
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly dispersed Rh single atoms over graphitic carbon nitride as a robust catalyst for the hydroformylation reaction†
Lole
Jurado
 a,
Jerome
Esvan
b,
Ligia A.
Luque-Álvarez
c,
Luis F.
Bobadilla
c,
José A.
Odriozola
c,
Sergio
Posada-Pérez
d,
Albert
Poater
d,
Aleix
Comas-Vives
ef and
M. Rosa
Axet
*a
a,
Jerome
Esvan
b,
Ligia A.
Luque-Álvarez
c,
Luis F.
Bobadilla
c,
José A.
Odriozola
c,
Sergio
Posada-Pérez
d,
Albert
Poater
d,
Aleix
Comas-Vives
ef and
M. Rosa
Axet
*a
aCNRS, LCC (Laboratoire de Chimie de Coordination), UPS, INPT, Université de Toulouse, 205 Route de Narbonne, F-31077 Toulouse Cedex 4, France. E-mail: rosa.axet@lcc-toulouse.fr
bCIRIMAT, CNRS-INPT-UPS, Université de Toulouse, 4 Allée Emile Monso, 31030 Toulouse, France
cDepartamento de Química Inorgánica e Instituto de Ciencia de Materiales de Sevilla, Centro Mixto CSIC-Universidad de Sevilla, Av. Américo Vespucio 49, 41092 Sevilla, Spain
dInstitut de Química Computacional i Catàlisi and Departament de Química, Universitat de Girona, c/ Maria Aurèlia Capmany 69, 17003 Girona, Catalonia, Spain
eInstitute of Materials Chemistry, TU Wien, 1060 Vienna, Austria
fDepartament de Química, Universitat Autònoma de Barcelona, 08193 Cerdanyola del Vallès, Catalonia, Spain
First published on 12th January 2023
Abstract
Rhodium-catalysed hydroformylation, effective tool in bulk and fine-chemical synthesis, predominantly uses soluble metal complexes. For that reason, the metal leaching and the catalyst recycling are still the major drawbacks of this process. Single-atom catalysts have emerged as a powerful tool to combine the advantages of both homogeneous and heterogeneous catalysts. Since using an appropriate support material is key to create stable, finely dispersed, single-atom catalysts, here we show that Rh atoms anchored on graphitic carbon nitride are robust catalysts for the hydroformylation reaction of styrene.
Introduction
Hydroformylation is an important catalysed process which produces aldehydes from olefins in the presence of syngas.1 Rh complexes modified with phosphines or phosphites are generally used as homogeneous catalysts, in both academic and industrial applications.2 In bulk industrial processes based on homogeneous catalysts, the catalyst recycling is usually implemented by phase separation, and leaching of Rh species is well managed. In order to provide an alternative solution to catalyst recycling, several strategies have been explored,3 including supported4 and non-supported5,6 clusters, supported/anchored monoatomic complexes,7 complexes on phosphine-functionalized polymers,8 and single-atom catalysts (SACs).9–17 SACs have emerged as a new approach that allows combining the use of isolated metal atoms, as in homogeneous catalysis, highly dispersed on a support, enabling recyclability as in heterogeneous catalysis.18 It has been shown that single-atoms can be solidly bonded to the support by strong interactions, thus form rather robust catalysts.19 Under hydroformylation conditions is very common to produce unmodified hydridorhodium carbonyl species from rhodium complexes,2 or clusters,5 and most probably from SACs.13,15,16 As these species are highly active catalysts for hydroformylation, to avoid leaching issues due to the formation of soluble species, they must display strong metal–support interactions. Recently, rhodium SACs have been applied as catalysts for hydroformylation reactions.9,12 Robust rhodium based SACs for hydroformylation reaction described so far involve mainly the use of oxides as supports. For instance, CoO-supported Rh single-atom catalyst (Rh/CoO)9 shows a high activity in the hydroformylation of propene at 373 K under 30 bar of syngas. 0.2% Rh/CoO displays a high selectivity towards linear products, up to 94.4% for butyraldehyde. Using high rhodium loaded catalysts (1% and 4.8% Rh/CoO), in which clusters were observed, performed worst in activity compared to the low loaded catalyst leading to a considerable decrease in the linear to branched (l/b) ratio. Recycling tests showing a rather robust catalyst. Rh–ReOx atomically dispersed on Al2O3 was used as catalysts for the hydroformylation of ethylene.12 The main products from this process were ethane and propanal; the activity and selectivity being governed by the coverage of ReOx and Rh, authors correlating this to the vicinity between Rh and ReOx species. Rh SACs supported on ZnO nanowires demonstrated to be competitive catalysts for the hydroformylation reaction of styrene, and other substrates.10 Several Rh loadings (0.3%, 0.03%,and 0.006%) were prepared by simple impregnation of RhCl3·3H2O over ZnO, followed by a treatment under H2 at 473 K. High loaded 0.3% Rh/ZnO catalysts displayed low catalytic performances in comparison to the other catalysts, which could be attributed to the presence of clusters in this sample. On the other hand, purely SACs 0.03% and 0.006% loaded materials displayed high efficiency for the hydroformylation of styrene at 373 K and 16 bar of CO/H2 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in which the corresponding aldehydes were synthesised with 99% of selectivity in an l/b ratio of 1. Recycling test demonstrating no Rh leaching. Recently, Rh SACs were studied both theoretically and experimentally for the hydroformylation of olefins.17 The calculated stability of the adsorption of HRh(CO)2 species onto ZnO, CeO2, and MgO correlated well with the catalytic activity and stability of the synthesised materials. Thus, Rh/ZnO and Rh/CeO2 displayed the highest adsorption energies for the hydridocarbonyl species and were remarkably efficient for the hydroformylation reaction. In contrast, atomically dispersed Rh on MgO, with lower affinity for the support, displayed the lowest activity. CeO2-supported Rh SACs are versatile since they catalyse the styrene hydroformylation and its derivatives by coupling hydroformylation with the low-temperature water-gas shift reaction. Overall, promoting a high selectivity toward the linear products.13 Coordinated Rh species on phosphorus functionalised nanodiamonds have been described as catalysts for the hydroformylation of styrene and related compounds.14 The presence of the phosphorus moieties allows to obtain high regioselectivity towards the branched aldehyde, b/l ratio up to 13 for styrene, with a TOF of 95 h−1. Porous organic polymers containing phosphorus ligands are able to accommodate Rh single atoms, which are active catalysts for the hydroformylation reaction.11 The presence of the phosphorus allowing to modulate the regioselectivity of the reaction. The nature of the ionic liquids has been also used on Rh based SAC in order to have a control over the regioselectivity in the HF reaction.15,16 Ru based SACs displaying the presence of clusters of different sizes have been also explored as hydroformylation catalysts, needing, however, harsher reaction conditions than the ones used with Rh based catalysts, i.e. 423 K and 40 bar of CO/H2 1:1.20
:1) in which the corresponding aldehydes were synthesised with 99% of selectivity in an l/b ratio of 1. Recycling test demonstrating no Rh leaching. Recently, Rh SACs were studied both theoretically and experimentally for the hydroformylation of olefins.17 The calculated stability of the adsorption of HRh(CO)2 species onto ZnO, CeO2, and MgO correlated well with the catalytic activity and stability of the synthesised materials. Thus, Rh/ZnO and Rh/CeO2 displayed the highest adsorption energies for the hydridocarbonyl species and were remarkably efficient for the hydroformylation reaction. In contrast, atomically dispersed Rh on MgO, with lower affinity for the support, displayed the lowest activity. CeO2-supported Rh SACs are versatile since they catalyse the styrene hydroformylation and its derivatives by coupling hydroformylation with the low-temperature water-gas shift reaction. Overall, promoting a high selectivity toward the linear products.13 Coordinated Rh species on phosphorus functionalised nanodiamonds have been described as catalysts for the hydroformylation of styrene and related compounds.14 The presence of the phosphorus moieties allows to obtain high regioselectivity towards the branched aldehyde, b/l ratio up to 13 for styrene, with a TOF of 95 h−1. Porous organic polymers containing phosphorus ligands are able to accommodate Rh single atoms, which are active catalysts for the hydroformylation reaction.11 The presence of the phosphorus allowing to modulate the regioselectivity of the reaction. The nature of the ionic liquids has been also used on Rh based SAC in order to have a control over the regioselectivity in the HF reaction.15,16 Ru based SACs displaying the presence of clusters of different sizes have been also explored as hydroformylation catalysts, needing, however, harsher reaction conditions than the ones used with Rh based catalysts, i.e. 423 K and 40 bar of CO/H2 1:1.20
Graphitic carbon nitride (g-C3N4 or ECN) has demonstrated to be a promising support for SACs, being able to host single atoms in its s-triazine rings, as well as integrate other heteroatoms.21–25 The stability of metallic atoms in this support makes it an excellent candidate to support atomically dispersed Rh. Applying the resulting material as an olefin hydroformylation catalyst could prevent the formation of clusters or/and nanoparticles harmful to the activity of the reaction. In this work, we have synthesised Rh/ECN using two straightforward procedures to obtain highly dispersed Rh atoms in ECN. Styrene was hydroformylated using these SACs, displaying high chemoselectivity towards the aldehyde with an l/b of 1.5. Density functional calculations further support the experimental results, in terms of the speciation of Rh species present on the SACs and the stability of these materials under hydroformylation conditions.
