DOI:
10.1039/D2RA06635A
(Review Article)
RSC Adv., 2022,
12, 33365-33402
Annulations involving 1-indanones to access fused- and spiro frameworks
Received
20th October 2022
, Accepted 16th November 2022
First published on 22nd November 2022
Abstract
Indanones are prominent motifs found in number of natural products and pharmaceuticals. Particularly, 1-indanones occupy important niche in chemical landscape due to their easy accessibility and versatile reactivity. In the past few years, significant advancement has been achieved regarding cyclization of 1-indanone core. The present review focuses on recent (2016–2022) annulations involving 1-indanones for the construction of fused- and spirocyclic frameworks. In this context, new strategies for synthesis of various carbocyclic as well as heterocyclic skeletons are demonstrated. Mechanistic aspects of representative reactions are illustrated for better understanding of reaction pathways. A large number of transformations described in this review offer stereoselective formation of desired polycyclic compounds. Importantly, several reactions provide biologically relevant compounds and natural products, such as, plecarpenene/plecarpenone, swinhoeisterol A, cephanolides A–D, diptoindonesin G and atlanticone C.
 Suven Das | Suven Das obtained his BSc, MSc and PhD Degree (2007) in Chemistry from the University of Calcutta, India. In 2007 he joined as Lecturer in Chemistry at Rishi Bankim Chandra College for Women, Naihati, India. After his post doctoral research at the National Tsing Hua University, Taiwan (2009) he joined as Assistant Professor in the same college to start his independent research career. He has published several research articles including some review articles in journals of international repute. His research interests focus on synthetic methodology, catalysis, indanone chemistry, and heterocycles. |
 Arpita Dutta | Arpita Dutta received her BSc, MSc and PhD Degree (2010) in Chemistry from the University of Calcutta, India. In 2010 she joined as Assistant Professor in Chemistry at Rishi Bankim Chandra Evening College, Naihati, India. She has published several research articles in journals of international repute. Her main research interests cover organic synthesis including peptide chemistry. |
1. Introduction
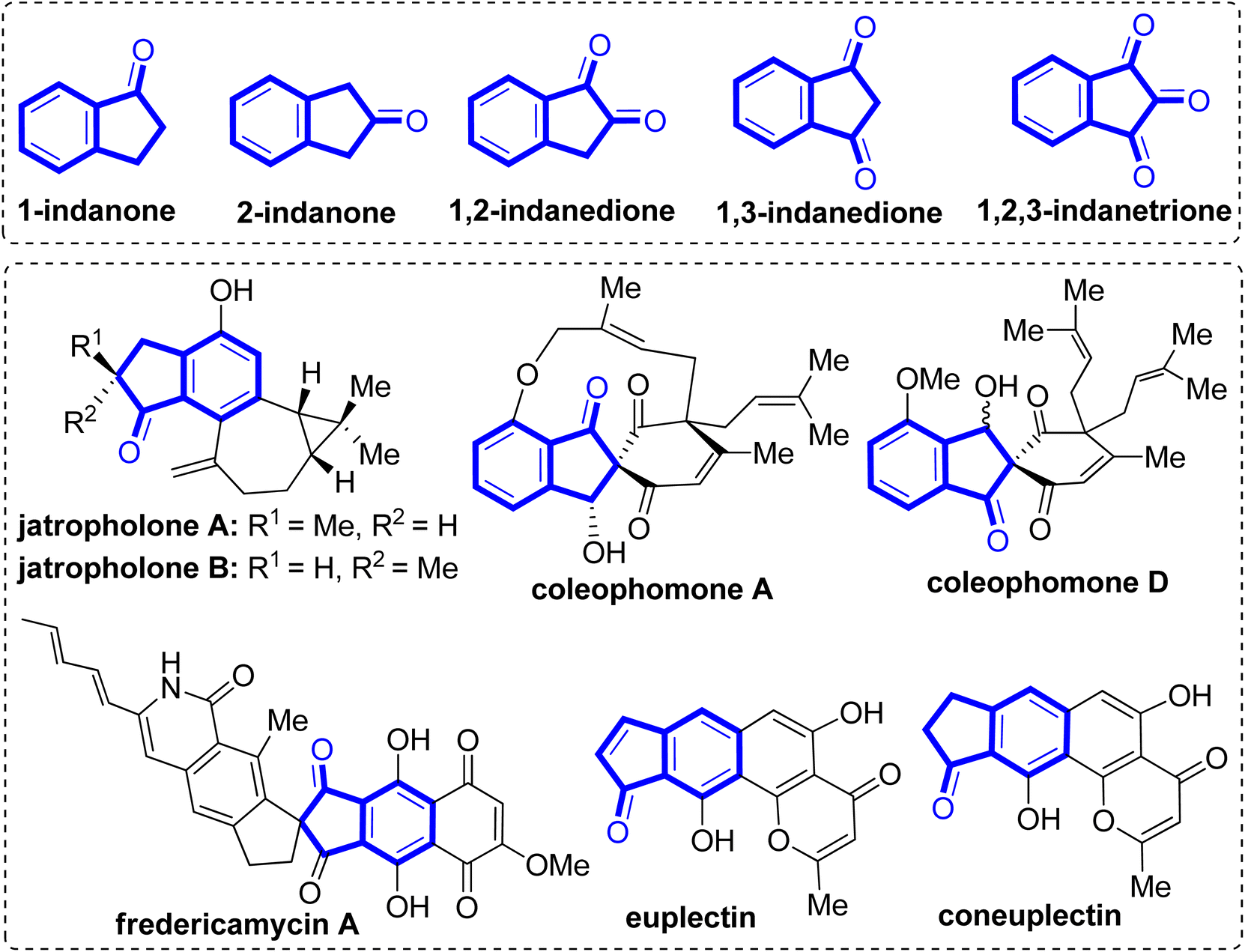
Indanones are privileged structural motifs frequently found in numerous natural products and synthetically bioactive molecules.1–6 In particular, annulated indanone scaffolds constitute key structural components of many bioactive natural products (Fig. 1). For example, jatropholone A and B isolated from the roots of Jatropha integerrima, exhibit antiplasmodial and cytotoxic activities.7,8 Coleophomone A extracted from a fungus Coleophoma sp. displays bacterial transglycosylase activity,9 whereas coleophomone D isolated from Stachybotrys cylindrospora reveal antifungal activity.10 Fredericamycin A obtained from Streptomyces griseus displays antitumor/anticancer activity.11,12 Euplectin (and coneuplectin), derived from the Lichen Flavoparmelia euplecta was found to exhibit promising cytotoxic and other biological activities.13 Likewise, several spirobenzylisoquinoline alkaloids containing indanone motif, viz. sibiricine, corydaine, raddeanine and yenhusomidine are well known for their potent pharmacological relevance.14,15
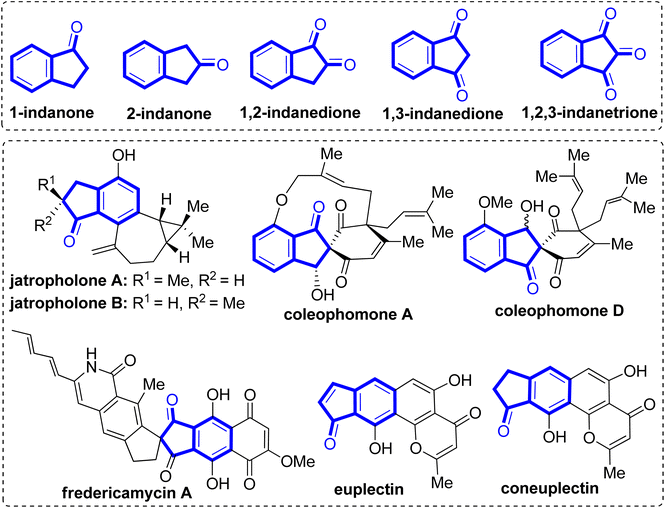 |
| | Fig. 1 Examples of various indanones and bioactive natural products containing indanone motif. | |
In addition to their profound biological profile, indanones have played significant role in the development of catalytic asymmetric synthesis.16–18 Moreover, indanone derivatives are largely employed as organic functional materials,19–21 OLEDs,22 dyes and fluorophores.23–27 Due to their ample appliances in different fields, various metal-catalyzed or metal-free methodologies have been adopted to develop 1-indanone core.28,29 Previously, Chanda and Singh published a review article on synthesis and application of 3-hydroxyindanone scaffolds covering the literature until 2015.30 However, the last few years have witnessed the emergence of efficient protocols for the target-oriented synthesis of novel annulation products involving 1-indanone moieties. This review emphasizes recent (2016–2022) applications of 1-indanones in cyclization to build up various fused- and spiro carbo-/heterocyclic compounds.
2. Synthesis of fused scaffolds
2.1. Fused carbocycles
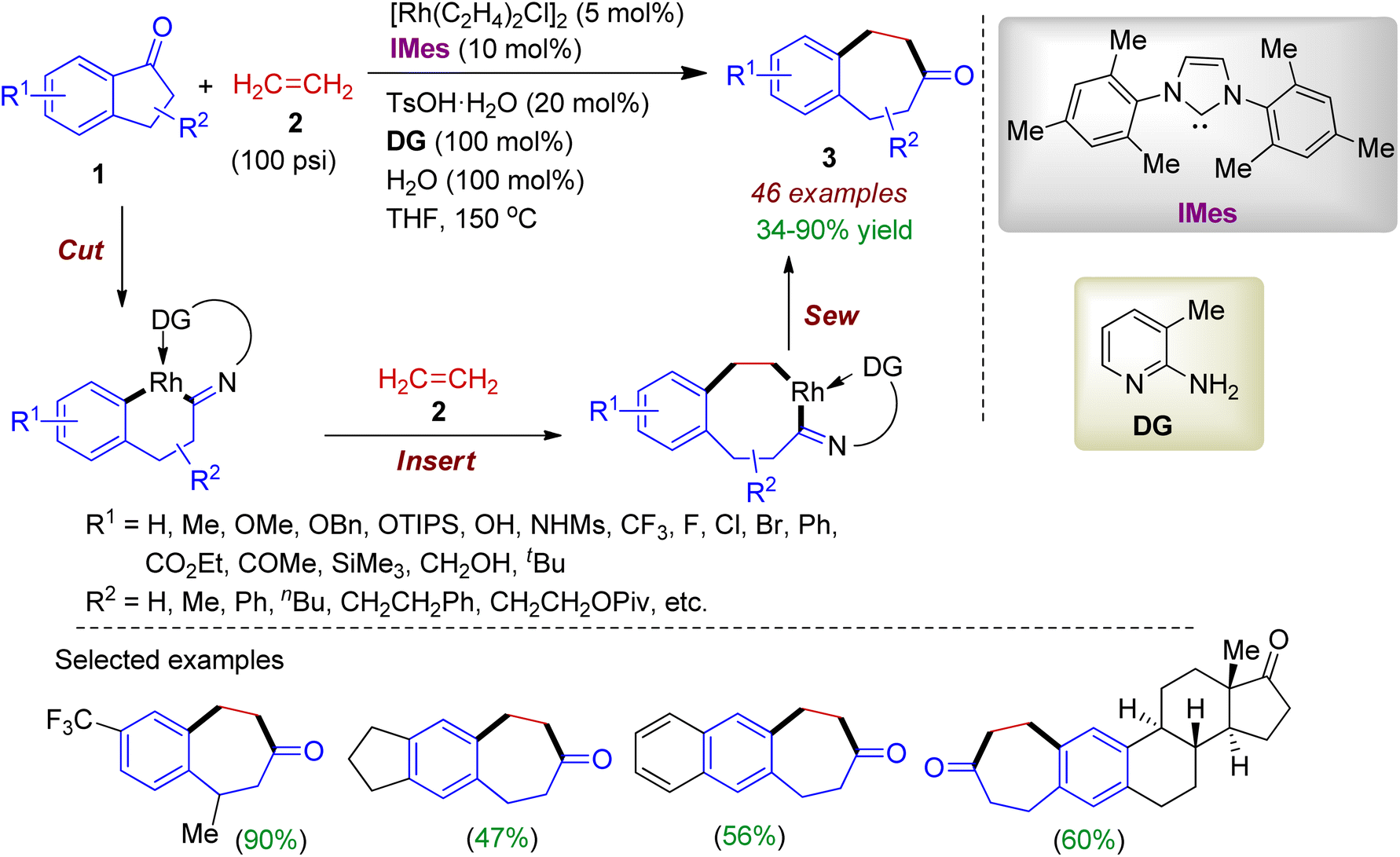
2.1.1. Benzannulated carbocycles via ring expansion. Ring expansion reactions of carbonyl compounds are frequently employed in organic transformations for the construction of complex molecular scaffolds. In this regard, 1-indanone 1 could serve as an effective cyclic substrate for various two-carbon ring expansion reactions. In 2019, Dong group carried out a rhodium-catalyzed direct insertion of ethylene 2 into relatively unstrained C–C bonds in 1-indanones 1 to form benzocycloheptenone skeleton 3.31 The reaction occurred in the presence of 5 mol% of catalyst [Rh(C2H4)2Cl]2, 10 mol% 1,3-bis(2,4,6-trimethylphenyl)imidazole-2-ylidene (IMes, ligand), 20 mol% p-toluenesulfonic acid monohydrate (TsOH·H2O), 100 mol% 2-amino-3-picoline (donor group) and 100 mol% H2O in THF to afford the desired compound. Substituted 1-indanones bearing electron-donating or -withdrawing substituents smoothly reacted with ethylene gas (100 psi) resulting benzannulated carbocycles in good yields (up to 90%). This two carbon ring-expansion reaction proceeded through “cut-insert-sew” fashion involving ethylene as a 2π unit (Scheme 1). The reaction is scalable, and also applied to natural product-derived or tethered indanones. Overall, the transition-metal catalyzed transformation is chemoselective, byproduct-free, redox-neutral, and applicable for straightforward synthesis of fused medium ring systems which are valuable synthetic intermediates for bioactive compounds.
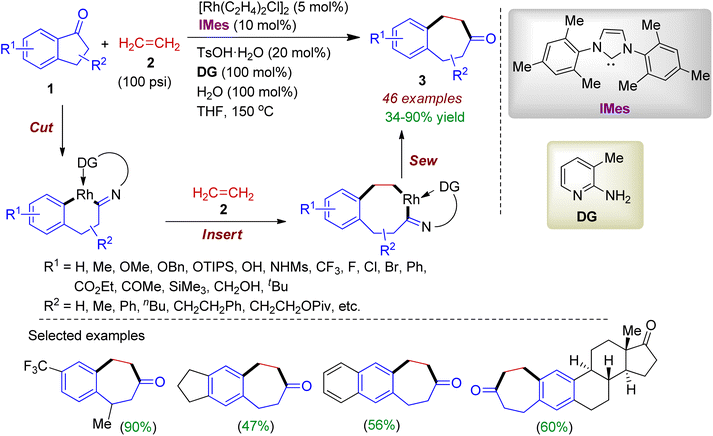 |
| | Scheme 1 Ring expansion of 1-indanones via insertion of ethylene to access fused seven-membered carbocycles. | |
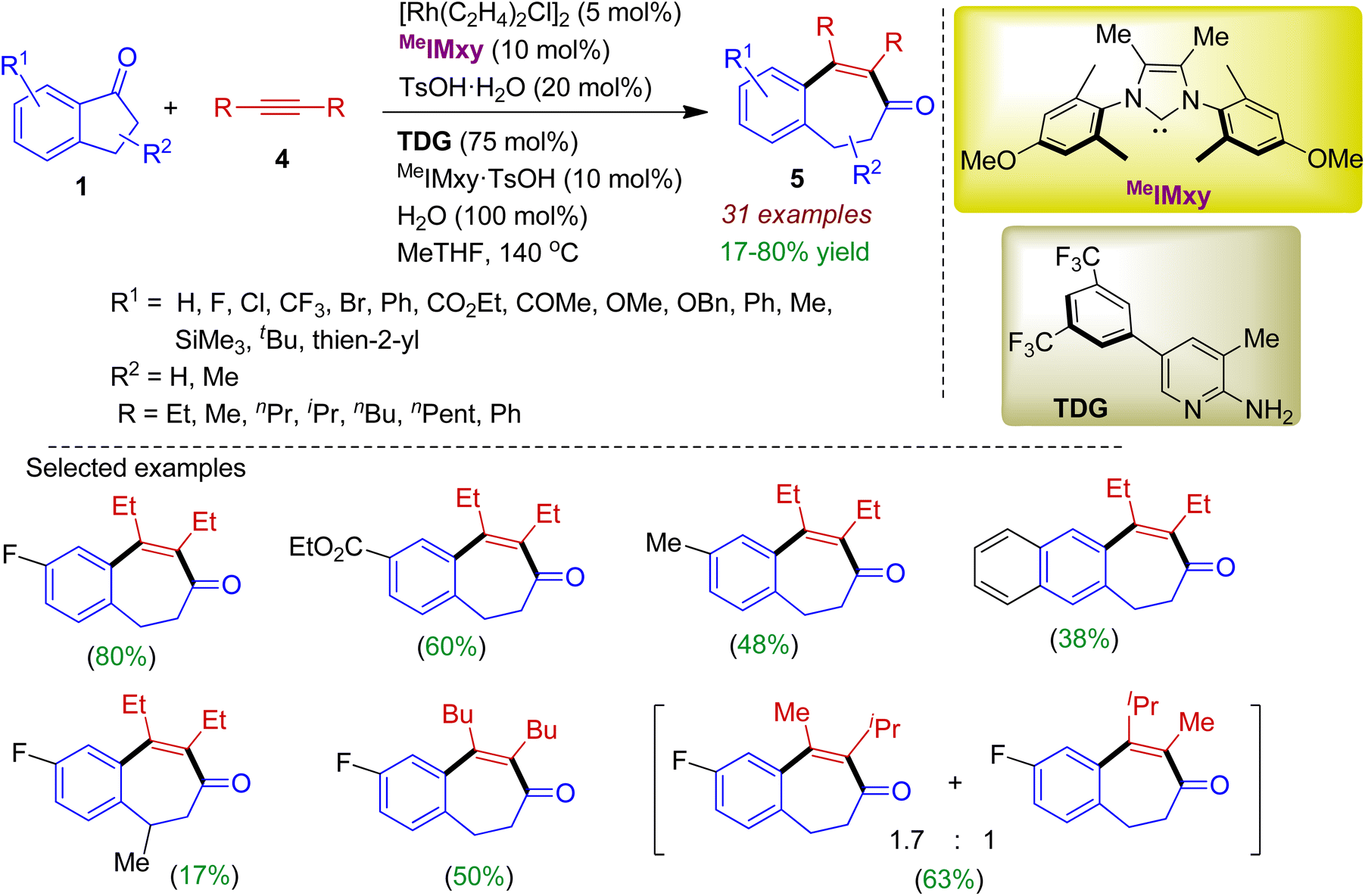
In 2021, the same author synthesized similar type of ring expansion products employing internal alkynes as the reaction partner.32 In fact, alkynes have better affinity with transition metals compared to alkenes due to smaller HOMO/LUMO gap, thereby facilitating the 2π-insertion process. The intermolecular [5+2] cycloaddition reaction between indanones 1 and internal alkynes 4 proceeded via the Rh-catalyzed C–C activation resulting richly decorated benzocycloheptenones 5 in moderate to good yields. The reaction was enabled by a strongly σ-donating NHC ligand (MeIMxy) in the presence of a temporary directing group (TDG) containing an electron-deficient 3,5-ditrifluoromethylphenyl moiety as depicted in Scheme 2. As expected, 1-indanones bearing halogen, ester, ketone, trimethylsilyl, methoxy, phenyl, thienyl functionalities were compatible with this protocol. The reaction worked well for alkynes with different alkyl substituents, however, diaryl-substituted alkynes and terminal alkynes failed to provide desired carbocycles.
