
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Coordination properties of Cu(II) ions towards a phosphorylated fragment from the R1 domain of the tau protein and the effect of Ser phosphorylation on Cu(II) binding affinity†
Dimitra
Kyriakou
a,
Eleni
Bletsa
b,
Vassilios
Moussis
c,
Yiannis
Deligiannakis
 b and
Gerasimos
Malandrinos
*a
b and
Gerasimos
Malandrinos
*a
aLaboratory of Inorganic Chemistry, Department of Chemistry, University of Ioannina, Ioannina, Greece. E-mail: gmalandr@uoi.gr
bLaboratory of Physical Chemistry of Materials & Environment, Department of Physics, University of Ioannina, Ioannina, Greece
cLaboratory of Protein and Peptide Chemistry, Department of Chemistry, University of Ioannina, Ioannina, Greece
First published on 16th November 2022
Abstract
Tau hyperphosphorylation plays a key role in Alzheimer's disease, mediating tau protein aggregation and deposition as neurofibrillary tangles in the intracellular space of neurons. The potential implications of Cu(II) ions on the disease have been studied intensively, focusing both on Cu(II)–amyloid and on Cu(II)–tau fragment interactions. Nevertheless, there is still a lack of information concerning the metal binding properties and the affinity of phosphorylated fragments of tau. In this work, the coordination properties of Cu(II) ions toward the peptides Ac-GSTENLKH-NH2 (R1) and Ac-GS(P)TENLKH-NH2 (R1P) (Ser phosphorylated analogue) were studied using potentiometric and spectroscopic (vis-EPR) methods. The above sequence belongs to the 261–268 segment of the R1 pseudorepeat of the microtubule-binding domain of the longest tau isoform. It includes both the required metal anchor site (His268) and the residue Ser262, a well-known tau phosphorylation site. There was no surprise in the coordination process of both peptides, which form 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 metal:ligand complexes and follow the already well-known binding trend (i.e., His imidazole as an anchor site followed by the deprotonation and subsequent coordination of 1–3 amide donors). The additional participation of the Glu-γ-COOH in the equatorial plane was evident for the species detected in acidic solutions, while in the pH range of 7–10, the same group occupies an axial position. The comparison of the Cu(II) binding affinities, revealed that the phosphorylated peptide (R1P) is a more effective ligand than R1 in acidic media. Above pH ∼ 7.9, the order is reversed, highlighting the role of the phosphate inter- or intra-molecular interactions in Cu(II) binding.
:1 metal:ligand complexes and follow the already well-known binding trend (i.e., His imidazole as an anchor site followed by the deprotonation and subsequent coordination of 1–3 amide donors). The additional participation of the Glu-γ-COOH in the equatorial plane was evident for the species detected in acidic solutions, while in the pH range of 7–10, the same group occupies an axial position. The comparison of the Cu(II) binding affinities, revealed that the phosphorylated peptide (R1P) is a more effective ligand than R1 in acidic media. Above pH ∼ 7.9, the order is reversed, highlighting the role of the phosphate inter- or intra-molecular interactions in Cu(II) binding.
Introduction
Alzheimer's disease (AD), a chronic neurodegenerative disorder of the brain, is the most common form of dementia, affecting about 10% of the population over age 65 and about 40% over 85 years, respectively.1–3 The disease is clinically characterized by the non-reversible and progressive loss of memory and other cognitive domains including the patient's behaviour, thinking and personality.1,4,5 In a brain suffering from Alzheimer's disease, two main pathological hallmarks are evident: extracellular senile plaques composed of aggregated amyloid beta (Aβ) peptides and intracellular neurofibrillary tangles (NFTs) composed of hyperphosphorylated tau protein. Both plaques and tangles are responsible for neuronal dysfunction, degeneration and eventually the death of brain neuron cells.6,7Tau is a microtubule-associated protein linked to the microtubules, which in turn are formed by the polymerization of a group of proteins called tubulins, primarily located in the axons. Microtubules are one of the most important parts of the cytoskeleton of neurons and are involved in maintaining the structure of the cells, intracellular transport, movement of secretory vesicles and organelles, etc. Strong tau protein-microtubule association is vital to the stabilization of dendritic microtubule structures. The binding of tau protein to the microtubules is controlled by the coordinated action of kinases and phosphatases.8–10 Phosphorylation is the most common post-translational modification of the tau protein and it plays a key role in regulating its normal function, including binding to the microtubules and thus stabilizing and assembling the latter.9
Nevertheless, abnormal phosphorylation (hyperphosphorylation) is expected to alter tau's normal structure and function and trigger the formation of paired helical filaments (PHFs), which may further aggregate to NFTs, eventually causing neurodegeneration.8,9 Among the 85 possible phosphorylation sites of tau protein, only 55 have been so far delineated under pathological conditions. Most of these are located in the microtubule-binding domain (MBD) that consists of three or four pseudorepeats (R1–R4) and in the proline-rich region (PRR) and the C-terminal of the protein.11–13 From a structural point of view, phosphorylated tau is more unfolded compared to tau and R1–R4 repeats mainly adopt a random coil conformation.14 Very recently, a progressive model for the disease was suggested, in which a sequence of events take place at different stages of the disease, (initial phosphorylation, C-terminal cleavage, increased phosphorylation in the PRR domain and acetylation–ubiquitination in the MBD), eventually leading to tau fibril formation.12,15
The bioinorganic chemistry of AD has been recently highlighted after the observation that biologically essential metal ions (i.e., Zn(II), Fe(III), and Cu(II)) were enriched both in the neuritic plaques of AD brains and in biological fluids.16 The potential implication of Cu(II) ions on the disease has been investigated more extensively, because of its biological importance as a cofactor for various enzymes and proteins, its strong interaction with proteins and in particular with histidine residues etc. Most of the research groups working in this area have studied the Cu(II)–Aβ peptide coordination chemistry, Cu(II) mediated reactive oxygen species (ROS) production, and/or Aβ-amyloid aggregation17,18 It should be mentioned that oxidative stress is another pathological hallmark of the disease.19,20
Cu(II)–tau interaction was also probed using various spectroscopic–physicochemical techniques. Full-length tau and short peptide fragments located mainly at the MBD (repeats R1–R4) and the N-terminal regions were used to determine the Cu(II) binding modes and affinity differentiation, the effect of the metal ion on tau aggregation, and ROS production mediated by Cu(II)–tau binding.21–31
The main outcomes of all these studies are that (a) the histidyl imidazole (as an anchor site) and deprotonated amide nitrogen donor atoms govern the coordination process. The dominant species are the mononuclear (b) Cys residues in the R2 and R3 repeats that are unlikely to become involved in Cu(II) binding.23 (c) The N-terminal tau peptide models (tau(9–16) and tau(26–33))27 and the R3 domain model (tau(326–333))30 present different Cu(II) binding affinities at physiological pH (His32 site present in the fragment 26–33 predominates over the others).30 (d) ROS formation and aggregation were detected as a consequence of Cu(II) binding to the full-length microtubule-binding repeats R1–R4.26
While the literature concerning the coordination chemistry of Cu(II)–tau and the implication of metal in ROS formation and aggregation are still increasing, there is still a lack of information about the metal binding properties and affinity of phosphorylated fragments of the tau protein. An investigation in this field might be of interest to reveal the probable effect of Cu(II) interaction in the phosphorylation–dephosphorylation process, governing the binding of tau protein to the microtubules. The catalytic effect of Cu(II)-phosphorylated fragments on ROS production and aggregation could be checked as well.
An electrochemical study of the Cu(II)-immobilized tau interaction revealed that tau phosphorylation affected Cu(II) binding.24 In another experiment by the same group, it was shown that Cu(II) could be displaced only by Zn(II) in phosphorylated tau films. This might be a sign that phosphorylation results in Cu(II) weaker binding. Finally, in a recent ESI-MS study by S. Ahmadi et al., the observed difference in Cu(II) binding affinity was attributed to protein phosphorylation.28
In this paper, we present a complete potentiometric–spectroscopic study of the interaction of Cu(II) with the peptide Ac-GSTENLKH-NH2 and its Ser-O-phosphorylated analogue Ac-GS(P)TENLKH-NH2. The studied sequence belongs to the 261–268 segment of the R1 pseudorepeat of the MBD of the longest tau isoform. It includes both the required metal anchor site (His268) and the residue Ser262, a well-known tau phosphorylation site.11–13 To the best of our knowledge, this is the first thermodynamic–spectroscopic study of a phosphorylated tau segment and might be a small step towards the elucidation of the Cu(II) ion role in tau hyperphosphorylation.
Experimental
Materials
Trifluoroacetic acid (TFA), D2O, Cu(NO3)2·6H2O (atomic absorption standard solution), piperidine, 99% diethyl ether, and dichloromethane (analytical grade) were purchased from Sigma-Aldrich Chemical Co. HNO3, acetic anhydride, and acetonitrile (HPLC grade) were obtained from Honeywell Riedel-de Haen. KNO3 and KOH were obtained from E. Merck (Darmstadt, Germany). n-Hexane and dimethylformamide (DMF) were purchased from Carlo Erba Reagents. The resin Rink Amide AM, 1-hydroxybenzotriazole (1-HOBt), O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate (TBTU), and the Fmoc-protected amino acids were purchased from GL Biochem, Shanghai, China and CBL Chemicals Ltd (Patras, Greece). N,N′-Diisopropylcarbodiimide (DIC), triisopropylsilane (TIS) and N,N-diisopropylethylamine (DIPEA) 98% were obtained from Fluka, Switzerland. Glycerol was purchased from Ferak. The reagents required for the phosphate detection assay were purchased from Sigma-Aldrich.Peptide synthesis
The synthesis of the N- and C-terminal protected peptides Ac–Gly–Ser–Thr–Glu–Asn–Leu–Lys–His–NH2 and Ac–Gly–Ser(P)–Thr–Glu–Asn–Leu–Lys–His–NH2 (Ser-O phosphorylated analogue) was carried out manually, in the solid-phase, on a Rink Amide AM resin, using the Fmoc/tBu strategy (Fmoc: 9-fluorenylmethyloxy-carbonyl).32 1-hydroxybenzotriazole (1-HOBt) and O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate (TBTU) were used as coupling reagents. Fmoc protecting groups were removed before each coupling step using 20% (v/v) piperidine in DMF. The coupling reactions were performed using an amino acid/TBTU/HOBt/DIEA solution (molar ratio of 3:3:3:6) in DMF. The N-terminal acetylation was accomplished by treatment of the resin with a solution of acetic anhydride and DIPEA in DMF. A solution of TFA/TIS/H2O (95:2.5:2.5, v/v/v) was used to cleave the peptides from the resin and remove the protecting groups. The crude peptides were purified by semipreparative or preparative reverse-phase HPLC (RP-HPLC) on a C18 column. The purity of the peptides was checked employing 1- and 2D 1H-NMR techniques (500 MHz) and the correct molecular masses were confirmed by ESI-MS.