Experimental section
Synthesis of [Rh(η3-C3H5)3]
[Rh(η3-C3H5)3] was synthesized by following the procedure previously reported by Fryzuk and Piers.26 An excess of allylmagnesium chloride (25 mL) was added dropwise into a cold (263 K) solution of RhCl3·3H2O (1.13 g) in THF (90 mL), turning from purple to yellowish-brown color. The solution was kept in the cold bath for 1 h. Later, the solution was warmed up to room temperature and vigorously stirred overnight. The THF was removed under reduced pressure and the Rh complex was extracted from the precipitated salts with pentane, resulting in a yellowish solution. The extraction was repeated at least three times and the yellowish extracts were filtered through celite. pentane was fully evaporated under reduced pressure and the resulting yellow powder was sublimated at 333 K under static vacuum for 2 h, yielding 320 mg (32% yield) of yellow crystals of [Rh(η3-C3H5)3].Synthesis of exfoliated carbon nitride (ECN)
ECN was synthesised following a modified procedure described elsewhere.24 A bulk carbon nitride was synthesized by pyrolyzing dicyandiamide (4 g) in a crucible partially covered at 823 K for 4 h under nitrogen flow (200 mL min−1) using a heating ramp of 2.3 K min−1. The bulk carbon nitride was successively submitted to a calcination in static air at 773 K for 5 h, with a heating ramp of 5 K min−1, giving rise to a yellow powder. The resulting ECN was washed with distilled water, ethanol, and pentane, and dried overnight at 383 K.Synthesis of Rh/ECN
Rh/ECN(0.1) was carried out through wetness impregnation using [Rh(η3-C3H5)3] complex as Rh precursor and ECN as support. 1 g of ECN was dispersed into THF (40 mL) and sonicated for 1 h at room temperature. [Rh(η3-C3H5)3] (9 mg) was added to the dispersion and kept overnight under vigorous stirring at room temperature. The yellow powder was filtered under argon using a cannula, subsequently washed three times with pentane and lastly dried under reduced pressure overnight. Yield: 900 mg. ICP anal.: 0.1% Rh.Synthesis of Rh-ECN
Rh-ECN catalysts were synthesised by direct incorporation of Rh during ECN synthesis using RhCl3·3H2O as Rh precursor. RhCl3·3H2O was dissolved in distilled water (50 mL) at room temperature, giving rise to a purple/red solution. Subsequently, the dicyandiamide (3 g) was added and the resulting dispersion was heated up to 333 K under vigorous stirring in order to fully dissolve the dicyandiamide, after that the solution was kept for 2 h at this temperature under stirring. The excess of water was removed by increasing the temperature up to 393 K. The resulting orange solid was dried overnight at 383 K in an oven. The solid was pyrolyzed in a crucible partially covered at 823 K for 4 h under nitrogen flow (200 mL min−1) using a heating ramp of 2.3 K min−1. The resulting materials were washed with distilled water, ethanol, and pentane, and dried overnight at 383 K. Rh-ECN(0.1): RhCl3·3H2O 3.3 mg; dark yellow powder; yield: 1.49 g. ICP anal.: 0.1% Rh. Rh-ECN(0.4): RhCl3·3H2O 33.7 mg; dark yellow powder; yield: 1.42 mg. ICP anal.: 0.4% Rh. Rh-ECN(6.5): RhCl3·3H2O 367.0 mg; brown powder; yield: 1.71 g. ICP anal.: 6.5% Rh.The hydroformylation of styrene was carried out in a 42 mL stainless steel high-pressure batch Top Industrie reactor. In a typical experiment, a mixture of the catalyst (0.002 mmol of metal), octane (0.25 mmol) as internal standard, and styrene (1 mmol) as substrate in 5 mL of THF were loaded into the autoclave inside the glovebox. The autoclave was purged three times using syngas (CO/H2 = 1) to remove the inert atmosphere. The reactor was fed with the syngas mixture up to 5 bar of pressure, heated up to the targeted reaction temperature and then the total pressure was fitted up to 20 bar. The stirring rate was fixed at 1200 rpm. Samples of the reaction mixture were taken at different time intervals and quantitative analyses of the reaction mixtures were performed via GC using calibration solutions of commercially available products. Recycling tests were performed following the same procedure using: 171.7 mg of catalyst (0.05 mmol of metal), octane (0.25 mmol) as internal standard, and styrene (0.6 mmol) as substrate in 5 mL of THF, 20 bar of syngas (CO/H2 = 1), 343 K, stirring rate of 1200 rpm. After 20 h of reaction the autoclave was cooled down and depressurized. Inside a glovebox the reactor was opened, and 10 ml of pentane were added to the vessel. The catalyst suspension was centrifugated. The solution was kept for GC and ICP analyses and the catalyst reused for a new catalytic reaction. The procedure was repeated 4 times. The homogeneous hydroformylation of styrene using [Rh(η3-C3H5)3] as metal precursor was evaluated following the same procedure using: several Rh loadings (0.04, 0.09, 0.22, 0.31 and 0.44 mM of Rh), octane (0.25 mmol) as internal standard, and styrene (1 mmol) as substrate in 5 mL of THF, 20 bar of syngas (CO/H2 = 1), 343 K, stirring rate of 1200 rpm for 5 h. Quantitative analyses of the reaction mixtures were performed via GC.
Characterization and computational details are available in ESI.†
Results and discussion
The addition Rh over ECN was carried out straightforwardly through wetness impregnation using [Rh(η3-C3H5)3] complex as Rh precursor. The Rh complex was added to a previously sonicated dispersion of ECN24 in THF targeting a 0.1% Rh loading. After maintaining a vigorous stirring overnight the yellow powder was filtered and cleaned three times with pentane. The yellow powder, Rh/ECN(0.1), was further dried under reduced pressure. The detailed procedure is described in the experimental section and in the ESI.† The synthesized catalyst was characterized by transmission electron microscopy (TEM), X-ray diffraction (XRD), thermogravimetric analysis (TGA), X-ray photoelectron spectrometry (XPS), attenuated total reflection infrared (ATR-IR), inductively coupled plasma optical emission spectroscopy (ICP-OES), elemental analysis, and N2 physisorption.TEM images (Fig. 1a) and S1†) display the characteristic sheets of ECN, without evidencing the presence of any Rh nanoparticle.27 HAADF-STEM-EDX (high-angle annular dark field-scanning transmission electron-energy-dispersive X-ray spectroscopy) analyses in this sample confirmed a high dispersion of Rh atoms on ECN (Fig. 1b) and S1†).
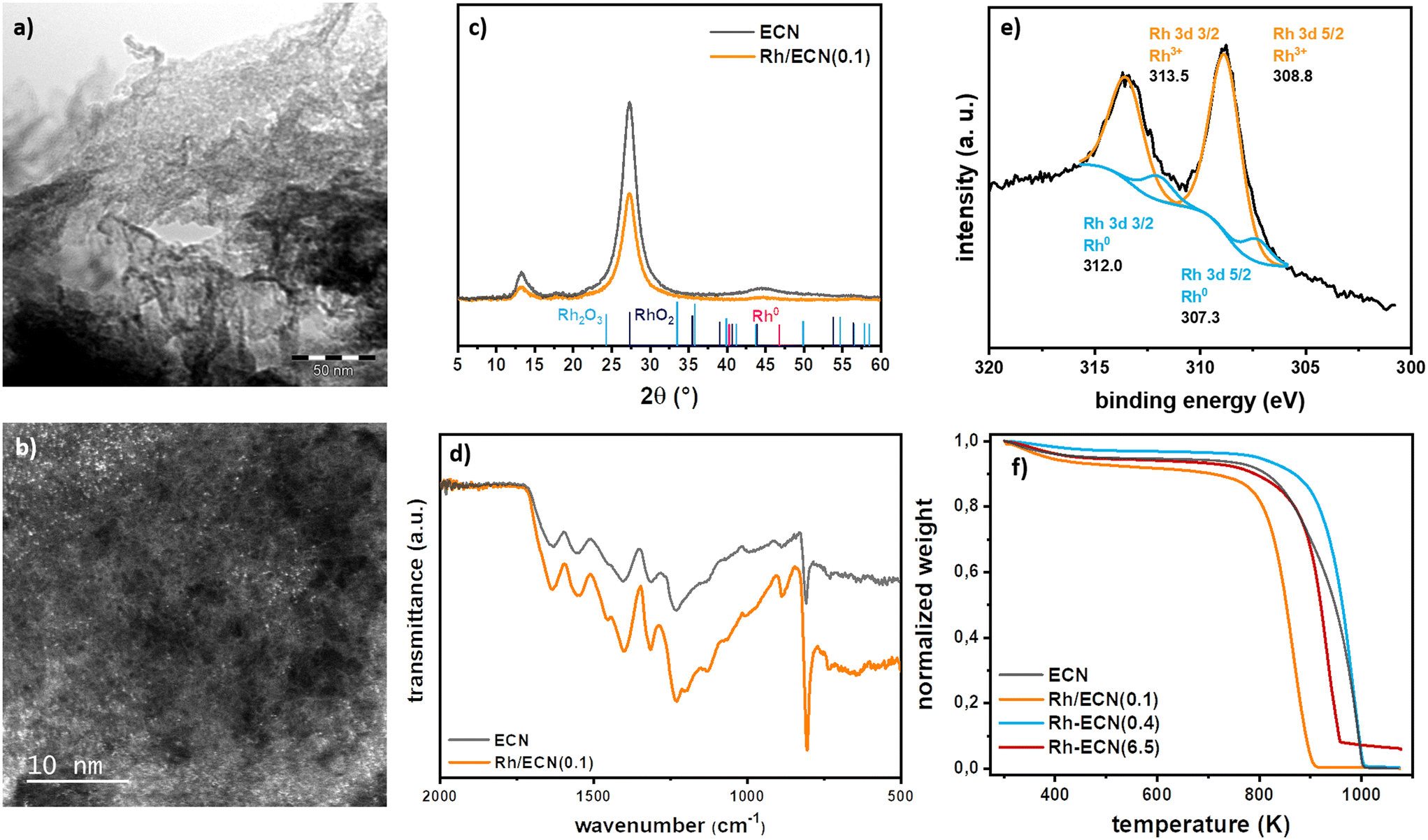 | ||
| Fig. 1 a) TEM and b) HAADF-STEM images of Rh/ECN(0.1) (scale bar = 50 and 10 nm, respectively); c) XRD diffractograms of ECN and Rh/ECN(0.1); d) ATR-IR spectra of ECN and Rh/ECN(0.1); e) high-resolution XPS spectra of Rh 3d Rh/ECN(0.1); f) thermogravimetric analysis curves under N2 for ECN, Rh/ECN(0.1), Rh-ECN(0.4), and Rh-ECN(6.5). | ||
