DOI:
10.1039/D3GC00219E
(Perspective)
Green Chem., 2023,
25, 4222-4233
Using earth abundant materials for long duration energy storage: electro-chemical and thermo-chemical cycling of bicarbonate/formate†
Received
18th January 2023
, Accepted 17th March 2023
First published on 29th March 2023
Abstract
Using hydrogen to store energy in chemical bonds is a key component of the global strategy to achieving a sustainable future and ameliorating climate change. The challenges associated with handling molecular hydrogen can be solved by using liquid hydrogen carriers. In this perspective, we discuss the concept of bicarbonate–formate cycle, where aqueous solutions of formate ions (HCO2−) are used as hydrogen and energy carriers. Such solutions are composed by earth abundant elements and, in contrast to common liquid organic carriers, are non-flammable, and readily convert to the oxide forms (HCO3−) under reaction with water to release hydrogen, or electrons, at moderate temperatures. We discuss thermodynamic aspects of the bicarbonate–formate cycle and show how it offers the opportunity of combining electrochemical and thermochemical operations, as well as of coupling CO2 capture with energy/hydrogen storage. We emphasize the potential role of electrochemistry in the generation of formate and in the release of energy in the form of electricity. At present, more information on both the fundamental and systems level, is needed to identify the feasible scenarios for using formate/bicarbonate salts for hydrogen/energy storage. It is likely, however, that several strategies, including hybrid electrochemical–thermochemical approaches, will suit different applications. It is also clear that more integration between the disciplines of electrochemistry and heterogeneous catalysis is needed to overcome the challenges for advancing the HCO3−–HCO2− system as a feasible green alternative for storing and transporting energy.
Introduction
The deployment of renewable electricity sources has increased significantly in the past few years to provide carbon-neutral energy and an opportunity to address carbon emissions and climate change. This trend is expected to continue. However, as renewable electricity supplies increase, relative to traditional supplies, their intermittency may result in instability and fluctuations in power availability and prices, without approaches to store energy at large scales. Energy storage, particularly at medium- and long-term duration, and reconversion to grid electricity will be needed to resolve the supply-demand balancing requirement. Batteries are convenient to store power for short durations, but when storing energy for prolonged periods of time, i.e., more than twelve hours, hydrogen quickly becomes an attractive option.1
There is a growing interest in the use of hydrogen carriers to store and transport hydrogen to avoid the high pressures and low temperatures required for compressed and liquified hydrogen, respectively. For example, Chiyoda Corporation is using methylcyclohexane to transport hydrogen from Brunei to Japan, Hydrogenious LOHC Technologies is scaling up a benzyltoluene-based system to advance the commercial use of hydrogen carriers.2 On the other hand, several groups have suggested the use of alkali formate salts, i.e., cesium, potassium, and sodium formate, as an attractive alternative to store hydrogen.3,4 While they do store less hydrogen on a volumetric basis, compared to conventional hydrogen carriers, formate salts provide several benefits for stationary storage applications. For instance, they are composed by earth abundant elements, they are non-toxic and non-flammable, and readily cycle between hydrogen lean and rich forms at temperatures below 80 °C and pressures <30 bar. Specifically, the oxidation of aqueous formate salts produces bicarbonate (HCO3−) salts, which are also safe to handle even if suspensions form upon oxidation (bicarbonates are less soluble than formate salts in water). Although we focus on alkali metal salts, it worth noting the interest in the corresponding ammonium salts that have also been investigated.5
A bicarbonate–formate (HCO3−–HCO2−) cycle has the thermodynamic advantage of having ΔG0r ∼ 0 kJ mol−1, eqn (1). Thus, one could store and release hydrogen by performing moderate pressure swings of H2 or by applying external electrical potential. The latter approach can make use of direct formate fuel cells. This cycle would further offer the opportunity to utilize large quantities of captured CO2 and use it to store large quantities of hydrogen – providing a carbon negative approach to store and transport energy. Note, to illustrate this point, that 1 MW h of electricity can be delivered by a fuel cell using the hydrogen released from formate derived from, i.e., 4 metric tons of CO2 captured in a potassium salt (Table 1).
| | | HCO3− + H2 ⇌ HCO2− + H2O | (1) |
Table 1 Amount of salt, which could be derived from CO2 (i.e., CO2 + OH− ⇌ HCO3−); amount of H2 that could be release from formate salts upon their conversion to bicarbonates (HCO2− + H2O ⇌ HCO3− + H2); amount of H2 stored from captured CO2; energy stored from captured CO2
| |
Salt from CO2 (kgSalt/kgCO2) |
H2 from salt (gH2/kgSalt) |
H2 from CO2 (gH2/kgCO2) |
Energya,b (kW h/kgCO2) |
|
Based on the lower heating value of hydrogen (33.3 kW h kg−1) and efficiency of a PEM fuel cell (60%), ca. 20 kW h produced from 1 kg H2.
Equivalent to MW h/MTCO2.
|
| NH4HCO2 |
0.69 |
31.7 |
22 |
0.44 |
| NaHCO2 |
0.65 |
29.4 |
19 |
0.38 |
| KHCO2 |
0.52 |
23.8 |
12 |
0.25 |
| CsHCO2 |
0.25 |
11.2 |
2.8 |
0.06 |
The increasing interest on the practical realization of the HCO3−–HCO2− cycle is evident in the large number of reviews appeared in recent years. A recent review, oriented towards application, compared catalysts and kinetics reported in literature with thermodynamic limitations.6 Previous perspectives had discussed the conversion of mixtures of formic acid and formate salts, as well as the performances of several materials and the corresponding challenges to advance towards application.3,4 More details of thermodynamics, reaction mechanisms, and of effects of reaction conditions on reaction have been covered in ref. 7 and 8. All these contributions were focused exclusively on thermochemical conversion. Another article highlights the flexibility of liquid compounds that can be hydrogenated electrochemically, provides an overview of them, as well as metrics to evaluate their potential applications.9 Complementing these views, our perspective discusses two approaches, i.e., thermochemical or electrochemical interconversion of formate and bicarbonate, to provide an outlook for opportunities and synergies ranging from fundamental understanding to catalyst design and applications.
The concept of bicarbonate–formate cycle
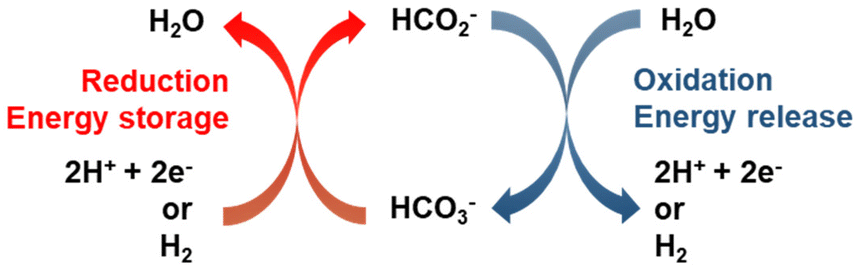
Fig. 1 illustrates the HCO3−–HCO2− cycle: catalytic reduction of bicarbonate by molecular hydrogen or electrochemically, and the reverse step, reaction of formate salts with water to yield bicarbonate and H2 or a pair of electrons. The thermo-catalytic cycle was proposed over thirty years ago for heterogeneous catalyst systems10,11 and has been revisited for homogenous catalyst systems over the past decade.12–14 The same catalysts are used to catalyze both formate synthesis by reduction of bicarbonate and H2 release from formate to yield bicarbonate, the reaction is reversible and operates under equilibrium conditions. This advantageous feature of the formate/bicarbonate cycle, as well as the abundance of the salts and their low toxicity have renewed the interest for their study.3,4
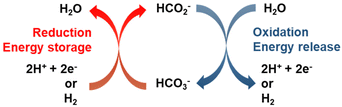 |
| | Fig. 1 Energy storage cycle based on the transformation between formate and bicarbonate. Adapted from ref. 3 with permission from Elsevier, copyright 2020. | |
Strictly speaking, formate salts are not hydrogen carriers but electron storage compounds, i.e., hydrogenating HCO3− to HCO2− does not add hydrogen to the carrier but changes the oxidation state of carbon from +4 to +2. This makes the bicarbonate/formate cycle comparable to aqueous redox flow batteries. The storage of electrons rather than hydrogen adds versatility to the bicarbonate/formate cycle and underscores the possibility of driving the cycle to store electrons generated from intermittent, renewable sources (e.g., solar, hydro, and wind) to complement or substitute a thermal process for seasonal storage, grid balancing or capture of stranded power. For instance, a saturated solution of KHCO2 (77 wt% in water at 20 °C) contains 18.3 mol e− per kg solution. This estimate takes into account the density of the saturated solution as described in Table 2.
Table 2 Solubility of selected formate salts in water at 20 °C, the corresponding maximum concentration of electrons per mass of solution, and H2 released upon full conversion to bicarbonate salts (HCO2− + H2O ⇌ HCO3− + H2) in a liter of saturated solution
| |
Solubilitya (g/kgH2O) |
Mol saltb (mol/kgSolution) |
Mol e− (mol/kgSolution) |
H2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c (g H2/LSolution) c (g H2/LSolution) |
|
Solubility of the salt in water taken from ref. 15.
Content of salt in one kilogram of solution calculated as the mol of salt in a saturated solution divided by the sum of the masses of water and salt in the solution.