Potentiometric measurements
The potentiometric titrations were carried out at 25 °C using a total volume of 2.0 mL. The titrations were performed in the presence of 0.1 M KNO3 over the pH range of 2.5–12 in a MOLSPIN pH-meter system (Molspin automatic titrator, Molspin Ltd, Newcastle-upon-Tyne, UK), using a 0.500 mL micrometre syringe and a combined glass–silver chloride electrode calibrated to hydrogen concentration using HNO3.33 A carbonate-free 0.1 M potassium hydroxide solution was used as the titrant. During the titration, argon was bubbled through the samples to ensure the absence of oxygen and carbon dioxide and to stir the solutions. The concentration of the peptides was between 2–3 mM at a metal:peptide molar ratio of 1:1.2. The overall stability constants (logβijk for MiLjHk) and the protonation constants were calculated utilizing the HYPERQUAD computer program.34 Standard deviations computed by HYPERQUAD refer to random errors.
Spectroscopic measurements
NMR experiments were performed using a Bruker Avance spectrometer operating at 500.13 MHz. Both one and two dimensional experiments (1H, 13C, 1H–1H-TOCSY, 1H–1H COSY, 1H–1H NOESY,1H–13C-HMBC, and 1H–13C-HSQC) were acquired in 9:1, v/v H2O:D2O solutions at a peptide concentration of 5 mM (pH = 2.5 at 25 °C). The interaction of Cu(II) with the phosphorylated peptide (2 mM) was studied at pH = 6, in 9:1, v/v H2O:D2O solutions. 1D (1H) and 2D NMR experiments were performed in both the free peptide and in the presence of 0.05 eq. of Cu(II) at the same pH value.
UV-visible and EPR spectroscopy were performed with samples having the same concentration as used for pH potentiometry. The pH was altered, from 5 to 12, by adding small portions of 0.01–0.1 M aqueous KOH to the solutions. Changes in the pH were monitored by a combined glass–silver chloride electrode. The absorption spectra of the complexes were recorded using a Shimadzu UVPC-2401 spectrophotometer in the spectral range of 400–850 nm using 1 cm cuvettes. The used Cu(II):peptides ratio was 1:1.2. The X-band EPR spectra were recorded at 77 K using a Bruker ER200D spectrometer equipped with an Agilent 5310A frequency counter. The samples consisted of 5 mM of peptides with a Cu(II):peptide molar ratio 1:1.2. A glycerol aqueous solution (10% v/v) was used as a solvent to ensure the homogeneity of the frozen samples. The CD spectra (250–800 nm) were recorded using a Jasco J-815 CD spectrometer under the same conditions as used for the absorption spectra.
Phosphate detection assay
The phosphomolybdenum blue assay was used to check the presence of phosphate anions in a mixture containing Cu(II)–phosphorylated peptide in a molar ratio of 1:1.2 (C = 2 mM, pH = 12). The sample was incubated for 2 h at 298 K, acidified and mixed with the reaction reagent prepared as described in the literature.35
Results and discussion
Characterization and protonation constants of the free peptides
The terminally blocked peptides Ac-GS(P)TENLKH-NH2 (R1P) and Ac-GSTENLKH-NH2 (R1) (Scheme 1) were characterized by 1H-NMR spectroscopy. The 1H chemical shifts (δ, ppm) are cited in Tables 1 and 2 in the ESI† while the corresponding 1 and 2D spectra, used for the assignment, are presented in Fig. S1–S11 (ESI†). | ||
| Scheme 1 Structural formulas of the studied peptides. | ||
The peptide Ac-GS(P)TENLKH-NH2 may be considered as an H4L type ligand due to the presence of four ionizable groups in the measurable pH range (2–12) (i.e., Glu γ-COOH, Ser-OP(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)(–O)(OH) phosphate group, His imidazole and the Lys ε-NH3+). The analogue Ac-GSTENLKH-NH2 lacks the phosphate group (H3L). The overall protonation constants of the free peptides were calculated by utilizing the potentiometric titrations and are collected in Table 1. The pKa value of 5.64 could be assigned to the ionization of the Ser-O-phosphate group according to the literature data,36–38 while the rest are in good agreement with the reported ones for the peptides containing Glu, Lys, and His residues in their sequence.29,39–42 It is worth mentioning that Ser phosphorylation has a small impact (ca. 0.3 log units) on the pKa values of both Glu-γ-COOH and His-imidazole (Table 1).
O)(–O)(OH) phosphate group, His imidazole and the Lys ε-NH3+). The analogue Ac-GSTENLKH-NH2 lacks the phosphate group (H3L). The overall protonation constants of the free peptides were calculated by utilizing the potentiometric titrations and are collected in Table 1. The pKa value of 5.64 could be assigned to the ionization of the Ser-O-phosphate group according to the literature data,36–38 while the rest are in good agreement with the reported ones for the peptides containing Glu, Lys, and His residues in their sequence.29,39–42 It is worth mentioning that Ser phosphorylation has a small impact (ca. 0.3 log units) on the pKa values of both Glu-γ-COOH and His-imidazole (Table 1).
| HL | H2L | H3L | H4L | |
|---|---|---|---|---|
Protonation constants (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) β)a β)a
|
||||
| R1P | 10.25(1) | 16.99(1) | 22.63(1) | 26.39(2) |
| R1 | 10.23(1) | 16.69(2) | 20.74(3) | — |
| pKLys | pKHis | pKSer-(HPO3−) | pKGlu | |
|---|---|---|---|---|
| a β = [HjL]/([H]j[L]), standard deviations of the last digit are given in parenthesis. | ||||
| pK a values | ||||
| R1P | 10.25 | 6.74 | 5.64 | 3.76 |
| R1 | 10.23 | 6.46 | — | 4.05 |
Cu(II)–R1P peptide interaction
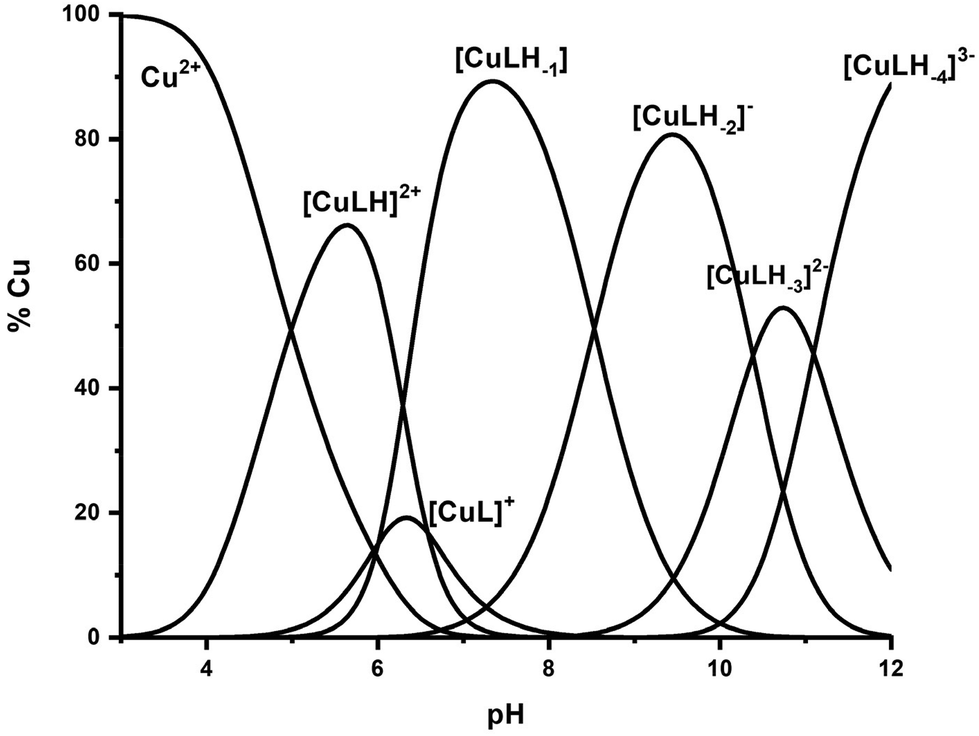
The interaction of the peptide Ac-GS(P)TENLKH-NH2 (R1P) with Cu(II) ions was investigated in aqueous solutions with a Cu(II):peptide 1:1.2 molar ratio. The best fit of the potentiometric data was achieved assuming the existence of seven Cu(II) species of 1:1 Cu:peptide stoichiometry, namely [CuLH2]+, [CuLH], [CuL]−, [CuLH−1]2−, [CuLH−2]3−, [CuLH−3]4− and [CuLH−4]5−. The species stoichiometry, selected thermodynamic data (logβ, pKa, logK*) and the distribution curves are shown in Table 2 and Fig. 1, respectively. For the assignment of the detected complexes, visible (Vis), EPR and CD spectra were recorded over the pH range 5–12 (Fig. 2, 3 and S12†). The relative Vis and EPR spectroscopic data are collected in Table 3.