The Rh loading was measured by ICP-EOS analysis and the C, N and H content by elemental analysis; Rh/ECN(0.1), displaying a Rh content of 0.1% and a C/N of 0.67 typical for ECN materials presenting residual uncondensed amino groups (Table S1†).28 The textural properties such as specific surface area (SSA) and pore volume (Vpore) are listed in Table S2.† The bare ECN exhibits a SSA of 5.4 m2 g−1 and a Vpore of 0.04 m3 g−1. No modifications of the textural properties with respect to the bare ECN are observed for Rh/ECN(0.1). Infrared spectra (Fig. 1d)) suggest that the presence of Rh does not cause any structural modification on the ECN support. Thus, several intense and broad bands are discerned in the 1700–1000 cm−1 range, ascribed to the stretching vibration of the CN heterocycles,21,22,29 where the bands at 1232, 1317 and 1401 cm−1 belong to the aromatic C–N stretching.21 The narrow band at 807 cm−1, characteristic of carbon nitride structure, is widely ascribed to the breathing mode of the tri-s-triazine rings. A weaker and broader band can be observed in the range between 3500–3000 cm−1, which might be associated to the N–H stretching mode of the residual uncondensed amino groups, in agreement with the C/N ratio determined by elemental analysis. TGA curves performed under N2 presented in Fig. 1e) show that bare ECN has a loss of mass at 948 K while for Rh/ECN(0.1) the decomposition starts at much lower temperature, 867 K, implying that ECN decomposition is promoted through the presence of Rh. X-ray diffractograms of Rh/ECN(0.1) and ECN are presented in Fig. 1c). A crystalline structure with two characteristic diffraction peaks is shown for the bare ECN. The broader peak with lower relative intensity at 2θ of 13.3°, related to the (001) reflection, is ascribed to the in-plane structural packing of the tri-s-triazine ring units. The main peak at 2θ of 27.3°, related to the reflection (002), is associated to the interplanar-stacking peak of the aromatic rings, indicating the formation of a layered structure for the ECN.30 The crystalline structure of ECN remained stable after Rh impregnation as the characteristic peaks of the support did not show any significative change. The crystallite size of the support (dsupport), the tri-s-triazine ring size (a) and the interlayer spacing (b) are listed in Table S2.† The Rh addition does not modify the crystallite size of the ECN, acquiring values of about 4 nm. The size of the tri-s-triazine ring and the interlayer spacing of the bare ECN are 6.615 Å and 3.261 Å, respectively. These values are in agreement with those found in literature for graphitic carbon nitrides.23,31 An increase in the size of the tri-s-triazine ring, from 6.615 Å to 6.660 Å, after Rh impregnation (Table S2,† ECN vs. Rh/ECN(0.1)) was observed. Furthermore, no additional diffraction peaks associated to Rh phases were detected, indicating the absence of Rh nanoparticles, as observed by TEM. Ex situ XPS under ultrahigh vacuum (UHV) analyses were performed on Rh/ECN(0.1). Survey spectra and high-resolution scan spectra together with the fitting peaks of the corresponding components are presented in Fig. 1e) and S3.† Table S3† summarizes the binding energies together with the concentration of the fitted peaks in atomic %. The XPS spectrum of Rh/ECN(0.1) displays the same characteristic peaks of ECN32 plus a new set of peaks of low intensity corresponding to Rh. The fitting of Rh peaks indicates a Rh3+ species as the main component with binding energies of 308.8 eV for Rh 3d5/2 and 313.5 for Rh 3d3/2, whereas traces of Rh0 are visible as well (307.1 and 311.8 for Rh 3d5/2 and Rh 3d3/2, respectively). The Rh3+ species is attributed to a Rh-centre coordinated to N atoms.33 DFT calculations were performed to find the lowest energy adsorption site of Rh on ECN, concluding that Rh atoms can be stably adsorbed in the surface (TOP) or in the subsurface (INT) (see below and Table S4 and Fig. S8 and S9†). The most stable adsorption sites depend on the Rh coverage; highly dispersed Rh (low coverage) prefers adsorbing on the TOP position occupying the hexagonal site bonded to two N atoms of the surface, while high coverages reveal subsurface adsorption. Considering the impregnation method has been used for the synthesis, and the XPS analysis suggesting Rh directly incorporates during the ECN synthesis, see below, Rh atoms are likely occupying a characteristic 6-fold cavity of ECN in Rh/ECN(0.1).
Rh/ECN(0.1) was synthesised through a straightforward methodology, which allowed obtaining the targeted materials, i.e. a low Rh content single-atom catalyst. A series of experiments was set up to explore these materials and create stronger metal–support interactions. We introduced Rh into ECN using several Rh/dicyandiamide ratios during the polymerization process. This methodology allowed incorporating a wide range of Rh content, from 0.03 to 6.50%, without forming Rh nanoparticles. Thus, we obtained uniformly dispersed Rh atoms on the support, as described below. The synthesis was carried out by pyrolyzing a RhCl3·3H2O/dicyandiamide solid mixture following the same pyrolyzing procedure described for the synthesis of ECN (see experimental section for details), without the exfoliation step. The incorporation of Rh onto the structure was readily visible by the characteristic change of colour of the material, from bright yellow for ECN, to an increasing brown colour while increasing the Rh loading (Fig. S10†). This phenomenon has already been described for other doped-ECN materials due to a change in the band gap energy.34 TEM analyses (Fig. 2a) and S11–S13†) show no formation of Rh nanoparticles and the characteristic formation of sheets of ECN for samples Rh-ECN(0.03), Rh-ECN(0.1), Rh-ECN(0.4) and Rh-ECN(6.5), in which the number of the samples indicates the Rh loading determined by ICP. HAADF-STEM images of Rh-ECN further confirmed the high Rh dispersion in the matrix (Fig. 2b) and S12 and S13†). Elemental analyses of the samples are summarized in Table S1.†
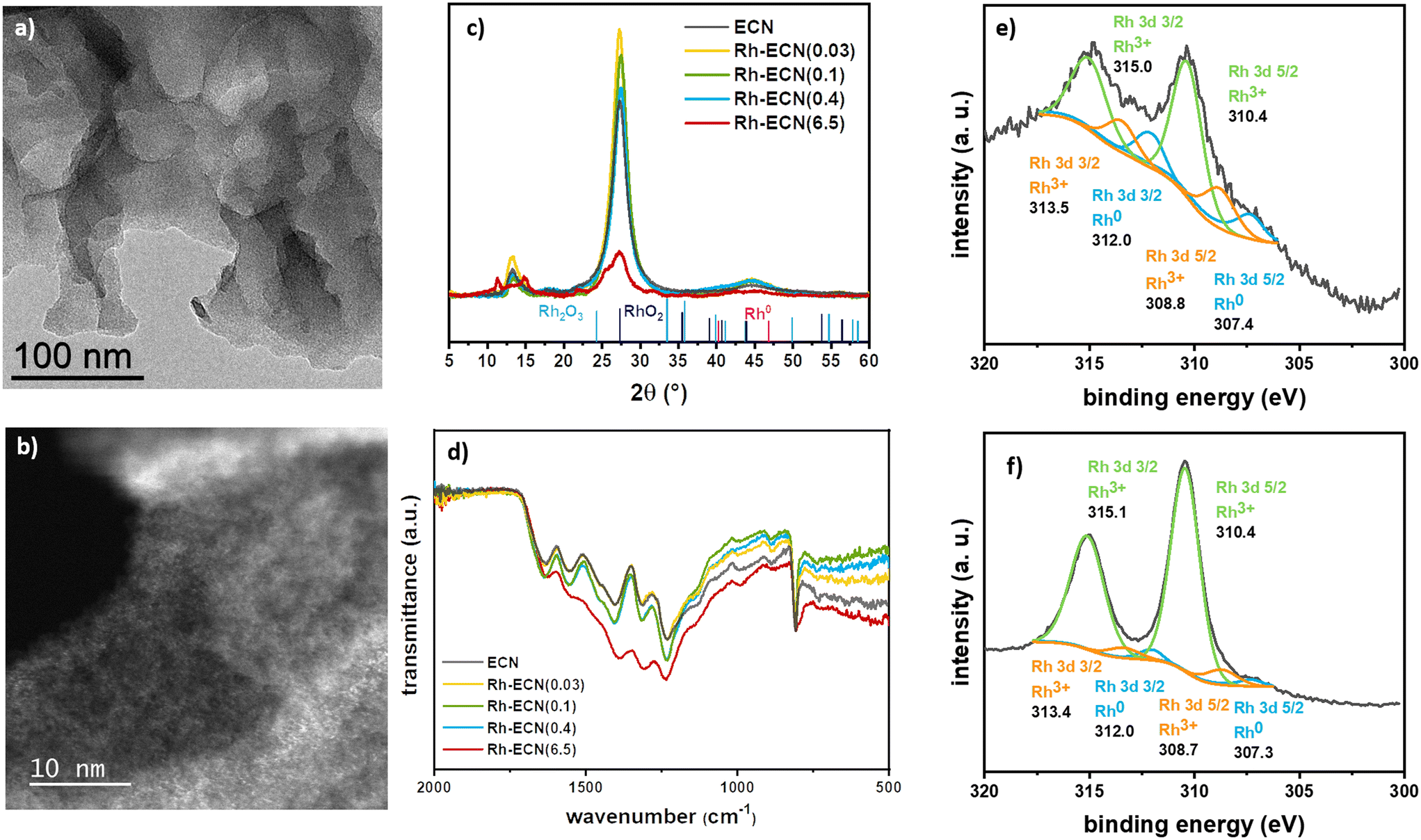 | ||
| Fig. 2 a) TEM and b) HAADF-STEM images of Rh-ECN(0.4) (scale bar = 50 and 10 nm, respectively); c) XRD diffractograms of ECN, Rh-ECN(0.03), Rh-ECN(0.1), Rh-ECN(0.4), and Rh-ECN(6.5); d) ATR-IR spectra of ECN, Rh-ECN(0.03), Rh-ECN(0.1), Rh-ECN(0.4), and Rh-ECN(6.5); e) and f) high-resolution XPS spectra of Rh 3d of Rh-ECN(0.4), and Rh-ECN(6.5), respectively. | ||
Fig. 2c) displays the XRD diffractograms of the incorporated Rh materials. In low Rh loading materials Rh-ECN(0.03), Rh-ECN(0.1), and Rh-ECN(0.4), the crystalline structure of ECN remained stable, suggesting that the ECN structure is well formed after the thermal treatment for the Rh incorporated ECN materials. In contrast to the impregnated sample, Rh/ECN(0.1), the peak position linked to the reticular plane (002) of the ECN structure are slightly shifted for the Rh-ECN(0.03), Rh-ECN(0.1), and Rh-ECN(0.4). The main peak slightly shifts to lower 2θ (27.2°) for the Rh/ECN(0.03) compared to the bare ECN (27.3°), which reveals the expansion of the interlayer stacking spacing (Fig. 2c)), suggesting that the Rh3+ cations are mainly located in the interlayers of the ECN. A similar shift to lower 2θ was found by Nam et al. for Rh incorporated ECN when using moderately low Rh loading (1 wt%).35 Contrary, a shift to higher 2θ (27.5°) is obtained for Rh-ECN(0.1) and Rh-ECN(0.4) compared to the bare ECN, indicating a contraction of the interlayer distance. A weaker and broader peak at 27.3° is observed