
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSmall molecules and conjugates as theranostic agents
Sumon
Pratihar†
,
Krithi K.
Bhagavath†
and
Thimmaiah
Govindaraju
 *
*
Bioorganic Chemistry Laboratory, New Chemistry Unit, and School of Advanced Materials (SAMat), Jawaharlal Nehru Centre for Advanced Scientific Research (JNCASR), Jakkur P.O., Bengaluru 560064, Karnataka, India. E-mail: tgraju@jncasr.ac.in
First published on 2nd September 2023
Abstract
Theranostics, the integration of therapy and diagnostics into a single entity for the purpose of monitoring disease progression and treatment response. Diagnostics involves identifying specific characteristics of a disease, while therapeutics refers to the treatment of the disease based on this identification. Advancements in medicinal chemistry and technology have led to the development of drug modalities that provide targeted therapeutic effects while also providing real-time updates on disease progression and treatment. The inclusion of imaging in therapy has significantly improved the prognosis of devastating diseases such as cancer and neurodegeneration. Currently, theranostic treatment approaches are based on nuclear medicine, while nanomedicine and a wide diversity of macromolecular systems such as gels, polymers, aptamers, and dendrimer-based agents are being developed for the purpose. Theranostic agents have significant roles to play in both early-stage drug development and clinical-stage therapeutic-containing drug candidates. This review will briefly outline the pros and cons of existing and evolving theranostic approaches before comprehensively discussing the role of small molecules and their conjugates.
 Sumon Pratihar | Sumon Pratihar received his BSc (Hons.) in Chemistry from Midnapore College, West Bengal, India in 2013, and MSc in Chemistry from Indian Institute of Technology Madras in 2015. He obtained his PhD under the supervision of Prof. T. Govindaraju, Bioorganic Chemistry Laboratory, New Chemistry Unit, JNCASR, Bengaluru, India. His research interests are in employing synthetic organic chemistry to understand the structure and functions of nucleic acids with diagnostic and therapeutic applications. |
 Krithi K. Bhagavath | Krithi K. Bhagavath received her BSc degree from Mangalore University, Karnataka, India in 2018 and subsequently MSc in Chemistry from the University of Mysore, Karnataka, India in 2020. Currently, she is pursuing her PhD under the supervision of Prof. T Govindaraju, Bioorganic Chemistry Laboratory, New Chemistry Unit, JNCASR, Bengaluru, India. Her research interests include developing small molecule probes for diagnostic and therapeutic applications in cancer and Alzheimer's disease. |
 Thimmaiah Govindaraju | Thimmaiah Govindaraju is a Professor at the Bioorganic Chemistry Laboratory, New Chemistry Unit, JNCASR, Bengaluru, Karnataka, India. He received his MSc (2000) from Bangalore University and PhD in Chemistry (2006) from the National Chemical Laboratory and University of Pune, India. He carried out postdoctoral research at the University of Wisconsin-Madison, USA (2005–2006) and the Max Planck Institute of Molecular Physiology, Dortmund, Germany (2006–2008) as an Alexander von Humboldt postdoctoral fellow. His research interests are at the interface of chemistry, biology, and biomaterials science, including Alzheimer's disease, peptide chemistry, molecular probes, theranostics, molecular architectonics, nanoarchitectonics, and silk and cyclic dipeptide derived biomimetics. |
Introduction
Theranostics is a biomedical approach that combines therapy and diagnostics into one entity. This allows for the monitoring of disease progression and treatment response.1 Diagnostics involves identifying specific characteristics of a disease, while therapeutics refers to the treatment that follows to cure the disease. The term “theranostics” was coined in 2002 by Funkhouser to describe the combination of therapeutic and diagnostic modalities in one entity.1 However, the concept has existed since 1964 when isotope of iodine (131I) was used in the treatment and diagnosis of thyroid cancer.2 The advantages of combining therapy and imaging include providing a clear picture of the lesion, etiological cause in disease, and spatiotemporal distribution of a drug in the patient's body. It also allows for monitoring the therapeutic effect of the drug in a dose-dependent manner. The inclusion of imaging in therapy has greatly improved the prognosis of diseases such as cancer and neurodegeneration. Theranostic-based treatment approaches are currently achieved using nuclear medicine, nanomedicine, and a wide diversity of macromolecular systems such as gels,3 polymers,4 aptamers,5 and dendrimer-based agents.6 These different theranostic approaches have been covered in a number of reviews and extensively discussed in numerous reviews and readers are encouraged to refer to these sources for further information. This review will briefly outline the advantages and disadvantages of existing and emerging theranostic approaches. It will then exclusively focus the discussion on the role of small molecules and their conjugates in advancing precision medicine. Most of the case studies discussed in this review include cancer, neurodegeneration such as Alzheimer's disease, and pathogenic infections.The ultimate goal of theranostics is to enable the imaging and monitoring of disease tissue, drug delivery kinetics, and drug efficacy. This information can provide unprecedented control over therapy and dosage, leading to personalized medicine. This review presents a brief overview of therapeutic and diagnostic strategies that can be integrated into the concept of theranostics. This will help readers understand the advantages and drawbacks associated with the evolution and development of these strategies in general (Fig. 1). While therapeutic approaches are relatively limited (i.e., chemical, photochemical, or radiation-based), diagnostic approaches are diverse and continue to evolve with advances in science and technology.
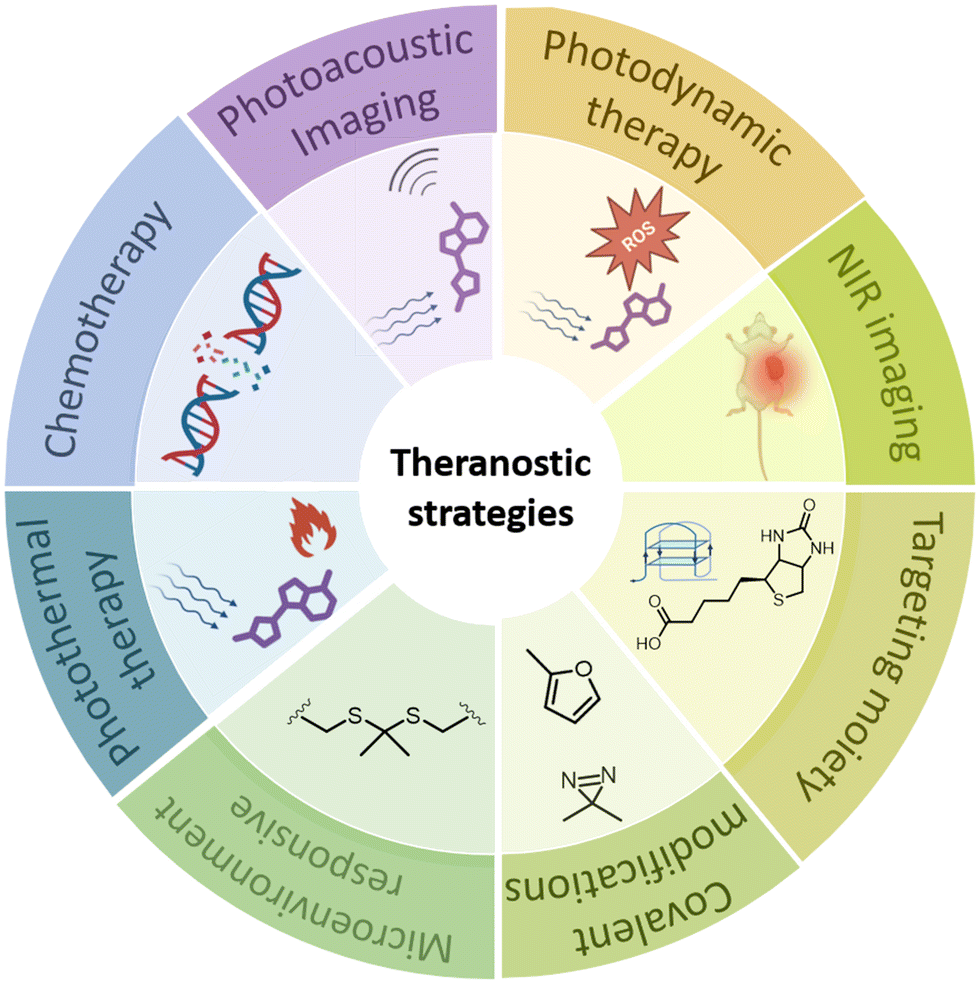 | ||
| Fig. 1 Illustration of the strategies used in small molecule and conjugate based theranostics. Created using https://BioRender.com. | ||
Imaging techniques in medicine
A critical evaluation of the various diagnostic techniques is necessary to understand and implement the most suitable approach in designing a strategy for a specific disease. Radiolabelling involves identifying distinct radio-emissions detected from diseased tissues, with minimal signal from normal tissues. Alternatively, the electronic spectroscopy properties or energy response to light are desirable properties in molecular design for diagnostic utility. The key to superior diagnostic strategies lies in tapping and modulating energy dissipation pathways of small molecules. The interaction of molecules with light provides a wealth of diagnostic insights into diseases. The emission property of a molecule has immense value and includes fluorescence in visible and NIR range, phosphorescence, luminescence, and afterglow luminescence among others.7,8 A desirable diagnostic modality must be a synergy of sensitivity, spatiotemporal resolution, penetration depth, and field of view.Positron emission tomography (PET) is a technique that maps the distribution of positron (β+)-emitting radionuclides from radiolabelled organic or inorganic theranostic agents.9 However, the use of radioactive elements and exposure to radiation pose potential hazards. Optical imaging, on the other hand, relies on light emitted from molecules and materials upon excitation at a suitable wavelength. PET is commonly used in cancer imaging to detect and locate tumors, assess disease severity, and monitor treatment response.10 In neurodegenerative diseases, PET can detect changes in brain metabolism or neurotransmitter systems, providing a means to diagnose and monitor disease progression. Despite its usefulness, PET has limitations such as high cost and limited availability, as well as potential risks associated with radiation exposure.
Magnetic resonance imaging (MRI) is a powerful medical imaging technique that utilizes a magnetic field and radio waves to produce detailed images of internal body structures. It is a non-invasive and painless procedure that can provide valuable information about the presence, location, and extent of cancer or neurodegenerative disease.11 In cancer cases, MRI can identify and locate tumors, determine their size and shape, and assess their involvement with nearby tissues and organs. It can also monitor treatment response and detect disease recurrence. In neurodegenerative diseases such as Alzheimer's or Parkinson's disease, MRI can provide detailed brain images to aid in diagnosis and monitoring disease progression. It can also identify changes in brain structure or function associated with the disease. However, MRI has limitations in cancer and neurodegenerative disease diagnosis, such as high cost, the need for specialized equipment and trained personnel, and unsuitability for patients with pacemakers or metallic implants.
Ultrasound imaging, also known as sonography, employs high-frequency sound waves to generate images of the body.12,13 It is frequently used in cancer imaging to detect and diagnose tumors and guide biopsies or other procedures. In neurodegenerative diseases, ultrasound can visualize changes in brain structure or blood flow, providing a non-invasive means of assessing disease progression. However, its usefulness in cancer imaging can be limited by factors such as tumor size and location, while its ability to visualize changes in neurodegenerative diseases may be constrained by brain tissue depth and resolution of the equipment.
Diverse molecular probes have been designed to emit in the visible range (380–700 nm) to accurately report the location and progression of disease. However, the potential of fluorescence-based imaging in the visible range is hindered by autofluorescence from biological milieu and poor penetration depth. Utilizing longer wavelengths in the near-infrared regions has helped to avoid cellular autofluorescence and improve tissue penetration depth in diagnosis.14 Significant improvements in NIR emissive probes through systematic molecular engineering have resulted in a paradigm shift in diagnostic sensitivity. Although optical imaging has high sensitivity, its low penetration depth of a few millimeters in fluorescence and a few centimeters in NIR imaging requires further improvement. Photoacoustic imaging (PAI) involves exciting organic molecules with light pulses, which release heat upon relaxation, creating ultrasound waves detected by an acoustic transducer.7,15 PAI offers improved sensitivity (picomolar range), high spatial resolution (sub-millimolar), and deep tissue imaging (several centimeters). Often, organic PAI agents are also compatible with NIR optical imaging.
Therapeutic approaches in theranostic medicine
Chemotherapy involves chemical interactions with DNA, proteins, enzymes, or specific cellular pathways. The development of photochemical techniques, which induce chemical changes in cells or tissues upon irradiation with specific light, and photothermal techniques have ushered in a new era of drug discovery.Photodynamic therapy (PDT) involves the use of chemical agents with photosensitizing properties upon exposure to light of a suitable wavelength, typically in the presence of oxygen.16 Common organic photosensitizer (PS) scaffolds include phenothiazine, methylene blue, porphyrin analogues, and their transition metal complexes. Light irradiation excites an electron in the PS to a singlet state. Subsequent intersystem crossing leads to a triplet state where energy transfer from the PS to triplet oxygen converts it to reactive singlet oxygen, capable of triggering a cascade of cell damage responses leading to apoptosis and necrosis. Targeting and preferential localization of PS agents in disease-associated tissues is leveraged to achieve desired therapeutic outcomes in cancer and neurodegeneration.17 There has been growing interest in increasing efficiency and incorporating two-photon (TP)-based strategies for improved therapeutic efficacy and biocompatibility. Current limitations of PDT include limited stability of photosensitizing molecules, high oxygen dependence, and limited NIR absorption.
Photothermal therapy (PTT), primarily used in cancer treatment, involves the excitation of NIR-absorbing molecules to generate heat, which aids in the thermal ablation of tumors.17 An ideal PTT agent would have absorption in the NIR window, low toxicity, biocompatibility, and the potential for easy functionalization. The deep tissue penetration ability of NIR light in this non-invasive approach, coupled with minimal side effects on surrounding healthy tissue, makes it a useful therapeutic approach, often used in combination with chemotherapy.
Nuclear medicine as theranostic agents
Nuclear medicine is a well-established multidisciplinary field that uses radioactive nuclides for both diagnostic and therapeutic purposes.18 Radiopharmaceuticals fall into two main categories: metal-based and organic molecules. Metal-based tracers rely on coordination chemistry, while radiopharmaceuticals based on organic compounds incorporate nonmetal radionuclides such as 18F, 11C, 13N, 15O, and 123I through covalent bond formation. Radiopharmaceuticals consist of two components: a radionuclide that transmits its mechanism of action through decay and a targeting biomolecule or organic ligand that dictates the localization of the radiopharmaceutical.19 They can be used as therapies to deliver a toxic payload to a tumor location or as diagnostics for non-invasive imaging of disease.20 The most common imaging modalities for diagnostic purposes are single-photon emission computed tomography (SPECT) and PET. Therapeutic radiometals exert their cytotoxic effect by irreparably damaging DNA, causing chromosomal abnormalities, cell death, and deletions. DNA damage can be caused by the release of β-particles, α-particles, or low-energy electrons. Radiotherapy as theranostic approach includes both imaging and therapy. These two objectives require radionuclides with different decay rates.21 Both imaging and therapy require radionuclides to transfer their energy in the target tissue, typically in a malignant tumor. Imaging relies on radionuclides that emit high energy photons, either rays (99mTc) or the annihilation photons produced by positron (+) decay (55Co), with minimal interaction with intervening tissue. Some radiometals such as 47Sc and 177Lu are naturally theranostic due to therapeutic nuclides that have an γ-emission for imaging as well as therapy.The concept of theranostics combines the use of an imaging radionuclide (64Cu) to identify disease sites and a therapeutic radionuclide (67Cu) to treat the disease, which can also be achieved by single radionuclide (188Re).21 Although SPECT is significantly less expensive than PET due to lower equipment costs and greater radionuclide availability, PET offers superior sensitivity and resolution. Both PET and SPECT provide high-quality morphological data but lack anatomical clarity. Radiolabeled nanomaterials have better labeling efficiency and enhanced blood circulation compared to radiolabeled small molecule probes.22 Radiolabeling can be achieved through various techniques such as surface labeling, encapsulation, and radiochemical doping.9 Conventional cancer therapies include radiation therapy and radiopharmaceutical therapy (RPT), where radionuclides are systematically delivered as therapeutic formulations to the tumor site. These mainly include a and β-particle-emitting radionuclides as radiation-delivering agents to induce cytotoxicity. Radionuclides can be administered alone or in conjugation with peptides, small molecules, antibodies, or nano constructs.19 One traditional method for detecting and characterizing tumors and etiological protein aggregates of neurodegenerative disorders such as Alzheimer's disease is SPECT/PET imaging of organs or tissues using radiotracers administered to the patient's body in the form of labeled small molecules or labeled nanomaterials.23 However, this method has drawbacks such as poor in vivo stability of labeled nanomaterials, leading to shedding of the diagnostic agent and false-positive detection of tumors. Chronic radiation exposure risk, high diagnosis cost, and toxicity of radiolabeled agents are among the other disadvantages.