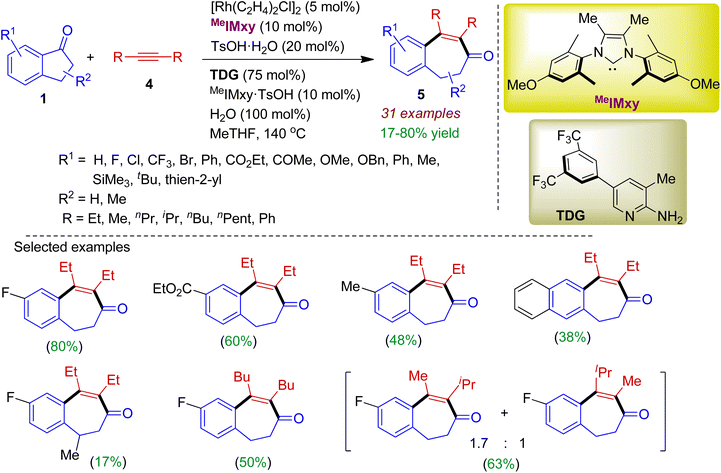 |
| | Scheme 2 Ring expansion of 1-indanones via insertion of alkynes to form fused seven-membered carbocycles. | |
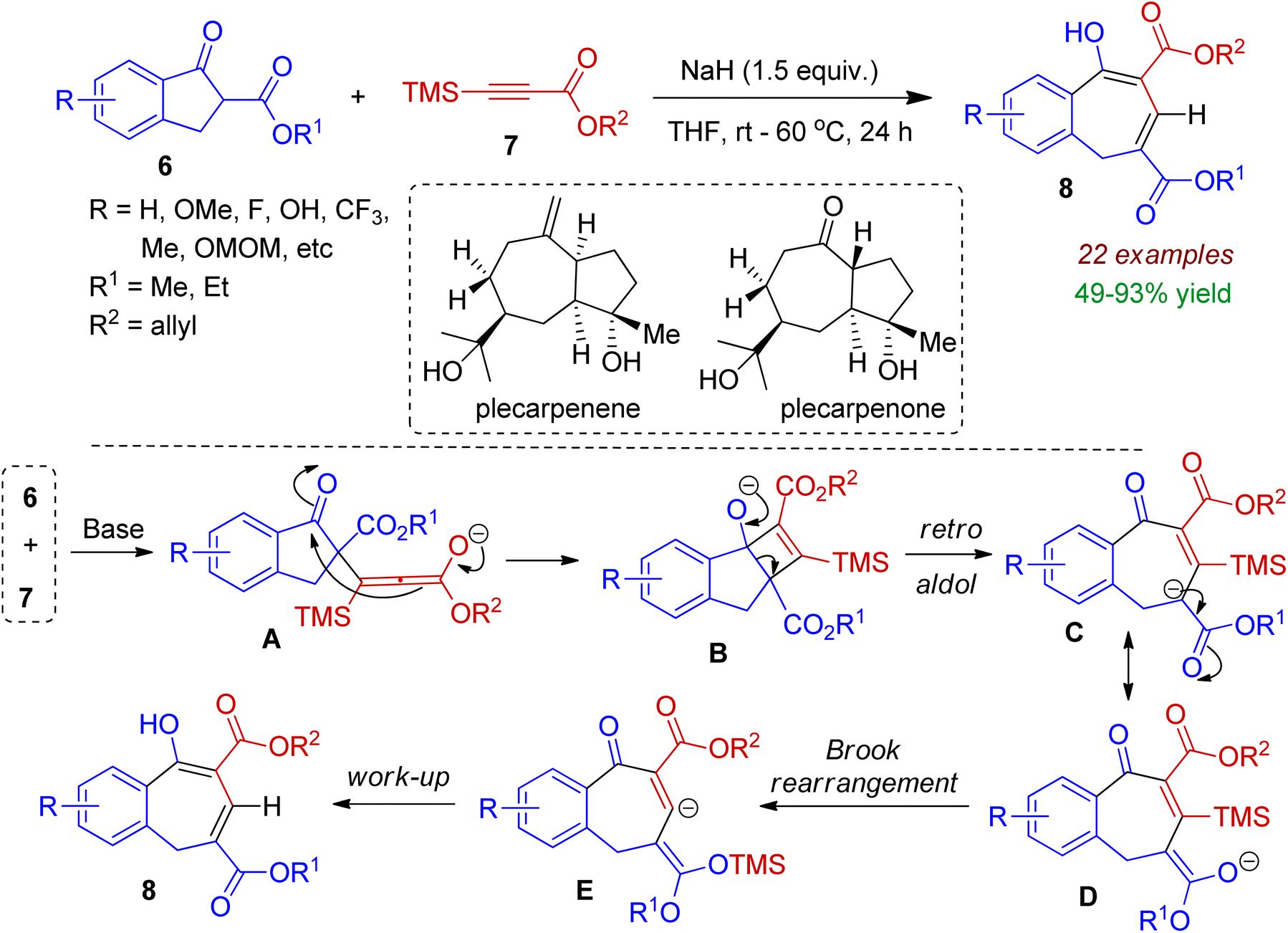
Very recently, Xie, Wang and co-workers devised a base-promoted ring expansion strategy to prepare benzocycloheptene systems from 2-substituted 1-indanone 6.33 In this reaction tetramethylsilyl (TMS)-substituted alkyne 7 was chosen to mimic the terminal alkyne. Among various organic and inorganic bases (such as DABCO, Na2CO3, K2CO3, NaOH, NaH etc.) NaH was found to offer best result in THF medium. A plausible mechanism is outlined in Scheme 3. In the presence of base, nucleophilic attack of carbanion (generated from indanone 6) to alkyne 7 produced intermediate A, which underwent intramolecular nucleophilic addition to form intermediate B. Retro-aldol reaction led to intermediate C, which remained in equilibrium with tautomer D. Subsequently the C–Si bond of the intermediate D was broken via 1,4-Brook rearrangement to afford intermediate E, which could be transformed to the desired product 8 during work-up process. Notably, the authors also synthesized two sesquiterpenoid natural products plecarpenene and plecarpenone on the basis of this protocol.
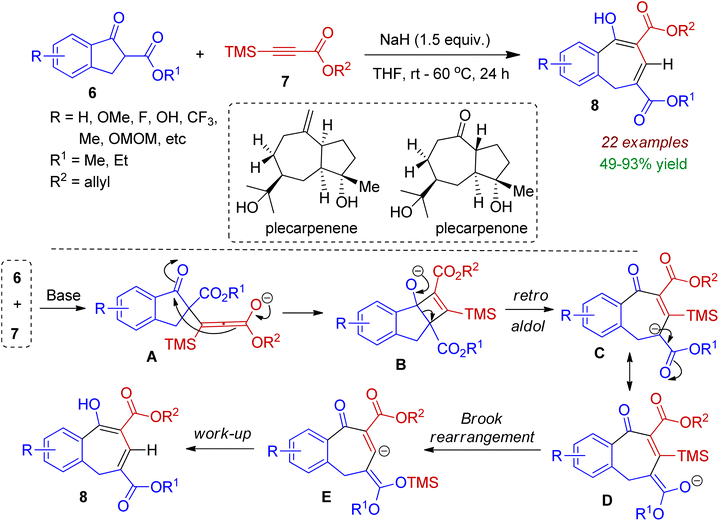 |
| | Scheme 3 Ring expansion of 1-indanones via reaction of alkynes towards fused seven-membered carbocycles. | |
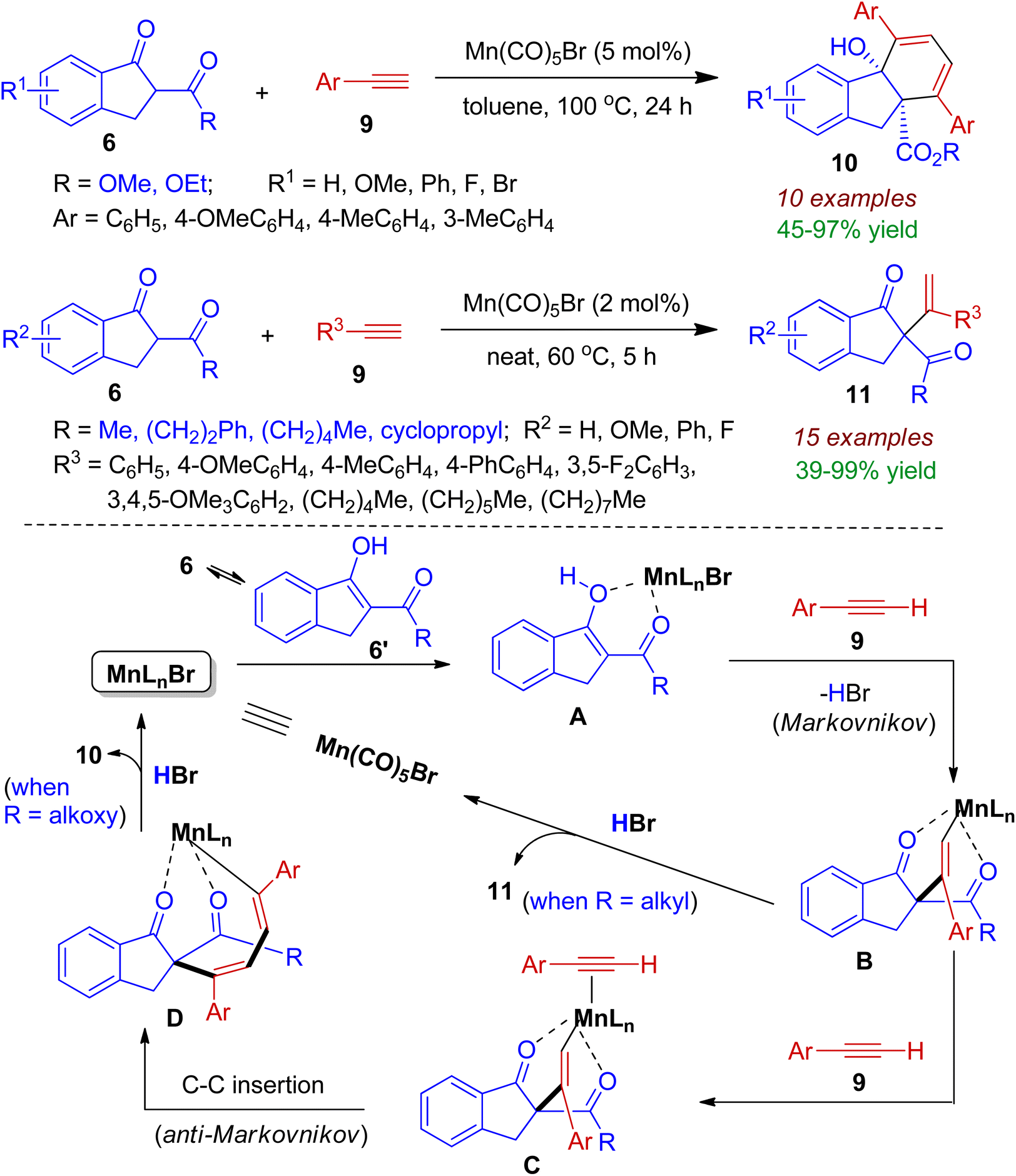
2.1.2. Indeno-fused carbocycles. Indanone derivatives acting as β-ketoester are good synthons for indeno-fused carbocycles. Maji et al. realized regioselective addition of indanone derivatives to terminal alkynes with the aid of Mn(CO)5Br catalyst.34 The indanones 6 (R = OMe, OEt) efficiently combined with aryl acetylene 9 via domino Markovnikov–anti-Markovnikov fashion to afford fused tricyclic scaffold 10 containing two all-carbon quaternary centers (Scheme 4). However, reaction of 2-carbonyl-1-indanones (6, R = alkyl) with alkyl/aryl acetylene 9 led to the formation of Markovnikov addition product 11. The proposed mechanism for the regioselective reaction is shown in Scheme 4. In the presence of Mn-catalyst, the keto–enol tautomerization (6→6′) forms an enolate A, which subsequently reacts with alkyne 9 to generate Markovnikov adduct B. For 1,3-diketone (6, R = alkyl), protonation of B delivered the desired product 11 along with regeneration of catalyst. In case of β-ketoester (6, R = OMe, OEt), complex B get further stabilized by stronger coordination with another equivalent of alkyne to form intermediate C. Next, alkyne insertion in an anti-Markovnikov manner affords intermediate D. Intramolecular cyclization of D by nucleophilic attack at the more electrophilic carbonyl carbon and protonation produces fused tricyclic compound 10 with regeneration of the catalyst. This regio-/stereoselective transformation is highly atom-economic and environmentally benign.
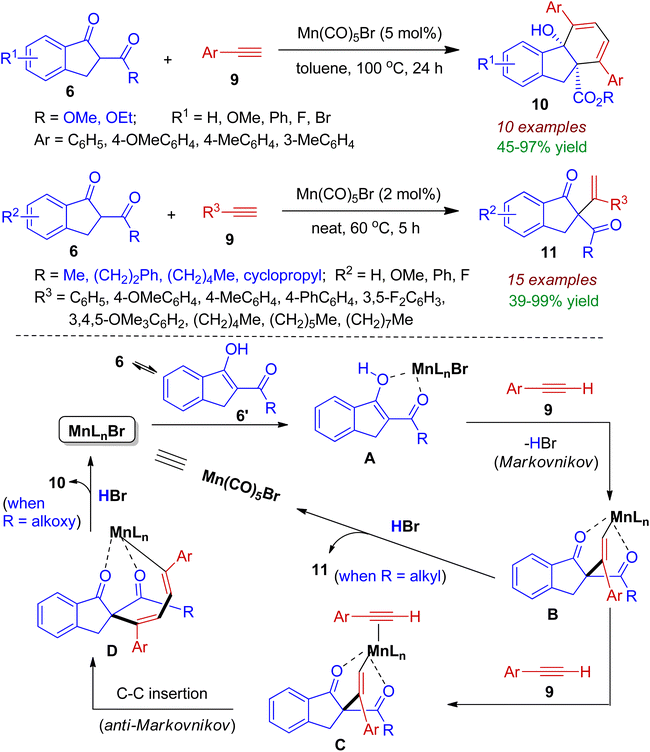 |
| | Scheme 4 Regio-/stereoselective reaction of 2-carbonyl-1-indanones with terminal alkynes to afford fused carbocycles. | |
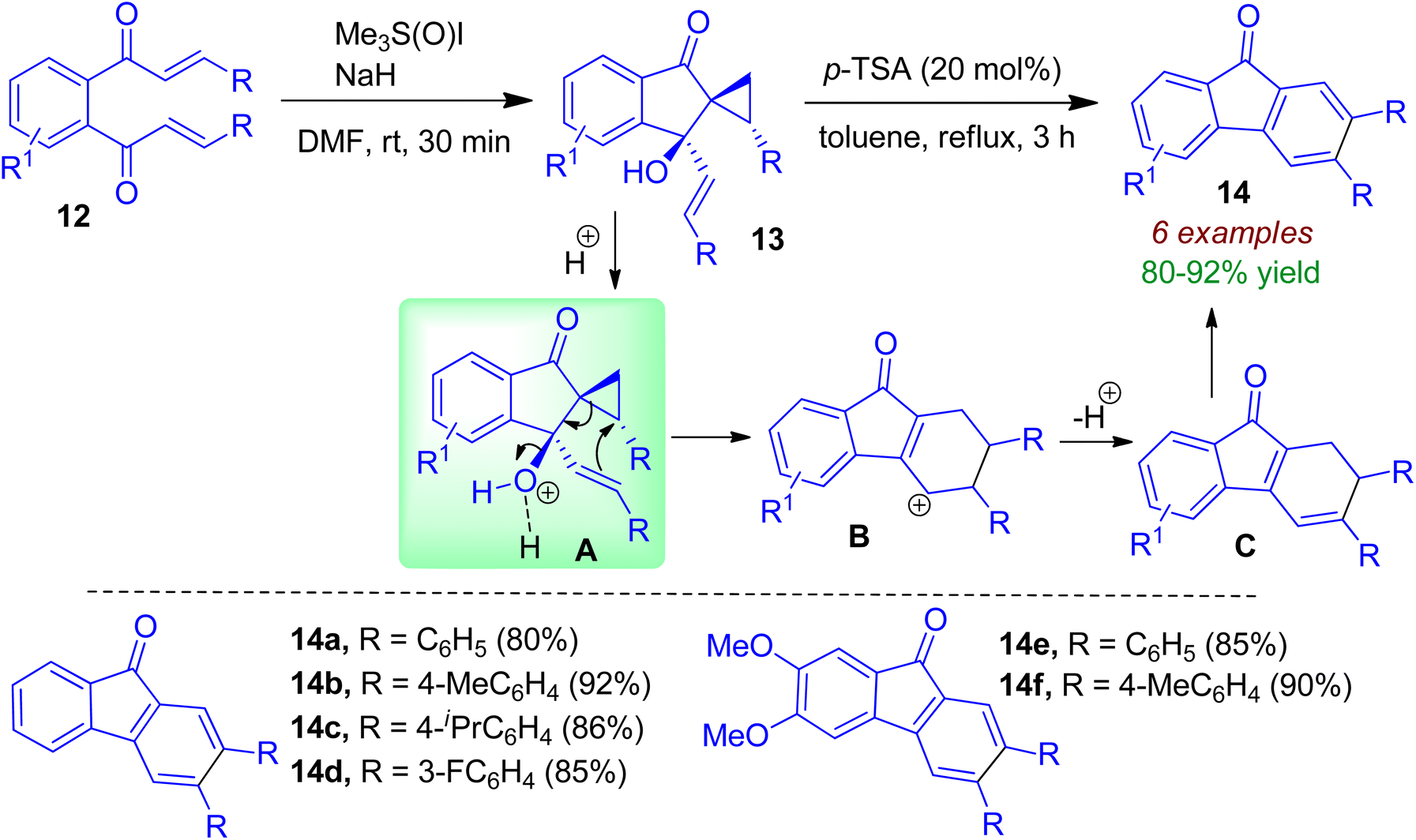
During the course of their study, Ramasastry and co-workers synthesized chiral indanone scaffolds 13 from symmetrical enone compounds 12.35 This diastereoselective transformation was facilitated by the Corey–Chaykovsky reagent, namely dimethyloxosulfonium methylide (DOSM) through initial Michael addition followed by aldol-type reaction. The indanone containing allylic-benzylic tertiary alcohol moiety 13 could be converted to fluorenone 14 in excellent yields with catalytic amount of p-TSA. Aromatic moieties possessing electron-donating and -withdrawing groups smoothly underwent this rearrangement. As shown in Scheme 5, a formal homo-Nazarov-type cyclization of vinyl-cyclopropyl cationic system (via intermediate A) leads to the formation of tetrahydrofluorenyl cation B. Intermediate B then undergoes deprotonation to form intermediate C, which is followed by aromatization to form fluorenone 14.
