
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEmerging investigator series: aqueous oxidation of isoprene-derived organic aerosol species as a source of atmospheric formic and acetic acids†
Kelvin H. Bates‡
 ab,
Daniel J. Jacobb,
James D. Copea,
Xin Chen§
c,
Dylan B. Milletc and
Tran B. Nguyen*a
ab,
Daniel J. Jacobb,
James D. Copea,
Xin Chen§
c,
Dylan B. Milletc and
Tran B. Nguyen*a
aDepartment of Environmental Toxicology, University of California Davis, Davis, CA 95616, USA. E-mail: kelvin.bates@colorado.edu
bSchool of Engineering and Applied Sciences, Harvard University, Cambridge, MA 02138, USA
cDepartment of Soil, Water, and Climate, University of Minnesota, St. Paul, MN 55108, USA
First published on 4th October 2023
Abstract
Atmospheric chemistry models generally assume organic aerosol (OA) to be photochemically inert. Recent mechanisms for the oxidation of biogenic isoprene, a major source of secondary organic aerosol (iSOA), produce excessive OA in the absence of subsequent OA reactivity. At the same time, models underestimate atmospheric concentrations of formic and acetic acids for which OA degradation could provide a source. Here we show that the aqueous photooxidation of an isoprene-derived organosulfate (2-methyltriolsulfate or MTS), an important iSOA component, produces formic and acetic acids in high yields and at timescales competitive with deposition. Experimental data are well fit by a kinetic model in which three sequential oxidation reactions of the isoprene organosulfate produce two molar equivalents of formic acid and one of acetic acid. We incorporate this chemistry and that of 2-methyltetrol, another ubiquitous iSOA component, into the GEOS-Chem global atmospheric chemistry model. Simulations show that photooxidation and subsequent revolatilization of this iSOA may account for up to half of total iSOA loss globally, producing 4 Tg a−1 each of formic and acetic acids. This reduces model biases in gas-phase formic acid and total organic aerosol over the Southeast United States in summer by ∼30% and 60% respectively. While our study shows the importance of adding iSOA photochemical sinks into atmospheric models, uncertainties remain that warrant further study. In particular, improved understanding of reaction dependencies on particle characteristics and concentrations of particle-phase OH and other oxidants are needed to better simulate the effects of this chemistry on the atmospheric budgets of organic acids and iSOA.
Environmental significanceAtmospheric chemistry models overestimate the production of organic aerosol (OA) from isoprene while simultaneously underestimating gas-phase formic and acetic acid concentrations in isoprene-rich regions. We perform aqueous photooxidation experiments on an isoprene-derived organosulfate – a key isoprene-OA constituent – and show that its oxidation produces formic and acetic acids in high yields on atmospherically relevant timescales. We integrate this chemistry, along with experimental work on other isoprene-derived OA species, into a global atmospheric chemistry model and demonstrate that these reactions improve both model overestimates of isoprene-derived OA and underestimates of gas-phase organic acids. Constraining reactive fates of other OA species and reducing uncertainties in modeling aqueous atmospheric OH will further improve our understanding of global budgets of both OA and organic acids. |
Introduction
Formic and acetic acids are ubiquitous and abundant in Earth's troposphere, and play important roles in determining the acidity of cloudwater and rainfall.1–6 By affecting the acidity of aerosol particles as well, they influence cloud nucleation7 and the aqueous chemistry of other aerosol constituents.8,9 Formic and acetic acids are directly emitted to the atmosphere by plants,10–12 fires,13–15 soils,16–18 ice,19 and anthropogenic activities,20–23 and can also be produced in the atmosphere through the gas-phase chemistry of biogenic,5,24–29 pyrogenic,30–33 and anthropogenic23,34–36 organic compounds. However, atmospheric modeling studies have repeatedly shown that these known sources are insufficient to account for the observed abundances of formic and acetic acids, especially in areas with strong biogenic influence.5,37–40One route to organic acid formation not generally included in models is the aqueous chemistry of biogenic organic compounds in aerosol and cloudwater (Fig. 1).41–47 Isoprene is an important primary precursor to biogenic secondary organic aerosol (SOA), especially via the intermediate formation of isoprene epoxydiols (IEPOX).48,49 Experimental evidence has shown that IEPOX efficiently produces 2-methyltetrols (1,2,3,4-tetrahydroxy-2-methylbutane; 2-MT) and their sulfate esters (2-MTS) following its uptake into sulfate-containing deliquesced particles,48–51 both of which can subsequently form dimers and oligomers.48,51–54 These compounds have been observed as significant fractions of organic aerosol in biogenically influenced regions around the world.55–59 Many models treat isoprene-derived SOA (iSOA) as inert, with deposition as its only loss, which may explain why the most up-to-date model mechanisms tend to overestimate its atmospheric abundance.60,61 Photochemical sinks of aqueous SOA, such as particle-phase oxidation reactions followed by revolatilization of fragmentation products, have been suggested as a possible mechanism to help balance the SOA budget.62–65 If these fragmentation products include formic and acetic acids, such photochemical sinks of SOA could simultaneously help correct the model underestimates for these acids.
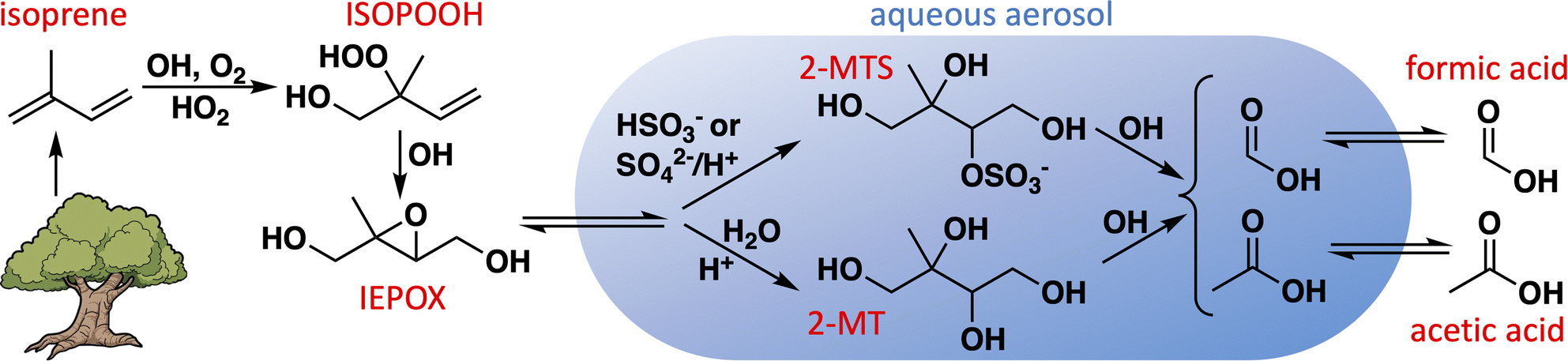 | ||
| Fig. 1 Route to aqueous production of formic and acetic acids via aqueous uptake of isoprene-derived epoxydiol (IEPOX). | ||
We recently showed using nuclear magnetic resonance (NMR) spectroscopy that the aqueous oxidation of 2-MT by OH produces large yields of formic and acetic acids.66 Here we extend that analysis to a synthetic 2-MTS isomer with similar experimental techniques and use a kinetic model to constrain organic acid yields from its oxidation. We then implement the aqueous reactions of 2-MT and 2-MTS into GEOS-Chem, a global chemical transport model, to assess the contributions of these pathways both to the global iSOA and organic acid budgets.
Experimental
Photochemical experiments
NMR experiments to determine formic and acetic acid yields from the aqueous photochemistry of 2-MTS followed the procedures described by Cope et al.66 for the study of 2-MT. In each of four experiments, 80–400 mM of 2-MTS was mixed with 0.8–4 M H2O2 (50% in H2O, Sigma Aldrich) in D2O, and the solution was irradiated directly in the quartz NMR tube using a UV-B broadband fluorescent light (peak emission at 310 nm; photon flux shown in Fig. S1†) within a photochemical enclosure. At regular intervals, the tube was removed from the photochemical enclosure and 1H NMR data were collected on a 400 MHz Bruker Avance III HD instrument (Fig. S2†). Data were analyzed using TopSpin, with suppression of the H2O peak using standard WATERSUP parameters. Control experiments without H2O2 showed no significant loss of 2-MTS.Experiments were performed with two methods to test the dependence of measured organic acid yields on potential gas–liquid partitioning, as in Cope et al.66 In the first, cyclohexane in CDCl3 was placed in a narrow glass capillary, which was flame sealed and dropped into the NMR tube containing 2-MTS and H2O2 in D2O prior to the experiment to act as an internal standard. In the second, cyclohexane in CDCl3 was instead placed in the NMR tube itself, and the 2-MTS reaction mixture was sealed in a glass capillary and dropped into the tube. In the former experiments, the 2-MTS and H2O2 solution is exposed to air in the headspace of the NMR tube during the reaction and products can partition between phases, while in the latter experiments the glass capillary contains only an aqueous phase.
2-Methyl-1,2,3-trihydroxy-4-butylsulfate, the isomer of 2-MTS investigated in this work, was synthesized according to previously published methods.67,68 All reagents were purchased from Sigma Aldrich. Briefly, 2-methylvinyloxirane (95%) was converted to 1,2-dihydroxy isoprene (DHI) by acid-catalyzed hydrolysis, which was then epoxidized with 3-chloroperbenzoic acid (≤77%) in acetonitrile at 0 °C to yield d1-IEPOX. Reaction of d1-IEPOX with tetrabutylammonium hydrogen sulfate (97%) then afforded 2-methyl-1,2,3-trihydroxy-4-butylsulfate, the purity of which (>95%) was confirmed by 1H NMR.