Nanoformulations as theranostic agents
Recently, there has been a surge of interest in the utilization of nanomaterials due to their numerous physical imaging modalities and potential biomedical utility, particularly for early and accurate biosensing and efficient therapy of various diseases.24 Theranostic nanomedicine is a promising option as it can quickly and non-invasively identify specific cancer biomarkers while simultaneously destroying cancer cells.25 Nanomedicine, a treatment based on nanoparticles, employ a variety of organic or inorganic nanomaterials to treat, diagnose, monitor, and regulate biological systems. These nanomaterials possess inherent anticancer therapeutic properties or can be loaded with various medication modalities. Additionally, they can be loaded with multimodal imaging contrast agents and target surface-bound compounds on cancer cells.26 The ability to produce nanoparticles with customizable size, shape, and surface chemistry, as well as their high surface-to-volume ratio and high surface-loading capacity has attracted researchers to develop cancer nanomedicines. Nanoparticles also possess desirable physicochemical characteristics such as electrical, optical, and magnetic properties. Nanotheranostics holds significant promise for the prevention, detection, monitoring, and treatment of illnesses including malignancies. Active sub-disciplines within nanomedicine include drug delivery, bioimaging, diagnostic agents and biosensors. The functionalization of nanoparticles with a range of biomolecules renders them effective agents for imaging, treatment, and medication administration.27,28 Despite several nanomedicines have received FDA-approval, there remains significant room for advancement in the pursuit of intelligent nanotherapeutics. Over the years, several nanoparticle formulations have been developed for cancer diagnostics and therapeutics. These are primarily categorized as inorganic nanoparticles, organic nanoparticles, and nanocomposites.24 Examples include quantum dots, nano emulsions, micelles, and proteins.29,30 However, there are challenges associated with nanomedicines such as the interaction of the nanoparticle formulation with systemic circulatory components that may result in charge and size variability, off-target binding of cells and receptors, among others.Polymer and dendrimer-based theranostic agents
Polymer–drug conjugates are pharmacologically active macromolecular constructs that consist of one or more therapeutic drugs such as small molecules, peptides, proteins, and aptamers are covalently attached to a polymeric carrier.31 In polymer-based theranostics, four different types of therapeutic treatments are investigated: drug delivery, gene delivery, photodynamic therapy, and hyperthermia treatment. These are used in conjunction with various imaging agents such as magnetic resonance imaging agents, fluorescent agents, and microbubbles for ultrasound imaging.32 Adagen, the first approved polymer–protein conjugate, is an enzyme replacement medication for adenosine deaminase deficiency in individuals.33 A polymer-based theranostic typically comprises a therapeutic modality, an imaging modality, and a biocompatible polymer. In some cases, the same chemical group or portion of the polymer conjugate can provide both imaging and therapeutic components.4,32 Theranostic probes are constructed from various classes of polymeric materials including micelles, liposomes, and dendrimers. Polymeric micelle candidates have certain drawbacks related to their critical micelle concentrations (CMC). Below their CMC, the micelles tend to disaggregate and cause systemic toxicity. As a result, conventional micelles are being replaced by unimolecular micelles.34 Liposomes are vesicular lipid bilayers with an aqueous core compartment for enclosing therapeutic and diagnostic modalities.35 Dendrimers are synthetic macromolecules with a highly branched spherical structure. They typically have repeated branches of a dendron around a central core forming a three-dimensional geometrical shape. Dendrimers offer several benefits including improved bioavailability, reduced premature release, tissue-specific accumulation, high payload, and enhanced pharmacodynamic and pharmacokinetic characteristics of diagnostic, therapeutic, and targeted agents.6 Numerous studies have been conducted on polymer-based platforms for cancer treatment and have demonstrated several advantages. In the body, biopolymers gradually degrade into harmless substances. Polymers such as polyethylene glycol (PEG) and poly(-caprolactone) have received approval for use in therapeutic macroformulations.36 Recently, polymer nanocomposites based on aggregation-induced emission luminogens (AIEgens) have shown potential in imaging-guided photothermal therapy (PTT) for cancer theranostics.37Antibody therapy
Targeted antibody therapy emerged as an alternative approach to address the non-specificity of chemotherapeutic drugs. Several monoclonal antibodies (mAbs) have been developed to inhibit tumor invasion by blocking the growth factors of cancer cells.38 Subsequently, efforts were made to develop antibody-based drugs that target specific cell surface receptors. For instance, most breast cancers are known to overexpress the HER2 gene and its receptor, which promotes cellular proliferation. Herceptin is an FDA-approved mAb that targets the HER2 receptor in breast cancers and halts the cell cycle in the G-phase.39 Avastin is another antibody-based drug candidate that functions by blocking the VEGF receptor on cancer cells, thereby inhibiting angiogenesis.Targeted gene therapy based theranostic agents
A significant challenge in the use of macromolecular systems is achieving efficient delivery to cells or disease areas within a patient's body. Nucleic acid-based theranostics are particularly relevant for genetic diseases, where specific nucleic acid sequences are administered to silence or enhance gene expression for a specific therapeutic outcome. Several classes of delivery vectors have been explored to achieve and enhance efficient and targeted delivery of nucleic acids. However, their delivery remains a limiting factor for nucleic acid-based theranostics.40 The classical form of nucleic acid-based therapeutics involves short antisense oligonucleotide (AO) sequences that bind to disease-associated mRNA and alter downstream protein expressions. In addition to AO, other approaches include small interfering RNA (siRNA), DNAzymes, and ribozymes. RNAi technology is most commonly used for sequence-specific gene silencing by the administration of short stretches of dsRNA which forms RISC (RNA induced silencing complex) which contains ribonuclease enzymes that cleaves the dsRNA to generate a single strand RNA that sequence specifically binds to a target mRNA to either cleave or silence its expression. RNAi therapy has been successfully used to silence antiapoptotic genes such a Bcl-2. Aptamers are short oligonucleotides with defined conformation in three dimensions and serve as high fidelity ligands to biomolecules, proteins, or cells. Inception of Systematic Evolution of Ligands by EXponential enrichment (SELEX) in 1990s have dramatically accelerated the development of aptamers with required selectivity and affinity.5 Rapid clearance and short half-life in biological milieu are the major limitations of aptamer-based approach.The success of macromolecules and nanoparticles has largely been limited to proof-of-concept studies due to two primary factors: (1) the lengthy journey from bench to bedside, and (2) non-adherence to the Lipinski rule and deviation from drug-like properties.41 In light of these disadvantages, there has been a resurgence in the field of theranostics where small molecules and their conjugates are the lead players. Theranostic entities involve the structural integration of therapeutic and diagnostic entities in a minimalistic fashion, often appended to a suitable targeting or localizing group. The concept of theranostics is gaining importance as it paves the way for precision medicine. In subsequent sections, we will discuss the disease conditions, existing clinical solutions/approaches, and recent progress in small molecule and conjugate-based theranostic strategies for cancer, Alzheimer's disease, and pathogenic infections.
Cancer
Cancer is the second leading cause of death after heart diseases. The number of cases has been steadily increasing due to factors such as genetics, environmental pollutants, and lifestyle. Despite this, only 2% of drugs are recommended for further development each year.42 The primary challenge in using anticancer drugs in clinical settings is their toxicity to healthy cells, which can result in acute, chronic, or life-threatening side effects.43 Cytotoxic chemotherapeutic agents delivered systemically are unable to differentiate between cancerous and healthy cells, leading to severe side effects that often limit their clinical use.Chemotherapeutic agents are categorized into several classes, including DNA alkylating agents, topoisomerase inhibitors, mitosis inhibitors/microtubule stabilizers, and antimetabolites.44 Cancer cells exhibit rapid DNA synthesis, enabling malignant behaviors such as invasion and metastasis. These cells also proliferate rapidly and uncontrollably. Chemotherapeutic agents interfere with DNA synthesis and target mitosis and DNA replication in cells to inhibit the growth and spread of tumors.45 Cisplatin, a clinically approved drug, forms DNA adducts at the N7 position of purine residues that block cell division and subsequently leading to apoptosis.46 DOX intercalates nucleic acids and inhibits topoisomerase II activity to prevent DNA and RNA synthesis. Daunorubicin inhibits topoisomerase II activity leading to single and double stranded DNA breaks. Etoposide, too works in a similar fashion resulting in mitotic inhibition, leading to cell death. Paclitaxel disrupts mitotic spindles by binding to microtubules and preventing their normal dissolution during cell division, ultimately leading to cellular apoptosis. However, most chemotherapeutic drugs exhibit toxicity to normal tissues and lack selectivity,47 causing severe toxicity to skin, hair, gastrointestinal cells, among other organs.48
This review briefly discusses small molecule-prodrug based conjugates developed as theranostics for various cancers. PTT and PDT have emerged as promising alternatives to minimize toxicity and ensure precise tumor targeting.49 Hyperthermia-based cancer treatment, which involves heating the tumor area, has gained interest as an alternative or complement to current chemotherapies.50 Hyperthermia (41–45 °C) damages localised intracellular proteins related to cellular survival and proliferation, resulting in minimal to no damage to normal tissues. Thermoablation (46–56 °C) destroys tumors through direct cell necrosis. PDT utilizes ROS, often singlet oxygen species (1O2), generated by photosensitizer molecules upon laser irradiation at specific wavelengths. PDT using ROS has attracted interest as a complementary, non-invasive cancer treatment with few side effects.51 Excessive ROS damages DNA, RNA, and protein, destroying cancer cells and causing tumor regression. Near-infrared (NIR) light is preferred over UV or visible light for its enhanced light penetration depth, making the therapy minimally invasive.
Small molecule theranostics for cancer
Small molecule theranostic agents are organic molecules without appended moieties for targeting or imaging. Few chemotherapeutic anticancer drugs, such as DOX, have detectable emission but with poor quantum yield. Molecules that specifically interact with oncogenic nucleic acid sequences to inhibit transcription and display turn-on fluorescence are promising for cancer theranostic strategies. DNA can adopt several secondary conformations besides the well-known double helical B form, with G-quadruplex (GQ) and i-motif being the most established noncanonical conformations.52 Targeting the binding pockets of these GQ or i-motif structures, located at the promoter region of oncogenes, forms a unique therapeutic strategy. GQ is a noncanonical DNA conformation where guanine-rich DNA sequences interact via hydrogen bonding to form four-stranded architecture. GQ sequences are abundant in the promoter regions of oncogenes, making them attractive targets for cancer treatment. The binding pockets in GQ are sequence and topology-dependent and form unique interaction sites for fluorogenic and therapeutic small molecules.52,53 While many fluorogenic small molecules are designed to recognize GQ sequences based on their topology and/or sequence, and another class of molecules stabilizes GQ, the amalgamation of the two to achieve both therapeutic and diagnostic properties has been rare in literature. Careful and systematic assessment is required in the choice of molecular scaffolds to merge the stabilizing (therapeutic) scaffolds while making their electronic-structure to extract required electronic-delocalization and desired emission property.Small molecules based on benzothiazole motifs exhibit promising fluorescence selectivity for a wide range of DNA secondary conformations. Molecules-based on the carbazole scaffold demonstrate remarkable pharmacological activity.54 The large aromatic scaffold favors strong π-stacking interaction with the G-quartet plane of the GQ due to its considerable π-interactions. A GQ-targeting theranostic molecule, 3,6-bis(1-methyl-4-vinylpyridinium) carbazole diiodide (BMVC) based on carbazole moiety showed promising anticancer activity in human cancer cell lines (Fig. 2A).55 BMVC stabilizes telomeric GQ and lights up GQ DNA in vitro. Long-term treatment with BMVC uncaps telomeric ends, shortens telomeres, and induces DNA damage foci leading to senescence and apoptosis. BMVC treatment represses tumor progression in a mouse xenograft model through inhibitory pathways, both telomere-dependent and independent. Although BMVC selectively images GQ in cellulo, its green emission limits deep tissue imaging. We have developed a small molecule-based theranostic approach for lung cancer that combines GQ-targeted detection of cancer tissue in the far-red window and transcription inhibition-mediated anti-cancer activity.53 The theranostic molecule TGP18 has a methylated neocuproine-based aromatic core conjugated to a coumarin unit bearing a diethylamine donor group (Fig. 2A). The fluorogenic molecule TGP18 selectively recognizes the oncogenic BCL2 GQ DNA sequence through a hybrid binding mode where the molecule stacks on the G-quartet and engages in specific interactions with loop nucleotides (Fig. 2B). For the first time, we have rationally exploited and executed the hybrid loop stacking and groove binding mode to achieve selectivity among the myriad of GQ conformational architectures and a number of other conventional and non-conventional DNA conformations. Although the G-quartet plane in GQ forms a suitable site for π-stacking interaction with small molecules with planar aromatic core, the nucleotide (nt) composition and unique three-dimensional orientation of the loops, and the grooves play a deciding role to impart binding specificity. Interestingly, BCL-2 GQ adopts a distinct mixed hybrid topology consisting of a single nucleotide double chain reversal loop, and two lateral loops of length 3 and 7 nt respectively. Computational studies revealed a significant contribution of 7 nt loop to ensure high affinity and selective binding of TGP18 to BCL-2 GQ. The dual effect of stabilization and selective far-red fluorescence light-up forms the basis of the small molecule-based theranostic approach. Among a set of cancer cell lines MCF-7, HeLa, A549, and MDA-MB-231 and non-cancerous cell line (HEK293T), TGP18 demonstrated remarkable antiproliferative activity for breast (MDA-MB-231) and lung (MCF-7) cancer cell lines. Interestingly, TGP18 was efficient in penetration and retarding the growth of A549 tumor 3D spheroid. The stabilizing interaction of TGP18 with BCL2 GQ reduces mRNA expression levels in the A549 cell line. The anti-tumor efficacy of TGP18 was further evaluated in mouse xenograft models of human breast cancer cell line MDA-MB-231 and lung carcinoma line A549 (Fig. 2C). Administration with maximum tolerated dosage (MTD) of 0.5 mg kg−1 of body weight showed marginal inhibitory effect in breast cancer model, while excellent antiproliferative activity after 14 days treatment, at a 200-fold lesser dose than gemcitabine (reference drug, Fig. 2D). The far-red emission of TGP18 enabled deep tissue imaging of tumor tissue sections of A549 xenografts (Fig. 2E). This minimalistic design strategy combines therapeutic and diagnostic modalities without the need for additional conjugates. Treatment of the A549 lung cancer cell line reduced BCL-2 protein expression, with associated cellular/replication stress responses evident from S-phase arrest, DNA damage response, nucleolar and oxidative stress. TGP18-mediated cellular stress response upregulated VEGF expression. Analysis of TGP18-treated cellular lysate for Caspase 3 activity indicated an apoptotic pathway. The probe's commendable far-red fluorescence (680 nm) was evident from fluorescence studies, gel electrophoresis, and lifetime measurement with BCL-2 GQ. With minimal autofluorescence in the far-red region enabled preferential imaging of tumor tissues in xenograft tumor mice, TGP18 forms the first example of GQ targeting small molecule-based theranostic drug candidate for lung cancer. In the same direction, a set of monocyanine ligands consisting of carbazole moiety conjugated to electron withdrawing benzothiazole were rationally designed to impart selective stabilization and turn on fluorescence with oncogenic GQ DNA sequences.56 Judicious tuning of the structure activity relationship resulted in two small molecules TCZ (Fig. 2A) and TCA (Fig. 2A) which selectively stabilized c-MYC and BCL-2 promoter GQ sequences respectively and supressed the relative mRNA and protein expression evaluated in human breast cancer cell line (MDA-MB-231). Molecular dynamics (MD) simulation revealed a preference for stacking interaction between BCL-2 and TCA, while TCZ binds to the groves of c-MYC GQ. Further they impede the recruitment of relevant transcription factors at the promoter region of the oncogenes supressing downstream processes. Fluorescence emission of the ligands confirm nucleolar uptake and nucleolar stress. TCZ supressed the abundance of Sp1 and NM23-H2 at the c-MYC-NHE III1, evident from Chip assay (Fig. 2F and G). The colocalization of the molecules with GQ specific antibody BG4 supported the GQ-small molecular interaction as the deciding factor for in celllulo anti-cancer activity (Fig. 2H). This study provides a useful roadmap for structural tuning to achieve binding selectivity among a set of structurally similar oncogenic GQs. Assessment of ribosomal biogenesis inhibition revealed competence of both TCA and TCZ in disrupting the interaction of rDNA GQs with nucleolin protein, although better efficiency was observed for TCZ. Small molecular drug candidates regulating/interrupting the translation, transcription, or ribosomal RNA biogenesis, compared to the mode of action of conventional chemotherapeutic agents, leading to reduced cancer cell viability surely opens promising avenue for cancer treatment (Fig. 3A).
