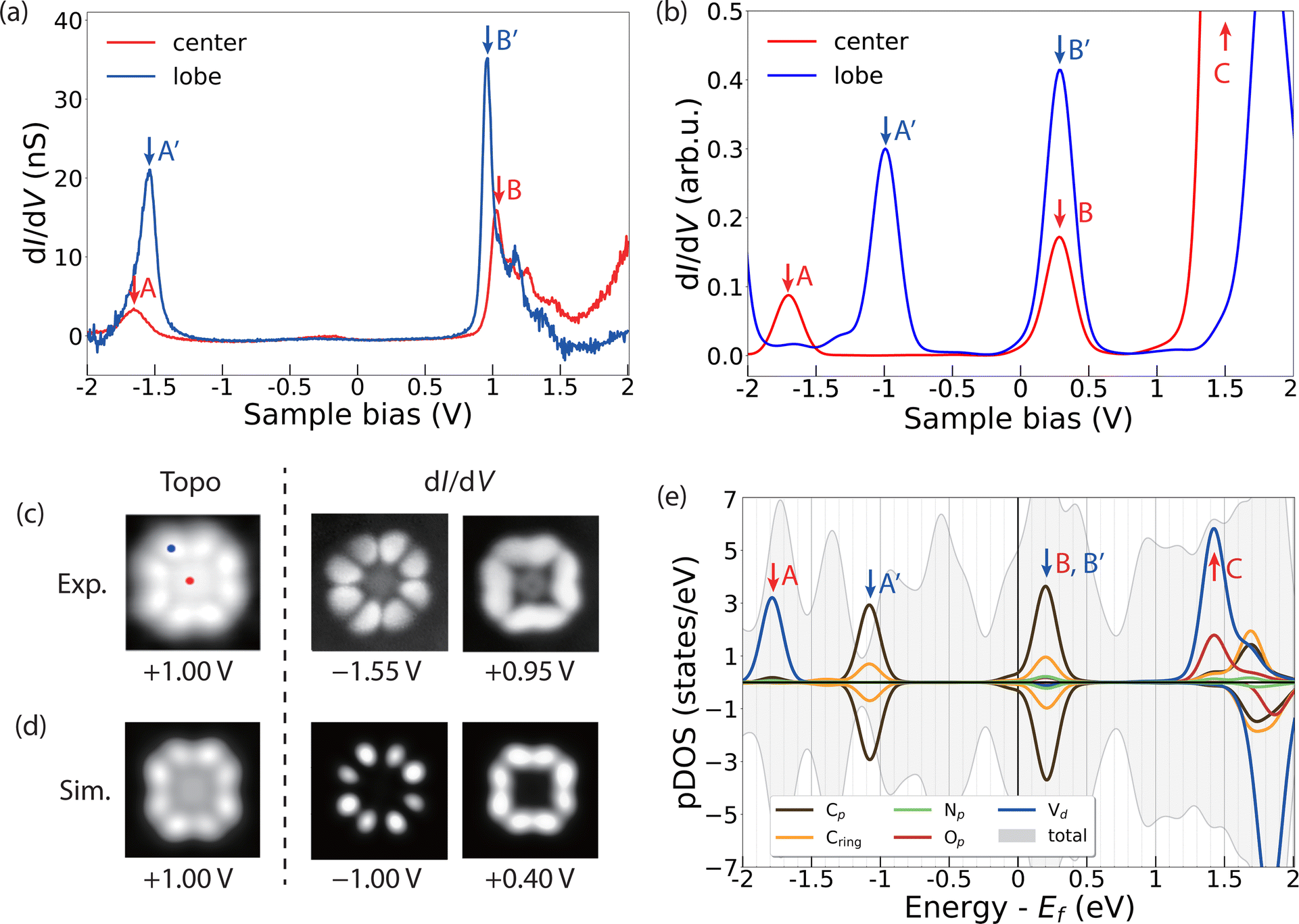 | ||
| Fig. 3 Orbital structure of a single VOPcA molecule. (a) dI/dV curves measured at the centre (red) and periphery (blue) of VOPcA (VDC = 1 V, Iset = 50 pA), and (b) simulated dI/dV curves at corresponding sites. (c) Experimental and (d) simulated constant current topography and dI/dV maps at bias voltages VDC, corresponding to the resonance peaks of the respective dI/dV curves (image size: 2.3 × 2.3 nm2, Iset = 100 pA). The tip positions while measuring dI/dV curves in (a) are marked as red and blue dots in the STM image. (e) pDOS plot of VOPcA on TiOPc slab projected on the orthogonalized atomic basis set. | ||
To assign the orbital contributions to the spectral features, we record conductance maps at the corresponding biases of the HOMO and the LUMO (Fig. 3c). The conductance maps reveal a well-defined distribution of electronic states, preferentially located on the organic molecular scaffold, which are reproduced by our simulated differential conductance maps at the corresponding resonance energies (Fig. 3d). To unravel the origin of these states, we project the density of states on the orthogonalized atomic basis set as shown in Fig. 3e. The partial density of states (pDOS) of these features reveals a dominant contribution of the C π and π* states to the frontier molecular orbitals, with minor contributions from N states. The singly occupied V 3dxy state, which contributes to the dI/dV measured at the centre of the molecule (peak A in Fig. 3a and b), is localized on a lower energy level (peak A in Fig. 3e). This calculated electronic configuration agrees well with that inferred from X-ray absorption measurements and multiplet calculations (see the inset in Fig. 2b).
Our calculations further show that the molecular orbitals of the VOPc on TiOPc/Ag(100) are essentially identical to those of an isolated free-standing molecule, with the TiOPc layer and Ag(100) substrate only affecting their relative energy with respect to the Fermi level of the system (Fig. S14 and Table S2, ESI†). Together with the sharp dI/dV features and well-resolved conductance maps, this indicates that the TiOPc layer effectively decouples the VOPc states from the Ag(100) substrate electrons.
