
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical characterization of prescribed burn emissions from a mixed forest in Northern Michigan†
Jamy Y.
Lee
 a,
Conner
Daube
b,
Ed
Fortner
b,
Nicholas
Ellsworth
a,
Nathaniel W.
May
a,
Jason
Tallant
c,
Scott
Herndon
b and
Kerri A.
Pratt
*ad
a,
Conner
Daube
b,
Ed
Fortner
b,
Nicholas
Ellsworth
a,
Nathaniel W.
May
a,
Jason
Tallant
c,
Scott
Herndon
b and
Kerri A.
Pratt
*ad
aDepartment of Chemistry, University of Michigan, Ann Arbor, MI, USA. E-mail: prattka@umich.edu
bAerodyne Research Inc., Billerica, MA, USA
cDepartment of Ecology and Evolutionary Biology and Biological Station, University of Michigan, Ann Arbor, Pellston, MI, USA
dDepartment of Earth & Environmental Sciences, University of Michigan, Ann Arbor, MI, USA
First published on 2nd November 2022
Abstract
A prescribed burn was conducted in October 2017 at the University of Michigan Biological Station located in Pellston, Michigan. Approximately 0.025 km2 of a temperate forest, primarily composed of red and white pine, red oak, bigtooth aspen, and red maple, were burned. The resulting smoke was sampled with a combination of real-time trace gas and aerosol instrumentation aboard the Aerodyne Mobile Laboratory. The resulting data were segmented into six plume periods, and the gas and particle concentration and composition measurements were characterized relative to modified combustion efficiency (MCE), which reflected both smoldering and flaming combustion. Emission factors for C2H2, C2H6, CH4, and HCN were inversely related to MCE. The bulk submicron particle composition was characterized as mostly organic by mass (>92%). The majority of the bulk organic mass was within individual biomass burning particles (>93%, by number) in the accumulation mode. Analysis of the mass spectral ion peaks of individual biomass burning particles reveals two noteworthy signatures. First, red pine smoke contained combustion products of eugenol, released during the early stages of lignin combustion. Second, the combustion of northern hardwoods (e.g., oak, aspen, maple) exhibited polycyclic aromatic hydrocarbons peaks corresponding to the combustion of furfural. The results from this study provide a detailed assessment of the composition of smoke emissions from biomass common to the understudied north-central United States.
Environmental significancePrescribed burns remove dry and dead vegetation that may contribute to high-severity wildfires and provide the opportunity for researchers to measure smoke emissions within a controlled environment. This study measured the gases and particles emitted from a prescribed burn in Michigan. In the north-central United States, prescribed burns occur more frequently than wildfires, but they are not commonly studied. The gases emitted followed trends previously observed in laboratory and field studies of pine wood. Key chemical differences were observed between the smoke particles emitted from softwood and hardwood combustion, highlighting the need for more studies in this region. These results can be incorporated into emission inventories used to inform future air quality and climate models and policies. |
1. Introduction
Wildfires have become more frequent and severe in the past three decades.1,2 Moreover, North American fire activity is projected to climb due to rising global temperatures and drought.2–5 The multi-year trend in total area burned during wildfires has increased by 3.5% per year in California. Emissions from fires impact air quality,5,7–9 health,1,10 and climate.11,12 Prescribed burns are used to minimize risks of high severity wildfires through staged controlled burns of excess dry fuel sources.1,13,14 As of 2019, the number of acres burned from prescribed burns exceeded the total amount of acres burned due to wildfires in the United States.16Prescribed burns provide a controlled environment to study atmospheric emissions from fires. The total area burned during prescribed fires in the central and southeastern United States is approximately equal to the total area burned during western United States wildfires.1 Prescribed burns, however, typically emit lower concentrations of PM2.5 (particulate matter below 2.5 μm in diameter) than wildfires.1 PM2.5 and gases emitted from fires vary widely by region, fuel type, density, and burning conditions.19–22 Gas and particle emissions from prescribed burns in the western20,23,24 and southeastern15,17,20,25 regions of the United States have been characterized by comparing fuel types (i.e. chaparral, montane, pine trees) and burn conditions (smoldering or flaming combustion). Despite the vast number of studies on fire emissions, the mixed temperate forest located in the central United States is understudied.1,21
Emissions based on fuel type and burn conditions have been studied extensively in laboratory settings. Studies of over 300 vegetation species commonly found in the southeastern and southwestern United States have identified over 200 different gases emitted in fresh smoke.17 To compare gas emissions from different vegetation and fires, emission factors (EFs) are calculated to estimate the mass of emitted species per mass of dry biomass consumed.27 The modified combustion efficiency (MCE) quantifies differences in combustion efficiency based on emitted CO2 and CO levels.28 Generally, flaming combustion emits fully combusted species, such as CO2, SO2, and nitrogen containing species (NO, NO2, and N2O), whereas the relative amounts of CO, CH4, and NH3 are greater during smoldering combustion.19,29 Furthermore, aerosols produced from fresh biomass burning vary in size and composition.12 While carbonaceous aerosols dominate the PM2.5 emissions from biomass burning, the type of combustion impacts aerosol chemical composition. Smoldering combustion generates organic carbon aerosol, while flaming combustion produces elemental carbon aerosol.6
Comprehensive EF and MCE values are essential for evaluating and predicting atmospheric impacts from fires.29–31 These values depend on vegetation, location, and meteorological conditions underscoring the importance of extending laboratory measurements to a wide range of fire environments.19–21,32 Field smoke measurements have been carried out through stationary15,17 and mobile33,34 ground-based methods, as well as airborne studies.20,23,35,36 Stationary measurements have primarily occurred in the western and southeastern United States, integrating multiple types of vegetation representative of different forest ecosystems, with results that differed from laboratory measurements of similar vegetation.20,23,35,36 Mobile platforms can measure pollutants directly downwind of fresh biomass burning plumes from prescribed burns,34 and map the distribution of gas and particle emissions across local communities.37 Results from stationary and mobile field campaigns have been compiled in emission inventories, aiding in the development of fire and smoke models.38 Overall, field sampling of prescribed burns provide insights into how pre- and post-fire conditions, including fuel moisture, fire behavior, and meteorology, affect the resulting gas and particle emissions,15 allowing connections to wildfires.
In this study, we used a combination of gas and particle instrumentation to characterize fresh biomass burning emissions from a prescribed burn near Pellston, Michigan. The burn occurred in a small section of a temperate forest (∼41 km2 total) composed of both deciduous and coniferous trees, representative of fuels common to the north-central United States.39–41 Using real-time measurements on a mobile laboratory platform, we documented the varying atmospheric trace gas and aerosol concentrations as the prescribed burn progressed. Both bulk aerosol and single-particle measurements were conducted for a detailed chemical characterization of the aerosols emitted from the mixed forest.
2. Methods
2.1 Prescribed burn sampling
A prescribed burn and subsequent sampling were conducted on October 10, 2017 at the University of Michigan Biological Station (UMBS) (45°35′N 84°43′W) near Pellston, Michigan. Weather conditions were recorded at the Pellston Regional Airport of Emmett County Station (KPLN), approximately 7 km west of the prescribed burn area, and meteorological conditions were retrieved from Weather Underground (https://www.wunderground.com/weather/us/mi/brutus/KPLN, last accessed: 02/25/2022). During the measurement period (11:45–18:00 Eastern Daylight Time (EDT), local time), the meteorological conditions remained relatively constant with moderate temperatures (11–15 °C), moderate wind speeds (3–5 m s−1 from the north-northwest), relative humidity values of 44–61%, and fair skies. Gas and aerosol emissions were sampled with a suite of instrumentation installed within the Aerodyne Mobile Laboratory (AML) that drove <1 km downwind of the three burn plots, as shown in Fig. 1. The AML is a custom modified box truck designed to serve as a platform to enable real-time ambient air sampling while stationary or in motion.42,43 On-board the mobile lab, instrument infrastructure, such as vacuum, networking, compressed zero air, and data storage, was powered via generators. Real-time data collection and visualization were used to direct the measurement activity. Instrument racks were shock mounted minimizing impacts from vibration and motion on instrument noise performance. Various inlets continuously sampled air at the front of the vehicle, separated from lofted exhaust pipes (generator and truck motor) at the rear. On-board GPS and anemometer units were used to calculate true wind speed while in motion. Data potentially influenced by AML exhaust emissions were not included in any analysis presented herein.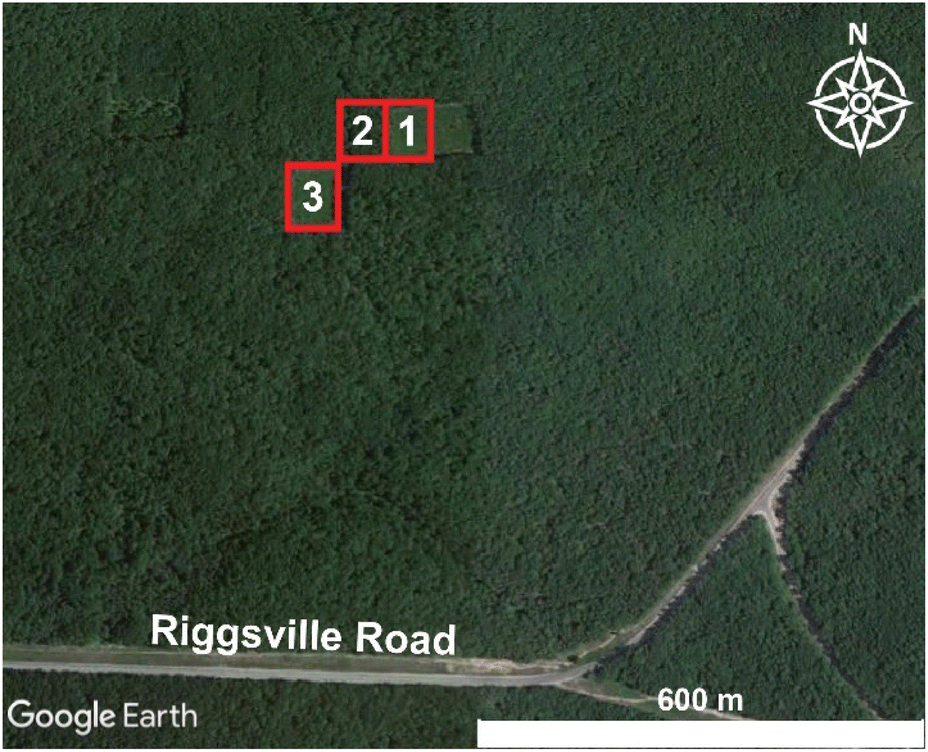 | ||
| Fig. 1 Map depicting the locations of the three plots burned during the prescribed burn. Sampling occurred along Riggsville Road. Imagery and map data were adapted from Landsat/Copernicus and Google Earth, respectively. Copyright 2021. | ||
The prescribed burn occurred on a long-term chronosequence research plot.41,44 These research plots were within a regrown forest after a cut and burn treatment in 1911. Since 1911, these plots have been cut and burned at a successional timescale, providing researchers with long-term datasets linking the effects of climate, ecological, and biogeochemical controls to an upper Michigan forest. Approximately 0.025 km2 of land containing both deciduous (red oak, red maple, bigtooth aspen, and paper birch) and coniferous trees (red and white pine) at the University of Michigan Biological Station were separated into three plot sections (Fig. 1, Table 1) and ignited by drip torches containing a mix of diesel and unleaded gasoline.