Kinetic modelling
To constrain the organic acid yields and multigenerational aqueous chemistry of 2-MTS, we use a simple kinetic model (Mechanism S1†) run on Matlab (MathWorks, Inc.). The mechanism uses light flux as measured in the photochemical reaction chamber (Fig. S1†) to initiate OH radical production via photolysis of H2O2, and includes the reactions OH + H2O2, HO2 + HO2, and OH + HO2.69 We also include the aqueous reactions of formic and acetic acid with OH,70 and use the OH + 2-MTS oxidation rate constant (kMTS+OH = 1.52 × 109 M−1 s−1) recently measured by Abellar et al.68 for the 2-MTS isomer used here. We initialize the model with aqueous concentrations of 2-MTS and H2O2 used in each experiment and vary the direct organic acid yields from 2-MTS, the number and OH oxidation rates of intermediate stable products of 2-MTS + OH, and the organic acid yields from those stable products to find the best fit to measured organic acid yields.Global modelling
We use the atmospheric chemistry model GEOS-Chem (http://geos-chem.org) to simulate the aqueous chemistry of 2-MT and 2-MTS on a global scale. GEOS-Chem is driven by assimilated meteorological observations from the NASA Modern-Era Retrospective analysis for Research and Applications (MERRA-2). We use model version 12.8 (https://doi.org/10.5281/zenodo.3860693) with complex SOA chemistry71 as a base and perform global simulations at 2° × 2.5° horizontal resolution with 47 vertical layers. Regional results reported for the Amazon (53.75 to 76.25°W, 11°S to 3°N), Southeast United States (81.25 to 93.75°W, 31 to 39°N), and Congo Basin (11.25 to 28.75°E, 5°S to 5°N) are from the same global simulations. Transport and convection are calculated at 15 min intervals, including advection based on the TPCORE algorithm,72 nonlocal boundary layer mixing,73 and convective mass fluxes from MERRA-2. Emissions and chemistry are calculated at intervals of 30 min. We use the standard Harmonized Emission Component (HEMCO) configuration in GEOS-Chem for emissions,74 which includes biogenic emissions calculated from the Model of Emissions of Gases and Aerosols from Nature (MEGAN) version 2.1 inventory with isoprene emissions scaled to 535 Tg a−1,75 anthropogenic emissions from the Community Emissions Data System (CEDS) inventory,76 and biomass burning emissions from the Global Fire Emissions Database (GFED4s).77 CEDS emissions in Africa are overwritten with the Diffuse and Inefficient Combustion Emissions inventory (DICE-Africa).78GEOS-Chem uses a recently compiled mechanism of isoprene chemistry61 based on the Reduced Caltech Isoprene Mechanism described by Wennberg et al.79 The model also includes the reactive uptake of IEPOX into aqueous aerosol as implemented by Marais et al.,80 with an aqueous-phase first order rate coefficient dependent on particle acidity and on the concentrations of sulfate, bisulfate, and nitrate ions (kaq = k1[H+] + k2 [SO42−][H+] + k3[HSO4−] + k4[NO3−][H+]).81,82 In the base model, IEPOX reactive uptake produces a single inert SOA species (SOAIE); here, we separate the reactive uptake into one acid-dependent reaction producing 2-MT (k1 = 3.6 × 10−2 M−1 s−1), one sulfate- and bi-sulfate-dependent reaction producing 2-MTS (k2 = 2.0 × 10−4 M−2 s−1, k3 = 7.3 × 10−4 M−1 s−1), and one nitrate-dependent reaction producing 2-methyltetrol nitrates (2-MTN; k4 = 2.0 × 10−4 M−2 s−1), where the individual rate coefficients k1–4 are identical to those used in the base model. We then add 2-MT + OH aqueous chemistry under acidic conditions from Cope et al.66 and 2-MTS + OH aqueous chemistry from this study (Mechanism S1†) to the GEOS-Chem mechanism. All formic and acetic acid produced from 2-MT and 2-MTS chemistry is allowed to partition instantaneously back to the gas phase. 2-MTN, which accounts for <1% of IEPOX uptake in our simulations, is still treated as inert. For aqueous OH, we use the parameterization described by Jacob et al.83 in which [OH]aq = (1 × 10−19 M cm3 per molecule) [OH]g.
We further update the “base” GEOS-Chem mechanism with additional formic acid sources as described by Chen et al.,84 building on the prior work of Paulot et al.5 and Millet et al.39 These updates include phototautomerization of acetaldehyde,85 oceanic emissions of alkene precurors to secondary formic acid,5,39 agricultural and soil emissions of formic acid scaled to ammonia and nitrogen oxide fluxes,5 and uptake of formic acid on ice and dust.5 We also include the newly implemented gas-phase chemistry of DHI as described by Bates et al.,86 which incorporates DHI formation from isoprene hydroxynitrate hydrolysis,87 and the reaction of isoprene hydroxy-hydroperoxide with aqueous bisulfite.88,89 All results are for annual or seasonal simulations using meteorology from 1 December 2015 to 30 November 2016, following a six-month model initialization.
Results and discussion
Photochemical experiments and kinetic models
Fig. 2 shows the combined results of the aqueous-phase NMR experiments in which 2-MTS was oxidized by OH along with results from the kinetic model. In order to compare results across experiments, the reaction time coordinate was converted to reaction extent (equivalent to kMTS+OH × [OH] × time) by taking the natural log of the inverse of the fraction of 2-MTS remaining in the NMR tube. When 2-MTS concentrations were too low to quantify (<10 mM), reaction extent was extrapolated as simple exponential decay from previous points. The kinetic model could then be fit to the combined data from all experiments. Measurements are binned by reaction extent in Fig. 2 for ease of interpretation; full plots of each experiment are shown in Fig. S3.† The kinetic model satisfactorily captures the acid yield trends and timing in individual experiments (Fig. S3†) in addition to the binned experimental results as a function of reaction extent.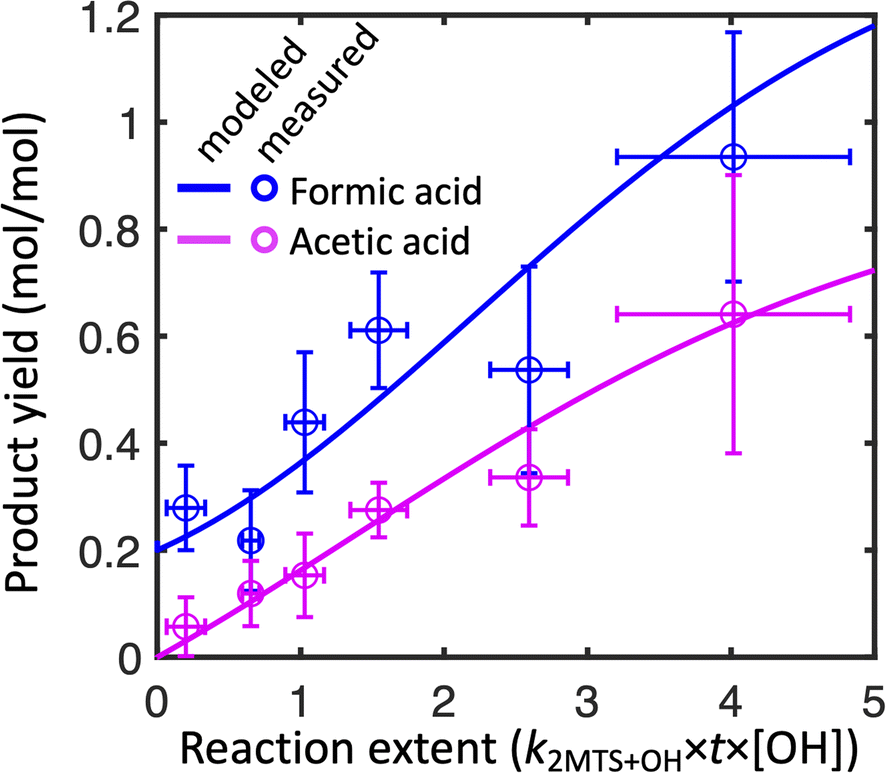 | ||
| Fig. 2 Modeled and measured yields of formic (blue) and acetic (pink) acids from 2-MTS + OH oxidation in aqueous-phase NMR experiments. Points represent observed product yields from all NMR experiments binned by reaction extent, with vertical and horizontal error bars representing standard deviations of yields and reaction extent respectively among the observations in each bin. Lines represent the simulated yields of formic and acetic acid from the kinetic model. | ||
Observed yields (moles of acid in solution per mole of 2-MTS reacted) of formic and acetic acid from 2-MTS oxidation are high, generally exceeding 100% and 30% respectively by the time 2-MTS is below detection limits. Actual yields are higher owing to the simultaneous oxidative losses of formic and acetic acids during the experiment. Yields are not constant with time but rather rise sharply with reaction extent, implying that the acids are produced in multigenerational oxidation processes. Production of formic acid starts promptly upon irradiation of the solution, with an initial yield of 20–35%, while observed acetic acid yields start near zero, suggesting that formic acid is made both in the initial and later oxidation steps while acetic acid is only produced in a multistep reaction mechanism. While the yields and identities of other products in the reaction could not be determined in these experiments, the temporal evolution of the NMR spectra closely resembles that shown by Cope et al. for 2-MT,66 suggesting that C4–C5 hydroxy-carbonyl compounds similar to those identified from 2-MT (but potentially retaining the sulfate group) might serve as intermediates in the aqueous oxidation of 2-MTS.
In experiments in which the 2-MTS and H2O2 solution was sealed in the capillary, no remaining 2-MTS was detected after 2–3 hours; in contrast, experiments with the 2-MTS and H2O2 solution outside the capillary required up to 8 hours to reach a point at which 50% of the 2-MTS had been oxidized. For experiments in the former category, because the capillary is sealed without headspace, organic acids cannot partition to the gas phase. Other differences between experiments include initial 2-MTS concentrations, distance between the NMR tube and light source, and attenuation of some UV-B light by the glass capillary in the quartz NMR tube. Despite differences in experimental design, no statistically significant difference in yields as a function of effective OH exposure ([OH] × time) was observed between the two experimental methods, so we aggregate results from all experiments together in our analysis.
To best fit experimental yields while accounting for simultaneous oxidative losses of the organic acids, the 2-MTS oxidation mechanism used in the kinetic model (Mechanism S1†) requires overall yields of 2 moles of formic acid and 1 mole of acetic acid per mole of 2-MTS oxidized, implying that four of the five carbon atoms in 2-MTS are converted to small organic acids during its aqueous oxidation. In the best-fit mechanism, these yields are spread over three oxidative steps, which is necessary to account for the prompt production of formic acid and then allow both yields to rise as the reaction progresses. The optimized fit also requires that the second and third oxidation steps progress a factor of two more slowly than the initial reaction of 2-MTS with OH. The kinetic mechanism for 2-MTS is similar to that proposed for 2-MT by Cope et al.66 (1.5 moles of formic acid and 1.25 moles of acetic acid over three oxidative steps per mole of 2-MT reacted), but with a higher formic![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :acetic acid ratio and faster later-generation oxidation reactions.
:acetic acid ratio and faster later-generation oxidation reactions.
Fig. 3 shows the temporal evolution of aqueous atmospheric 2-MTS and 2-MT chemistry in the optimized mechanism under representative conditions as implemented in GEOS-Chem. To properly track aerosol mass and oxidation state in the model, we assign chemical formulae to the oxidative intermediates of both precursors; these formulae (see Mechanism S1†) are not meant to represent actual species, but rather the approximate elemental ratios of aqueous products remaining at each reaction step. Based on the small fraction of mass lost to formic and acetic acids in the first oxidation step and the likelihood that the reaction adds oxygen to the molecule, we assume that the first-generation intermediates from both 2-MT and 2-MTS are slightly more massive than their parent compounds, resulting in an initial increase in both molar mass and oxidation state. We assume that the final carbon from 2-MTS that does not form formic or acetic acid remains in the particle phase as a C1 organosulfate, resulting in a high particulate O:C ratio of 5 and a final organic mass 58% as large as the initial mass. In contrast, the remaining carbon from 2-MT is presumed to form CO2, removing all organic mass from the particle phase.66 The optimized mechanism is meant as a simple parameterization of the reaction progress for modeling; for example, it cannot rule out the possibility that a larger number of faster oxidation steps are involved in the overall reaction process, and the chemical formulae assigned to intermediates are not constrained by our experimental results.