Stoichiometric amounts of H2 produced from one liter of solution calculated using the molar mass of H2 and the specific gravity of the saturated solutions.
|
| NH4HCO2 |
1430 |
9.3 |
18.7 |
22.4 |
| NaHCO2 |
812 |
6.6 |
13.2 |
16.7 |
| KHCO2 |
3370 |
9.2 |
18.3 |
29.3 |
| CsHCO2 |
4500 |
4.6 |
9.2 |
21.2 |
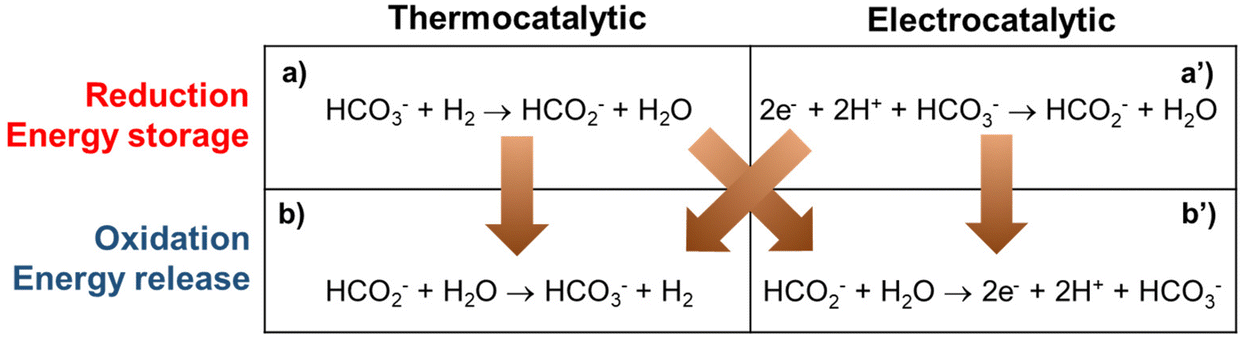
There are four conceptual scenarios for energy storage using the bicarbonate–formate cycle (Fig. 2). Starting with the reduction, i.e., the generation of formate: thermocatalytic reversible cycle with pressure swing (a–b in Fig. 2); hybrid with thermocatalytic reduction of bicarbonate – and electrochemical oxidation of formate (a–b′ in Fig. 2); hybrid with electrochemical reduction and thermocatalytic release of H2 (a′–b in Fig. 2); reversible fuel cell, i.e., electrochemical reduction of bicarbonate and oxidation of formate (a′–b′ in Fig. 2). Scenarios a–b and a′–b produce H2 as the product, which can either be used to produce energy when oxidized in a fuel cell, or the H2 can be used in a chemical reaction as a reductant, e.g., iron ore reduction to facilitate the decarbonatization of steel industry.16 In scenarios a–b′ and a′–b′ ‘electrons’ are the end product that can be used directly as a source of energy to enable long duration energy storage. In the electrocatalytic steps, complementary half reactions will be required, e.g., water oxidation to enable carbonate reduction. Studies are needed to understand the practicability and feasibility of these routes. A factor to consider is the source of the reduction equivalents. That is, e.g., H2 gas or electrons from a renewable source. The source of H2 will also be important for life-cycle assessment and carbon emissions, i.e., from fossil sources (grey if generated via steam methane reforming, black if generated via coal gasification, blue if the generation is supported by CO2 capture, or turquoise hydrogen if generated via pyrolysis), or from water electrolysis (green if generated using renewable electricity, purple if generated using nuclear power, or yellow hydrogen if generated using grid electricity from multiple sources).
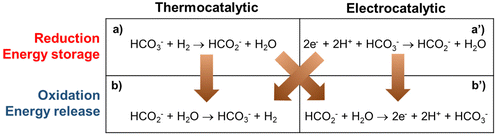 |
| | Fig. 2 Possible routes to store energy in formate and to release it generating bicarbonate by thermocatalytic and electrocatalytic methods. | |
Note that the electrochemical oxidation of formate (b′ in Fig. 2), or formic acid in equilibrium (HCOOH ⇄ HCOO− + H+), favors the formation of CO2 in acid (eqn (2)).17 Thus, in practice, the electrooxidation of formate is performed under basic reaction conditions, which yields carbonate as shown in eqn (3). Next, we describe in detail some key aspects of the thermal and electrochemical steps involved in the scenarios described above, considering eqn (3) instead of the reaction “b’” in Fig. 2.
| | | HCO2− → CO2 + 2e− + H+ | (2) |
| | | HCO2− + 3OH− → CO32− + 2H2O + 2e− | (3) |
Thermodynamic aspects of the thermochemical bicarbonate–formate cycle
The dissolution of CO2 or (bi)carbonate salts in water produces an equilibrium of dissolved CO2, carbonic acid, bicarbonate, and carbonate. Reactions on eqn (4)–(7) show the transitions among these species (other possible reactions are described below). Overall, aqueous carbon dioxide is the predominate species in equilibrium with carbonic acid under acidic conditions. The bicarbonate ion is prevalent in weakly basic conditions, while the carbonate ion predominates in strongly basic conditions.18| | | HCO3− + OH− ⇌ H2O + CO32− | (7) |
Most studies in (electro)catalytic conversion of CO2, and related solvated species, downplay the effect of these equilibria, whose balance is sensitive to pH and temperature and on the kinetics of (de)hydrogenation reactions. In thermocatalytic hydrogenation, it is generally assumed that bicarbonate is the precursor of the species that undergoes reduction at the catalyst surface, whereas in electrochemical reactions carbonate is considered the species participating in the oxidation/reduction with formate.19,20 These assumptions are likely derived from the expected most abundant species in solution at the conditions typically practiced in thermocatalytic and electrocatalytic reduction.
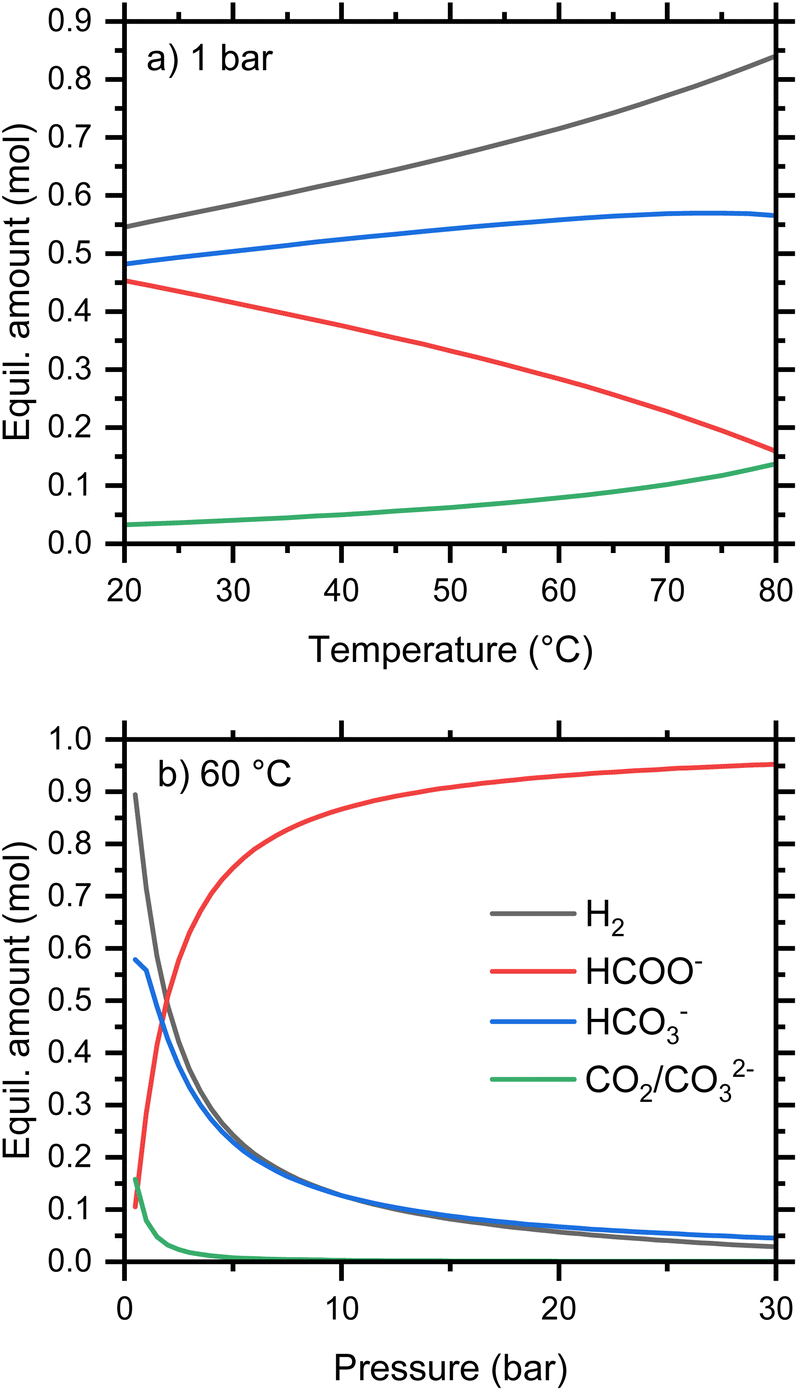
Concerning the thermodynamics of the formate-bicarbonate transformation (eqn (1)), the Gibbs free energy of reaction, ΔGr, for the release of hydrogen from formate and water is close to 0 kJ mol−1 under ambient conditions, which suggests that both reduction and oxidation can be accomplished under moderate conditions of temperature or pressure. The experimentally determined value for ΔG of the oxidation reaction is 0.72 kJ mol−1.21 This is in good agreement with the calculated value from standard parameterizations of reactants and products is 0.99 kJ mol−1 at 25 °C. We used the Gibbs Energy Minimizer module of HSC Chemistry (Metso Outotec, Finland) to calculate the quantities of aqueous, gaseous, and solid species in equilibrium, i.e., the maximum degree of reduction and oxidation expected under various conditions. The temperature dependence of the equilibrium composition starting from 1 mol KHCO2 and 1 kg water is shown in Fig. 3a for a pressure of 1 bar. When all relevant species are considered, a little more than 50 mol% formate is oxidized to bicarbonate at 20 °C, and this rises to around 85 mol% at 80 °C. At low temperature, the oxidized anion is mostly bicarbonate, but an increasing proportion of carbonate is predicted with increasing temperature as a result of the equilibrium shown in eqn (8).
| | | 2HCO3− ⇌ CO32− + CO2 + H2O | (8) |
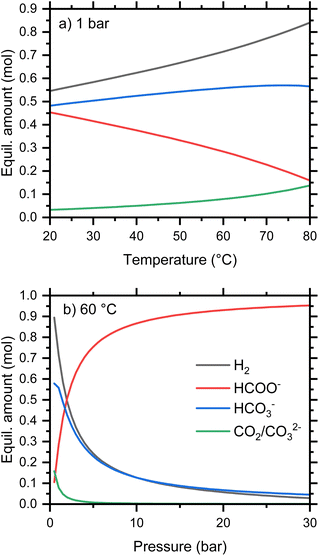 |
| | Fig. 3 Calculated equilibrium amounts of selected species from 1 mol KHCO2 in 1 kg water as a function of (a) pressure, and (b) temperature. | |
Consequently, the quantity of molecular CO2 is equal to the quantity of aqueous carbonate. The equilibria are calculated at a constant total pressure and will therefore be affected by the presence of gases other than that generated by the reaction. For example, the partial pressure of CO2 in these calculations will be altered by an initial gas atmosphere, even of H2.