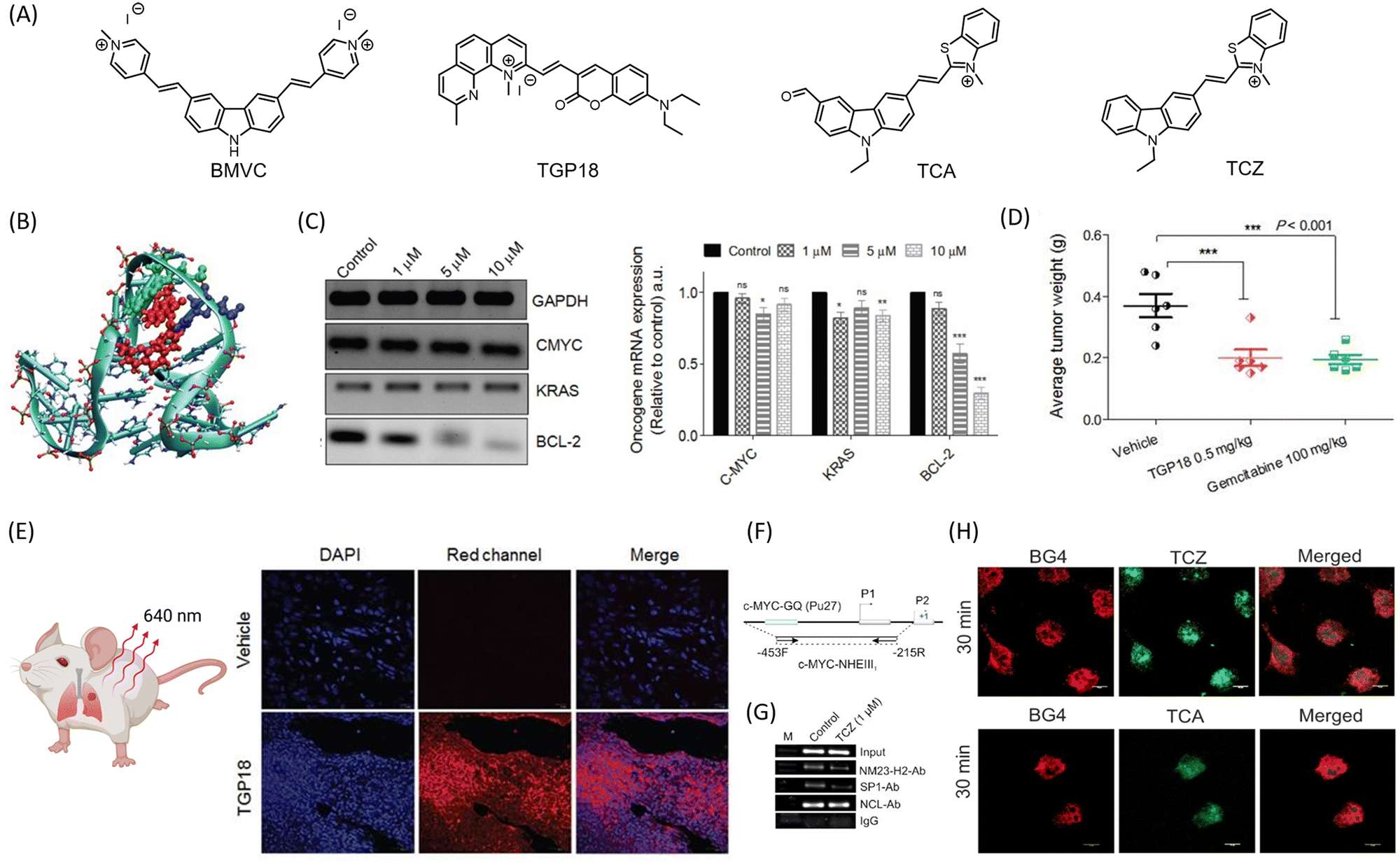 | ||
| Fig. 2 (A) Molecular structures of the donor–acceptor (D–A) based noncanonical nucleic acids targeted theranostic agents for cancer. (B) Hybrid mode of interaction of TGP18 with BCL-2 GQ involving loop stacking and groove binding. (C) mRNA expression profiles of BCL-2, CMYC, KRAS and GAPDH upon treatment with increasing concentration of TGP-18 (1, 5 and 10 μM, and relative mRNA expression relative to GAPDH. (D) Reduction in tumor weight in BALB/c mice upon treatment of TGP 18, compared to anticancer drug gemcitabine, and (E) images of tissue sections of A549 tumor with and without treatment with TGP 18, imaged in blue channel (DAPI) for nuclei and red channel for TGP 18. (B)–(E) are reproduced from ref. 53, with permission from IVYSPRING international publisher, copyright 2020. (F) Schematic representation of the c-MYC promoter harbouring the GQ-forming sequence in the NHE III1 region. (G) ChIP experiment validates the occupancy of NM23-H2-, and Sp1- at the c-MYC promoter in MDAMB-231 cells upon treatment with 1 μM TCZ for 24 h. (H) Selective staining of cellular GQ using theranostic agents TCZ and TCA, and colocalized with GQ specific BG4 antibody. Reproduced from ref. 56, with permission from American Chemical Society copyright 2021. Illustrative image in (E) was created using https://BioRender.com. | ||
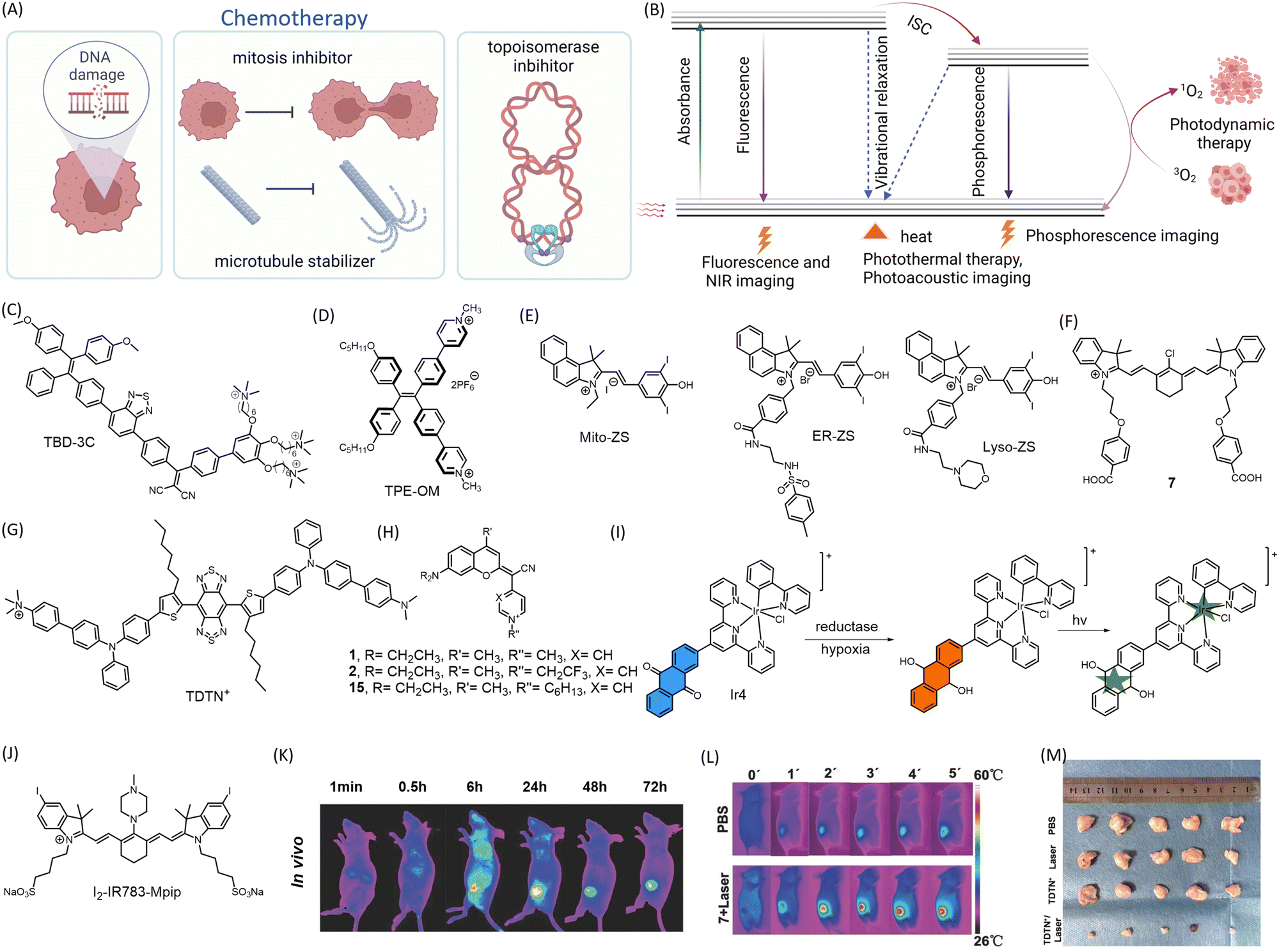 | ||
| Fig. 3 Representation of mechanism of action of (A) chemotherapeutic drugs and (B) PDT, PTT, optical imaging, and photoacoustic imaging modality. Chemical structure of (C) TBD-3c, (D) TPE-OM, (E) Mito-ZS, ER-ZS, Lyso-ZS (F) 7, (G) TDTN+, (H) COUPY coumarins, (I) Ir4 undergoing reduction to generate carbon radicals upon irradiation, (J) I2-IR783-Mpip (K) preferential accumulation of 7 in subcutaneous A549 mice model visualized by in vivo NIR fluorescence imaging (L) PTT by 7 as seen by IR thermal imaging of subcutaneous A549 mice xenograft model after 5 min irradiation at 808 nm, (K) and (L) are reproduced from ref. 62 with permission from Wiley VCH, copyright 2016 (M) reduction in tumor volume in 4T1 tumor bearing mice upon treatment with TDTN+ followed by subsequent irradiation at 808 nm, compared to control groups without TDTN+ or without irradiation. Reproduced from ref. 63 with permission from The Royal Society of Chemistry, copyright 2023. (A and B) Were created using https://Biorender.com. | ||
Recent advancements in the field of small molecule-based anticancer agents have primarily focused on PDT and PTT (Fig. 3B). These therapies target overexpressed proteins or oncogenes. A common characteristic of such molecules is that they exhibit a fluorescence response only upon aggregation.57 While aggregation of fluorogenic molecules can pose challenges in theranostic applications, careful tuning of the aggregation process can result in enhanced photophysical properties that are advantageous for biomedical imaging. The modulation of photophysical properties in the aggregated state has altered molecular motion, energy gaps, the nature of the excited state, and modes of de-excitation. These changes provide opportunities for tuning fluorescence, phosphorescence, photoacoustic imaging, PDT, and PTT. Porfimer sodium (Photofrin), a PDT-based anticancer agent, has proven to be a successful treatment strategy for cancer and has received approval from the U.S. Food and Drug Administration (FDA).58 However, there is a continuous search for small molecule-based PDT drugs that offer better biocompatibility, reduced toxicity, and the ability to treat a wider variety of cancers.
A series of photosensitizers based on aggregation-induced emission (AIE) (TBD-1C to 3C) with cationic pendant groups have been reported for use in light-activated cancer cell ablation through pyroptosis.59 These photosensitizers, known as 1,1,2,2-tetraphenylethene-benzo[c][1,2,5]thiadiazole-2-(diphenyl methylene)malononitrile (TBD-R) photosensitizers, were produced by conjugating TBD and phenyl rings with cationic chains. This allowed for dual functionality: fluorescence turn-on imaging at 650 nm (a weak red emission upon excitation in the range of 300–500 nm) and photosensitization for PDT. The cationic chains also enabled the probes to anchor onto cell membranes. The membrane anchoring capacity increased with the number of cationic chains and TBD-3C (Fig. 3C) with three cationic chains had the strongest membrane anchoring capacity when compared to TBD-1C and TBD-2C, which had one and two cationic chains, respectively. When exposed to white light at an intensity of 40 mW cm−2, membrane-anchored small molecules produce ROS, which trigger the release of lactate dehydrogenase and activate inflammatory pathways in tumor cells, ultimately leading to cell death. In another study utilizing an AIE-based approach, a pyridinium-functionalized donor–acceptor AIE luminogen (TPE-OM) (Fig. 3D) is developed as a theranostic candidate for colon cancer.60 TPE-OM displayed a strong emission at 590 nm in the aggregated state, upon excitation at 350 nm. TPE-OM was found to localize in the mitochondria and proved cytotoxic to HCT116 colorectal cancer cells by inhibiting cell-cycle regulatory genes. This effect could be traced in vivo with a high signal-to-noise ratio in mice tumor xenograft models. Further, RNA sequencing of control and treated mice revealed upregulation of genes that negatively regulate the cell cycle, while cell-cycle promoting genes were suppressed.
The development of small molecule photosensitizers that specifically target organelles has been reported.61 These photosensitizers can induce ROS-mediated damage to subcellular organelles, leading to membrane rupture, inflammatory responses, and pyroptosis. ROS-mediated damage to subcellular organelles can activate specific caspases, such as caspase 1, triggering a cascade of events that ultimately leads to the rupture of the plasma membrane and the release of intracellular contents. This process, known as pyroptosis, is characterized by the release of pro-inflammatory molecules that can promote an immune response. Based on the cyanine core, three molecules were developed to target mitochondria (Mito-ZS), endoplasmic reticulum (ER-ZS), and lysosomes (Lyso-ZS) (Fig. 3E). These molecules were designed with variable groups and different heavy atom counter ions to achieve organelle specificity and enhanced ROS production. The localization modules for mitochondria, lysosomes, and endoplasmic reticulum were indolyl cation, morpholine group, and methylbenzenesulfonamide moieties, respectively. When the treated tissue was exposed to light at a wavelength of 580 nm, it emitted light in the range of 600–750 nm, with the highest intensity at around 620 nm. This finding suggests that these emissions have promising applications in self-tracking and theranostic uses. Pyroptosis was induced in HeLa and 4T1 cells through organelle-targeting PDT by irradiation at 580 nm (24 J cm−2). This effect was also observed in an in vivo 4T1 triple-negative breast cancer mouse model and validated through substantial RNA-seq analysis.
An NIR photosensitizer with mitochondrial localization for synchronous PDT and PTT has been developed.62 A library of 27 heptamethine cyanine dyes with different N-alkyl side chains was synthesized for mitochondrial localization, achieved due to the cationic heptamethine cyanine core. The lead compound 7 (Fig. 3F) raised the temperature of the tumor to over 50 °C in 2 min, resulting in tumor tissue damage. As mitochondria are essential energy-producing cellular organelles that are vulnerable to both hyperthermia and excessive ROS, compound 7 demonstrated synchronous PDT/PTT effects, significantly increasing phototherapeutic efficacy (Fig. 3L). Near-infrared fluorescence (NIRF) imaging 24 h post-injection showed preferential accumulation of compound 7 in the tumor both in vivo and ex vivo (Fig. 3K). A mitochondria-targeting NIR-II AIEgen (emission at 1052 nm) named TDTN+ has been reported (Fig. 3G).63 The AIEgen was equipped with a benzobisthiadiazole and a triphenylamine moiety as acceptor and donor units, respectively, with long alkyl chains that hamper π–π interactions. It also included a cationic trimethylamonium unit for mitochondrial localization. The designed molecule was amphiphilic and could self-assemble to form TDTN+ nanoparticles displaying excellent PDT/PTT effects upon 808 nm laser irradiation (0.6 W cm−2) with a photothermal conversion efficiency of 66.7% (Fig. 3M). A library of small molecules was developed in which the lactone of the coumarin moiety was replaced with a cyano(4-pyridine/pyrimidine) methylene moiety (COUPYs).64 These N-alkylated COUPY coumarins (Fig. 3H) selectively accumulate in mitochondria and produce singlet oxygen upon irradiation, leading to PDT. COUPY coumarins, which combine imaging and therapy in a single compound, offer tremendous promise for the creation of novel theranostic agents.
An oxygen-independent photosensitizer has been developed that generates carbon radicals through an iridium(III)-anthraquinone complex (Ir4), which targets the mitochondria of hypoxic tumors (Fig. 3I).65 This metal complex has a unique TP excitation property (λex = 730 nm, λem = 585 nm) that can be used for imaging mitochondrial dynamics. It exerts its therapeutic action by producing carbon radicals that induce loss of mitochondrial membrane potential, proving cytotoxic to cells. In hypoxic conditions, the reductase enzyme can reduce the anthracene group of Ir4 to anthracene diol (Ir4-red), activating the complex's TP emissive characteristics. This reduction does not occur in normoxia. Subsequent irradiation of Ir4 produces a carbon radical, allowing the agent to perform both imaging and therapeutic tasks. The agent shows a significant decrease in tumor volume in A549 xenograft mice upon irradiation with TP excitation at 730 nm.
A heptamethine cyanine dye-based pH-responsive photosensitizer for cancer theranostics is reported.66 This probe, I2-IR783-Mpip (Fig. 3J), consists of an N-methylpiperazine moiety on the cyanine backbone. This probe showed an absorption maximum at 860 nm in pH 6–7, which was blue-shifted in basic pH 8–12 and red-shifted in acidic pH 3–5. Under acidic conditions, as is prevalent in tumor microenvironments, this probe exhibits excellent NIR absorption and efficiently generates singlet oxygen for tumor ablation. In the molecular design, the PDT effect is enhanced by the introduction of two iodine substituents on the 5-position of the indole rings. The N-methylpiperazine moiety is pH-responsive and exhibits fluorescence quenching under neutral pH conditions due to photoinduced electron transfer (PeT) from the lone pair of electrons on nitrogen. Under acidic conditions, protonation of the nitrogen atom stops PeT and results in NIR fluorescence in the HepG2 cell line. The PDT drug shows intrinsic tumor binding and can eliminate specific tumors in an acidic environment when irradiated by an 850 nm NIR light (30 mW cm−2) for 30 min.