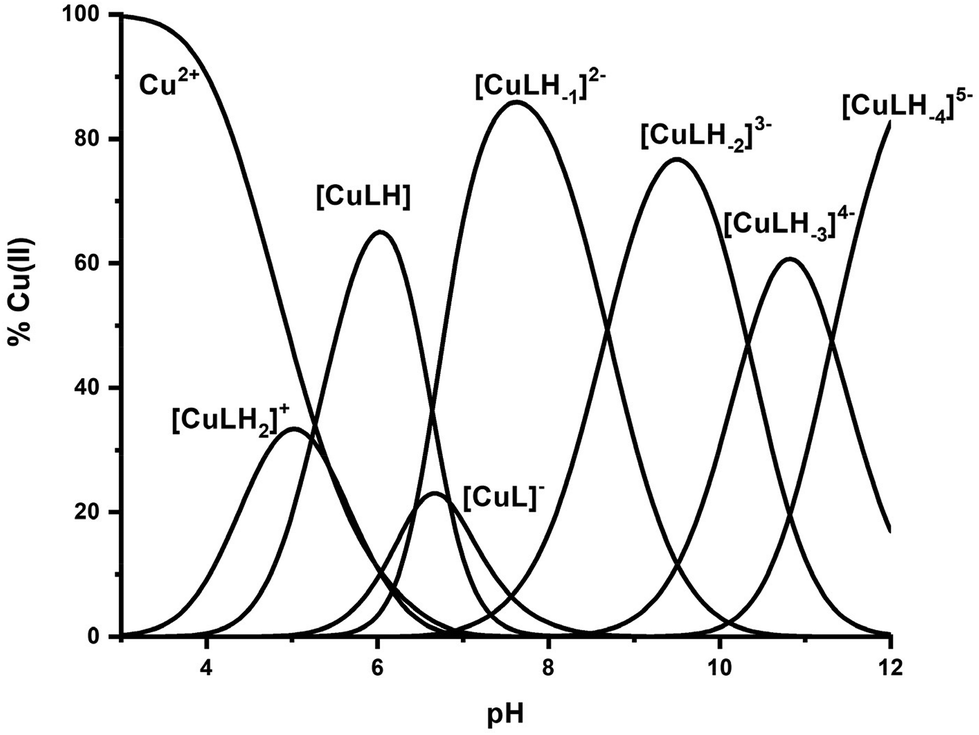 | ||
| Fig. 1 Species distribution diagram of the system Cu(II)–Ac-GS(P)TENLKH-NH2 at a molar ratio of 1:1.2 (C = 2 mM). | ||
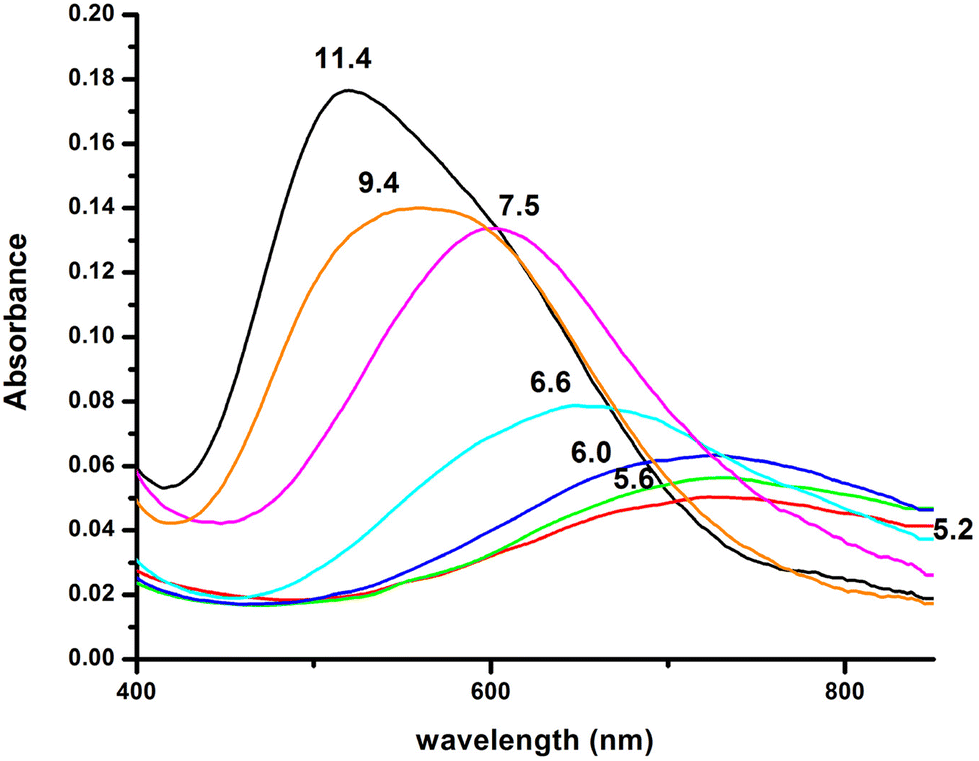 | ||
| Fig. 2 Visible absorption spectra of the solutions of Cu(II) and R1P (Cu(II):R1P = 1:1.2, T = 298 K, I = 0.1 (KNO3)), recorded at the indicated pH values. | ||
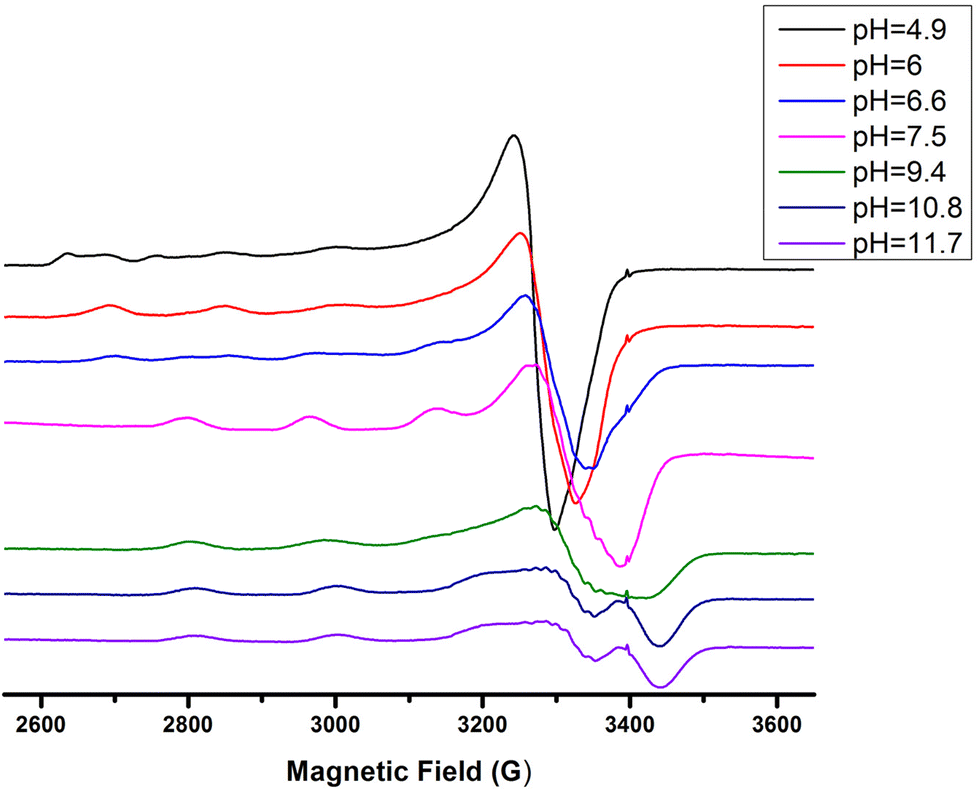 | ||
| Fig. 3 EPR spectra of the Cu(II)–R1P system in a molar ratio of 1:1.2 at various pH values. | ||
| 1N{NIm} | 1N{NIm} | 2N{NIm, N−} | 3N{NIm, 2N−} | 4N{NIm, 3N−} | |
|---|---|---|---|---|---|
|
a
β = [MHjL]/([M][H]j[L]), standard deviations of the last digit are given in parenthesis.
b pKa = logβ(MLHj) − logβ(MLHj−1).
c logK* = logβ(MLHj) − logβ(HkL) k = 2, 3 (protonation corrected stability constants).
d Value for 1NIm coordination with a deprotonated phosphate group.
|
|||||
|
logK*
|
|||||
| R1P | −2.12 | −1.70d | −8.54 | −14.98 | −23.66 |
| R1 | −2.12 | −8.70 | −14.70 | −23.23 | |
:1.2)
| Species | UV/Vis | EPR | |
|---|---|---|---|
| λ max (nm), ε (M−1 cm−1) | g ‖ | A ‖ (G) | |
| Cu2+ | 732, (30) | (a) 2.41 | 122 |
| [CuLH2]+ | (b) 2.37 | 134 | |
| [CuLH] | (c) 2.32 | 162 | |
| [CuLH] | 728, (41) | 2.32 | 158 |
| [CuLH−1]2− | 602, (97) | 2.22 | 170 |
| [CuLH−2]3− | 558, (109) | 2.20 | 188 |
| [CuLH−3]4− | — | 2.19 | 194 |
| [CuLH−4]5− | 520, (149) | 2.19 | 196 |
Cu(II) coordination with the R1P peptide starts at pH 3.0 with the formation of [CuLH2]+ species whose concentration reaches a maximum at pH ∼ 5. The logK* value calculated for the [CuLH2]+ complex (−2.12) is close to those reported for peptides with monodentically bound imidazole nitrogen (Nim), as in the cases of the peptides H2B32–62 (−2.66), H2B63–93 (−1.58), H2B94–125 (−1.94), –LAHYNK– (−2.73), –ELAKHA– (−2.13),43 –EVMEDHAG– (−1.72), –QGGYTMHQ– (−2.24),27 –IKQHT– (−2.55),44 and –KTNFKHVAG– (−2.54).45
Moreover, the value λmax = 732 nm, obtained from the absorption spectrum, confirms the specific binding mode. The blue shift of this value compared to that predicted by the equation of Prenesti46 (λmax = 760 nm), assuming {NIm, 3 H2O} coordination, is an indication of equatorial Glu γ-COOH binding. The theoretically expected λmax value in the case of a {NIm, COO−, 2H2O} donor set is 729 nm.
Increasing the pH of the solution results in the formation of the [CuLH] complex in the pH range of 4–8. The pKa value (5.23) of the reaction [CuLH2]+ ↔ [CuLH] + H+ (Table 2) cannot be assigned to the ionization and coordination of the first amide donor as in terminally protected peptides containing His residues this value is higher than 6.0.39,43,47–51 Moreover, the recorded (at pH 6.0, where [CuLH] predominates) λmax value (728 nm) does not support amide binding and is almost the same as the one (732 nm) previously assigned to [CuLH2]+ {NIm, COO−, 2H2O}. On the other hand, the EPR parameters (A‖ = 158 G, g‖ = 2.32) match perfectly with the ones assigned to the [CuL] species of the system Cu(II)–EVMEDHAG– (A‖ = 160 G, g‖ = 2.315).27 In the latter case, the involvement of both the imidazole nitrogen atom and Glu carboxylate was suggested.
A plausible explanation is that [CuLH] adopts the same coordination mode as [CuLH2]+ {NIm, COO−, 2H2O}. In that case, the pKa value of 5.23 should correspond to the ionization of the phosphate group in the complexed peptide (pKa = 5.6 in the free one). The extra thermodynamic stability gained due to the deprotonation of this group in the complexed ligand is reflected in both the thermodynamic data for [CuLH] and the distribution diagram. [CuLH] at pH ∼ 6 is responsible for binding 65% of the total Cu(II) concentration (Fig. 1), while the calculated logK value of 5.03 (logK = logβ([CuLH]) − logβ(HL)) is higher (0.5–1 logarithmic units) than the values (4–4.5) reported for complexes presenting the same coordination type {NIm, COO−, 2H2O}.41,42,52 In addition, the logK* value of −1.70 (Table 2) shows the same trend, being higher than the corresponding value of [CuLH2]+ (−2.12), in the same system, similar to the values calculated for the 30 peptides H2B63–93 (−1.58), H2B94–125 (−1.94)43 and almost identical for–EVMEDHAG– (−1.72).27
The 1H-NMR spectra of the peptide recorded at pH = 6 in the absence and in the presence of 0.05 eq. of Cu(II) (Fig. 4 and 5) provide additional evidence for the [CuLH] binding proposal. Paramagnetic Cu(II) ions broaden the 1H-NMR signals assigned to protons which are located close to the coordination sites.53,54 Inspection of both the aromatic and aliphatic regions of the NMR spectra revealed (a) a general line broadening and signal reduction (b) that the most affected resonances (almost washed out) belong to His(NH), His (Hε1 + Hδ2) and Glu (β- and γ-CH2). The results of NMR spectroscopy are in line with the thermodynamic and spectroscopic data (Vis and EPR) indicating the presence of the donor set {NIm, COO−, 2H2O} in the equatorial plane of Cu(II). The equatorial binding of the phosphate group should be then ruled out.