The pressure dependence of the 1 molal equilibria at 60 °C is shown in Fig. 3b. This indicates that relatively modest pressures are sufficient to reduce bicarbonate to formate, with 95% conversion at 30 bar. This pressure is within the range attainable using commercial water electrolyzers,22 simplifying the engineering requirements.
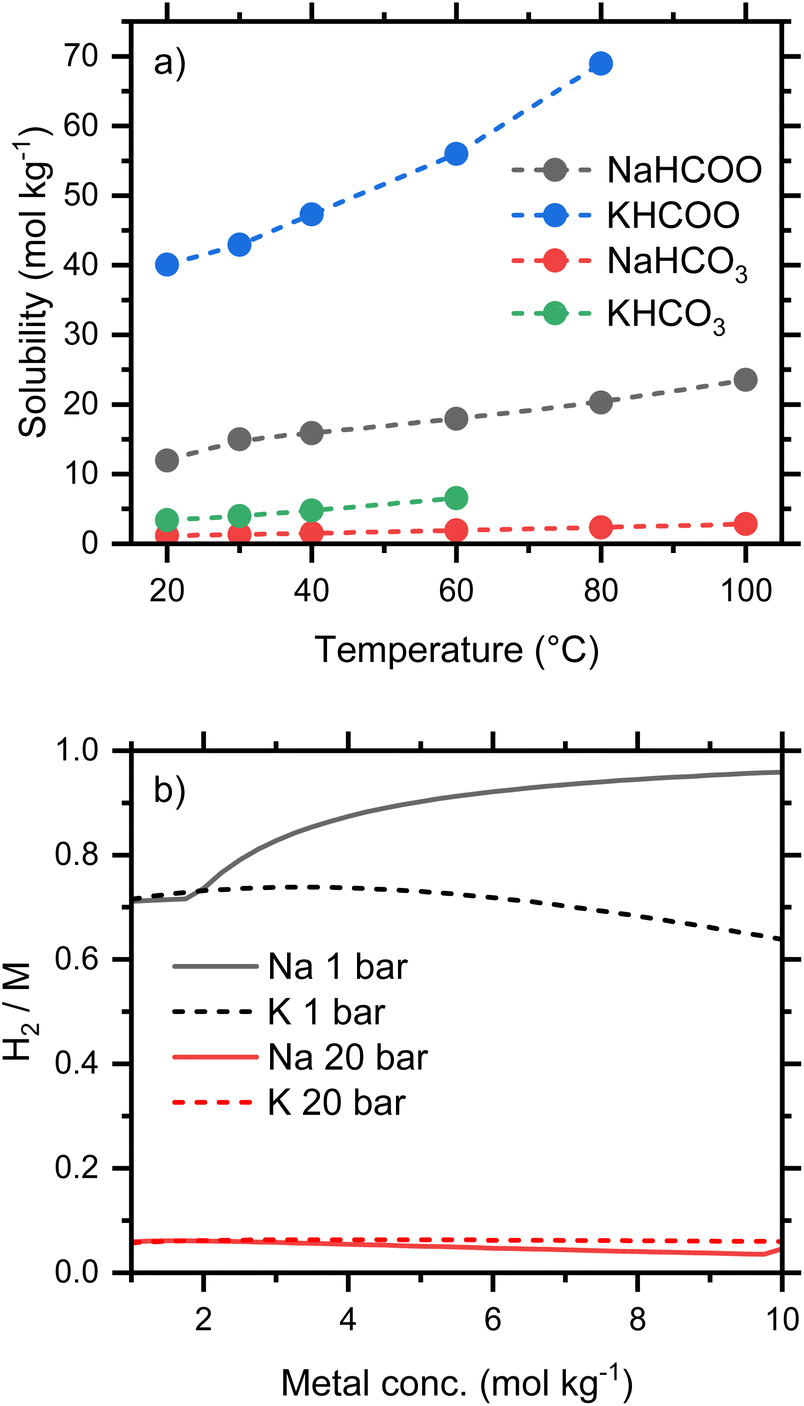
Conversion is weakly dependent on concentration, provided the salts remain soluble. A tabulation of solubilities at temperatures between 20 and 100 °C has been compiled by Grubel et al.4 which shows that formates are significantly more soluble than the corresponding carbonates and bicarbonates, and that sodium bicarbonate is the least soluble of the common salts investigated. The solubilities for Na and K salts are plotted in Fig. 4a, which illustrates the stark difference in solubility at 20 °C between NaHCO3 (1.1 mol kg−1) and KHCO2 (40 mol kg−1). Since oxidation involves water as a reactant, it is important to recognize that the concentration of bicarbonate may be higher than the starting formate concentration. For example, a fully converted 5 mol kg−1 formate solution would give an approximately 5.5 mol kg−1 bicarbonate solution due to the consumption of water in the process.
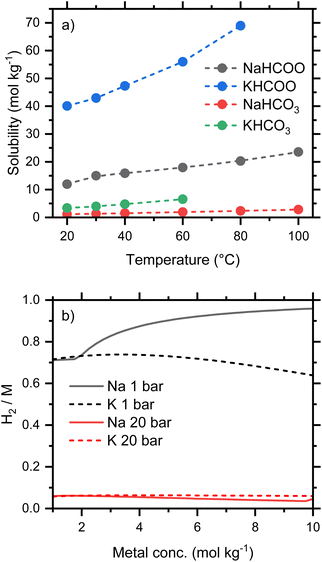 |
| | Fig. 4 (a) Solubilities of Na and K salts; (b) equilibrium conversion, expressed as the molar ratio of H2 gas to metal, of aqueous Na and K formate/bicarbonate solutions between 1 and 10 mol kg−1 at 60 °C. | |
The predicted conversion equilibria for Na and K salts of varying concentration are compared in Fig. 4b. The ratios H2/M (M is the alkali metal) indicate the conversion from fully reduced (H2/M = 0) to fully oxidized salt (H2/M = 1). Both potassium formate and bicarbonate remain soluble at the conditions considered, and this system shows conversions of ca. 70% and 6% at 1 and 20 bar respectively. At the lower pressure the conversion drops from a maximum of 74% at 3 mol kg−1 to 64% at 10 mol kg−1. Oxidation at 60 °C and 1 bar limits the usable storage capacity of this system, as H2, to a maximum of 70% of stoichiometric; larger capacities would require oxidation at lower pressure or sweeping away the gaseous products with an inert gas. Both of these properties, due to reversibility, will add engineering complexity and may result in a higher concentration of CO2 in the hydrogen stream because CO2 is favored at low pressures (Fig. 3b). Increased temperature (Fig. 3a) may be a more effective strategy for improving usable capacity.
The low solubility of NaHCO3 increases the equilibrium conversion at higher concentrations, since the removal of HCO3− from solution by precipitation will drive the equilibrium in eqn (1) further to the left and generate more H2. This increases the amount of H2 released from a 10 mol kg−1 sodium formate solution to 96%, a substantial capacity improvement compared to the 64% equilibrium conversion of potassium formate. The smaller HCO3− concentrations engendered by NaHCO3 precipitation also affects the bicarbonate/carbonate equilibrium, eqn (8), and reduces the predicted concentration of CO2 in the hydrogen stream. At 60 °C and 1 bar, the calculated equilibria from 1 mol kg−1 solutions of either potassium or sodium predict 10% CO2 in the H2 gas. This increases to 17% CO2 in the H2 gas for 5 mol kg−1 KHCO2. However, for NaHCO2, the fraction of CO2 in the H2 gas drops with the precipitation of NaHCO3 and reaches a predicted value of 4% at 10 mol kg−1. The precipitation can make a hydrogen storage system using a flow through reactor problematic but could provide an advantage to a small batch reactor storage system.