 | ||
| Fig. 3 Temporal evolution of aqueous atmospheric chemistry of the 2-MTS (solid lines) and 2-MT (dashed lines) systems in the optimized mechanism (Mechanism S1†) as implemented in GEOS-Chem for [OH]g = 106 molecule per cm3. | ||
Global modeling
Fig. 4 shows the change in annually averaged surface IEPOX-derived secondary organic aerosol (IEPOX-SOA) loadings when the reactions from the kinetic model (Mechanism S1†) used to simulate the photochemical experiments of both 2-MT (Cope et al.66) and 2-MTS (this work) are incorporated into GEOS-Chem. The initial reactions of 2-MT and 2-MTS with OH are fast compared with deposition; as a result, the simulated total tropospheric mass burden of 2-MT + 2-MTS is reduced by 90% when their oxidation by OH is included in GEOS-Chem. Their aqueous-phase oxidation products, however, have longer lifetimes, so the total tropospheric burden of IEPOX-SOA only decreases by 67% with the new mechanism. Global surface values, which generally reflect more recently formed SOA with lower OH exposure, exhibit smaller declines – an 85% total reduction in 2-MT + 2-MTS and 47% total reduction in IEPOX-SOA. Results for the free troposphere (Fig. S4†) are spatially similar to those shown in Fig. 4, but with much lower total burdens and slightly greater fractional decreases in 2-MT, 2-MTS, and IEPOX-SOA due to longer OH exposures. Seasonal variability in these changes, shown in Fig. S5–S10,† predominantly reflects the seasonal patterns of isoprene emissions.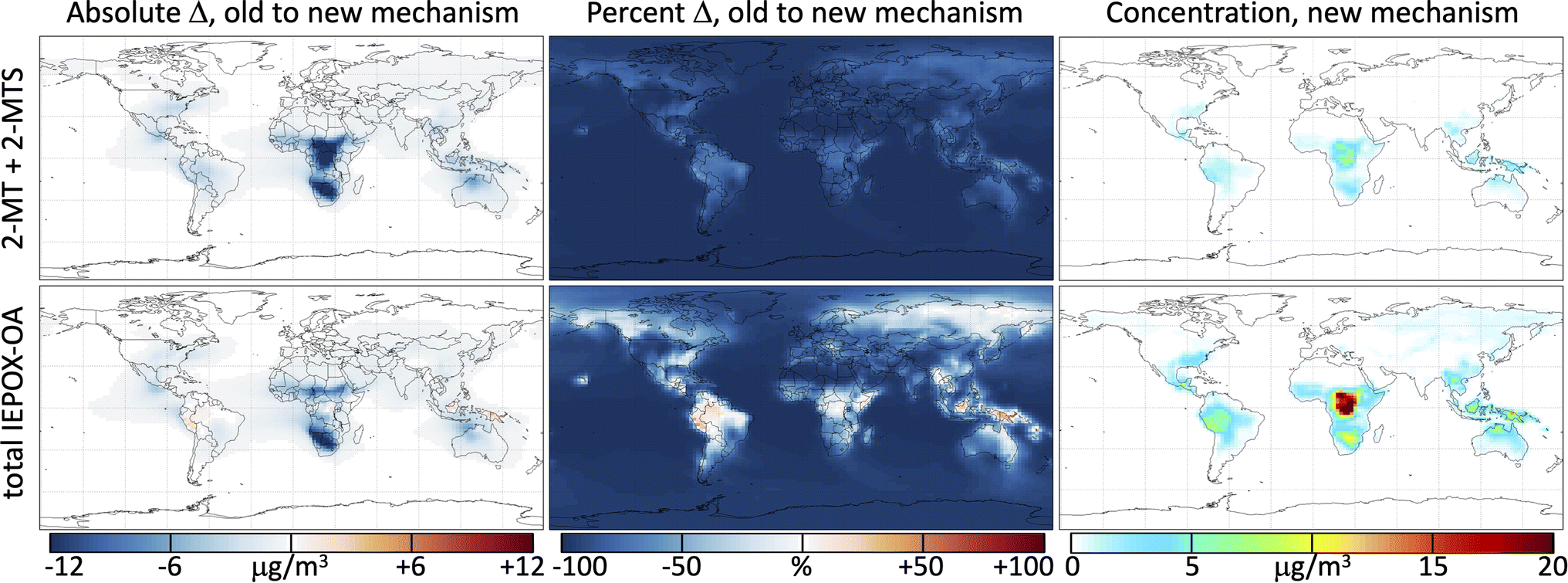 | ||
| Fig. 4 IEPOX-derived secondary organic aerosol (IEPOX-SOA) and changes in IEPOX-SOA concentrations between the base GEOS-Chem mechanism and the updated mechanism with 2-MT and 2-MTS oxidation. Panels show the absolute change between the mechanisms (left), the percent changes between the mechanisms (center), and the absolute concentrations in the updated mechanism (right) for both 2-MT + 2-MTS concentrations (top) and total IEPOX-SOA concentrations (bottom). All maps show annual averages at 0–1 km altitude. | ||
Regional variability in the effects of the added 2-MT and 2-MTS chemistry reveals the extent of oxidation within particles, i.e., the number of oxidative generations away from the C5 parent compounds. The slightly more massive first-generation oxidative intermediates (see Fig. 3 and Mechanism S1†) can lead to a slight increase in total IEPOX-SOA mass relative to the base mechanism in locations close to IEPOX source regions. For example, in the Amazon, where OH is low due to titration by biogenic hydrocarbons, annual average surface IEPOX-SOA mass increases by 11% in simulations with the new 2-MT and 2-MTS chemistry. The second and third oxidation steps of 2-MT and 2-MTS lead to rapid organic aerosol mass loss and production of formic and acetic acids, which return to the gas phase. Therefore, in other isoprene-rich regions like the Congo basin and Southeast United States, where higher OH concentrations mean that more IEPOX-SOA oxidation is accomplished close to the source, annual average surface IEPOX-SOA mass decreases by 5% (Congo Basin) and 24% (Southeast US) in simulations with the new mechanism. Over the Southeast United States in the summer, this reduces the simulated boundary layer organic aerosol burden from 7.6 to 6.5 μg m−3, improving the GEOS-Chem bias relative to atmospheric observations of OA by 60%.71 Even sharper relative decreases in IEPOX-SOA burdens are evident in regions far from IEPOX-SOA sources such as over the oceans, after multiple generations of photooxidation have occurred, where positive model biases have also been noted.71,90
As shown in Fig. 5, this added photochemical sink of 2-MT and 2-MTS in GEOS-Chem drastically changes the simulated IEPOX-SOA budget. Where the model previously predicted that all particle-phase 2-MT and 2-MTS (of which 16 Tg of carbon mass is produced annually) deposited unreacted, the updated model suggests that 92% of 2-MT and 2-MTS instead react with OH, forming more oxidized organic aerosol components and fragmenting to produce formic and acetic acids. These oxidized intermediates continue to react in the aqueous phase, and as a result, half of all carbon that enters the particle phase as IEPOX eventually becomes formic or acetic acids. The reactive flux through these oxidation pathways can also act as a major local sink of OH; the updated simulation shows decreased boundary layer OH of up to 15% compared to the base simulation over major isoprene-emitting regions (Fig. S11†).
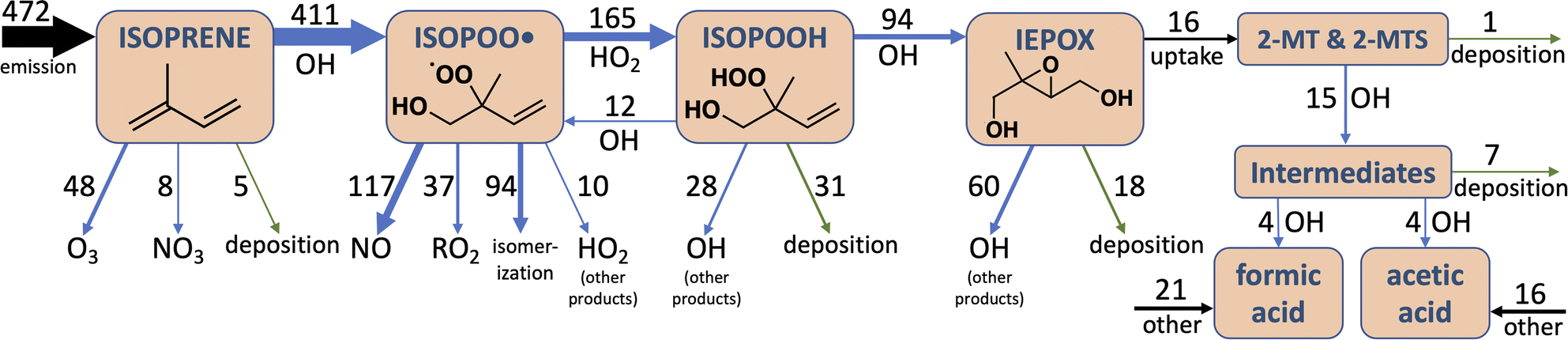 | ||
| Fig. 5 Schematic of isoprene oxidation to formic and acetic acids via IEPOX-derived organic aerosol (OA). Blue arrows denote chemical reactions, green arrows deposition, and black arrows other processes. Numbers denote troposphere-wide annual average carbon fluxes through each pathway in TgC a−1 from a GEOS-Chem simulation with the updated 2-MT and 2-MTS oxidation chemistry. The small direct formic and acetic acid yields from 2-MT & 2-MTS + OH are not shown. In the base GEOS-Chem mechanism, the deposition arrow from 2-MT and 2-MTS has a flux of 16 TgC a−1, and the pathways extending downward from the 2-MT & 2-MTS box have no flux. | ||
Fig. 6 shows the global distribution of formic acid produced by 2-MT and 2-MTS oxidation and the contribution of this source to the total atmospheric burden. As implemented in GEOS-Chem, the oxidation of IEPOX-SOA produces 14 Tg a−1 of formic acid – approximately equivalent to direct emissions from fires, anthropogenic sources, and plants combined (see Table S1†) – increasing the global atmospheric source (emissions plus secondary production) by 18%. This in turn increases the tropospheric formic acid burden by 15%. These changes are spatially heterogeneous, with the largest absolute and fractional contributions of formic acid from IEPOX-SOA chemistry occurring over or downwind of isoprene source regions. Regionally, annual average surface formic acid from IEPOX-SOA chemistry can exceed 500 ppt, contributing over 50% to the local burden.