The dI/dV spectral features of VOPcB are remarkably different from those of VOPcA as shown in Fig. 4a. The spectrum shows two broad features at about ±0.5 V, indicating a rearrangement of the orbitals with respect to the Fermi level possibly induced by a charge transfer to the molecules. As previously discussed, the XAS measurements at the C, N, and O K edges, as well as at the V and Ti L2,3 edges can rule out a chemical modification of the organic scaffold (Fig. 2c and Fig. S8–S11, ESI†). In addition, our DFT calculations show that such a change of the electronic structure cannot be induced by the sole azimuthal rotation of the VOPc molecule (Fig. S15, ESI†). Conversely, a charge transfer of 0.1 electrons occurs after inserting an additional Ag atom between the Ag surface and the O atom of VOPc (Fig. 4b). The coordination of a molecule to an additional surface metal atom can occur after thermal treatment, as observed in previous experiments.45–47 In support of this assumption, we note that the VOPcB molecules show a stronger anchoring to the surface compared to VOPcA and cannot be displaced using STM manipulation. The calculated dI/dV and STM images obtained using this model (Fig. 4c and Fig. S13, ESI†) are in excellent agreement with the experimental data. According to DFT, the charge transfer to VOPc partially fills up the molecular orbital localized on the molecular scaffold and aligns it with the Fermi level. The electronic configuration of V does not change and essentially remains 3d1 (Fig. 4d), in agreement with XMCD data and multiplet calculations (Fig. 2b). Finally, the spin polarization remains essentially localized on the V atoms, as indicated by the green isosurface in Fig. 4b. Therefore, both VOPcA and VOPcB preserve a spin-1/2 character, but only the latter additionally offers a long-range molecular order.