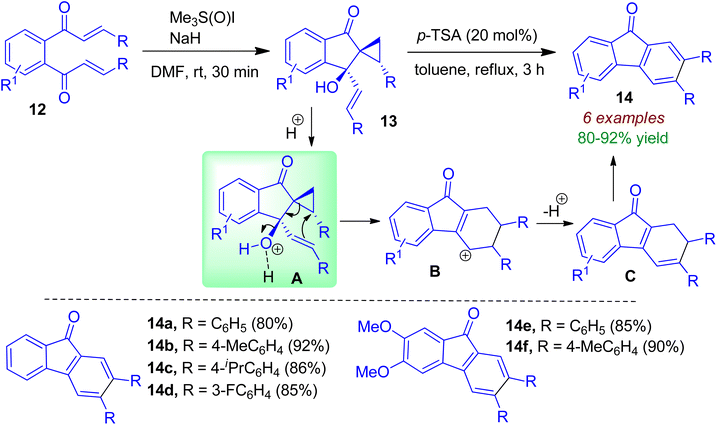 |
| | Scheme 5 Synthesis of chiral indanones and acid-catalyzed transformations to fluorenones. | |
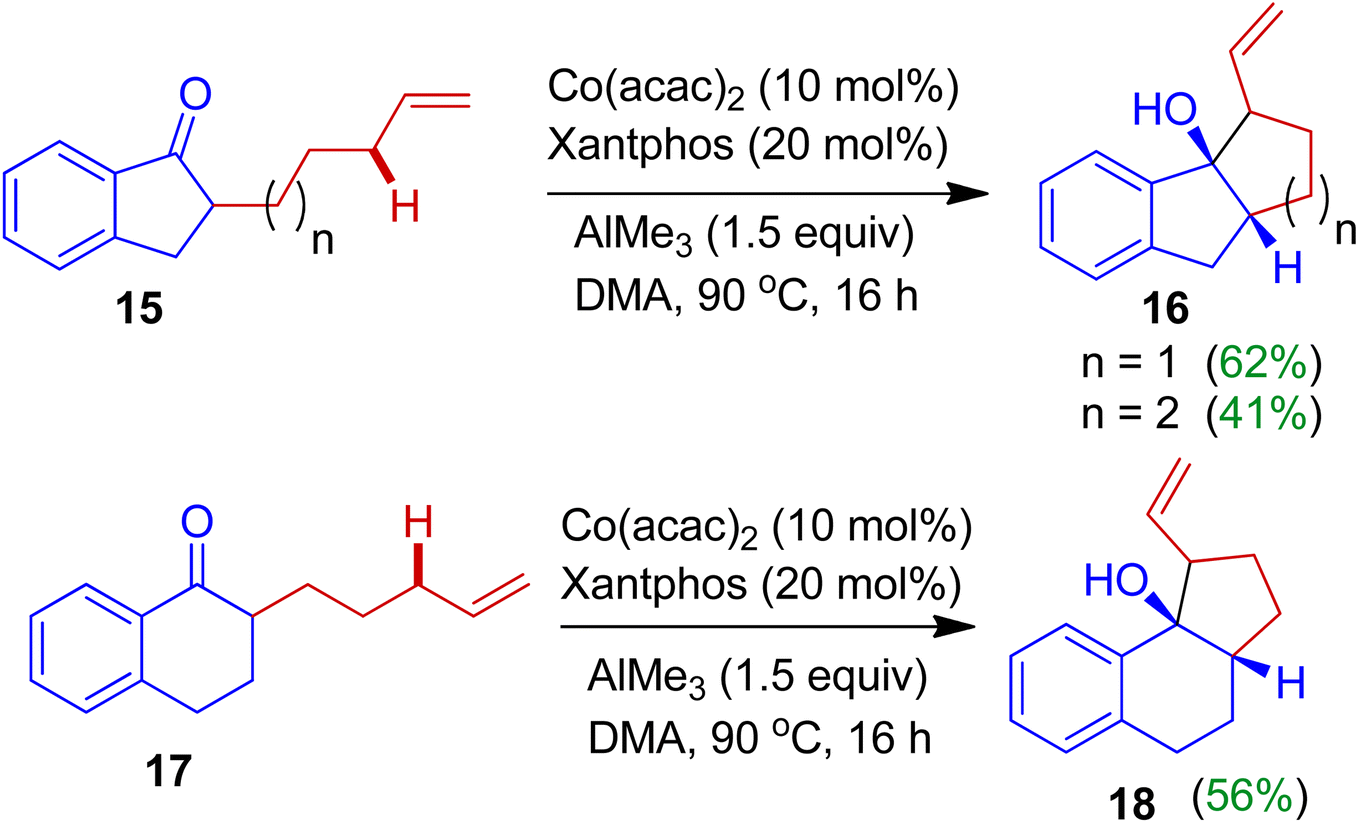
A fascinating cobalt-catalyzed intramolecular cyclization of alkylated indanones was investigated by Mita, Uchiyama and Sato.36 The authors found that alkylated indanones 15 in the presence of cobalt(II)acetylacetone/Xantphos combined catalyst system and trimethylaluminium (AlMe3) afforded fused carbocyclic compounds 16 with good regio- and stereoselectivity (Scheme 6). The reaction of was triggered by allylic C(sp3)–H bond activation. Under similar reaction conditions, cyclohexanone derivatives 17 delivered corresponding annulated product 18 as single diastereoisomer. Importantly, this strategy offers efficient approach of the synthesis of bi- and tricarbocyclic derivatives initiated by C(sp3)–H activation without a gem-disubstituent on the tethered carbon atoms (without the Thorpe–Ingold effect).37
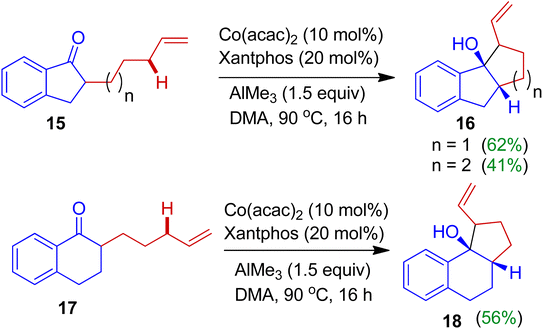 |
| | Scheme 6 Cobalt-catalyzed intramolecular cyclization of alkylated indanones towards fused tricarbocycles. | |
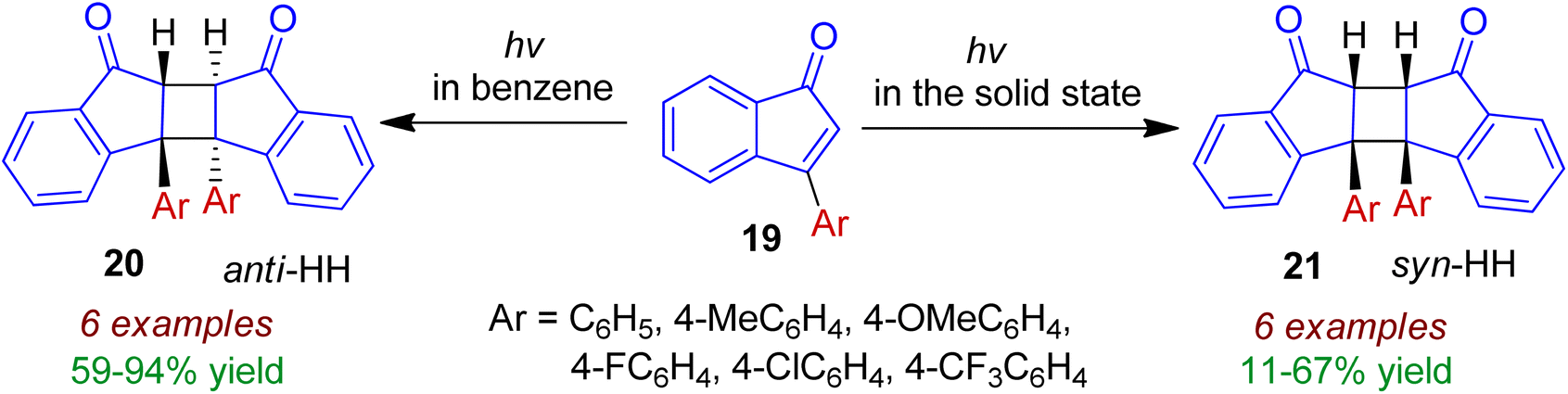
Photodimerization of 3-arylindenone derivatives in both solution and in the solid state was examined by Sakamoto and co-workers.38 The photoreaction of 3-arylindenones 19 using 365 nm line in benzene solution led to efficient dimerization, resulting in anti-HH dimers 20 as the exclusive stereoisomer (Scheme 7). In contrast to the solution photochemistry, the solid-state photoreaction furnished syn-HH cyclobutane dimers 21 in moderate yields. The latter is formed probably by the influence of molecular rearrangement, which was affected by π–π-stacking in the crystal lattices. Various para substituted 3-arylindenones (Ar = 4-MeC6H4, 4-OMeC6H4, 4-FC6H4, 4-ClC6H4, 4-CF3C6H4) were well tolerated for these transformations.
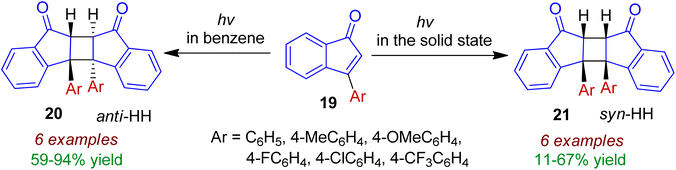 |
| | Scheme 7 Photodimerization of 3-arylindenone derivatives in solution and in the solid state. | |
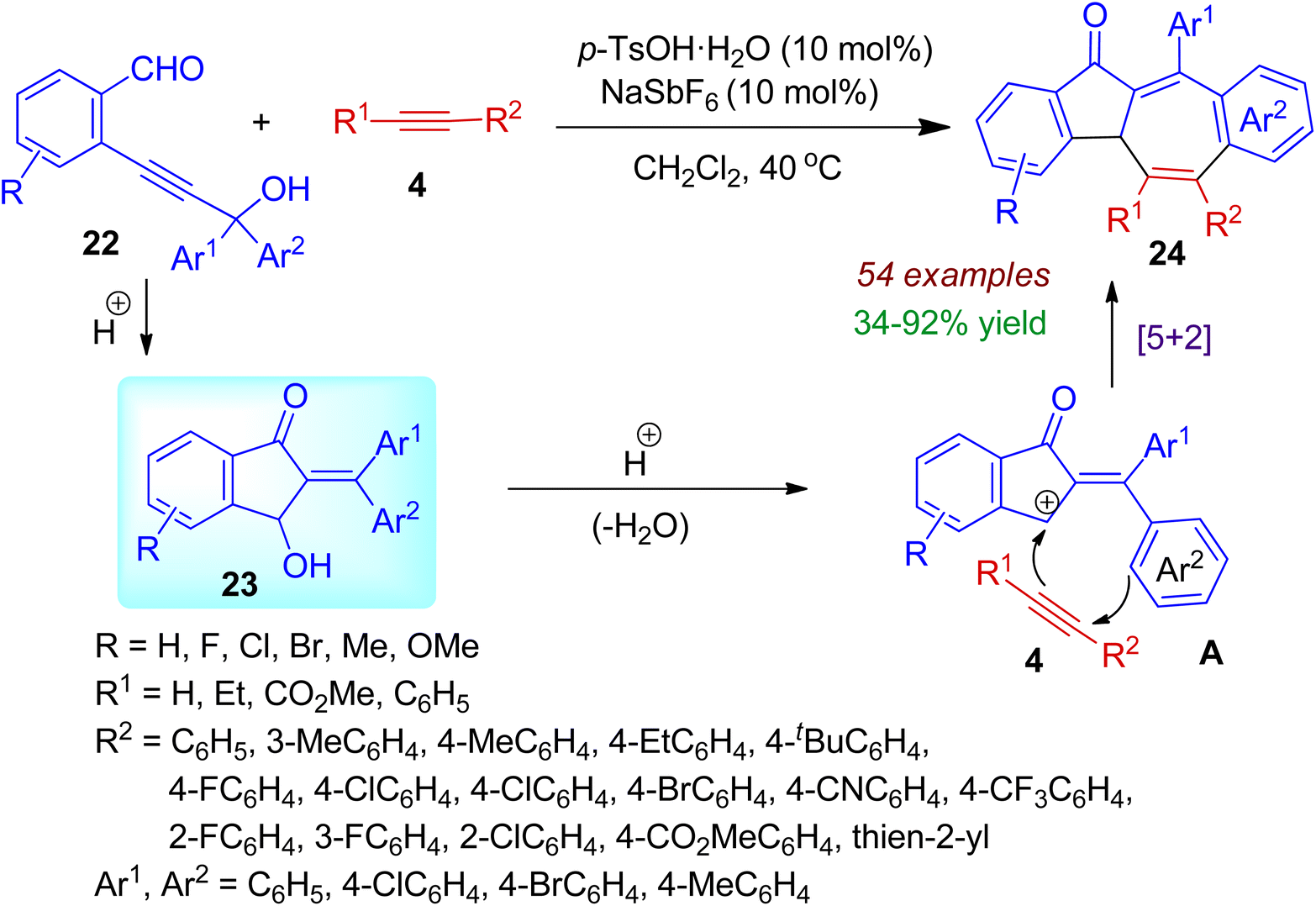
Fused azulenone architectures are important structural motifs often found in bioactive natural products. In 2020, Zhou et al. successfully designed and synthesized dibenzo[a,f]azulene-12-one derivatives 24 from readily available o-propargyl alcohol benzaldehydes 22 and alkynes 4 with the help of p-TsOH catalyst (Scheme 8).39 Various tertiary/secondary aryl propargyl alcohols participated in the annulation process. The cycloaddition reaction was relevant for both terminal as well as internal alkynes with a variety of substituents such as alkyl, eater, aryl, heteroaryl, etc. Under the acidic conditions, 3-hydroxy-1-indanones 23 are formed through intramolecular cyclization of aldehydes 22. The in situ generated indanone 23 then undergoes acid-catalyzed dehydration to form cationic intermediate A. Then formal [5+2] cycloaddition with alkyne yields final polycarbocyclic scaffolds 24. The reaction is regioselective and applicable for wide range of substrates (54 examples). Significantly, this annulation strategy comprised high atom-economy, resulting three C–C and one C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds under mild conditions.
O bonds under mild conditions.
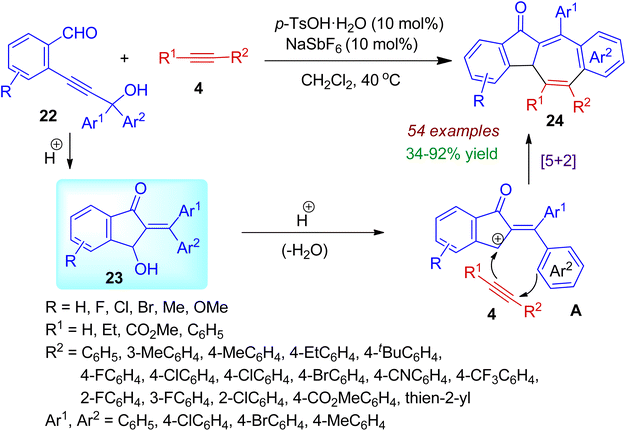 |
| | Scheme 8 Acid-catalyzed cyclization of o-propargyl alcohol benzaldehydes and alkynes resulting dibenzo[a,f]azulene-12-ones. | |
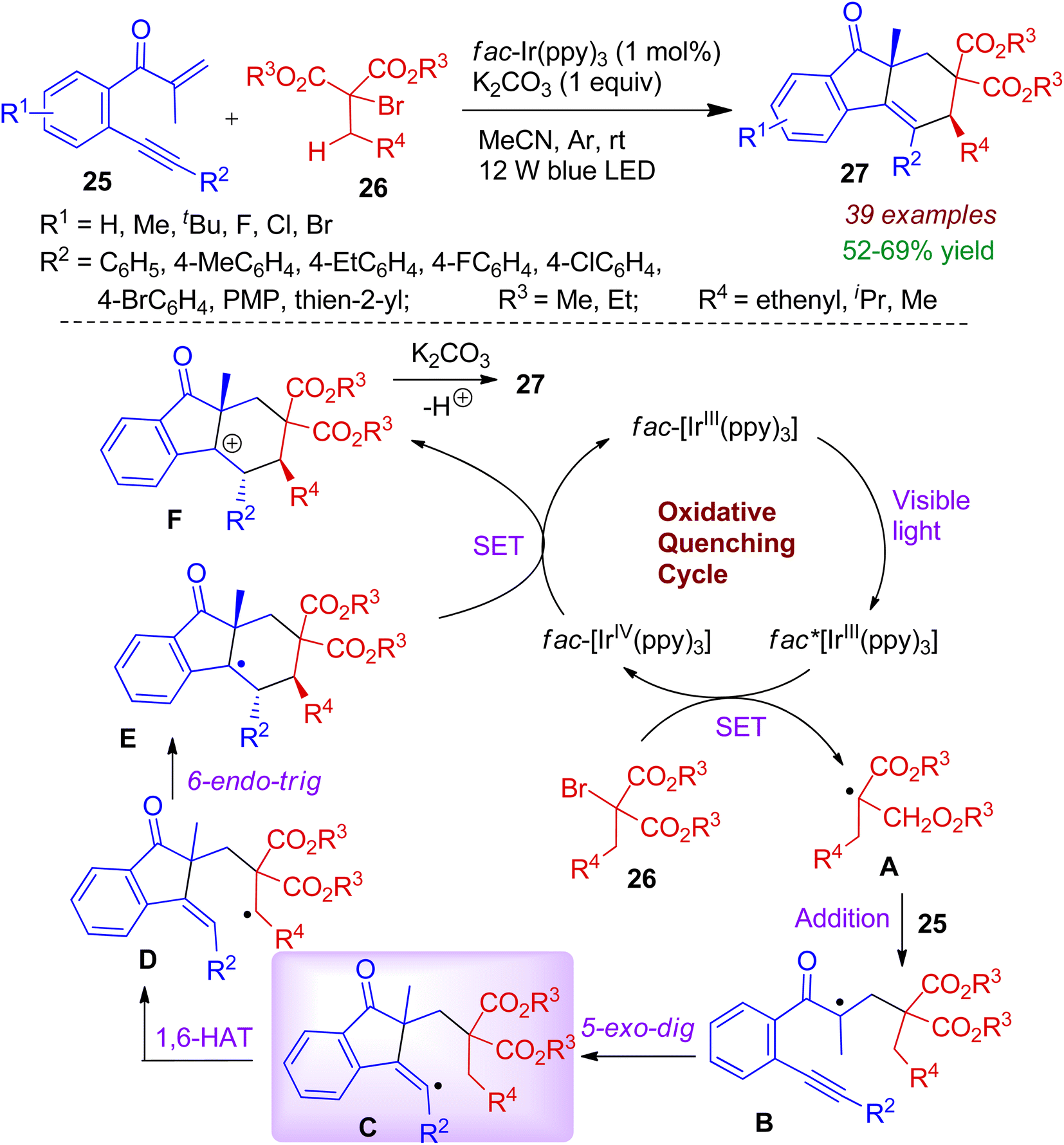
Starting from β-alkynylpropanones 25 and α-bromomalonates 26, an interesting visible-light photocatalytic conversion was realized by Jiang and co-workers.40 This photocatalytic approach accommodates high diastereoselectivity with broad substrate scope resulting substituted syn-fluoren-9-ones 27 in the presence of iridium-photocatalyst. The startegy comprises formation of two new rings via radical-induced C(sp3)–H bond cleavage. The reaction takes place through single electron transfer (SET) process (Scheme 9). Initially, visible light triggers [fac-IIIIr(ppy)3] photocatalyst to the excited state [fac-IIIIr(ppy)3]*, which reduces α-bromomalonate 26 to form a C-centered radical A along with [fac-IVIr(ppy)3] via SET process. The radical addition of intermediate A to CC of β-alkynylpropanones 25 affords quaternary carbon radical B. Intermediate B then experiences 5-exo-dig cyclization (intermediate C) and 1,6-hydrogen atom transfer (HAT) to generate intermediate D, which subsequently undergoes 6-endo-trig cyclization leading to trans-intermediate E. Meanwhile, a crucial oxidation (SET) step between E and [fac-IVIr(ppy)3] occurs, regenerating the photocatalyst and the cationic intermediate F. Finally, deprotonation delivers desired syn-products 27.
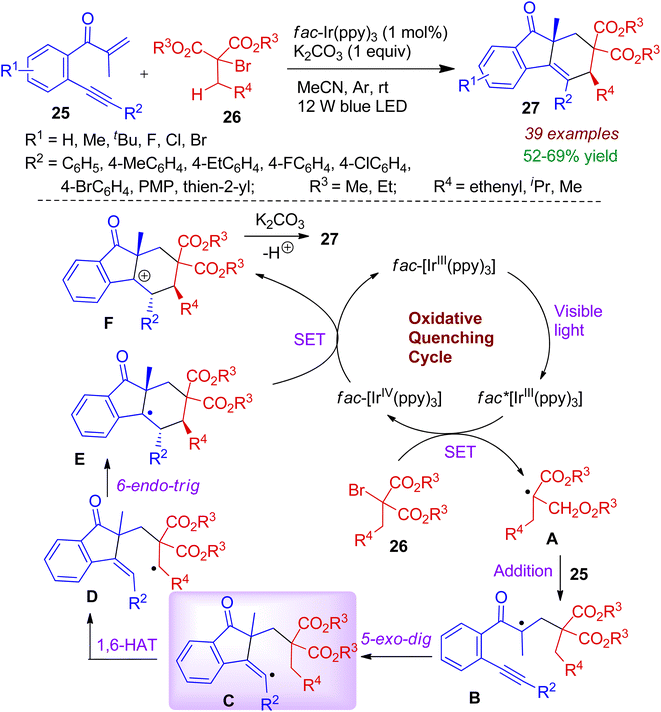 |
| | Scheme 9 Visible-light photocatalytic cyclization between β-alkynylpropenones and α-bromomalonates towards syn-fluoren-9-ones. | |
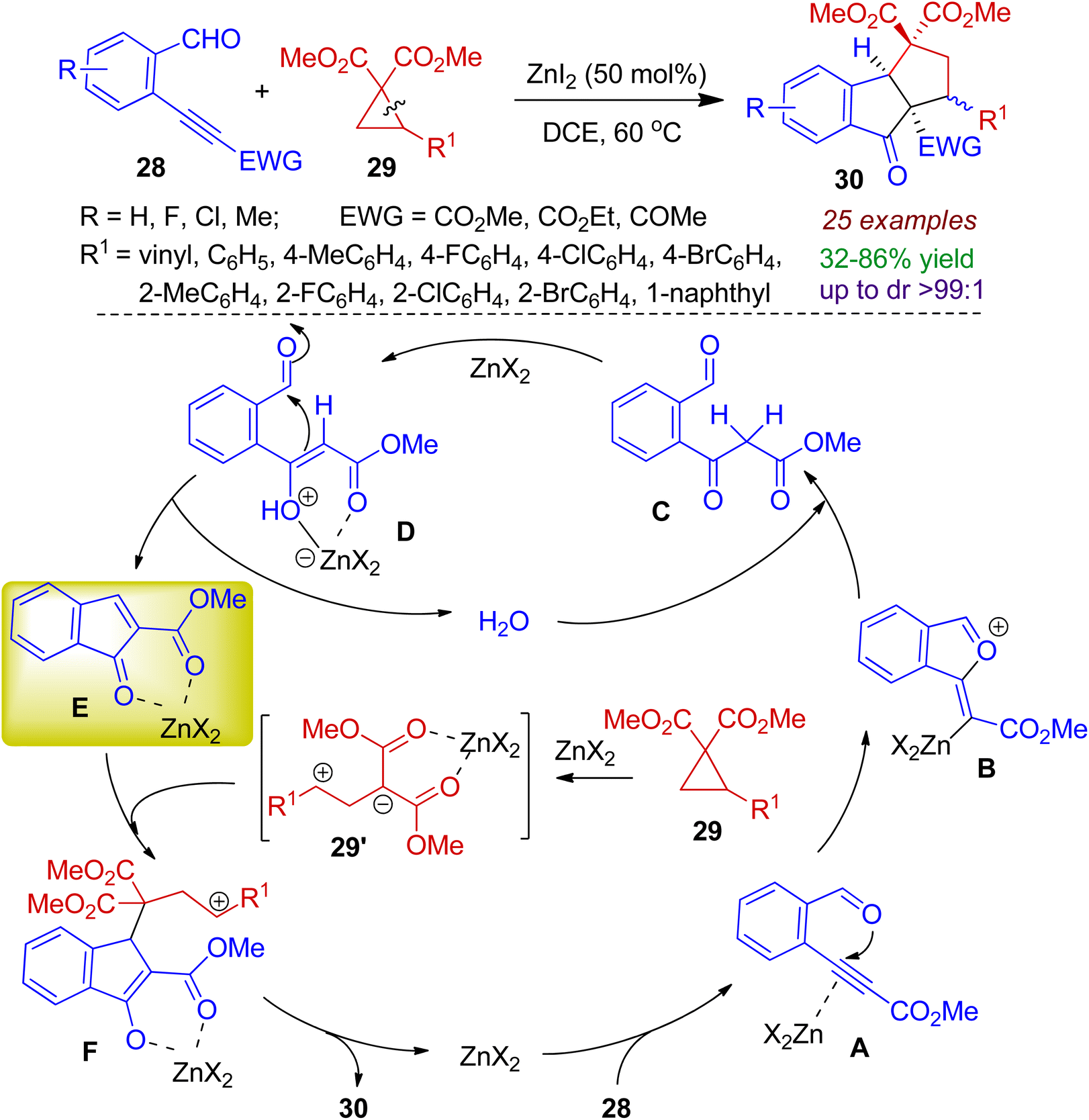
Zhu group exploited donor–acceptor cyclopropanes as efficient 1,3-dipoles to trap the in situ generated electron deficient indenones through [3+2] cycloaddition reaction for synthesizing indanone fused scaffolds.41 The assembly of enynals 28 and donor–acceptor cyclopropanes 29 in the presence of Zn-catalyst successfully generated indanone fused cyclopentanes 30. Cycloprapanes bearing vinyl and aromatic moieties responded the reaction well, however, the reaction failed for heteroaromatic cyclopropanes. A plausible mechanism for cascade cyclization is depicted in Scheme 10. At first, activation of alkyne moiety through a [Zn]-π complex A generates the 5-exo-dig intermediate B. Hydrolysis of B produces keto ester C in the presence of catalytic amount of water. Keto–enol tautomerism/Knoevenagel condensation sequence affords key intermediate E (via intermediate D) and water is liberated for the next catalytic cycle. In the mean time, the 1,3-dipole 29′ is formed in situ via ring opening of the cyclopropane 29 with the aid of Zn-salt. Next, [3+2] cycloaddition reaction between the indenone E and 1,3-dipole 29′ accomplishes final product 30.