for the material with the highest Rh content, Rh-ECN(6.5), proving a poorer crystallinity compared to the ECN (Fig. 2c)). Additionally, the peak at 13.3° no longer appears, suggesting the lack of periodicity in the layer when a high amount of Rh inserts into the ECN structure.36 Wang et al. also reported a loss of the crystallinity at high iron content associated to the host–guest interaction and to the inhibition of the polymeric condensation when an excess of iron was inserted.37 Nam et al. achieved the same results by increasing the amount of inserted Rh into the g-C3N4 structure.35 The additional peaks found for Rh-ECN(6.5) relate to some intermediate polymers formed during the ECN synthesis, as shown in Fig. S14,† indicating that the ECN polymerization is partially hampered at high Rh content following this procedure. For low Rh loading samples (0.03, 0.1, and 0.4 wt% Rh), diffraction peaks associated to Rh phases such as metallic rhodium, rhodium oxides, or rhodium carbides are not distinguished. It is worth mentioning the absence of diffraction peaks, ascribed to Rh related phases for Rh-ECN(6.5), despite the high Rh content (6.5 wt%). This absence points out that a highly dispersed Rh is achieved through direct incorporation. The crystallite size of the support (dsupport), the tri-s-triazine ring size (a), and the interlayer spacing (b) are listed in Table S2† for the synthesized materials. In agreement with the XRD results discussed above, a rather higher interplanar distance is displayed for the Rh-ECN(0.03) (3.274 Å) than for ECN (3.261 Å), attributed to the distance expansion by the Rh species incorporation between layers. Besides, same ring size as for Rh/ECN(0.1), larger than for ECN, is shown for Rh-ECN(0.03) (6.660 Å), revealing that part of the Rh could also be stabilized by the nitrogen pots. The incomplete insertion of carbon into ECN structure when incorporating a Rh loading of 0.1 and 0.4 wt% lead to a strong decrease of the interplanar distance, being lower for Rh-ECN(0.1) and Rh-ECN(0.4) (3.244 Å) than for ECN (3.261 Å). TGA analyses also point to a different structure between samples prepared by impregnation or pyrolysis (Fig. 1e) and S2†). The decomposition curves of samples with low Rh loading prepared by pyrolysis show that the temperature of Rh-ECN decomposition is delayed at high values compared to the bare ECN and Rh/ECN(0.1). These changes in the decomposition temperature could provide a notion of the metal–support interaction reached through each Rh catalyst synthetic pathway. The high temperature required for decomposing the ECN when Rh is incorporated suggests a stronger metal–support interaction. For the catalysts displaying higher Rh content, Rh-ECN(6.5) presents a lower decomposition temperature than ECN, which agrees with the presence of intermediate polymerisation products detected by XRD (Fig. S14†). FTIR spectra of Rh incorporated species (Fig. 2d)) do not show any significant change with respect IR spectrum of ECN. XPS spectra of the Rh 3d region for Rh-ECN(0.4) and Rh-ECN(6.5), displayed in Fig. 2e) and f), respectively, show that both samples contain similar Rh species, one of them distinct from the observed for the Rh impregnated sample, Rh/ECN(0.1). The main peaks are attributed to Rh3+ species, 310.0 and 315.1 for Rh 3d5/2 and Rh 3d3/2, respectively, which we have attributed to the intercalated Rh atoms. Moreover, a set of peaks at 308.8 and 313.5 eV for Rh 3d5/2 and Rh 3d3/2, respectively, are also visible, attributed to Rh3+ species on the 6-fold cavity of ECN. Traces of Rh0 are also observed. Survey and high-resolution scan spectra together with the fitting peaks of the corresponding components are presented in Fig. S4 and S5, and Table S3† summarizes binding energies.
In order to provide additional insights into the bonding of CO with specific Rh single-atom sites, in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) measurements were carried out first adsorbing CO and then purging with argon both at 50 °C. Fig. 3a) shows the temporal evolution of the IR spectra recorded during the CO adsorption and the subsequent Ar purging on Rh-ECN(6.5), Rh-ECN(0.4) and Rh/ECN(0.1) samples, from left to right. As can be noticed, both Rh-ECN(6.5) and Rh-ECN(0.4) catalysts hardly showed CO adsorbed species and only two lobules centered at 2143 cm−1 ascribed to gaseous CO were detected. These features disappeared totally after 5 min of Ar purging. By contrast, Rh/ECN(0.1) catalyst shows clearly the formation of two bands in the 2100–2000 cm−1 region that are typical of CO adsorbed species on Rh sites, Fig. 3a) and b). It can be stressed that these species remain still adsorbed after Ar purging showing that CO is chemisorbed on Rh sites.
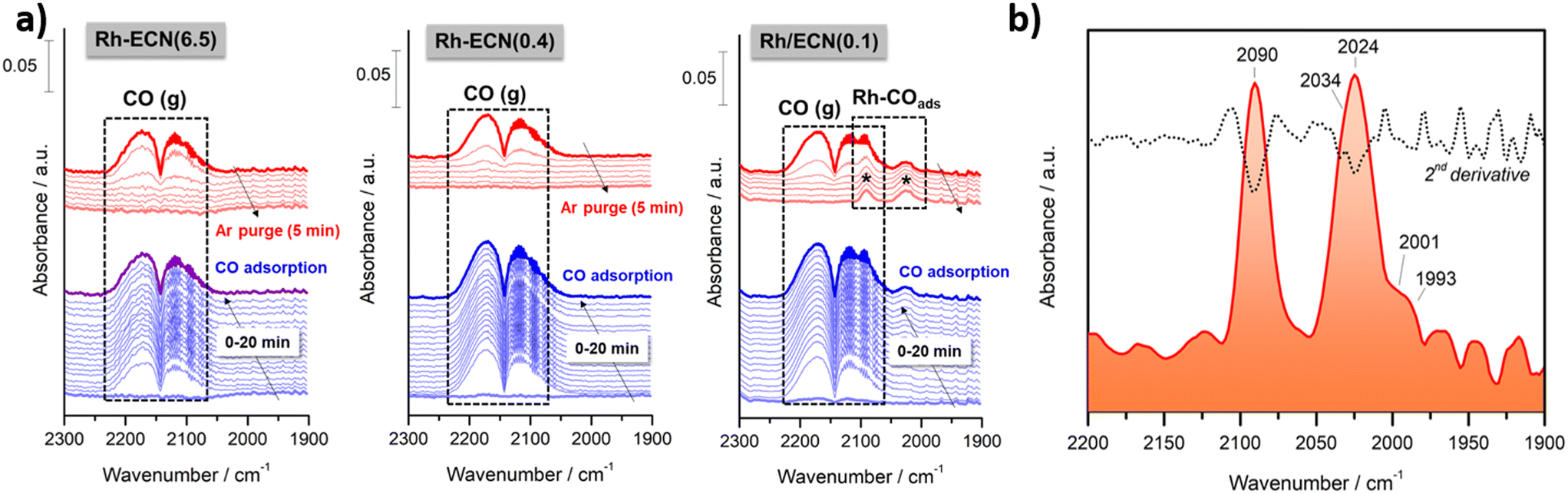 | ||
| Fig. 3 a) Evolution of in situ DRIFT spectra during CO adsorption (20 min) and subsequent Ar purging (5 min) at 50 °C on Rh-ECN(6.5), Rh-ECN(0.4) and Rh/ECN(0.1) samples. b) DRIFT spectra recorded on Rh/ECN(0.1) sample after CO adsorption (20 min) and Ar purging (5 min) at 50 °C. The inclusion of second derivative curve facilitates the identification of the bands. | ||
In order to identify the bands positions related to each CO adsorbed species on different Rh sites we performed the second derivative of the last spectrum recorded after Ar purging. As shown in Fig. 3b), five bands at 2090, 2034, 2024, 2001 and 1993 cm−1 were identified. As expected, the absence of bands in the 1900–1800 cm−1 suggests that Rh particles are very small and bridge carbonyls cannot be formed. According to literature, the features at 2090 and 2024 cm−1 correspond to the symmetric and asymmetric vibration of gem-dicarbonyl Rhδ+(CO)2 species in which atoms of rhodium are partially charged.13,14,38 On the other side, the band overlapped at 2034 cm−1 could be ascribed to CO linearly adsorbed on isolated Rh0 sites14,39 suggesting that very small particles of reduced rhodium are coexisting with oxidized rhodium atoms, which agrees with the traces of Rh0 detected by XPS. It cannot be fully discarded the presence of small clusters of rhodium, although the high intensity of bands related to gem-dicarbonyl Rhδ+(CO)2 species indicates that rhodium is fundamentally dispersed as single-atoms. We believe that these species are the main responsible of the catalytic activity favoring the activation of CO on the perimetral Rh/support interfacial sites. On the other hand, both bands at 2001 and 1993 cm−1 can be assigned to Rh(CO)3 species according to the DFT calculations performed (Table S5†). The frequency at 2001 cm−1 corresponds to the stretching of the CO ligand at 2.07 Å of Rh atom, whereas the stretching is asymmetric for the other 2 CO ligands at 1.87 Å (less intense). The frequency at 1993 cm−1 is the asymmetric vibration of the two closest CO ligands (intense) and the less intense stretching of the far CO. On the basis of these observations, we can conclude that isolated Rh single atoms are highly dispersed and exposed on the surface support bringing to light the catalytic activity showed by Rh/ECN(0.1) catalyst, which is discussed below.