| Plot # | Fuel description | First ignition time (EDT) | Last ignition time (EDT) |
|---|---|---|---|
| 1 | Red pine with minor contributions of red oak, red maple, white pine, bigtooth aspen, and paper birch | 13:25 | 15:30 |
| 2 | Leafy debris and red pine needles with minor contributions of red oak leaves and bracken fern leaves | 13:45 | 14:30 |
| 3 | Northern hardwood forest primarily composed of red oak, big tooth aspen, white pine, and red maple as mixed slash | 14:52 | 16:15 |
2.2 Gas analysers
Nitrous oxide (NO) was measured with 1 s resolution by a model 42i NO–NO2–NOx analyzer (Thermo Scientific, Waltham, MA, USA). Since interferences from additional nitrogen species can occur when using a molybdenum oxide catalyst,45 the instrument was operated in “NO-mode”. O3 was measured with 2 s resolution by a model 202 ozone monitor (2B Technologies, Boulder, CO, USA). CO2 was measured at 1 s resolution using a LI-6262 CO2/H2O analyzer (LI-COR Biosciences, Lincoln, NE, USA). Three compact tunable infrared laser differential absorption spectrometers (TILDAS, Aerodyne Research Inc., Billerica, MA, USA) collectively measured HCN, C2H2, CH4, C2H6, N2O, and CO with 1 s resolution.46,47 Gas-phase instruments sampled from a forward-facing inlet (10 mm I.D. PFA) with a flow rate of ∼8 L min−1. All trace gas measurements were averaged to 20 s for analysis.2.3 Real-time measurements of aerosols
Total particle number concentrations were measured with an ultrafine condensation particle counter (UCPC, model 3776, TSI, Inc., Shoreview, MN, USA). The UCPC measured particles ranging from 0.04 to 2.5 μm aerodynamic diameter with 1 s time resolution at a flow rate of 1.5 L min−1. The particle number concentration and a Couette Centrifugal Particle Mass Analyzer were used to calibrate the mass concentration for refractory black carbon (rBC) measured with the Soot Particle Aerosol Mass Spectrometer (SP-AMS).![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 117 individual particles were chemically analyzed by the ATOFMS during the ∼7 h sampling period aboard the AML. 5894 particles (primarily ranging from 0.15–0.60 μm in vacuum aerodynamic diameter) out of the 20117 total particles measured occurred during the six smoke plume periods, defined in Section 2.5.
117 individual particles were chemically analyzed by the ATOFMS during the ∼7 h sampling period aboard the AML. 5894 particles (primarily ranging from 0.15–0.60 μm in vacuum aerodynamic diameter) out of the 20117 total particles measured occurred during the six smoke plume periods, defined in Section 2.5.
2.4 Calculations of modified combustion efficiency (MCE) and emission factors (EFs)
To evaluate burn conditions during sampling, the MCE was calculated, based on the equation defined by Ward and Radke,28 for each plume sampling period using eqn (1):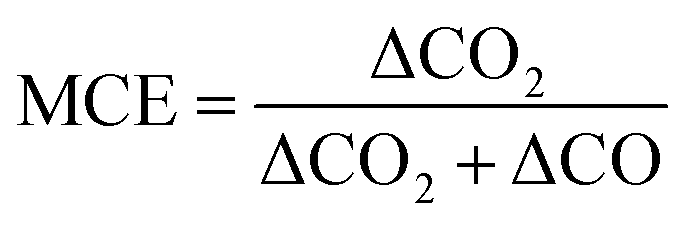 | (1) |
To quantify the variations in trace gas emissions, EFs were calculated in units of grams of compound per kilogram of dry biomass burned for CO, CO2, C2H6, C2H2, CH4, HCN, NO, and N2O. EFs were calculated based on the carbon mass balance method,27 assuming that the mass fraction of carbon in the dry biomass was 0.50,27,29 and that the majority of volatized carbon was emitted as CO2, CO, and CH4, since other carbon-containing compounds contribute less than 5% of total carbon in EF calculations.14,21,27 Brief (20 s average) time points with lower CO2 or CO mole ratios compared to background levels were omitted from EF calculations, since they do not reflect smoke plume sampling.
2.5 Smoke plume identification
Prior to the ignition of the first plot (13:25 EDT), all instruments sampled ambient air while the AML was parked 2 km east of the burn site. The period from 11:45–12:44 EDT was identified as background, based on constant signals for the trace gas and particle measurements (except HCN and C2H2), and lack of contribution from local combustion emissions. HCN and C2H2 data were not available for this period due to an instrument error. Background concentrations for HCN and C2H2 were instead defined to be during 17:53–18:00 EDT, after plume sampling had concluded. Smoke plume periods, when the AML sampled smoke from the prescribed burn, were identified by an increase in total particle number concentrations, plus an increase in CO or CO2 molar ratios, compared to the average background levels (11:45–12:44 EDT), for at least two consecutive minutes. Data that was potentially contaminated by vehicle and/or generator emissions, determined from observational notes and large concentration fluctuations, were excluded from the analysis.3. Results and discussion
3.1 Prescribed burn conditions and trace gas emissions
Gas and particle measurements were made ∼1 km south to southeast of the burn plots Fig. 1 when wind speeds were 3–5 m s−1. Therefore, the smoke emissions for the measured plumes were ∼3–5 min in age. The total particle number concentrations and trace gas mole ratios measured during the six smoke plume sampling periods are shown in Fig. 2 and Table 2.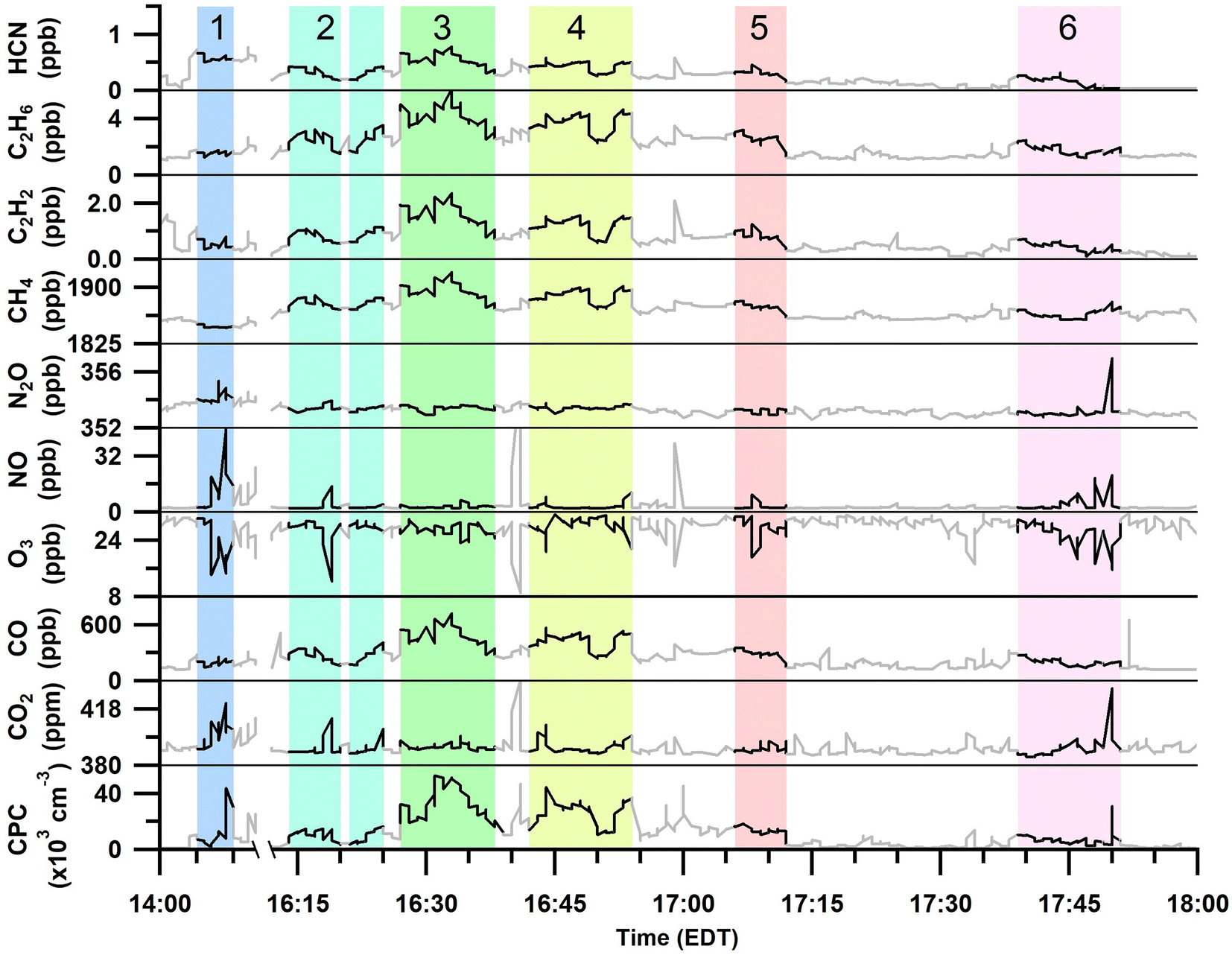 | ||
| Fig. 2 Time series of 20 s averaged HCN, C2H6, C2H2, CH4, N2O, NO, O3, CO, and CO2 mole ratios and total particle (CPC) number concentrations over the full prescribed burn sampling period. Identified periods of smoke plume sampling are highlighted and numbered. Areas that are greyed out were not included in the plume analyses (Table 2). | ||
| Plume 1 | Plume 2 | Plume 3 | Plume 4 | Plume 5 | Plume 6 | |
|---|---|---|---|---|---|---|
| Start time (EDT) | 14:05:40 | 16:14:00 | 16:27:20 | 16:42:40 | 17:06:00 | 17:39:20 |
| End time (EDT) | 14:09:40 | 16:25:00 | 16:38:20 | 16:53:40 | 17:12:20 | 17:51:00 |
| Plot # | 1 & 2 | 3 | 3 | 3 | 3 | 3 |
| MCE | 0.993 ± 0.002 | 0.98 ± 0.02 | 0.87 ± 0.04 | 0.92 ± 0.04 | 0.95 ± 0.05 | 0.98 ± 0.01 |
| Carbon dioxide (CO2) | 1821 ± 5 | 1780 ± 50 | 1570 ± 80 | 1670 ± 80 | 1740 ± 100 | 1790 ± 30 |
| Carbon monoxide (CO) | 8 ± 3 | 30 ± 30 | 150 ± 50 | 90 ± 50 | 60 ± 50 | 20 ± 20 |
| Methane (CH4) | N/A | 4 ± 5 | 17 ± 5 | 10 ± 5 | 6 ± 6 | 4 ± 5 |
| Ethane (C2H6) | 0.03 ± 0.03 | 0.2 ± 0.2 | 0.8 ± 0.3 | 0.4 ± 0.2 | 0.3 ± 0.3 | 0.07 ± 0.04 |
| Acetylene (C2H2) | 0.06 ± 0.04 | 0.1 ± 0.2 | 0.6 ± 0.2 | 0.3 ± 0.2 | 0.2 ± 0.2 | 0.13 ± 0.08 |
| Hydrogen cyanide (HCN) | 0.09 ± 0.06 | 0.05 ± 0.06 | 0.21 ± 0.07 | 0.12 ± 0.06 | 0.09 ± 0.08 | 0.05 ± 0.04 |
| Nitric oxide (NO) | 1.6 ± 0.4 | 0.8 ± 0.3 | 1.7 ± 0.7 | 1.1 ± 0.4 | 1.2 ± 0.7 | 4 ± 2 |
| Nitrous oxide (N2O) | 0.2 ± 0.1 | 0.07 ± 0.02 | 0.19 ± 0.07 | 0.14 ± 0.07 | 0.03 ± 0.02 | 0.3 ± 0.2 |
Sampling of plume 1 occurred approximately 40 min after the first ignition of plot 1 and 20 min after the first ignition of plot 2 (Tables 1 and 2). The first burned plot (plot 1, Fig. 1) was primarily composed of red pine, with minor contributions from other mixed fuels (red oak, red maple, white pine, bigtooth aspen, and paper birch), and was ignited in the southeastern corner at 13:25 EDT; ignition of fuels continued intermittently until 15:30 EDT. The burning of the second plot overlapped in time with the burning of the first plot and consisted of similar vegetation as the first section except that the vegetation in the second plot was broken down as leafy debris and pine needles. Plot 2 failed to stay aflame beyond the initial ignition from the drip torches at 13:45 EDT and attempts to re-ignite the second plot stopped by 14:30 EDT. In general, plots 1 and 2 burned with low to moderate severity, consuming less than 40% of the available fuel.