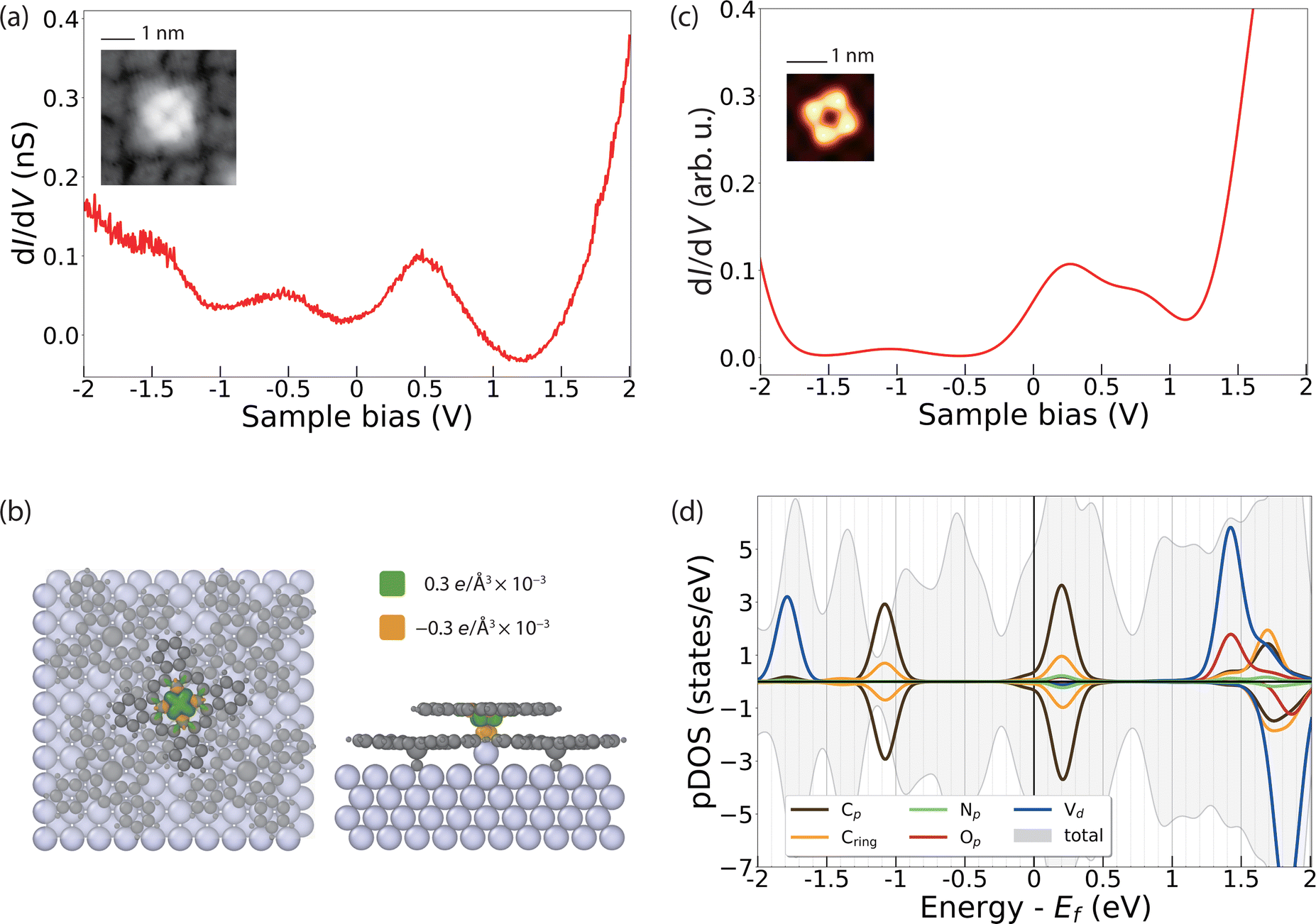 | ||
| Fig. 4 Orbital structure of a single VOPcB molecule. (a) dI/dV curve measured at the centre of VOPcB (VDC = 1 V, Iset = 50 pA). (b) Top view (left) and side view (right) of DFT calculation on spin density of VOPcB molecule. The distribution of positive (green) and negative (orange) spin polarization is overlapped with the molecular structure. (c) Simulated dI/dV curve of VOPcB molecules with one silver atom added. (d) pDOS plot of VOPcB molecules projected on the orthogonalized atomic basis set. Inset in (a): STM topography of VOPcB (scan size of 3.9 × 3.9 nm2, VDC = 100 mV, Iset = 100 pA). Inset in (c): simulated topography of VOPcB (Image size of 1.44 × 1.44 nm2, VDC = 300 mV) | ||
Our results indicate that the nanoscale patterning of VOPcB is owing to a combination of three factors: (i) the structural modification occurring on the Ag surface, (ii) local charges of the molecular layer, and (iii) strain induced in the TiOPc layer after accommodating the additional Ag adatoms. In particular, the accumulation of the strain in the TiOPc layer is expected to prevent the formation of closely packed VOPcB, resulting in a minimum separation of 3 molecular unit cells as a requirement for the formation of the highly ordered array of VOPcB on TiOPc. Remarkably, increasing the coverage of VOPc beyond the 0.25 ML suppresses the formation of the VOPcB type (see Fig. S4, ESI†). In this case, due to the increased number of closely packed molecules to be accommodated on the surface, the charge neutral VOPcA type on top of undistorted TiOPc layer appears to be the most favourable configuration.