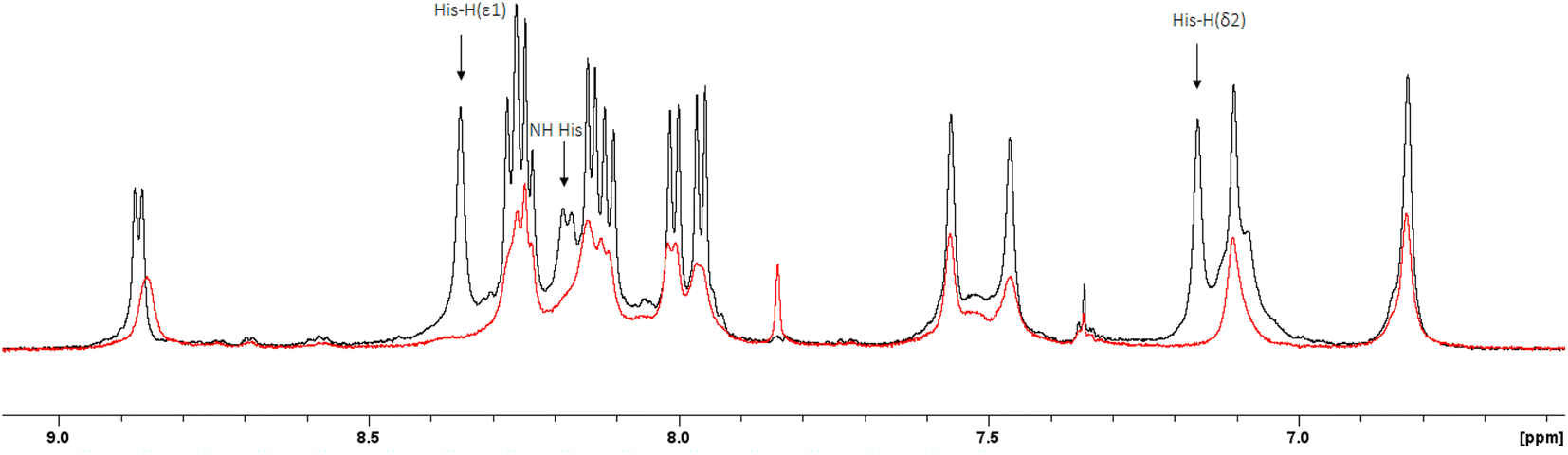 | ||
| Fig. 4 Superposition of the 1H-NMR spectra (aromatic region) of the peptide R1P in the presence (red trace) and absence (black trace) of 0.05 eq. of Cu(II) (Cpeptide = 2 mM, H2O:D2O 9:1, v/v, pH = 6). The most affected resonances have been indicated by arrows. | ||
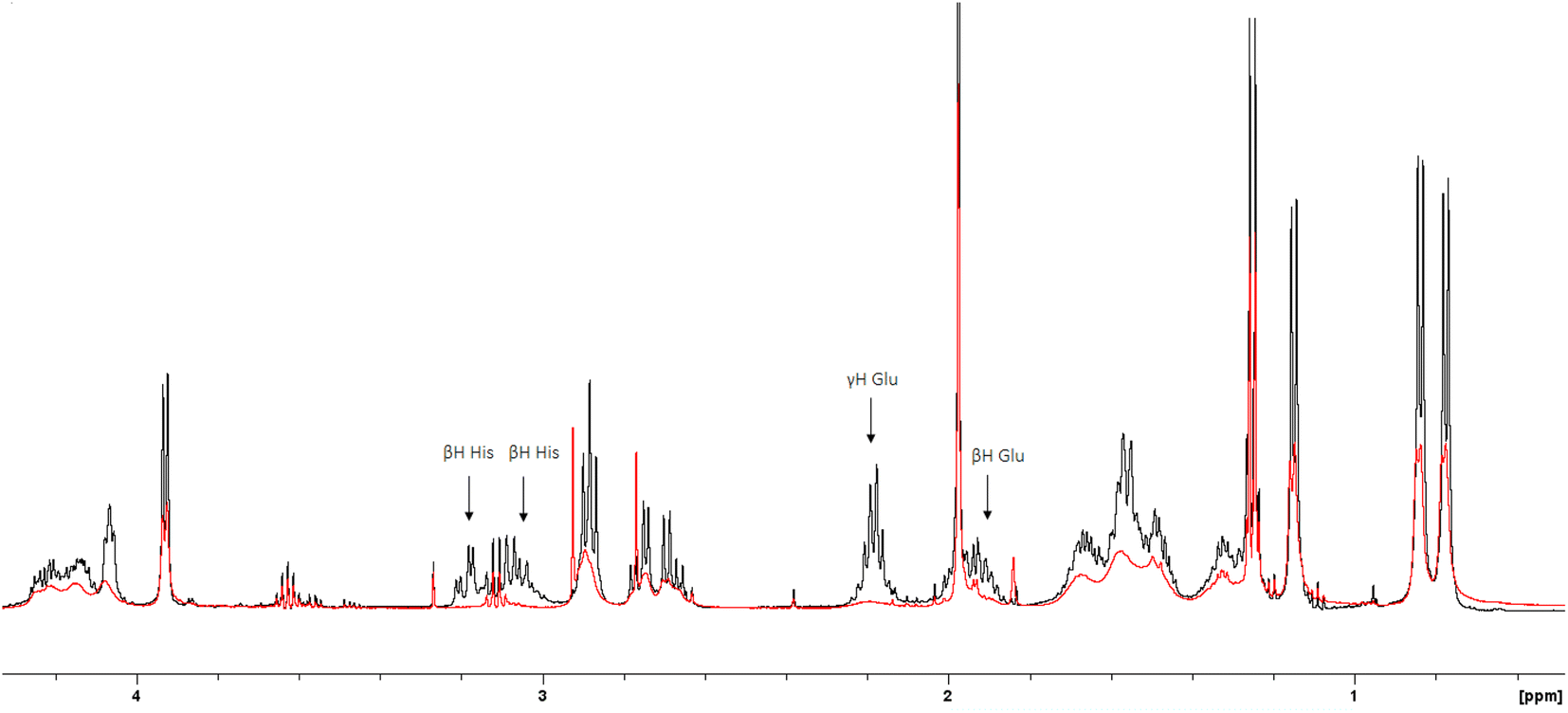 | ||
| Fig. 5 Superposition of the 1H-NMR spectra (aliphatic region) of the peptide R1P in the presence (red trace) and absence (black trace) of 0.05 eq. of Cu(II) (Cpeptide = 2 mM, H2O:D2O 9:1, v/v, pH = 6). The most affected resonances have been indicated by arrows. | ||
In light of these outcomes, a question arose: Could the reason for this extra stabilization be a direct Cu(II)–phosphate interaction? As we will additionally see, in the next section, in which we will discuss the experimental data for the non-phosphorylated analogue, the same binding mode is suggested, although the calculated logK and logK* values are smaller. Taking into account the observations up to this point, it is reasonable to assume that the phosphate group does not interact with Cu(II) directly and the small extra stabilization observed for the phosphorylated peptide in the presence of copper is possibly ascribed to the participation of the anionic phosphate group in non-covalent interactions or hydrogen bonding.55
At slightly higher pH values, the deprotonation and subsequent coordination of the first amide nitrogen donor atom (pKa = 6.84) occur, resulting in the formation of [CuL]−. Although the cooperative deprotonation and binding of two amide donors were expected, [CuL]− inclusion in the model improves the fitting of the titration curves. The low concentration of the complex and overlap with [CuLH] and [CuLH−1]2− prevent its spectroscopic study. This species can be characterized by stoichiometry and the stability constant. The value logK* = −8.54 is typical of 2N {NIm, N−amide} binding, leading to the formation of a five-membered chelate ring.56,57
The next ionization process that takes place (pKa = 6.44) results in the formation of the [CuLH−1]2− species. This is the major complex formed at physiological pH values, being responsible for the binding of 85% of the total copper concentration, while its existence covers the pH-metric range 5.5–10.5. The value logK* = −14.98 is comparable to those of the Cu(II)–peptide complexes, where the metal ion binds with both the imidazole nitrogen (N3) of His and two amide nitrogen atoms of the peptide backbone,27,39,44,45,56,58 suggesting the same coordination type. The spectroscopic data corroborate the above proposition. More specifically, in the Vis spectrum, the λmax value recorded equals 602 nm. This is within the reported range (λmax = 560–610 nm) for chromophores that include three nitrogen atoms of the {NIm, 2N−amide} type.27,44,45,47 Additional confirmation is obtained from the EPR spectrum recorded at pH 7.5. The value of the g factor in the parallel region (g‖ = 2.22) and the hyperfine coupling constant (A‖ = 170 G) is typical of divalent copper complexes with three nitrogen and one oxygen donor atoms.27,44,45,59 Apical interactions of either the γ-COO of the Glu residue or the phosphate group (SerOP) cannot be ruled out since the experimental λmax value (602 nm) is red-shifted by about 20 nm compared to that theoretically predicted for the donor set {NIm, 2N−amide, H2O} (584 nm).46,60 The presence of axial donors may also be inferred from the examination of the perpendicular region of the EPR spectrum registered at pH 7.5 (Fig. S12†). More than the seven expected (for 3N binding) lines are present, implying isotropy disturbance at the x, y region caused by the distortion of the tetragonal geometry.57 This effect may also explain the relatively low value of A‖ (170 G).61,62 [CuLH−1]2− species is also CD active (Fig. 6).
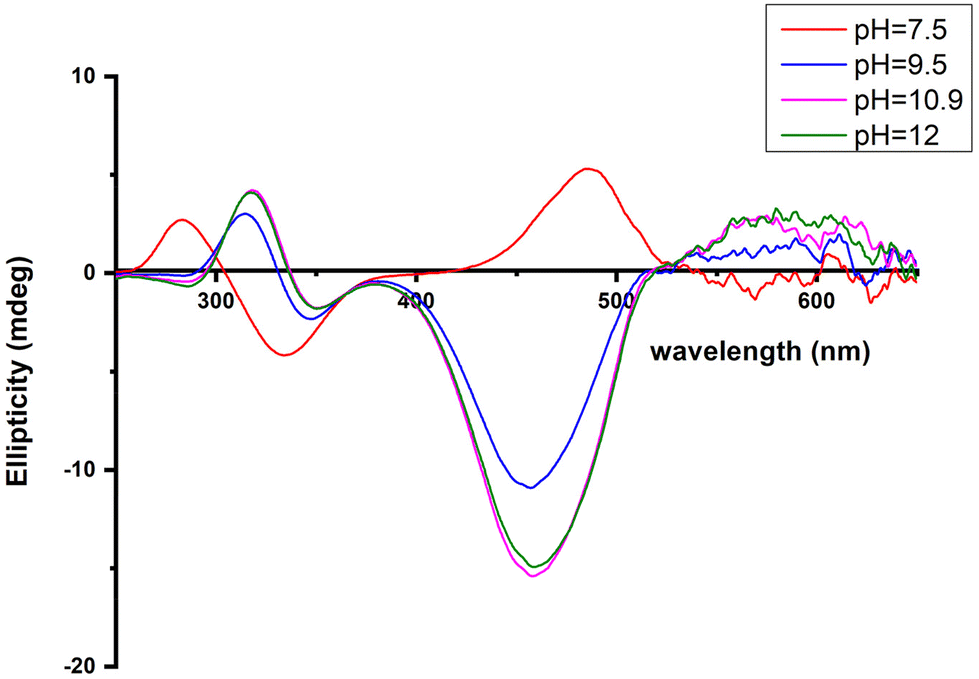 | ||
| Fig. 6 Circular dichroism spectra of the solutions of Cu(II) and R1P (Cu(II):R1P = 1:1.2), recorded at the indicated pH values. | ||
The CT bands due to Nim → Cu(II) and N−amide → Cu(II) (334 and 284 nm, respectively) indicate that a 3N donor set {NIm, 2N−amide, H2O} occupies the equatorial plane.44,63
Above pH 7, the formation of the [CuLH−2]3− complex (pH range of existence 7–12) is evident (pKa = 8.68). The value of logK* = −23.66 calculated for this species is close to the values of Cu(II) complexes with peptides in which 4N {NIm, 3N−amide} coordination is proposed (∼−21 to −23).27,39,43,44,56–58 The presence of an extra amide nitrogen donor atom is also supported by both the Vis and EPR data. The λmax of 558 nm, the values g‖ = 2.20 and A‖ = 188 G and the nine lines present (AN = 15 G) in the perpendicular region of the EPR spectrum (Fig. S12†) as expected for four equivalent nitrogen donors in the Cu(II) coordination plane, indicate the equatorial binding of one NIm and three amide nitrogen atoms {NIm, 3N−amide}.41,42,64–67 However, it should be noted that the theoretically predicted λmax for the above coordination type is 523 nm,46 featuring a 35 nm red shift compared to the experimental value. This observation and the broadening of both the absorption band and the EPR resonance lines at pH ∼ 9.4 (Fig. 2 and 3) possibly indicate the presence of coordination isomers, which share the same donor set in the equatorial plane, while differing in the apical substituent (i.e., H2O vs. Glu-carboxylate).