Small molecule conjugates for cancer
Conventional therapeutic agents often lack diagnostic properties, making it necessary to conjugate them with suitable imaging modalities for theranostic applications. Additionally, most chemotherapeutic agents are toxic to healthy tissues and have poor biodistribution.67 It is therefore imperative to design theranostic agents that can specifically target cancer cells, release the drug in the tumor microenvironment, or activate the fluorogenic moiety in the tumor microenvironment. To achieve this, various small molecular (prodrug) conjugates have been designed that leverage the specific differences between cancer and normal cells and their microenvironments.68 Cancer cells possess distinct physiochemical properties that can be preferentially targeted. Stimuli such as pH, ROS, hypoxic conditions, and increased nitroreductase concentration can be leveraged to uncage and activate prodrugs or active molecular probes or diagnostic modules.51 In the presence of appropriate environmental stimuli, the triggering component of the theranostic agent is released. Among the receptors overexpressed in cancer cells, β-galactosidase forms a unique identification moiety. Aberrant proliferation of cancer cells is associated with increased ROS production and low oxygen availability. These factors can be targeted by designing theranostic candidates that include a targeting moiety, drug, diagnostic agent, and stimuli-responsive masking agent. Combination therapy, where multiple modes of cell death are integrated to synergistically enhance antitumor activity, has inspired the development of smart theranostic conjugates. First, we will discuss conjugates of conventional chemotherapeutic agents before delving into PDT and PTT based conjugates.Traditional drug-based conjugates
One notable characteristic of cancer cells is the overexpression of glutathione. To leverage this factor, a multifunctional gemcitabine-coumarin-biotin conjugate 5 was developed as a theranostic anticancer prodrug (Fig. 4A).69 In this conjugate, the nucleoside analogue anticancer drug gemcitabine is linked to coumarin as a fluorescence reporter via a disulfide linkage that is susceptible to cleavage by intracellular thiols (GSH). The biotin moiety facilitates receptor-mediated preferential uptake in the lung cancer cell line A549, compared to fibroblast cells WI38, where the biotin receptor is absent. The analogue lacking the biotin moiety showed lower anticancer effects than the biotin-bearing conjugate. TP excitation of coumarin at 750 nm and an emission range of 420–600 nm was utilized for fluorescence imaging while minimizing cellular damage. A prodrug based on topoisomerase I inhibitor SN-38 was linked to biotin for cancer localization and piperazine rhodol for fluorescence monitoring to obtain a theranostic agent 4a (Fig. 4B).70 The stimuli for releasing the prodrug were designed to vary, with one option being self-immolative cleavage in response to cellular glutathione (GSH). The agent 4a showed antitumor activity compared to the biotin-excluded analogue in HeLa tumor in mice (Fig. 4I). The fluorescence of both SN-38 and piperazine rhodol were imaged ex vivo (Fig. 4J). Using a similar approach, gemcitabine and other anticancer drugs have been tested in cellular and in vivo models. On similar lines, an imidazolyl-naphthalene-diimide conjugate-based topoisomerase inhibitor has been reported with promising anticancer activity (Fig. 4C).71 The chemotherapeutic drug SN-38 was attached to an endogenous H2S-responsive azide moiety linked to rhodol to evaluate its anticancer effect in colon and lung cancer cell lines via apoptosis.72 An activatable folate-doxorubicin conjugate connected by a disulfide bond (Doxo-S-S-Fol) masks both fluorescence and cytotoxicity (Fig. 4D).73 The activation of this conjugate by glutathione specifically in tumors induces its transformation into an active state, leading to both cytotoxic effects and the fluorescence of doxorubicin (λex = 497 nm, λem = 594 nm). This probe can selectively internalize into folate receptor-expressing cancer cells and can be visualized by the action of glutathione on the conjugate inside the cells. The conjugate becomes fluorescent upon cleavage of the disulfide bond, along with concomitant cytotoxic effects exerted by doxorubicin, as demonstrated in A549 cells.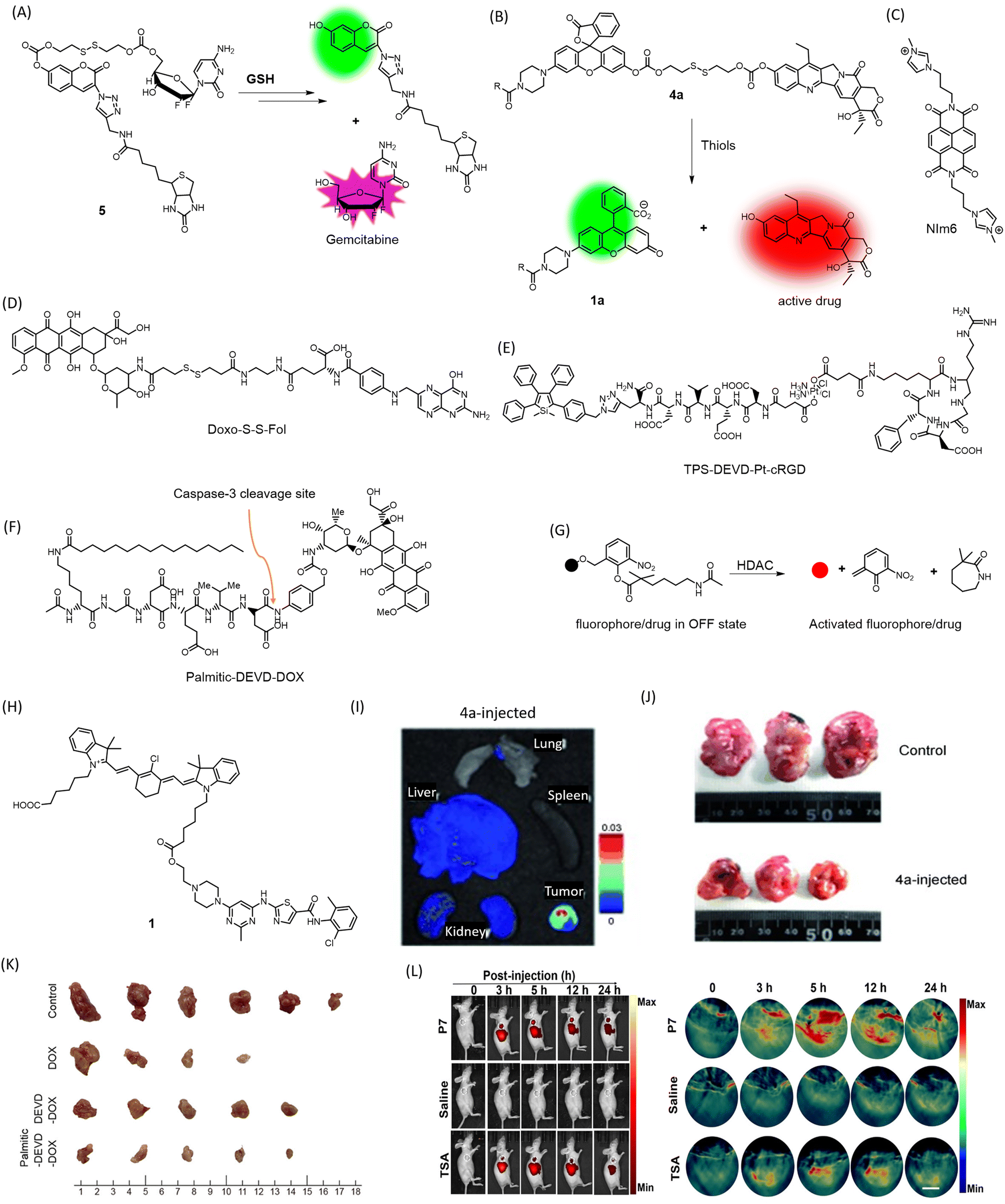 | ||
| Fig. 4 Mechanism of cleavage of (A) 5 and (B) 4a by GSH to release fluorophore and the drug Gemcitabine and SN-38 respectively, structures of (C) NIm6, (D) Doxo-S-S-Fol, (E) TPS-DEVD-Pt-cRGD, (F) Palmitic-DEVD-DOX, (G) mechanism of HDAC-responsive cleavage of conjugate to activate the drug/fluorophore, (H) Structure of conjugate 1, (I) ex vivo fluorescence imaging of organs to visualize preferential accumulation of 4a in tumor site (J) reduction in tumor volume in HeLa mice xenografts treated with 4a for 35 days. (I) and (J) Are reproduced from ref. 70 with permission from Wiley VCH, copyright 2014, (K) chemotherapeutic efficacy of Palmitic-DEVD-DOX conjugate in CT26 bearing mice in comparison to unconjugated counterparts. Reproduced from ref. 75 with permission from American Chemical Society, copyright 2023. (L) In vivo NIRF/PA imaging of MDA-MB-231 tumor-bearing mice administered with HDAC responsive P7 and HDAC inhibitor trichostatin A (TSA). Reproduced from ref. 76 with permission from Wiley VCH, copyright 2022. | ||
A Pt(IV) prodrug TPS-DEVD-Pt-cRGD (Fig. 4E) with an axially functionalized cyclic arginine–glycine–aspartic acid tripeptide specific to αvβ3 membrane integrins overexpressed in cancers is reported.74 The prodrug also includes an AIE fluorophore tetraphenylsilole (TPS), with a caspase-3 enzyme-specific DEVD peptide (λex = 365 nm, λem = 480 nm). The TPS fluorophore acts as a marker for apoptosis caused by the Pt(IV) drug, which activates caspase-3 activity, leading to cleavage of DEVD and releasing the AIE fluorophore that can be traced by fluorescence light-up due to aggregation. Selective uptake was evident in αvβ3 membrane integrin-positive U87-MG cells compared to MCF-7 as a negative control.
A novel, carrier-free amphiphilic prodrug Palmitic-DEVD-DOX conjugate (Fig. 4F) has been designed composed of a hydrophobic palmitic moiety (n-hexadecane chain), a hydrophilic group, a caspase-3-specific cleavable peptide (Asp-Glu-Val-Asp, DEVD), and a cytotoxic drug to address issues with nanocarriers and conventional cytotoxic drugs (DOX).75 The DEVD peptide is apoptosis-specific, leading to cleavage and release of doxorubicin at the tumor site. In its prodrug form, doxorubicin eliminates the chances of cytotoxicity to healthy tissues. The palmitic moiety facilitates self-assembly, which has been shown to enhance antitumor activity (Fig. 4K). This prodrug was superior to its earlier counterpart that lacked the palmitic moiety and required a carrier while lacking tumor specificity. The NIR Cy5.5 tag attached to the conjugate allowed the visualization of the tumor in CT26 tumor-bearing mice. Treated mice showed a drastic reduction in excised tumor tissue size, proving the therapeutic efficacy of the novel conjugate.
Histone deacetylase (HDAC) is an enzyme that removes acetyl groups from histone proteins, causing DNA to become more tightly packed and leading to gene repression.76 HDAC inhibitors have shown potential in cancer therapy as they can reverse the repressive effects of HDAC activity on gene expression, leading to tumor cell death and/or differentiation. A self-immolative conjugate for NIRF or photoacoustic (PA) dual imaging and HDAC-responsive prodrug release has been developed, bearing a tunable ester bond that allows for efficient release of caged fluorophores or drugs upon deacetylation (Fig. 4G). A xanthene-based fluorophore with ring-opening ability served for PA imaging, while HDAC-dependent prodrug release facilitated tumor therapy in a mice model of HDAC-overexpressed triple-negative breast cancer (Fig. 4L). Furthermore, the probe exhibited excellent specificity to HDAC against common carboxylesterases.
Kinase inhibitors (KIs) are a widely used class of anticancer drugs. However, they can suffer from off-target effects and reduction of platelet count, among other disadvantages such as immediate efflux from cells. A novel KI (dasatinib)-heptamethine cyanine dye conjugate 1 (Fig. 4H) was developed for cancer theranostics (λex = 796 nm, λem = 815 nm).77 The cyanine dye addresses the issue of off-target effects due to its well-reported tumor specificity and enables optical imaging of mitochondria in cancer cells for up to 24 h. The dasatinib conjugate was proved to be much more efficient at inducing cytotoxicity in HepG2 tumor cells compared to dasatinib alone.
Phototherapeutic approaches
Overexpressed receptors on cancer cells, such as biotin and folate receptors, can serve as suitable targets for tumor-specific delivery. In this vein, a biotin-attached cyclocyanine bearing an aza installation acts as a TP photosensitizer (ACC-B) (Fig. 5A) for spatiotemporal NIR cancer therapy for colorectal/colon cancer (Fig. 5G).78 This molecule's robust ROS generation ability upon 770 nm TP treatment (emission range: 590–640 nm), spatial selectivity, low dark toxicity, and appreciable selectivity towards cancer cells make it a promising candidate for TP-induced phototoxicity towards HeLa and A549 cancer cells. It can also distinguish human colon cancer tissue from normal colon tissue with appreciable accuracy.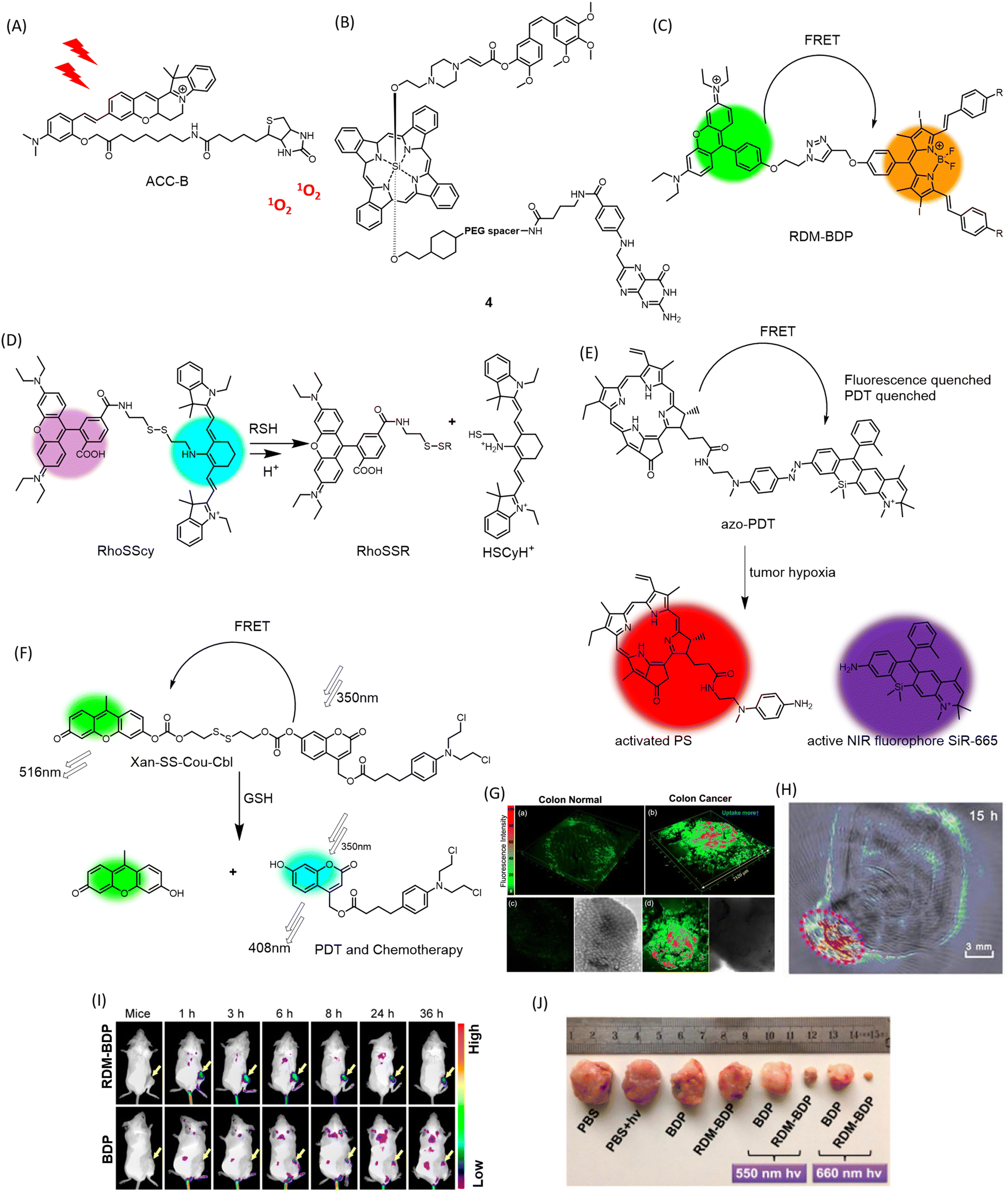 | ||
| Fig. 5 Chemical structures of (A) ACC-B, (B) conjugate 4, (C) mechanism of dual response of RhoSSCy to pH and thiols, (D) hypoxia triggered activation of FRET-quenched azo-PDT, releasing active PS for PDT and NIR fluorophore for imaging, (E) chemical structure of FRET-based theranostic RDM-BDP, (F) GSH-responsive multimodal PDT and chemotherapeutic activity of FRET theranostic Xan-SS-Cou-Cbl, (G) 3D fluorescence imaging of colon cancer tissue stained with ACC-B. Reproduced from ref. 78 with permission from American Chemical Society, copyright 2021. (H) PA imaging of tumor-bearing mice administered with RhoSSCy. Reproduced from ref. 81 with permission from IVYSPRING Insternational Publisher, copyright 2023. (I) Tumor preferential accumulation of RDM-BDP compared to BDP alone in 4T1 tumor-bearing mice visualized by in vivo fluorescence imaging. (J) Changes in tumor volume in RDM-BDP injected mice, upon irradiation. (I) and (J) Are reproduced from ref. 80 with permission from American Chemical Society, copyright 2018. | ||
Overexpression of the folate receptor (FR) is a characteristic of a broad spectrum of cancer cells. Conjugation of FR with anticancer agents such as taxol has been explored for targeted delivery of anticancer drugs to minimize collateral damage to normal cells. Strategies involving conjugation of folic acid with prodrugs and imaging modalities have shown promising anticancer activity. You and colleagues designed an aminoacrylate bond as a singlet oxygen-sensitive cleavable moiety.79 A prodrug 4 (Fig. 5B) bearing combretastatin A-4 propiolate was designed with a photodynamic-responsive moiety activated by singlet oxygen. The folate receptor enhances the bioaccumulation of the moiety in folate receptor-positive colon 26 cancer cells. The molecule effectively reduced tumor volume in colon 26 tumors in BALB/c mice as a combined effect of PDT and chemotherapy, while the core-modified porphyrin and phthalocyanine moiety allowed for live imaging of biopsy samples upon irradiation at 690 nm.