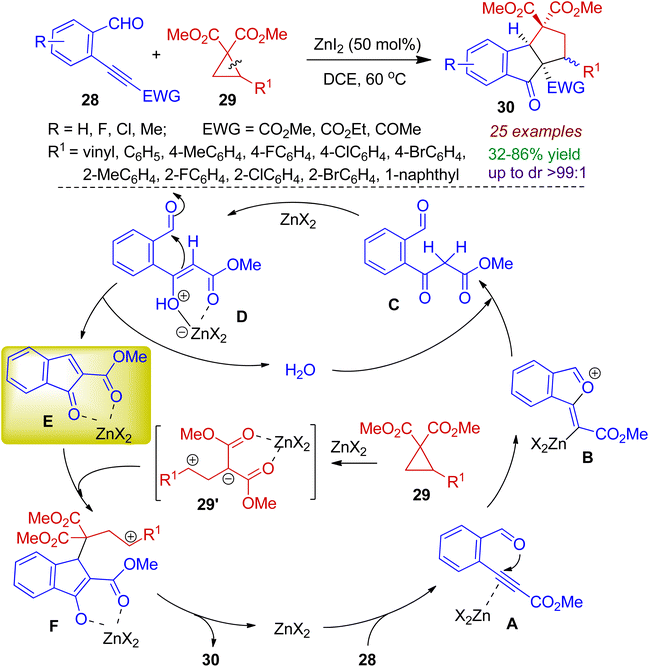 |
| | Scheme 10 Reaction of donor–acceptor cyclopropanes and enynals to obtain indanone fused cyclopentanes. | |
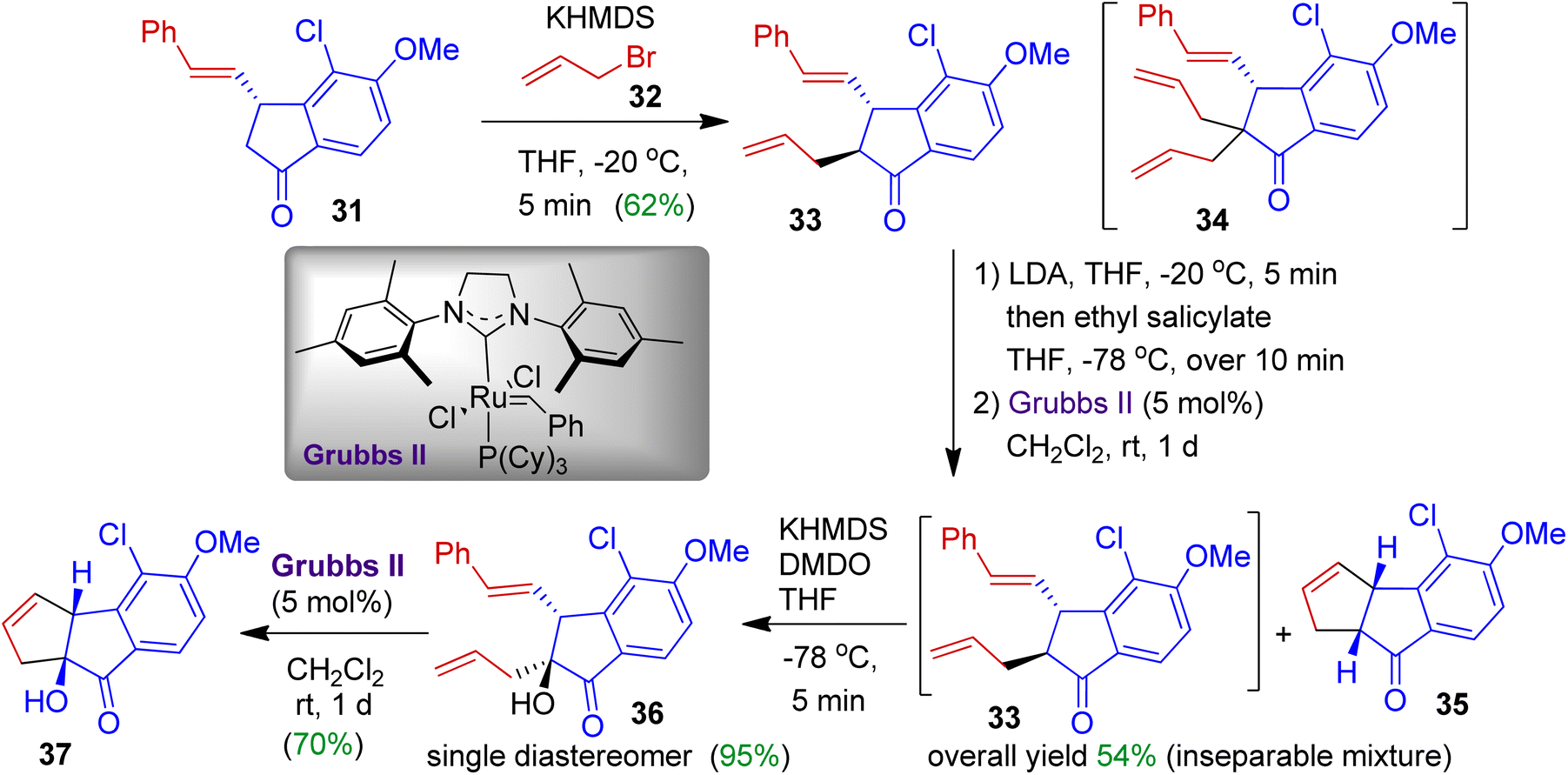
Benzo-dihydropentalenes are unique structural motifs usually found in many naturally occurring compounds. As part of their total synthesis programme, Koert et al. constructed cyclopentane fused indanone skeleton via a multistep approach (Scheme 11).42 The authors employed styryl substituted indanone 31 and installed an allyl substitution at α-position using allyl bromide 32 to obtain trans-indanone 33. At this stage, doubly allylated compound 34 was formed as a side product. The trans-indanone 33, after ring closing metathesis (RCM) with Grubbs II catalyst gives inseparable mixture of tricyclic compounds 35 and 33. The introduction of the hydroxyl group at C9 position of compound 33 could be achieved by treatment of potassium enolate with dimethyldioxirane (DMDO). This strategy offered alcohol 36 as a single diastereoisomer in 95% yield. In the next step, ring closing metathesis successfully yielded hydroxylated tricycle 37 (70% yield).
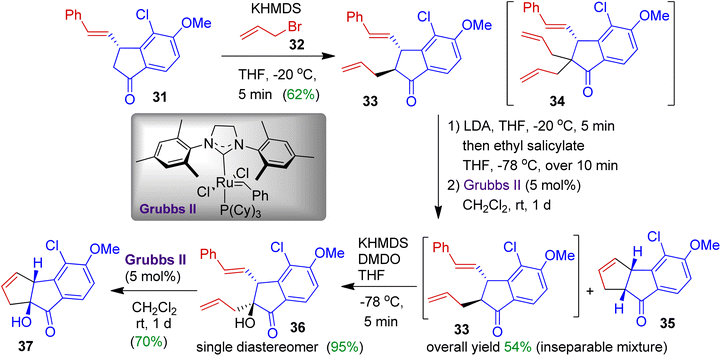 |
| | Scheme 11 Multistep synthesis of cyclopentane fused indanone from indanone motif. | |
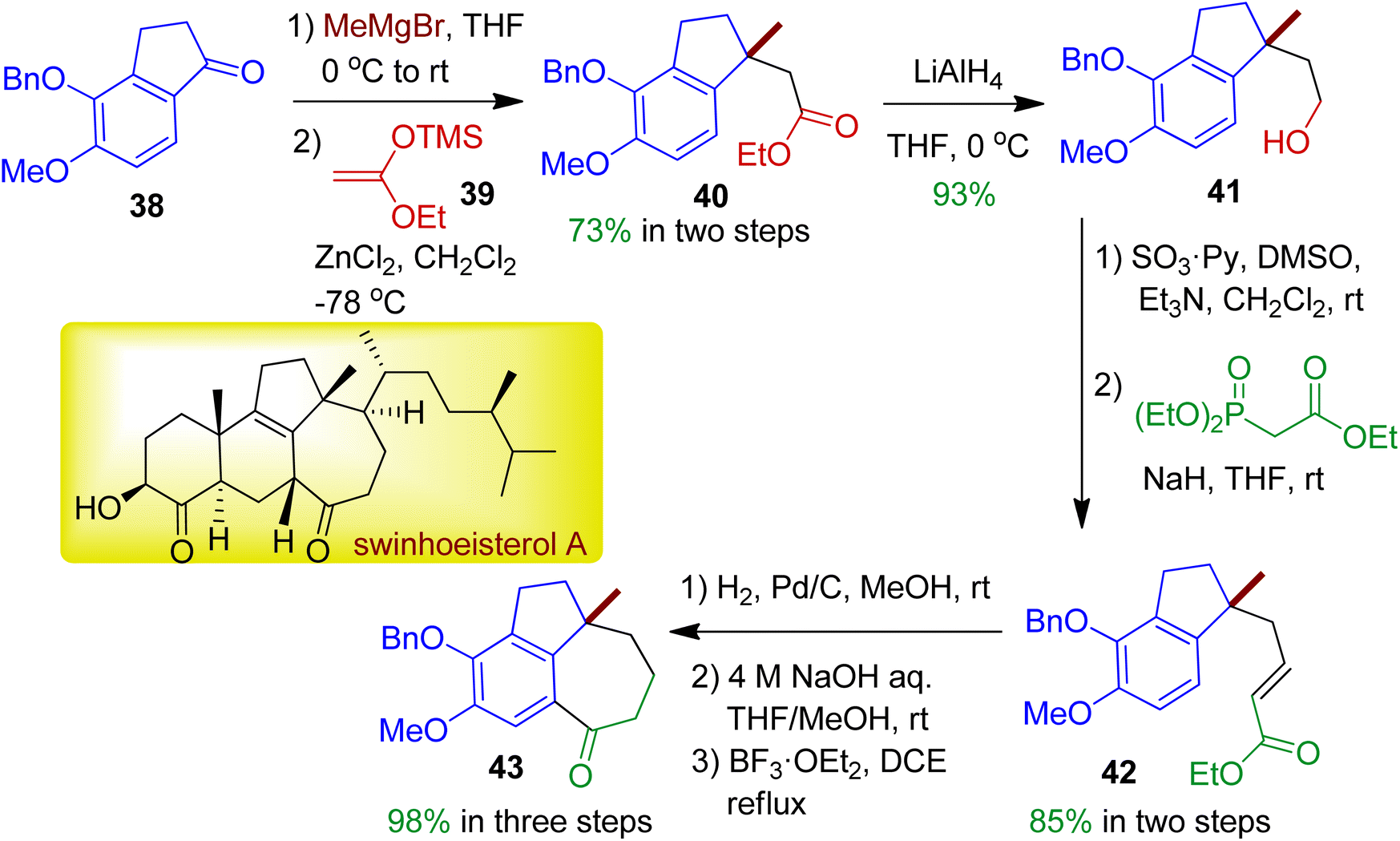
In 2019, Kigoshi's group employed indanone derivative 38 towards novel fused tricyclic compound 43 which has core structural similarity with natural product swinhoeisterol A.43 The reaction sequence is outlined in Scheme 12. Initially, methylation of indanone 38 by MeMgBr and substitution of the resultant benzylic tertiary alcohol with ketene silyl acetal 39 afforded ester 40 (73% in two steps). Reduction of the ester with LiAlH4 resulted corresponding alcohol 41. Subsequent oxidation followed by Horner–Wadsworth–Emmons reaction accomplished unsaturated ester 42, which could be converted into desired tricyclic scaffold 43 with 6/5/7 ring via three step reaction sequence (98% in three steps). The relative configuration of the target compound was determined by NOE experiments.
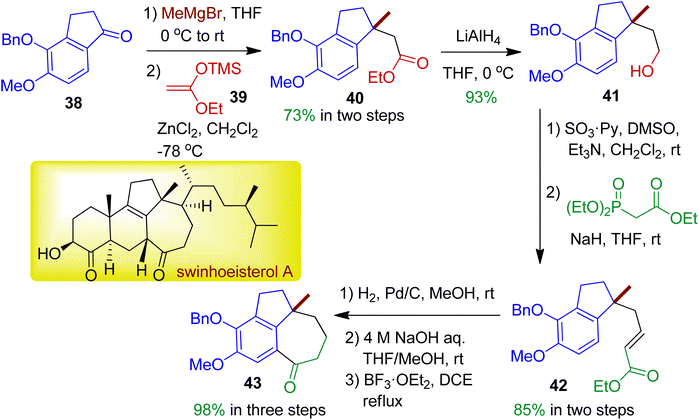 |
| | Scheme 12 Multistep synthesis of novel fused tricyclic skeleton (6/5/7) starting from indanone derivative. | |
2.2. Fused heterocycles
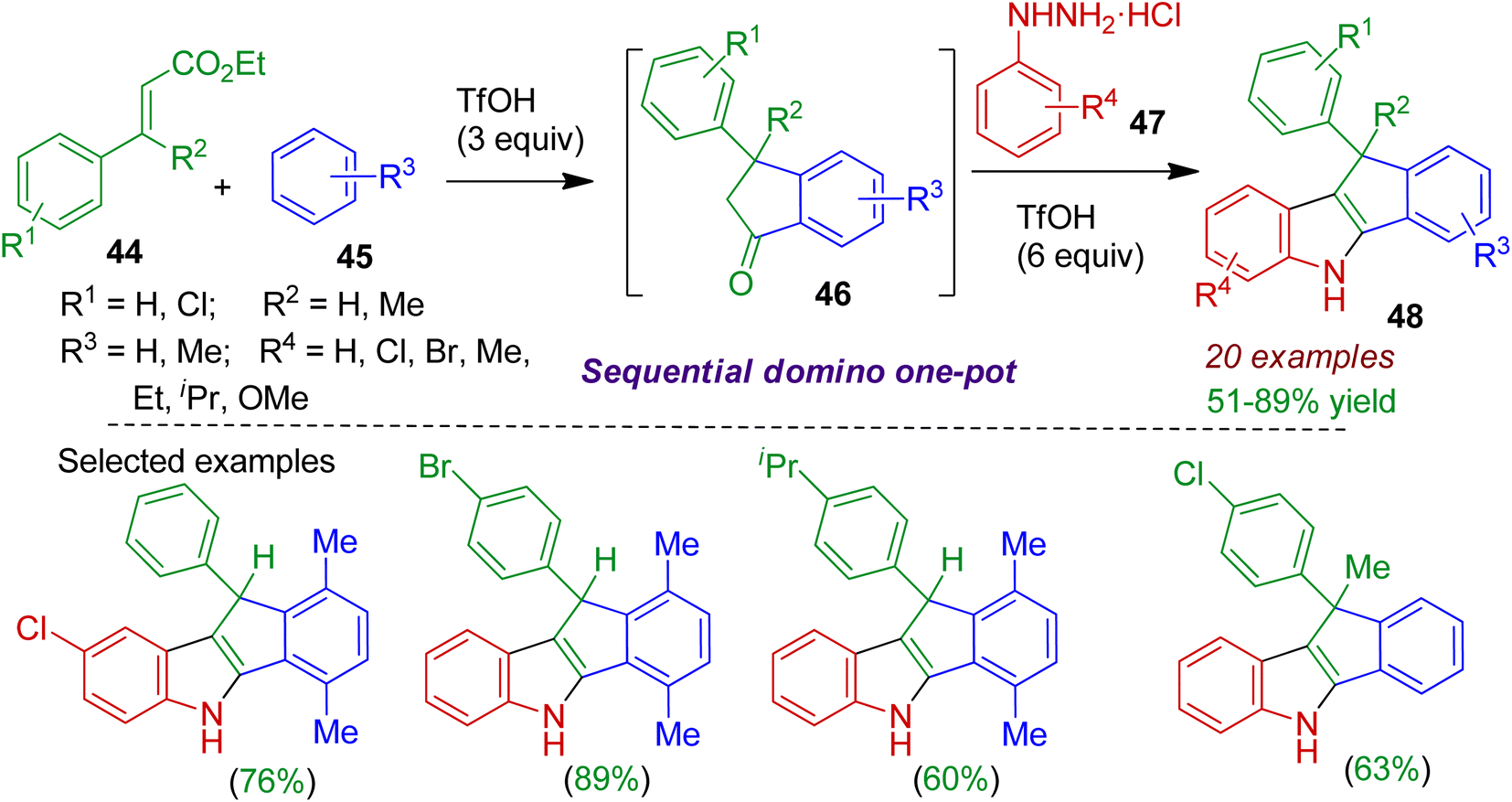
2.2.1. N-Containing fused heterocycles. Nitrogenous heterocycles fused with indane substructure are commonly encountered in alkaloids and several useful pharmaceuticals.44 An efficient sequential domino one-pot protocol to build fused tetracyclic indole skeleton was realized by Reddy and Satyanarayana.45 The reaction between ethyl cinnamates 44 and arenes 45 in the presence of superacidic triflic acid (TfOH) afforded indanone derivative 46, which after reaction with aryl hydrazines 47 led to indenoindoles 48 in good yields (Scheme 13). The reaction proceeded via a domino intermolecular Friedel–Crafts alkylation and intramolecular acylation of ethyl cinnamates to form indanones 46, followed by the Fischer indole reaction under acidic conditions. The conversion was enabled for large number of substrates and comprised formation of three C–C and one C–N bonds. Significantly, synthesis of dihydroindeno[1,2-b]indoles containing quaternary carbon at 10 position could also be achieved using this methodology.