The succeeding complexes [CuLH−3]4− and [CuLH−4]5− are formed in a highly alkaline solution. Their spectroscopic parameters are similar, suggesting the same coordination mode. The experimental data cited in Table 3 are compatible with {NIm, 3N−amide} binding, already proposed for the previously discussed [CuLH−2]3− species. On the other hand, the small decrease in both the λmax value (520 nm) and g‖ (2.19) and the higher value of the hyperfine coupling constant (A‖ = 194–196 G) compared to the same spectroscopic parameters for [CuLH−2]3− may imply the lack of axial interactions in both [CuLH−3]4− and [CuLH−4]5−. The CD spectra of the last three species do not differ significantly (Fig. 6) and share the following common features that are observable in Cu(II)–N-protected one His containing peptides adopting the {NIm, 3N−amide} coordination mode:30,44,68,69 (a) charge transfer bands assigned to Nim → Cu(II) (347–350 nm) and N−amide → Cu(II) (314–318 nm)44,63 and (b) negative and positive Cotton effects around 460 and 570 nm, respectively. The slight shift in the position of CT bands and the intensity increment of the negative d–d band passing from the [CuLH−2]3− to [CuLH−3]4− reveals that the deprotonation process yielding the latter affects the 4N donor set strength. The pKa values obtained for the deprotonation and the subsequent formation of the above-mentioned species are 10.33 and 11.31, respectively. The first is similar to the one calculated for the ionization of the Lys-ε-NH3+ in the free peptide (10.25) and it is therefore assigned to the deprotonation of the same group in the complexed peptide. Concerning the last base consuming process (pKa = 11.31), there are four candidates (a) an extra amide donor (requires the detachment of the imidazole nitrogen atom from the equatorial plane), (b) an apically coordinated water molecule (complex hydrolysis), (c) R1P hydrolysis, and (d) the pyrrole N(1) atom of the imidazole ring. In the first case, the EPR g‖ value and the intensity of the negatively signed d–d band in the CD spectrum are expected to decrease significantly,70 while in the second, an axial donor would entail a small red shift in the UV-vis spectra and a decrease in the A‖ value (EPR).62,71 Based on the available spectroscopic data (unchanged vis-EPR-CD parameters for [CuLH−2]3− and [CuLH−3]4−), the amide hypothesis and complex hydrolysis should be ruled out. To check the possibility of Cu(II)-mediated R1P hydrolysis, a sample consisting of Cu(II)–R1P at a molar ratio of 1:1.2 (C = 2 mM, pH = 12) was incubated at 298 K for 2 h and then tested for the presence of o-phosphate anions using the phosphomolybdenum blue method. The test was negative. Concluding, the only reasonable explanation remaining is the ionization and no coordination of the pyrrole N(1) atom of the imidazole ring.57,72–79
Cu(II)–R1 peptide interaction
The potentiometric study of the system Cu(II):Ac-GSTENLKH-NH2 (R1) was performed in a 1:1.2 molar ratio at the pH range of 3–11. The species distribution diagram is provided in Fig. 7, while the calculated stability data are presented in Table 2. The Vis, EPR, and CD spectra and the spectroscopic parameters at the pH range 5–12 are presented in Fig. 8, 9 (and S13) and S14,† and Table 4 respectively.
 | ||
| Fig. 7 Species distribution diagram of the system Cu(II)–Ac-GSTENLKH-NH2 (R1) in a molar ratio of 1:1.2 (C = 2 mM). | ||
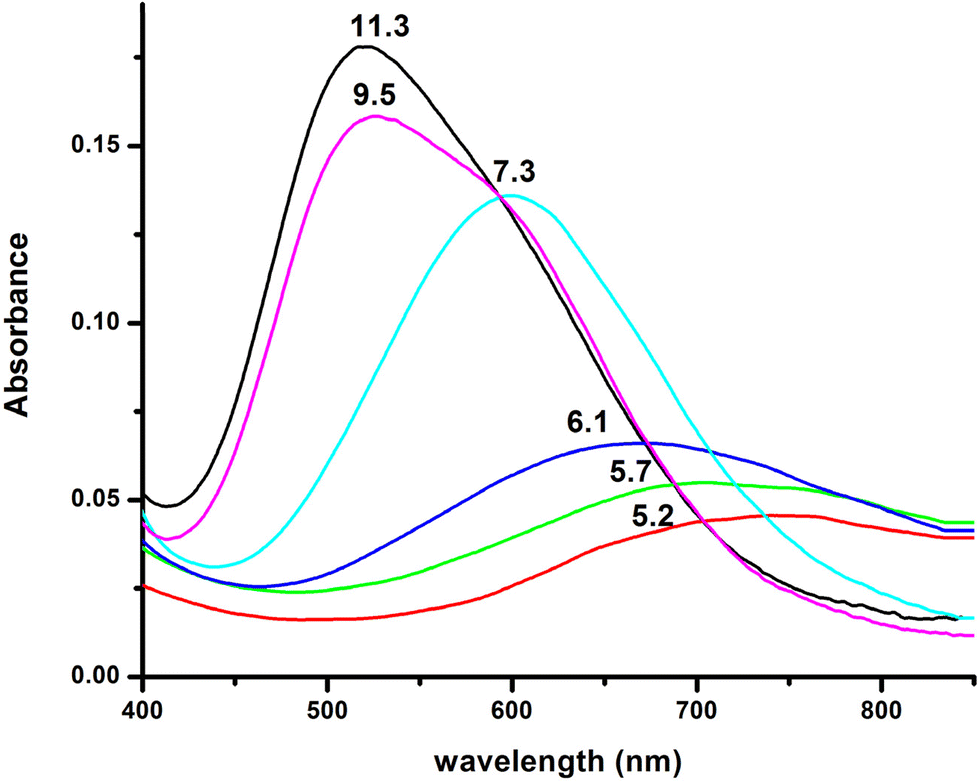 | ||
| Fig. 8 Visible absorption spectra of the solutions of Cu(II) and R1 (Cu(II):R1 = 1:1.2, T = 298 K, I = 0.1 (KNO3)), recorded at the indicated pH values. | ||
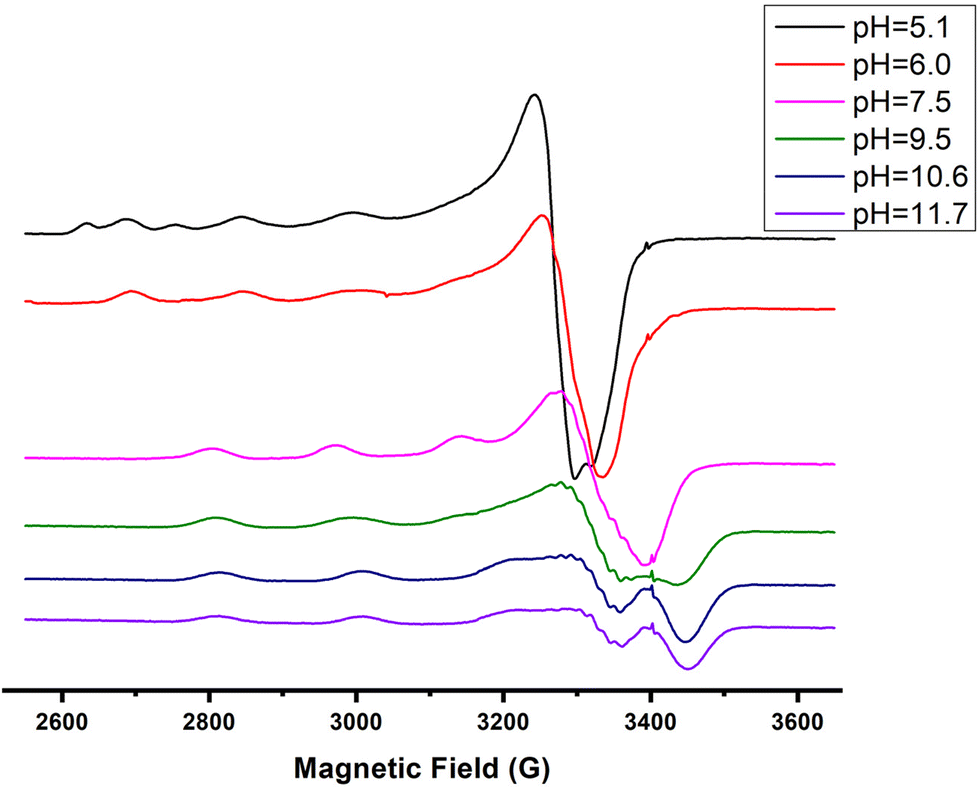 | ||
| Fig. 9 EPR spectra of the Cu(II)–R1 system in a molar ratio of 1:1.2 at various pH values. | ||
:1.2)
| Species | UV/Vis | EPR | |
|---|---|---|---|
| λ max (nm), ε (M−1 cm−1) | g ‖ | A ‖ (G) | |
| Cu2+ | — | (a) 2.41 | 120 |
| [CuLH]2+ | 705, (33) | (b) 2.33 | 152 |
| [CuLH−1] | 599, (99) | 2.22 | 168 |
| [CuLH−2]− | (a) 527, (122) | 2.20 | 188 |
| (b) 560 (sh) | |||
| [CuLH−3]2− | — | 2.19 | 196 |
| [CuLH−4]3− | 520, (140) | 2.18 | 200 |
Six copper complexes were detected in the pH range 4–12, namely [CuLH]2+, [CuL]+, [CuLH−1], [CuLH−2]−, [CuLH−3]2−, and [CuLH−4]3−. As can be seen from the distribution diagram (Fig. 7), the Cu(II)–R1 interaction starts at pH 3.5, yielding the [CuLH]2+ complex, the concentration of which is maximized at pH = 5.7. The value logK* = −2.12 is close to those reported in the literature for His containing peptides monodentically bound to Cu(II) through the N3 atom of the imidazole,27,43–45,56,67 implying a similar coordination mode.