Fluorescence resonance energy transfer (FRET) is an effective way to keep a fluorogenic probe or PDT agent in a latent state. Linking FRET-compatible multiple chromophores or probe-PDT agent pairs using cleavable linkages has been exploited as a useful strategy to conditionally activate PDT in tumors. Fan and colleagues incorporated a novel and innovative FRET-based strategy into the development of structure-inherent-targeting (SIT) phototheranostics for cancer.80 Conventional PSs are hydrophobic and can easily aggregate in physiological environments, leading to limited membrane permeability and cell uptake, which can limit their effectiveness as photosensitizers for PDT. They also have issues with singlet-to-triplet intersystem crossing (ISC), which is necessary for efficient generation of singlet oxygen. This can be due to improper triplet state energy levels or short triplet lifetimes, hindering their ability to generate singlet oxygen and limiting their effectiveness as photosensitizers for PDT. In the context of PDT, FRET can be used to enhance the efficiency of singlet oxygen generation by transferring excitation energy from a donor chromophore to an acceptor chromophore with a higher triplet state energy level. This leads to increased photon utilization and more efficient singlet oxygen generation. In this case, the FRET process was induced by pairing a typical cationic rhodamine moiety (RDM) with a known photosensitizer, diiododistyrylbodipy (BDP), and providing an energy donor. The resulting SIT theranostic (RDM-BDP) (Fig. 5C) showed strong tumor-targeting ability, exhibiting rapid mitochondrial localization compared to BDP alone, with a high NIR signal-to-background ratio (Fig. 5I). Notably, the RDM-BDP conjugate achieved significantly broader absorption and enhanced singlet oxygen generation due to the FRET effect upon irradiation with a 660 nm light (12 J cm−2) in 4T1 cells (Fig. 5J). Similar outstanding results were obtained in BALB/c mice bearing 4T1 tumors, demonstrating the theranostic efficiency of the FRET probe RDM-BDP.
A multifunctional theranostic probe, RhoSSCy (Fig. 5D) conjugated by 5′-carboxyrhodamine (Rho) and heptamethine cyanine IR765 (Cy) interconnected by a disulphide linker and an amino group was designed for cancer PDT and NIRF/PA imaging (Fig. 5H).81 This probe was designed to show thiol/pH dual sensing property specific to tumor microenvironments. When stimulated at 480 nm, RhoSSCy exhibits two emission peaks at 580 nm and 765 nm. Due to good spectral overlap of the emission spectrum of the Rho dye (550–630 nm) with absorption spectrum of the Cy (580–700 nm), there is the occurrence of FRET from the excited state of Rho to Cy. When left in its natural state, RhoSSCy can react with thiols (RSH) to generate RhoSSR and HSCyH or with H+ to form RhoSSCyH+. It could potentially react simultaneously with H+ and thiols to produce RhoSSR and HSCyH2+. As the concentration of thiols in the solution grew in response to RhoSSCy, the fluorescence emission intensity of the probe at 580 nm (ex = 480 nm) increased. This occurred because the disulfide bond of RhoSSCy could be cleaved by decreased GSH, and as a result, the FRET effect vanished. RhoSSCy showed excellent tumor visualizing properties in ex vivo NIRF imaging of tumor tissue and non-invasive PA imaging of tumor-bearing mice. The photosensitizing ability of RhoSSCy robustly increased the survival rate of mice irradiated for PDT, from 10% to 80%. The authors claim that this is the first report of a four-in-one theranostic probe for sensing, targeting, multi-modal imaging and therapy of cancers.
A new chemical compound called azo-PDT (Fig. 5E) was created by combining a photosensitizer and a NIR fluorophore with an azo group as a trigger for hypoxia.82 This compound demonstrated the ability to specifically target hypoxic tumor cells for ablation and detect solid tumors through its hypoxia-activated fluorescent response. Azo-PDT is composed of pyropheophorbide α (Pyro) as the PS and SiR-665 as the NIR fluorophore, connected by an azo group. In hypoxic conditions, the azo group is reduced, releasing and activating both the fluorophore for imaging and the photosensitizer for PDT upon laser irradiation at 670 nm to produce 1O2. The azo group also quenches the fluorescence of the conjugate, while FRET between Pyro and SiR-665 prevents PDT under normoxic conditions. This makes azo-PDT a highly effective pro-PS that is non-toxic to normal tissues.
A new approach to cancer treatment has been developed that combines chemotherapy and PDT in a single probe activated by specific stimuli.83 In this context, GSH-responsive activatable FRET based prodrug conjugate, Xan-SS-Cou-Cbl (Fig. 5F) has been designed. The FRET donor–acceptor pair is composed of a 7-hydroxy-(4-methyl) coumarin-xanthene derivative. The prodrug is “locked” and unable to exhibit therapeutic action when FRET occurs successfully between the coumarin donor and the xanthene acceptor, connected by a disulfide linker. When the prodrug encounters GSH, it “unlocks” and releases the coumarin-chlorambucil conjugate and the xanthene dye, leading to simultaneous PDT and photoinduced drug delivery.
Tumour microenvironment-responsive theranostics
The targeted delivery and spatiotemporal mapping of chemotherapeutic agents is often a challenging task and a major concern that limits the success of anticancer drugs. To ensure the targeted accumulation of drugs, ligands corresponding to overexpressed receptors are appended to the drug molecule.84 Alternatively, the drugs are masked as prodrugs, where the active drug molecule is released in the cancer micro-environment. The acidity and increased ROS in the cancer microenvironment have also been leveraged to incorporate diagnostic modalities. Boronate ester has been demonstrated and extensively used as a sensor for H2O2 for stimuli responsive imaging of tumors. A quinone–cyanine based dye (QCy-BA) (Fig. 6A) has been developed, functionalized with a cleavable phenyl boronic acid linker that opens up in response to H2O2 and specifically stains the nuclear AT-rich DNA in live cells (λex = 400 nm, λem = 565 nm).85 A bifunctional probe for fluorescence and photoacoustic imaging of cancer has been reported.86 The probe (JW41) (Fig. 6B) bears a boronate ester group for sensing H2O2 while the 2-deoxyglucose moiety assists in preferential accumulation in cancer cells. | ||
| Fig. 6 Structures of (A) Qcy-BA, (B) JW-41, (C) nitroreductase activated Se-NO2, structures of (D) conjugate 1, (E) PTO-Biotin Nps, (F) ENBS-B, (G) SORgenTAM, (H) hCES2 responsive Benz-AP which exhibits PDT in its amide form under low hCES2 conditions while PDT is quenched in amine form due to high hCES2 concentrations, (I) structure of compound 2, (J) PA imaging of 4T1-tumor xenograft with PTO-Biotin Nps. (K) IR thermal images of 4T1 tumor-bearing mice administered with PTO-Biotin Nps and irradiated with 808 nm laser showing PTT. (J) and (K) Are reproduced from ref. 89 with permission from American Chemical Society, copyright 2023. (L) PDT demonstrated in HeLa tumor-bearing mice treated with compound 2 and irradiated at 590 nm LED. Reproduced from ref. 93 with permission from American Chemical Society, copyright 2019. | ||
The overexpression of glutathione (GSH) in cancer cells has been exploited to develop an AIE fluorophore-chlorambucil prodrug conjugate 1 (Fig. 6C) with mitochondria targeting properties.87 GSH cleaves the disulfide bond to release chlorambucil and simultaneously trigger the fluorescence of the AIE counterpart. This conjugate showed excellent cytotoxicity to GSH overexpressing colon and cervical cancer cells, with traceable mitochondrial localization. A small molecule with photo-redox activity to delineate a new approach, namely conditionally activatable photoredox catalysis (ConAPC) has been reported.88 Unlike conventional metal complexes-based photo-redox catalysts, which suffer from poor cellular tolerability or deployability, an organic molecule has been used. The approach relies on the oxygen-deprived tumor microenvironment for uncaging and activation of the metal-free photocatalyst Se-NO2 (Fig. 6D). In the quest to design a biologically benign photoredox catalyst, they substituted the central oxygen atom of Nile red with selenium, enabling photocatalytic oxidation of coenzyme NADH triggering the reductive transformation of cytochrome c. A photoredox-catalysis-induced collapse of NADH and cyt c (Fe3+) states elicit and facilitate cell death. A nitroreductase selective trigger was deployed in the PC which releases under hypoxic tumor microenvironment resulting in a 165-fold fluorescence increment in the NIR region in cellular imaging (λex = 660 nm, λem = 710 nm). Intratumoral (i.t.) administration of Se-NH2 in a xenograft mice model resulted in 95% tumor growth inhibition (TGI) compared to continued tumor growth in the non-treated group. As a successful realization of ConAPC, administration of Se-NO2 followed by irradiation enabled precise visualization of the tumor and resulted in significantly decreased tumor volume after 28 days of treatment.
A oxazine-based conjugate has been developed with activatable intrinsic fluorescence upon protonation in the lysosomes while triggering ferroptosis via lysosomal dysfunction in tumors.89 The judicious molecular engineering design ensures spatiotemporal control over the intracellular Fenton reaction. The molecular conjugate self-assembles into a vesicle imparting lysosomotropism. The molecular conjugate phenothiazine-fused oxazine biotinylation nanoparticles, abbreviated as PTO-Biotin Nps (Fig. 6E), localizes in the lysosomes possibly via protonation and imparts a NIR photothermal effect upon irradiation with 808 nm, triggering lysosomal dysfunction and leakage of protons and irons into the cytoplasm promoting the Fenton reaction and evoking ferroptosis (Fig. 6K). The photothermal effect upon light irradiation using 808 nm triggers the Fenton reaction. The model has impressive therapeutic efficacy in cellulo (HeLa and 4T1 cell line) and in vivo 4T1-tumor models with minimal off-target effect. Incorporation of the biotin ligand imparts significant cancer cell selectivity. The cell death was proved to be independent of apoptosis or necroptosis. Lysosome membrane potential (LMP) is disturbed only upon irradiation. PA imaging (PA755 and PA680) enabled deep tissue imaging (λem = 848 nm) proved substantial enrichment and prolonged retention of the PTO-Biotin Nps in the tumor, providing an effective theranostic strategy (Fig. 6J).
The inherent hypoxic environment of many solid tumors forces cancer cells to adopt self-protective mechanisms for survival and growth. Conventional oxygen-dependent PDT is ineffective in these conditions. To overcome this limitation, a theranostic conjugate (ENBS-B) (Fig. 6F) based on a benzophenothiazine core capable of generating ROS upon irradiation with infrared light has been developed.90 The appended biotin moiety enhances uptake in cancer cells by 87-fold in vitro. Irradiation with far-red light (660 nm) results in enhanced NIR fluorescence in the range 680–720 nm (Φf = 0.21), making it suitable for non-invasive imaging and guiding PDT. Injection of tumor-bearing BALB/c mice with the biocompatible probe ENBS-B enabled visualization of the tumor with a high signal-to-noise ratio of 7 ± 0.54. The antitumor activity arises from abundant O2−˙ generation and subsequent transformation to toxic ˙OH via superoxide dismutase (SOD)-mediated pathway, disrupting lysosomal and nuclear integrity leading to apoptosis. To combat hypoxia resistance, the same group designed a small molecule conjugate SORgenTAM (Fig. 6G) consisting of a Nile Blue analogue (SORgen) appended to tamoxifen (TAM), an antiestrogenic drug via “click” reaction (λex = 660 nm, λem = 694 nm).91 This design strategy aims to economize physiological oxygen levels by blocking cellular respiration, oxygen consumption, and decreasing HIF-1α expression. The TAM subunit effectively disrupts the electron transport chain in mitochondria, translating well in both in vitro and in vivo experiments.
An enzymatically deactivating PS, named (6-Aminophenalenone) Benz-AP (Fig. 6H) was developed, that is selective to cancer cells expressing low levels of human carboxylesterase 2 (hCSE2).92 This PS functions as a ratiometric fluorescent chemosensor for low hCSE2 environments and generates singlet oxygen to activate PDT to ablate cancer cells. Benz-AP is structurally based on a singlet oxygen-generating moiety, phenalone. Its amine derivative, AP, is red-fluorescent and unable to generate singlet oxygen or reactive oxygen species (ROS), while its amide derivative, Benz-AP, simultaneously emits singlet oxygen and displays yellow fluorescence when exposed to radiation. Low hCSE2 levels in cancer cells maintain the photosensitizer in its amide form, thus activating PDT. Benz-AP demonstrated excellent phototoxicity to tumor cells within 1 h of injection into tumor spheroids and proved to be an efficient photosensitizer for combating resistance of conventional prodrug-based therapies for low-hCSE2 tumors. A fluorescein derivative inspired theranostic probe 2 (Fig. 6I) displaying thermally activated delayed fluorescence (TADF) has been designed with nitroreductase (NTR) activatable moiety.93 NTR in mild hypoxic tumor microenvironments could activate the probe to the fluorescent emitting parent fluorescein derivative (λex = 488 nm, λem = 640 nm), which efficiently produces singlet oxygen due to its long triplet state lifetime. Additionally, this smart photosensitizer selectively localized in lysosomes, making the photodynamic therapy (PDT) process even more efficient. In vivo imaging using a 590 nm LED (16 mW cm−2) demonstrated preferential visualization of HeLa tumor in SCID/BALB/c-nude mice (Fig. 6L).
Covalent conjugation
Covalent conjugation strategy is adopted in theranostics for improved antitumor activity and prolonged retention. Many current small molecular probes used in cancer diagnosis and therapy have limitations such as poor specificity for tumors, insufficient accumulation at targeted sites, and rapid excretion by the kidneys, limiting their effectiveness in clinical applications. To overcome these challenges, new strategies are needed to efficiently immobilize conventional probes at targeted sites, leading to enhanced accumulation and prolonged retention in tumors. A red light-initiated probe-RNA cross-linking (RLIPRC) method has been described for development of a theranostic probe, f-CR (Fig. 7A).94 This NIR fluorescent probe comprises a cyclic-(arginine–glycine–aspartic acid) (cRGD) peptide targeting ανβ3 integrin receptor overexpressed on cancer cell membrane, a singlet oxygen-sensitive furan moiety for RNA cross-linking, and an NIR dye (Cy7) as a signal reporter. This probe was capable of both passive and active tumor targeting and produced strong NIR/photoacoustic signals, enabling sensitive and accurate dual-modality tumor imaging in vivo. The cycloaddition reaction between furan and nucleobases such as adenine, cytosine, or guanine under the oxidation of singlet oxygen generated in situ by irradiation of methylene blue (MB) using 660 nm laser light allowed the probe f-CR to cross-link to cytoplasmic RNAs specifically and covalently (Fig. 7B). This effectively blocked the probes’ exocytosis, resulting in improved tumor accumulation and retention, eventually leading to apoptotic death of 4T1 tumor cells (Fig. 7C). The system achieved 70% tumor suppression. Covalent linking with cytoplasmic RNA interferes with normal functions and results in abnormal protein expression, leading to apoptotic cell death. The same group further developed the concept by creating a smart conjugate called ALARM. They attached an ALP responsive moiety to an NIR probe (f-RCP) capable of generating singlet oxygen (Fig. 7A).95 The overexpressed ALP on the cancer cell membrane cleaves the phosphate group, stimulating NIR fluorescence and PA response for tumor imaging. Irradiation of the NIR probe at 660 nm induces ring opening in furan and covalently labels nucleobases of cytoplasmic RNAs for prolonged localization. Transcriptomic analysis revealed downregulation of several genes linked to signalling pathways such as p53, carcinoma, TNF, MAPK, and TNF in HepG cells (Fig. 7D). Upregulation of several apoptosis-inducing genes supports the mode of tumor cell death by ALARM. A control probe RCP, without the furan moiety, failed to induce changes in apoptosis-inducing genes. For in vivo evaluation of the antitumor effect, HepG cells injected into BALB/c mice were administered with f-RCP and control RCP via i.v. injection followed by irradiation with a 660 nm laser (0.1 W cm−2) for 3 minutes, 4 hours post-injection. The mice group treated with f-RCP + 600 nm irradiation showed up to a 70% reduction in tumor volume with low systemic toxicity (Fig. 7E). This small molecule conjugate-based system provides promising insight towards precision medicine, a primary objective of theranostics.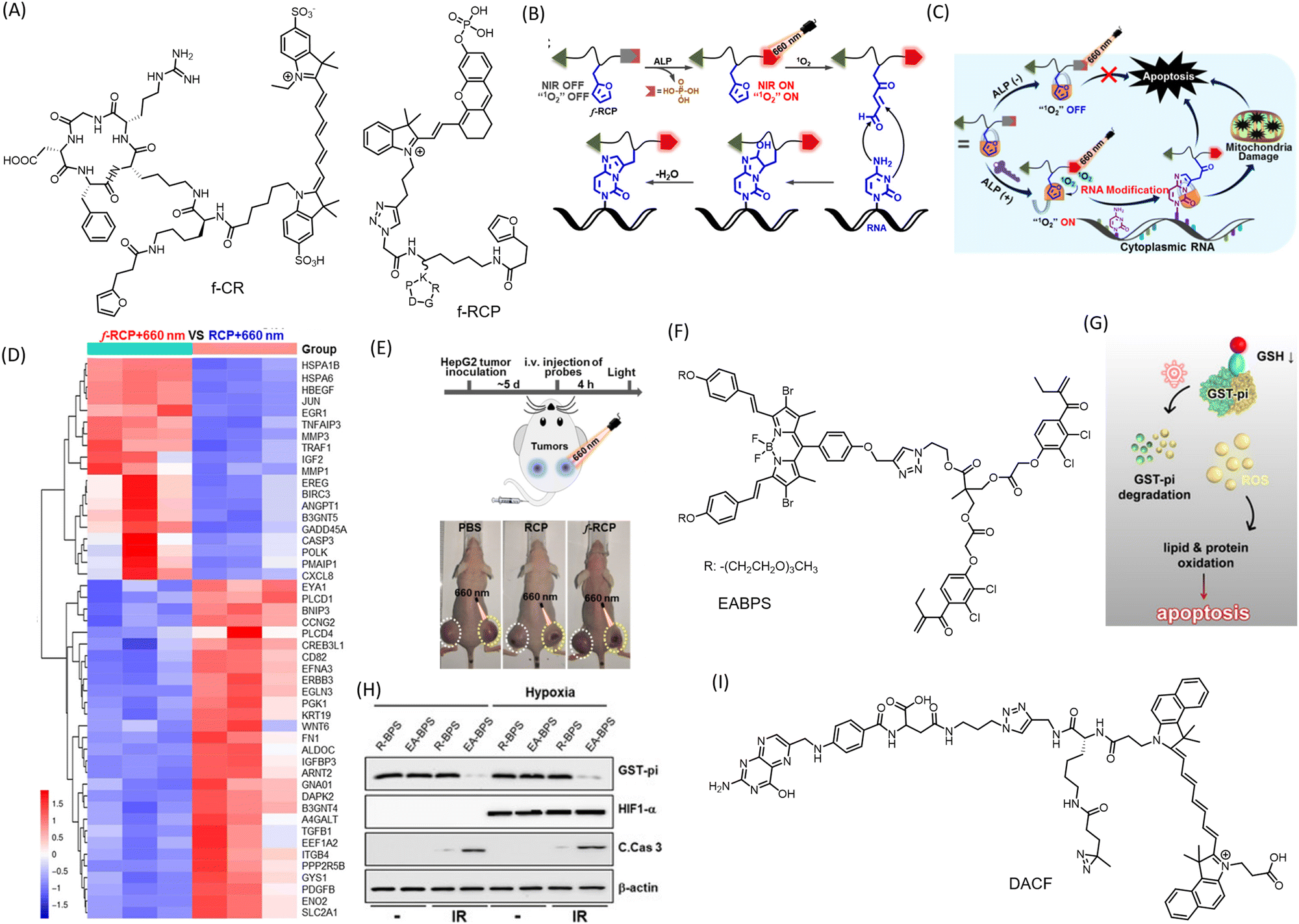 | ||
| Fig. 7 Small molecule conjugates with covalent conjugation strategies. (A) Molecular structures of conjugates fCR and fRCP (B) Mechanism of chemical conjugation of the furan moiety in fRCP with cellular RNA, and (C) mechanism of anticancer activity of the fRCP. (D) Transcriptomic analysis of HepG cells upon treatment with fRCP and with control conjugate. (E) Schematic representation of protocol for PDT and reduction in HepG2 tumor in mice injected with fRCP. (B)–(E) Are reproduced from ref. 95 with permission from American Chemical Society. (F) Molecular structure of EABPS possessing ethacrynic acid (EA) for Michael addition. (G) Mechanism of action of the theranostic conjuagate in inducing apoptosis mediated cell death. (H) Western blots showing decreased protein expression of GST-pi upon treatment with EA-BPS. (G) and (H) Are reproduced from ref. 97 with permission from Wiley-VCH, copyright 2020. (I) Molecular structure of DACF, with aziridine moiety for covalent conjugation with biomolecules. | ||
A novel BODIPY (boron-dipyrromethene) based agent with ROS-independent photothermal (PTT) activity for cancer treatment has been reported.96 The probe exhibits an excellent phototoxicity index (PI) value of 360![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 under oxygen-deprived conditions, indicating oxygen-independent activity. Its far-red emission makes it a promising agent for in vivo imaging applications. Upon irradiation at 630 nm, the molecule causes photocytotoxicity and cellular toxicity via disruption of MMP in HeLa cells, resulting in apoptosis or necrosis. With the extensive exploration of BODIPY's excellent emissive properties, a large library of molecules can be assessed for their photothermal and therapeutic efficacy. A molecular scaffold (EABPS) (Fig. 7F) combining brominated BODIPY and ethacrynic acid (EA) as a PS and glutathione S-transferase-pi (GST-pi) inhibitor respectively was successfully used to overcome hypoxia-induced resistance in PDT (Fig. 7G and H).97 EA undergoes covalent conjugation with reduced glutathione via Michael addition.