Conclusions
Our combined experimental and theoretical investigation outlines a scalable approach for building a highly ordered spin-1/2 system on a surface by introducing a molecular templating layer between the spin carrying molecule and the metallic substrate. The surface modification obtained after thermal treatment allows partial charging of the molecular layer without modifying the spin configuration of the V centre. Our results suggest that the electrostatic repulsion among partially charged molecular units and the strain induced in the TiOPc interlayer building up upon the Ag surface modification are key factors to attain the regular spacing between the molecular spin qubits. We expect that a further increase of the yield of ordered molecules could be attained by using molecular scaffolds with a stronger chemically active apical point to enhance the anchoring effect to the surface or, alternatively, by introducing other adatoms that can offer more efficient electron donation to the molecular layer.The indicated mechanism can be potentially used to induce long-range order in a wide variety of molecular spin systems. By fine-tuning the intermolecular and molecule–substrate interactions, it will be possible to additionally tailor the spacing and symmetry of the molecular spin qubit array while preserving the desired spin configuration. Our work offers a viable direction in the quest for the scalability of molecular qubits, further highlighting the potential of on-surface molecular spin arrays for the study of quantum information processing.
Author contributions
KN, LC, PD, and YB performed the STM characterizations. X-ray experiments were carried out by KN, JL, DK, and FD with the support of AD, and interpreted using multiplet calculations performed by JL and FD. DFT calculations were carried out by CU and CW. The manuscript was written by KN, LC, CU, CW, FD, and YB, and finalized through the contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Institute for Basic Science, Korea (Grant No. IBS-R027-D1). K. N., C. W., and Y. B. acknowledge support from Asian Office of Aerospace Research and Development (Grant No. FA2386-20-1-4052).Notes and references
- A. Gaita-Ariño, F. Luis, S. Hill and E. Coronado, Nat. Chem., 2019, 11, 301–309 CrossRef PubMed.
- M. Atzori and R. Sessoli, J. Am. Chem. Soc., 2019, 141, 11339–11352 CrossRef CAS PubMed.
- M. J. Graham, J. M. Zadrozny, M. S. Fataftah and D. E. Freedman, Chem. Mater., 2017, 29, 1885–1897 CrossRef CAS.
- E. Coronado, Nat. Rev. Mater., 2020, 5, 87–104 CrossRef.
- L. Yang and N. R. Champness, Functional Supramolecular Materials: From Surfaces to MOFs, The Royal Society of Chemistry, 2017, pp. 1–36 10.1039/9781788010276-00001.
- J. V. Barth, G. Costantini and K. Kern, Nature, 2005, 437, 671–679 CrossRef CAS PubMed.
- J. A. Elemans, S. Lei and S. De Feyter, Angew. Chem., Int. Ed., 2009, 48, 7298–7332 CrossRef CAS PubMed.
- N. Ishikawa, M. Sugita, T. Ishikawa, S.-y Koshihara and Y. Kaizu, J. Am. Chem. Soc., 2003, 125, 8694–8695 CrossRef CAS PubMed.
- M. A. AlDamen, J. M. Clemente-Juan, E. Coronado, C. Martí-Gastaldo and A. Gaita-Ariño, J. Am. Chem. Soc., 2008, 130, 8874–8875 CrossRef CAS PubMed.