Plumes 2–6 primarily overlapped with the burning of plot 3 and with contributions from the smoldering of plots 1 and 2. Plot 3 was ignited at 14:52 EDT and burned with higher severity than plots 1 and 2, consuming approximately 60% of the available fuels. This third and final burned plot contained a diverse hardwood slash mixture composed of red maple, red oak, and bigtooth aspen, as well as softwood white pine (Table 1, Fig. 1). Plume 2 began at the time of the final ignition of plot 3 (16:15 EDT), and subsequently, was likely most influenced by the ignition of the mixed slash from plot 3. Plumes 3 to 6 corresponded to emissions after all plots had been ignited and followed the spread of the burn as it rekindled and/or self-extinguished. Plot 3 continued to burn throughout the night, after air sampling had concluded.
The range of MCEs measured during the smoke sampling periods was 0.87–0.993 (Table 2). MCE values ranging from ∼0.65–0.85 correspond to smoldering combustion, and a MCE near 0.99 describes a fire with primarily flaming combustion.30 A fire with roughly equal contributions from smoldering and flaming combustion has a MCE near 0.9.30 All six plume periods had average MCEs greater than 0.85, implying that all plumes had some or majority influence from flaming combustion. Plume 1 had the highest MCE (0.993 ± 0.002, Table 2), corresponding to primarily flaming emissions from the first and second plots. Plume 2 corresponded with the last ignition of plot 3, and this is reflected by a high MCE of 0.98 ± 0.02. Plumes 3 and 4 occurred after the last ignition as the fire intensity decreased, resulting in lower MCE values (0.87 ± 0.04 and 0.92 ± 0.04, respectively). Plumes 5 and 6 coincided with flare-ups, observed as the fire spread throughout plot 3 and as reflected by the increase in MCE values (0.95 ± 0.05 and 0.98 ± 0.01, respectively).
MCE values were inversely related with the average EFs for C2H6, C2H2, CH4, and HCN shown in Fig. 3, as well as the total particle number concentrations (Table S1†). Average trace gas mole ratios and particle number concentrations for the six smoke plumes are provided in Table S1†. As described by Ward and Radke,28 smoldering combustion produces higher total particle emissions, while flaming combustion produces fewer total particles. This trend can often be observed in plume particle concentrations when sampling in close proximity to the fire source. Plumes 3, 4, and 5 had the lowest MCE values of all plume periods (0.87 ± 0.04, 0.92 ± 0.04, and 0.95 ± 0.05, respectively) and were characterized by having the largest total particle number concentrations (33000 ± 4000 particles per cm3, 26000 ± 3000 particles per cm3, and 14000 ± 1000 particles per cm3, respectively). Conversely, plumes 1, 2, and 6 had almost pure flaming MCE values (0.993 ± 0.002, 0.98 ± 0.02, and 0.98 ± 0.01, respectively) and lower total particle number concentrations (11000 ± 7000 particles per cm3, 10000 ± 1000 particles per cm3, and 7000 ± 2000 particles per cm3, respectively). Not surprisingly, these total particle number concentrations that were significantly higher than background concentrations measured prior to the prescribed burn (1500 ± 900 particles per cm3), which were similar to previous summertime measurements at this site (2000 ± 1000 particles per cm3).60 The particle concentrations during the lower MCE plume periods were on par with typical near-source particle concentrations for smoke plumes (>20000 particles per cm3).61
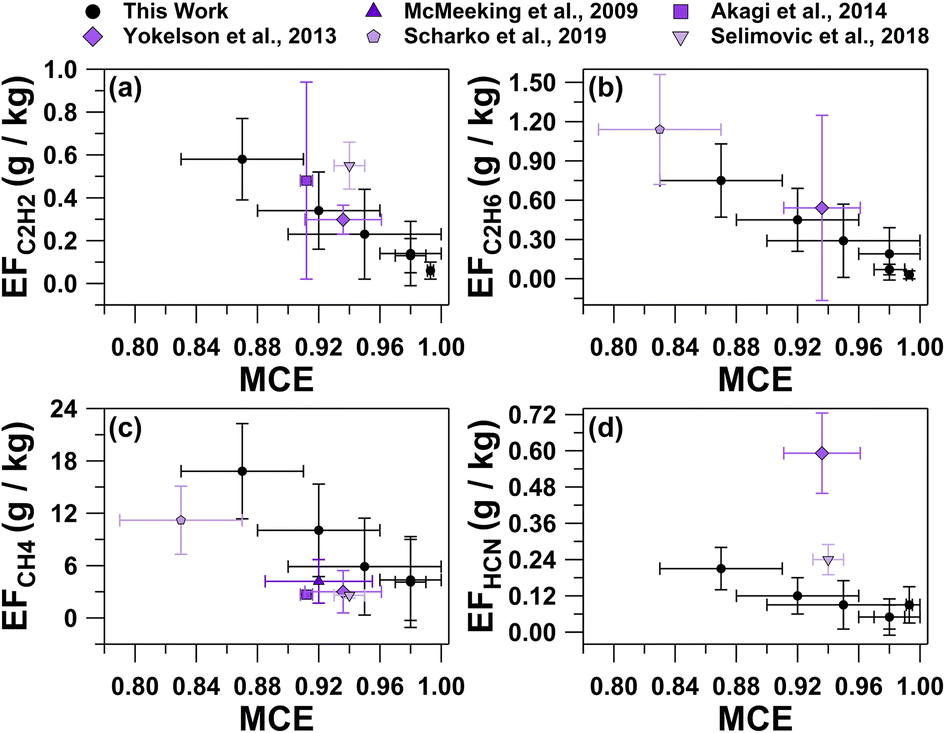 | ||
| Fig. 3 Average emission factors for (a) C2H2, (b) C2H6, (c) CH4, and (d) HCN versus MCE values for the six smoke plume periods, as well as previous studies. Error bars represent the 95% confidence interval for each calculated value. Previous values are from McMeeking et al.,6 Akagi et al.,15 Yokelson et al.,17 Scharko et al.,18 and Selimovic et al.26 | ||
The range of ethane (C2H6) EFs (0.03–0.75 g kg−1, Table 2) were similar to the average C2H6 EF calculated based on a mixture of lab and field measurements of a pine understory (0.5 ± 0.7 g kg−1) collected from the southeastern and the southwestern United States.17 Similarly, the acetylene (C2H2) EFs ranged from 0.06–0.58 g kg−1 (Table 2), which is consistent with previous prescribed burn measurements from pine forests in the southeastern United States (0.3 ± 0.1 g kg−1, Akagi et al.30 and 0.5 ± 0.5 g kg−1, Akagi et al.15). However, the C2H2 EF can vary widely, as previously observed from a pine forest prescribed burn in the southeastern United States (7 ± 3 g kg−1)18 and laboratory measurements of lodgepole pine collected from the western United States (1.9 ± 0.4 g kg−1)26 that were significantly greater than the range measured in this study. The lower C2H2 EFs in this study compared to what was observed by Scharko et al.18 and Selimovic et al.26 may be associated with a difference in the pyrolysis temperature of the burn, as C2H2 EFs increase with higher burn temperature, as discussed by Scharko et al.18 and references therein.