Overall, the solubility of bicarbonate salts will pose important operational constrains in the two directions of the bicarbonate–formate cycle. During reduction, it will clearly limit the concentration of bicarbonates in aqueous solutions for formate production if precipitation is to be avoided. In the energy release step, the concentration of formate solutions, or their conversion level, will have to be limited so that the formed (bi)carbonate does not precipitate. For temperature-driven conversions, the low solubility of bicarbonate and the concomitant precipitation could be handled by hydrogenating slurries in batch or semicontinuous reactors.23 This approach, however, will be difficult to implement during electrochemical conversions and thus, concentration of the salts will be constrained within the operational limits of, e.g., current direct formate fuel cells (vide infra). The choice of Na+ or K+ cations will depend on engineering considerations and the ability to handle slurries containing precipitated solids. If solids do not clog catalyst beds, pumps, etc., then the increased conversion of the Na formate salts favors both H2 production and gas purity. However, if slurries cannot be tolerated, more concentrated solutions with higher energy densities will be achieved using K salts. Thus, the solubility of potassium salts gives them a clear advantage over the more common sodium salts. Potassium bicarbonate, moreover, is reported to have higher reactivity towards hydrogenation than sodium bicarbonate.7,24
Illustration of the thermocatalytic bicarbonate–formate transformation
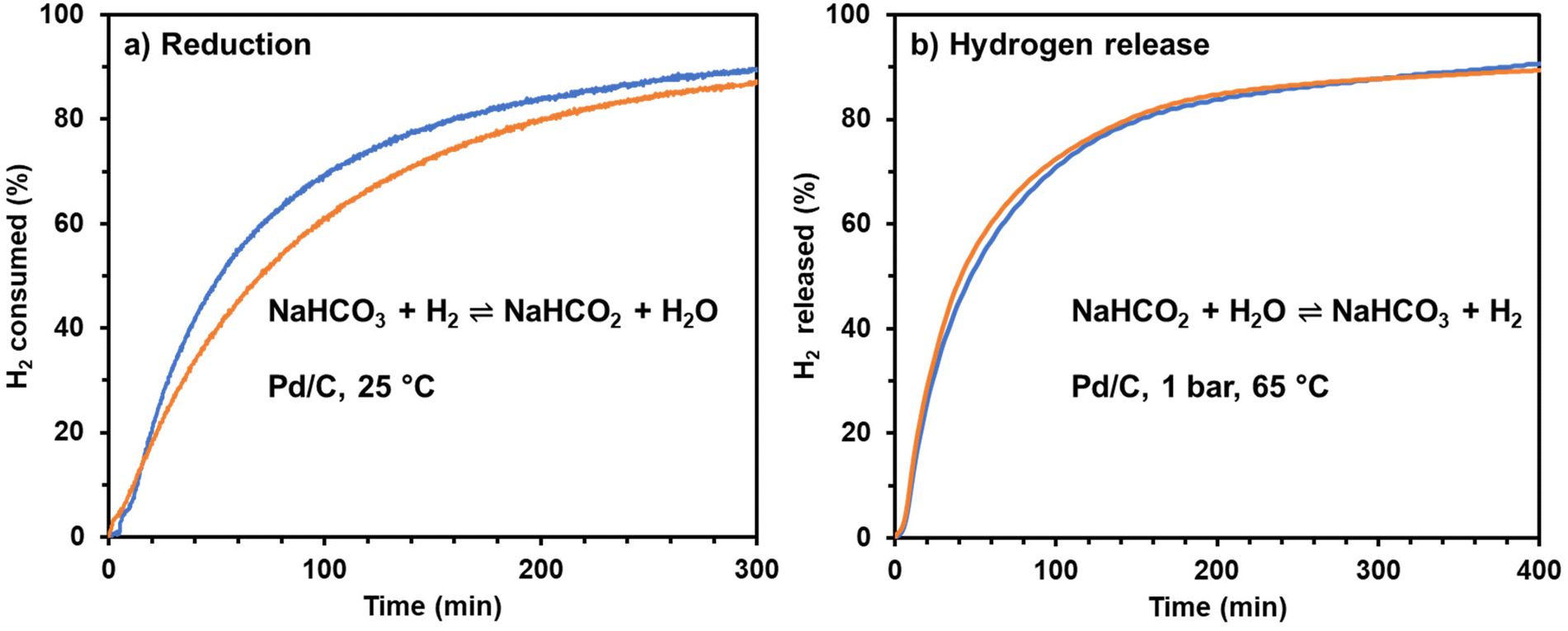
A simple experiment was performed to illustrate the convenience and simplicity of using sodium bicarbonate (baking soda) to store and release hydrogen using a carbon-supported Pd catalyst. Baking soda, food-grade, purchased from the grocery store was used in place of reagent-grade sodium bicarbonate. Tap water from the laboratory sink was used in place of laboratory de-ionized water. This simple experiment demonstrated that both hydrogenation and hydrogen release will occur at rates comparable to the rates observed using reactant grade solutions, Fig. 5.
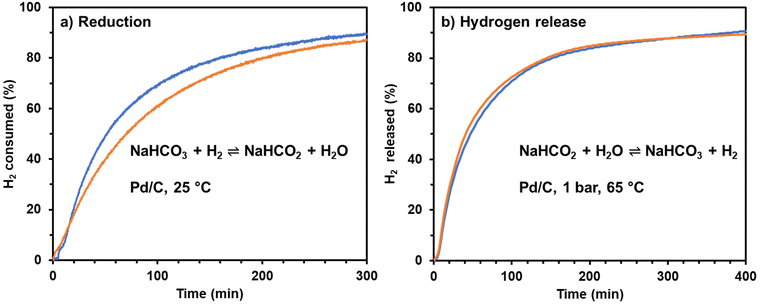 |
| | Fig. 5 Thermochemical bicarbonate–formate cycle for hydrogen storage: (a) Blue line: reduction of 1 M NaHCO3 in ultra-pure water in the presence of commercial 3 wt% Pd/C under 40 bar H2 (initial pressure); orange line: reduction of baking soda in sea water in the presence of commercial 3 wt% Pd/C catalyst under 40 bar H2 (initial pressure). (b) Blue line: oxidation of in situ formed sodium formate from 1 M NaHCO3 in ultra-pure water; orange line: oxidation of in situ formed sodium formate from baking soda in tap water. In both plots 100% represents the maximum amount of H2 that the system can uptake or release. | |
Notably, the Pd/C catalyst used to hydrogenate the bicarbonate was used to release the hydrogen without any special treatment between the two steps. That is, ultra-pure water and salts are not required in the bicarbonate–formate cycle if used as a hydrogen carrier. The reversible hydrogen storage can be achieved by simple temperature and pressure swings.
Analysis of a hybrid bicarbonate–formate cycle
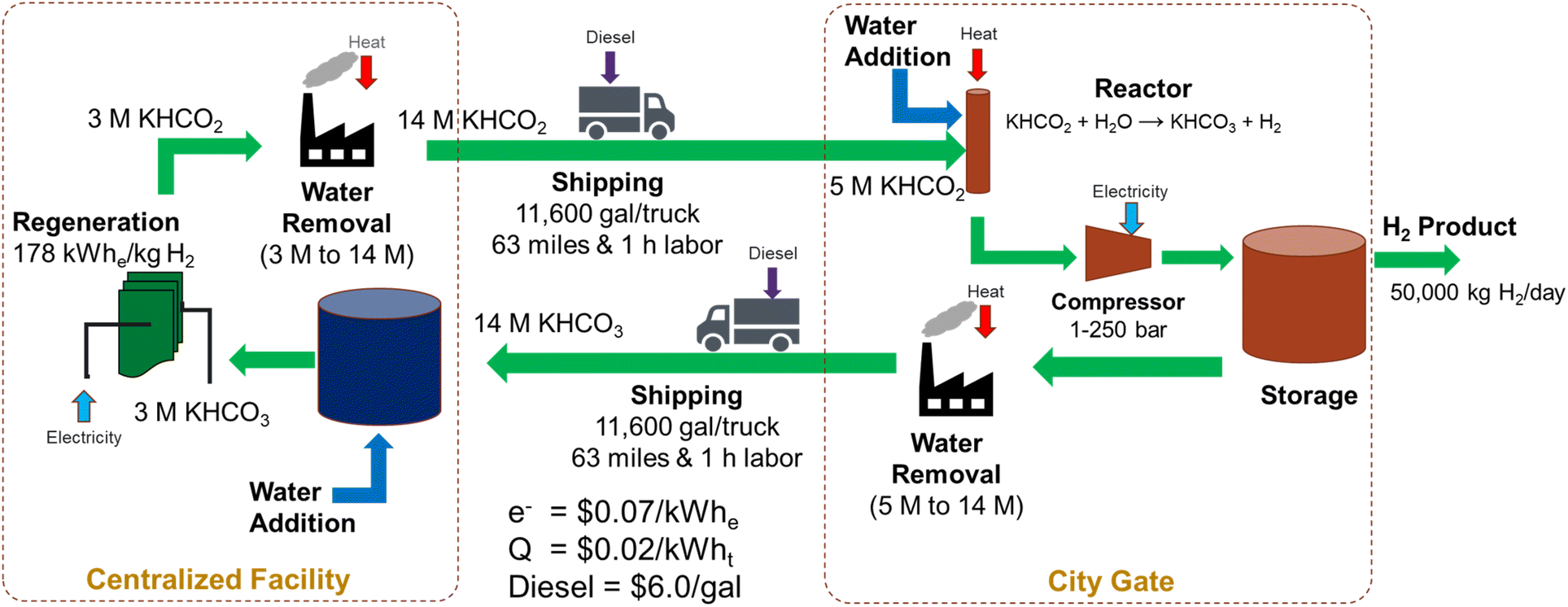
Let us evaluate the hybrid bicarbonate/formate cycle, i.e., an electrochemical reduction of bicarbonate followed by a catalytic oxidation of formate to bicarbonate. For this exercise, we evaluated a regional refueling depot that would transport potassium formate from a centralized facility to the city gate to supply 50 metric tonnes of hydrogen per day to refueling stations in a community. The regeneration of the bicarbonate to formate would be performed at a centralized facility and the hydrogen carrier would be shipped between these facilities as a liquid/slurry rather than as a high-pressure gas. A diagram of this process is shown in Fig. 6.