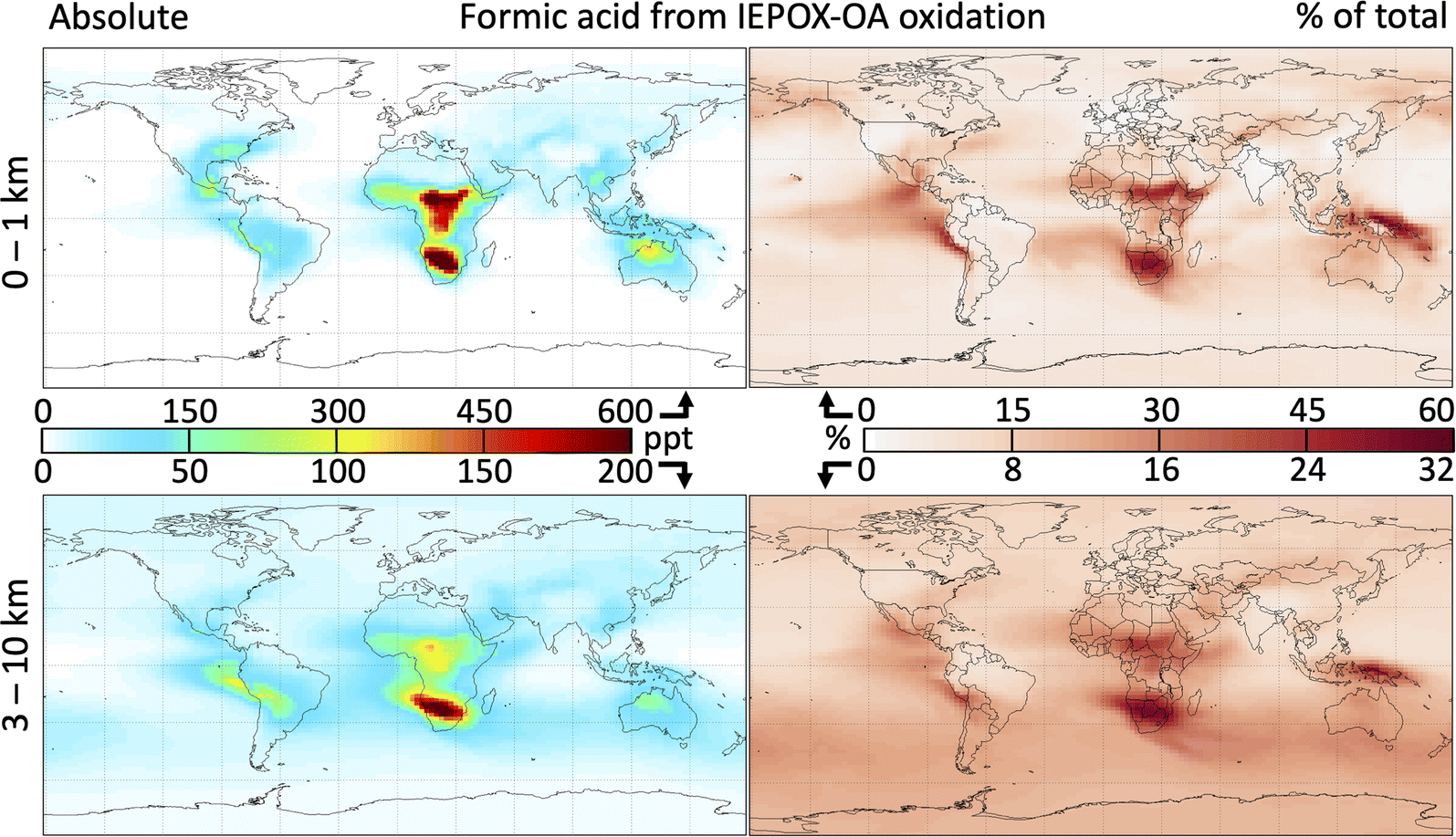 | ||
| Fig. 6 Formic acid from the multi-generational oxidation of 2-MT and 2-MTS. Panels show the absolute mixing ratios of formic acid produced from IEPOX-derived organic aerosol (IEPOX-OA) oxidation (left) and the fraction of the total formic acid mixing ratios that this IEPOX-OA-derived formic acid comprises (right) at altitudes of 0–1 km (top) and 3–10 km (bottom). All maps show annual averages from a GEOS-Chem simulation with the updated 2-MT and 2-MTS oxidation mechanism. | ||
Because the majority of acid production takes place only after multiple aqueous oxidation steps, source areas with low OH such as the Amazon see minimal formic acid production from this mechanism; instead, most acid production occurs downwind. In contrast, the Southeast United States and Congo Basin sustain sufficiently high OH for 2-MT and 2-MTS to undergo multiple oxidation steps, resulting in high local formic acid production. Over the Southeast United States, where Millet et al.39 showed that simulated near-surface formic acid is underestimated in GEOS-Chem by 1.5 ppb in the summer, the chemistry of 2-MT and 2-MTS produces an additional 500 pptv. IEPOX-SOA oxidation contributes to forested tropical Africa exhibiting some of the highest total formic acid mixing ratios in the world (Fig. S12†) sustained year-round (Fig. S13†), consistent with hotspots of both isoprene and formic acid observed by satellite,91,92 although previous comparisons between models and satellite observations of formaldehyde have concluded that MEGAN overestimates central African isoprene emissions,93 and that total African isoprene emissions should be reduced by 26%.94
Acetic acid production from IEPOX-SOA oxidation is approximately concurrent with that of formic acid; its distribution (Fig. S14–S16†) therefore looks nearly identical to that shown in Fig. 5. Although 2-MT and 2-MTS oxidation produces slightly less acetic acid (9.5 Tg a−1) than formic acid, this makes up a larger fraction of the total atmospheric acetic acid source. The effect of this chemistry on the tropospheric acetic acid burden – a 19% increase – is therefore larger than the effect for formic acid, although the additional 300 pptv of acetic acid simulated from this chemistry over the Southeast United States in the summer is insufficient to make up the 1.1 ppb shortfall previously identified in GEOS-Chem.39
The modeling results described above rely on a number of assumptions. First, we assume that the distribution of 2-MTS isomers present in the atmosphere reacts similarly to the one synthesized and studied here, which is likely only a minor isomer in the ambient distribution.52 Second, our experiments were performed in dilute aqueous solution, whereas the atmospheric aerosols in which these reactions occur frequently have high ionic strength and likely contain many other possible coreactants. Third, we assume that 2-MT and 2-MTS and their IEPOX precursors do not undergo other reactions in aerosol, such as dimerization and oligomerization, that are not included in GEOS-Chem, although these processes have been shown to occur.51–54 While such oligomers could likely fragment upon OH-initiated oxidation as readily as their C5 precursors, they may produce formic and acetic acids in different yields or timescales. Further mechanistic insights into the chemical pathways to formic and acetic acid formation in particle liquid water will be helpful for modeling.
A large source of uncertainty in these modeling results is the representation of particle-phase OH. GEOS-Chem simulates aqueous OH in aerosols as though it exists in rapid equilibrium with the gas phase, using a parameterization developed for cloud water.83 Although this is certainly an oversimplification, aqueous atmospheric OH is notoriously difficult both to measure and to accurately model.95,96 Ambient particles are generally highly concentrated solutions containing many other species with which OH can react, in addition to numerous aqueous-phase sources of OH,97–100 none of which are explicitly represented here. While aqueous OH concentrations in GEOS-Chem (∼10−13 M) are comparable to those in some other models,101–105 few measurements exist for comparison, and only for marine aerosol106,107 and larger droplets.95 Improving model representation of aerosol-phase OH will be critical for accurately simulating the fate of IEPOX-SOA in the atmosphere.
In addition to IEPOX-SOA, terpene- and aromatic-derived SOA have been shown to lose mass when exposed to OH or light on atmospherically relevant timescales.64,108,109 While the gas-phase products of these reactions are not characterized, their incorporation into GEOS-Chem would further accelerate losses of organic aerosol and may lead to OA underestimates in parts of the world, which would need to be balanced by larger sources. Indeed, the source strength of IEPOX-SOA was tuned in GEOS-Chem (via the effective Henry's Law constant, H*) to match summertime IEPOX-SOA observations from aerosol mass spectrometry (AMS) in the Southeast United States,80,110 meaning that the model likely underestimates IEPOX-SOA there with the new loss pathways added. Retuning the model's H* to accelerate IEPOX-SOA formation would counterbalance the oxidative loss, and would further increase formic and acetic production from IEPOX-SOA oxidation; however, more recent analyses suggest that IEPOX-SOA mass loadings derived from AMS measurements may be overestimated in the Southeast United States,111–113 so we do not increase the IEPOX-SOA source here. Future incorporation of oxidative OA losses into models should also reassess OA sources to ensure that burdens remain well represented.
This study, with its noted assumptions, shows that adding photochemical sinks to SOA species in atmospheric models may have significant impacts on atmospheric composition. Although the cumulative effects of our assumptions on the atmospheric IEPOX-SOA oxidation rate and the magnitude of acid production cannot be straightforwardly determined, the results presented here provide a starting point from which to consider the potential impacts of aqueous-phase SOA photooxidation on the organic aerosol and organic acid budgets. In addition to this homogeneous (one phase) aging process, heterogeneous aging of IEPOX-SOA – the direct reaction of SOA with gas-phase OH – may also contribute. Studies of heterogeneous IEPOX-SOA aging using oxidation flow reactors (OFRs) measured much longer lifetimes (∼2 weeks) of SOA against oxidative mass loss.114–116 However, such methods require their own assumptions to generalize from laboratory to ambient conditions; for example, the OH reactive-diffusive distance in particles is small,117 and the high OH and short residence time in OFRs can make surface reactions self-limiting when the particle-phase organics cannot rapidly reach the surface,118,119 an effect exacerbated when the particles are highly organic.120,121 The actual atmospheric reactivity of IEPOX-SOA will depend on both gas- and particle-phase sources and sinks of OH, and on the aerosol phase, viscosity, water content, and chemical composition. Further studies of aerosol oxidative aging in particle water at different particle phases and viscosities, and with aqueous OH sources and conditions relevant to the atmosphere, would facilitate an improved extrapolation from the lab to the ambient and more accurately inform future modeling studies.
Conclusions
We have shown using a synthetic standard in NMR-monitored reaction experiments that the aqueous-phase OH-initiated oxidation of 2-MTS, a ubiquitous product of IEPOX uptake into aerosol, rapidly produces formic and acetic acids in high yields in a multistep process. When incorporated into a global atmospheric model, simulations suggest that this oxidation (along with the analogous chemistry of 2-MT) is the dominant loss process of 2-MTS and 2-MT, reducing their combined tropospheric burden by 90%. These reactions may produce more oxidized aqueous intermediates that can locally increase SOA mass loadings, most notably over the Amazon, but ultimately lead to a 67% reduction in the tropospheric burden of total IEPOX-derived aerosol. The mass is removed from particles via conversion to gas-phase formic and acetic acids, increasing the tropospheric burdens of these species by 14.8% and 19.4% respectively. Aqueous-phase oxidation of IEPOX-SOA can also contribute significantly to local reactivity and depletion of OH, particularly over African forests.While the modeling herein relies on many assumptions that introduce uncertainty to our results, we have shown that OH-initiated oxidation of aqueous IEPOX-SOA could substantially alter the global budgets of both organic aerosol and organic acids, and have provided a benchmark for quantifying these effects. Furthermore, the direction and magnitude of the changes to aerosol and acid burdens we simulate are in line with what previous studies suggest is needed to remedy model biases. Recent modeling studies suggest that incorporating all known sources of SOA from isoprene would lead to a severe overestimate of organic aerosol and that additional losses like those proposed here are needed to balance the SOA budget.36,60 Our simulated 1.1 μg m−3 reduction in IEPOX-SOA over the Southeast United States in summer due to 2-MT and 2-MTS chemistry would reduce the current model-measurement bias in GEOS-Chem by 60%.71 The increased tropospheric formic and acetic acid burdens from IEPOX-SOA oxidation also serve to reduce model-measurement biases; while the additional 500 ppt of formic acid and 300 ppt of acetic acid we simulate from 2-MT and 2-MTS chemistry over the Southeast United States in summer only makes up ∼30% of the model shortfall,39 similar oxidative chemistry of other particle-phase organics could further contribute. Extending the methods herein to other common aqueous aerosol constituents, improving model representation of aerosol-phase OH, and minimizing the other uncertainties from assumptions used in simulating gas–aerosol interactions will further improve our ability to simulate the burdens and budgets of organic aerosol and organic acids in the atmosphere.
Author contributions
KHB prepared and performed GEOS-Chem simulations with assistance from DJJ, XC, and DBM; JDC performed photooxidation experiments; KHB, JDC, and TBN analysed experimental data; KHB prepared the manuscript with assistance from all authors.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
KHB and DJJ acknowledge support from the EPA STAR program (R840014). The views and opinions expressed in this article are those of the authors and do not represent the official views of the U.S. Environmental Protection Agency (EPA).References
- J. N. Galloway, G. E. Likens, W. C. Keene and J. M. Miller, The composition of precipitation in remote areas of the world, J. Geophys. Res., 1982, 87, 8771–8786 CrossRef CAS.