The methane (CH4) EFs for plumes 2–6, which sampled smoke emissions after the ignition from all three plots, ranged from 4–17 g kg−1. The CH4 EFs were greater than previous laboratory and field measurements of pine tree fires with MCEs greater than 0.9 (CH4 EFs of 2.68 ± 0.05 g kg−1, Akagi et al.;15 4 ± 3 g kg−1, McMeeking et al.;6 2.6 ± 0.3 g kg−1, Selimovic et al.26). Methane molar ratios during plume 1, which correspond to the smoke released from plots 1 and 2, were not greater than background values. Plume 1 had the greatest MCE with near pure flaming contributions (0.993 ± 0.002), which may have contributed to the limited CH4 emissions.62
The EFs for HCN (0.05–0.21 g kg−1, Table 2) were significantly lower than previously reported (0.6–0.7 g kg−1),17,30,63 potentially related to HCN being primarily emitted during smoldering combustion.15,64 For NO and N2O, average EFs previously reported for temperate forests (3 ± 1 g kg−1 and 0.2 ± 0.2 g kg−1, respectively)30 fall within range of this study for NO and N2O (0.8–4 g kg−1 and 0.03–0.2 g kg−1, respectively, Table 2). Variability in these measurements is expected because the nitrogen content of the biomass fuel can influence the emissions of nitrogen-containing compounds.23,65 Compared to the commonly studied vegetation (ponderosa pine, lodgepole pine, grass, peat, etc.) in previous studies from the southeastern and southwestern United States, the primary fuel burned here, red pine, has a lower nitrogen content.66
3.2 Characterization of particle composition from resulting smoke plumes
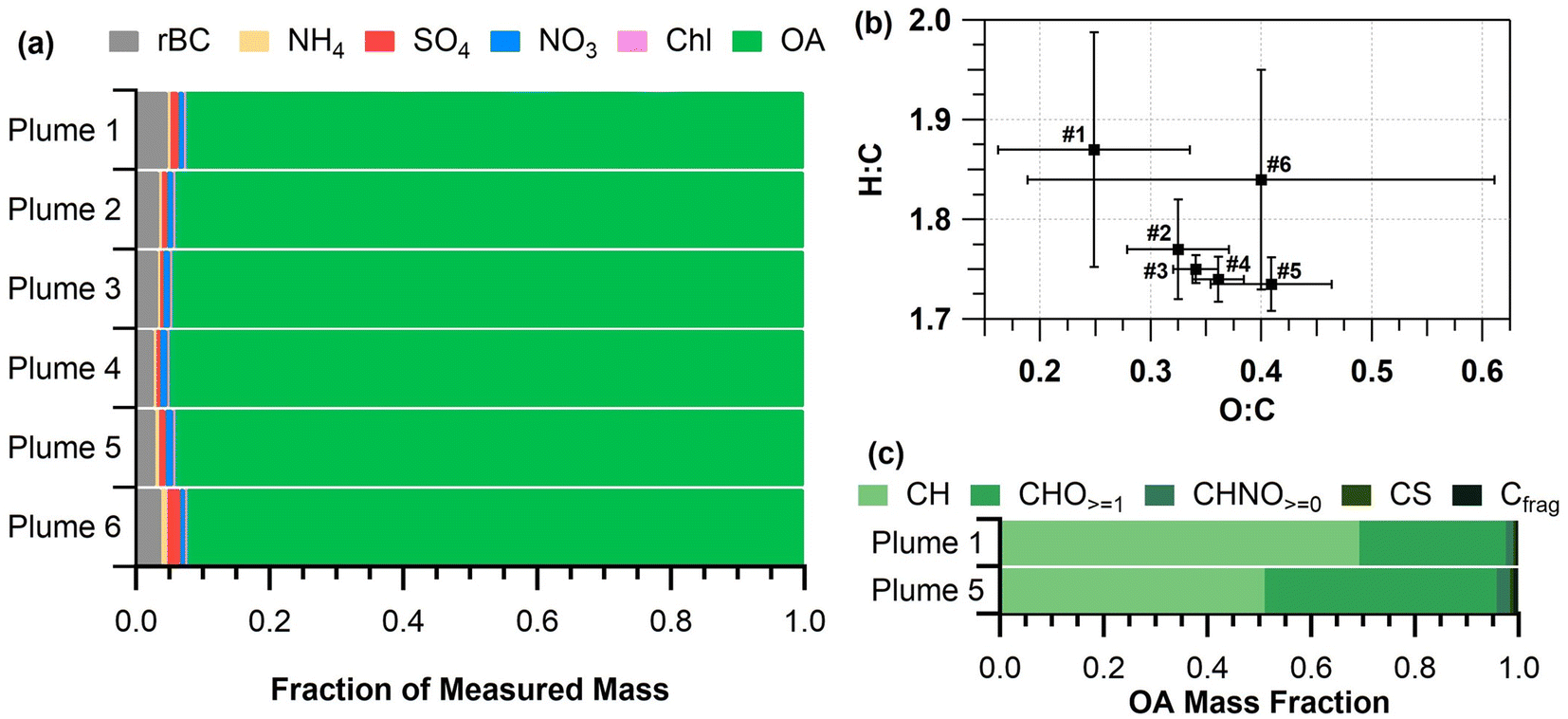 | ||
| Fig. 4 (a) Mass fractions of submicron refractory black carbon (rBC), as well as non-refractory organic aerosol (OA), ammonium (NH4), sulfate (SO4), nitrate (NO3), and chloride (Chl) for the six smoke plume periods during the prescribed burn, (b) Van Krevelen diagram depicting the average H:C and O:C ratios for each smoke plume, (c) fractional contributions to OA from hydrocarbon (CH), oxygenated (CHO≥1), nitrogen-containing (CHNO≥0), sulfur-containing (CS), and carbon fragments (Cfrag). | ||
Chemically-resolved submicron aerosol ERs for the six smoke plumes are listed in Table S2†. Laboratory-produced PM1 ERs are generally 0.5–14 times greater than PM1 ERs from prescribed burns.20,33,35,70 Similarly, the ERs for non-refractory OA and rBC were 10 and 3 times lower, respectively, than OA and rBC ERs from laboratory measurements of similar vegetation.20 However, the range of OA ERs for this study (50–120 mg g−1) was in line with previous mixed conifer forest prescribed burn studies from California (95–104 mg g−1).20 The range of rBC ERs for this study (2.2–4.5 mg g−1) was lower than observed in the previous mixed conifer forest prescribed burn (5.5–5.8 mg g−1),20 most likely due to this low severity burn producing lower mass concentrations of rBC. Differences in PM1, OA, and rBC ERs between laboratory and field measurements were attributed to fuel type, fuel moisture, and location.20,33 ERs for sulfate, nitrate, ammonium, and chloride for the six plume periods were all below 1.3 mg g−1 (Table S2†).
Both O:C and H:C ratios for all plume periods were similar (ranges of 0.25–0.41 and 1.74–1.87, respectively, Fig. 4b) with no significant MCE dependence, in contrast to the linear MCE relationships observed by Heringa et al.71 Fresh biomass burning emissions for various fuel types (e.g., pine, spruce, etc.) have been previously observed to have similar O:C (0.18–0.48) and H:C ratios (1.34–1.7).32,67,71 Plumes 1 and 6 had larger variations in O:C and H:C ratios (0.25 ± 0.09 and 1.8 ± 0.1 for plume 1 and 0.4 ± 0.2 and 1.8 ± 0.1 for plume 6, respectively) compared to plumes 2–5 (Fig. 4b). This is likely an indication of more intense burning during these two plumes compared to plumes 2–5. Sampling for plume 1 occurred while plots 1 and 2 were continually being ignited, resulting in an increase in non-oxygenated (higher H:C, lower O:C) organics from higher intensity burning. The average O:C ratio gradually increased through plumes 2–5 as the plots burned (Fig. 4b). In contrast, plume 6 sampled smoke while plot 3 flared-up part way through the measurement period, resulting in a large range of H:C and O:C ratios.
Plume 1 had a lower O:C ratio compared to plumes 2–6, as reflected in the higher OA mass fraction contribution of hydrocarbon (CH) ion signal shown in Fig. 4c. Fig. 4c shows the OA signal percentage by ion type (CH, CHO≥1, CHNO≥0, CS, and carbon fragments (Cfrag) measured by the SP-AMS) normalized to the OA mass fraction (Fig. 4a), for plumes 1 and 5. Plumes 2–6, which all included the emissions from plot 3, were similar in OA composition to plume 5 (Fig. 4c and S2†), and thus plume 5 is discussed here as representative of plumes 2–6. Plume 5 had a higher contribution from oxygenated (CHO≥1) and nitrogen-containing (CHNO≥0) organics compared to plume 1 (Fig. 4c). The OA for plume 1 was composed of 69% hydrocarbon ion signal, 28% oxygenated species, and 1.5% nitrogen-containing organic species, by mass, whereas plume 5 was composed of 51% hydrocarbon ion signals, 45% oxygenated species, and 2.7% nitrogen-containing organic species, by mass. Sulfur-containing (CS) and carbon mass fragments (Cfrag) made up fewer than 1% of the organic species, by mass, for both plumes. The average organic mass spectra for plumes 1–3, 5, and 6 are shown in Fig. S3.† A difference plot of the average OA mass spectra from plumes 1 and 5 is shown in Fig. S4,† illustrating an increased presence of oxygenated and nitrogen-containing compounds in plume 5. The oxygenated and nitrogen-containing organic species in plume 5 could be related to phenolic and nitrophenolic species.72 This difference in chemical composition may be related to vegetation, as hardwoods (red oak, big tooth aspen, and red maple burned and sampled in plume 5) generally have higher nitrogen content,73 and produce more phenolic compounds when combusted74 than softwoods. Note that red pine was burned and sampled in plume 1. Previous studies have suggested that phenolic compounds contribute to secondary organic aerosol production as smoke plumes age.75,76 Notably, nitrophenolic compounds from fires make up a significant portion of brown carbon.72
The average ATOFMS single-particle mass spectrum for all measured biomass burning particles during the prescribed burn is shown in Fig. 5. This is consistent with previously observed biomass burning particles measured by ATOFMS.57–59,77 The mass spectra are characterized by dominant potassium (m/z 39 (K+)), sulfate (m/z −97 (HSO4−)), and nitrate (m/z −46 (NO2−) and −62 (NO3−)) peaks.57,59 Potassium accounts for less than 5% of smoke particle mass,61 but is a common tracer of biomass burning particles6,77,78 that is easily ionized by laser desorption/ionization.79 These freshly emitted biomass burning particles also included elemental carbon (m/z 12 (C+), 24 (C2+), Cn+, …; −24 (C2−), −36 (C3−), Cn−, …), consistent with soot from flaming combustion.80 In addition, organic aerosol markers59,81,82 characteristic of smoldering combustion were present, including organic carbon (m/z 27 (C2H3+), 37 (C3H+), and 43 (C2H3O+/C3H7+)) and organic nitrogen (m/z 27 (NCH+), 43 (CHNO+), −26 (CN−), and −42 (CNO−)) markers. The average negative ion mass spectrum contains fresh biomass burning markers including levoglucosan fragments (m/z −73 (C3H5O2−), −71 (C3H3O2−), −59 (C2H3O2−), −45 (CHO2−)),58,59 and potassium salt clusters (m/z −163 (K(NO3)2−), −137 (KSO4−), and −109 (KCl2)).57 Fewer than half of the individual biomass burning particles, by number, (Fig. S6†) contained polycyclic aromatic hydrocarbons (PAHs)83,84 and aromatics measured as ion peaks at m/z 51, 63, 77, 87, 91, 102, 115, 128, 139, 152, 165, 178, 189, and 202.80,85–87