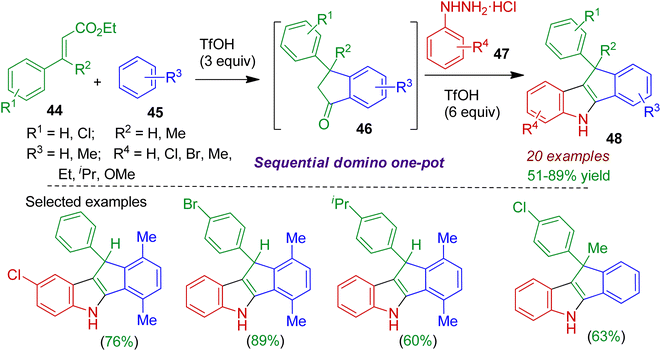 |
| | Scheme 13 TfOH-promoted sequential domino one-pot synthesis of indenoindoles from ethyl cinnamate, arene and aryl hydrazine. | |
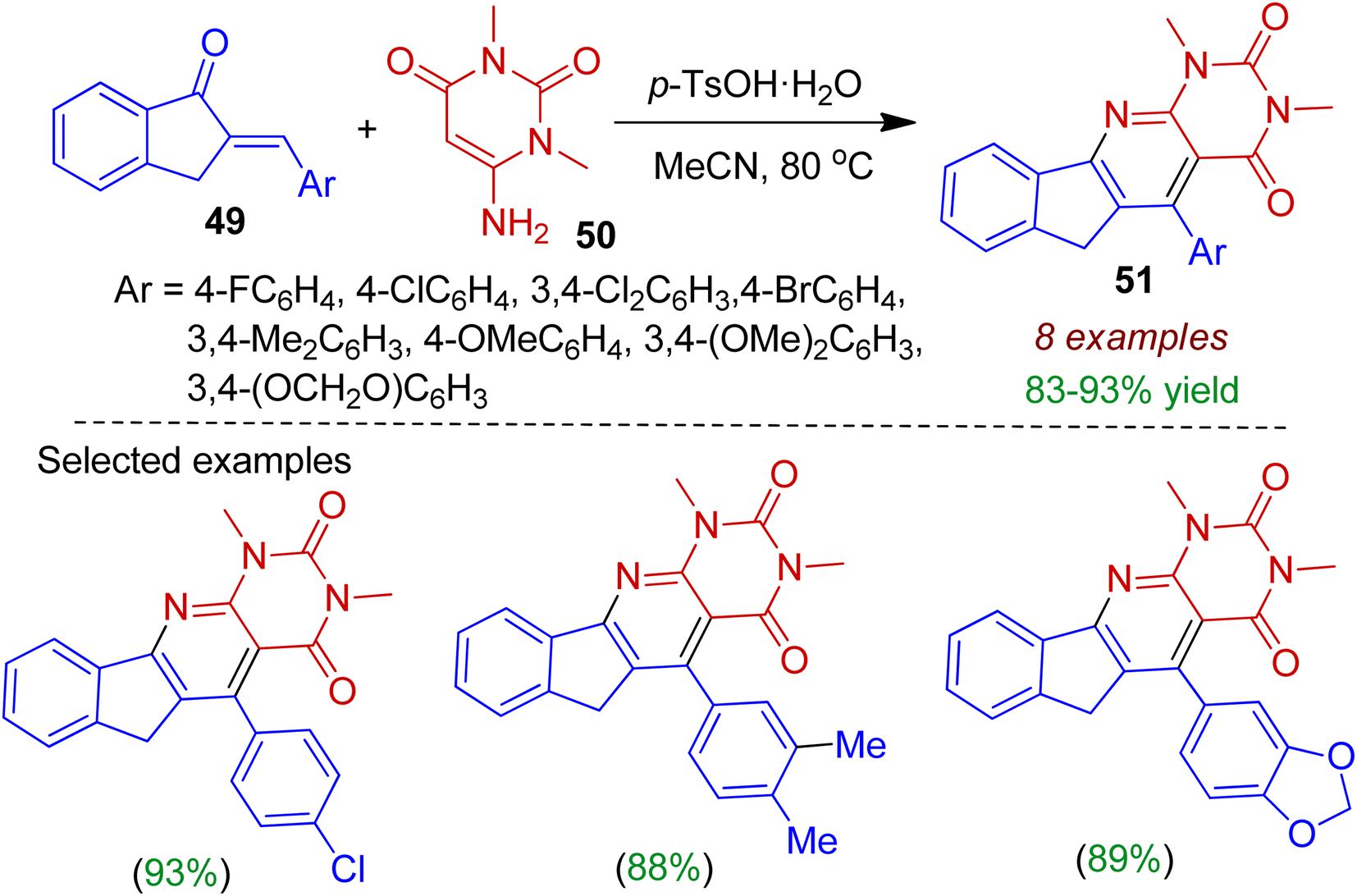
Rong et al. carried out the one-step synthesis of indeno-fused pyridopyrimidine scaffolds 51 involving readily accessible 2-arylidene 1-indanone system under mild reaction conditions.46 The authors employed 6-amino-1,3-dimethylpyrimidine 50 as the annulation partner to react with 2-arylidene 1-indanone 49 in the presence of catalytic amount of p-TsOH in refluxing acetonitrile resulting polyheterocyclic compounds 51 in excellent yields (Scheme 14). The reaction was also applicable for 2-arylidene dihydronaphthalenone systems.
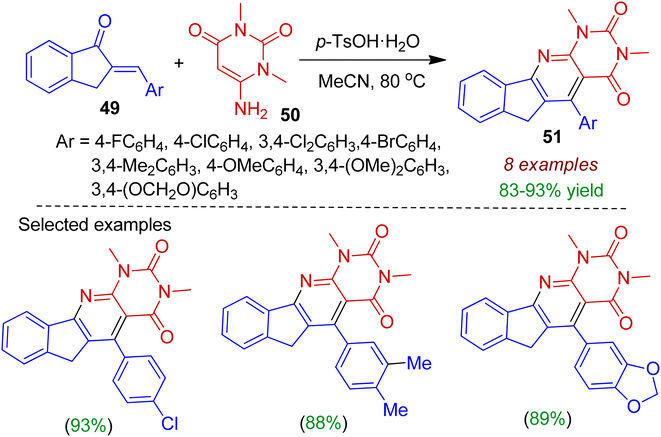 |
| | Scheme 14 Synthesis of indeno-fused pyridopyrimidine scaffolds from 2-arylidedene indanones and 6-aminopyrimidine. | |
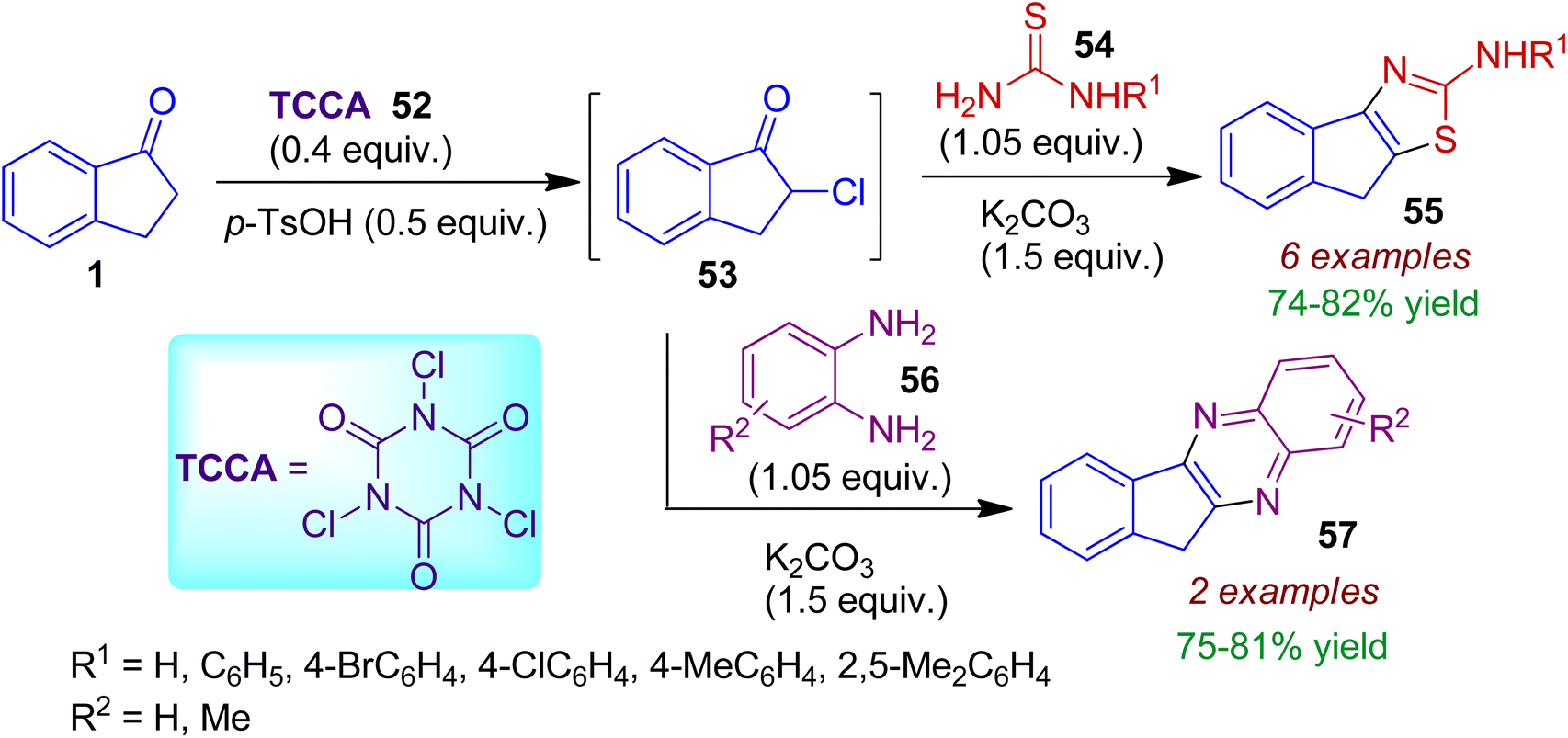
α-Chlorination reaction of indanone 1 with trichloroisocyanuric acid (TCCA) 52 under mechanochemical ball-milling condition may be applied for synthesis of fused heterocycles.47 The intermediary α-chloroindanone 53 formed in this process was directly subjected to base-mediated condensation with thiourea/N-arylthiourea 54 resulting 2-indenothiazoles 55 in 74–82% yields (Scheme 15). In the similar sequential manner, o-phenylenediamines 56 could be treated with α-chloroketone 53 to accomplish corresponding indenoquinoxaline derivatives 57. This strategy is environmentally benign, comprising one-pot sequential acid- and base-mediated reactions in the solid state resulting biologically relevant heterocycles.
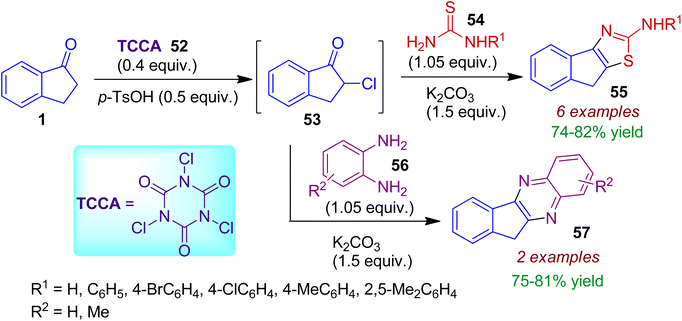 |
| | Scheme 15 Solid-state synthesis of 2-indenothiazoles and indenoquinoxalines exploiting α-chloroindanone. | |
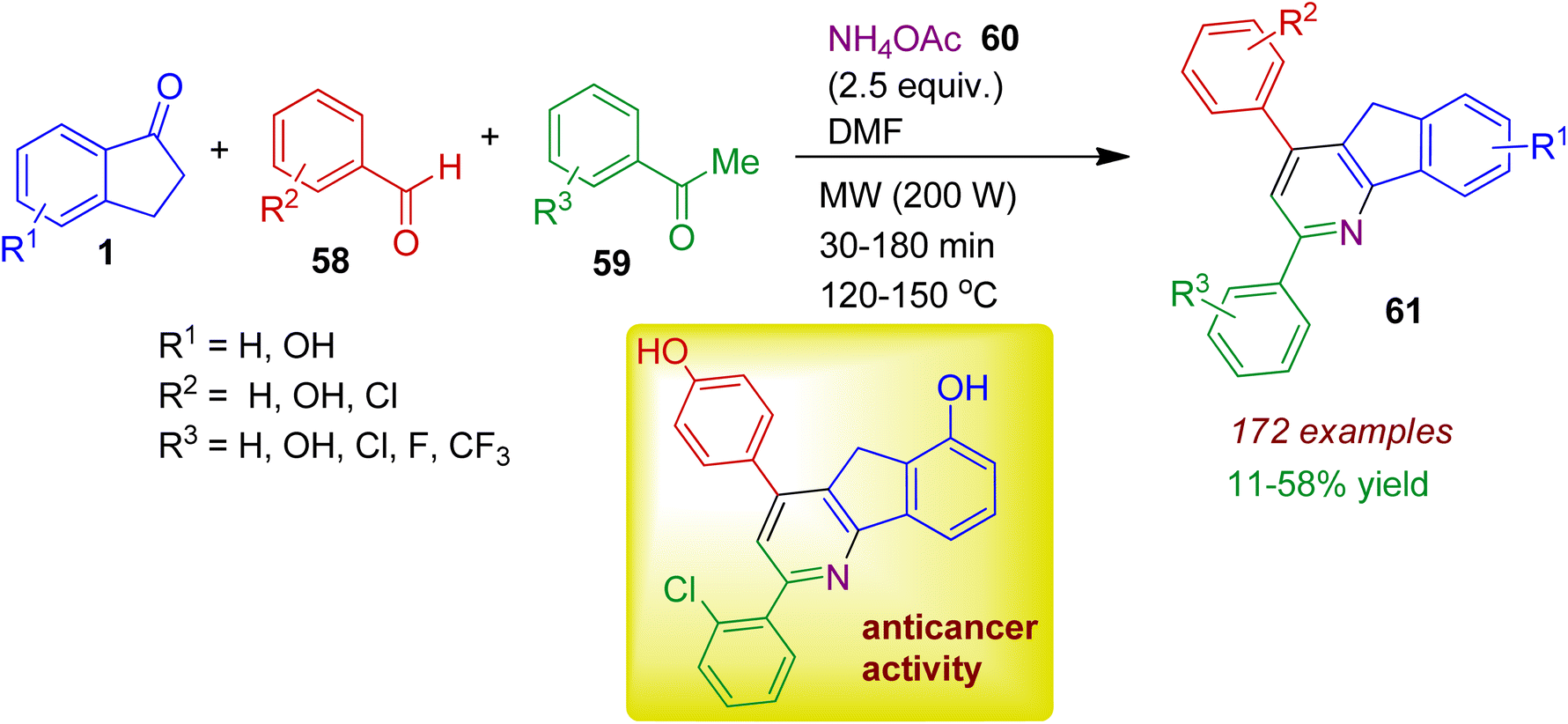
Starting from readily available substrates Kwon, Lee and co-workers synthesized indeno pyridine derivatives under microwave irradiation.48 This multicomponent strategy involved the assembly of 1-indanones 1, aromatic aldehydes 58, acetophenones 59, and NH4OAc 60 to obtain a library of hydroxy- and halogenated 2,4-diphenylindeno[1,2-b]pyridinols 61 in acceptable yields (Scheme 16). The authors fruitfully investigated structure–activity relationships of the synthesized compounds. It has been found that the majority of compounds with chlorophenyl group at 2-position and phenol moiety at the 4-position of the indeno[1,2-b]pyridinols revealed potent antiproliferative activity and topoisomerase IIα-selective inhibition against human breast cancer cell lines.
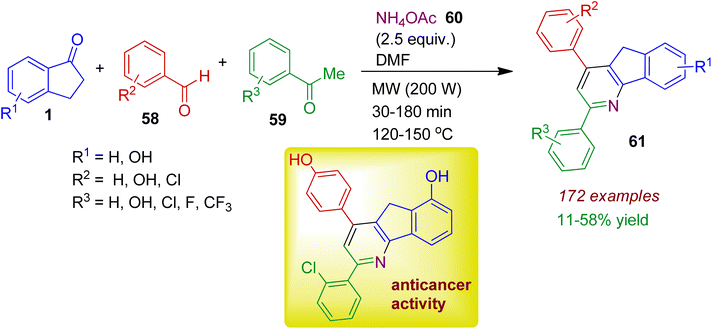 |
| | Scheme 16 MW-assisted multicomponent synthesis of indeno pyridines from 1-indanones, aromatic aldehydes, acetophenones and NH4OAc. | |
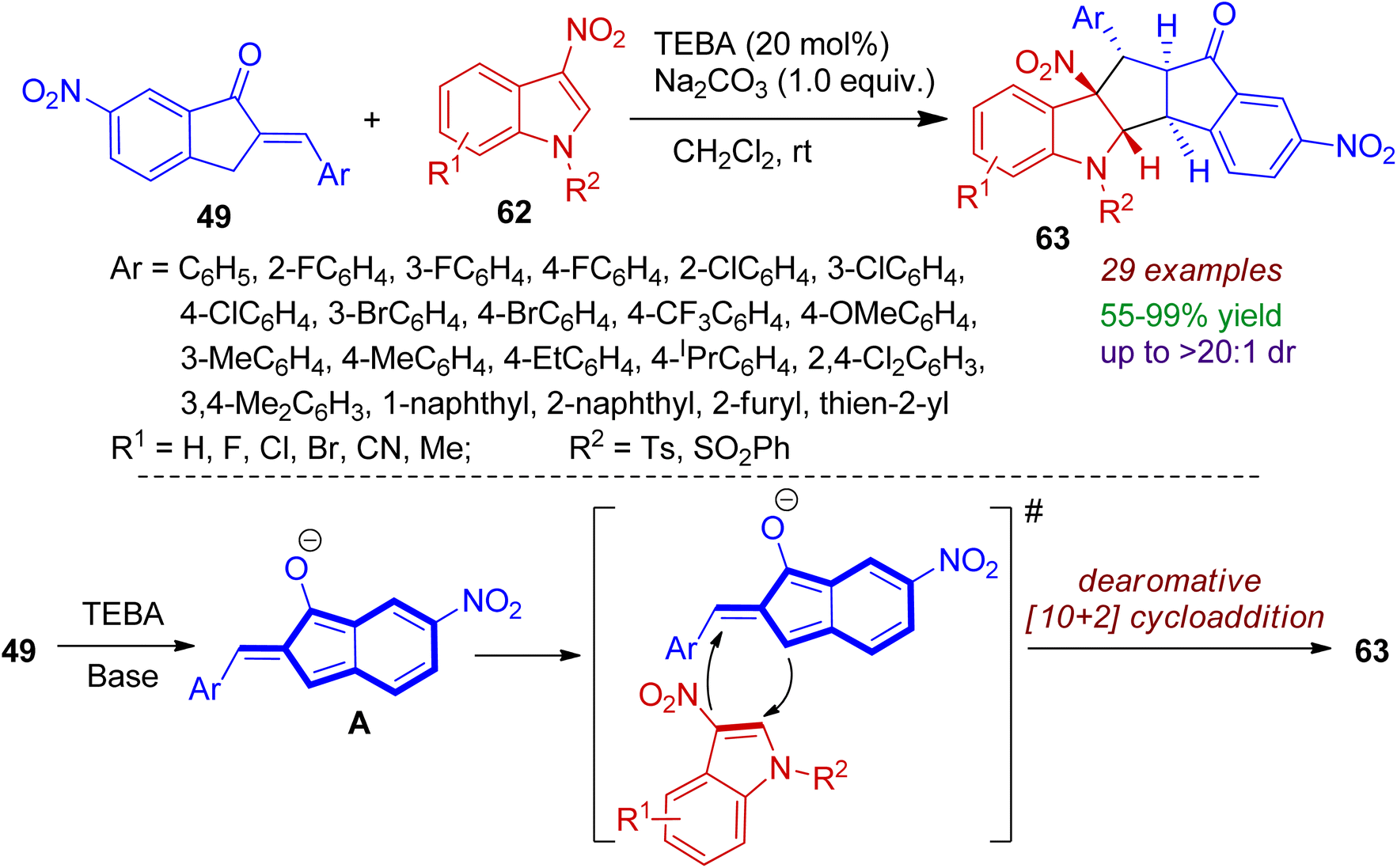
Higher order cycloaddition strategy is an important tool for constructing polycyclic architectures in a single step. Very recently, dearomative [10+2] cycloaddition of 2-arylidene-1-indanones 49 and 3-nitroindoles 62 at ambient temperature was developed.49 This transformation was attained using triethyl benzyl ammonium chloride (TEBA) as phase transfer catalyst under basic conditions (Na2CO3) to afford wide range of polycyclic cyclopenta[b]indolines 63 in excellent yields and diastereoselectivity (up to 99% yield and >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr). 3-Nitroindoles bearing electron-withdrawing and donating substituents (F, Cl, Br, CN, Me) were well tolerated with the catalytic system. A plausible mechanism is illustrated in Scheme 17. In the presence of TEBA and base, 2-arylidene-1-indanone 49 is first converted to 1-hydroxyl isobenzofulvene anion intermediates A, which is highly nucleophilic in nature. After that, higher order [10+2] cycloaddition takes place between in situ generated intermediate A and 3-nitroindoles 62 to accomplish dearomatized annulation products 63. Notably, the reaction is amenable for gram-scale synthesis of desired products.