- W. C. Keene and J. N. Galloway, Organic acidity in precipitation of North America, Atmos. Environ., 1984, 18, 2491–2497 CrossRef CAS.
- M. O. Andreae, R. W. Talbot, T. W. Andreae and R. C. Harriss, Formic and acetic acid over the central Amazon region, Brazil: 1. Dry season, J. Geophys. Res., 1988, 93, 1616–1624 CrossRef CAS.
- P. Khare, N. Kumar, K. M. Kumari and S. S. Srivastava, Atmospheric formic and acetic acids: An overview, Rev. Geophys., 1999, 37(2), 227–248 CrossRef CAS.
- F. Paulot, D. Wunch, J. D. Crounse, G. C. Toon, D. B. Millet, D. P. F. DeCarlo, C. Vigouroux, N. M. Deutscher, G. Gonzalez Abad, J. Notholt, T. Warneke, J. W. Hannigan, C. Warneke, J. A. de Gouw, E. J. Dunlea, M. De Maziere, D. W. T. Griffith, P. Bernath, J. L. Jimenez and P. O. Wennberg, Importance of secondary sources in the atmospheric budgets of formic and acetic acids, Atmos. Chem. Phys., 2011, 11(5), 1989–2013 CrossRef CAS PubMed.
- V. Shah, D. J. Jacob, J. M. Moch, X. Wang and S. Zhai, Global modeling of cloud water acidity, precipitation acidity, and acid inputs to ecosystems, Atmos. Chem. Phys., 2020, 20, 12223–12245 CrossRef CAS.
- S. C. Yu, Role of organic acids (formic, acetic, pyruvic and oxalic) in the formation of cloud condensation nuclei (CCN): a review, Atmos. Res., 2000, 53(4), 185–217 CrossRef CAS.
- J. D. Surratt, M. Lewandowski, J. H. Offenberg, M. Jaoui, T. E. Kleindienst, E. O. Edney and J. H. Seinfeld, Effect of acidity on secondary organic aerosol formation from isoprene, Environ. Sci. Technol., 2007, 41, 5363–5369 CrossRef CAS PubMed.
- B. Ervens, B. J. Turpin and R. J. Weber, Secondary organic aerosol formation in cloud droplets and aqueous particles (aqSOA): a review of laboratory, field and model studies, Atmos. Chem. Phys., 2011, 11, 11069–11102 CrossRef CAS.
- J. Kesselmeier, K. Bode, C. Gerlach and E. M. Jork, Exchange of atmospheric formic and acetic acids with trees and crop plants under controlled chamber and purified air conditions, Atmos. Environ., 1998, 32(10), 1765–1775 CrossRef CAS.
- A. Guenther, C. Geron, T. Pierce, B. Lamb, P. Harley and R. Fall, Natural emissions of non-methane volatile organic compounds, carbon monoxide, and oxides of nitrogen from North America, Atmos. Environ., 2000, 34, 2205–2230 CrossRef CAS.
- U. Kuhn, S. Rottenberger, T. Biesenthal, C. Ammann, A. Wolf, G. Schebeske, S. T. Oliva, T. M. Tavares and J. Kesselmeier, Exchange of short-chain monocarboxylic acids by vegetation at a remote tropical forest site in Amazonia, J. Geophys. Res.: Atmos., 2002, 107, 8069 CrossRef.
- A. Ito and J. E. Penner, Global estimates of biomass burning emissions based on satellite imagery for the year 2000, J. Geophys. Res.: Atmos., 2004, 109, D14S05 Search PubMed.
- P. Veres, J. M. Roberts, I. R. Burling, C. Warneke, J. de Gouw and R. J. Yokelson, Measurements of gas-phase inorganic and organic acids from biomass fires by negative-ion proton-transfer chemical-ionization mass spectrometry, J. Geophys. Res.: Atmos., 2010, 115, D23302 CrossRef.
- S. Chaliyakunnel, D. B. Millet, K. C. Wells, K. E. Cady-Pereira and M. W. Shephard, A large underestimate of formic acid from tropical fires: Constraints from space-borne measurements, Environ. Sci. Technol., 2016, 50, 5631–5640 CrossRef CAS PubMed.
- E. Sanhueza and M. O. Andreae, Emission of formic and acetic acids from tropical Savanna soils, Geophys. Res. Lett., 1991, 18, 1707–1710 CrossRef CAS.
- B. T. Jones, J. Muller, S. O'Shea, A. Bacak, G. Allen, M. Gallagher, K. Bower, M. Le Breton, T. J. Bannan, S. Bauguitte, J. Pyle, D. Lowry, R. Fisher, J. France, E. Nisbet, D. Shallcross and C. Percival, Are the Fenno-Scandinavian arctic wetlands a significant regional source of formic acid?, Atmosphere, 2017, 8, 112 CrossRef.
- A. Mielnik, M. Link, J. Mattila, S. R. Fulgham and D. K. Farmer, Emission of formic and acetic acid from two Colorado soils, Environ. Sci.: Processes Impacts, 2018, 20, 1537–1545 RSC.
- J. E. Dibb and M. Arsenault, Shouldn't snowpacks be sources of monocarboxylic acids?, Atmos. Environ., 2002, 36, 2513–2522 CrossRef CAS.
- K. Kawamura, L. L. Ng and I. R. Kaplan, Determination of organic acids (C1-C10) in the atmosphere, motor exhausts, and engine oils, Environ. Sci. Technol., 1985, 19, 1082–1086 CrossRef CAS PubMed.
- T. J. Bannan, A. Bacak, J. B. A. Muller, A. M. Booth, B. Jones, M. Le Breton, K. E. Leather, M. Ghalaieny, P. Xiao, D. E. Shallcross and C. J. Percival, Importance of direct anthropogenic emissions of formic acid measured by a chemical ionisation mass spectrometer (CIMS) during the Winter ClearfLo Campaign in London, January 2012, Atmos. Environ., 2014, 83, 301–310 CrossRef CAS.
- T. A. Crisp, J. M. Brady, C. D. Cappa, S. Collier, S. D. Forestieri, M. J. Kleeman, T. Kuwayama, B. M. Lerner, E. J. Williams, Q. Zhang and T. H. Bertram, On the primary emission of formic acid from light duty gasoline vehicles and ocean-going vessels, Atmos. Environ., 2014, 98, 426–433 CrossRef CAS.
- J. Liggio, S. G. Moussa, J. Wentzell, A. Darlington, P. Liu, A. Leithead, K. Hayden, J. O'Brien, R. L. Mittermeier, R. Staebler, M. Wolde and S.-M. Li, Understanding the primary emissions and secondary formation of gaseous organic acids in the oil sands region of Alberta, Canada, Atmos. Chem. Phys., 2017, 17, 8411–8427 CrossRef CAS.
- B. R. Larsen, D. Di Bella, M. Glasius, R. Winterhalter, N. Jensen and J. Hjorth, Gas-phase OH oxidation of monoterpenes: Gaseous and particulate products, J. Atmos. Chem., 2001, 38(3), 231–276 CrossRef CAS.
- A. Lee, A. H. Goldstein, J. H. Kroll, N. L. Ng, V. Varutbangkul, R. C. Flagan and J. H. Seinfeld, Gas-phase products and secondary aerosol yields from the photooxidation of 16 different terpenes, J. Geophys. Res., 2006, 111, D17305 CrossRef.
- T. B. Nguyen, G. S. Tyndall, J. D. Crounse, A. P. Teng, K. H. Bates, R. H. Schwantes, M. M. Coggon, L. Zhang, P. Feiner, D. O. Milller, K. M. Skog, J. C. Rivera-Rios, M. Dorris, K. F. Olson, A. Koss, R. J. Wild, S. S. Brown, A. H. Goldstein, J. A. de Gouw, W. H. Brune, F. N. Keutsch, J. H. Seinfeld and P. O. Wennberg, Atmospheric fate of Criegee intermediates in the ozonolysis of isoprene, Phys. Chem. Chem. Phys., 2016, 18(15), 10241–10254 RSC.
- H. M. Allen, J. D. Crounse, K. H. Bates, A. P. Teng, M. P. Krawiec-Thayer, J. C. Rivera-Rios, F. N. Keutsch, J. M. St. Clair, T. F. Hanisco, K. H. Møller, H. G. Kjaergaard and P. O. Wennberg, Kinetics and product yields of the OH initiated oxidation of hydroxymethyl hydroperoxide, J. Phys. Chem. A, 2018, 122, 6292–6302 CrossRef CAS PubMed.
- H. D. Alwe, D. B. Millet, X. Chen, J. D. Raff, Z. C. Payne and K. Fledderman, Oxidation of volatile organic compounds as the major source of formic acid in a mixed forest canopy, Geophys. Res. Lett., 2019, 46, 2940–2948 CrossRef CAS PubMed.
- M. F. Link, T. B. Nguyen, K. H. Bates, J.-F. Müller and D. K. Farmer, Can isoprene oxidation explain high concentrations of atmospheric formic and acetic acid over forests?, ACS Earth Space Chem., 2020, 4(5), 730–740 CrossRef CAS.
- J. G. Goode, R. J. Yokelson, D. E. Ward, R. A. Susott, R. E. Babbitt, M. A. Davies and W. M. Hao, Measurements of excess O3, CO2, CH4, C2H4, C2H2, HCN, NO, NH3, HCOOH, CH3COOH, HCHO, and CH3OH in 1997 Alaskan biomass burning plumes by airborne Fourier transform infrared spectroscopy (AFTIR), J. Geophys. Res., 2000, 105, 22147–22166 CrossRef CAS.
- S. Gao, D. Hegg, P. Hobbs, T. Kirchstetter, B. Magi and M. Sadilek, Water-soluble organic components in aerosols associated with savanna fires in southern Africa: Identification, evolution, and distribution, J. Geophys. Res., 2003, 108, 8491 Search PubMed.
- R. J. Yokelson, I. T. Bertschi, T. J. Christian, P. V. Hobbs, D. E. Ward and W. M. Hao, Trace gas measurements in nascent, aged, and cloud-processed smoke from African savanna fires by airborne Fourier transform infrared spectroscopy (AFTIR), J. Geophys. Res., 2003, 108, 8478 CrossRef.
- S. C. Herndon, M. S. Zahniser, D. D. Nelson, J. Shorter, J. B. McManus, R. Jiménez, C. Warneke and J. A. de Gouw, Airborne measurements of HCHO and HCOOH during the New England Air Quality Study 2004 using a pulsed quantum cascade laser spectrometer, J. Geophys. Res. - Atmos, 2007, 112, D10S03 CrossRef.
- B. Yuan, P. R. Veres, C. Warneke, J. M. Roberts, J. B. Gilman, A. Koss, P. M. Edwards, M. Graus, W. C. Kuster, S.-M. Li, R. J. Wild, S. S. Brown, W. P. Dubé, B. M. Lerner, E. J. Williams, J. E. Johnson, P. K. Quinn, T. S. Bates, B. Lefer, P. L. Hayes, J. L. Jimenez, R. J. Weber, R. Zamora, B. Ervens, D. B. Millet, B. Rappenglück and J. A. de Gouw, Investigation of secondary formation of formic acid: urban environment vs. oil and gas producing region, Atmos. Chem. Phys., 2015, 15, 1975–1993 CrossRef CAS.