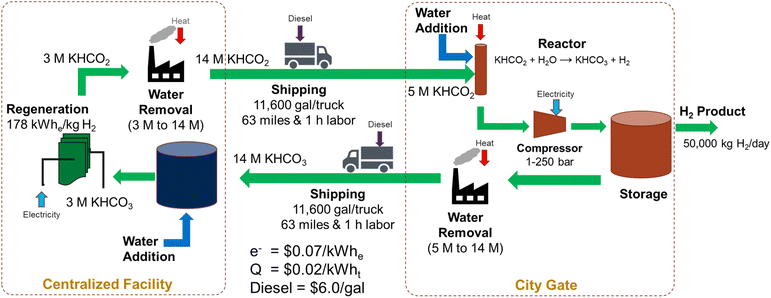 |
| | Fig. 6 Material flow diagram used for an initial cost analysis of the hybrid bicarbonate/formate cycle (electrochemical reduction of bicarbonate coupled with catalytic oxidation of formate to bicarbonate) for a regional refueling depot use case. | |
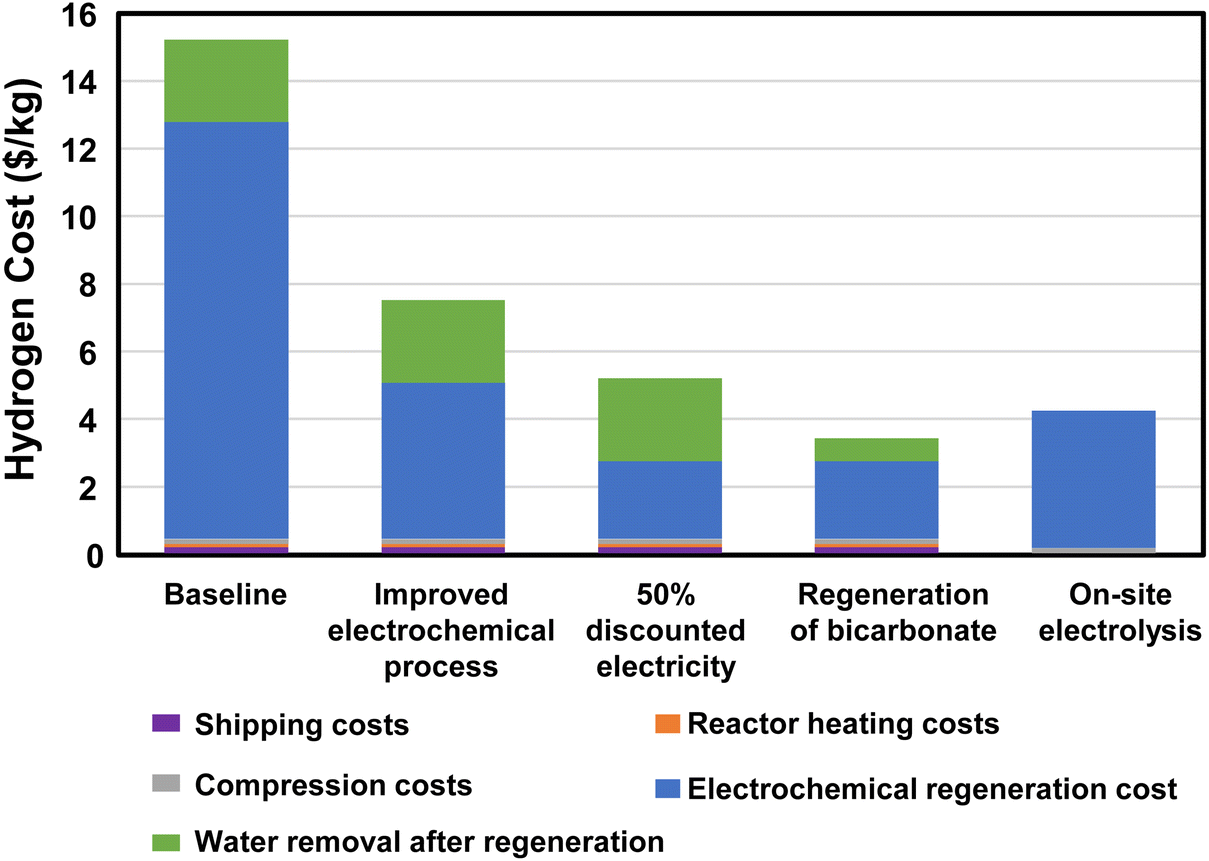
The electrochemical regeneration of bicarbonate is based on experimental data reported by Li, et al. that achieved 60% faradaic efficiency at a cell voltage of 4 V with a 3M KHCO3 solution.25 To reduce the number of trips required between the two facilities, the concentration of formate is increased to 75% (14 M) prior to shipping at 11600 gallons of trailer capacity. This results in 41 trips per day to transport 50000 kg H2. The cost of electricity and heat for regeneration, concentration, dehydrogenation, and H2 compression and the labor and fuel cost of transportation were included in this cost estimate (see assumptions in Fig. 6 and the legend of Fig. 7). The results of the initial baseline conditions as well as scenarios to improve this cost are shown in Fig. 7. Based on the assumptions above, the cost of hydrogen would be over $15 per kg. This cost is dominated by the electrochemical regeneration of formate. The next most significant cost is water removal after regeneration. Shipping, compression, and heating costs are comparatively minor.
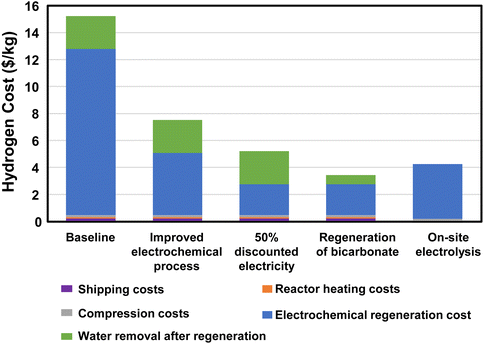 |
| | Fig. 7 Hydrogen cost for hybrid bicarbonate/formate cycle for a regional refueling depot use case compared to on-site electrolysis of water. Assumptions: fuel consumption = 5.29 miles per gallon, labor cost = $24 per h, compression cost = 2.11 kW he kg−1 H2, and water electrolysis = 59 kW he kg−1 H2. | |
For this baseline case, the electrochemical regeneration is not optimized. If the cell voltage could be reduced to 2 V and the faradaic efficiency raised to 80%, the total cost of hydrogen would be nearly cut in half (“Improved electrochemical process” case in Fig. 7). Further improvements could be made if the regeneration process could use during times of discounted electricity or by using renewables (“50% discounted electricity” case in Fig. 7). Furthermore, if the regeneration and reaction could be performed at 8 M rather than 3 M concentration along with an improved process and discounted electricity, the cost of hydrogen could drop to just over $3 per kg. Only then would the cost of the hydrogen be reduced below the cost of on-site electrolysis with full-priced electricity (“Regeneration of bicarbonate” case in Fig. 7). These results indicate a strong need to further develop the electrochemical regeneration process, especially at high formate concentrations.
Electrochemical bicarbonate–formate cycle
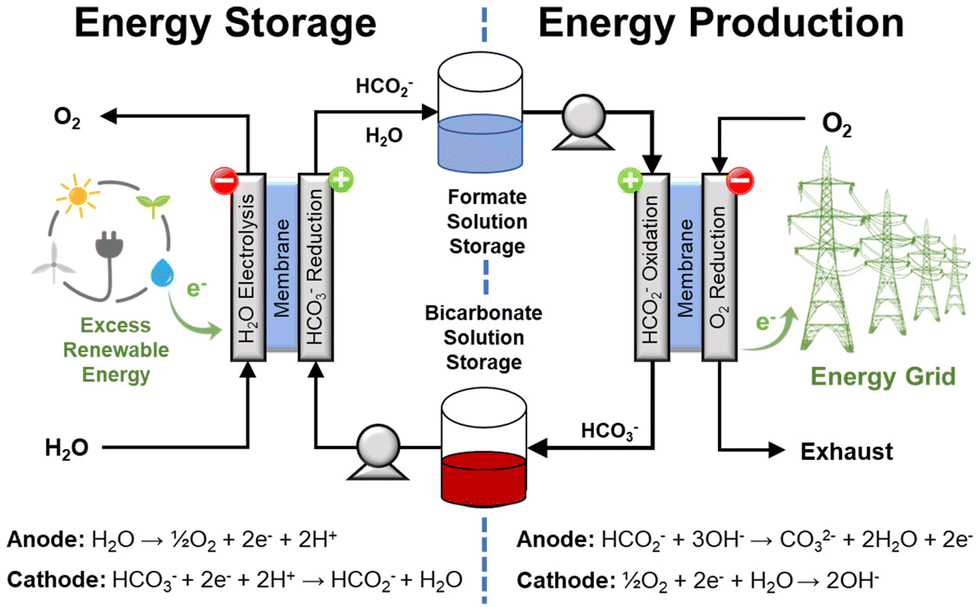
In an electrochemical cycle (Fig. 8), the electrochemical reduction of bicarbonate to formate, shown in the half reaction in eqn (9), requires the application of an external potential. However, the overpotential needed to drive the reaction could be close to the thermodynamic potential, even without optimized catalysts.13,26 Moreover, this electrochemical generation of formate from bicarbonate can be coupled with the oxidation of H2 or of water, eqn (10), or any other half reaction that is convenient for the intended application, e.g., oxidation of organic compounds or ammonia, eqn (11)–(13).| | | HCO3− + 2e− + 2H+ → HCO2− + H2O E ∼ 0 V vs. RHE | (9) |
| | | H2O → 2e− + 2H+ + ½O2 E = 1.23 V vs. RHE | (10) |
| | | CH3COOH + 2H2O → 8e− + 8H+ + 2CO2 E = −0.02 V vs. RHE | (11) |
| | | CH3OH + H2O → 6e− + 6H+ + CO2 E = −0.03 V vs. RHE | (12) |
| | | 2NH3 → 6e− + 6H+ + N2 E = −0.77 V vs. RHE | (13) |
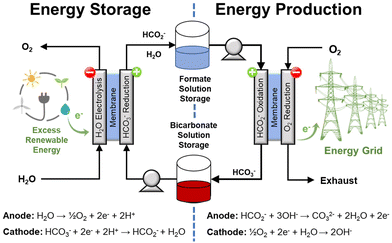 |
| | Fig. 8 Conceptual depiction of the electrochemical formate-bicarbonate cycle to store and to release energy. This approach does not require gaseous H2. | |
Indeed, the possibility of coupling a suitable oxidation reaction with the reduction of bicarbonate is one of the main advantages of an electrochemical approach. This implies that the hydrogenation step does not necessarily require external H2 because another reaction can be used to provide the electrons to reduce bicarbonate. For instance, the oxidation of organic compounds in wastewaters (e.g., acetic acid, methanol, and ammonia) is thermodynamically more favorable than H2O electrolysis as they require lower potential and, in principle, lower operation voltage. This example of coupling bicarbonate reduction with reactions less demanding than water oxidation would increase the operation efficiency and integrate an additional process (water clean-up) into the same unit.27 Another potential advantage is that reduction and oxidation could be performed in the same electrochemical cell operating either as electrolyzer or as a fuel cell; hence reducing the number of equipment needed for the electrochemical process. Thus, this cycle can be realized adapting available fuel cell/electrolysis technology, which operates at conditions much milder than those typical of current H2 compression technology.