Rh SACs were tested as catalysts for the hydroformylation of styrene. Results are summarized in Table 1. Impregnated Rh/ECN(0.1) performed well as catalyst for this reaction with less of 1% mol of Rh leaching with respect to the initial Rh amount and without any noticeable change on the morphology of the sample in the TEM images, as no nanoparticles were detected and the ECN sheets displayed same textural aspect (Fig. 4 and S16†).
| Catalyst | T (K) | Rh/styrene | Conv.b (%) | Selectivityb (%) | l/b | Rh leaching | |
|---|---|---|---|---|---|---|---|
| (mmol) | (%) | ||||||
| a Reaction conditions: 2 × 10−3 mmol of metal, 1 mmol of styrene, 0.25 mmol of octane (internal standard), 20 bar of syngas (CO/H2 = 1), 5 mL of THF. b Determined by GC using an internal standard technique. c 48h of reaction. d 0.4 mmol of styrene for Rh/styrene 1/200. n. d. non detected. | |||||||
| Rh/ECN(0.1) | 343 | 1/500 | 64 | 99 | 1.2 | 1.6 × 10−5 | 0.8 |
| 363 | 1/500 | 97 | 82 | 1.6 | 1.6 × 10−5 | 0.8 | |
| Rh-ECN(0.4) | 343 | 1/500 | 3 | 93 | 1.2 | n. d. | 0 |
| 363 | 1/500 | 7 | 94 | 1.4 | n. d. | 0 | |
| 363c | 1/500 | 13 | 87 | 1.6 | 8.5 × 10−7 | <0.1 | |
| 363d | 1/200 | 34 | 96 | 1.4 | 7.0 × 10−7 | <0.1 | |
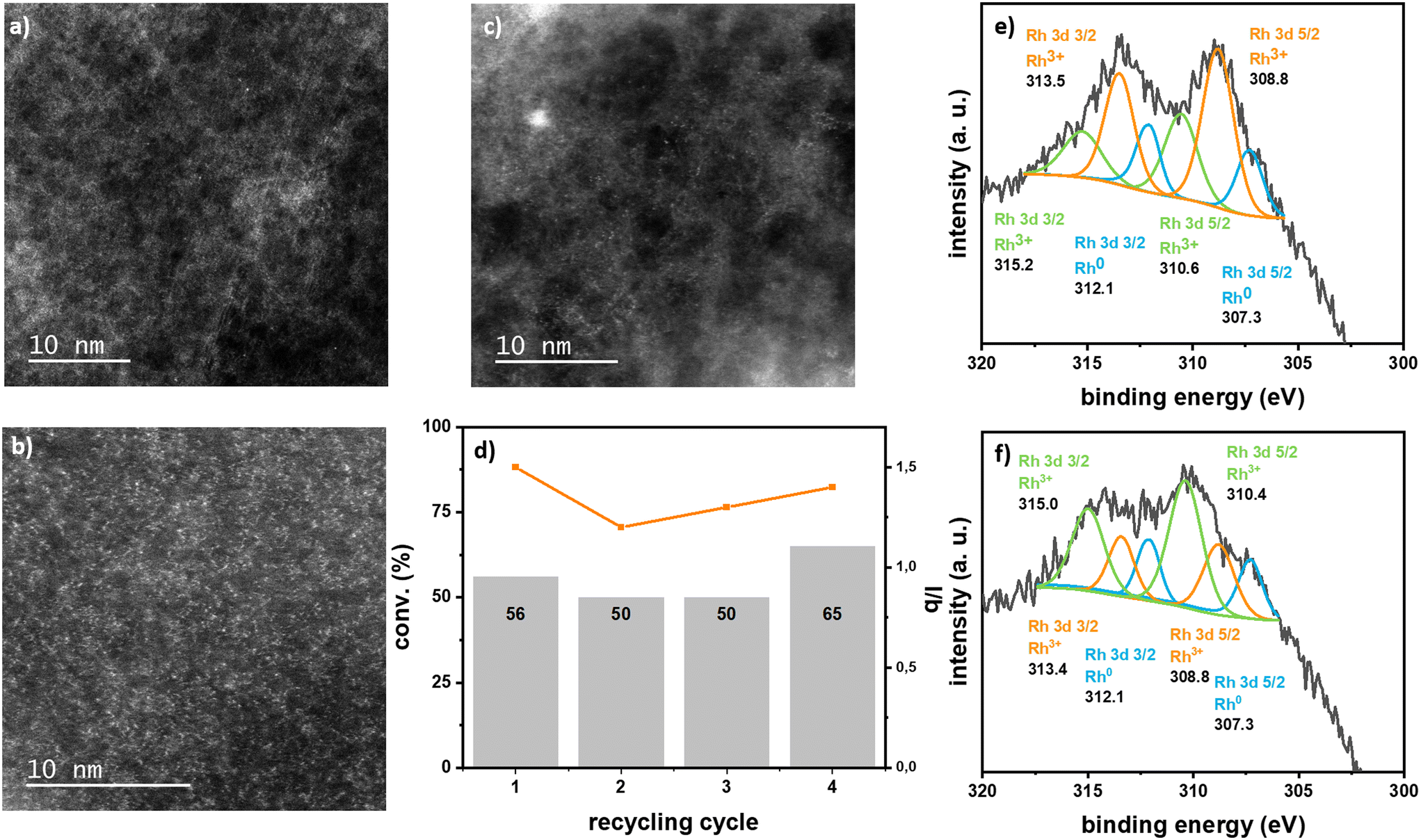 | ||
| Fig. 4 HAADF-STEM images of a) Rh/ECN(0.1) and b) Rh-ECN(0.4) after catalysis (scale bar = 10 nm); c) HAADF-STEM image of Rh/ECN(0.1) after 4 catalytic cycles; d) conversion (grey bars) and l/b ratio (orange line) of recycling tests performed with Rh/ECN(0.1) (5 × 10−2 mmol of metal, 0.6 mmol of styrene, 0.25 mmol of octane (internal standard), 20 bar of syngas (CO/H2 = 1), 5 mL of THF, 20 h, 343 K, 1200 rpm); e) and f) high-resolution XPS spectra of Rh 3d of Rh/ECN(0.1), and Rh-ECN(0.4) after catalysis, respectively. | ||
Rh/ECN(0.1) fully converted styrene after 24 h reaction at 363 K using a Rh/styrene ratio of 1/500 displaying a 82% selectivity towards the aldehydes with an l/b ratio of 1.6 and a TOF of 125 h−1 at 2 h of reaction. Lowering the temperature to 343 K did not significantly affect the Rh leaching, while it decreased the conversion to 64%, increased the aldehyde selectivity to 99% and slightly lowered the l/b ratio to 1.2. Bare ECN did not show any conversion of styrene when submitted to reaction at 343 K and 20 bar of syngas for 24 h, discarding any reactivity due to the presence of the support. In terms of activity, Rh/ECN(0.1) is comparable to other SACs applied to the styrene HF,17 yet other reported Rh-SACs displaying much higher TOFs,10,16 usually at expenses of selectivity. On the other hand, the l/b ratio displayed by Rh/ECN(0.1) is substantially higher than that previously reported when using this kind of catalysts or homogeneous systems,10,16,17 in which usually the l/b ratios are ranged between 0.1 to 1; only catalysts based in Rh-single atoms on phosphorus modified nanodiamonds,14 or Rh/CeO2 SAC in which hydrogen is generated in situ through water-gas shift reaction13 displayed higher production of the linear aldehyde, reaching l/b ratios of 3. Rh incorporated on ECN catalyst, Rh-ECN(0.4), displayed significantly lower activity. At 343 K only 3% of conversion was attained, and 7% at 363 K, after 24 h of reaction, in contrast with the activity displayed by Rh/ECN(0.1) (Table 1). The lower activity of Rh-ECN(0.4) is in line with its structure, as Rh is less accessible for catalysis, since it is mainly located in the interlayer space; and its diffusion is energetically unfavoured according to DFT calculations. It is worth mentioning that the presence of CO can promote the Rh displacement from the subsurface to the TOP site, although the diffusion energy barrier for Rh is at least of 0.57 eV (see Table S4†). On the other hand, the Rh leaching was almost completely suppressed. Longer reaction time and lowering the styrene content (Rh/styrene 1/200) allowed increasing the conversion, while keeping the Rh leaching significantly low. L/b ratio was close to the one observed with Rh/ECN(0.1) catalyst. As Rh soluble species are highly active in this reaction, a series of catalysed hydroformylation reactions were performed by using [Rh(η3-C3H5)3] as precatalyst at low concentrations (Fig. S15†). Considering the Rh leaching detected by ICP, ca. 5% of the total conversion could be due to the presence of Rh soluble species. It is worth noting that the l/b ratio for the homogeneous catalysis was 0.9, which is significatively different than the one observed for the heterogeneous systems, pointing to a different catalyst nature.