000 under oxygen-deprived conditions, indicating oxygen-independent activity. Its far-red emission makes it a promising agent for in vivo imaging applications. Upon irradiation at 630 nm, the molecule causes photocytotoxicity and cellular toxicity via disruption of MMP in HeLa cells, resulting in apoptosis or necrosis. With the extensive exploration of BODIPY's excellent emissive properties, a large library of molecules can be assessed for their photothermal and therapeutic efficacy. A molecular scaffold (EABPS) (Fig. 7F) combining brominated BODIPY and ethacrynic acid (EA) as a PS and glutathione S-transferase-pi (GST-pi) inhibitor respectively was successfully used to overcome hypoxia-induced resistance in PDT (Fig. 7G and H).97 EA undergoes covalent conjugation with reduced glutathione via Michael addition.
A small molecule conjugate named DACF (Fig. 7I) using diaziridine chemistry has been reported.98 It consists of three motifs: an NIR dye cypate for in vivo NIR/PA dual imaging, a folic acid unit for tumor targeting, and a diazirine group for crosslinking with available biomolecules upon irradiation. One of the most notable features of this probe is its ability to undergo a photo-crosslinking reaction with surrounding biomolecules through an insertion reaction upon illumination with a 405 nm laser (1 W cm−2). This reaction results in the covalent immobilization of the probe within tumor cells, leading to enhanced tumor accumulation and prolonged retention. This significantly improves the imaging and PTT efficacy of the tumor in vivo. Localization by folic acid-mediated anchoring followed by photo-initiated in situ immobilization of the theranostic conjugate in tumors proved excellent for image-guided PTT to ablate tumor tissue in vivo. DACF proved to be an excellent contrast agent for tumor PA imaging in 4T1 tumor-bearing mice. For PTT, irradiation with an 808 nm laser significantly raised the temperature by ∼17.2 °C.
Antibody–drug conjugate theranostics
Targeting specific disease antigens is an emerging strategy for achieving target specificity in therapeutic intervention. The conjugation of an antibody with a drug molecule, known as an antibody–drug conjugate (ADC), is of great importance in the realization of precision medicine. While antibodies provide highly specific targeting of cancer cells, small molecules have the advantage of targeting specific cellular pathways to slow cancer progression. The combination of precise targeting and effective therapeutic action makes ADCs a promising tool for precision medicine. The use of FDA-approved molecules in the design of ADCs can reduce the lengthy approval process for drug candidates. An ADC design strategy incorporates three components: a disease antigen-specific antibody, a linker, and a drug candidate (payload). The chosen antibody should be highly specific, efficiently internalized and retained, and exhibit high specificity towards tumor antigens while minimizing immunogenicity. The early success of this strategy is evident from the approval of 14 ADCs, mostly for the treatment of different types of cancer, with around 60 ADCs currently in clinical trials. A comprehensive summary of the design and application of ADCs has been covered elsewhere.99 Current developments in ADCs have considered the incorporation of theranostic modalities into the design strategy.A recent strategy has employed the use of a cathepsin B-sensitive 7-amino-3-hydroxyethyl-coumarin (7-AHC) self-elimination and turn-on fluorescence (at 471 nm).100 The 7-AHC, attached to a valine–alanine dipeptide, serves as a linker connecting the anti-Her2 antibody and the payload monomethyl auristatin E (MMAE), an antimitotic anticancer drug. The Her-2 receptors facilitate selective targeting and internalization of the ADC into tumor cells, where CTSB induces cleavage of the dipeptide bond in Mil40-E-15C, releasing MMAE and 7-AHC. The ADC Mil40-E-15C (Fig. 8A) had a long half-life of 5–7 days in mice and showed an effective and sustained tumor inhibition rate of 54.3% upon administration of 5 mg kg−1 ADC in SKOV3 xenograft mice (Fig. 8B). Selective accumulation of the ADC in the tumor was evident from ex vivo imaging of the tumor and organs collected from treated mice (Fig. 8C). A photocaging strategy was incorporated for the ADC, where irradiation with IR light initiated a cascade of chemical reactions resulting in the uncaging of the drug molecule.101 Combretastatin A4 (CA4), a microtubule-binding antiangiogenic drug, was conjugated to an anti-EGFR mAb Panitumumab (Pan) and a cyanine photocage using IR-783. CY-Pan-CA4 (Fig. 8D) showed excellent tumor accumulation in an A431 (EGFR+) xenograft tumor model with emission at 800 nm. Irradiation with a 690 nm laser (500 mW cm−2) resulted in reduced tumor volume, while no therapeutic effect was observed in the absence of light (Fig. 8E).
 | ||
| Fig. 8 (A) Chemical structure of theranostic ADC Mil40-E-15C. (B) Change in tumor volume upon administration of Mil40-E-15C in SKOV3 xenograft mice. (C) Ex vivo fluorescence image of tumor and major organs in mice upon administration of the ADC. (B) and (C) Are reproduced from ref. 100, with permission from IVYSPRING international publisher, copyright 2021. (D) Design of photocleavable heptamethine cyanine-based ADC CY-Pan-CA4. (E) Optical and fluorescence images of A431 tumors, and effect of dose-dependent irradiation of right tumor with near-IR light two days after administration with CY-Pan-CA4. Reproduced from ref. 101 with permission from Wiley-VCH, copyright 2015. (F) Molecular design of CyEt-Pan-Duo. | ||
In order to improve stability and in vivo efficacy, systematic modifications were introduced to the linker domain and heterocyclic moieties in the cyanine dye, refining the structure–function relationship of the ADC.102 The optimized conjugate CyEt-Pan-Duo (Fig. 8F) showed selective binding and internalization in MDA-MB-468 (EGFR+) cells, while no internalization was observed in MCF-7 (EGFR−) cells. Treatment with CyEt-Pan-Duo for 24 h followed by irradiation using a 690 nm light (20 J cm−2) resulted in reduced cell viability comparable to duocarmycin DM after 72 h. The increased absorption coefficient and red-shifted absorbance band allowed irradiation at longer wavelength (780 nm) with lower energy (5 J cm−2), making CyEt-Pan-Duo an attractive tool for in vivo applications.
Although ADC-based theranostics is still in its infancy, rigorous and systematic optimization of various ADC components, such as antigen/antibody combinations, linkers, and payloads, could bring about a revolution in the field of precision medicine.
Combating pathogens
The emergence of new pathogens and their rapid spread across the world poses a major challenge to global health infrastructure. Targeting specific pathogens by nucleic acid or protein recognition is an attractive approach for tackling pathogen infections. Malaria is a life-threatening disease caused by the parasite called Plasmodium falciparum and transmitted by anopheles’ mosquito. Often associated with life threatening complications, early diagnosis and effective treatment of malaria is of utmost importance. Plasmodium falciparum is the etiological agent for malaria. Interestingly, ∼80% of its genome is composed of AT-rich DNA, which forms characteristic narrow binding pockets at the minor grooves. A crescent-shaped NIR probe recognizing AT-rich dsDNA with high fidelity was developed (Fig. 8A), exploiting the abundance of AT nucleobases in the parasite genome.103 The probe consists of two methylated benzothiazole groups as acceptor and donor phenol moiety designed to undergo internal charge transfer, transforming it into a turn-on NIR-probe for AT-rich DNA (Fig. 9A). Quinone cyanine–dithiazole (QCy-DT) binds specifically with the minor grooves of AT-rich DNA, eliciting ∼200-fold fluorescence enhancement at 650 nm (Fig. 9B and C). A striking sequence specificity was observed among AT-rich DNA, with the highest emission for the 5′-AAATTT-3′ sequence compared to other variations in DNA sequences (Fig. 9D). The QCy-DT probe could unambiguously stain the malarial parasite genome and effectively arrest it while exhibiting minimal toxicity in human cells lines (Fig. 9E). | ||
| Fig. 9 (A) Chemical structure of QCy-DT, and mechanism of transformation of QCy-DT into donor–acceptor based Cy7 system. (B) AT rich dsDNA minor groove recognition of QCy-DT by turn-on fluorescence emission. (C) Fluorescence spectra of QCy-DT and 200 folds turn-on emission upon addition of Drew-AT sequence. (D) Emission intensities of QCy-DT upon interaction with variable)(A/T)4 sequences. (E) Selective fluorescence detection of live Plasmodium falciparum parasite in human red blood cells upon incubation with QCy-DT. (F) Determination of IC50 value of Qcy-DT in Plasmodium falciparum. Reproduced from ref. 103 with permission from Oxford University Press, copyright 2015. | ||
Notably, QCy-DT is the first of its kind NIR-probe to recognize minor groove of AT-rich DNA with sequence selectivity and sensitivity. The malarial parasite at the trophozoite stage can be arrested by QCy-DT in the trophozoite stage with an IC50 value of <4 μM and failed to form rings in subsequent cycle at increased concentration of the theranostic probe, possibly via abrogation of DNA replication (Fig. 9F). Further, structure–activity relationship (SAR) on QCy-DT can be undertaken to develop potential drug candidate for malaria.
Noncanonical conformations like GQ have been attractive targets for identifying pathogens. Molecular probes targeting the GQ conformation in pathogens such as HCV and SARS-CoV-2 have been reported. A small molecule probe with specificity to GQ conformations has been developed for detecting hepatitis C virus (HCV) in living cells by fluorescence light up of viral RNA.104 We have recently reported GQ targeted reliable conformational polymorphism (GQ-RCP) platform based for selective and sensitive detection of SARS-CoV-2.105,106 These highly selective and sensitive designer probes based on benzothiazole and benzobisthiazole scaffolds detect specific nucleic acid sequences of SARS-CoV-2 that adapt GQ conformation. Further, exploiting the therapeutic efficacy of these probes in their present form and through structural fine-tuning would open avenues for more precise treatment of viral infections.