The electrochemical generation of H2, associated with the conversion of formate to bicarbonate, eqn (14), is close to spontaneous under standard conditions but would require an overpotential under electrochemical operation. This scenario, an electrochemical bicarbonate–formate cycle with H2 release, would require one electrolyzer to reduce bicarbonate to formate (energy storage) and one to oxidize formate (H2 release). This approach, however, will be redundant if the objective of H2 release is to generate power with a fuel cell; especially if some conditioning is needed (i.e., pressurization) prior utilization of H2 in the fuel cell. A more general option could be to use the energy stored in formate with a direct formate fuel cell to generate electricity as shown by the half reaction shown in eqn (15). At present, there are few scenarios, where H2 may be needed to be produced electrochemically instead of power. However, H2 evolution is possible through the reaction shown in eqn (16) at the neutral-basic conditions associated with formate oxidation to carbonate. A possible scenario, where H2 would be produced electrochemically may be a use case of using electrons to make formate at a central facility, then transport it to a ‘filling’ station for a heavy duty vehicle, use electrons to release H2 and put it into the H2 tank.
| | | HCO2− + H2O ⇌ HCO3− + H2 ΔG0 ∼ 1 kJ mol−1 | (14) |
| | | HCO2− + 3OH− → CO32− + 2H2O + 2e− E = −1.05 V vs. RHE | (15) |
| | | H2O + e− → ½H2 + OH− E = −0.83 V vs. RHE | (16) |
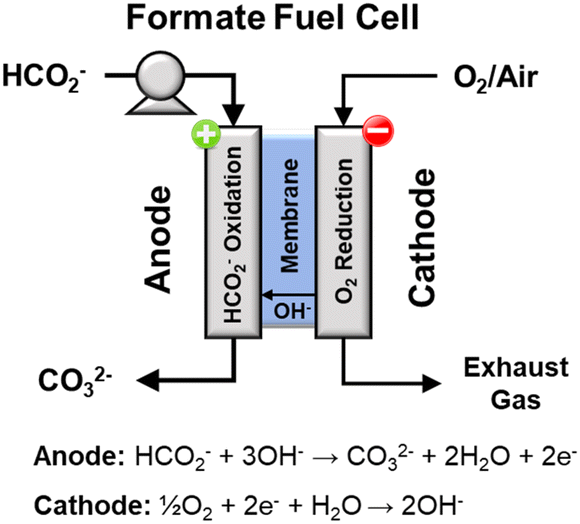
Direct formate fuel cells
A direct fuel cell running on formate has a theoretical potential as high as 1.45 V (with oxygen as oxidant), which is 0.24 V and 0.31 V higher than the fuel cells running on methanol and ethanol, respectively.28,29 A direct formate fuel cell (Fig. 9)30 is composed of an anode and a cathode separated by an anion exchange membrane. On the anode, the solution containing formate salts and alkali dissolved in water flows into the anode flow channel to the anode catalyst layer. There, formate ions are oxidized to generate electrons, water, and carbonate ions. Water in the fuel solution diffuses through the membrane to the cathode catalyst, while the produced electrons pass through an external circuit. On the cathode channel, oxygen is transported to the cathode catalyst, where oxygen reduction produces hydroxide ions according to eqn (17).| | | ½O2 + H2O + 2e− → 2OH− E = 0.40 V vs. RHE | (17) |
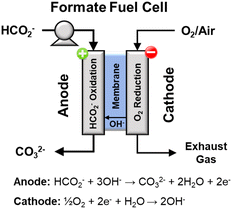 |
| | Fig. 9 Schematic representation of a direct formate fuel cell. | |
The hydroxide ions generated by the reaction in eqn (17) are conducted through the membrane to the anode to react viaeqn (15). The overall reaction for this fuel cell system is shown in eqn (18). The current state of the art for direct formate fuel cells, according to a review from 2016 is 591 mW cm−2 at 60 °C.30
| | | HCO2− + ½O2 + OH− → CO32− + H2O E = 1.45 V | (18) |
Pd-based materials have been, by far, the most widely investigated electrocatalysts for oxidation of formates in direct formate fuel cells.30 Pd catalysts are claimed to be more active than Pt-based catalysts although direct comparisons between Pt and Pd are difficult to find. Combining both Pd and Pt increased the performance of Pt electrodes, but it is not clear whether the bimetallic formulation is also more active than Pd alone.31 Adding a second metal to Pt and Pd catalysts is reported to increase their reactivity. However, studies on bimetallic catalysts are relatively scarce, even for Pd-based formulations. Regardless, the combination of Cu and Pd is particularly successful for oxidation of formate, which is often attributed to electronic interactions between Pd and Cu.32,33 Note however, that intrinsic reactivities, either turnover frequencies (TOFs), or rates normalized to the electrochemical active surface area (ECSA) are usually not reported. This makes it difficult to make definitive conclusions on the origins of improvements produced by using bimetallic formulations.
It worth noting that direct formate fuel cells are still not commercial, whereas both alkaline water splitting and fuel cells are mature technologies. Thus, although redundant, we do not discard that a combination of an electrolyzer that generates H2 (while oxidizing formate) and a fuel cell that generates power could still be more practicable than a direct formate fuel cell to utilize the energy stored in formate salts.
Electrochemical conversion of bicarbonate to formate
The concept of an inverse formate fuel cell, i.e., an electrolyzer that hydrogenates bicarbonate to formate has attracted attention in recent years. Let us first describe the context of such concept, i.e., the relevance of electrochemical CO2 hydrogenation as benchmark technology. The state of the art to electrochemically convert CO2 to CO, the transformation that requires the least number of electrons, is an operation with current density of 200–500 mA cm−2 and faradaic efficiency >95%. There are multiple companies offering CO2 electrolyzers (e.g., OPUS 12, Liquid Light Inc., Dioxide Materials, Siemens, and Sunfire) to produce syngas and fuel, although the energy efficiency of such processes is still limited by: (1) the use of water electrolysis at the anode, (2) the CO2 reduction in aqueous electrolytes with low CO2 solubility, and (3) expensive ionic liquid solvents. According to a recent perspective,34 pilot units for electrochemical CO2 conversion to CO are already being tested. Units to convert CO2 to formic acid, a popular hydrogen carrier,35 were projected to reach pilot scale in 2022.
The conversion of bicarbonate solutions instead of gaseous CO2 has interesting potential benefits. For example, bicarbonates and formates have higher solubility than CO2 in aqueous alkaline media, resulting in an enhanced single pass conversion and selectivity.25,36 Another motivation to study the electrochemical conversion of bicarbonate is the possibility of coupling this step with CO2 capture using caustic solutions as shown in eqn (19) and (20).25,37 In principle, this approach does not require an energy-intensive steps of CO2 recovery from a capture medium and of compression of gaseous CO2. Atwater and co-workers38 directly addresses the integration of CO2 capture and electrochemical hydrogenation of (bi)carbonate to syngas. The analysis shows that the energy required to produce CO via this coupled approach is lower, i.e., 0.7 MJ mol−1 CO than the energy needed in commercial processes to produce CO from coal (1.5–3.7 MJ mol−1 CO). This analysis is based on the performance of electrolysis cells that can convert carbonate solutions to syngas achieving complete conversion per pass.39
| | | CO2(g) + 2KOH(aq) ⇄ K2CO3(aq) + H2O(l) | (19) |
| | | CO2(g) + K2CO3(aq) + H2O(l) ⇄ 2KHCO3(aq) | (20) |
The electroreduction of CO2via carbonate to CO can achieve 150 mA cm−2 and the conversion of bicarbonate to formate has been reported at 100 mA cm−2.25,36,39 This makes the conversion of carbonate commensurate with electrochemical CO2 conversion technology. Therefore, perspectives of carbonate hydrogenation are promising. However, analyses on the productivity of such systems, their energy efficiency in a wide context, and economic feasibility of coupling both carbon capture and electrochemical conversion of bicarbonate will benefit from further research.
There is little work on catalyst development for electrochemical reduction of bicarbonate. Electrocatalysts containing Ag and Bi were selective to produce CO and formate, respectively, which coincides with the observations from electrocatalytic conversion of CO2.40,41 This is not surprising as both CO2 and bicarbonate participate in the mechanisms towards reduced products, as discussed below. Yet, developing electrocatalysts with optimum activity under the conditions of bicarbonate conversion, e.g., in the absence of CO2 in the bulk liquid and gas phases, would improve further the perspectives of realizing the scenarios, where electrochemical conversion of bicarbonate is relevant.