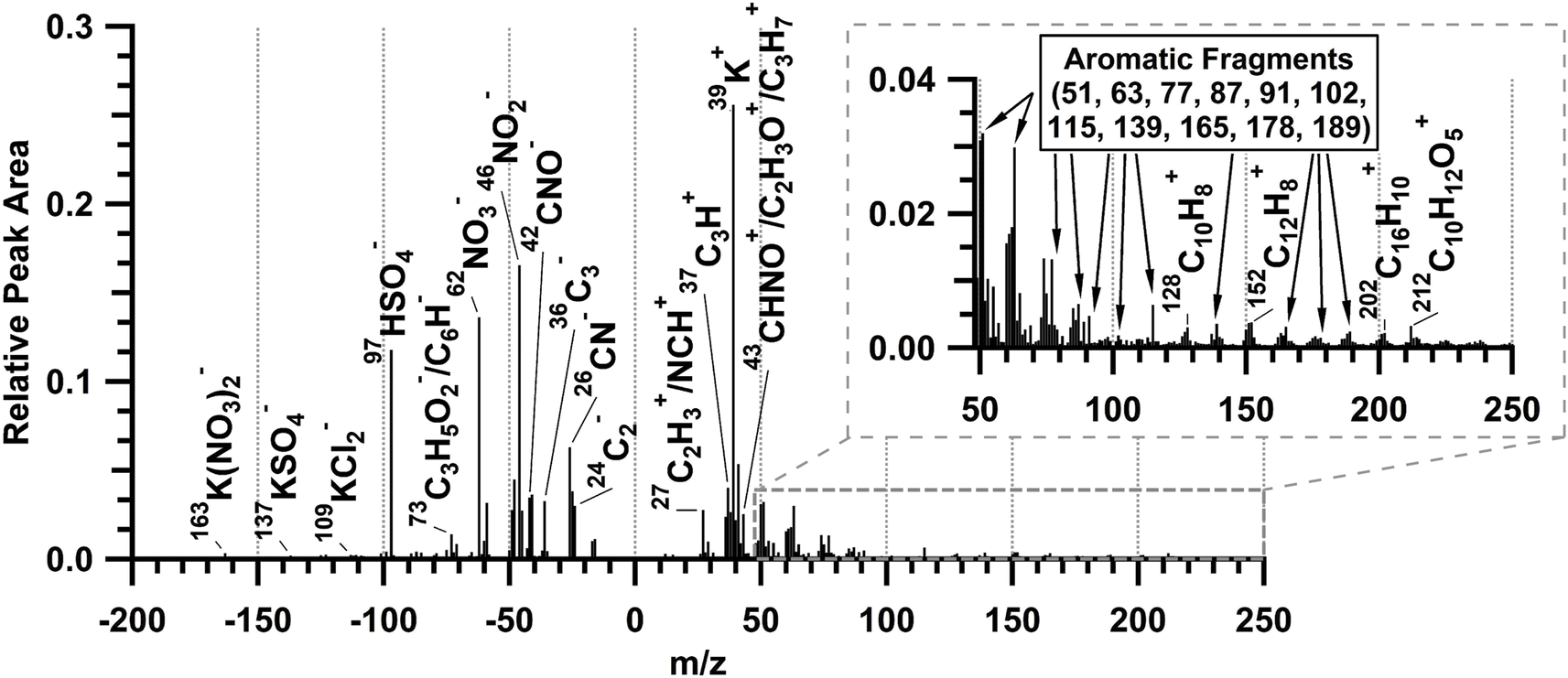 | ||
| Fig. 5 ATOFMS average positive and negative ion mass spectrum of 5500 freshly emitted biomass burning particles sampled during plumes 1–6. | ||
The average ATOFMS individual biomass burning particle mass spectra for each plume period are shown in Fig. S7,† with similar mass spectra for plumes 2–6, and differing composition for plume 1, similar to that observed for the SP-AMS organic aerosol mass spectra. Therefore, the average biomass burning single-particle mass spectrum from plume 5 was compared to the average biomass burning particle mass spectrum from plume 1 and shown in Fig. 6. Plume 1 corresponded with the burn of plots 1 and 2, which contained primarily red pine, a softwood, whereas plume 5 corresponded with the burn of plot 3, containing a mixture of northern hardwood forest (i.e., red oak, bigtooth aspen, and red maple, Table 1) with minor contributions from softwood species (i.e., white pine, Table 1). Molecular ion peaks corresponding to the PAHs80,85–89 naphthalene (m/z 128 (C10H8+)), acenaphtylene (m/z 152 (C12H8+)), and pyrene/fluoranthene (m/z 202 (C16H10+)), as well as aromatic ion fragments (m/z 51, 63, 77, 87, 102, 115, 139, 165, 189), were more prominent in plume 5, compared to plume 1. The abundance of aromatic ion fragments observed by ATOFMS is consistent with the phenolic and nitrophenolic species observed in the SP-AMS spectrum for plume 5. As observed in previous single-particle mass spectrometry studies, naphthalene, acenaphtylene, and pyrene/fluoranthene are common PAHs released from the combustion of furfural, a common component of hardwoods.88 High-density hardwoods are expected to burn longer and slower, leaving behind more unreacted wood mass compared to pine, a low-density softwood, when burned at the same pyrolysis temperature.22,90,91 This is consistent with the increased presence of PAHs in the individual biomass burning particles measured during plume 5 (Table 1). In addition, the higher peak intensities for nitrate (m/z −46 (NO2−) and −62 (NO3−)) in the plume 5 biomass burning particles compared to plume 1 is consistent with the nitrate mass fractions measured by the SP-AMS (0.8 and 1.1% for plumes 1 and 5, respectively, Fig. 4a).
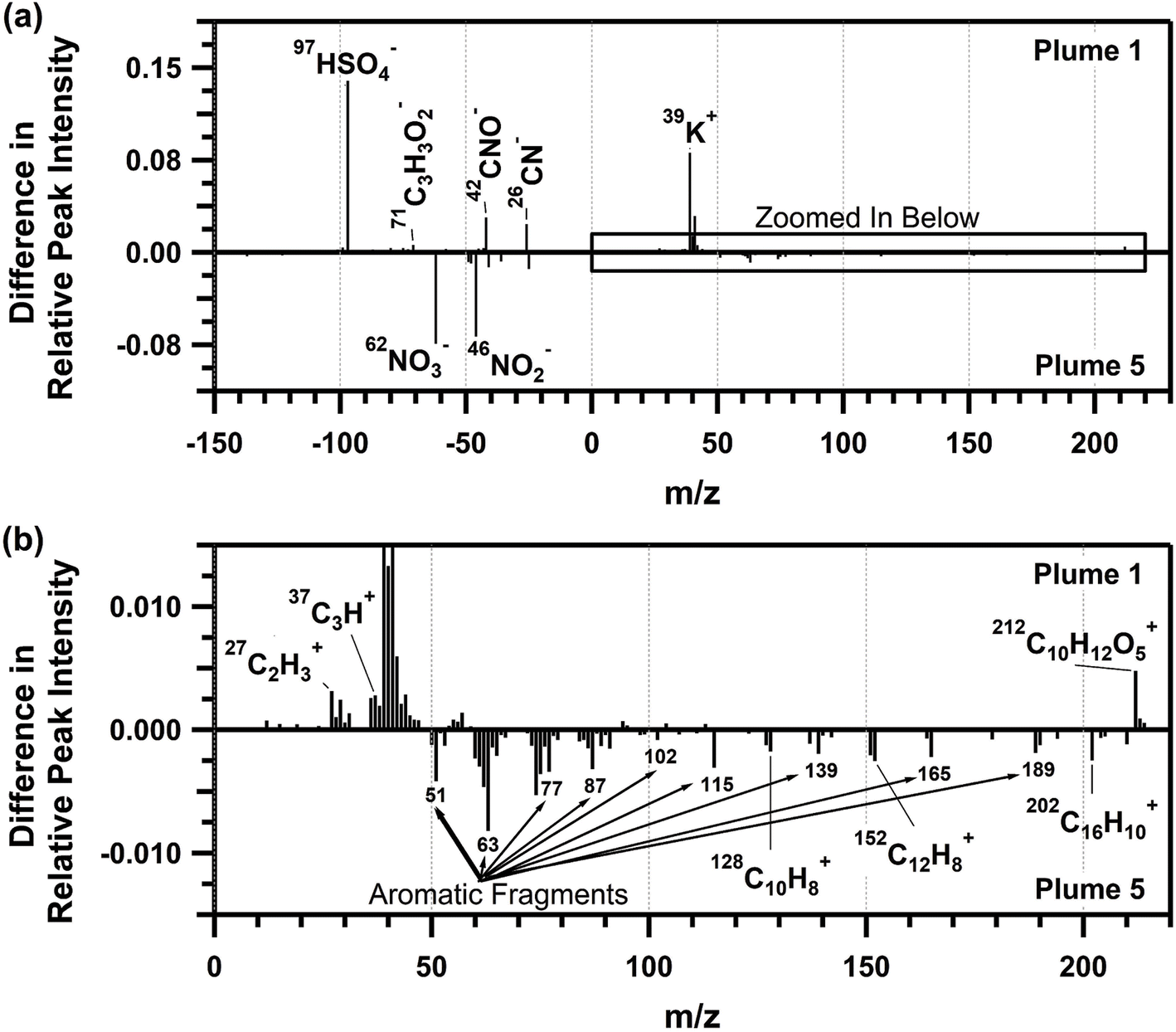 | ||
| Fig. 6 (a) ATOFMS positive and negative ion mass average spectrum and (b) expanded positive ion mass spectrum for biomass burning particles during plume 5 subtracted from biomass burning particles during plume 1. Positive relative peak intensities correspond to greater abundance during plume 1; negative relative peak intensities correspond to greater abundance during plume 5. Only ion peaks above 95% confidence limits following subtraction are shown. | ||
As shown in Fig. 6, the plume 1 biomass burning particles showed higher signals for potassium (m/z 39 (K+)), organic carbon (m/z 27 (C2H3+), 29 (C2H5+), and 37 (C3H+)), sulfate (m/z −97 (HSO4−)), organic nitrogen (m/z −26 (CN−), −42 (CNO−)), levoglucosan (m/z −73 (C3H5O2−), −71 (C3H3O2−), −59 (C2H3O2−), −45 (CHO2−)), and an aromatic peak at m/z 212 (C10H12O5+)89 that results from the combustion of eugenol.83,88 The higher signals for the organic carbon fragments within plume 1, compared to plume 5, also agrees with the greater CH mass fraction observed by the SP-AMS (Fig. 4c). The larger sulfate signal for plume 1 compared to plume 5 aligns with the sulfate mass fractions measured by the SP-AMS (1.3 and 1.0% for plumes 1 and 5, respectively, Fig. 4a). With respect to m/z 212, eugenol is released during the early stages of lignin combustion and has a resulting mass spectrum similar to pine wood soot.83,88 The higher contribution of this ion in plume 1 compared to plume 5 reflects the increased contribution of pine wood combustion during plume 1 (Table 1) and further highlights the influence fuel type has on the chemical composition of the emitted biomass burning particles.
4. Conclusions
Trace gas and aerosol particle emissions from a prescribed burn in Pellston, Michigan were measured within ∼3–5 min following emission. Three plots, two containing primarily red pine and the third containing a mixture of northern hardwood (red oak, bigtooth aspen, and red maple) and white pine were burned sequentially. The measured emission factors for CH4, C2H6, C2H2, and HCN were inversely related to MCE, as previously determined in laboratory pine studies. Lower CH4 and C2H2 emission factors, compared to previous field measurements, may be explained by the low pyrolysis temperature of this burn.18 CO2, CO, C2H6, HCN, NO, and N2O EFs did not significantly vary between plots, suggesting minimal fuel type influence, and followed trends explained by MCE.The majority (93.0 ± 0.7%, by number) of the individual particles analysed by ATOFMS corresponded to biomass burning and were determined by SP-AMS to be mainly OA by mass. Throughout the prescribed burn, over 92% of the submicron aerosol mass was OA, consistent with laboratory and field measurements of pine wood combustion. OA/rBC ratios (25–37) were consistent with a lower severity burn that produced less rBC, by mass, compared to previous laboratory studies.67 Biomass burning particles measured from the combustion of primary red pine displayed markers corresponding to eugenol soot. Particles measured from the combustion of the northern hardwoods (red oak, bigtooth aspen, and red maple) and white pine displayed PAH markers consistent with the combustion of furfural, and showed greater mass fractions of oxygenated and nitrogen-containing organic compounds. The prominence of PAH markers in the latter plume periods may be associated with the incomplete combustion of higher-density woods.22,90,91 Previous field measurements in the western and southeastern United States had higher emission ratios of particulate OA and rBC than what was emitted in this Northern Michigan prescribed burn. However, the first two plots burned in this study occurred at low to moderate severity, which may have contributed to the lower rBC emission ratios. Additional biomass burning trace gas and aerosol particle studies are required to better understand the impacts of fires across different seasons, severity, and in this area of the United States.