Analyses of the spent catalysts show that Rh supported on ECN are stable. TEM images clearly show the ECN sheets and HRTEM in HAADF-STEM display isolated Rh single atoms without any formation of nanoparticles (Fig. 3 and S16†). XPS analyses of the spent catalysts Rh/ECN(0.1) and Rh-ECN(0.4) show a clear evolution of the Rh3+ species from the fresh catalyst. After catalysis, the XPS spectrum of Rh/ECN(0.1) displays a new set of peaks in the Rh 3d region with binding energies of 310.6 and 315.2 eV for Rh 3d5/2 and Rh 3d3/2, respectively, while the peaks attributed to Rh3+ coordinated to N in the 6-fold cavity are still the main species present on the sample. The new peaks present the same binding energies as the species attributed to Rh3+ intercalated in the ECN layers, see discussion above and Fig. 2e) and f). These results agree with the DFT calculations, which predict the Rh3+ diffusion is possible from the 6-fold cavity towards the first interlayer of the ECN. Similarly, the XPS spectrum of Rh 3d of Rh-ECN(0.4) after catalysis shows a new set of peaks, 308.8 and 313.4 for Rh 3d5/2 and Rh 3d3/2, respectively, which correspond to the Rh3+ located onto the 6-fold cavity, thus suggesting that the diffusion of Rh can take place in both directions. Furthermore, recycling tests suggest a highly stable catalyst (see Fig. 4d)), as the conversion remains stable for four cycles, and no Rh leaching was detected in the solution by ICP analyses. HRTEM in HAADF-STEM shows mainly Rh isolated atoms on the material, and barely a few Rh nanoparticles after four catalytic runs (Fig. S16†). This observation could explain the slight increase in activity in the last catalytic run of the recycling test, as it has been demonstrated that Rh nanoparticles can produce Rh soluble species highly active in hydroformylation.5
Then, we evaluated the stability of Rh SACs under hydroformylation conditions at the computational level since single Rh atom desorption, and leaching are critical for this reaction. To assess the catalyst stability, the formation energy of the supported Rh species has been computed referenced to the lowest energy Rh/ECN configuration at low θRh (1/24) and the Gibbs energy of CO and H2 molecules in the gas phase at 363 K (see ESI† for further details). The Rh/ECN with the Rh placed on the TOP site was selected because it is the lowest energy configuration at low coverage regime. Rh deposition at the subsurface is predicted stable at higher metal coverage, although at hydroformylation reaction conditions the Rh is displaced upwards (with an energy cost) from the subsurface to the TOP position due to the interaction with CO molecule, being this position the most relevant for the catalytic purpose.
Table 2 collects the formation Gibbs energy for the different Rh species containing both CO and H coordinated to the metal center, which are likely to be active species during the hydroformylation reaction.2,17 The related structures are depicted in Fig. S17.† A previous computational study shows that the H1Rh(CO)2 complex is the most stable when supported on MgO surfaces.17 DFT calculations confirm one configuration, H1Rh(CO)4/ECN, as the most favored Rh complex, while three other systems, namely H1Rh(CO)3/ECN, Rh(CO)4/ECN, and Rh(CO)3/ECN, are between 0.30 and 0.33 eV higher in energy. It is observed that the adsorption of CO and H ligands to the metal center promotes the Rh displacement upwards with respect to the ECN support. The more ligands anchored to the metal center, the higher the Rh displacement, leading to the Rh placed on top of the surface atoms instead of being embedded on the support. Nevertheless, the Rh–N bond length only increases ∼0.1–0.3 Å for complexes with three or fewer carbonyl ligands (Table 2). In these scenarios, the Rh metal center is still formally chemisorbed on the ECN support. However, upon successive CO coordination, the binding of the Rh–CO ligand is energetically favored, reducing the metal–support interactions.
| System | ΔGads (eV) | d (Å) Rh–N |
|---|---|---|
| Rh/ECN | 0.00 | 1.98 |
| Rh(CO)/ECN | −1.47 | 2.02 |
| Rh(CO)2/ECN | −2.65 | 2.14 |
| Rh(CO)3/ECN | −2.85 | 2.16 |
| Rh(CO)4/ECN | −2.87 | 2.86 |
| H1Rh(CO)/ECN | −1.93 | 2.15 |
| H1Rh(CO)2/ECN | −2.26 | 2.25 |
| H1Rh(CO)3/ECN | −2.84 | 2.29 |
| H1Rh(CO)4/ECN | −3.17 | 4.35 |
| H1Rh(CO)4 + ECN | −4.01 | — |
| H1Rh(CO)(C2H4)/ECN | −2.39 | 2.21 |
| H1Rh(CO)2(C2H4)/ECN | −2.93 | 2.31 |
As shown in Fig. 5a) for H1Rh(CO)3/ECN, in this structure Rh is not embedded in the support but Rh remains coordinated to the N surface atoms. The formation of HxRh(CO)y (x = 0, 1, y = 1–3) complexes does increase the Rh single atom distance from the ECN surface. Our simulations show that the insertion of the fourth CO ligand is thermodynamically favorable, being the H1Rh(CO)4/ECN system 0.33 eV lower in energy with respect to H1Rh(CO)3/ECN. The fifth coordination site around Rh might be occupied by the support or by a CO molecule, although the coordination of the fourth CO ligand changes the geometry of the Rh complex. The Rh atom is not adsorbed on the surface in this structure since the Rh/ECN distance is longer than 4 Å. As illustrated in Fig. 5b), the support–cluster interaction is made via two of the CO ligands. In addition, DFT simulations suggest that the formation of the H1Rh(CO)4 complex would promote its spontaneous desorption due to the high stability of the gas-phase complex with respect to the physiosorbed one: H1/Rh(CO4)/ECN, −4.01 vs. −3.17 eV; i.e. the desorbed specie is 0.85 eV more stable than the physiosorbed one. The desorption of the active Rh complexes is considered the main drawback of this reaction, and contrasts with the experimental observations, where very low leaching and excellent catalytic activity were shown. Even though the formation of H1Rh(CO)4/ECN is thermodynamically favored, the fourth carbonyl coordination is likely to be sterically hindered. The insertion of the fourth CO would imply a high reorganization of the metal coordination sphere via a repulsive square pyramidal configuration having H on top. Thus, to proceed, the cleavage of the Rh–N bond is needed with an energy cost of 1.44 eV, energy required to desorb the H1Rh(CO)3 complex. Moreover, from the catalytic point of view, the active species should have free Rh coordination sites to facilitate the styrene addition to the complex to promote hydroformylation. Thus, to obtain further insights into the stability of supported Rh complexes and rationalize the absence of leaching we evaluated the competition between CO ligand and the reactant (styrene) coordination to the Rh using ethylene molecule as styrene model. Ethylene was used instead of styrene as a substrate for the sake of simplicity.40 As reported in Table 2, the coordination of ethylene to H1Rh(CO)1/ECN (Fig. 5c) is slightly favored (0.13 eV) with respect to the coordination of the second CO ligand. As depicted in Fig. 5c), the Rh atom is coordinated with two N atoms of the ECN surface, showing that Rh tends to have five coordination sites. The insertion of a second CO ligand to H1Rh(CO)(C2H4)/ECN (Fig. 5d)) is favored by 0.54 eV. On the other hand, the insertion of ethylene or CO on H1Rh(CO)2/ECN configuration is competitive, being the interaction with ethylene only 0.09 eV more favorable with respect to the coordination of the third CO ligand. As expected, the coordination of ethylene molecule to H1Rh(CO)3/ECN is hampered due to steric reasons. After the optimization process, one of the CO ligands is ejected and physisorbed on the ECN support, far away from the Rh complex, to allow the ethylene insertion. Therefore, the supported Rh complexes containing three or more carbonyl ligands are not catalytically active due to the impossibility of the olefin to coordinate to the Rh atom.
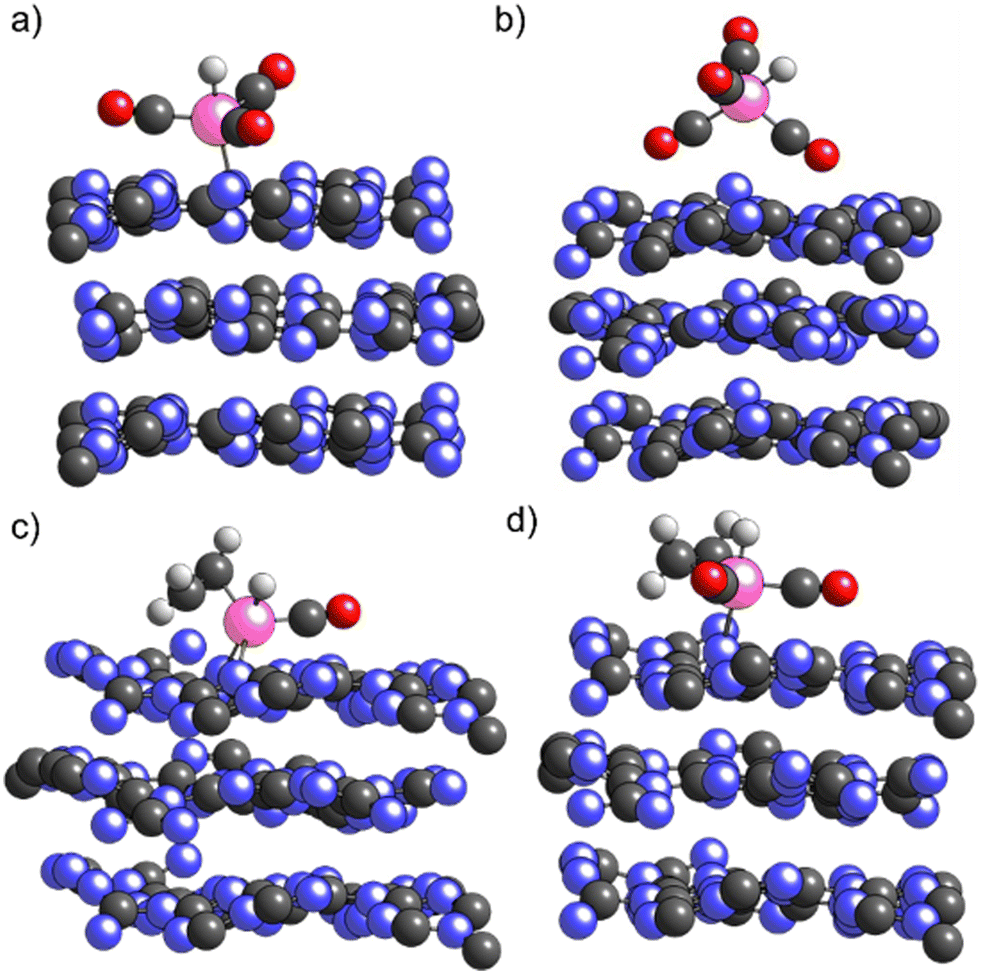 | ||
| Fig. 5 Sketches of the optimized a) H1Rh(CO)3, b) H1Rh(CO)4, c) H1Rh(CO)(C2H4) and d) H1Rh(CO)2(C2H4) species supported on ECN (grey, blue, pink, red, and white balls represent carbon, nitrogen, rhodium, oxygen, and hydrogen atoms respectively). | ||
In summary, DFT simulations suggest the reactant (styrene) can block the formation of H1Rh(CO)4, avoiding the Rh leaching, in agreement with the low leaching observed by experiments, and highlighting the role of the support in the hydroformylation process. Moreover, it proves the slightly lower energy coordination of the olefin on free coordination sites of Rh supported complex with respect to CO.