- S. Wang, M. J. Newland, W. Deng, A. R. Rickard, J. F. Hamilton, A. Munoz, M. Rodenas, M. M. Vazquez, L. Wang and X. Wang, Aromatic photo-oxidation, a new source of atmospheric acidity, Environ. Sci. Technol., 2020, 54(13), 7798–7806 CrossRef CAS PubMed.
- K. H. Bates, D. J. Jacob, K. Li, P. D. Ivatt, M. J. Evans, Y. Yan and J. Lin, Development and evaluation of a new compact mechanism for aromatic oxidation in atmospheric models, Atmos. Chem. Phys., 2021, 21, 18351–18374 CrossRef CAS.
- T. Stavrakou, J.-F. Müller, J. Peeters, A. Razavi, L. Clarisse, C. Clerbaux, P.-F. Coheur, D. Hurtmans, M. D. Mazière, C. Vigouroux, N. M. Deutscher, D. W. T. Griffith, N. Jones and C. Paton-Walsh, Satellite evidence for a large source of formic acid from boreal and tropical forests, Nat. Geosci., 2012, 5, 26–30 CrossRef CAS.
- K. E. Cady-Pereira, S. Chaliyakunnel, M. W. Shephard, D. B. Millet, M. Luo and K. C. Wells, HCOOH measurements from space: TES retrieval algorithm and observed global distribution, Atmos. Meas. Tech., 2014, 7, 2297–2311 CrossRef CAS PubMed.
- D. B. Millet, M. Baasandorj, D. K. Farmer, J. A. Thornton, K. Baumann, P. Brophy, S. Chaliyakunnel, J. A. de Gouw, M. Graus, L. Hu, A. Koss, B. H. Lee, F. D. Lopez-Hilfiker, J. A. Neuman, F. Paulot, J. Peischl, I. B. Pollack, T. B. Ryerson, C. Warneke, B. J. Williams and J. Xu, A large and ubiquitous source of atmospheric formic acid, Atmos. Chem. Phys., 2015, 15(11), 6283–6304 CrossRef CAS.
- X. Chen, D. B. Millet, H. B. Singh, A. Wisthaler, E. C. Apel, E. L. Atlas, D. R. Blake, I. Bourgeois, S. S. Brown, J. D. Crounse, J. A. de Gouw, F. M. Flocke, A. Fried, B. G. Heikes, R. S. Hornbrook, T. Mikoviny, K. E. Min, M. Muller, J. A. Neuman, D. W. O'Sullivan, J. Peischl, G. G. Pfister, D. Richter, J. M. Roberts, T. B. Ryerson, S. R. Shertz, C. R. Thompson, V. Treadaway, P. R. Veres, J. Walega, C. Warneke, R. A. Washenfelder, P. Weibring and B. Yuan, On the sources and sinks of atmospheric VOCs: an integrated analysis of recent aircraft campaigns over North America, Atmos. Chem. Phys., 2019, 19(14), 9097–9123 CrossRef CAS PubMed.
- D. J. Jacob, Chemistry of OH in remote clouds and its role in the production of formic acid and peroxymonosulfate, J. Geophys. Res., 1986, 91, 9807–9826 CrossRef CAS.
- T. L. Eliason, S. Aloisio, D. J. Donaldson, D. J. Cziczo and V. Vaida, Processing of unsaturated organic acid films and aerosols by ozone, Atmos. Environ., 2003, 37, 2207–2219 CrossRef CAS.
- M. J. Molina, A. V. Ivanov, S. Trakhtenberg and L. T. Molina, Atmospheric evolution of organic aerosol, Geophys. Res. Lett., 2004, 31, L22104 Search PubMed.
- M. L. Walser, J. Park, A. L. Gomez, A. R. Russell and S. A. Nizkorodov, Photochemical aging of secondary organic aerosol particles generated from the oxidation of d-limonene, J. Phys. Chem. A, 2007, 111, 1907–1913 CrossRef CAS PubMed.
- A. Vlasenko, I. J. George and J. P. D. Abbatt, Formation of volatile organic compounds in the heterogeneous oxidation of condensed-phase organic films by gas-phase OH, J. Phys. Chem. A, 2008, 112, 1552–1560 CrossRef CAS PubMed.
- X. Pan, J. S. Underwood, J.-H. Xing, S. A. Mang and S. A. Nizkorodov, Photodegradation of secondary organic aerosol generated from limonene oxidation by ozone studied with chemical ionization mass spectrometry, Atmos. Chem. Phys., 2009, 9, 3851–3865 CrossRef CAS.
- B. Franco, T. Blumenstock, C. Cho, C. Clarisse, C. Cierbaux, P.-F. Coheur, M. de Maziere, I. de Smedt, H.-P. Dorn, T. Emmerichs, H. Fuchs, G. Gkatzelis, D. W. T. Griffith, S. Gromov, J. W. Hannigan, F. Hase, T. Hohaus, N. Jones, A. Kerkweg, A. Kiendler-Scharr, E. Lutsch, E. Mahieu, A. Novelli, I. Ortega, C. Paton-Walsh, M. Pommier, A. Pozzer, D. Reimer, S. Rosanka, R. Sander, M. Schneider, K. Strong, R. Tillmann, M. van Roozendael, L. Vereecken, C. Vigouroux, A. Wahner and D. Taraborrelli, Ubiquitous atmospheric production of organic acids mediated by cloud droplets, Nature, 2021, 593, 233–237 CrossRef CAS PubMed.
- J. D. Surratt, A. W. H. Chan, N. C. Eddingsaas, M. N. Chan, C. L. Loza, A. J. Kwan, S. P. Hersey, R. C. Flagan, P. O. Wennberg and J. H. Seinfeld, Reactive intermediates revealed in secondary organic aerosol formation from isoprene, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6640–6645 CrossRef CAS PubMed.
- T. B. Nguyen, M. M. Coggon, K. H. Bates, X. Zhang, R. H. Schwantes, K. A. Schilling, C. L. Loza, R. C. Flagan, P. O. Wennberg and J. H. Seinfeld, Organic aerosol formation from the reactive uptake of isoprene epoxydiols (IEPOX) onto non-acidified inorganic seeds, Atmos. Chem. Phys., 2014, 14, 3497–3510 CrossRef.
- J. D. Surratt, J. H. Kroll, T. E. Kleindienst, E. O. Edney, M. Claeys, A. Sorooshian, N. L. Ng, J. H. Offenberg, M. Lewandowski, M. Jaoui, R. C. Flagan and J. H. Seinfeld, Evidence for organosulfates in secondary organic aerosol, Environ. Sci. Technol., 2007, 41, 517–527 CrossRef CAS PubMed.
- Y. H. Lin, Z. F. Zhang, K. S. Docherty, H. F. Zhang, S. H. Budisulistiorini, C. L. Rubitschun, S. L. Shaw, E. M. Knipping, E. S. Edgerton, T. E. Kleindienst, A. Gold and J. D. Surratt, Isoprene epoxydiols as precursors to secondary organic aerosol formation: Acid-catalyzed reactive uptake studies with authentic compounds, Environ. Sci. Technol., 2012, 46, 250–258 CrossRef CAS PubMed.
- T. Cui, Z. Zeng, E. O. dos Santos, Z. Zhang, Y. Chen, Y. Zhang, C. A. Rose, S. H. Budisulistiorini, L. B. Collins, W. M. Bodnar, R. A. F. de Souza, S. T. Martin, C. M. D. Machado, B. J. Turpin, A. Gold, A. P. Ault and J. D. Surratt, Development of a hydrophilic interaction liquid chromatography (HILIC) method for the chemical characterization of water-soluble isoprene epoxydiol (IEPOX)-derived secondary organic aerosol, Environ. Sci.: Processes Impacts, 2018, 20, 1524–1536 RSC.
- Y.-H. Lin, S. Budisulistiorini, K. Chu, R. A. Siejack, H. Zhang, M. Riva, Z. Zhang, A. Gold, K. E. Kautzman and J. D. Surratt, Light-Absorbing Oligomer Formation in Secondary Organic Aerosol from Reactive Uptake of Isoprene Epoxydiols, Environ. Sci. Technol., 2014, 48, 12012–12021 CrossRef CAS PubMed.
- S. J. Stropoli, C. R. Miner, D. R. Hill and M. J. Elrod, Assessing Potential Oligomerization Reaction Mechanisms of Isoprene Epoxydiols on Secondary Organic Aerosol, Environ. Sci. Technol., 2018, 53, 176–184 CrossRef PubMed.
- I. Kourtchev, T. Ruuskanen, W. Maenhaut, M. Kulmala and M. Claeys, Observation of 2-methyltetrols and related photo-oxidation products of isoprene in boreal forest aerosols from Hyytiälä, Finland, Atmos. Chem. Phys., 2005, 5(10), 2761–2770 CrossRef CAS.
- A. L. Clements and J. H. Seinfeld, Detection and quantification of 2-methyltetrols in ambient aerosol in the southeastern United States, Atmos. Environ., 2007, 41(9), 1825–1830 CrossRef CAS.
- L. L. Liang, G. Engling, F. K. Duan, Y. Cheng and K. B. He, Characteristics of 2-methyltetrols in ambient aerosol in Beijing, China, Atmos. Environ., 2012, 59, 376–381 CrossRef CAS.
- J. Liao, K. D. Froyd, D. M. Murphy, F. N. Keutsch, G. Yu, P. O. Wennberg, J. M. St. Clair, J. D. Crounse, A. Wisthaler, T. Mikoviny, J. L. Jimenez, P. Campuzano-Jost, D. A. Day, W. Hu, T. B. Ryerson, I. B. Pollack, J. Peischl, B. E. Anderson, L. D. Ziemba, D. R. Blake, S. Meinardi and G. Diskin, Airborne measurements of organosulfates over the continental US, J. Geophys. Res.: Atmos., 2015, 120(7), 2990–3005 CrossRef CAS PubMed.
- D. D. Hughes, M. B. Christiansen, A. Milani, M. P. Vermeuel, G. A. Novak, H. D. Alwe, A. F. Dickens, R. B. Pierce, D. B. Millet, T. H. Bertram, C. O. Stanier and E. A. Stone, PM2.5 chemistry, organosulfates, and secondary organic aerosol during the 2017 Lake Michigan Ozone Study, Atmos. Environ., 2021, 244, 117939 CrossRef CAS.
- S. Stadtler, T. Kühn, S. Schröder, D. Taraborrelli, M. G. Schultz and H. Kokkola, Isoprene-derived secondary organic aerosol in the global aerosol–chemistry–climate model ECHAM6.3.0–HAM2.3–MOZ1.0, Geosci. Model Dev., 2018, 11, 3235–3260 CrossRef CAS.
- K. H. Bates and D. J. Jacob, A new model mechanism for atmospheric oxidation of isoprene: global effects on oxidants, nitrogen oxides, organic products, and secondary organic aerosol, Atmos. Chem. Phys., 2019, 19, 9613–9640 CrossRef CAS.
- A. Hodzic, P. S. Kasibhatla, D. S. Jo, C. D. Cappa, J. L. Jimenez, S. Madronich and R. J. Park, Rethinking the global secondary organic aerosol (SOA) budget: stronger production, faster removal, shorter lifetime, Atmos. Chem. Phys., 2016, 16, 7917–7941 CrossRef CAS.
- A. Hodzic, S. Madronich, P. S. Kasibhatla, G. Tyndall, B. Aumont, J. L. Jimenez, J. Lee-Taylor and J. Orlando, Organic photolysis reactions in tropospheric aerosols: effect on secondary organic aerosol formation and lifetime, Atmos. Chem. Phys., 2015, 15, 9253–9269 CrossRef CAS.