Alzheimer's disease
Alzheimer's disease (AD) is a severe neurological disorder with no early diagnosis or effective therapeutic tools.89,107 The accumulation of protein aggregation of Aβ and tau forms the major hallmarks of AD and are attractive targets for diagnosis and treatment.106,108 Development of tools for early detection and effective drug candidates to cure AD is a challenging task. Existing treatments for AD target N-methyl-D-aspartate (NMDA) receptor agonists like memantine and AChE inhibitors such as rivastigmine, galantamine, and donepezil, which provide temporary relief from certain symptoms of AD. Monoclonal antibody-based therapy has been designed to solubilize and clear Aβ aggregates, but the high cost and need for multiple dosages serve as a bottleneck, in addition to severe side effects. Small molecules have been found to be effective in preventing and sensing Aβ and tau aggregates (Fig. 10A), considering protein aggregation as the major hallmark of AD.109,110 Highly sensitive and specific molecular probes have been developed to image protein aggregation species in vivo. Reliable theranostic strategies for AD are imperative for early detection and treatment. However, the discovery of suitable probes is impeded by the inability to cross the blood–brain-barrier (BBB) and selectivity in binding and recognition of protein aggregates. Thioflavin T (ThT) (Fig. 8B) is a green-emitting probe that shows enhanced emission with protein aggregates and GQ DNA without any selectivity. Further, probes that can differentiate the different protein aggregates are rare.111 Our group has developed highly sensitive far-red emissive molecular probes, TC112 and CQ113 (Fig. 10B), for selective and sensitive detection of Aβ aggregates with nanomolar affinity. The CQ probe has a unique characteristic in that it can detect Alzheimer's disease and also differentiate it from other neurodegenerative diseases. This is especially useful in cases of mixed dementia. We have also developed a far-red fluorescent probe, Mito-TG, to track changes in mitochondrial dynamics during mitophagy and Aβ-induced mitochondrial changes, which has theranostic implications.114 Molecular probes for detecting tau neurofibrillary tangles (NFTs) have also been developed (Fig. 10B).115–118 However, theranostic agents for tau and Aβ are limited.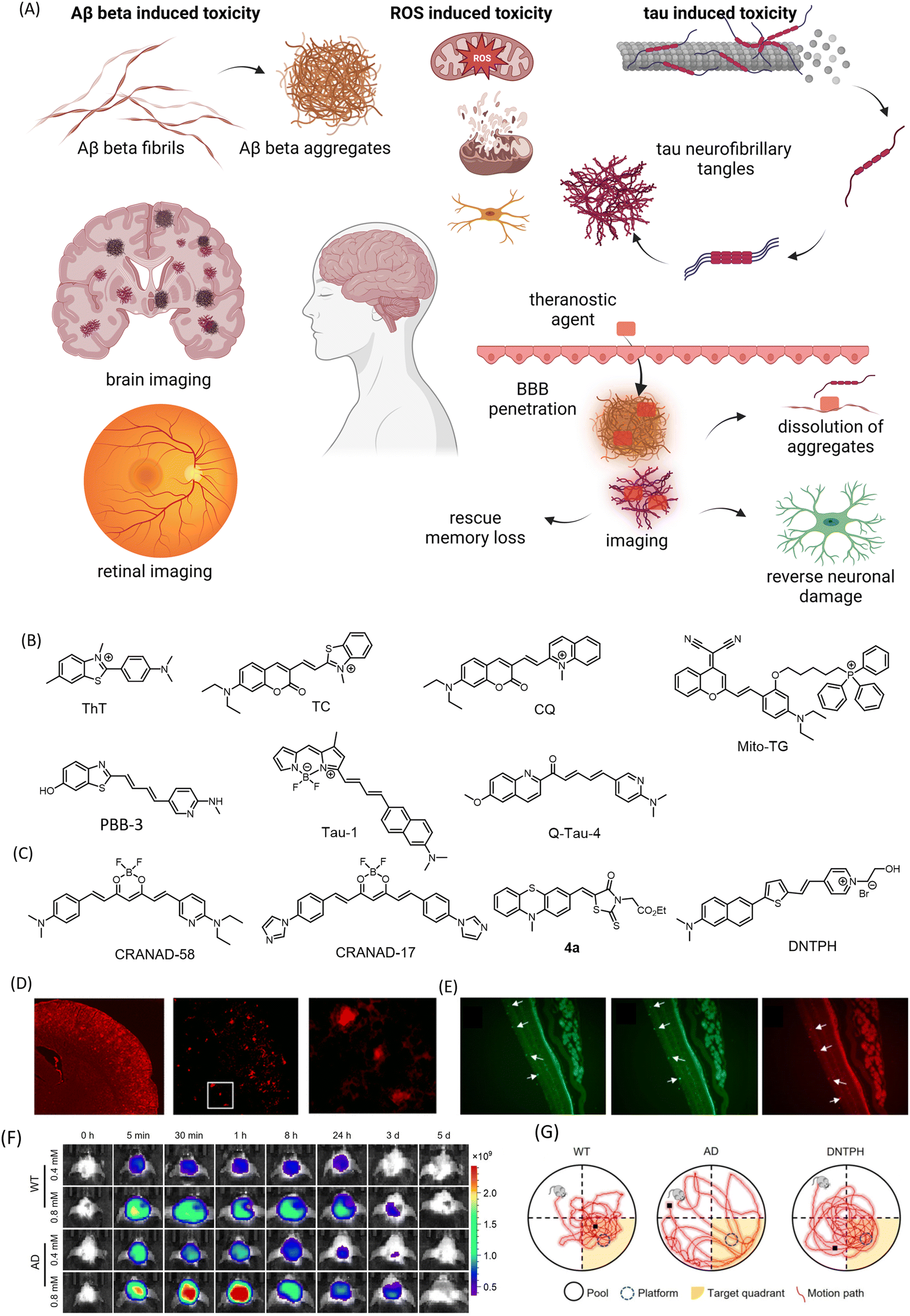 | ||
| Fig. 10 (A) Multifaceted toxicity, diagnosis, and treatment strategies in Alzheimer's disease. (B) turn-on fluorescent probes for Aβ (ThT, TC, and CQ), mitochondria (Mito-TG) and tau (PBB-3, Tau-1, Q-Tau-4, 2e) aggregates. (C) Molecular structures of theranostic candidates for Alzheimer's disease. (D) Fluorescence images of brain slices from 18 months old APP/PS1 mice stained with CRANAD-58. Reproduced from ref. 119 with permission from American Chemical Society, copyright 2013. (E) Fluorescence images of retinal tissue slices stained with ThT, Aβ antibody ab2454, and 4a1 in double Tg mice. Reproduced from ref. 120 with permission from American Chemical Society, copyright 2017. (F) In vivo fluorescence imaging of wild type (WT) and AD mice brain (7 months old) treated with DNTPH and (G) representative swimming track showing improvement in recognition/cognitive ability in AD mice treated with DNTPH, evident from Morris water maze (MWM) test. (F) and (G) Are reproduced from ref. 122 with permission from Wiley-VCH, copyright 2023. (A) Was created using https://BioRender.com. | ||
A curcumin-based small molecular theranostic agent has been designed where the diketo group was derivatized with difluoroboron for NIR imaging of Aβ deposits.119 Subsequent exploration of structure–activity led to the development of probes with increased sensitivity in fluorescent imaging of Aβ aggregates and attenuation of Aβ crosslinking induced by copper. CRANAD-58 (Fig. 10C) was successfully used to differentiate transgenic and wild-type mice (4 months old) via in vivo NIR imaging (Fig. 10D). CRANAD-17 (Fig. 10C), bearing imidazole groups, competitively coordinates with copper and prevents Aβ crosslinking via histidine 13 and 14. Owing to curcumin's ability to penetrate the BBB, the natural product holds great promise as a theranostic candidate for AD. Curcumin exhibits therapeutic potential against the multifaceted toxicity of AD such as amyloid accumulation, oxidative stress, inflammation, β-secretase, and even has a role in alleviating neurodegeneration-induced cognitive deficits. The retina and optic nerves share many commonalities with the brain. The presence of Aβ and tau has been established in retinal cells. A small molecule probe has been developed as a theranostic candidate for AD to visualize Aβ plaques in the brain by NIRF imaging and simultaneously dissolve and disaggregate the formed fibrils and inhibit its fibrillation.120 This phenothiazine-based theranostic probe, 4a1 (Fig. 10C), showed excellent binding affinity towards Aβ plaques in the nanomolar range (Kd = 7.5 nM). It was used to selectively visualize plaques in the brain slices as well as retinal tissue of double transgenic mice model (Fig. 10E). The probe displayed remarkable in vitro aggregation inhibition and dissolution compared to curcumin, a known therapeutic candidate for AD. To improve the probe characteristics, they designed 5a1 and 6a with NIR emission and improved affinity towards Aβ plaques.121 These molecules showed strong inhibitory effects on Aβ aggregation, promoted disaggregation, and reduced cytotoxicity brought on by Aβ in human neuroblastoma cells (SH-SY5Y), demonstrating their high theranostic potential for AD.
A multifunctional small molecule called DNTPH (Fig. 10C) has been designed with moieties to balance lipophilicity and hydrophilicity, resulting in a clogP value of 1.21.122 DNTPH showed around an 18-fold enhancement in fluorescence with Aβ fibrils. Its emission in the NIR region upon aggregation allowed for in vivo imaging of Aβ fibrils with good penetration depth (Fig. 10F). The theranostic agent exhibited a Kd value of 143 nM, which is 6 times higher than ThT. The agent also showed remarkable disaggregation of Aβ fibrils at sub-micromolar concentration (0.19 μM), resulting in the rescue of PC12 cells. Its BBB permeability allowed for in vivo imaging of fibrils in APP/PS1 transgenic mice. Administration of DNTPH resulted in a promising decline in Aβ plaques and improved memory impairments (Fig. 10G). Our group has reported small molecule TGR63 with excellent therapeutic effects and improvement in memory and behavioral deficits.123 TGR 63 shows remarkable dissolution of oligomers and fully formed fibrils. Improvising molecular structures to accommodate therapeutic and imaging modalities would be an exciting avenue for precise diagnosis and treatment. Besides imaging the aggregates, other reliable biomarkers for AD may complement or provide leads for early diagnosis of AD. In this direction, the spatiotemporal localization/deposition of specific metal ions or reactive intermediate species (RIS) may provide useful insight into AD progression. We recently discovered the localization of HOCl (an RIS) near Aβ deposits in APP/PSN mice.124 Changes in the morphological and chemical features of cells and organelles (mitochondria and lysosome) might prove to be reliable indicators of AD.
Conclusions and outlook
Theranostic agents based on small molecules and conjugates offer several advantages, including seamless delivery, precise targeting, imaging capabilities, and the ability to easily monitor drug action and treatment. These agents can also be easily tuned to enhance their therapeutic or diagnostic properties. Compared to other theranostic agents such as nuclear medicine, nanomaterials, polymers, and macromolecules, small molecule-based theranostic agents are preferred due to their small size and excellent pharmacological properties in terms of delivery and therapeutic effects. This review has discussed the emerging field of small molecules and conjugates as theranostic agents, which has the potential to pave the way for personalized medicine.Last decade has witnessed tremendous advancements in terms of innovation and improvisation in the theranostic approaches to combat deadly diseases like cancer, pathogenic infections, and neurodegeneration. Although a large volume of work has been done yielding diverse classes of theranostic agents, the majority of modalities are limited to proof of concept. The sensitivity, specificity, and quantifiability of imaging/diagnosis with existing theranostics platforms need careful evaluation and refinement. System-specific challenges in translation to clinical setup need to be considered in the design strategy. Exploring the therapeutic potential of currently explored molecular probes and the diagnostic potential of approved drugs would accelerate the identification of theranostic agents. Similarly, minimalistic blending of chemical moieties modulating cellular processes into emissive scaffolds would lead the way forward to purely small molecular theranostic agents. As understanding of various cellular degradation (autophagy) and programmed cell death pathways (apoptosis, necroptosis, pyroptosis, ferroptosis, cuproptosis, etc.) unfolds, tapping into such mechanisms using small molecular tools would be a powerful tool in theranostics. Another aspect that would significantly revolutionize the field of theranostics would be state-of-the-art technologies used for diagnosis. A synergy of advancements in synthetic organic chemistry, biology, and biomedical engineering would lead to the development of small molecular theranostics in light of precision medicine.
The discovery and validation of accessible markers for specific cancer types, and new reliable triggers for drug release and activation of imaging modalities would greatly enhance the development of theranostic tools. To realize and utilize the versatility of theranostics, careful trade-offs between molecular complexity/molecular weight and optimal efficiency and developing synergy between dose response and diagnostic requirements needs utmost attention. Along with the development of new chemical approaches to theranostics, extensive rigorous interrogation of the effect of drugs in a dose and time-dependent way on the level of various genes and proteins needs to be studied to achieve tangible progress in the number of theranostic candidates transitioning from bench to bedside. Synthetic organic chemistry and biorthogonal chemistry have much to offer in the realization of precision medicine.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors JNCASR (JNC/JNCASR/D0018/2021/01178 and TRC II: JNC/2022/01653) and DST, Govt. of India for grants to T. G. and UGC and DST-INSPIRE for research fellowships to S. P. and K. K. B.References
- S. S. Kelkar and T. M. Reineke, Bioconjugate Chem., 2011, 22, 1879–1903 CrossRef CAS PubMed.
- H. Amro, S. J. Wilderman, Y. K. Dewaraja and P. L. Roberson, J. Nucl. Med., 2010, 51, 654–659 CrossRef CAS PubMed.
- D. Xia, P. Wang, X. Ji, N. M. Khashab, J. L. Sessler and F. Huang, Chem. Rev., 2020, 120, 6070–6123 CrossRef CAS PubMed.
- B. T. Luk and L. Zhang, ACS Appl. Mater. Interfaces, 2014, 6, 21859–21873 CrossRef CAS PubMed.
- Y. H. Lao, K. K. L. Phua and K. W. Leong, ACS Nano, 2015, 9, 2235–2254 CrossRef CAS PubMed.
- H. Moorthy and T. Govindaraju, ACS Appl. Bio Mater., 2021, 4, 1115–1139 CrossRef CAS PubMed.
- W. Choi, B. Park, S. Choi, D. Oh, J. Kim and C. Kim, Chem. Rev., 2023, 123, 7379–7419 CrossRef CAS PubMed.
- Y. Liu, Y. Li, S. Koo, Y. Sun, Y. Liu, X. Liu, Y. Pan, Z. Zhang, M. Du, S. Lu, X. Qiao, J. Gao, X. Wang, Z. Deng, X. Meng, Y. Xiao, J. S. Kim and X. Hong, Chem. Rev., 2022, 122, 209–268 CrossRef CAS PubMed.
- P. J. Gawne, F. Man, P. J. Blower and R. T. M. De Rosales, Chem. Rev., 2022, 122, 10266–10318 CrossRef CAS PubMed.
- Z. Hu, W. Yang, H. Liu, K. Wang, C. Bao, T. Song, J. Wang and J. Tian, Mol. Pharmaceutics, 2014, 11, 3798–3809 CrossRef CAS PubMed.
- J. Wahsner, E. M. Gale, A. Rodríguez-Rodríguez and P. Caravan, Chem. Rev., 2019, 119, 957–1057 CrossRef CAS PubMed.
- G. Zhang, H. R. Ye, Y. Sun and Z. Z. Guo, ACS Sens., 2022, 7, 2857–2864 CrossRef CAS PubMed.
- C. Guo, Y. Jin and Z. Dai, Bioconjugate Chem., 2014, 25, 840–854 CrossRef CAS PubMed.
- J. S. Kim, M. Li, Y. Xu and X. Peng, Acc. Chem. Res., 2022, 55, 3253–3264 CrossRef PubMed.
- Y. Liu, L. Teng, B. Yin, H. Meng, X. Yin, S. Huan, G. Song and X. B. Zhang, Chem. Rev., 2022, 122, 6850–6918 CrossRef CAS PubMed.
- A. P. Castano, P. Mroz and M. R. Hamblin, Nat. Rev. Cancer, 2006, 6, 535–545 CrossRef CAS PubMed.
- X. Li, J. F. Lovell, J. Yoon and X. Chen, Nat. Rev. Clin. Oncol., 2020, 17, 657–674 CrossRef PubMed.
- T. I. Kostelnik and C. Orvig, Chem. Rev., 2019, 119, 902–956 CrossRef CAS PubMed.
- G. Sgouros, L. Bodei, M. R. McDevitt and J. R. Nedrow, Nat. Rev. Drug Discovery, 2020, 19, 589–608 CrossRef CAS PubMed.
- C. S. Cutler, H. M. Hennkens, N. Sisay, S. Huclier-Markai and S. S. Jurisson, Chem. Rev., 2013, 113, 858–883 CrossRef CAS PubMed.
- E. Boros and A. B. Packard, Chem. Rev., 2019, 119, 870–901 CrossRef CAS PubMed.
- T. A. Lemaître, A. R. Burgoyne, M. Ooms, T. N. Parac-Vogt and T. Cardinaels, ACS Appl. Nano Mater., 2022, 5, 8680–8709 CrossRef.
- S. M. Ametamey, M. Honer and P. A. Schubiger, Chem. Rev., 2008, 108, 1501–1516 CrossRef CAS PubMed.
- J. Shi, P. W. Kantoff, R. Wooster and O. C. Farokhzad, Nat. Rev. Cancer, 2017, 17, 20–37 CrossRef CAS PubMed.
- X. Y. Wong, A. Sena-Torralba, R. Álvarez-Diduk, K. Muthoosamy and A. Merkoçi, ACS Nano, 2020, 14, 2585–2627 CrossRef CAS PubMed.
- E. K. Lim, T. Kim, S. Paik, S. Haam, Y. M. Huh and K. Lee, Chem. Rev., 2015, 115, 327–394 CrossRef CAS PubMed.
- L. Cheng, C. Wang, L. Feng, K. Yang and Z. Liu, Chem. Rev., 2014, 114, 10869–10939 CrossRef CAS PubMed.
- S. S. Lucky, K. C. Soo and Y. Zhang, Chem. Rev., 2015, 115, 1990–2042 CrossRef CAS PubMed.
- H. Chen, W. Zhang, G. Zhu, J. Xie and X. Chen, Nat. Rev. Mater., 2017, 2, 17024 CrossRef CAS PubMed.
- X. Xue, A. Lindstrom and Y. Li, Bioconjugate Chem., 2019, 30, 1585–1603 CrossRef CAS PubMed.
- I. Ekladious, Y. L. Colson and M. W. Grinstaff, Nat. Rev. Drug Discovery, 2019, 18, 273–294 CrossRef CAS PubMed.
- T. Krasia-Christoforou and T. K. Georgiou, J. Mater. Chem. B, 2013, 1, 3002–3025 RSC.
- S. Mignani, X. Shi, J. Rodrigues, R. Roy, Á. Muñoz-Fernández, V. Ceña and J. P. Majoral, Bioconjugate Chem., 2020, 31, 2060–2071 CrossRef CAS PubMed.
- J. Guo, H. Hong, G. Chen, S. Shi, T. R. Nayak, C. P. Theuer, T. E. Barnhart, W. Cai and S. Gong, ACS Appl. Mater. Interfaces, 2014, 6, 21769–21779 CrossRef CAS PubMed.
- H. Xing, K. Hwang and Y. Lu, Theranostics, 2016, 6, 1336–1352 CrossRef CAS PubMed.
- A. Ediriwickrema and W. M. Saltzman, ACS Biomater. Sci. Eng., 2015, 1, 64–78 CrossRef CAS PubMed.
- J. Wang, J. Li, L. Wang, T. Han, D. Wang and B. Z. Tang, ACS Appl. Polym. Mater., 2020, 2, 4306–4318 CrossRef CAS.
- K. Haupt, P. X. Medina Rangel and B. T. S. Bui, Chem. Rev., 2020, 120, 9554–9582 CrossRef CAS PubMed.
- M. R. Gordon, M. Canakci, L. Li, J. Zhuang, B. Osborne and S. Thayumanavan, Bioconjugate Chem., 2015, 26, 2198–2215 CrossRef CAS PubMed.
- Z. Saadatpour, G. Bjorklund, S. Chirumbolo, M. Alimohammadi, H. Ehsani, H. Ebrahiminejad, H. Pourghadamyari, B. Baghaei, H. R. Mirzaei, A. Sahebkar, H. Mirzaei and M. Keshavarzi, Cancer Gene Ther., 2016, 1–5 Search PubMed.
- F. M. Haque, J. S. A. Ishibashi, C. A. L. Lidston, H. Shao, F. S. Bates, A. B. Chang, G. W. Coates, C. J. Cramer, P. J. Dauenhauer, W. R. Dichtel, C. J. Ellison, E. A. Gormong, L. S. Hamachi, T. R. Hoye, M. Jin, J. A. Kalow, H. J. Kim, G. Kumar, C. J. Lasalle, S. Liffland, B. M. Lipinski, Y. Pang, R. Parveen, X. Peng, Y. Popowski, E. A. Prebihalo, Y. Reddi, T. M. Reineke, D. T. Sheppard, J. L. Swartz, W. B. Tolman, B. Vlaisavljevich, J. Wissinger, S. Xu and M. A. Hillmyer, Chem. Rev., 2022, 122, 6322–6373 CrossRef CAS PubMed.
- A. C. Therapy, Nat. Cancer, 2021, 2, 245–246 CrossRef PubMed.
- R. C. Fitzgerald, A. C. Antoniou, L. Fruk and N. Rosenfeld, Nat. Med., 2022, 28, 666–677 CrossRef CAS PubMed.
- B. A. Chabner and T. G. Roberts, Nat. Rev. Cancer, 2005, 5, 65–72 CrossRef CAS PubMed.
- L. Kelland, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef CAS PubMed.
- Z. H. Siddik, Oncogene, 2003, 22, 7265–7279 CrossRef CAS PubMed.