Furthermore, a comparison of the logK value of 4.34 calculated for the reaction Cu(II) + LH ↔ [CuLH]2+ {logK = logβ([CuLH]2+) − logβ(LH)} with the literature data for the same type of reactions in similar systems27,41,42,52 demonstrates, possibly, the additional interaction of Glu γ-COOH in the equatorial plane.
The EPR spectrum recorded at pH = 5.4 shows the presence of two species (a and b in Table 4) in accordance with the speciation diagram (Fig. 7). The former (a) is allocated to free copper ions, while the other one (b) belongs to [CuLH]2+. The EPR parameters (A‖ = 152 G, g‖ = 2.33) are typical of 1N {NIm} binding,27,39,50,51 while the absorption maximum centered at 705 nm is indicative of the {NIm, COO−, 2H2O} coordination mode.27,50
The next species, [CuL]+, can only be characterized by the thermodynamic data due to its low concentration and heavy overlapping with other species. The pKa = 6.58 calculated for the deprotonation reaction [CuLH]2+ ↔ [CuL]+ + H+ characterizes the ionization and subsequent coordination of the first backbone amide nitrogen atom in peptides containing a histidine residue at the C-terminus or intermediate positions.39,43,47–51 On the other hand, the value logK* = −8.70 is similar to the values found in the literature for the 2N {NIm, N−amide} species of analogous copper–peptide complexes.43,56,57
The next deprotonation process (pKa = 6.0) leads to the formation of [CuLH−1]. The latter prevails at physiological pH value and binds about 90% of total Cu(II). The protonation corrected stability constant value logK* = −14.70 is close to the reported values for a 3N {NIm, 2N−amide} binding type (i.e., −14.2 to −14.8)27,39,43–45,56,58 and almost the same as the value calculated (−14.35) for the recently studied Cu(II)–Ac-VKSKIGSTENLKHQPGGG-am system29 in which a longer R1 peptide segment containing our sequence was used. The latter logK* comparison also implies that the stability of the 3N Cu(II) complexes (octadecapeptide vs. octapeptide) does not differ significantly, despite the presence of secondary effects in the longer peptide, which may contribute to its stability.
The spectroscopic data (Table 4) confirm the proposed binding mode. The value λmax = 599 nm in the visible spectrum is comparable to the theoretically predicted value46 (λmax = 583 nm), assuming the presence of a {NIm, 2N−amide, H2O} donor set. It is also within the range of the λmax values (560–610 nm) that characterize the {NIm, 2N−amide} type chromophores.27,29,47 The small redshift (16 nm) could be attributed to a weak apical interaction of the Glu γ-COO− group.60 Finally, the values of the parameters g‖ and A‖ (2.22, 168 G), extracted from the EPR spectrum at pH 7.5, are characteristic of the presence of three nitrogen and one oxygen donor atoms in the equatorial plane.27,44,45,59 The relatively small value of the hyperfine coupling constant A‖ = 168 G and the x, y region anisotropy indicated by the presence of more than the expected 7 lines in the perpendicular region of the spectrum (Fig. S13†) provides further evidence for the tetragonal distortion caused by the axial Glu γ-COO− interaction.57,61
Over pH 9, the [CuLH−2]− species predominate (Fig. 4). The value pKa = 8.53 should correspond to the deprotonation and binding of the last amide nitrogen atom. The value logK* = −23.23 is comparable to the values obtained for Cu(II)–peptide complexes with a 4N {NIm, 3N−amide} coordination mode (i.e., −21 to −23).27,29,39,43,44,56–58 This is further sustained by the Vis and EPR spectroscopy. The value λmax = 527 nm of the visible spectrum recorded at pH 9.5 matches perfectly the theoretically predicted value46 (λmax = 522 nm) for the proposed binding type. The presence of an additional “shoulder” (Fig. 8) in the spectrum (λmax = 560 nm) could indicate the presence of coordination isomers, adopting the same coordination mode in the equatorial plane, while in one of them, the apical binding of the Glu γ-COO− group is taking place. On the other hand, in the EPR spectrum, the observed decrease in the g‖ factor value (2.20) and the increase in A‖ (188 G) compared to the same values for [CuLH−1] and the presence of a 4N confirmatory superhyperfine pattern consisting of 9 lines (AN = 15.5 G) (Fig. S14†), demonstrate the presence of species in which Cu(II) coordination involves the His imidazole and three backbone amide nitrogen donors: (g‖ = 2.19–2.20, A‖ = 195–205 G).29,30,64,66 The relatively smaller value of A‖ (188 G) manifests a slightly distorted coordination environment due to the already mentioned Glu–carboxylate apical interaction.
Two more base-consuming processes are observed in the pH range 10–12, yielding the last two complexes, [CuLH−3]2− and [CuLH−4]3−, respectively. The spectroscopic parameters (Table 4) for these are almost identical to those of [CuLH−2]−, implying the same coordination mode. Moreover, in the CD spectra (Fig. S14†) of the last three species, the position of both the CT and d–d bands implying 4N {NIm, 3N−amide}30,44,63,68,69 binding does not change. On the other hand, the slight decrease in g‖ (2.18–2.19) and the increase in A‖ (196–200) indicate a less distorted coordination environment in which no apical interaction should occur. The pKa value for the ionization reaction that yields [CuLH−3]2− is 10.38, close to the calculated one for the Lys ε-NH3+ in the free peptide (10.23) and is therefore assigned to this group. For the last dissociable proton ([CuLH−4]3−), the pKa value of 11.09 was calculated. Based on the above observations, the relative data for the R1P analogue and literature findings,72–79 we propose as the source of this extra proton the pyrrole N(1) atom of the imidazole ring. A schematic representation of the Cu(II) coordination environment in [CuLH−1] and [CuLH−2]− for both the peptides is provided in Scheme 2.
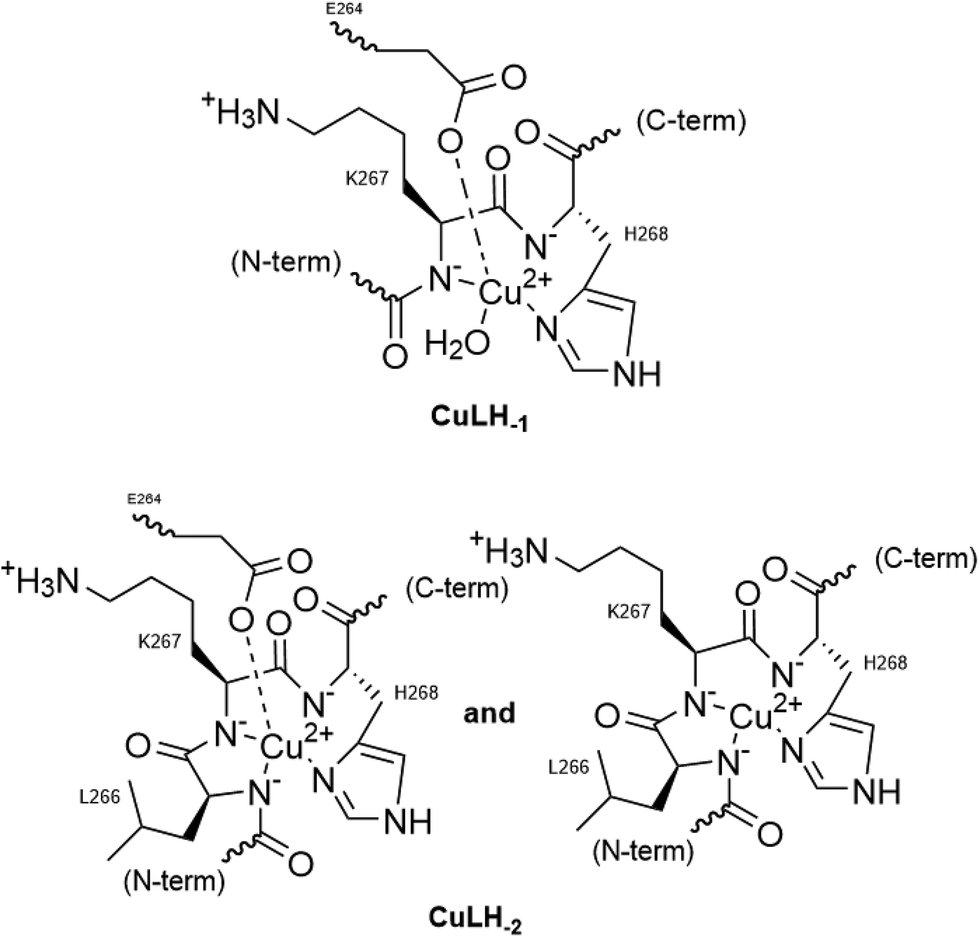 | ||
| Scheme 2 The proposed Cu(II) coordination environment in CuLH−1 and CuLH−2 (L = R1, R1P). | ||
The copper binding affinity of the peptides
We have so far discussed the coordination behaviour of a small segment of the R1 repeat of tau (261–268) and its Ser-phosphorylated analogue towards Cu(II) ions. Both peptides, R1P and R1, interact efficiently with Cu(II), forming thermodynamically stable 1:1 Cu:peptide species. The next step was to check if there is a difference in the Cu(II) binding affinity in the presence of the phosphate at Ser262, a well-known phosphorylated residue of tau. In this context, we will recall the logK* values cited in Table 2 for both peptides. In addition, as shown in Fig. 10, Cu(II) distribution in a hypothetical system containing Cu(II), R1 and R1P at equimolar concentrations is presented.