- M. A. Zawadowicz, B. H. Lee, M. Shrivastava, A. Zelenyuk, R. A. Zaveri, C. Flynn, J. A. Thornton and J. E. Shilling, Photolysis controls atmospheric budgets of bioogenic secondary organic aerosol, Environ. Sci. Technol., 2020, 54(7), 3861–3870 CrossRef CAS PubMed.
- S. Lou, M. Shrivastava, R. C. Easter, Y. Yang, P.-L. Ma, H. Wang, M. J. Cubison, P. Campuzano-Jost, J. L. Jimenez, Q. Zhang, P. J. Rasch, J. E. Shilling, A. Zelenyuuk, M. Dubey, P. Cameron-Smith, S. T. Martin, J. Schneider and C. Schulz, New SOA treatments within the Energy Exascale Earth System Model (E3SM): Strong production and sinks govern atmospheric SOA distributions and radiative forcing, J. Adv. Model. Earth Syst., 2020, 12, e2020MS002266 CrossRef.
- J. D. Cope, K. A. Abellar, K. H. Bates, X. Fu and T. B. Nguyen, Aqueous photochemistry of 2-methyltetrol and erythritol as sources of formic acid and acetic acid in the atmosphere, ACS Earth Space Chem., 2021, 5(6), 1265–1277 CrossRef CAS.
- A. L. Bondy, R. L. Craig, Z. Zhang, A. Gold, J. D. Surratt and A. P. Ault, Isoprene-Derived Organosulfates: Vibrational Mode Analysis by Raman Spectroscopy, Acidity-Dependent Spectral Modes, and Observation in Individual Atmospheric Particles, J. Phys. Chem. A, 2018, 122(1), 303–315 CrossRef CAS PubMed.
- K. A. Abellar, J. D. Cope and T. B. Nguyen, Second-order kinetic rate coefficients for the aqueous-phase hydroxyl radical (OH) oxidation of isoprene-derived secondary organic aerosol compounds at 298 K, Environ. Sci. Technol., 2021, 55(20), 13728–13736 CrossRef CAS PubMed.
- J. B. Burkholder, S. P. Sander, J. Abbatt, J. R. Barker, C. Cappa, J. D. Crounse, T. S. Dibble, R. E. Huie, C. E. Kolb, M. J. Kurylo, V. L. Orkin, C. J. Percival, D. M. Wilmouth, and P. H. Wine, Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies, Evaluation No. 19, JPL Publication 19-5, Jet Propulsion Laboratory, Pasadena, 2019, http://jpldataeval.jpl.nasa.gov Search PubMed.
- M. Chin and P. H. Wine, A temperature-dependent compete-tive kinetics study of the aqueous-phase reactions of OH Radicals with Formate, Formic-Acid, Acetate, Acetic-Acid, and Hydrated Formaldehyde, Aquat. Surf. Photochem., 1994, 85–96 CAS.
- S. J. Pai, C. L. Heald, J. R. Pierce, S. C. Farina, E. A. Marais, J. L. Jimenez, P. Campuzano-Jost, B. A. Nault, A. M. Middlebrook, H. Coe, J. E. Shilling, R. Bahreini, J. H. Dingle and K. Vu, An evaluation of global organic aerosol schemes using airborne observations, Atmos. Chem. Phys., 2020, 20, 2637–2665 CrossRef CAS.
- S.-J. Lin and R. B. Rood, Multidimensional flux-form semi-Lagrangian transport schemes, Mon. Weather Rev., 1996, 124, 2046–2070 CrossRef.
- J.-T. Lin and M. B. McElroy, Impacts of boundary layer mixing on pollutant vertical profiles in the lower troposphere: Implications to satellite remote sensing, Atmos. Environ., 2010, 44, 1726–1739 CrossRef CAS.
- C. A. Keller, M. S. Long, R. M. Yantosca, A. M. Da Silva, S. Pawson and D. J. Jacob, HEMCO v1.0: a versatile, ESMF-compliant component for calculating emissions in atmospheric models, Geosci. Model Dev., 2014, 7, 1409–1417 CrossRef.
- A. Guenther, X. Jiang, C. L. Heald, T. Sakulyanontvittaya, T. Duhl, L. K. Emmons and X. Wang, The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN 2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 2012, 5, 1471–1492 CrossRef.
- R. M. Hoesly, S. J. Smith, L. Feng, Z. Klimont, G. Janssens-Maenhout, T. Pitkanen, J. J. Seibert, L. Vu, R. J. Andres, R. M. Bolt, T. C. Bond, L. Dawidowski, N. Kholod, J.-I. Kurokawa, M. Li, L. Liu, Z. Lu, M. C. P. Moura, P. R. O'Rourke and Q. Zhang, Historical (1750-2014) anthropogenic emissions of reactive gases and aerosols from the Community Emissions Data System (CEDS), Geosci. Model Dev., 2018, 11, 369–408 CrossRef CAS.
- G. R. van der Werf, J. T. Randerson, L. Giglio, G. J. Collatz, M. Mu, P. S. Kasibhatla, D. C. Morton, R. S. DeFries, Y. Jin and T. T. van Leeuwen, Global fire emissions and the contribution of deforestation, savanna, forest, agricultural, and peat fires (1997-2009), Atmos. Chem. Phys., 2010, 10, 11707–11735 CrossRef CAS.
- E. A. Marais and C. Wiedinmyer, Air Quality Impact of Diffuse and Inefficient Combustion Emissions in Africa (DICE-Africa), Environ. Sci. Technol., 2016, 50, 10739–10745 CrossRef CAS PubMed.
- P. O. Wennberg, K. H. Bates, J. D. Crounse, L. G. Dodson, R. C. McVay, L. A. Mertens, T. B. Nguyen, E. Praske, R. H. Schwantes, M. D. Smarte, J. M. St. Clair, A. P. Teng, X. Zhang and J. H. Seinfeld, The gas-phase oxidation of isoprene and its first-generation products, Chem. Rev., 2018, 118(7), 3337–3390 CrossRef CAS PubMed.
- E. A. Marais, D. J. Jacob, J. L. Jimenez, P. Campuzano-Jost, D. A. Day, W. Hu, J. Krechmer, L. Zhu, P. S. Kim, C. C. Miller, J. A. Fisher, K. Travis, K. Yu, T. F. Hanisco, G. M. Wolfe, H. L. Arkinson, H. O. T. Pye, K. D. Froyd, J. Liao and V. F. McNeill, Aqueous-phase mechanism for secondary organic aerosol formation from isoprene: application to the southeast United States and co-benefit of SO2 emission controls, Atmos. Chem. Phys., 2016, 16, 1603–1618 CrossRef CAS PubMed.
- N. C. Cole-Filipiak, A. E. O'Connor and M. J. Elrod, Kinetics of the hydrolysis of atmospherically relevant isoprene-derived hydroxy epoxides, Environ. Sci. Technol., 2010, 44, 6718–6723 CrossRef CAS PubMed.
- N. C. Eddingsaas, D. G. VanderVelde and P. O. Wennberg, Kinetics and products of the acid-catalyzed ring-opening of atmospherically relevant butyl epoxy alcohols, J. Phys. Chem. A, 2010, 114, 8106–8113 CrossRef CAS PubMed.
- D. J. Jacob, B. D. Field, Q. Li, D. R. Blake, J. de Gouw, C. Warneke, A. Hansel, A. Wisthaler, H. B. Singh and A. Guenther, Global budget of methanol: Constraints from atmospheric observations, J. Geophys. Res., 2005, 110, D08303 Search PubMed.
- X. Chen, D. B. Millet, J. A. Neuman, P. R. Veres, E. A. Ray, R. Commane, B. C. Daube, K. McKain, J. P. Schwarz, J. M. Katich, K. D. Froyd, G. P. Schill, M. J. Kim, J. D. Crounse, H. M. Allen, E. C. Apel, R. S. Hornbrook, D. R. Blake, B. A. Nault, P. Campuzano-Jost, J. L. Jimenez and J. E. Dibb, HCOOH in the Remote Atmosphere: Constraints from Atmospheric Tomography (ATom) Airborne Observations, ACS Earth Space Chem., 2021, 5(6), 1436–1454 CrossRef CAS PubMed.
- M. F. Shaw, B. Sztáray, L. K. Whalley, D. E. Heard, D. B. Millet, M. J. T. Jordan, D. L. Osborn and S. H. Kable, Phototautomer-ization of acetaldehyde as a photochemical source of formic acid in the troposphere, Nat. Commun., 2018, 9, 2584 CrossRef PubMed.
- K. H. Bates, J. D. Cope and T. B. Nguyen, Gas-phase oxidation rates and products 1,2-dihydroxy isoprene, Environ. Sci. Technol., 2021, 55, 14294–14304 CrossRef CAS PubMed.
- K. T. Vasquez, J. D. Crounse, B. C. Schulze, K. H. Bates, A. P. Teng, L. Xu and P. O. Wennberg, Rapid hydrolysis of tertiary isoprene nitrate efficiently removes NOx from the atmosphere, Proc. Natl. Acad. Sci. U. S. A., 2020, 117(52), 33011–33016 CrossRef CAS PubMed.
- E. Dovrou, J. C. Rivera-Rios, K. H. Bates and F. N. Keutsch, Sulfate formation via cloud processing from isoprene hydroxyl hydroperoxides (ISOPOOH), Environ. Sci. Technol., 2019, 53(21), 12476–12484 CrossRef CAS PubMed.
- E. Dovrou, K. H. Bates, J. C. Rivera-Rios, J. L. Cox, J. D. Shutter and F. N. Keutsch, Towards a chemical mechanism of the oxidation of aqueous sulfur dioxide via isoprene hydroxyl hydroperoxides (ISOPOOH), Atmos. Chem. Phys., 2021, 21, 8999–9008 CrossRef CAS.
- C. Y. Gao, C. L. Heald, J. M. Katich, G. Luo and F. Yu, Remote aerosol simulated during the Atmospheric Tomography (ATom) campaign and implications for aerosol lifetime, J. Geophys. Res.: Atmos., 2022, 127, e2022JD036524 CrossRef CAS PubMed.
- A. Razavi, F. Karagulian, L. Clarisse, D. Hurtmans, P. F. Coheur, C. Clerbaux, J.-F. Müller and T. Stavrakou, Global distributions of methanol and formic acid retrieved for the first time from the IASI/MetOp thermal infrared sounder, Atmos. Chem. Phys., 2011, 11, 857–872 CrossRef CAS.
- K. C. Wells, D. B. Millet, V. H. Payne, J. Deventer, K. H. Bates, J. A. de Gouw, M. Graus, C. Warneke, A. Wisthaler and J. D. Fuentes, Satellite isoprene retrievals constrain emissions and atmospheric oxidation, Nature, 2020, 585, 225–233 CrossRef CAS PubMed.
- E. A. Marais, D. J. Jacob, T. P. Kurosu, K. Chance, J. G. Murphy, C. Reeves, G. Mills, S. Casadio, D. B. Millet, M. P. Barkley, F. Paulot and J. Mao, Isoprene emissions in Africa inferred from OMI observations of formaldehyde columns, Atmos. Chem. Phys., 2012, 12, 6219–6235 CrossRef CAS PubMed.