:1 dr). 3-Nitroindoles bearing electron-withdrawing and donating substituents (F, Cl, Br, CN, Me) were well tolerated with the catalytic system. A plausible mechanism is illustrated in Scheme 17. In the presence of TEBA and base, 2-arylidene-1-indanone 49 is first converted to 1-hydroxyl isobenzofulvene anion intermediates A, which is highly nucleophilic in nature. After that, higher order [10+2] cycloaddition takes place between in situ generated intermediate A and 3-nitroindoles 62 to accomplish dearomatized annulation products 63. Notably, the reaction is amenable for gram-scale synthesis of desired products.
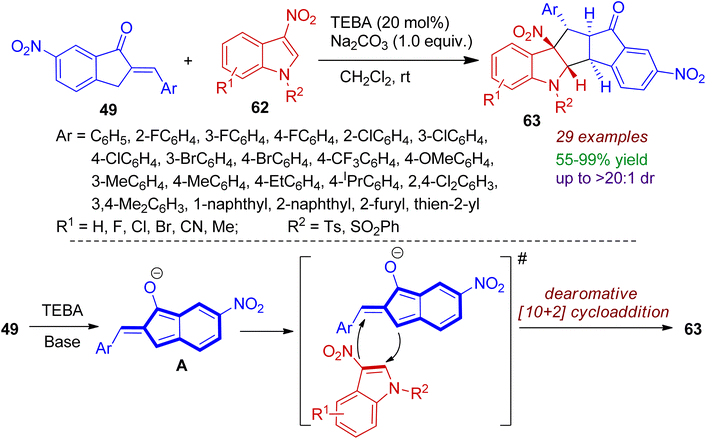 |
| | Scheme 17 Higher-order [10+2] cycloaddition of 2-arylidene-1-indanones and 3-nitroindoles to access polycyclic cyclopenta[b]indolines. | |
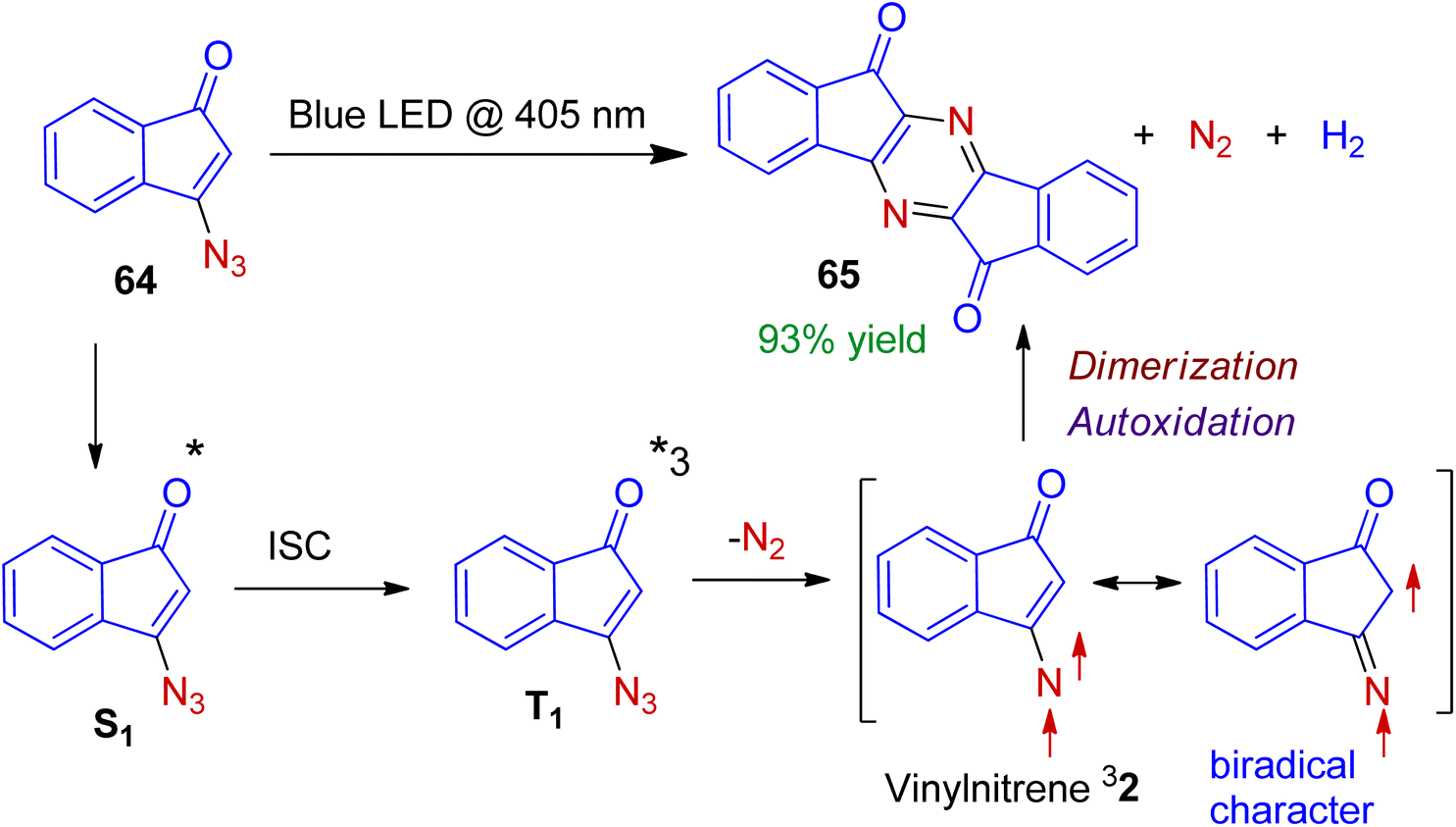
The mission for sustainable chemistry using sunlight or low energy light-emitting diodes (LED) has sparked interest in the recent synthetic research. Gudmundsdottir et al. realized photolysis of 3-azido-1-indenone 64 with light-emitting diode (LED, λ = 405 nm) or mercury arc lamp to access heterocyclic dimer 65 in excellent yield.50 Mechanistically it is conceivable that (Scheme 18), the irradiation of azido-1-indenone 64 forms its first singlet excited state S1. Intersystem crossing leads to the triplet configuration T1, which is followed by extrusion of an N2 molecule to generate vinylnitrene 32. The vinylnitrene 32 is sufficiently stable to dimerize (not decayed by intramolecular rearrangement) to produce N-heterocyclic dimer 65. Importantly, due to the significant 1,3-biradical character of vinylnitrene 32 (which is confirmed by ESR spectroscopy), it dimerizes to form C–N bond, rather than N–N bond. In this methodology, the molecular architecture is finely tuned to control the reactivity of triplet vinylnitrene.
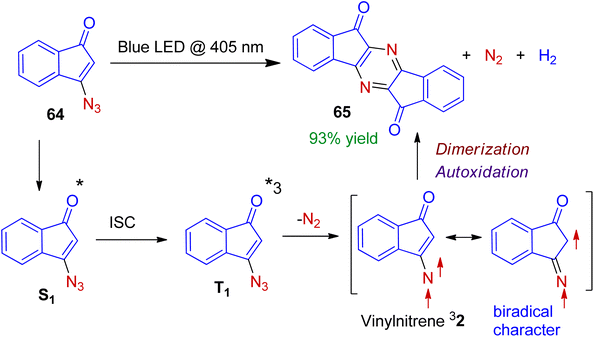 |
| | Scheme 18 Photodimerization of 3-azido-1-indenone to form N-heterocyclic dimer. | |
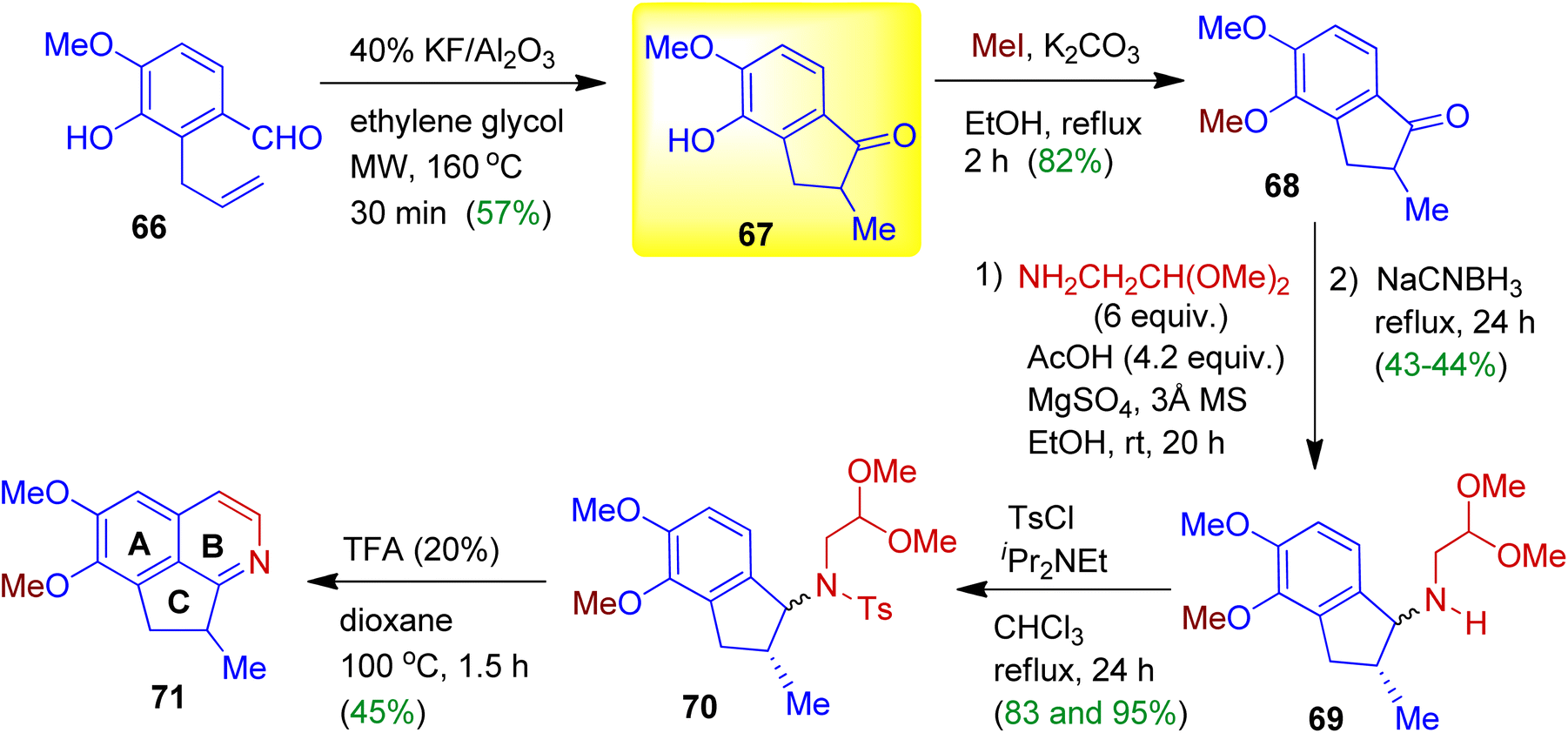
During their synthetic programme, Vargas, Larghi and Kaufman derived cyclopenta[ij]isoquinoline 71 starting from readily accessible 2-allylbenzaldehyde 66 (Scheme 19).51 When compound 66 was exposed to 40% w/w KF/Al2O3 in ethylene glycol under microwave irradiation, 5-exo-trig cyclised product, viz. indanone 67 obtained in 57% yield. Then alkylation of the phenolic moiety was carried out using MeI/K2CO3 in ethanol (68, yield 82%). The indanone 68 was subjected to a reductive amination with aminoacetaldehyde dimethyl acetal in the presence of anhydrous MgSO4 and 3 Å MS. To promote condensation of the intermediate imine and further iminium ion formation AcOH might be added. After reduction with NaCNBH3 in refluxing EtOH, the expected amine 69 was produced as diastereomers in good yields. The amines 69 then allowed to react with tosyl chloride to give corresponding tosylated diastereomers 70. Treatment with 20% trifluoroacetic acid (TFA) in refluxing dioxane delivered cyclopenta[ij]isoquinoline 71 in 45% yield (Pomeranz–Fritsch cyclization). It should be mentioned that ABC-ring system of this type are ubiquitous in alkaloids, such as, azafluoranthene, tropoisoquinoline and proaporphine.
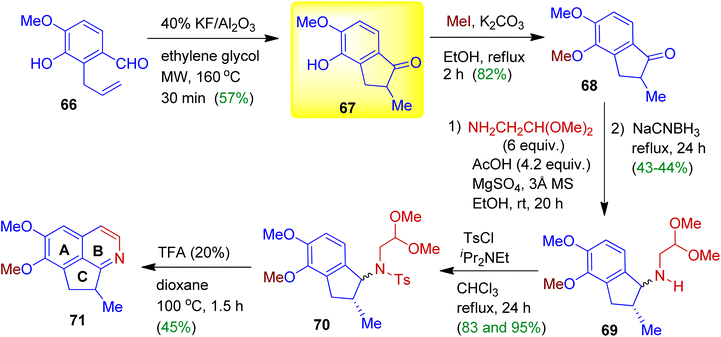 |
| | Scheme 19 Sequential synthesis of cyclopenta[ij]isoquinoline involving 1-indanone scaffold. | |
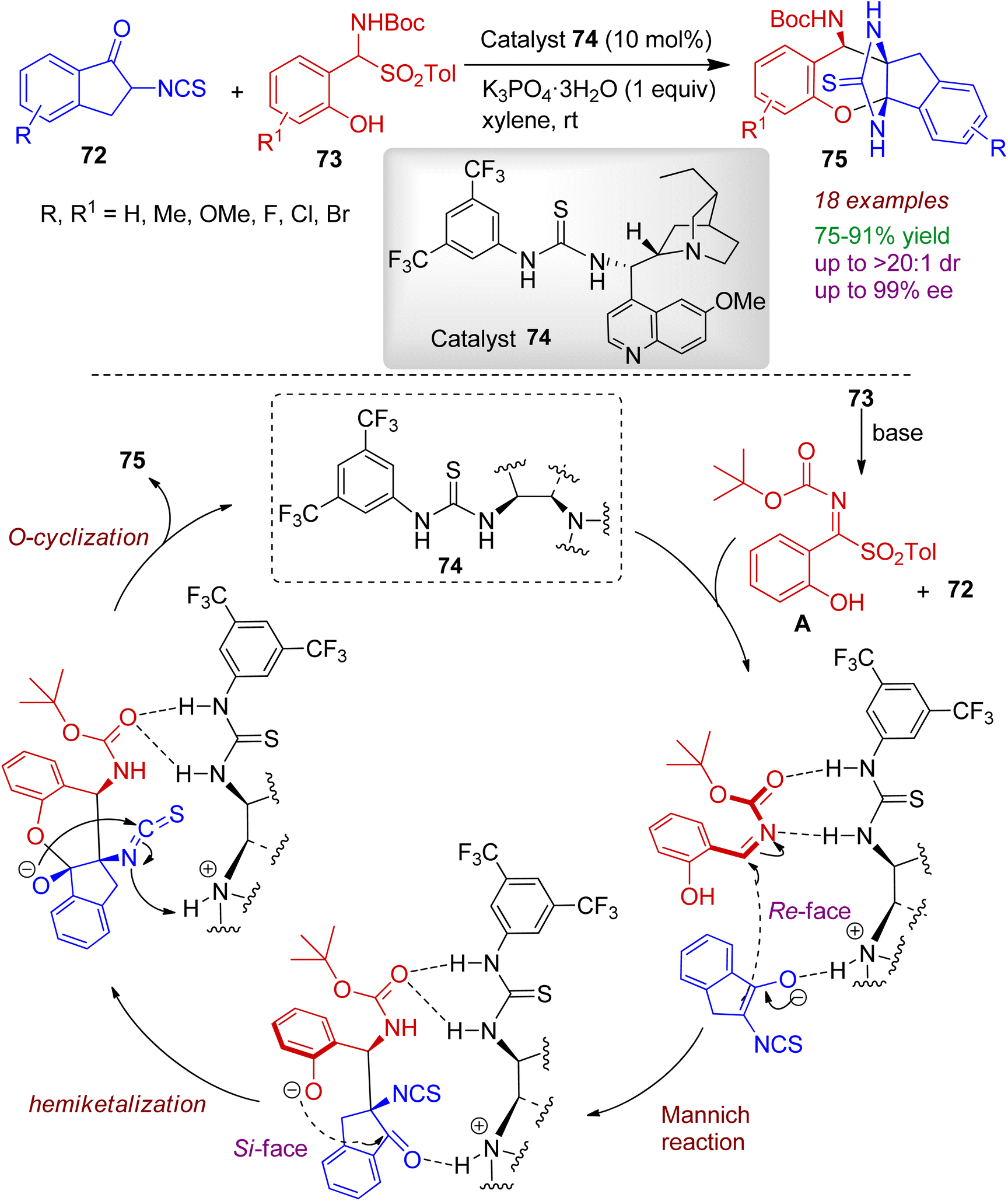
2.2.2. O-Containing fused heterocycles. Isothiocyanato-1-indanones can be employed as building blocks in the heteroannulation reactions. In 2021, Du et al. developed a one-pot asymmetric domino annulation of 2-isothiocyanato-1-indanones 72 with 2-hydroxyaryl-substituted α-amido sulfones 73 using thiourea derived tertiary amine organocatalyst 74.52 This reaction prescribes an efficient protocol for preparing fused ring heterocycles 75 with three adjacent stereogenic centers in good yields (up to 91%) with excellent stereoselectivities (up to 99% ee and >20:1 dr). A plausible reaction mechanism is illustrated in Scheme 20. Firstly, the salicyl N-carbamoyl imine intermediate A is generated from 73 in the presence of a base. In the mean time, nitrogen atom of quinine in organocatalyst 74 promotes the enolization of indanone moiety. The imine is activated by double hydrogen bonding between the thiourea moiety, and carbonyl oxygen and the nitrogen atom form the N-Boc protected imine. Subsequently, the enolate of indanone attacks imine moiety from the Re-face (Mannich addition), and the phenoxide ion (generated by deprotonation of hydroxyl group) attacks the carbonyl of indanone from Si-face (hemiketalization step). The bridged fused heterocyclic product 75 is formed via intramolecular O-cyclization reaction with regeneration of the organocatalyst. The reaction is a rare example of asymmetric catalytic hemiketalization reaction involving indanone system.