- G. Cavaletti and P. Marmiroli, Nat. Rev. Neurol., 2010, 6, 657–666 CrossRef PubMed.
- A. Lin, C. J. Giuliano, A. Palladino, K. M. John, C. Abramowicz, M. Lou Yuan, E. L. Sausville, D. A. Lukow, L. Liu, A. R. Chait, Z. C. Galluzzo, C. Tucker and J. M. Sheltzer, Sci. Transl. Med., 2019, 11, eaaw8412 CrossRef CAS PubMed.
- T. C. Pham, V. N. Nguyen, Y. Choi, S. Lee and J. Yoon, Chem. Rev., 2021, 121, 13454–13619 CrossRef CAS PubMed.
- Y. Zhu, Q. Li, C. Wang, Y. Hao, N. Yang, M. Chen, J. Ji, L. Feng and Z. Liu, Chem. Rev., 2023, 123, 7326–7378 CrossRef CAS PubMed.
- B. Perillo, M. Di Donato, A. Pezone, E. Di Zazzo, P. Giovannelli, G. Galasso, G. Castoria and A. Migliaccio, Exp. Mol. Med., 2020, 52, 192–203 CrossRef CAS PubMed.
- Y. V. Suseela, N. Narayanaswamy, S. Pratihar and T. Govindaraju, Chem. Soc. Rev., 2018, 47, 1098–1131 RSC.
- Y. V. Suseela, P. Satha, N. Arul Murugan and T. Govindaraju, Theranostics, 2020, 10, 10394–10414 CrossRef CAS PubMed.
- D. Müller, P. Saha, D. Panda, J. Dash and H. Schwalbe, Chem. – Eur. J., 2021, 27, 12726–12736 CrossRef PubMed.
- Y. L. Tsai, Z. F. Wang, W. W. Chen and T. C. Chang, Nucleic Acids Res., 2011, 39, 1–12 CrossRef PubMed.
- Y. Venkata Suseela, P. Sengupta, T. Roychowdhury, S. Panda, S. Talukdar, S. Chattopadhyay, S. Chatterjee and T. Govindaraju, ACS Bio Med Chem Au, 2022, 2, 125–139 CrossRef CAS PubMed.
- J. Dai, X. Wu, S. Ding, X. Lou, F. Xia, S. Wang and Y. Hong, J. Med. Chem., 2020, 63, 1996–2012 CrossRef CAS PubMed.
- S. Singh, A. Aggarwal, N. V. S. D. K. Bhupathiraju, G. Arianna, K. Tiwari and C. M. Drain, Chem. Rev., 2015, 115, 10261–10306 CrossRef CAS PubMed.
- M. Wu, X. Liu, H. Chen, Y. Duan, J. Liu, Y. Pan and B. Liu, Angew. Chem., Int. Ed., 2021, 60, 9093–9098 CrossRef CAS PubMed.
- H. T. Feng, S. Zou, M. Chen, F. Xiong, M. H. Lee, L. Fang and B. Z. Tang, J. Am. Chem. Soc., 2020, 142, 11442–11450 CrossRef CAS PubMed.
- S. Zeng, C. Chen, L. Zhang, X. Liu, M. Qian, H. Cui, J. Wang, Q. Chen and X. Peng, Bioact. Mater., 2022, 25, 580–593 CrossRef PubMed.
- S. Luo, X. Tan, S. Fang, Y. Wang, T. Liu, X. Wang, Y. Yuan, H. Sun, Q. Qi and C. Shi, Adv. Funct. Mater., 2016, 26, 2826–2835 CrossRef CAS.
- Y. Li, M. He, Z. Liu, C. Chuah, Y. Tang, Y. Duo and B. Z. Tang, J. Mater. Chem. B, 2023, 2700–2705 RSC.
- E. Ortega-Forte, A. Rovira, A. Gandioso, J. Bonelli, M. Bosch, J. Ruiz and V. Marchán, J. Med. Chem., 2021, 64, 17209–17220 CrossRef CAS PubMed.
- S. Kuang, L. Sun, X. Zhang, X. Liao, T. W. Rees, L. Zeng, Y. Chen, X. Zhang, L. Ji and H. Chao, Angew. Chem., Int. Ed., 2020, 59, 20697–20703 CrossRef CAS PubMed.
- S. Siriwibool, N. Kaekratoke, K. Chansaenpak, K. Siwawannapong, P. Panajapo, K. Sagarik, P. Noisa, R. Y. Lai and A. Kamkaew, Sci. Rep., 2020, 10, 1–10 CrossRef PubMed.
- J. Rautio, H. Kumpulainen, T. Heimbach, R. Oliyai, D. Oh, T. Järvinen and J. Savolainen, Nat. Rev. Drug Discovery, 2008, 7, 255–270 CrossRef CAS PubMed.
- I. R. Vlahov and C. P. Leamon, Bioconjugate Chem., 2012, 23, 1357–1369 CrossRef CAS PubMed.
- S. Maiti, N. Park, J. H. Han, H. M. Jeon, J. H. Lee, S. Bhuniya, C. Kang and J. S. Kim, J. Am. Chem. Soc., 2013, 135, 4567–4572 CrossRef CAS PubMed.
- S. Bhuniya, S. Maiti, E. J. Kim, H. Lee, J. L. Sessler, K. S. Hong and J. S. Kim, Angew. Chem., Int. Ed., 2014, 53, 4469–4474 CrossRef CAS PubMed.
- Y. V. Suseela, S. Das, S. K. Pati and T. Govindaraju, ChemBioChem, 2016, 17, 2162–2171 CrossRef CAS PubMed.
- K. N. Bobba, G. Saranya, P. T. Sujai, M. M. Joseph, N. Velusamy, A. Podder, K. K. Maiti and S. Bhuniya, ACS Appl. Bio Mater., 2019, 2, 1322–1330 CrossRef CAS PubMed.
- S. Santra, C. Kaittanis, O. J. Santiesteban and J. M. Perez, J. Am. Chem. Soc., 2011, 133, 16680–16688 CrossRef CAS PubMed.
- Y. Yuan, R. T. K. Kwok, B. Z. Tang and B. Liu, J. Am. Chem. Soc., 2014, 136, 2546–2554 CrossRef CAS PubMed.
- S. Bin Yang, D. N. Lee, J. H. Lee, M. Seo, D. W. Shin, S. Lee, Y. H. Lee and J. Park, Bioconjugate Chem., 2023, 34, 333–344 CrossRef PubMed.
- F. Liu, X. Ding, X. Xu, F. Wang, X. Chu and J. H. Jiang, Angew. Chem., Int. Ed., 2022, 61, e202203243 CrossRef CAS PubMed.
- S. M. Usama, B. Zhao and K. Burgess, Bioconjugate Chem., 2019, 30, 1175–1181 CrossRef CAS PubMed.
- D. J. Lee, V. Juvekar, H. W. Lee, E. S. Kim, C. K. Noh, S. J. Shin and H. M. Kim, Anal. Chem., 2021, 93, 16821–16827 CrossRef CAS PubMed.
- G. Nkepang, M. Bio, P. Rajaputra, S. G. Awuah and Y. You, Bioconjugate Chem., 2014, 25, 2175–2188 CrossRef CAS PubMed.
- M. Li, S. Long, Y. Kang, L. Guo, J. Wang, J. Fan, J. Du and X. Peng, J. Am. Chem. Soc., 2018, 140, 15820–15826 CrossRef CAS PubMed.
- X. Meng, Y. Yang, L. Zhou, L. Zhang, Y. Lv, S. Li, Y. Wu, M. Zheng, W. Li, G. Gao, G. Deng, T. Jiang, D. Ni, P. Gong and L. Cai, Theranostics, 2017, 7, 1781–1794 CrossRef CAS PubMed.
- C. Wang, S. Wang, Y. Wang, H. Wu, K. Bao, R. Sheng and X. Li, Sci. Rep., 2020, 10, 1–9 CrossRef CAS PubMed.
- M. Gangopadhyay, R. Mengji, A. Paul, Y. Venkatesh, V. Vangala, A. Jana and N. D. P. Singh, Chem. Commun., 2017, 53, 9109–9112 RSC.
- D. C. Singleton, A. Macann and W. R. Wilson, Nat. Rev. Clin. Oncol., 2021, 18, 751–772 CrossRef PubMed.
- N. Narayanaswamy, S. Narra, R. R. Nair, D. K. Saini, P. Kondaiah and T. Govindaraju, Chem. Sci., 2016, 7, 2832–2841 RSC.
- J. Weber, L. Bollepalli, A. M. Belenguer, M. Di Antonio, N. De Mitri, J. Joseph, S. Balasubramanian, C. A. Hunter and S. E. Bohndiek, Cancer Res., 2019, 79, 5407–5417 CrossRef CAS PubMed.
- W. S. Shin, S. K. Park, P. Verwilst, S. Koo, J. H. Lee, S. G. Chi and J. S. Kim, Chem. Commun., 2017, 53, 1281–1284 RSC.
- M. Li, K. H. Gebremedhin, D. Ma, Z. Pu, T. Xiong, Y. Xu, J. S. Kim and X. Peng, J. Am. Chem. Soc., 2022, 144, 163–173 CrossRef CAS PubMed.
- W. Li, S. Yin, Y. Shen, H. Li, L. Yuan and X. B. Zhang, J. Am. Chem. Soc., 2023, 145, 3736–3747 CrossRef CAS PubMed.
- M. Li, J. Xia, R. Tian, J. Wang, J. Fan, J. Du, S. Long, X. Song, J. W. Foley and X. Peng, J. Am. Chem. Soc., 2018, 140, 14851–14859 CrossRef CAS PubMed.
- M. Li, Y. Shao, J. H. Kim, Z. Pu, X. Zhao, H. Huang, T. Xiong, Y. Kang, G. Li, K. Shao, J. Fan, J. W. Foley, J. S. Kim and X. Peng, ACS Appl. Mater. Interfaces, 2020, 142, 5380–5388 CAS.
- K. Kailass, O. Sadovski, W. R. Zipfel and A. A. Beharry, J. Med. Chem., 2022, 65, 8855–8868 CrossRef CAS PubMed.
- Z. Liu, F. Song, W. Shi, G. Gurzadyan, H. Yin, B. Song, R. Liang and X. Peng, ACS Appl. Mater. Interfaces, 2019, 11, 15426–15435 CrossRef CAS PubMed.
- S. Ye, C. Cui, X. Cheng, M. Zhao, Q. Mao, Y. Zhang, A. Wang, J. Fang, Y. Zhao and H. Shi, J. Am. Chem. Soc., 2020, 142, 21502–21512 CrossRef CAS PubMed.
- J. Fang, Y. Feng, Y. Zhang, A. Wang, J. Li, C. Cui, Y. Guo, J. Zhu, Z. Lv, Z. Zhao, C. Xu and H. Shi, J. Am. Chem. Soc., 2022, 144, 23061–23072 CrossRef CAS PubMed.
- L. Schneider, M. Kalt, S. Koch, S. Sithamparanathan, V. Villiger, J. Mattiat, F. Kradolfer, E. Slyshkina, S. Luber, M. Bonmarin, C. Maake and B. Spingler, J. Am. Chem. Soc., 2023, 145, 4534–4544 CrossRef CAS PubMed.
- M. Won, S. Koo, H. Li, J. L. Sessler, J. Y. Lee, A. Sharma and J. S. Kim, Angew. Chem., Int. Ed., 2021, 60, 3196–3204 CrossRef CAS PubMed.
- R. Sun, Y. Zhang, Y. Gao, M. Zhao, A. Wang, J. Zhu, X. Cheng and H. Shi, Chem. Sci., 2023, 14, 2369–2378 RSC.
- Z. Fu, S. Li, S. Han, C. Shi and Y. Zhang, Signal Transduction Targeted Ther., 2022, 7, 93 CrossRef CAS PubMed.
- D. Xiao, L. Zhao, F. Xie, S. Fan, L. Liu, W. Li, R. Cao, S. Li, W. Zhong and X. Zhou, Theranostics, 2021, 11, 2550–2563 CrossRef CAS PubMed.
- R. R. Nani, A. P. Gorka, T. Nagaya, H. Kobayashi and M. J. Schnermann, Angew. Chem., Int. Ed., 2015, 54, 13635–13638 CrossRef CAS PubMed.
- R. R. Nani, A. P. Gorka, T. Nagaya, T. Yamamoto, J. Ivanic, H. Kobayashi and M. J. Schnermann, ACS Cent. Sci., 2017, 3, 329–337 CrossRef CAS PubMed.
- N. Narayanaswamy, S. Das, P. K. Samanta, K. Banu, G. P. Sharma, N. Mondal, S. K. Dhar, S. K. Pati and T. Govindaraju, Nucleic Acids Res., 2015, 43, 8651–8663 CrossRef CAS PubMed.
- J. Zhang, H. Li, B. Lin, X. Luo, P. Yin, T. Yi, B. Xue, X. L. Zhang, H. Zhu and Z. Nie, J. Am. Chem. Soc., 2021, 143, 19317–19329 CrossRef CAS PubMed.
- S. Pratihar, R. Agrawal, V. K. Pal, A. Singh and T. Govindaraju, ACS Sens., 2022, 7, 453–459 CrossRef CAS PubMed.
- S. Pratihar, M. N. Mattath and T. Govindaraju, Chem. Commun., 2023, 59, 5717–5720 RSC.
- Alzheimer's disease: recent findings in pathophysiology, diagnostic and therapeutic modalities, ed. T. Govindaraju, Royal Society of Chemistry, 2021 Search PubMed.
- H. Arora, M. Ramesh, K. Rajasekhar and T. Govindaraju, Bull. Chem. Soc. Jpn., 2020, 93, 507–546 CrossRef CAS.
- K. Rajasekhar, M. Chakrabarti and T. Govindaraju, Chem. Commun., 2015, 51, 13434–13450 RSC.
- K. Rajasekhar and T. Govindaraju, RSC Adv., 2018, 8, 23780–23804 RSC.
- J. Hatai, L. Motiei and D. Margulies, J. Am. Chem. Soc., 2017, 139, 2136–2139 CrossRef CAS PubMed.
- K. Rajasekhar, N. Narayanaswamy, N. A. Murugan, G. Kuang, H. Ågren and T. Govindaraju, Sci. Rep., 2016, 6, 23668 CrossRef CAS PubMed.
- K. Rajasekhar, N. Narayanaswamy, N. A. Murugan, K. Viccaro, H. G. Lee, K. Shah and T. Govindaraju, Biosens. Bioelectron., 2017, 98, 54–61 CrossRef CAS PubMed.
- M. Ramesh, K. Rajasekhar, K. Gupta, V. Babagond, D. K. Saini and T. Govindaraju, Org. Biomol. Chem., 2021, 19, 801–808 RSC.
- M. Maruyama, H. Shimada, T. Suhara, H. Shinotoh, B. Ji, J. Maeda, M. R. Zhang, J. Q. Trojanowski, V. M. Y. Lee, M. Ono, K. Masamoto, H. Takano, N. Sahara, N. Iwata, N. Okamura, S. Furumoto, Y. Kudo, Q. Chang, T. C. Saido, A. Takashima, J. Lewis, M. K. Jang, I. Aoki, H. Ito and M. Higuchi, Neuron, 2013, 79, 1094–1108 CrossRef CAS PubMed.
- P. Verwilst, H. R. Kim, J. Seo, N. W. Sohn, S. Y. Cha, Y. Kim, S. Maeng, J. W. Shin, J. H. Kwak, C. Kang and J. S. Kim, J. Am. Chem. Soc., 2017, 139, 13393–13403 CrossRef CAS PubMed.
- A. A. Elbatrawy, S. J. Hyeon, N. Yue, E. E. A. Osman, S. H. Choi, S. Lim, Y. K. Kim, H. Ryu, M. Cui and G. Nam, ACS Sens., 2021, 6, 2281–2289 CrossRef CAS PubMed.
- K. S. Park, M. K. Kim, Y. Seo, T. Ha, K. Yoo, S. J. Hyeon, Y. J. Hwang, J. Lee, H. Ryu, H. Choo and Y. Chong, ACS Chem. Neurosci., 2017, 8, 2124–2131 CrossRef CAS PubMed.
- X. Zhang, Y. Tian, Z. Li, X. Tian, H. Sun, H. Liu, A. Moore and C. Ran, J. Am. Chem. Soc., 2013, 135, 16397–16409 CrossRef CAS PubMed.
- P. Dao, F. Ye, Y. Liu, Z. Y. Du, K. Zhang, C. Z. Dong, B. Meunier and H. Chen, ACS Chem. Neurosci., 2017, 8, 798–806 CrossRef CAS PubMed.
- P. Dao, F. Ye, Z. Y. Du, Q. Chen, K. Zhang, C. Z. Dong, B. Meunier and H. Chen, Dyes Pigm., 2017, 147, 130–140 CrossRef CAS.
- T. Zhang, X. Chen, C. Yuan, X. Pang, P. Shangguan, Y. Liu, L. Han, J. Sun, J. W. Y. Lam, Y. Liu, J. Wang, B. Shi and B. Zhong Tang, Angew. Chem., Int. Ed., 2023, 62, e202211550 CrossRef CAS PubMed.
- S. Samanta, K. Rajasekhar, M. Ramesh, N. A. Murugan, S. Alam, D. Shah, J. P. Clement and T. Govindaraju, Adv. Ther., 2021, 4, 1–12 Search PubMed.
- S. Samanta and T. Govindaraju, ACS Chem. Neurosci., 2019, 10, 4847–4853 CrossRef CAS PubMed.
Footnote |
| † S. P and K. K. B. contributed equally to this manuscript. |
| This journal is © The Royal Society of Chemistry 2023 |