Data availability
Data measured by the SP-AMS, TILDAS, TSI, 2B Tech, LI-COR, and Thermo Scientific instruments are archived through the CSL FIREX-AQ archive (https://csl.noaa.gov/groups/csl7/measurements/2019firex-aq/AML/). ATOFMS data are archived through the University of Michigan Library's Deep Blue Repository (https://doi.org/10.7302/1dbr-2f25).Author contributions
CD, EF, NE, and NM conducted the measurements. JL, CD, EF, and JT conducted the formal analysis. KP and SH conceptualized and supervised the project. JL wrote the manuscript with contributions from KP, CD, and EF.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the University of Michigan Biological Station and PlantWise, LLC for leading the prescribed burn and for research logistical support. Photos used to create the Table of Contents Entry are credited to Roger Hart and Michigan Photography. The authors acknowledge the University of Michigan Department of Chemistry for funding and the Pratt Lab and Aerodyne Research Inc. for their support. The Aerodyne Mobile Laboratory was supported by National Oceanic and Atmospheric Administration (Grant number: NA16OAR4310104). We thank Rachel Hems for helpful discussions.References
- D. A. Jaffe, S. M. O'Neill, N. K. Larkin, A. L. Holder, D. L. Peterson, J. E. Halofsky and A. G. Rappold, Wildfire and prescribed burning impacts on air quality in the United States, J. Air Waste Manage. Assoc., 2020, 70, 583–615 CrossRef CAS PubMed.
- P. E. Dennison, S. C. Brewer, J. D. Arnold and M. A. Moritz, Large wildfire trends in the western United States, 1984–2011, Geophys. Res. Lett., 2014, 41, 2928–2933 CrossRef.
- M. Flannigan, A. S. Cantin, W. J. de Groot, M. Wotton, A. Newbery and L. M. Gowman, Global wildland fire season severity in the 21st century, For. Ecol. Manage., 2013, 294, 54–61 CrossRef.
- A. L. Westerling, Increasing western US forest wildfire activity: sensitivity to changes in the timing of spring, Philos. Trans. R. Soc., B, 2016, 371, 20150178 CrossRef PubMed.
- A. P. Williams, J. T. Abatzoglou, A. Gershunov, J. Guzman-Morales, D. A. Bishop, J. K. Balch and D. P. Lettenmaier, Observed Impacts of Anthropogenic Climate Change on Wildfire in California, Earth's Future, 2019, 7, 892–910 CrossRef.
- G. R. McMeeking, S. M. Kreidenweis, S. Baker, C. M. Carrico, J. C. Chow, J. L. Collett Jr, W. M. Hao, A. S. Holden, T. W. Kirchstetter, W. C. Malm, H. Moosmüller, A. P. Sullivan and C. E. Wold, Emissions of trace gases and aerosols during the open combustion of biomass in the laboratory, J. Geophys. Res.: Atmos., 2009, 114, D19210 CrossRef.
- S. P. Urbanski, M. C. Reeves, R. E. Corley, R. P. Silverstein and W. M. Hao, Contiguous United States wildland fire emission estimates during 2003–2015, Earth Syst. Sci. Data, 2018, 10, 2241–2274 CrossRef.
- S. L. Altshuler, Q. Zhang, M. T. Kleinman, F. Garcia-Menendez, C. T. Moore, M. L. Hough, E. D. Stevenson, J. C. Chow, D. A. Jaffe and J. G. Watson, Wildfire and prescribed burning impacts on air quality in the United States, J. Air Waste Manage. Assoc., 2020, 70, 961–970 CrossRef CAS PubMed.
- J. R. Laing and D. A. Jaffe, Wildfires are causing extreme PM concentrations in the western U.S, 2019, https://pubs.awma.org/flip/EM-June-2019/jaffe.pdf Search PubMed.
- K. O'Dell, K. Bilsback, B. Ford, S. E. Martenies, S. Magzamen, E. V. Fischer and J. R. Pierce, Estimated Mortality and Morbidity Attributable to Smoke Plumes in the United States: Not Just a Western US Problem, GeoHealth, 2021, 5, e2021GH000457 Search PubMed.
- S. O'Neill, S. Urbanski, S. Goodrick and S. Larkin, Smoke plumes: Emissions and effects, Fire Manag. Today, 2017, 75, 10–15 Search PubMed.
- W. T. Sommers, R. A. Loehman and C. C. Hardy, Wildland fire emissions, carbon, and climate: Science overview and knowledge needs, For. Ecol. Manage., 2014, 317, 1–8 CrossRef.
- C. C. Hardy, R. D. Ottmar, J. L. Peterson, J. E. Core and P. Seamon, Smoke management guide for prescribed and wildland fire: 2001 edition, PMS 420-2. NFES 1279, National Wildfire Coodination Group, Boise, ID, p. 226, 2001 Search PubMed.
- S. P. Urbanski, Combustion efficiency and emission factors for wildfire-season fires in mixed conifer forests of the northern Rocky Mountains, US, Atmos. Chem. Phys., 2013, 13, 7241–7262 CrossRef.
- S. K. Akagi, I. R. Burling, A. Mendoza, T. J. Johnson, M. Cameron, D. W. T. Griffith, C. Paton-Walsh, D. R. Weise, J. Reardon and R. J. Yokelson, Field measurements of trace gases emitted by prescribed fires in southeastern US pine forests using an open-path FTIR system, Atmos. Chem. Phys., 2014, 14, 199–215 CrossRef.
- NICC, National Interagency Coordination Center, Wildland Fire Summary and Statistics Annual Report 2019, 2019 Search PubMed.
- R. J. Yokelson, I. R. Burling, J. B. Gilman, C. Warneke, C. E. Stockwell, J. de Gouw, S. K. Akagi, S. P. Urbanski, P. Veres, J. M. Roberts, W. C. Kuster, J. Reardon, D. W. T. Griffith, T. J. Johnson, S. Hosseini, J. W. Miller, D. R. Cocker III, H. Jung and D. R. Weise, Coupling field and laboratory measurements to estimate the emission factors of identified and unidentified trace gases for prescribed fires, Atmos. Chem. Phys., 2013, 13, 89–116 CrossRef.
- N. K. Scharko, A. M. Oeck, T. L. Myers, R. G. Tonkyn, C. A. Banach, S. P. Baker, E. N. Lincoln, J. Chong, B. M. Corcoran, G. M. Burke, R. D. Ottmar, J. C. Restaino, D. R. Weise and T. J. Johnson, Gas-phase pyrolysis products emitted by prescribed fires in pine forests with a shrub understory in the southeastern United States, Atmos. Chem. Phys., 2019, 19, 9681–9698 CrossRef CAS.
- I. R. Burling, R. J. Yokelson, D. W. T. Griffith, T. J. Johnson, P. Veres, J. M. Roberts, C. Warneke, S. P. Urbanski, J. Reardon, D. R. Weise, W. M. Hao and J. de Gouw, Laboratory measurements of trace gas emissions from biomass burning of fuel types from the southeastern and southwestern United States, Atmos. Chem. Phys., 2010, 10, 11115–11130 CrossRef CAS.
- A. A. May, G. R. McMeeking, T. Lee, J. W. Taylor, J. S. Craven, I. Burling, A. P. Sullivan, S. Akagi, J. L. Collett Jr, M. Flynn, H. Coe, S. P. Urbanski, J. H. Seinfeld, R. J. Yokelson and S. M. Kreidenweis, Aerosol emissions from prescribed fires in the United States: A synthesis of laboratory and aircraft measurements, J. Geophys. Res.: Atmos., 2014, 119, 11826–11849 CrossRef CAS.
- S. Urbanski, Wildland fire emissions, carbon, and climate: Emission factors, For. Ecol. Manage., 2014, 317, 51–60 CrossRef.
- M. Fawaz, A. Avery, T. B. Onasch, L. R. Williams and T. C. Bond, Technical note: Pyrolysis principles explain time-resolved organic aerosol release from biomass burning, Atmos. Chem. Phys., 2021, 21, 15605–15618 CrossRef CAS.
- I. R. Burling, R. J. Yokelson, S. K. Akagi, S. P. Urbanski, C. E. Wold, D. W. T. Griffith, T. J. Johnson, J. Reardon and D. R. Weise, Airborne and ground-based measurements of the trace gases and particles emitted by prescribed fires in the United States, Atmos. Chem. Phys., 2011, 11, 12197–12216 CrossRef CAS.
- C. N. Jen, L. E. Hatch, V. Selimovic, R. J. Yokelson, R. Weber, A. E. Fernandez, N. M. Kreisberg, K. C. Barsanti and A. H. Goldstein, Speciated and total emission factors of particulate organics from burning western US wildland fuels and their dependence on combustion efficiency, Atmos. Chem. Phys., 2019, 19, 1013–1026 CrossRef CAS.
- S. K. Akagi, R. J. Yokelson, I. R. Burling, S. Meinardi, I. Simpson, D. R. Blake, G. R. McMeeking, A. Sullivan, T. Lee, S. Kreidenweis, S. Urbanski, J. Reardon, D. W. T. Griffith, T. J. Johnson and D. R. Weise, Measurements of reactive trace gases and variable O3 formation rates in some South Carolina biomass burning plumes, Atmos. Chem. Phys., 2013, 13, 1141–1165 CrossRef.
- V. Selimovic, R. J. Yokelson, C. Warneke, J. M. Roberts, J. de Gouw, J. Reardon and D. W. T. Griffith, Aerosol optical properties and trace gas emissions by PAX and OP-FTIR for laboratory-simulated western US wildfires during FIREX, Atmos. Chem. Phys., 2018, 18, 2929–2948 CrossRef CAS.
- R. J. Yokelson, J. G. Goode, D. E. Ward, R. A. Susott, R. E. Babbitt, D. D. Wade, I. Bertschi, D. W. T. Griffith and W. M. Hao, Emissions of formaldehyde, acetic acid, methanol, and other trace gases from biomass fires in North Carolina measured by airborne Fourier transform infrared spectroscopy, J. Geophys. Res.: Atmos., 1999, 104, 30109–30125 CrossRef CAS.
- D. E. Ward and L. F. Radke, in Fire in the Environment: The Ecological, Atmospheric, and Climatic Importance of Vegetation Fires. Dahlem Workshop Reports: Environmental Sciences Research Report 13, ed. P. J. Crutzen and J. G. Goldammer, John Wiley & Sons., Chichester, England, 1993, pp. 53–76 Search PubMed.