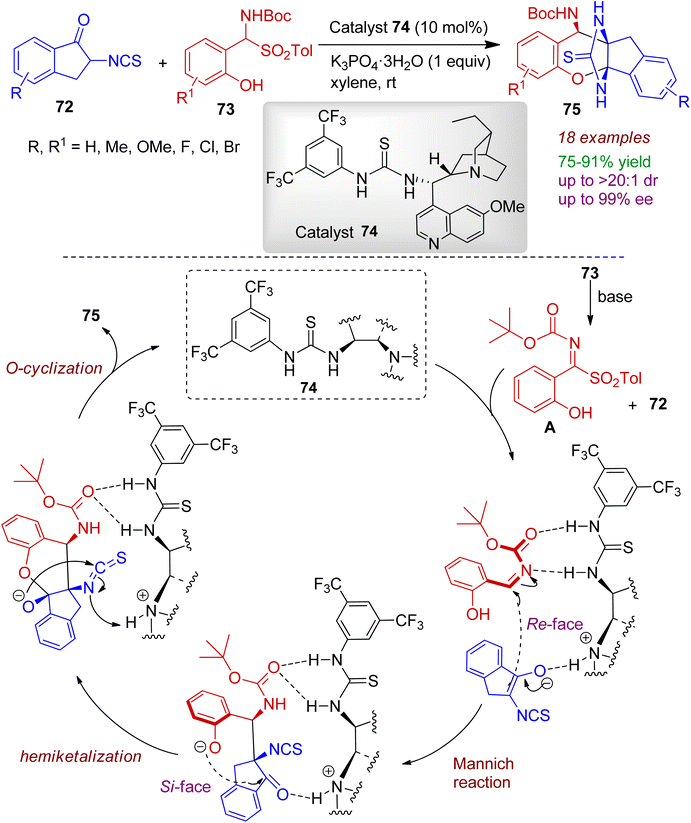 |
| | Scheme 20 Stereoselective construction of bridged fused heterocycles from 2-isothiocyanato-1-indanones and 2-hydroxyaryl-α-amido sulfones. | |
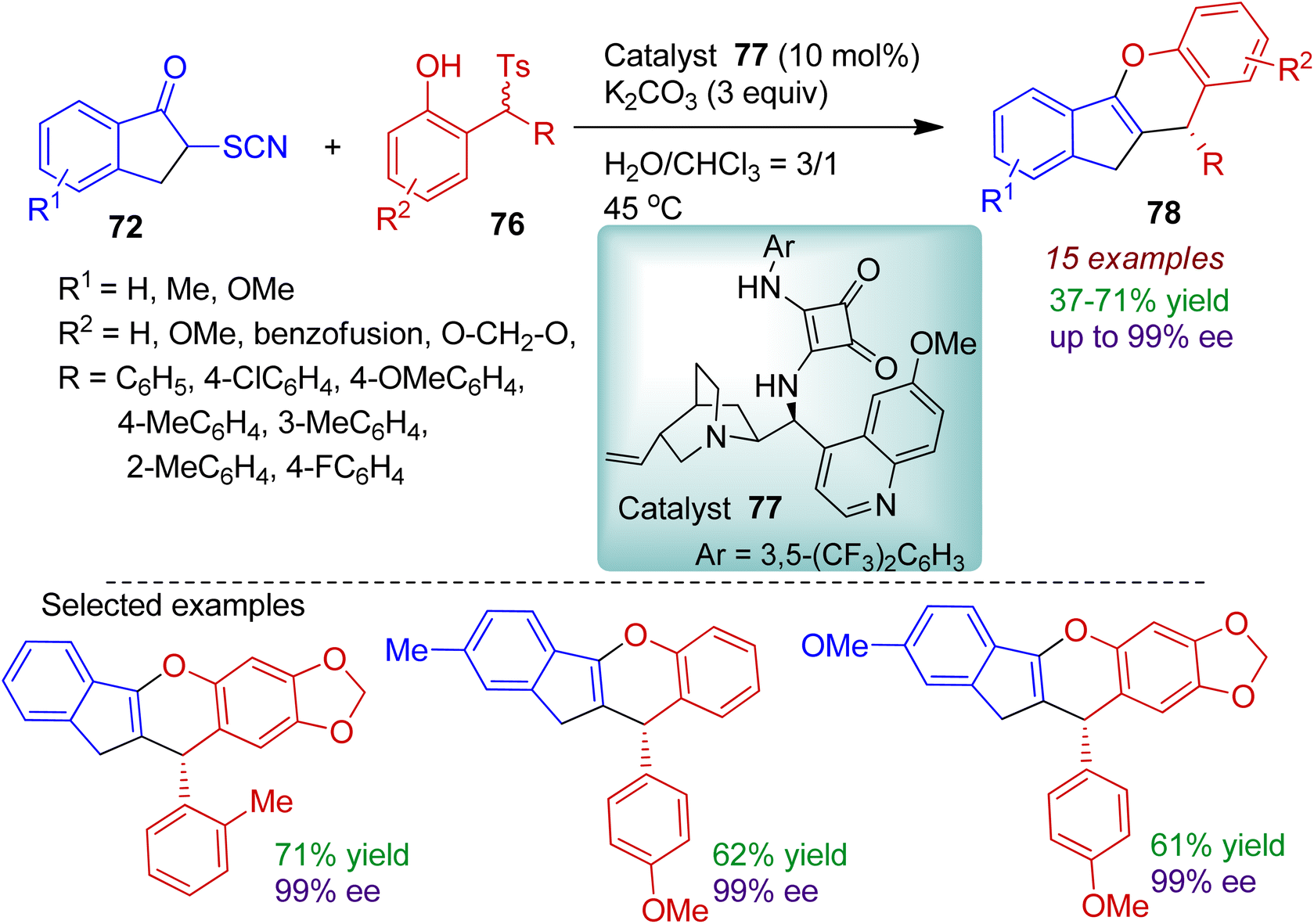
An interesting water-triggered chemodivergent stereoselective cyclization of α-thiocyanato indanone was investigated by Li's group.53 The reaction of α-thiocyanato indanone 72 with 2-(tosylmethyl)phenols 76 catalyzed by quinine-derived squaramide catalyst 77 in H2O:CHCl3 (3:1) system conveniently delivered dihydroindeno[1,2-b]chromene compound 78 (Scheme 21). The products were formed with excellent enantioselectivity (up to 99% ee) and the absolute configuration was determined with the help of X-ray crystallographic study. The α-thiocyanato indanone 72 containing methyl and methoxy substituents were amenable for the catalytic process. Notably, 2-(tosylmethyl)-sesamols bearing different aryl substituents smoothly afforded corresponding cascade products in acceptable yields with excellent chemo- and enantioselectivity.
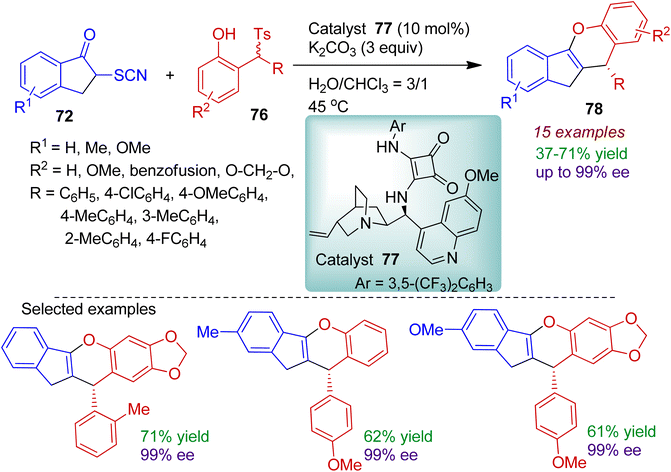 |
| | Scheme 21 Water-triggered formation of oxygen-containing fused polycyclic compounds from α-thiocyanato indanone and 2-(tosylmethyl)phenols. | |
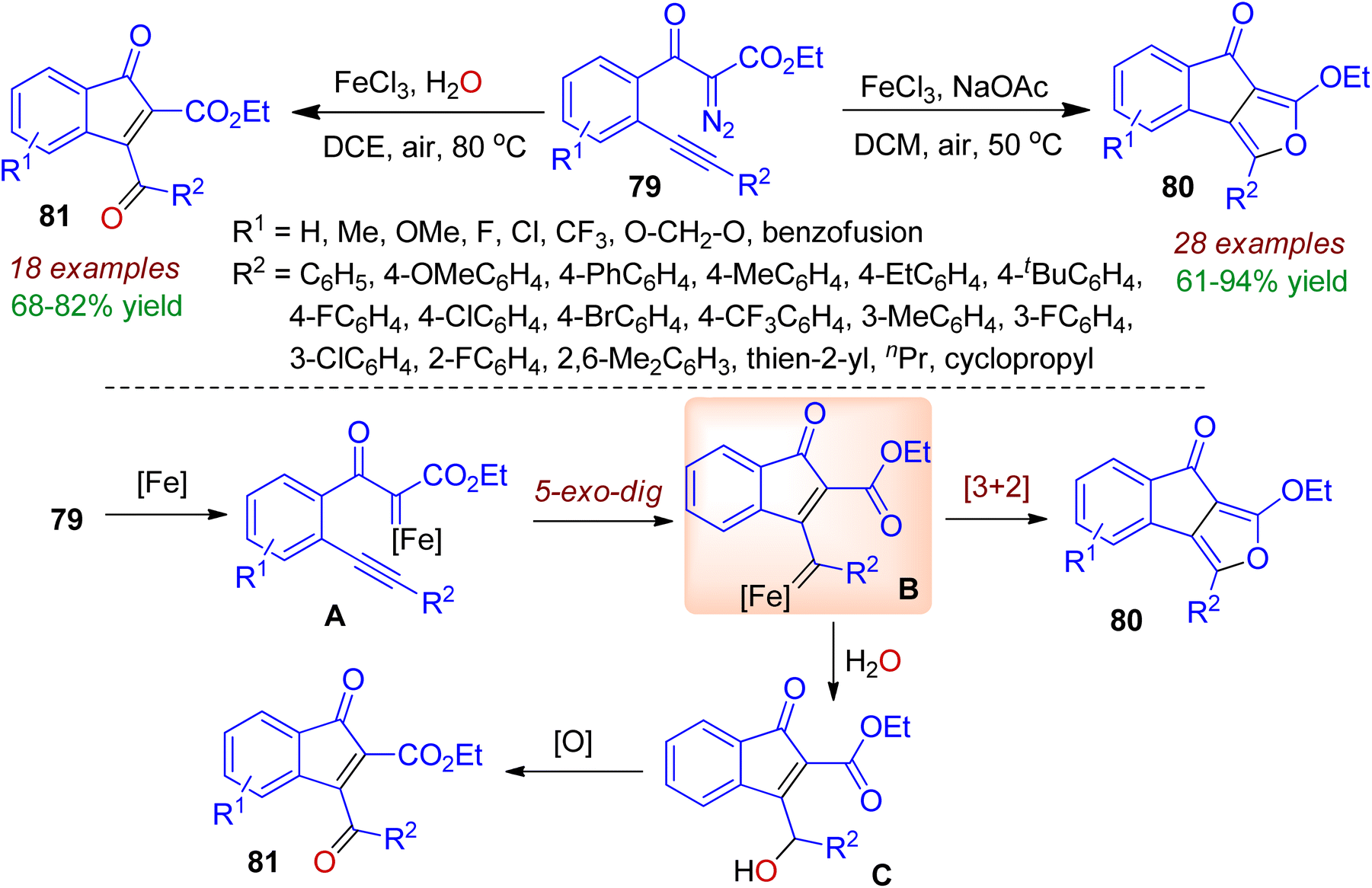
o-Alkynylaryl α-diazoester may be used in metal-catalyzed carbonylation reaction for building indanone skeleton. In 2021, Li, Fan and co-workers reported the FeCl3-catalyzed carbene/alkyne metathesis (CAM) reaction of o-alkynylbenzoyl diazoacetates 79 for the synthesis of indeno[1,2-c]furan core 80.54 The reaction readily occurred in dichloromethane with NaOAc and air at 50 °C. However, the presence of water in the reaction medium resulted in 3-benzoylindenone derivatives 81. The different types of product formation could be realized by mechanism (Scheme 22). Initially, iron-catalyzed dinitrogen elimination from o-alkynylbenzoyl diazoacetates 79 afforded iron carbene intermediate A. Next, 5-exo-dig carbocyclization process produced vinyl iron carbene B which might be the key intermediate. Subsequently intramolecular [3+2] cycloaddition directed the formation of fused indenofuran core 80. On the other hand, in the presence of H2O, B is terminated with O–H insertion to give intermediate C, which underwent aerial oxidation resulting indenone motif 81.
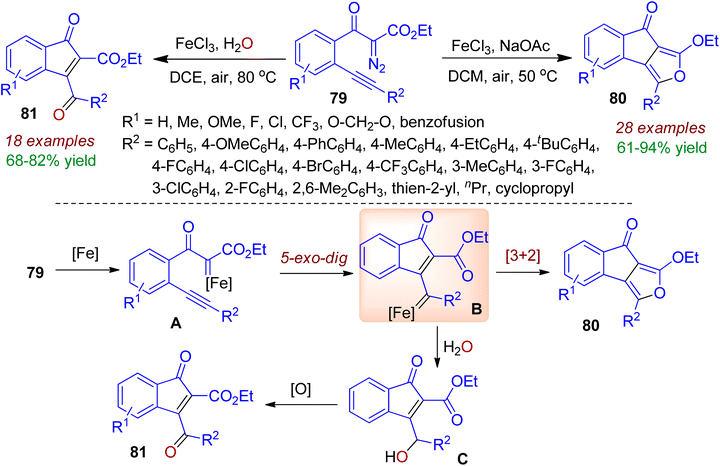 |
| | Scheme 22 FeCl3-catalyzed carbene/alkyne metathesis reaction of o-alkynylbenzoyl diazoacetates to obtain indeno[1,2-c]furans. | |
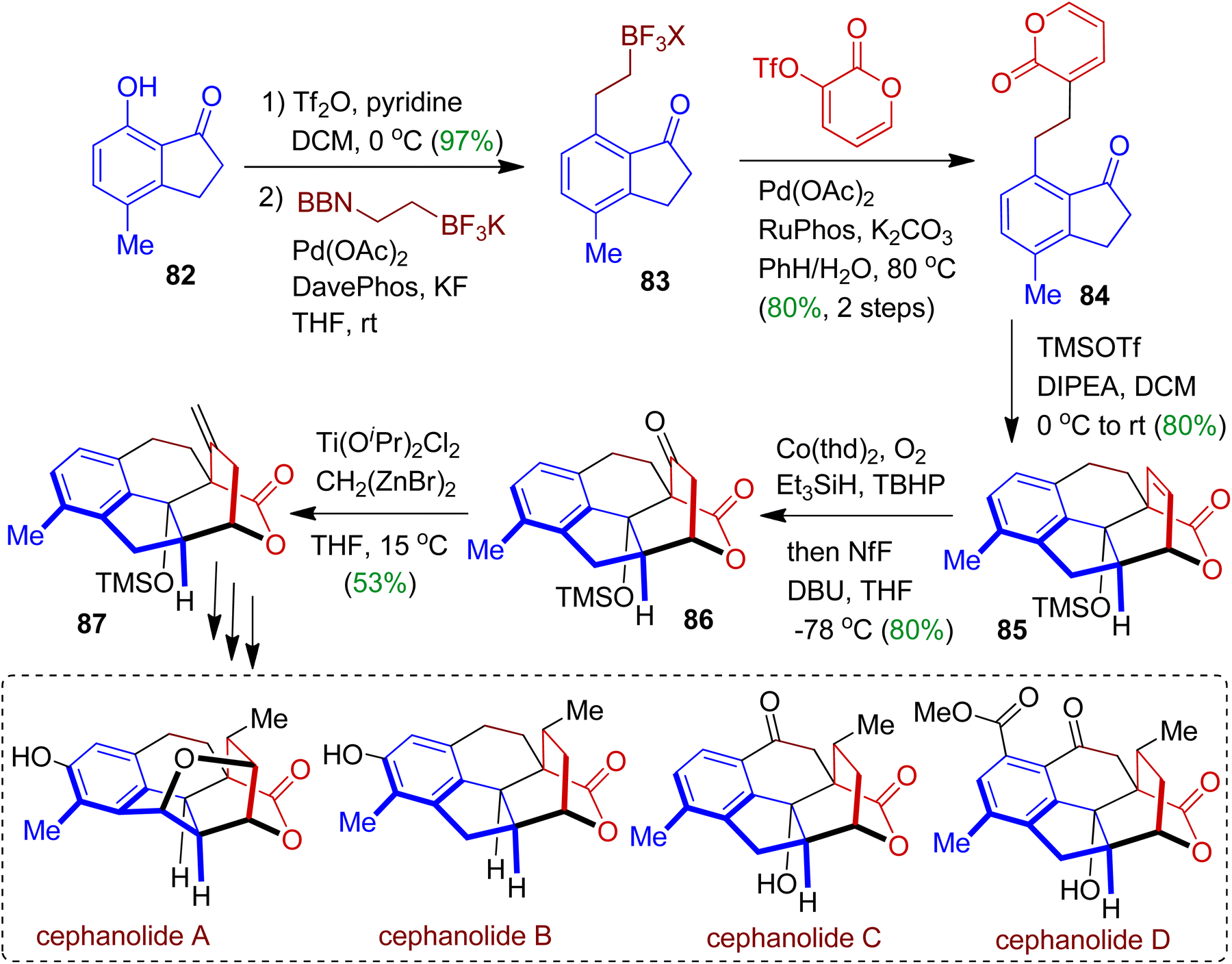
Sarpong et al. employed commercially available 7-hydroxy-4-methylindanone 82 and assembled successfully to synthesize naturally occurring cephanolides A–D via multistep approach.55 At the outset, the triflation product of indanone 82 was subjected to react with BF3K-ethylene-9BBN to obtain boron compound 83. Iterative sp3–sp3 Suzuki cross-coupling with pyrone triflate accomplished indanone derivative 84 (80% yield). In the next step, [4+2] cycloaddition reaction was performed by using TMSOTf/DIPEA to afford corresponding cycloadduct 85 as sole diastereoisomer. The cycloadduct 85 was then converted to ketone 86 in one-pot via hydrocobaltation process. Olefination of 86 using Ti(OiPr)2Cl2 led to exo-methylene product 87 (53% yield) which is the key synthon for cephanolide core. This reaction was generally performed on a 500 mg scale. Compound 87 could be transformed into cephanolides A–D through multistep synthesis and structural diversification (Scheme 23).
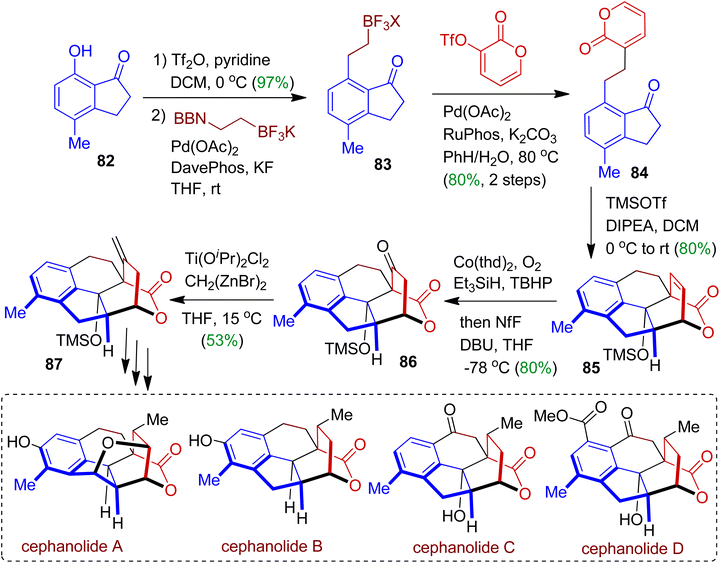 |
| | Scheme 23 Multistep synthesis of cephanolides A–D starting from 7-hydroxy-4-methylindanone. | |
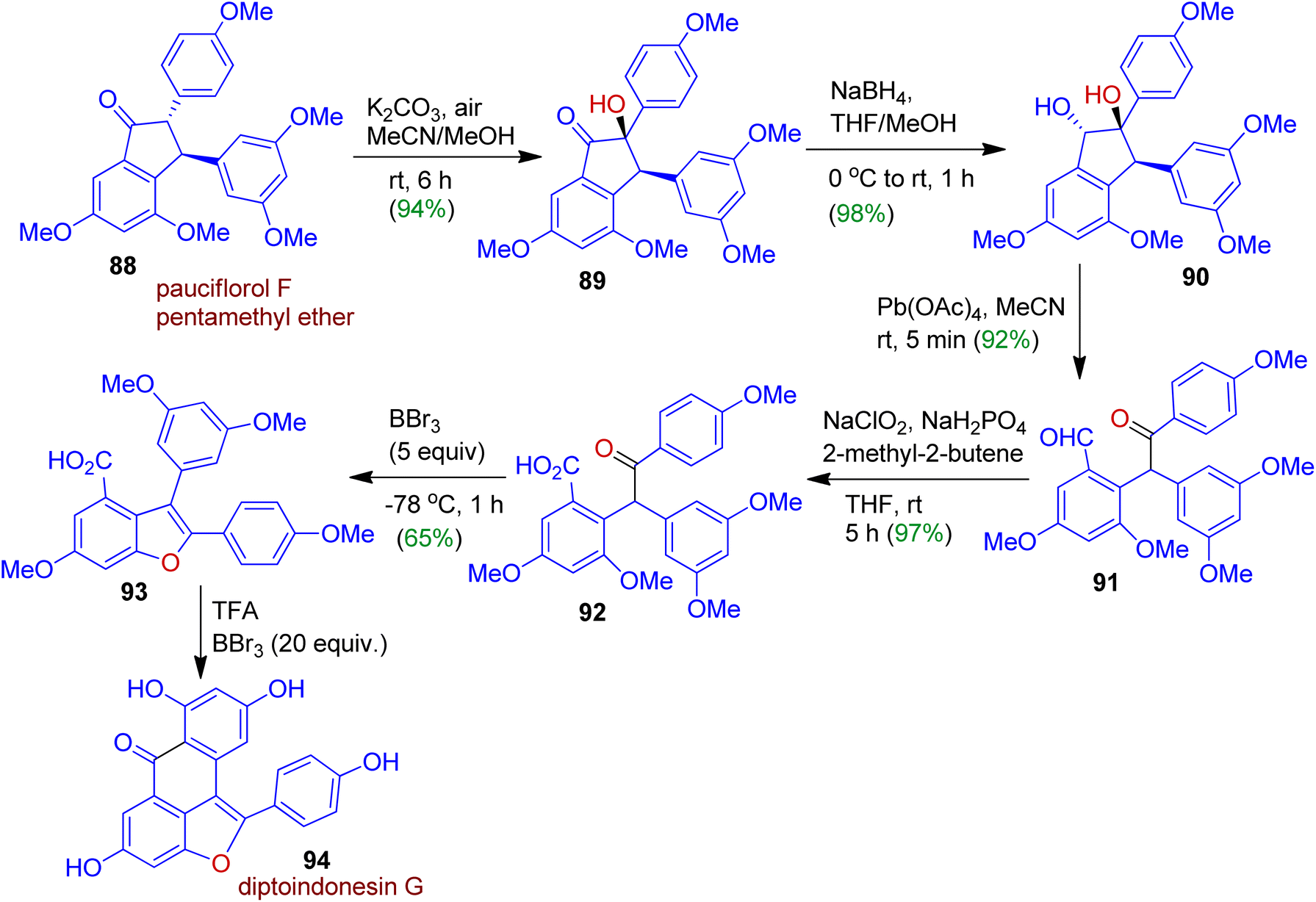
Singh and Kim devised a novel synthetic approach towards diptoindonesin G, a potent anticancer natural product starting from readily accessible pauciflorol F pentamethyl ether 88. The transformation could be achieved through skeletal reconstruction adapting oxidative ring-opening and sequential ring closure strategy (Scheme 24).56 As expected, the α-hydroxylation of indanone 88 in the presence of K2CO3 and air at room temperature afforded hydroxy keto compound 89 (94% yield), which was then reduced with sodium borohydride for the diastereoselective formation of trans-diol dreivative 90. The conversion of trans-diol 90 into ketoaldehyde 91 was carried out with Pb(OAc)4. Subsequently, Pinnick–Kraus oxidation of 91 with NaClO2/NaH2PO4 gave corresponding acid 92 in 97% yield. Exposure of acid 92 to BBr3 led to 65% of benzofuran 93 which was finally converted to target tetracyclic skeleton 94 (diptoindonesin G) using TFA/BBr3 system.