Conclusions
Carbon nitride can accommodate Rh single atoms robustly. The interactions are strong enough to endure the severe conditions of the hydroformylation reaction. The catalysts were synthetized by impregnation or direct incorporation. The resulting materials are active in the hydroformylation reaction of styrene in high selectivity towards the aldehydes, ranging from 82 to 99%, and an l/b ratio of up to 1.6. The l/b ratio being higher of that described for other Rh SAC which do not contain phosphorus ligands. The most active catalyst is the one in which the Rh3+ species are impregnated as the metal is more accessible to catalyze the reaction, being located in the characteristic 6-fold cavity of ECN. DFT calculations confirmed that low Rh coverage facilitates the metal embedded on the surface, while large Rh coverages stabilize the adsorption between layers. In addition, the formation of HxRh(CO)y/ECN species are stably adsorbed onto the ECN surface, being the complexes with less than three carbonyl ligands the responsible of the catalytic activity, preventing the Rh leaching in line with the low leaching observed experimentally and the robustness of the catalysts after four recycling cycles.Author contributions
Lole Jurado: investigation, experiments, data curation, analysis, writing – original draft. Jerome Esvan: investigation, experiments, analysis, writing. Ligia A. Luque-Álvarez: investigation, experiments, analysis. Luis F. Bobadilla: investigation, experiments, analysis, writing. José A. Odriozola: analysis, writing. Sergio Posada-Pérez: investigation, experiments, analysis, writing. Albert Poater: investigation, experiments, analysis, writing. Aleix Comas-Vives: investigation, experiments, analysis, writing. M. Rosa Axet: conceptualization, funding acquisition, writing – review & editing, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the CNRS, which we gratefully acknowledge. The authors acknowledge the financial support from EMERGENCE@INC2021 for L. J. grant. A. P. is a Serra Húnter Professor and thanks ICREA Academia 2019. A. P. and A. C.-V. Thank the Ministerio de Ciencia e Innovación for the projects PID2021-127423NB-I00 and PGC2018-100818-A-I00, respectively. A. C.-V. also thanks the Spanish MEC and the European Social Fund (grant no. RyC-2016-19930). S. P.-P. thanks Marie Curie fellowship (H2020-MSCA-IF-2020-101020330) and “Juan de la Cierva Formación” fellowship ref. FJC2019-039623-I. Ligia A. Luque-Álvarez thanks VI-PPITUS (University of Sevilla) for her current predoctoral contract.References
- B. Cornils, A. Börner, R. Franke, B. Zhang, E. Wiebus and K. Schmid, Hydroformylation, in Applied Homogeneous Catalysis with Organometallic Compounds, ed. B. Cornils, W. A. Herrmann, M. Beller and R. Paciello, 2017 Search PubMed.
- Rhodium Catalyzed Hydroformylation, in Catal. Met. Complexes, ed. P. W. N. M. van Leeuwen and C. Claver, Kluwer, 2000, vol. 22 Search PubMed.
- L. Luo, H. Li, Y. Peng, C. Feng and J. Zeng, Rh-based nanocatalysts for heterogeneous reactions, ChemNanoMat, 2018, 4(5), 451–466, DOI:10.1002/cnma.201800033.
- (a) Y. Shi, G. Ji, Q. Hu, Y. Lu, X. Hu, B. Zhu and W. Huang, Highly uniform Rh nanoparticles supported on boron doped g-C3N4 as a highly efficient and recyclable catalyst for heterogeneous hydroformylation of alkenes, New J. Chem., 2020, 44(1), 20–23, 10.1039/c9nj05385a; (b) J. Zhang, P. Sun, G. Gao, J. Wang, Z. Zhao, Y. Muhammad and F. Li, Enhancing regioselectivity via tuning the microenvironment in heterogeneous hydroformylation of olefins, J. Catal., 2020, 387, 196–206, DOI:10.1016/j.jcat.2020.03.032.
- M. R. Axet, S. Castillon, C. Claver, K. Philippot, P. Lecante and B. Chaudret, Chiral diphosphite-modified rhodium(0) nanoparticles: catalyst reservoir for styrene hydroformylation, Eur. J. Inorg. Chem., 2008, 3460–3466, DOI:10.1002/ejic.200800421.
- A. J. Bruss, M. A. Gelesky, G. Machado and J. Dupont, Rh(0) nanoparticles as catalyst precursors for the solventless hydroformylation of olefins, J. Mol. Catal. A: Chem., 2006, 252(1–2), 212–218, DOI:10.1016/j.molcata.2006.02.063.
- S. Shylesh, D. Hanna, A. Mlinar, X.-Q. Kong, J. A. Reimer and A. T. Bell, In situ formation of Wilkinson-type hydroformylation catalysts: insights into the structure, stability, and kinetics of triphenylphosphine- and Xantphos-modified Rh/SiO2, ACS Catal., 2013, 3(3), 348–357, DOI:10.1021/cs3007445.
- Y. Wang, L. Yan, C. Li, M. Jiang, Z. Zhao, G. Hou and Y. Ding, Heterogeneous Rh/CPOL-BP&P(OPh)3 catalysts for hydroformylation of 1-butene: the formation and evolution of the active species, J. Catal., 2018, 368, 197–206, DOI:10.1016/j.jcat.2018.10.012.
- L. Wang, W. Zhang, S. Wang, Z. Gao, Z. Luo, X. Wang, R. Zeng, A. Li, H. Li and M. Wang, et al., Atomic-level insights in optimizing reaction paths for hydroformylation reaction over Rh/CoO single-atom catalyst, Nat. Commun., 2016, 7, 14036, DOI:10.1038/ncomms14036.
- R. Lang, T. Li, D. Matsumura, S. Miao, Y. Ren, Y.-T. Cui, Y. Tan, B. Qiao, L. Li and A. Wang, et al., Hydroformylation of olefins by a rhodium single-atom catalyst with activity comparable to RhCl(PPh3)3, Angew. Chem., Int. Ed., 2016, 55(52), 16054–16058, DOI:10.1002/anie.201607885.
- (a) C. Li, L. Yan, L. Lu, K. Xiong, W. Wang, M. Jiang, J. Liu, X. Song, Z. Zhan and Z. Jiang, et al., Single atom dispersed Rh-biphephos&PPh3@porous organic copolymers: highly efficient catalysts for continuous fixed-bed hydroformylation of propene, Green Chem., 2016, 18(10), 2995–3005, 10.1039/c6gc00728g; (b) K. Zhao, H. Wang, X. Wang, T. Li, X. Dai, L. Zhang, X. Cui and F. Shi, Confinement of atomically dispersed Rh catalysts within porous monophosphine polymers for regioselective hydroformylation of alkenes, J. Catal., 2021, 401, 321–330, DOI:10.1016/j.jcat.2021.08.004.
- I. Ro, M. Xu, G. W. Graham, X. Pan and P. Christopher, Synthesis of heteroatom Rh-ReOx atomically dispersed species on Al2O3 and their tunable catalytic reactivity in ethylene hydroformylation, ACS Catal., 2019, 9(12), 10899–10912, DOI:10.1021/acscatal.9b02111.
- T. Li, F. Chen, R. Lang, H. Wang, Y. Su, B. Qiao, A. Wang and T. Zhang, Styrene hydroformylation with in situ hydrogen: regioselectivity control by coupling with the low-temperature water-gas shift reaction, Angew. Chem., 2020, 59(19), 7430–7434, DOI:10.1002/anie.202000998.
- P. Gao, G. Liang, T. Ru, X. Liu, H. Qi, A. Wang and F.-E. Chen, Phosphorus coordinated Rh single-atom sites on nanodiamond as highly regioselective catalyst for hydroformylation of olefins, Nat. Commun., 2021, 12(1), 4698, DOI:10.1038/s41467-021-25061-0.
- S. Ding, M. J. Hulsey, H. An, Q. He, H. Asakura, M. Gao, J.-Y. Hasegawa, T. Tanaka and N. Yan, Ionic liquid-stabilized single-atom Rh catalyst against leaching, CCS Chem., 2021, 3(10), 1814–1822, DOI:10.31635/ccschem.021.202101063.
- X. Wei, Y. Jiang, Y. Ma, J. Fang, Q. Peng, W. Xu, H. Liao, F. Zhang, S. Dai and Z. Hou, Ultralow-loading and high-performing ionic liquid-immobilizing rhodium single-atom catalysts for hydroformylation, Chem. – Eur. J., 2022, 28, e202200374, DOI:10.1002/chem.202200374.
- J. Amsler, B. B. Sarma, G. Agostini, G. Prieto, P. N. Plessow and F. Studt, Prospects of heterogeneous hydroformylation with supported single atom catalysts, J. Am. Chem. Soc., 2020, 142(11), 5087–5096, DOI:10.1021/jacs.9b12171.