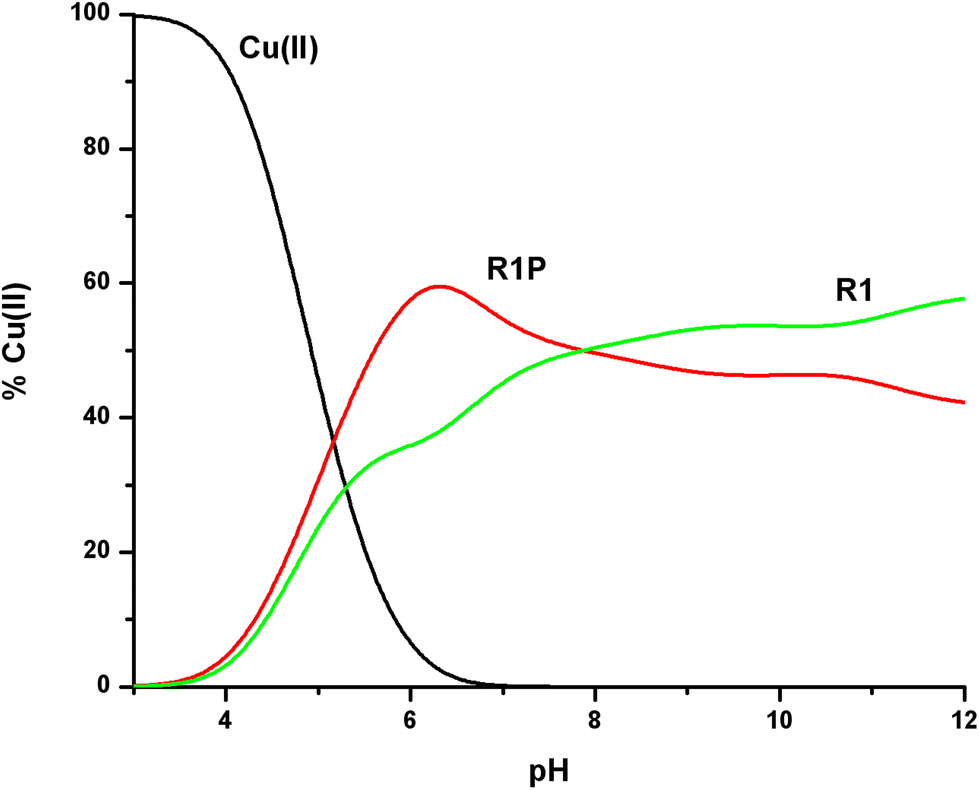 | ||
| Fig. 10 Cu(II) ion distribution diagram in a model system of R1, R1P and Cu(II) in a 1:1:1 molar ratio (C = 1 mM). | ||
The first conclusion drawn by the comparison of the logK* values cited in Table 2 is that small differences exist (0.2–0.5 log units). At the same time, Fig. 10 reveals the Cu(II) binding affinity trend which is pH-dependent. The phosphorylated peptide (R1P) is a more effective ligand than R1 in acidic media, reaching its maximum Cu(II) binding affinity at pH ∼ 6.3, while after the intersection point (pH ∼ 7.9) the metal ion preference is reversed (R1). The presence of an extra negative charge (phosphate group) in R1P cannot solely explain the observed differences and especially the affinity order reversal. It is noteworthy that the pH value at which the maximum affinity differentiation is observed is the one at which the maximum concentration of the [CuLH] species (R1P) is observed (Fig. 1). Based on our data, we have already suggested that the phosphate group should not interact directly with the metal center whose coordination sphere involves the N3 imidazole nitrogen, the Glu γ-COOH, and water molecules. Taking into account these findings and the coordination proposal for both systems, we can tentatively ascribe the observed affinity differentiation to the participation of the anionic phosphate group in intramolecular and/or intermolecular interactions of the electrostatic nature or hydrogen bonding.55 The strength of these could be pH-dependent. This should account for the Cu(II) binding affinity order reverse in alkaline media.
As we have already mentioned in the Introduction, the binding of the tau protein to microtubules is regulated by the coordinated action of kinases and phosphatases.8–10 In the case of phosphatases,80–82 direct interaction of the substrate (phosphate) with the enzyme metal centers was proposed. Cu(II)–phosphate binding, or another kind of indirect phosphate interaction induced by Cu(II) coordination near the tau phosphorylation sites, might have a negative effect on the enzyme activity. Tau hyperphosphorylation due to decreased phosphatase activity could in turn induce tau detachment from the microtubules and finally aggregation and deposition as PHFs and NFTs.8,9 It is also interesting to note that the chemistry we presented (especially at pH values less than the physiological one) could be of relevance to tau pathology. In cases of Alzheimer's disease, neuronal acidosis is common,83 which affects tau phosphorylation,83,84 while glutathione (which can reduce the Cu(II) toxic species) depletion in the brain is a common finding in neurodegenerative diseases.85
Conclusions
In the present work, the first thermodynamic–spectroscopic study of the interaction of Cu(II) ion with a phosphorylated tau segment, namely Ac-GS(P)TENLKH-NH2 (R1P = L) was presented. The non-phosphorylated analogue Ac-GSTENLKH-NH2 (R1 = L′) was also synthesized and its Cu(II) complexing behaviour was checked as well to reveal the phosphorylation impact on metal ion binding affinity. The synthesized sequence belongs to the 261–268 segment of the R1 pseudorepeat of the MBD of the longest tau isoform. It includes both the required metal anchor site (His268) and the residue Ser262, a well-known tau phosphorylation site.11–13 It was found that both the peptides are effective ligands for copper binding, following the typical coordination trend for peptides containing a histidine residue at the C-terminus.86 As expected, the metal anchor site is the histidine imidazole N(3) atom with the additional involvement of the γ-carboxylate of Glu in the equatorial plane forming a macrochelate ring (species [CuLH2]+ and [CuL′H]2+). The first deprotonation process for Cu(II)–R1P leads to the formation of species [CuLH] which adopts the same coordination mode {NIm, COO−, 2H2O} as the former one and differs only in the protonation state of the Ser-phosphate group. The above process affects the thermodynamic stability of [CuLH] vs. [CuL′H]2+. Although both species share the same donor atoms in the equatorial plane, the stability of the former is higher (ΔlogK* ∼ 0.5). This relatively small differentiation was tentatively attributed to the participation of the phosphate group in non-covalent interactions or hydrogen bonding.50 In the next low concentration 2N species ([CuL]−, [CuL′]+), the first amide nitrogen atom is involved, while for the complexes present at physiological pH, a 3N {NIm, 2N−amide, H2O} coordination type was evident. A weak apical interaction of the Glu-carboxylate was also proposed. Either [CuLH−1]2− or [CuL′H−1] binds 85–90% of the total Cu(II) concentration, reflecting their great thermodynamic stability. In more basic solutions, a third proton is titrated (pKa's = 8.68 (L), 8.53 (L′)) and species with stoichiometry [CuLH−2]3− or [CuL′H−2]− are detected. For these, the thermodynamic–spectroscopic data are compatible with 4N {NIm, 3N−amide} binding, and the existence of coordination isomers differentiated by the presence in one of them, of an axially coordinated Glu γ-carboxylate. For the last two complexes, namely [CuLH−3]4−or [CuL′H−3]2− and [CuLH−4]5− or [CuL′H−4]3− predominating above pH ∼ 10, the same donor set {NIm, 3N−amide} occupies the equatorial plane. The two deprotonation processes leading to their formation were assigned to the ionization and no coordination of the Lys-ε-NH3+ and the pyrrole N(1) of the imidazole ring, respectively. The experimental data (EPR) also indicated a less distorted coordination environment in which no apical interactions should occur. The comparison of the Cu(II) binding affinity of the two peptides (Fig. 10) revealed that the phosphorylated peptide (R1P) is a more effective ligand than R1 in acidic media. This order is reversed above pH ∼ 7.9, highlighting the role of the phosphate inter- and/or intra-molecular interactions in the binding efficiency. These effects might have a negative impact on phosphatase activity, thus increasing the hyperphosphorylated tau levels. More research is needed to clarify the potential implication of Cu(II) in tau phosphorylation–dephosphorylation fine-tuning, thus affecting the hyperphosphorylated tau levels. In this context, we will continue our investigation, involving more peptide fragments located at the MBD.
Conflicts of interest
There are no conflicts to declare.References
- W. Thies and L. Bleiler, Alzheimer's Dementia, 2021, 17, 327 CrossRef.
- Z. K. Mathys and A. R. White, Adv. Neurobiol., 2017, 18, 199 CrossRef PubMed.
- Y. Ju and K. Tam, Neural Regener. Res., 2022, 17, 543 CrossRef CAS PubMed.
- B. B. Bendlin, C. M. Carlsson, C. E. Gleason, S. C. Johnson, A. Sodhi, C. L. Gallagher, L. Puglielli, C. D. Engelman, M. L. Ries, G. Xu, W. Wharton and S. Asthana, Maturitas, 2010, 65(2), 131 CrossRef CAS.
- A. I. Mot and P. J. Crouch, Biometals and Alzheimer's Disease, in Biometals in Neurodegenerative Diseases: Mechanisms and Therapeutics, ed. A. R. White, M. Aschner, L. G. Costa and A. I. Bush, 2017, pp. 1–17 Search PubMed.
- A. Lloret, T. Fuchsberger, E. Giraldo and J. Viña, Free Radicals Biol. Med., 2015, 83, 186 CrossRef CAS PubMed.
- K. G. Yiannopoulou and S. G. Papageorgiou, J. Cent. Nerv. Syst. Dis., 2020, 12, 1 Search PubMed.
- E. M. Mandelkow and E. Mandelkow, Cold Spring Harbor Perspect. Biol., 2012, 2, a006247 Search PubMed.
- M. Kolarova, F. García-Sierra, A. Bartos, J. Ricny and D. Ripova, Int. J. Alzheimer's Dis., 2012, 2012, 731526 Search PubMed.
- J. C. Polanco, C. Li, L. G. Bodea, R. Martinez-Marmol, F. A. Meunier and J. Götz, Nat. Rev. Neurol., 2018, 14, 22 CrossRef CAS.
- C. Alquezar, S. Arya and A. W. Kao, Front. Neurol., 2021, 11, 595532 CrossRef.
- H. Wesseling, W. Mair, M. Kumar, C. N. Schlaffner, S. Tang, P. Beerepoot, B. Fatou, A. J. Guise, L. Cheng, S. Takeda, J. Muntel, M. S. Rotunno, S. Dujardin, P. Davies, K. S. Kosik, B. L. Miller, S. Berretta, J. C. Hedreen, L. T. Grinberg, W. W. Seeley, B. T. Hyman, H. Steen and J. A. Steen, Cell, 2020, 183, 1699 CrossRef CAS PubMed.
- S. Wegmann, J. Biernat and E. Mandelkow, Curr. Opin. Neurobiol., 2021, 69, 131 CrossRef CAS PubMed.
- S. Ahmadi, B. Wu, R. Song, S. Zhu, A. Simpson, D. J. Wilson and H. B. Kraatz, J. Inorg. Biochem., 2020, 205, 110987 CrossRef CAS PubMed.
- R. Brandt, N. I. Trushina and L. Bakota, Front. Neurol., 2020, 11, 590059 CrossRef PubMed.
- M. Sastre, C. W. Ritchie and N. Hajji, JSM Alzheimer's Dis. Relat. Dementia, 2015, 2(1), 1014 Search PubMed.
- K. P. Kepp, Chem. Rev., 2012, 112(10), 5193 CrossRef CAS PubMed.
- K. P. Kepp, Coord. Chem. Rev., 2017, 351, 127 CrossRef CAS.
- Y. Christen, Am. J. Clin. Nutr., 2000, 71(2), 6215 CrossRef PubMed.
- S.-M. Alavi Naini and N. Soussi-Yanicostas, Oxid. Med. Cell. Longevity, 2015, 2015, 151979 Search PubMed.
- S. Lee, Y. Liu and M. H. Lim, ACS Chem. Biol., 2013, 8(5), 856 CrossRef PubMed.
- Q.-F. Ma, Y.-M. Li, J.-T. Du, K. Kanazawa, T. Nemoto, H. Nakanishi and Y.-F. Zhao, Biopolymers, 2005, 79, 74 CrossRef CAS PubMed.
- B. K. Shin and S. Saxena, J. Phys. Chem. B, 2011, 115, 15067 CrossRef CAS PubMed.
- S. Martic, M. K. Rains and H. B. Kraatz, Anal. Biochem., 2013, 442, 130 CrossRef CAS.
- G. Di Natale, F. Bellia, M. F. M. Sciacca, T. Campagna and G. Pappalardo, Inorg. Chim. Acta, 2018, 472, 82 CrossRef.
- S. Ahmadi, S. Zhu, R. Sharma, B. Wu, R. Soong, R. Dutta Majumdar, D. J. Wilson, A. J. Simpson and H. B. Kraatz, ACS Omega, 2019, 5, 5356 CrossRef PubMed.