- E. A. Marais, D. J. Jacob, A. Guenther, K. Chance, T. P. Kurosu, J. G. Murphy, C. E. Reeves and H. O. T. Pye, Improved model of isoprene emissions in Africa using Ozone Monitoring Instrument (OMI) satellite observations of formaldehyde: implications for oxidants and particulate matter, Atmos. Chem. Phys., 2014, 14, 7693–7703 CrossRef.
- Y. Rudich, N. M. Donahue and T. F. Mentel, Aging of Organic Aerosol: Bridging the Gap Between Laboratory and Field Studies, Annu. Rev. Phys. Chem., 2007, 58, 321–352 CrossRef CAS PubMed.
- A. Tilgner and H. Herrmann, Tropospheric Aqueous-Phase OH Oxidation Chemistry: Current Understanding, Uptake of Highly Oxidized Organics and Its Effect, in Multiphase Environmental Chemistry in the Atmosphere, ed. S. W. Hunt, A. Laskin, and S. A. Nizkorodov, 2018, pp. 49–85 Search PubMed.
- K. M. Badali, S. Zhou, D. Aljawhary, M. Antiñolo, W. J. Chen, A. Lok, E. Mungall, J. P. S. Wong, R. Zhao and J. P. D. Abbatt, Formation of hydroxyl radicals from photolysis of secondary organic aerosol material, Atmos. Chem. Phys., 2015, 15, 7831–7840 CrossRef CAS.
- H. Tong, A. M. Arangio, P. S. J. Lakey, T. Berkemeier, F. Liu, C. J. Kampf, W. H. Brune, U. Pöschl and M. Shiraiwa, Hydroxyl radicals from secondary organic aerosol decomposition in water, Atmos. Chem. Phys., 2016, 16, 1761–1771 CrossRef CAS.
- M. Gen, R. Zhang, D. D. Huang, Y. Li and C. K. Chan, Hetero-geneous SO2 oxidation in sulfate formation by photolysis of particulate nitrate, Environ. Sci. Technol. Lett., 2019, 6(2), 86–91 CrossRef CAS.
- T. Arakaki, Y. Kuroki, K. Okada, Y. Nakama, H. Ikota, M. Kinjo, T. Higuchi, M. Uehara and A. Tanahara, Chemical composition and photochemical formation of hydroxyl radicals in aqueous extracts of aerosol particles collected in Okinawa, Japan, Atmos. Environ., 2006, 40(25), 4764–4774 CrossRef CAS.
- H. Herrmann, D. Hoffmann, T. Schaefer, P. Bräuer and A. Tilgner, Tropospheric aqueous-phase free-radical chemistry: radical sources, spectra, reaction kinetics and prediction tools, ChemPhysChem, 2017, 11, 3796–3822 CrossRef PubMed.
- B. Ervens and R. Volkamer, Glyoxal processing by aerosol multiphase chemistry: towards a kinetic modeling framework of secondary organic aerosol formation in aqueous particles, Atmos. Chem. Phys., 2010, 10, 8219–8244 CrossRef CAS.
- A. Tilgner, P. Bräuer, R. Wolke and H. Herrmann, Modelling multiphase chemistry in deliquescent aerosols and clouds using CAPRAM3.0i, J. Atmos. Chem., 2013, 70, 221–256 CrossRef CAS.
- P. Bräuer, A. Tilgner, R. Wolke and H. Herrmann, Mechanism development and modelling of tropospheric multiphase halogen chemistry: The CAPRAM Halogen Module 2.0 (HM2), J. Atmos. Chem., 2013, 70, 19–52 CrossRef.
- B. Ervens, A. Sorooshian, Y. B. Lim and B. J. Turpin, Key parameters controlling OH-initiated formation of secondary organic aerosol in the aqueous phase (aqSOA), J. Geophys. Res.: Atmos., 2014, 119, 3997–4016 CrossRef CAS.
- C. Anastasio and J. T. Newberg, Sources and sinks of hydroxyl radical in sea-salt particles, J. Geophys. Res.: Atmos., 2007, 112(D10), D10306 CrossRef.
- T. Arakaki, C. Anastasio, Y. Kuroki, H. Nakajima, K. Okada, Y. Kotani, D. Handa, S. Azechi, T. Kimura, A. Tsuhako and Y. Miyagi, A general scavenging rate constant for reaction of hydroxyl radical with organic carbon in atmospheric waters, Environ. Sci. Technol., 2013, 47(15), 8196–8203 CrossRef CAS PubMed.
- J. H. Kroll, C. Y. Lim, S. H. Kessler and K. R. Wilson, Heterogeneous oxidation of atmospheric organic aerosol: kinetics of changes to the amount and oxidation state of particle-phase organic carbon, J. Phys. Chem. A, 2015, 119(44), 10767–10783 CrossRef CAS PubMed.
- M. A. Zawadowicz, B. H. Lee, M. Shrivastava, A. Zelenyuk, R. A. Zaveri, C. Flynn, J. A. Thornton and J. E. Shilling, Photolysis controls atmospheric budgets of biogenic secondary organic aerosol, Environ. Sci. Technol., 2020, 54(7), 3861–3870 CrossRef CAS PubMed.
- W. W. Hu, P. Campuzano-Jost, B. B. Palm, D. A. Day, A. M. Ortega, P. L. Hayes, J. E. Krechmer, Q. Chen, M. Kuwata, Y. J. Liu, S. S. de Sá, K. McKinney, S. T. Martin, M. Hu, S. H. Budisulistiorini, M. Riva, J. D. Surratt, J. M. St. Clair, G. Isaacman-Van Wertz, L. D. Yee, A. H. Goldstein, S. Carbone, J. Brito, P. Artaxo, J. A. de Gouw, A. Koss, A. Wisthaler, T. Mikoviny, T. Karl, L. Kaser, W. Jud, A. Hansel, K. S. Docherty, M. L. Alexander, N. H. Robinson, H. Coe, J. D. Allan, M. R. Canagaratna, F. Paulot and J. L. Jimenez, Characterization of a real-time tracer for isoprene epoxydiols-derived secondary organic aerosol (IEPOX-SOA) from aerosol mass spectrometer measurements, Atmos. Chem. Phys., 2015, 15, 11807–11833 CrossRef CAS.
- Y. Liu, M. Kuwata, B. F. Strick, F. M. Geiger, R. J. Thomson, K. A. McKinney and S. T. Martin, Uptake of epoxydiol isomers accounts for half of the particle-phase material produced from isoprene photooxidation via the HO2 pathway, Environ. Sci. Technol., 2015, 49, 250–258 CrossRef CAS PubMed.
- L. Xu, H. O. T. Pye, J. He, Y. Chen, B. N. Murphy and N. L. Ng, Experimental and model estimates of the contributions from biogenic monoterpenes and sesquiterpenes to secondary organic aerosol in the southeastern United States, Atmos. Chem. Phys., 2018, 18, 12613–12637 CrossRef CAS PubMed.
- H. Zhang, L. D. Yee, B. H. Lee, M. P. Curtis, D. R. Worton, G. Isaacman-VanWertz, J. H. Offenberg, M. Lewandowski, T. E. Kleindienst, M. R. Beaver, A. L. Holder, W. A. Lonneman, K. S. Docherty, M. Jaoui, H. O. T. Pye, W. Hu, D. A. Day, P. Campuzano-Jost, J. L. Jimenez, H. Guo, R. J. Weber, J. de Gouw, A. R. Koss, E. S. Edgerton, W. Brune, C. Mohr, F. D. Lopez-Hilfiker, A. Lutz, N. M. Kreisberg, S. R. Spielman, S. V. Hering, K. R. Wilson, J. A. Thornton and A. H. Goldstein, Monoterpenes are the largest source of summertime organic aerosol in the southeastern United States, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 2038–2043 CrossRef CAS PubMed.
- W. W. Hu, B. B. Palm, D. A. Day, P. Campuzano-Jost, J. E. Krechmer, Z. Peng, S. S. de Sa, S. T. Martin, M. L. Alexander, K. Baumann, L. Hacker, A. Kiendler-Scharr, A. R. Koss, J. A. de Gouw, A. H. Goldstein, R. Seco, S. J. Sjostedt, J. H. Park, A. B. Guenther, S. Kim, F. Canonaco, A. S. H. Prevot, W. H. Brune and J. L. Jimenez, Volatility and lifetime against OH heterogeneous reaction of ambient isoprene-epoxydiols-derived secondary organic aerosol (IEPOX-SOA), Atmos. Chem. Phys., 2016, 16, 11563–11580 CrossRef CAS.
- H. K. Lam, K. C. Kwong, H. Y. Poon, J. F. Davies, Z. Zhang, A. Gold, J. D. Surratt and M. N. Chan, Heterogeneous OH oxidation of isoprene-epoxydiol-derived organosulfates: kinetics, chemistry and formation of inorganic sulfate, Atmos. Chem. Phys., 2019, 19, 2433–2440 CrossRef CAS.
- Y. Chen, Y. Zhang, A. T. Lambe, R. Xu, Z. Lei, N. E. Olson, Z. Zhang, T. Szalkowski, T. Cui, W. Vizuete, A. Gold, B. J. Turpin, A. P. Ault, M. N. Chan and J. D. Surratt, Heterogeneous hydroxyl radical oxidation of isoprene-epoxydiol-derived methyltetrol sulfates: plausible formation mechanisms of previously unexplained organosulfates in ambient fine aerosols, Environ. Sci. Technol. Lett., 2020, 7(7), 460–468 CrossRef CAS.
- Y. Huang, K. M. Barraza, C. M. Kenseth, R. Zhao, C. Want, J. L. Beauchamp and J. H. Seinfeld, Probing the OH Oxidation of Pinonic Acid at the Air–Water Interface Using Field-Induced Droplet Ionization Mass Spectrometry (FIDI-MS), J. Phys. Chem. A, 2018, 122(31), 6445–6456 CrossRef CAS PubMed.
- F. A. Houle, A. A. Wiegel and K. R. Wilson, Predicting aerosol reactivity across scales: from the laboratory to the atmosphere, Environ. Sci. Technol., 2018, 52(23), 13774–13781 CrossRef CAS PubMed.
- F. A. Houle, W. D. Hinsberg and K. R. Wilson, Oxidation of a model alkane aerosol by OH radical: the emergent nature of reactive uptake, Phys. Chem. Chem. Phys., 2015, 17, 4412–4423 RSC.
- M. M. Chim, C. Y. Chow, J. F. Davies and M. N. Chan, Effects of Relative Humidity and Particle Phase Water on the Heterogeneous OH Oxidation of 2-Methylglutaric Acid Aqueous Droplets, J. Phys. Chem. A, 2017, 121(8), 1666–1674 CrossRef CAS PubMed.
- H. Guo, L. Xu, A. Bougiatioti, K. M. Cerully, S. L. Capps, J. R. Hite Jr, A. G. Carlton, S.-H. Lee, M. H. Bergin, N. L. Ng, A. Nenes and R. J. Weber, Fine-particle water and pH in the southeastern United States, Atmos. Chem. Phys., 2015, 15, 5211–5228 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional figures, tables, and mechanisms related to experimental and modelling results. See DOI: https://doi.org/10.1039/d3ea00076a |
| ‡ Now at: NOAA Chemical Sciences Laboratory, Earth System Research Laboratories, and Cooperative Institute for Research in Environmental Sciences, University of Colorado, Boulder, CO 80305, United States. |
| § Now at: Department of Civil and Environmental Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, United States. |
| This journal is © The Royal Society of Chemistry 2023 |