- A. Ardavan, O. Rival, J. J. L. Morton, S. J. Blundell, A. M. Tyryshkin, G. A. Timco and R. E. P. Winpenny, Phys. Rev. Lett., 2007, 98, 057201 CrossRef PubMed.
- C. Schlegel, J. van Slageren, G. Timco, R. E. P. Winpenny and M. Dressel, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 134407 CrossRef.
- K. Bader, D. Dengler, S. Lenz, B. Endeward, S.-D. Jiang, P. Neugebauer and J. van Slageren, Nat. Commun., 2014, 5, 5304 CrossRef CAS PubMed.
- M. Atzori, L. Tesi, E. Morra, M. Chiesa, L. Sorace and R. Sessoli, J. Am. Chem. Soc., 2016, 138, 2154–2157 CrossRef CAS PubMed.
- N. P. Kazmierczak, R. Mirzoyan and R. G. Hadt, J. Am. Chem. Soc., 2021, 143, 17305–17315 CrossRef CAS PubMed.
- P. J. Blowey, R. J. Maurer, L. A. Rochford, D. A. Duncan, J. H. Kang, D. A. Warr, A. J. Ramadan, T. L. Lee, P. K. Thakur, G. Costantini, K. Reuter and D. P. Woodruff, J. Phys. Chem. C, 2019, 123, 8101–8111 CrossRef CAS PubMed.
- H. Adler, M. Paszkiewicz, J. Uihlein, M. Polek, R. Ovsyannikov, T. V. Basova, T. Chassé and H. Peisert, J. Phys. Chem. C, 2015, 119, 8755–8762 CrossRef CAS.
- K. Eguchi, Y. Takagi, T. Nakagawa and T. Yokoyama, J. Phys. Chem. C, 2013, 117, 22843–22851 CrossRef CAS.
- K. Eguchi, Y. Takagi, T. Nakagawa and T. Yokoyama, J. Phys. Chem. C, 2014, 118, 17633–17637 CrossRef CAS.
- L. Malavolti, M. Briganti, M. Hänze, G. Serrano, I. Cimatti, G. McMurtrie, E. Otero, P. Ohresser, F. Totti, M. Mannini, R. Sessoli and S. Loth, Nano Lett., 2018, 18, 7955–7961 CrossRef CAS PubMed.
- I. Cimatti, L. Bondì, G. Serrano, L. Malavolti, B. Cortigiani, E. Velez-Fort, D. Betto, A. Ouerghi, N. B. Brookes, S. Loth, M. Mannini, F. Totti and R. Sessoli, Nanoscale Horiz., 2019, 4, 1202–1210 RSC.
- T. Niu, J. Zhang and W. Chen, J. Phys. Chem. C, 2014, 118, 4151–4159 CrossRef CAS.
- S. Colonna, G. Mattioli, P. Alippi, A. Amore Bonapasta, A. Cricenti, F. Filippone, P. Gori, A. M. Paoletti, G. Pennesi, F. Ronci and G. Zanotti, J. Phys. Chem. C, 2014, 118, 5255–5267 CrossRef CAS.
- W. Zhao, H. Zhu, H. Song, J. Liu, Q. Chen, Y. Wang and K. Wu, J. Phys. Chem. C, 2018, 122, 7695–7701 CrossRef CAS.
- G. B. McGaughey, M. Gagné and A. K. Rappé, J. Biol. Chem., 1998, 273, 15458–15463 CrossRef CAS PubMed.
- C. R. Martinez and B. L. Iverson, Chem. Sci., 2012, 3, 2191–2201 RSC.
- M. R. Battaglia, A. D. Buckingham and J. H. Williams, Chem. Phys. Lett., 1981, 78, 421–423 CrossRef CAS.
- C. Piamonteze, U. Flechsig, S. Rusponi, J. Dreiser, J. Heidler, M. Schmidt, R. Wetter, M. Calvi, T. Schmidt, H. Pruchova, J. Krempasky, C. Quitmann, H. Brune and F. Nolting, J. Synchrotron Radiat., 2012, 19, 661–674 CrossRef CAS PubMed.