- (a) S. Mitchell, E. Vorobyeva and J. Perez-Ramirez, The multifaceted reactivity of single-atom heterogeneous catalysts, Angew. Chem., Int. Ed., 2018, 57(47), 15316–15329, DOI:10.1002/anie.201806936; (b) L. Liu and A. Corma, Metal catalysts for heterogeneous catalysis: from single atoms to nanoclusters and nanoparticles, Chem. Rev., 2018, 118, 4981–5079, DOI:10.1021/acs.chemrev.7b00776; (c) X. Li, Y. Huang and B. Liu, Catalyst: single-atom catalysis: directing the way toward the nature of catalysis, Chem, 2019, 5(11), 2733–2735, DOI:10.1016/j.chempr.2019.10.004; (d) L. Li, X. Chang, X. Lin, Z.-J. Zhao and J. Gong, Theoretical insights into single-atom catalysts, Chem. Soc. Rev., 2020, 49(22), 8156–8178, 10.1039/d0cs00795a; (e) Z. Li, S. Ji, Y. Liu, X. Cao, S. Tian, Y. Chen, Z. Niu and Y. Li, Well-defined materials for heterogeneous catalysis: from nanoparticles to isolated single-atom sites, Chem. Rev., 2020, 120(2), 623–682, DOI:10.1021/acs.chemrev.9b00311; (f) M. K. Samantaray, V. D'Elia, E. Pump, L. Falivene, M. Harb, S. Ould Chikh, L. Cavallo and J.-M. Basset, The comparison between single atom catalysis and surface organometallic catalysis, Chem. Rev., 2020, 120(2), 734–813, DOI:10.1021/acs.chemrev.9b00238.
- Y. Tang, C. Asokan, M. Xu, G. W. Graham, X. Pan, P. Christopher, J. Li and P. Sautet, Rh single atoms on TiO2 dynamically respond to reaction conditions by adapting their site, Nat. Commun., 2019, 10(1), 1–10, DOI:10.1038/s41467-019-12461-6.
- F. J. Escobar-Bedia, M. Lopez-Haro, J. J. Calvino, V. Martin-Diaconescu, L. Simonelli, V. Perez-Dieste, M. J. Sabater, P. Concepcion and A. Corma, Active and regioselective Ru single-site heterogeneous catalysts for alpha-olefin hydroformylation, ACS Catal., 2022, 12(7), 4182–4193, DOI:10.1021/acscatal.1c05737.
- M. Zhu, S. Yu, R. Ge, L. Feng, Y. Yu, Y. Li and W. Li, Cobalt oxide spported on phosphorus-doped g-C3N4 as an efficient electrocatalyst for oxygen evolution reaction, ACS Appl. Energy Mater., 2019, 2(7), 4718–4729, DOI:10.1021/acsaem.9b00273.
- S. Kumar, M. B. Gawande, J. Kopp, S. Kment, R. S. Varma and R. Zboril, P- and F-co-doped carbon nitride nanocatalysts for photocatalytic CO2 reduction and thermocatalytic furanics synthesis from sugars, ChemSusChem, 2020, 13(19), 5231–5238, DOI:10.1002/cssc.202001172.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, A metal-free polymeric photocatalyst for hydrogen production from water under visible light, Nat. Mater., 2009, 8(1), 76–80, DOI:10.1038/nmat2317.
- Z. Chen, E. Vorobyeva, S. Mitchell, E. Fako, M. A. Ortuno, N. Lopez, S. M. Collins, P. A. Midgley, S. Richard and G. Vile, et al., A heterogeneous single-atom palladium catalyst surpassing homogeneous systems for Suzuki coupling, Nat. Nanotechnol., 2018, 13(8), 702–707, DOI:10.1038/s41565-018-0167-2.
- G. Vile, D. Albani, M. Nachtegaal, Z. Chen, D. Dontsova, M. Antonietti, N. Lopez and J. Perez-Ramirez, A stable single-site palladium catalyst for hydrogenations, Angew. Chem., Int. Ed., 2015, 54(38), 11265–11269, DOI:10.1002/anie.201505073.
- M. D. Fryzuk and W. E. Piers, in Organometallic Syntheses, ed. R. B. King and J. J. Eisch, Elsevier, 1986, vol. 3, p. 128 Search PubMed.
- M. R. Axet and K. Philippot, Catalysis with colloidal ruthenium nanoparticles, Chem. Rev., 2020, 120, 1085–1145, DOI:10.1021/acs.chemrev.9b00434.
- (a) Y. Bai, Y. Zheng, Z. Wang, Q. Hong, S. Liu, Y. Shen and Y. Zhang, Metal-doped carbon nitrides: synthesis, structure and applications, New J. Chem., 2021, 45(27), 11876–11892, 10.1039/d1nj02148f; (b) T. S. Miller, A. B. Jorge, T. M. Suter, A. Sella, F. Cora and P. F. McMillan, Carbon nitrides: synthesis and characterization of a new class of functional materials, Phys. Chem. Chem. Phys., 2017, 19(24), 15613–15638, 10.1039/c7cp02711g; (c) Y. Zhang, T. Mori, J. Ye and M. Antonietti, Phosphorus-doped carbon nitride solid: enhanced electrical conductivity and photocurrent generation, J. Am. Chem. Soc., 2010, 132(18), 6294–6295, DOI:10.1021/ja101749y.
- (a) Q. Zhu, B. Qiu, M. Du, J. Ji, M. Nasir, M. Xing and J. Zhang, Dopant-induced edge and basal plane catalytic sites on ultrathin C3N4 nanosheets for photocatalytic water reduction, ACS Sustainable Chem. Eng., 2020, 8(19), 7497–7502, DOI:10.1021/acssuschemeng.0c02122; (b) H. Li, S. Gan, H. Wang, D. Han and L. Niu, Intercorrelated superhybrid of AgBr supported on graphitic-C3N4-decorated nitrogen-doped graphene: high engineering photocatalytic activities for water purification and CO2 reduction, Adv. Mater., 2015, 27(43), 6906–6913, DOI:10.1002/adma.201502755.
- S. P. Pattnaik, A. Behera, R. Acharya and K. Parida, Green exfoliation of graphitic carbon nitride towards decolourization of Congo-Red under solar irradiation, J. Environ. Chem. Eng., 2019, 7(6), 103456, DOI:10.1016/j.jece.2019.103456.
- (a) J. Gracia and P. Kroll, Corrugated layered heptazine-based carbon nitride: the lowest energy modifications of C3N4 ground state, J. Mater. Chem., 2009, 19(19), 3013–3019, 10.1039/b821568e; (b) X. Ma, Y. Lv, J. Xu, Y. Liu, R. Zhang and Y. Zhu, A strategy of enhancing the photoactivity of g-C3N4 via doping of nonmetal elements: a first-principles study, J. Phys. Chem. C, 2012, 116(44), 23485–23493, DOI:10.1021/jp308334x.
- Q. Guo, Y. Xie, X. Wang, S. Lu, T. Hou and X. Liu, Characterization of well-crystallized graphitic carbon nitride nanocrystallites via a benzene-thermal route at low temperatures, Chem. Phys. Lett., 2003, 380(1,2), 84–87, DOI:10.1016/j.cplett.2003.09.009.
- G. H. Gunasekar, K. Park, H. Jeong, K.-D. Jung, K. Park and S. Yoon, Molecular Rh(III) and Ir(III) catalysts immobilized on bipyridine-based covalent triazine frameworks for the hydrogenation of CO2 to formate, Catalysts, 2018, 8(7), 295, DOI:10.3390/catal8070295.
- H. Starukh and P. Praus, Doping of graphitic carbon nitride with non-metal elements and its applications in photocatalysis, Catalysts, 2020, 10(10), 1119, DOI:10.3390/catal10101119.
- J. S. Nam, A. R. Kim, D. M. Kim, T. S. Chang, B. S. Kim and J. W. Bae, Novel heterogeneous Rh-incorporated graphitic-carbon nitride for liquid-phase carbonylation of methanol to acetic acid, Catal. Commun., 2017, 99, 141–145, DOI:10.1016/j.catcom.2017.06.007.
- Y. Chen, X. Liu, L. Hou, X. Guo, R. Fu and J. Sun, Construction of covalent bonding oxygen-doped carbon nitride/graphitic carbon nitride Z-scheme heterojunction for enhanced visible-light-driven H2 evolution, Chem. Eng. J., 2020, 383, 123132, DOI:10.1016/j.cej.2019.123132.
- X. Wang, X. Chen, A. Thomas, X. Fu and M. Antonietti, Metal-containing carbon nitride compounds: a new functional organic-metal hybrid material, Adv. Mater., 2009, 21(16), 1609–1612, DOI:10.1002/adma.200802627.
- Y. Kwon, T. Y. Kim, G. Kwon, J. Yi and H. Lee, Selective activation of methane on single-atom catalyst of rhodium dispersed on zirconia for direct conversion, J. Am. Chem. Soc., 2017, 139(48), 17694–17699, DOI:10.1021/jacs.7b11010.
- P. Malet, J. J. Benitez, M. J. Capitan, M. A. Centeno, I. Carrizosa and J. A. Odriozola, EXAFS and DRIFTS study of lanthanide doped rhodium catalysts, Catal. Lett., 1993, 18(1–2), 81–97, DOI:10.1007/bf00769501.
- (a) W. Natongchai, S. Posada-Perez, C. Phungpanya, J. A. Luque-Urrutia, M. Solà, V. D'elia and A. Poater, Enhancing the catalytic performance of group I, II metal halides in the cycloaddition of CO2 to epoxides under atmospheric conditions by cooperation with homogeneous and heterogeneous highly nucleophilic aminopyridines: Experimental and theoretical study, J. Org. Chem., 2022, 87(5), 2873–2886, DOI:10.1021/acs.joc.1c02770; (b) E. Pump, A. Poater, N. Bahri-Laleh, R. Credendino, L. Serra, V. Scarano and L. Cavallo, Regio, stereo and chemoselectivity of 2nd generation Grubbs ruthenium-catalyzed olefin metathesis, Catal. Today, 2022, 388–389, 394–402, DOI:10.1016/j.cattod.2020.04.071.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cy02094g |
| This journal is © The Royal Society of Chemistry 2023 |