- M. Lukacs, G. Szunyog, A. Grenacs, N. Lihi, C. Kallay, G. Di Natale, T. Campagna, V. Lanza, G. Tabbi, G. Pappalardo, I. Sovago and K. Varnagy, ChemPlusChem, 2019, 84, 1697 CrossRef CAS.
- S. Ahmadi, S. Zhu, R. Sharma, D. J. Wilson and H. B. Kraatz, J. Inorg. Biochem., 2019, 194, 44 CrossRef CAS.
- C. Bacchella, S. Gentili, D. Bellotti, E. Quartieri, S. Draghi, M. C. Baratto, M. Remelli, D. Valensin, E. Monzani, S. Nicolis, L. Casella, M. Tegoni and S. Dell'Acqua, Inorg. Chem., 2020, 59, 274 CrossRef CAS.
- B.-D. Balogh, B. Szakacs, G. Di Natale, G. Tabbì, G. Pappalardo, I. Sovago and K. Varnagy, J. Inorg. Biochem., 2021, 217, 111358 CrossRef CAS.
- B.-D. Balogh, G. Szunyog, M. Lukács, B. Szakács, I. Sóvágó and K. Várnagy, Dalton Trans., 2021, 50, 14411 RSC.
- M. Amblard, J. A. Fehrentz, J. Martinez and G. Subra, Mol. Biotechnol., 2006, 33, 239 CrossRef CAS PubMed.
- H. M. Irving, M. G. Miles and L. D. Pettit, Anal. Chim. Acta, 1967, 38, 475 CrossRef CAS.
- P. Gans, A. Sabatini and A. Vacca, Talanta, 1996, 43, 1739 CrossRef CAS.
- K. M. Giannoulis, G. Z. Tsogas, D. L. Giokas and A. G. Vlessidis, Talanta, 2012, 99, 62 CrossRef CAS PubMed.
- R. Jastrzab, J. Inorg. Biochem., 2009, 103, 766 CrossRef CAS PubMed.
- B. Liu, Y. Tian, Q. Yu, Q. Li, W. Mu, Z. Tan, F. Wu, D. Wang and X. Li, J. Solution Chem., 2017, 46, 2281 CrossRef CAS.
- R. Jastrzab and L. Lomozik, J. Coord. Chem., 2009, 62, 710 CrossRef CAS.
- K. Zavitsanos, A. M. P. C. Nunes, G. Malandrinos, C. Kállay, I. Sóvágó, V. Magafa, P. Cordopatis and N. Hadjiliadis, Dalton Trans., 2008, 6179 RSC.
- T. Kowalik-Jankowska, A. Rajewska, K. Wiśniewska, Z. Grzonka and J. Jezierska, J. Inorg. Biochem., 2005, 99, 2282 CrossRef CAS PubMed.
- M. Mylonas, A. Krezel, J. C. Plakatouras, N. Hadjiliadis and W. Bal, J. Mol. Liq., 2005, 118, 119 CrossRef CAS.
- M. Mylonas, A. Krezel, J. C. Plakatouras, N. Hadjiliadis and W. Bal, Inorg. Chim. Acta, 2002, 399, 60 CrossRef.
- G. Malandrinos and N. Hadjiliadis, Coord. Chem. Rev., 2014, 262, 55 CrossRef CAS.
- D. Grasso, G. Grasso, V. Guantieri, G. Impellizzeri, C. La Rosa, D. Milardi, G. Micera, K. Osz, G. Pappalardo, E. Rizzarelli, D. Sanna and I. Sovago, Chem. – Eur. J., 2006, 12, 537 CrossRef.
- G. Di Natale, G. Grasso, G. Impellizzeri, D. La Mendola, G. Micera, N. Mihala, Z. Nagy, K. Osz, G. Pappalardo, V. Rigo, E. Rizzarelli, D. Sanna and I. Sovago, Inorg. Chem., 2005, 44(20), 7214 CrossRef CAS PubMed.
- E. Prenesti, P. G. Daniele, M. Prencipe and G. Ostacoli, Polyhedron, 1999, 18, 3233 CrossRef CAS.
- T. Kowalik-Jankowska, M. Ruta-Dolejsz, K. Wisniewska, L. Lankiewicz and H. Kozlowski, J. Chem. Soc., Dalton Trans., 2000, 4511 RSC.
- T. Kowalik-Jankowska, M. Ruta, K. Wisniewska and L. Lankiewicz, J. Inorg. Biochem., 2001, 86, 535 CrossRef CAS.
- V. Joszai, Z. Nagy, K. Osz, D. Sanna, G. Natale, D. Mendola, G. Pappalardo, E. Rizzarelli and I. Sovago, J. Inorg. Biochem., 2006, 100, 1399 CrossRef CAS.
- T. Kowalik-Jankowska, A. Rajewska, E. Jankowska and Z. Grzonka, Dalton Trans., 2006, 5068 RSC.
- M. Orfei, M. C. Alcaro, G. Marcon, M. Chelli, M. Ginanneschi, H. Kozlowski, J. Brasun and L. Messori, J. Inorg. Biochem., 2003, 97, 299 CrossRef CAS.
- T. Karavelas, M. Mylonas, G. Malandrinos, J. C. Plakatouras, N. Hadjiliadis, P. Mlynarz and H. Kozlowski, J. Inorg. Biochem., 2005, 99, 606 CrossRef CAS PubMed.
- I. Bertini, C. Luchinat, G. Parigi and R. Pierattelli, ChemBioChem, 2005, 6, 1536 CrossRef CAS.
- M.-F. Peana, S. Medici, A. Ledda, V.-M. Nurchi and M.-A. Zoroddu, Sci. World J., 2014, 2014, 656201 Search PubMed.
- O. Yamauchi, A. Odani and M. Takani, J. Chem. Soc., Dalton Trans., 2002, 3411 RSC.
- M. A. Zoroddu, T. Kowalik-Jankowska, H. Kozlowski, K. Salnikow and M. Costa, J. Inorg. Biochem., 2001, 85, 47 CrossRef.
- M. A. Zoroddu, P. Massimiliano, T. Kowalik-Jankowska, H. Kozlowski and M. Costa, J. Chem. Soc., Dalton Trans., 2002, 458 RSC.
- C. Conato, W. Kamysz, H. Kozlowski, M. Luczkowski, Z. Mackiewicz, P. Mlynarz, M. Remelli, D. Valensin and G. Valensin, J. Chem. Soc., Dalton Trans., 2002, 3939 RSC.
- J. Peisach and W. E. Blumberg, Arch. Biochem. Biophys., 1974, 165, 691 CrossRef CAS.
- E. Prenesti, P.-G. Daniele, S. Berto and S. Toso, Polyhedron, 2006, 25, 2815 CrossRef CAS.
- U. Sakaguchi and A. W. Addison, J. Chem. Soc., Dalton Trans., 1979, 600 RSC.
- I. Sóvágó, D. Sanna, A. Dessí, K. Várnagy and G. Micera, J. Inorg. Biochem., 1996, 63(2), 99 CrossRef.
- P. G. Daniele, E. Prenesti and G. Ostacoli, J. Chem. Soc., Dalton Trans., 1996, 3269 RSC.
- M. A. Zoroddu, T. Kowalik-Jankowska, H. Kozlowski, H. Molinari, K. Salnikow, L. Broday and M. Costa, Biochim. Biophys. Acta, 2000, 1475, 163 CrossRef CAS PubMed.
- L. D. Pettit, S. Pyburn, W. Bal, H. Kozlowski and M. Bataille, J. Chem. Soc., Dalton Trans., 1990, 3565 RSC.
- K. Varnagy, J. Szado, I. Sovago, G. Malandrinos, N. Hadjiliadis, D. Sanna and G. Micera, J. Chem. Soc., Dalton Trans., 2000, 467 RSC.
- N. Camerman, A. Camerman and B. Sarkar, Can. J. Chem., 1976, 54, 1309 CrossRef CAS.
- M. Klewpatinond and J. H. Viles, FEBS Lett., 2007, 581, 1430 CrossRef CAS.
- H. F. Stanyon, X. Cong, Y. Chen, N. Shahidullah, G. Rossetti, J. Dreyer, G. Papamokos, P. Carloni and J. H. Viles, FEBS J., 2014, 281, 3945 CrossRef CAS.
- P. G. Daniele, E. Prenesti, O. Zerbinati, R. Aigotti and G. Ostacoli, Spectrochim. Acta, Part A, 1993, 49A(9), 1373 CrossRef CAS.
- H. Sigel and R. B. Martin, Chem. Rev., 1982, 82(4), 385 CrossRef CAS.
- R. J. Sundberg and R. B. Martin, Chem. Rev., 1974, 74(4), 471 CrossRef CAS.
- H. Aiba, A. Yokoyama and H. Tanaka, Bull. Chem. Soc. Jpn., 1974, 47(6), 1437 CrossRef CAS.
- P. J. Morris and R. B. Martin, J. Am. Chem. Soc., 1970, 92(6), 1543 CrossRef CAS.
- K. Panagiotou, M. Panagopoulou, T. Karavelas, V. Dokorou, A. Hagarman, J. Soffer, R. Schweitzer-Stenner, G. Malandrinos and N. Hadjiliadis, Bioinorg. Chem. Appl., 2008, 2008, 257038 Search PubMed.
- K. Zavitsanos, A. M. Nunes, G. Malandrinos and N. Hadjiliadis, J. Inorg. Biochem., 2011, 105, 102 CrossRef CAS.
- D. Sanna, G. Micera, C. Kállay, V. Rigó and I. Sóvágo, Dalton Trans., 2004, 2702 RSC.
- M. A. Zoroddu, T. Kowalik-Jankowska, S. Medici, M. Peana and H. Kozlowski, Dalton Trans., 2008, 6127 RSC.
- M. Łuczkowski, K. Wisniewska, L. Łankiewicz and H. Kozlowski, J. Chem. Soc., Dalton Trans., 2002, 2266 RSC.
- F. Guo, V. Stanevich, N. Wlodarchak, R. Sengupta, L. Jiang, K. A. Satyshur and Y. Xing, Cell Res., 2014, 24, 190 CrossRef CAS PubMed.
- M. Zhang, S. D. Yogesha, J. E. Mayfield, G. N. Gill and Y. Zhang, FEBS J., 2013, 280(19), 4739 CrossRef CAS.
- U.-S. Cho and W. Xu, Nature, 2007, 445, 53 CrossRef CAS PubMed.
- T. P. Silverstein, Front. Mol. Biosci., 2021, 8, 764099 CrossRef CAS.
- L. Schwartz, S. Pere, M. Jolicoeur and J. da Veiga Moreira, Biogerontology, 2020, 21, 683 CrossRef CAS.
- K. Aoyama, Int. J. Mol. Sci., 2021, 22, 5010 CrossRef CAS PubMed.
- I. Sovago and K. Osz, Dalton Trans., 2006, 32, 3841 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: 1 and 2D NMR, EPR and CD spectra, and NMR data for both peptides. See DOI: https://doi.org/10.1039/d2dt02838g |
| This journal is © The Royal Society of Chemistry 2023 |