Mechanistic aspects and implications
The electrochemical hydrogenation of CO2, and bicarbonate, has been abundantly documented and reviewed. Thus, here we just discuss the facts that are potentially relevant for the advancement of bicarbonate/formate energy cycle using a heterogeneous catalyst as outlined by Grubel.4 The reduction of bicarbonate can follow two overall routes, i.e., the indirect reduction (eqn (21) and (22)) and direct reduction eqn (23).| | | HCO3−(aq) + H+(aq) ⇄ CO2(aq) + H2O(l) | (21) |
| | | CO2(aq) + H2O(l) + 2e− ⇄ HCOO−(aq) + OH−(aq) | (22) |
| | | HCO3−(aq) + H2O(l) + 2e− ⇄ HCOO−(aq) + 2OH−(aq) | (23) |
There are several mechanistic proposals for the set of elementary steps that the two routes for formate production follow. However, the consensus in literature is that CO2 is the species that is reduced on the catalyst, i.e., the overall reaction follows the indirect reduction (eqn (21) and (22)).42–44 Other studies addressing the conversion of bicarbonate have reached the same conclusion.25 A particular investigation on the effect of the identity of the cation (Li+, Na+, K+, Cs+) on the reduction of bicarbonate to formate showed that the cation identity does not influence the rate of CO2 formation viaeqn (21). However, the conversion of CO2 to CO and the faradaic efficiency were enhanced with increasing size of the cation (i.e., from Li+ to Cs+).45
Accepting this literature consensus, a likely mechanism for reduction of (bi)carbonate will require the formation of CO2. In turn, the concentration of CO2 in the bulk of the solution will be given by the equilibria described in eqn (24)–(27) (this set of reactions complements eqn (4)–(7) above), and by the equilibration of CO2 in the gas and in the aqueous phases (depending on the conditions of the overall conversion).
| | | HCO3− + OH− ⇄ CO32− + H2O | (25) |
| | | HCO3− + H+ ⇄ CO2 + H2O | (26) |
The catalytic transformation would start from the adsorption of CO2 on the catalyst (eqn (28)), which then may be followed by hydrogen addition and reduction (eqn (29) and (30)). There are indications that the reduction equivalent that reacts with CO2 is indeed adsorbed hydrogen,4 which can be generated by proton reduction (eqn (31)). The electrons for the reduction steps are provided by the oxidation half reaction in an electrochemical cell. However, the mechanism could hold for thermocatalytic operation as well, with the neutrality of the process being kept by H2 dissociation, i.e., eqn (32) and the reverse of eqn (31).
| | | HCO2* + e− ⇄ HCO2− + * | (30) |
Understanding catalyst deactivation is arguably one of the most pressing issues (besides elucidation of the reaction mechanism) to enable development of a large scale formate oxidation process.
4 There has not been any systematic investigation of deactivation of the catalyst used in the bicarbonate/formate system, but several factors have been discussed as deactivators in other systems,
i.e., pore blocking (or fouling),
46 changes in metal particle size,
47 and chemical poisons (
e.g., reactants, by-products, or products themselves). One of the early investigation of the formate/formic acid mixtures pointed out to the deactivation of the catalyst with changing pH of solution.
48 A more recent review quotes the presence of CO as the catalytic poison for heterogeneous Pd catalyst;
7 however, there is little evidence in literature that this is an issue in a purely bicarbonate/formate system.
It has been shown that cycling between dehydrogenation of a formate salt and hydrogenation of resulting the corresponding bicarbonate minimizes deactivation of the catalyst.24,46 However, these reported cycles involve treatment of the catalyst after each cycle and the amount of the cycles was limited to only a few.
Work on the electrochemical conversion of CO2 and bicarbonate to formate has shown that there is a minor pathway to CO, which deactivates the catalyst.49Eqn (33) shows the overall conversion from adsorbed CO2 to adsorbed CO, which would be strongly adsorbed on the active site. Encouragingly, CO can be removed by oxidation, i.e., a brief air exposure suffices to recover the activity. In fuel cells (direct formic acid fuel cell), reactivation of Pd catalysts can be achieved by reversing the potential of the cell for some time.50
| | | CO2* + 2e− + 2H+ ⇄ CO* + H2O | (33) |
We have focused on heterogeneous catalysts because most of current hydrogen conversion technologies are based on them. We should note however, that homogeneous catalysts are reported to be long-lived and to do not require reactivation.51
Perspective on electrochemical synthesis of formate as hydrogen carrier
Electrochemical reduction of bicarbonate to formate is increasingly attracting attention and some groups are pushing the concept at both fundamental and applied levels. However, the main motivation of these studies is not to store energy but to improve the production of chemical compounds from CO2. This strategy of combining CO2 capture with electrochemical conversion has been practiced mainly using organic capture solvents.52 The interconversion of bicarbonate and formate is a special concept for energy storage because this couple stores electrons (making it close to flow batteries) instead of hydrogen, like common hydrogen carriers operate. Thus, for power generation, it seems preferable to release the energy stored in formate using direct formate fuel cells, rather than to combine the release of H2 with the subsequent operation of a fuel cell. In cases, where H2 is desired as reactant such as in chemical processing, formate could be directly used as donor of reduction equivalents. For instance, the oxidation of formate could be coupled with the synthesis of a chemical product via electrocatalytic reduction.
There are publications reporting the integrated electrochemical interconversion of carbonate and formate in the same device.20,53 In both cases, carbonate could be reduced to formate on Sn electrodes. Direct formate fuel cells, with Pd catalysts, were operated to oxidize formate to carbonate and to produce current. The electrolysis sections for formate production and the direct formate fuel cells were connected to reservoirs that maintain constant carbon balances and that, ideally, would allow for on-demand electricity production. These examples are, however, still isolated efforts without clear links with the concept of integrating CO2 capture and sequestration with its electrochemical conversion.38 The emphasis of current research on using bicarbonate to link CO2 capture/sequestration and electrochemical production of formate is still on the utilization of CO2, rather than on the character of formate as an energy storage medium.
Naturally, the reasonable question is, why use an electrochemical approach to hydrogenate bicarbonate instead of a thermocatalytic approach? There are two answers: (1) it provides an efficient integration with renewable electricity (by-passing the stage of water electrolysis) and, (2) it enables the development of compact, on demand, devices for energy storage. Most likely, the choice of an electrochemical or a thermocatalytic method will depend on its compatibility with CO2 capture, which in turn depends on the source of CO2.
Even if not coupled with CO2 utilization, the electrochemical reduction of bicarbonate, would be advantageous when the availability of molecular hydrogen is limited, and when the oxidation half reaction adds value to the electrochemical approach. Moreover, the reduction of bicarbonate combines several steps and could simplify the process of storing energy. Several operations, i.e., electrolysis, H2 compression, and thermochemical reduction of bicarbonate, are done in one step without handling of flammable, high pressure hydrogen. H2 compression is expensive, and compressors are unreliable.
Summary and outlook
The US Department of Energy released three Energy Earthshots Initiatives in 2021, Hydrogen Shot, Long Duration Storage Shot and Carbon Negative Shot challenging the scientific community to accelerate breakthroughs to reach the 2050 net-zero carbon goals.54 The bicarbonate-formate (HCO3−–HCO2−) cycle has the unique ability to make impacts in all three of these Earthshots. We have discussed the cycle as a means to store and release hydrogen – this will be critical to the success of the Hydrogen Earthshot. When the cost of producing H2 is decreased by 80% we will need a way to store and transport H2. Given the stability and economics of the earth abundant salts there is a strong connection to reducing the cost of grid-scale energy storage by 90% to enable energy storage in the medium-/long-term duration. Finally, as described above, the capture and utilization of CO2 in the bicarbonate formate cycle provides the potential for innovative technologies to remove CO2 from the atmosphere and use it to store renewal energy. In this perspective, we described the complexity of the equilibria involved in the HCO3−–HCO2− cycle in aqueous phase and commented on the thermodynamic limitations imposed by solubility of salts and the phase transitions (i.e., from aqueous phase to gas–liquid mixtures and vice versa). We discussed the state of the mechanistic understanding of an electrochemical HCO3−–HCO2− cycle, including related topics such as CO2 capture, and direct formate fuel cells.
We showed that the thermocatalytic reduction and oxidation steps are straightforward with a simple example utilizing commonly available salts, tap water, and a commercial Pd/C catalyst. We also showed that the cost of a process including an electrochemical cycle (i.e., electrochemical carbonate reduction and thermocatalytic formate oxidation) is highly sensitive to the cost of electricity and the efficiency of the electrochemical operation. This is a common feature with any new electrochemical process, which also indicates that there is plenty of opportunities for improving efficiency and abating costs tailoring catalyst formulations, electrode preparation, and cell design.
The interconversion of (bi)carbonate and formate ions in aqueous phase is at the core of several urgent scientific challenges. For instance, environmentally benign capture of CO2, electrochemical conversion of CO2, realization of direct formate fuel cells, and catalysis in aqueous phase. Thus, integrating concepts and knowledge of the areas of electrochemistry and heterogeneous catalysis will help answer fundamental questions and to solve application challenges. Fundamental questions that need to be answered are related with the specific mechanisms of formate production and oxidation and with the control of such mechanisms with the composition of the aqueous media and applied electrical potential. In terms of application challenges, if the chemistries under electrocatalytic and thermocatalytic operations are similar, the strategies to avoid (or reverse) catalyst deactivation, as well as to enhance reaction rates can be translated from one discipline to another.
A key, but often overlooked, variable to control is the local concentration of ions and molecular species. Assuming that molecular CO2 is the reactive intermediate, as many mechanistic studies propose, it is likely that the local availability of this species (at the catalyst surface or in the electric double layer) will depend on the bulk salt concentration, CO2 partial pressure and pH. The correlations between bulk and local concentrations have not been stablished. It is also unknown how the concentration and speciation of other species that are apparent spectators would affect the reactivity of species undergoing conversion. This also includes the reactivity of adsorbed hydrogen, i.e., its binding energy with the catalyst. For electrochemical operation, the effect of the electrical potential adds another level of complexity, and control parameters, to the interfacial phenomena involved in the reaction.