- M. O. Andreae, Emission of trace gases and aerosols from biomass burning – an updated assessment, Atmos. Chem. Phys., 2019, 19, 8523–8546 CrossRef CAS.
- S. K. Akagi, R. J. Yokelson, C. Wiedinmyer, M. J. Alvarado, J. S. Reid, T. Karl, J. D. Crounse and P. O. Wennberg, Emission factors for open and domestic biomass burning for use in atmospheric models, Atmos. Chem. Phys., 2011, 11, 4039–4072 CrossRef CAS.
- S. J. Prichard, S. M. O'Neill, P. Eagle, A. G. Andreu, B. Drye, J. Dubowy, S. Urbanski and T. M. Strand, Wildland fire emission factors in North America: synthesis of existing data, measurement needs and management applications, Int. J. Wildland Fire, 2020, 29, 132–147 CrossRef.
- Q. Chen, C. L. Heald, J. L. Jimenez, M. R. Canagaratna, Q. Zhang, L.-Y. He, X.-F. Huang, P. Campuzano-Jost, B. B. Palm, L. Poulain, M. Kuwata, S. T. Martin, J. P. D. Abbatt, A. K. Y. Lee and J. Liggio, Elemental composition of organic aerosol: The gap between ambient and laboratory measurements, Geophys. Res. Lett., 2015, 42, 4182–4189 CrossRef CAS.
- A. L. Holder, G. S. W. Hagler, J. Aurell, M. D. Hays and B. K. Gullett, Particulate matter and black carbon optical properties and emission factors from prescribed fires in the southeastern United States, J. Geophys. Res.: Atmos., 2016, 121, 3465–3483 CrossRef CAS.
- R. W. Long, A. Whitehill, A. Habel, S. Urbanski, H. Halliday, M. Colón, S. Kaushik and M. S. Landis, Comparison of ozone measurement methods in biomass burning smoke: an evaluation under field and laboratory conditions, Atmos. Meas. Tech., 2021, 14, 1783–1800 CrossRef CAS PubMed.
- J. Aurell, B. K. Gullett and D. Tabor, Emissions from southeastern U.S. Grasslands and pine savannas: Comparison of aerial and ground field measurements with laboratory burns, Atmos. Environ., 2015, 111, 170–178 CrossRef CAS.
- X. Liu, L. G. Huey, R. J. Yokelson, V. Selimovic, I. J. Simpson, M. Müller, J. L. Jimenez, P. Campuzano-Jost, A. J. Beyersdorf, D. R. Blake, Z. Butterfield, Y. Choi, J. D. Crounse, D. A. Day, G. S. Diskin, M. K. Dubey, E. Fortner, T. F. Hanisco, W. Hu, L. E. King, L. Kleinman, S. Meinardi, T. Mikoviny, T. B. Onasch, B. B. Palm, J. Peischl, I. B. Pollack, T. B. Ryerson, G. W. Sachse, A. J. Sedlacek, J. E. Shilling, S. Springston, J. M. St. Clair, D. J. Tanner, A. P. Teng, P. O. Wennberg, A. Wisthaler and G. M. Wolfe, Airborne measurements of western U.S. wildfire emissions: Comparison with prescribed burning and air quality implications, J. Geophys. Res.: Atmos., 2017, 122, 6108–6129 CrossRef CAS.
- F. Fachinger, F. Drewnick and S. Borrmann, How villages contribute to their local air quality – The influence of traffic- and biomass combustion-related emissions assessed by mobile mappings of PM and its components, Atmos. Environ., 2021, 263, 118648 CrossRef CAS.
- S. Prichard, N. S. Larkin, R. Ottmar, N. H. F. French, K. Baker, T. Brown, C. Clements, M. Dickinson, A. Hudak, A. Kochanski, R. Linn, Y. Liu, B. Potter, W. Mell, D. Tanzer, S. Urbanski and A. Watts, The Fire and Smoke Model Evaluation Experiment—A Plan for Integrated, Large Fire–Atmosphere Field Campaigns, Atmosphere, 2019, 10(2), 66 CrossRef PubMed.
- M. A. Carroll, S. B. Bertman and P. B. Shepson, Overview of the Program for Research on Oxidants: PHotochemistry, Emissions, and Transport (PROPHET) summer 1998 measurements intensive, J. Geophys. Res.: Atmos., 2001, 106, 24275–24288 CrossRef CAS.
- K. M. Bergen and I. Dronova, Observing succession on aspen-dominated landscapes using a remote sensing-ecosystem approach, Landsc. Ecol., 2007, 22, 1395–1410 CrossRef.
- C. M. Gough, B. S. Hardiman, L. E. Nave, G. Bohrer, K. D. Maurer, C. S. Vogel, K. J. Nadelhoffer and P. S. Curtis, Sustained carbon uptake and storage following moderate disturbance in a Great Lakes forest, Ecol Appl, 2013, 23, 1202–1215 CrossRef PubMed.
- C. E. Kolb, S. C. Herndon, J. B. McManus, J. H. Shorter, M. S. Zahniser, D. D. Nelson, J. T. Jayne, M. R. Canagaratna and D. R. Worsnop, Mobile Laboratory with Rapid Response Instruments for Real-Time Measurements of Urban and Regional Trace Gas and Particulate Distributions and Emission Source Characteristics, Environ. Sci. Technol., 2004, 38, 5694–5703 CrossRef CAS PubMed.
- S. C. Herndon, J. T. Jayne, M. S. Zahniser, D. R. Worsnop, B. Knighton, E. Alwine, B. K. Lamb, M. Zavala, D. D. Nelson, J. B. McManus, J. H. Shorter, M. R. Canagaratna, T. B. Onasch and C. E. Kolb, Characterization of urban pollutant emission fluxes and ambient concentration distributions using a mobile laboratory with rapid response instrumentation, Faraday Discuss., 2005, 130, 327–339 RSC.
- S. B. Wales, M. R. Kreider, J. Atkins, C. M. Hulshof, R. T. Fahey, L. E. Nave, K. J. Nadelhoffer and C. M. Gough, Stand age, disturbance history and the temporal stability of forest production, For. Ecol. Manage., 2020, 460, 117865 CrossRef.
- E. J. Dunlea, S. C. Herndon, D. D. Nelson, R. M. Volkamer, F. San Martini, P. M. Sheehy, M. S. Zahniser, J. H. Shorter, J. C. Wormhoudt, B. K. Lamb, E. J. Allwine, J. S. Gaffney, N. A. Marley, M. Grutter, C. Marquez, S. Blanco, B. Cardenas, A. Retama, C. R. Ramos Villegas, C. E. Kolb, L. T. Molina and M. J. Molina, Evaluation of nitrogen dioxide chemiluminescence monitors in a polluted urban environment, Atmos. Chem. Phys., 2007, 7, 2691–2704 CrossRef CAS.
- T. I. Yacovitch, S. C. Herndon, J. R. Roscioli, C. Floerchinger, R. M. McGovern, M. Agnese, G. Pétron, J. Kofler, C. Sweeney, A. Karion, S. A. Conley, E. A. Kort, L. Nähle, M. Fischer, L. Hildebrandt, J. Koeth, J. B. McManus, D. D. Nelson, M. S. Zahniser and C. E. Kolb, Demonstration of an Ethane Spectrometer for Methane Source Identification, Environ. Sci. Technol., 2014, 48, 8028–8034 CrossRef CAS PubMed.
- J. B. McManus, M. S. Zahniser, D. D. Nelson, J. H. Shorter, S. C. Herndon, D. Jervis, M. Agnese, R. McGovern, T. I. Yacovitch and J. R. Roscioli, Recent progress in laser-based trace gas instruments: performance and noise analysis, Appl. Phys. B: Lasers Opt., 2015, 119, 203–218 CrossRef CAS.
- A. K. Y. Lee, M. D. Willis, R. M. Healy, T. B. Onasch and J. P. D. Abbatt, Mixing state of carbonaceous aerosol in an urban environment: single particle characterization using the soot particle aerosol mass spectrometer (SP-AMS), Atmos. Chem. Phys., 2015, 15, 1823–1841 CrossRef.
- T. B. Onasch, A. Trimborn, E. C. Fortner, J. T. Jayne, G. L. Kok, L. R. Williams, P. Davidovits and D. R. Worsnop, Soot Particle Aerosol Mass Spectrometer: Development, Validation, and Initial Application, Aerosol Sci. Technol., 2012, 46, 804–817 CrossRef CAS.
- M. D. Willis, R. M. Healy, N. Riemer, M. West, J. M. Wang, C. H. Jeong, J. C. Wenger, G. J. Evans, J. P. D. Abbatt and A. K. Y. Lee, Quantification of black carbon mixing state from traffic: implications for aerosol optical properties, Atmos. Chem. Phys., 2016, 16, 4693–4706 CrossRef CAS.
- M. O. Andreae and P. Merlet, Emission of trace gases and aerosols from biomass burning, Global Biogeochem. Cycles, 2001, 15, 955–966 CrossRef CAS.
- K. A. Pratt, J. E. Mayer, J. C. Holecek, R. C. Moffet, R. O. Sanchez, T. P. Rebotier, H. Furutani, M. Gonin, K. Fuhrer, Y. Su, S. Guazzotti and K. A. Prather, Development and Characterization of an Aircraft Aerosol Time-of-Flight Mass Spectrometer, Anal. Chem., 2009, 81, 1792–1800 CrossRef CAS PubMed.
- M. J. Gunsch, R. M. Kirpes, K. R. Kolesar, T. E. Barrett, S. China, R. J. Sheesley, A. Laskin, A. Wiedensohler, T. Tuch and K. A. Pratt, Contributions of transported Prudhoe Bay oil field emissions to the aerosol population in Utqiaġvik, Alaska, Atmos. Chem. Phys., 2017, 17, 10879–10892 CrossRef CAS.
- C. M. Sultana, G. C. Cornwell, P. Rodriguez and K. A. Prather, FATES: a flexible analysis toolkit for the exploration of single-particle mass spectrometer data, Atmos. Meas. Tech., 2017, 10, 1323–1334 CrossRef.
- X.-H. Song, P. K. Hopke, D. P. Fergenson and K. A. Prather, Classification of Single Particles Analyzed by ATOFMS Using an Artificial Neural Network, ART-2A, Anal. Chem., 1999, 71, 860–865 CrossRef CAS.
- K. A. Pratt and K. A. Prather, Real-Time, Single-Particle Volatility, Size, and Chemical Composition Measurements of Aged Urban Aerosols, Environ. Sci. Technol., 2009, 43, 8276–8282 CrossRef CAS PubMed.
- K. A. Pratt, S. M. Murphy, R. Subramanian, P. J. DeMott, G. L. Kok, T. Campos, D. C. Rogers, A. J. Prenni, A. J. Heymsfield, J. H. Seinfeld and K. A. Prather, Flight-based chemical characterization of biomass burning aerosols within two prescribed burn smoke plumes, Atmos. Chem. Phys., 2011, 11, 12549–12565 CrossRef CAS.
- M. D. Zauscher, Y. Wang, M. J. K. Moore, C. J. Gaston and K. A. Prather, Air Quality Impact and Physicochemical Aging of Biomass Burning Aerosols during the 2007 San Diego Wildfires, Environ. Sci. Technol., 2013, 47, 7633–7643 CrossRef CAS PubMed.