- K. Eguchi, T. Nakagawa, Y. Takagi and T. Yokoyama, J. Phys. Chem. C, 2015, 119, 9805–9815 CrossRef CAS.
- M. O. Krause and J. H. Oliver, J. Phys. Chem. Ref. Data, 1979, 8, 329–338 CrossRef CAS.
- M. W. Haverkort, J. Phys.: Conf. Ser., 2016, 712, 012001 CrossRef.
- T. M. Willey, M. Bagge-Hansen, J. R. I. Lee, R. Call, L. Landt, T. V. Buuren, C. Colesniuc, C. Monton, I. Valmianski and I. K. Schuller, J. Chem. Phys., 2013, 139, 034701 CrossRef CAS PubMed.
- R. De Francesco, M. Stener and G. Fronzoni, J. Phys. Chem. A, 2012, 116, 2885–2894 CrossRef CAS PubMed.
- L. Floreano, A. Cossaro, R. Gotter, A. Verdini, G. Bavdek, F. Evangelista, A. Ruocco, A. Morgante and D. Cvetko, J. Phys. Chem. C, 2008, 112, 10794–10802 CrossRef CAS.
- A. Milev, K. Kannangara, N. Tran and M. Wilson, Int. J. Nanotechnol., 2007, 4, 516–522 CrossRef CAS.
- A. S. Milev, N. Tran, G. S. Kamali Kannangara, M. A. Wilson and I. Avramov, J. Phys. Chem. C, 2008, 112, 5339–5347 CrossRef CAS.
- K. Müller, M. Richter, D. Friedrich, I. Paloumpa, U. I. Kramm and D. Schmeißer, Solid State Ionics, 2012, 216, 78–82 CrossRef.
- F. Sedona, M. Di Marino, D. Forrer, A. Vittadini, M. Casarin, A. Cossaro, L. Floreano, A. Verdini and M. Sambi, Nat. Mater., 2012, 11, 970–977 CrossRef CAS PubMed.
- F. Petraki, H. Peisert, U. Aygül, F. Latteyer, J. Uihlein, A. Vollmer and T. Chassé, J. Phys. Chem. C, 2012, 116, 11110–11116 CrossRef CAS.
- R. Temirov, S. Soubatch, A. Luican and F. S. Tautz, Nature, 2006, 444, 350–353 CrossRef CAS PubMed.
- K. W. Hipps and U. Mazur, J. Phys. Chem. B, 2000, 104, 4707–4710 CrossRef CAS.
- D. E. Barlow and K. W. Hipps, J. Phys. Chem. B, 2000, 104, 5993–6000 CrossRef CAS.
- U. Mazur and K. W. Hipps, J. Chem. Phys., 1995, 99, 6684–6688 CrossRef CAS.
- A. J. Cohen, P. Mori-Sánchez and W. Yang, Science, 2008, 321, 792–794 CrossRef CAS PubMed.
- E. B. Linscott, D. J. Cole, M. C. Payne and D. D. O'Regan, Phys. Rev. B, 2018, 98, 235157 CrossRef CAS.
- P. Maksymovych, D. C. Sorescu and J. T. Yates, Phys. Rev. Lett., 2006, 97, 146103 CrossRef PubMed.
- P. J. Mousley, L. A. Rochford, P. T. P. Ryan, P. Blowey, J. Lawrence, D. A. Duncan, H. Hussain, B. Sohail, T.-L. Lee, G. R. Bell, G. Costantini, R. J. Maurer, C. Nicklin and D. P. Woodruff, J. Phys. Chem. C, 2022, 126, 7346–7355 CrossRef CAS PubMed.
- A. Della Pia, M. Riello, D. Stassen, T. S. Jones, D. Bonifazi, A. De Vita and G. Costantini, Nanoscale, 2016, 8, 19004–19013 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Methods, 2nd layer coverage calculation, characterization of a monolayer, characterization of a single component bilayer, coverage and annealing temperature dependence of ‘B’ molecule creation, and more dI/dV measurements are available free of charge via the internet. See DOI: https://doi.org/10.1039/d2nh00375a |
| This journal is © The Royal Society of Chemistry 2023 |