If electrochemical conversion is realized, the HCO3−–HCO2− cycle may not require molecular hydrogen at some of any point, i.e., energy generation, storage, and release. At present, however, foundational studies, aiming at obtaining proof of principle, have not triggered the efforts needed to evaluate the feasibility of the bicarbonate and formate interconversion for energy storage at scales relevant for application. This gap has probably hindered scaling up the concept beyond laboratory scale.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We acknowledge the Hydrogen Materials-Advanced Research Consortium (HyMARC), established as part of the Energy Materials Network under the U.S. Department of Energy, Office of Energy Efficiency and Renewable Energy, Hydrogen and Fuel Cell Technologies Office (Contract No. DE-AC36-08GO28308). Pacific Northwest National Laboratory is operated by Battelle for the U.S. Department of Energy.
References
- J.-P. Maton, L. Zhao and J. Brouwer, Int. J. Hydrogen Energy, 2013, 38, 7867–7880 CrossRef CAS.
- A. Tullo, Chem. Eng. News, 2022, 100, 14–16 Search PubMed.
- K. Grubel, H. Jeong, C. W. Yoon and T. Autrey, J. Energy Chem., 2020, 41, 216–224 CrossRef.
- K. Grubel, J. Su, J. Kothandaraman, K. Brooks, G. A. Somorjai and T. Autrey, J. Energy Power Technol., 2020, 2(4), 016 Search PubMed.
- K. Nakajima, M. Tominaga, M. Waseda, H. Miura and T. Shishido, ACS Sustainable Chem. Eng., 2019, 7, 6522–6530 CrossRef CAS.
- M. Calabrese, D. Russo, A. di Benedetto, R. Marotta and R. Andreozzi, Renewable Sustainable Energy Rev., 2023, 173, 113102 CrossRef CAS.
- A. Bahuguna and Y. Sasson, ChemSusChem, 2021, 14, 1258–1283 CrossRef CAS PubMed.
- D. Russo, M. Calabrese, R. Marotta, R. Andreozzi and A. Di Benedetto, Int. J. Hydrogen Energy, 2022, 47, 31370–31380 CrossRef CAS.
- M. L. Perry, ECS Trans., 2021, 104, 23–38 CrossRef CAS.
- H. Wiener, J. Blum, H. Feilchenfeld, Y. Sasson and N. Zalmanov, J. Catal., 1988, 110, 184–190 CrossRef CAS.
- B. Zaidman, H. Wiener and Y. Sasson, Int. J. Hydrogen Energy, 1986, 11, 341–347 CrossRef CAS.
- A. Boddien, F. Gartner, C. Federsel, P. Sponholz, D. Mellmann, R. Jackstell, H. Junge and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 6411–6414 CrossRef CAS PubMed.
- D. Chatterjee, N. Jaiswal and P. Banerjee, Eur. J. Inorg. Chem., 2014, 2014, 5856–5859 CrossRef CAS.
- A. Guerriero, H. Bricout, K. Sordakis, M. Peruzzini, E. Monflier, F. Hapiot, G. Laurenczy and L. Gonsalvi, ACS Catal., 2014, 4, 3002–3012 CrossRef CAS.
-
CRC handbook of chemistry and physics, CRC Press, Florida, 97th edn, 2016 Search PubMed.
- J. Andersson, Energies, 2021, 14, 1392 CrossRef CAS.
- J. Joo, T. Uchida, A. Cuesta, M. T. Koper and M. Osawa, J. Am. Chem. Soc., 2013, 135, 9991–9994 CrossRef CAS PubMed.
-
W. Mook, Chemistry of carbonic acid in water, in Environmental Isotopes in the Hydrological Cycle: Principles and Applications, INEA/UNESCO, Paris, 2000, pp. 143–165 Search PubMed.
- A. M. Bartrom and J. L. Haan, J. Power Sources, 2012, 214, 68–74 CrossRef CAS.
- S. Saric, B. Biggs, M. Janbahan, R. Hamilton, H. K. Do, S. Mayoral and J. L. Haan, Appl. Energy, 2016, 183, 1346–1350 CrossRef CAS.
- D. C. Engel, G. F. Versteeg and W. P. M. van Swaaij, Fluid Phase Equilib., 1997, 135, 109–136 CrossRef CAS.
- S. Shiva Kumar and V. Himabindu, Mater. Sci. Energy Technol., 2019, 2, 442–454 Search PubMed.
-
Y. Sasson, H. Wiener and A. Givant, A method for storage and release of hydrogen, US10207921B2, 2015.
- J. Su, L. Yang, M. Lu and H. Lin, ChemSusChem, 2015, 8, 813–816 CrossRef CAS PubMed.
- T. Li, E. W. Lees, Z. Zhang and C. P. Berlinguette, ACS Energy Lett., 2020, 5, 2624–2630 CrossRef CAS.
- C. J. Stalder, S. Chao and M. S. Wrighton, J. Am. Chem. Soc., 1984, 106, 3673–3675 CrossRef CAS.
- J. A. Lopez-Ruiz, Y. Qiu, E. Andrews, O. Y. Gutiérrez and J. D. Holladay, J. Appl. Electrochem., 2020, 51, 107–118 CrossRef.
- T. S. Zhao, C. Xu, R. Chen and W. W. Yang, Prog. Energy Combust. Sci., 2009, 35, 275–292 CrossRef CAS.
- L. An, T. S. Zhao and Y. S. Li, Renewable Sustainable Energy Rev., 2015, 50, 1462–1468 CrossRef CAS.
- L. An and R. Chen, J. Power Sources, 2016, 320, 127–139 CrossRef CAS.
- K. Ohyama, T. Sugino, T. Nitte, C. Kimura and H. Aoki, ECS Trans., 2011, 16, 1473–1477 Search PubMed.
- J. Noborikawa, J. Lau, J. Ta, S. Hu, L. Scudiero, S. Derakhshan, S. Ha and J. L. Haan, Electrochim. Acta, 2014, 137, 654–660 CrossRef CAS.
- W.-J. Wang, F. Roberts, S. Peterson, S. Ha, L. Scudiero, R. Coustel, M. Mallet, M. Abdelmoula and C. Ruby, Chem. Eng. J., 2022, 427, 131763 CrossRef CAS.
- R. I. Masel, Z. Liu, H. Yang, J. J. Kaczur, D. Carrillo, S. Ren, D. Salvatore and C. P. Berlinguette, Nat. Nanotechnol., 2021, 16, 118–128 CrossRef CAS PubMed.
- K. Müller, K. Brooks and T. Autrey, Energy Fuels, 2017, 31, 12603–12611 CrossRef.
- T. Li, E. W. Lees, M. Goldman, D. A. Salvatore, D. M. Weekes and C. P. Berlinguette, Joule, 2019, 3, 1487–1497 CrossRef CAS.
- M. Liu, A. Hohenshil and G. Gadikota, Energy Fuels, 2021, 35, 8051–8068 CrossRef CAS.
- A. J. Welch, E. Dunn, J. S. DuChene and H. A. Atwater, ACS Energy Lett., 2020, 5, 940–945 CrossRef CAS.
- Y. C. Li, G. Lee, T. Yuan, Y. Wang, D.-H. Nam, Z. Wang, F. P. García de Arquer, Y. Lum, C.-T. Dinh, O. Voznyy and E. H. Sargent, ACS Energy Lett., 2019, 4, 1427–1431 CrossRef CAS.
- E. V. Kondratenko, G. Mul, J. Baltrusaitis, G. O. Larrazábal and J. Pérez-Ramírez, Energy Environ. Sci., 2013, 6, 3112–3135 RSC.
- X. Zhang, S.-X. Guo, K. A. Gandionco, A. M. Bond and J. Zhang, Mater. Today Adv., 2020, 7, 1000742 Search PubMed.
- S. Zhu, B. Jiang, W. B. Cai and M. Shao, J. Am. Chem. Soc., 2017, 139, 15664–15667 CrossRef CAS PubMed.
- M. Dunwell, Q. Lu, J. M. Heyes, J. Rosen, J. G. Chen, Y. Yan, F. Jiao and B. Xu, J. Am. Chem. Soc., 2017, 139, 3774–3783 CrossRef CAS.
- A. Wuttig, Y. Yoon, J. Ryu and Y. Surendranath, J. Am. Chem. Soc., 2017, 139, 17109–17113 CrossRef CAS PubMed.
- A. G. Fink, E. W. Lees, Z. Zhang, S. Ren, R. S. Delima and C. P. Berlinguette, ChemElectroChem, 2021, 8, 2094–2100 CrossRef CAS.
- Y. J. Hwang, Y. Kwon, Y. Kim, H. Sohn, S. W. Nam, J. Kim, T. Autrey, C. W. Yoon, Y. S. Jo and H. Jeong, ACS Sustainable Chem. Eng., 2020, 8, 9846–9856 CrossRef CAS.
- J. Wang, H. Tan, D. Jiang and K. Zhou, Nano Energy, 2017, 33, 410–417 CrossRef CAS.
- S. P. Hill and J. M. Winterbottom, J. Chem. Technol. Biotechnol., 1988, 41, 121–133 CrossRef CAS.
- X. Min and M. W. Kanan, J. Am. Chem. Soc., 2015, 137, 4701–4708 CrossRef CAS PubMed.
- X. Yu and P. G. Pickup, J. Power Sources, 2009, 187, 493–499 CrossRef CAS.
- J. Kothandaraman, M. Czaun, A. Goeppert, R. Haiges, J. P. Jones, R. B. May, G. K. Prakash and G. A. Olah, ChemSusChem, 2015, 8, 1442–1451 CrossRef CAS PubMed.
- L. A. Diaz, N. Gao, B. Adhikari, T. E. Lister, E. J. Dufek and A. D. Wilson, Green Chem., 2018, 20, 620–626 RSC.
- T. Vo, K. Purohit, C. Nguyen, B. Biggs, S. Mayoral and J. L. Haan, ChemSusChem, 2015, 8, 3853–3858 CrossRef CAS PubMed.
-
https://www.energy.gov/policy/energy-earthshots-initiative
.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Katarzyna
Grubel
*a,
Katarzyna
Grubel