- P. J. Silva, D.-Y. Liu, C. A. Noble and K. A. Prather, Size and Chemical Characterization of Individual Particles Resulting from Biomass Burning of Local Southern California Species, Environ. Sci. Technol., 1999, 33, 3068–3076 CrossRef CAS.
- M. J. Gunsch, N. W. May, M. Wen, C. L. H. Bottenus, D. J. Gardner, T. M. VanReken, S. B. Bertman, P. K. Hopke, A. P. Ault and K. A. Pratt, Ubiquitous influence of wildfire emissions and secondary organic aerosol on summertime atmospheric aerosol in the forested Great Lakes region, Atmos. Chem. Phys., 2018, 18, 3701–3715 CrossRef CAS.
- J. S. Reid, R. Koppmann, T. F. Eck and D. P. Eleuterio, A review of biomass burning emissions part II: intensive physical properties of biomass burning particles, Atmos. Chem. Phys., 2005, 5, 799–825 CrossRef CAS.
- W. M. Hao and D. E. Ward, Methane production from global biomass burning, J. Geophys. Res.: Atmos., 1993, 98, 20657–20661 CrossRef.
- C. E. Stockwell, R. J. Yokelson, S. M. Kreidenweis, A. L. Robinson, P. J. DeMott, R. C. Sullivan, J. Reardon, K. C. Ryan, D. W. T. Griffith and L. Stevens, Trace gas emissions from combustion of peat, crop residue, domestic biofuels, grasses, and other fuels: configuration and Fourier transform infrared (FTIR) component of the fourth Fire Lab at Missoula Experiment (FLAME-4), Atmos. Chem. Phys., 2014, 14, 9727–9754 CrossRef.
- M. Becidan, Ø. Skreiberg and J. E. Hustad, NOx and N2O Precursors (NH3 and HCN) in Pyrolysis of Biomass Residues, Energy Fuels, 2007, 21, 1173–1180 CrossRef CAS.
- P. J. Crutzen, L. E. Heidt, J. P. Krasnec, W. H. Pollock and W. Seiler, Biomass burning as a source of atmospheric gases CO, H2, N2O, NO, CH3Cl and COS, Nature, 1979, 282, 253–256 CrossRef CAS.
- J. S. King, C. P. Giardina, K. S. Pregitzer and A. L. Friend, Biomass partitioning in red pine (Pinus resinosa) along a chronosequence in the Upper Peninsula of Michigan, Can. J. For. Res., 2007, 37, 93–102 CrossRef.
- E. Fortner, T. Onasch, M. Canagaratna, L. R. Williams, T. Lee, J. Jayne and D. Worsnop, Examining the chemical composition of black carbon particles from biomass burning with SP-AMS, J. Aerosol Sci., 2018, 120, 12–21 CrossRef CAS.
- L. A. Garofalo, M. A. Pothier, E. J. T. Levin, T. Campos, S. M. Kreidenweis and D. K. Farmer, Emission and Evolution of Submicron Organic Aerosol in Smoke from Wildfires in the Western United States, ACS Earth Space Chem., 2019, 3, 1237–1247 CrossRef CAS.
- S. Zhou, S. Collier, D. A. Jaffe, N. L. Briggs, J. Hee, A. J. Sedlacek Iii, L. Kleinman, T. B. Onasch and Q. Zhang, Regional influence of wildfires on aerosol chemistry in the western US and insights into atmospheric aging of biomass burning organic aerosol, Atmos. Chem. Phys., 2017, 17, 2477–2493 CrossRef CAS.
- J. Aurell and B. K. Gullett, Emission Factors from Aerial and Ground Measurements of Field and Laboratory Forest Burns in the Southeastern U.S.: PM2.5, Black and Brown Carbon, VOC, and PCDD/PCDF, Environ. Sci. Technol., 2013, 47, 8443–8452 CAS.
- M. F. Heringa, P. F. DeCarlo, R. Chirico, A. Lauber, A. Doberer, J. Good, T. Nussbaumer, A. Keller, H. Burtscher, A. Richard, B. Miljevic, A. S. H. Prevot and U. Baltensperger, Time-Resolved Characterization of Primary Emissions from Residential Wood Combustion Appliances, Environ. Sci. Technol., 2012, 46, 11418–11425 CrossRef CAS PubMed.
- B. B. Palm, Q. Peng, C. D. Fredrickson, B. H. Lee, L. A. Garofalo, M. A. Pothier, S. M. Kreidenweis, D. K. Farmer, R. P. Pokhrel, Y. Shen, S. M. Murphy, W. Permar, L. Hu, T. L. Campos, S. R. Hall, K. Ullmann, X. Zhang, F. Flocke, E. V. Fischer and J. A. Thornton, Quantification of organic aerosol and brown carbon evolution in fresh wildfire plumes, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 29469 CrossRef CAS PubMed.
- A. R. Martin, S. Gezahegn and S. C. Thomas, Variation in carbon and nitrogen concentration among major woody tissue types in temperate trees, Can. J. For. Res., 2015, 45, 744–757 CrossRef CAS.
- J. D. McDonald, B. Zielinska, E. M. Fujita, J. C. Sagebiel, J. C. Chow and J. G. Watson, Fine Particle and Gaseous Emission Rates from Residential Wood Combustion, Environ. Sci. Technol., 2000, 34, 2080–2091 CrossRef CAS.
- A. Akherati, Y. He, M. M. Coggon, A. R. Koss, A. L. Hodshire, K. Sekimoto, C. Warneke, J. de Gouw, L. Yee, J. H. Seinfeld, T. B. Onasch, S. C. Herndon, W. B. Knighton, C. D. Cappa, M. J. Kleeman, C. Y. Lim, J. H. Kroll, J. R. Pierce and S. H. Jathar, Oxygenated Aromatic Compounds are Important Precursors of Secondary Organic Aerosol in Biomass-Burning Emissions, Environ. Sci. Technol., 2020, 54, 8568–8579 CrossRef CAS PubMed.
- E. A. Bruns, I. El Haddad, J. G. Slowik, D. Kilic, F. Klein, U. Baltensperger and A. S. H. Prévôt, Identification of significant precursor gases of secondary organic aerosols from residential wood combustion, Sci. Rep., 2016, 6, 27881 CrossRef CAS PubMed.
- A. K. Y. Lee, M. D. Willis, R. M. Healy, J. M. Wang, C. H. Jeong, J. C. Wenger, G. J. Evans and J. P. D. Abbatt, Single-particle characterization of biomass burning organic aerosol (BBOA): evidence for non-uniform mixing of high molecular weight organics and potassium, Atmos. Chem. Phys., 2016, 16, 5561–5572 CrossRef CAS.
- J. Li, M. Pósfai, P. V. Hobbs and P. R. Buseck, Individual aerosol particles from biomass burning in southern Africa: 2, Compositions and aging of inorganic particles, J. Geophys. Res.: Atmos., 2003, 108, 8484 Search PubMed.
- P. K. Hudson, D. M. Murphy, D. J. Cziczo, D. S. Thomson, J. A. de Gouw, C. Warneke, J. Holloway, H.-J. Jost and G. Hübler, Biomass-burning particle measurements: Characteristic composition and chemical processing, J. Geophys. Res.: Atmos., 2004, 109, D23S27 CrossRef.
- J. Pagels, D. D. Dutcher, M. R. Stolzenburg, P. H. McMurry, M. E. Gälli and D. S. Gross, Fine-particle emissions from solid biofuel combustion studied with single-particle mass spectrometry: Identification of markers for organics, soot, and ash components, J. Geophys. Res.: Atmos., 2013, 118, 859–870 CrossRef CAS.
- E. Gard, J. E. Mayer, B. D. Morrical, T. Dienes, D. P. Fergenson and K. A. Prather, Real-Time Analysis of Individual Atmospheric Aerosol Particles: Design and Performance of a Portable ATOFMS, Anal. Chem., 1997, 69, 4083–4091 CrossRef CAS.
- M. T. Spencer and K. A. Prather, Using ATOFMS to Determine OC/EC Mass Fractions in Particles, Aerosol Sci. Technol., 2006, 40, 585–594 CrossRef CAS.
- M. T. Baeza-Romero, J. M. Wilson, E. M. Fitzpatrick, J. M. Jones and A. Williams, In Situ Study of Soot from the Combustion of a Biomass Pyrolysis Intermediate—Eugenol—and n-Decane Using Aerosol Time of Flight Mass Spectrometry, Energy Fuels, 2010, 24, 439–445 CrossRef CAS.
- E. M. Fitzpatrick, J. M. Jones, M. Pourkashanian, A. B. Ross, A. Williams and K. D. Bartle, Mechanistic Aspects of Soot Formation from the Combustion of Pine Wood, Energy Fuels, 2008, 22, 3771–3778 CrossRef CAS.
- M. Dall'Osto and R. M. Harrison, Urban organic aerosols measured by single particle mass spectrometry in the megacity of London, Atmos. Chem. Phys., 2012, 12, 4127–4142 CrossRef.
- D. A. Sodeman, S. M. Toner and K. A. Prather, Determination of Single Particle Mass Spectral Signatures from Light-Duty Vehicle Emissions, Environ. Sci. Technol., 2005, 39, 4569–4580 CrossRef CAS PubMed.
- D. S. Gross, M. E. Galli, P. J. Silva, S. H. Wood, D.-Y. Liu and K. A. Prather, Single Particle Characterization of Automobile and Diesel Truck Emissions in the Caldecott Tunnel, Aerosol Sci. Technol., 2000, 32, 152–163 CrossRef CAS.
- J. M. Wilson, M. T. Baeza-Romero, J. M. Jones, M. Pourkashanian, A. Williams, A. R. Lea-Langton, A. B. Ross and K. D. Bartle, Soot Formation from the Combustion of Biomass Pyrolysis Products and a Hydrocarbon Fuel, n-Decane: An Aerosol Time Of Flight Mass Spectrometer (ATOFMS) Study, Energy Fuels, 2013, 27, 1668–1678 CrossRef CAS.
- P. J. Silva and K. A. Prather, Interpretation of Mass Spectra from Organic Compounds in Aerosol Time-of-Flight Mass Spectrometry, Anal. Chem., 2000, 72, 3553–3562 CrossRef CAS PubMed.
- M. A. Bari, G. Baumbach, J. Brodbeck, M. Struschka, B. Kuch, W. Dreher and G. Scheffknecht, Characterisation of particulates and carcinogenic polycyclic aromatic hydrocarbons in wintertime wood-fired heating in residential areas, Atmos. Environ., 2011, 45, 7627–7634 CrossRef CAS.
- M. Elsasser, C. Busch, J. Orasche, C. Schön, H. Hartmann, J. Schnelle-Kreis and R. Zimmermann, Dynamic Changes of the Aerosol Composition and Concentration during Different Burning Phases of Wood Combustion, Energy Fuels, 2013, 27, 4959–4968 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ea00069e |
| This journal is © The Royal Society of Chemistry 2023 |