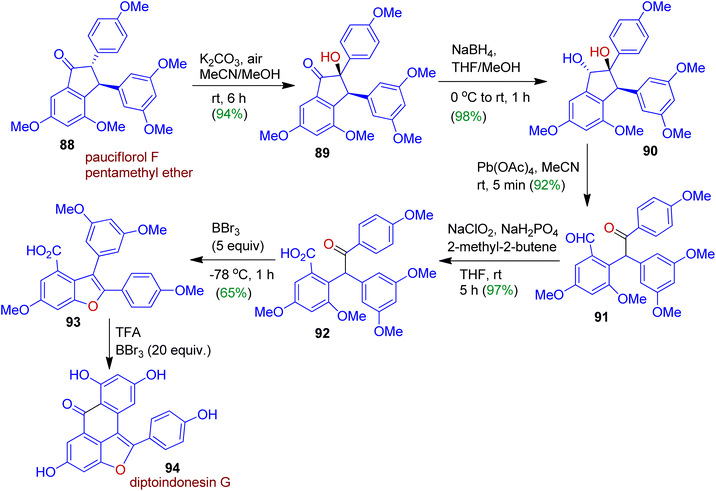 |
| | Scheme 24 Synthesis of diptoindonesin G strating from pauciflorol F pentamethyl ether via skeletal reorganization. | |
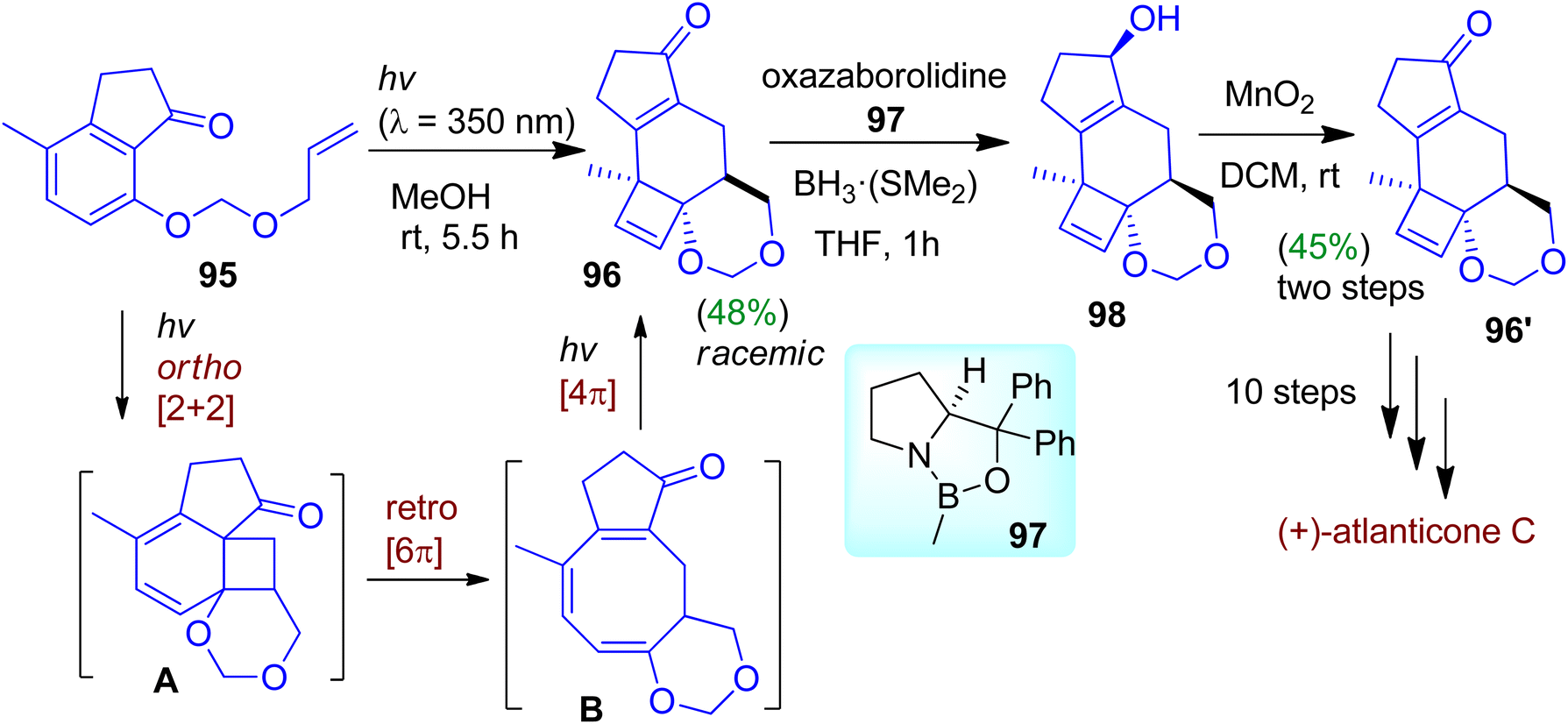
Recently, Bach's group designed and synthesized strained polycyclic frameworks based on photochemical reaction cascade exploiting an indanone precursor.57 The photochemical reaction cascade was started with ortho-photocycloaddition of the indanone substrate 95. The strained diene intermediate A formed by this process then underwent thermal disrotatory ring opening to give triene intermediate B (Scheme 25). Further irradiation of triene B afforded [4π]-photocyclized product 96 possessing three stereogenic centers as racemic mixtures (in 48% yield). The authors tactfully carried out the resolution employing Corey–Bakshi–Shibata (CBS) reduction process. Enantioselective CBS reduction of the racemic photoproduct (i.e. catalytic chiral resolution using oxazaborolidine 97 and BH3·SMe2) led to the formation of enantiopure alcohol 98. Subsequent oxidation accomplished enantiopure ketone 96′. The enantiomerically enriched product could be transformed into naturally occurring (+)-atlanticone C after 10 steps.
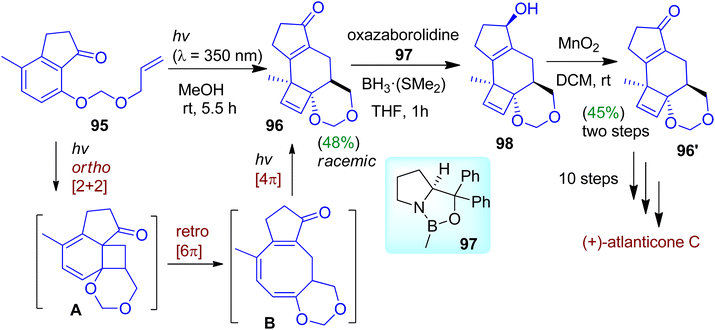 |
| | Scheme 25 Construction of complex polycyclic skeleton via photochemical reaction cascade of 1-indanone derivative. | |
3. Synthesis of spiro scaffolds
3.1. Spiro carbocycles
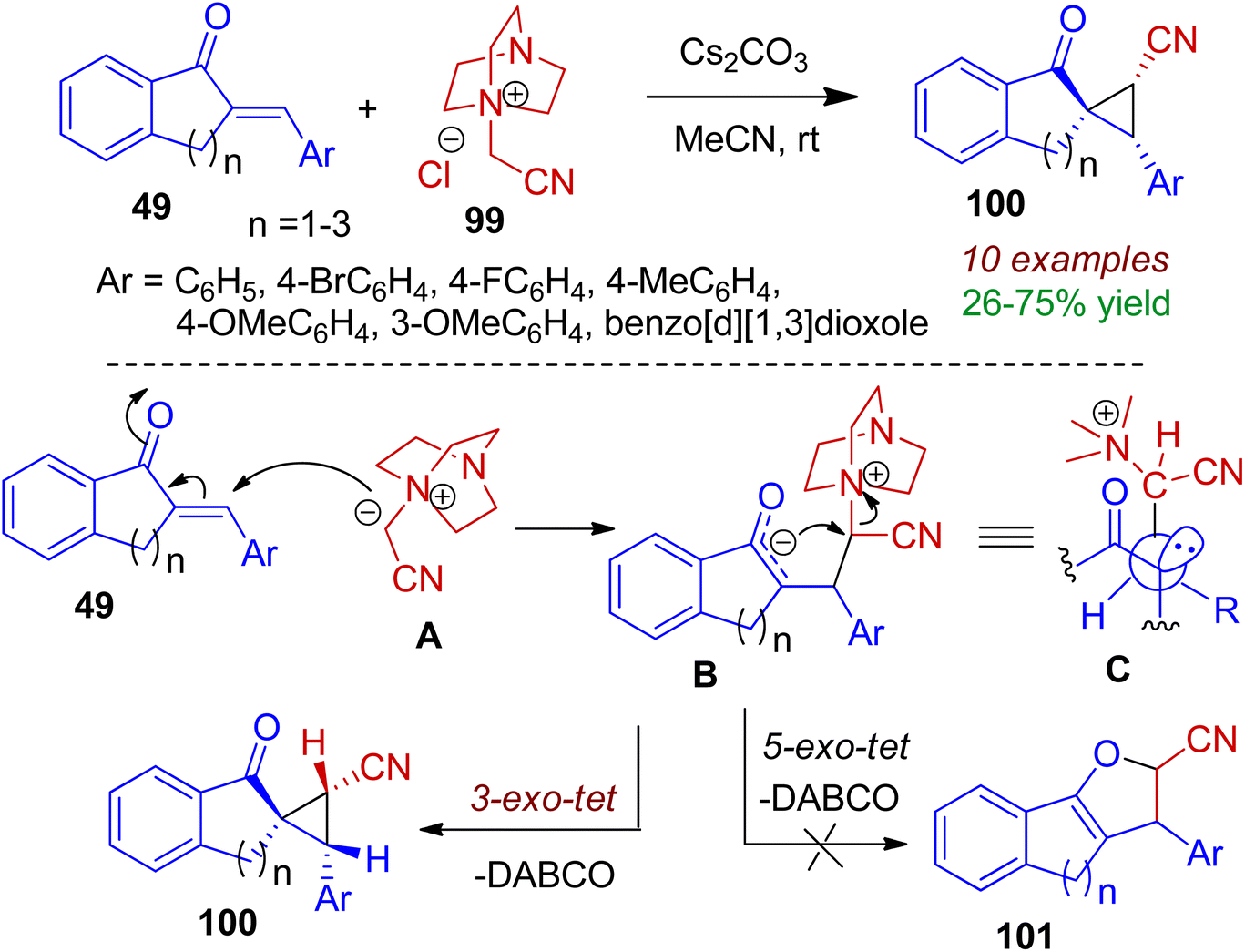
Spirocyclic compounds play an important role in drug development due to their three dimensionality and structural novelty. Particularly, spiroindanones are special class of compounds found in many natural products and pharmaceuticals. This type of molecules could be accomplished through cyclopropanation strategy. In 2017, Namboothiri et al. devised a new route towards spirocyclopropanes 100 via addition of 2-arylidene indanones 49 to DABCO derivative 99 at ambient temperature (Scheme 26).58 The reaction holds good for tetralone (n = 2) and benzosuberone (n = 3) system resulting corresponding spirocyclic analogues in moderate to good yields (up to 75%). Interestingly, in all the cases single diastereoisomers were obtained. According to the mechanism, initially ylide A is generated from 99 in the presence of base Cs2CO3. Michael type addition of intermediate A towards chalcone 49 gives enolate B which is stabilized in a conformation B (where the carbonyl and the R groups are anti to each other i.e., conformation C). Finally cyclization in 3-exo-tet fashion selectively produces spiroindeno cyclopropane 100 instead of fused product 101.
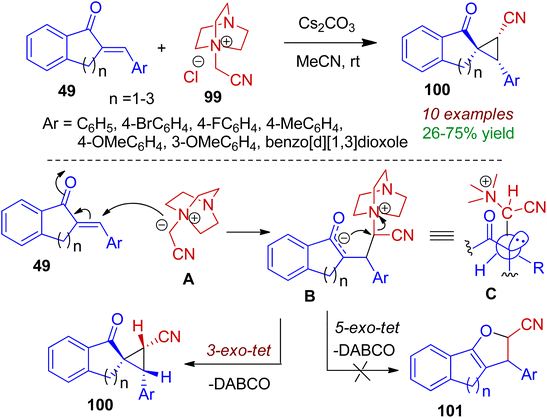 |
| | Scheme 26 Base-promoted synthesis of spiroindeno cyclopropanes from arylidene indanones and DABCO-derived N-ylides. | |
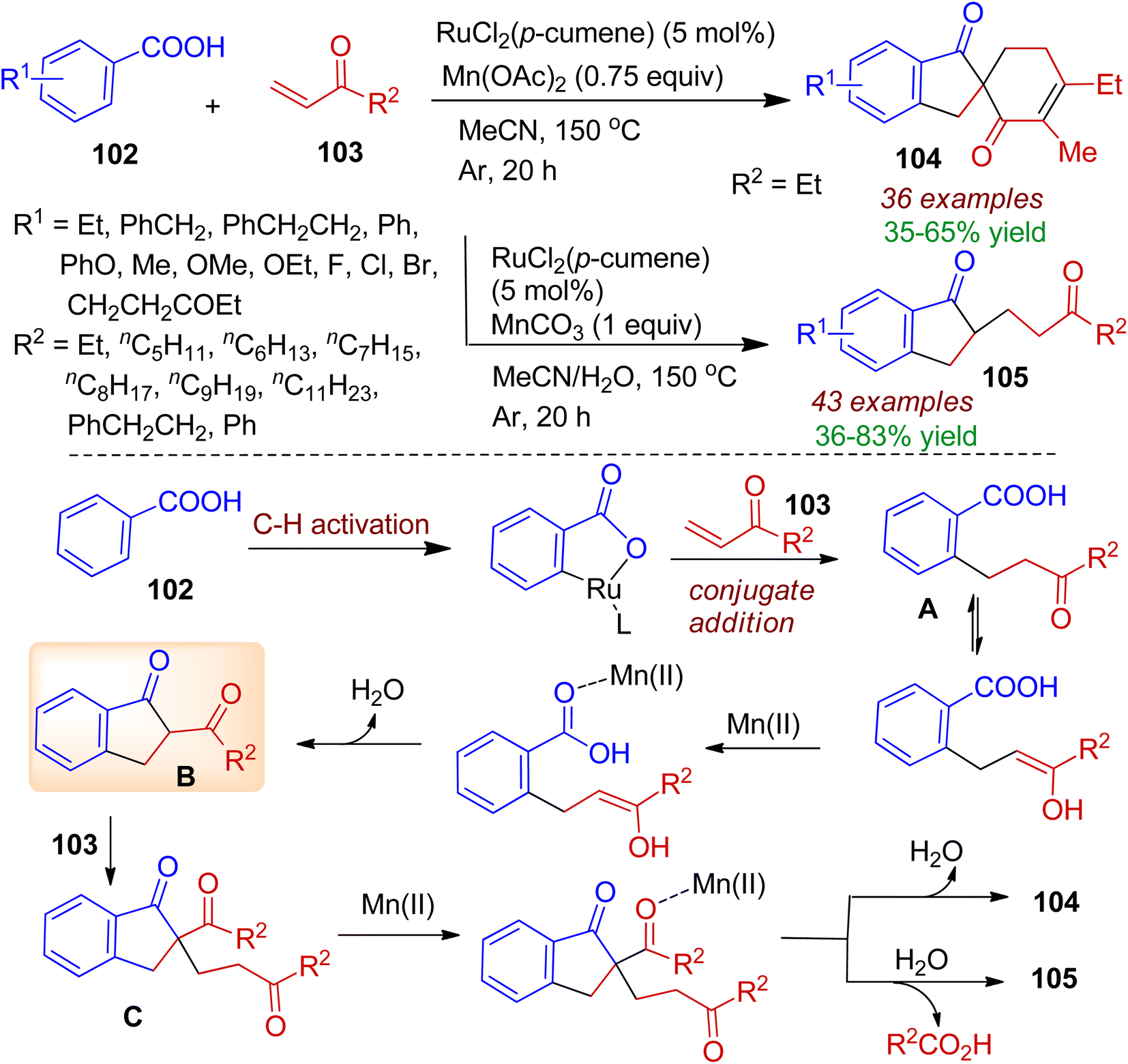
Starting from commercially available aromatic acids 102 and α,β-unsaturated ketones 103 Shi's group developed a ruthenium-catalyzed tandem coupling and cyclization reaction to obtain structurally diverse 1-indanone derivatives.59 In this case, switchable access to wide range of spiroindanones 104 and 2-substituted indanones 105 could be tuned by Mn(II) additive and water. A probable mechanism is illustrated in Scheme 27. Initially, a ruthenium-catalyzed and carboxyl group directed conjugate addition of C–H bond to the α,β-unsaturated ketones 103 gave ortho alkylated benzoic acid intermediate A. Subsequently, intermediate A underwent Dieckmann condensation reaction through enol attacking Mn(II)-activated carbonyl to form 2-acyl-1-indanone B. Michael addition of the intermediate B to the second α,β-unsaturated ketone resulted intermediate C. Finally, the intermediate C could proceed by two pathways depending upon chelation interactions of Mn(II) and oxygen atom. The intramolecular Aldol condensation directly furnished spiroindanone 104. Notably, presence of trace amount of water caused hydrolysis to give 2-substituted-1-indanones 105.
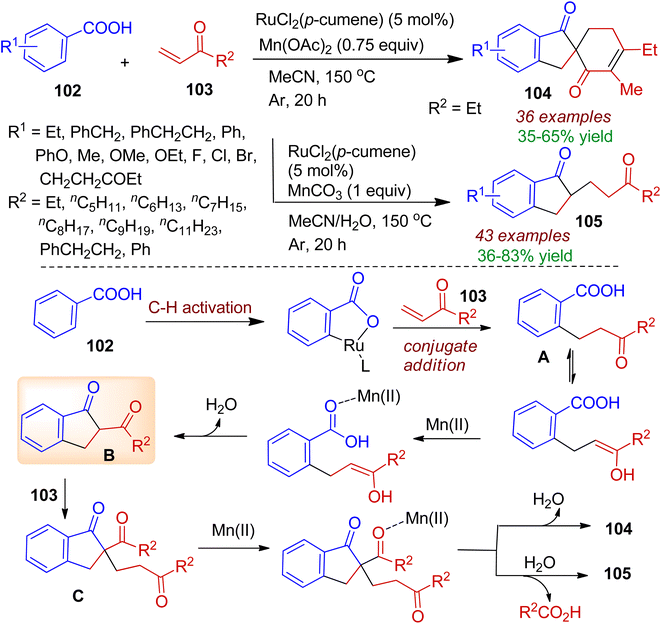 |
| | Scheme 27 Ru-catalyzed annulation of aromatic acids and α,β-unsaturated ketones to form spiroindanones. | |
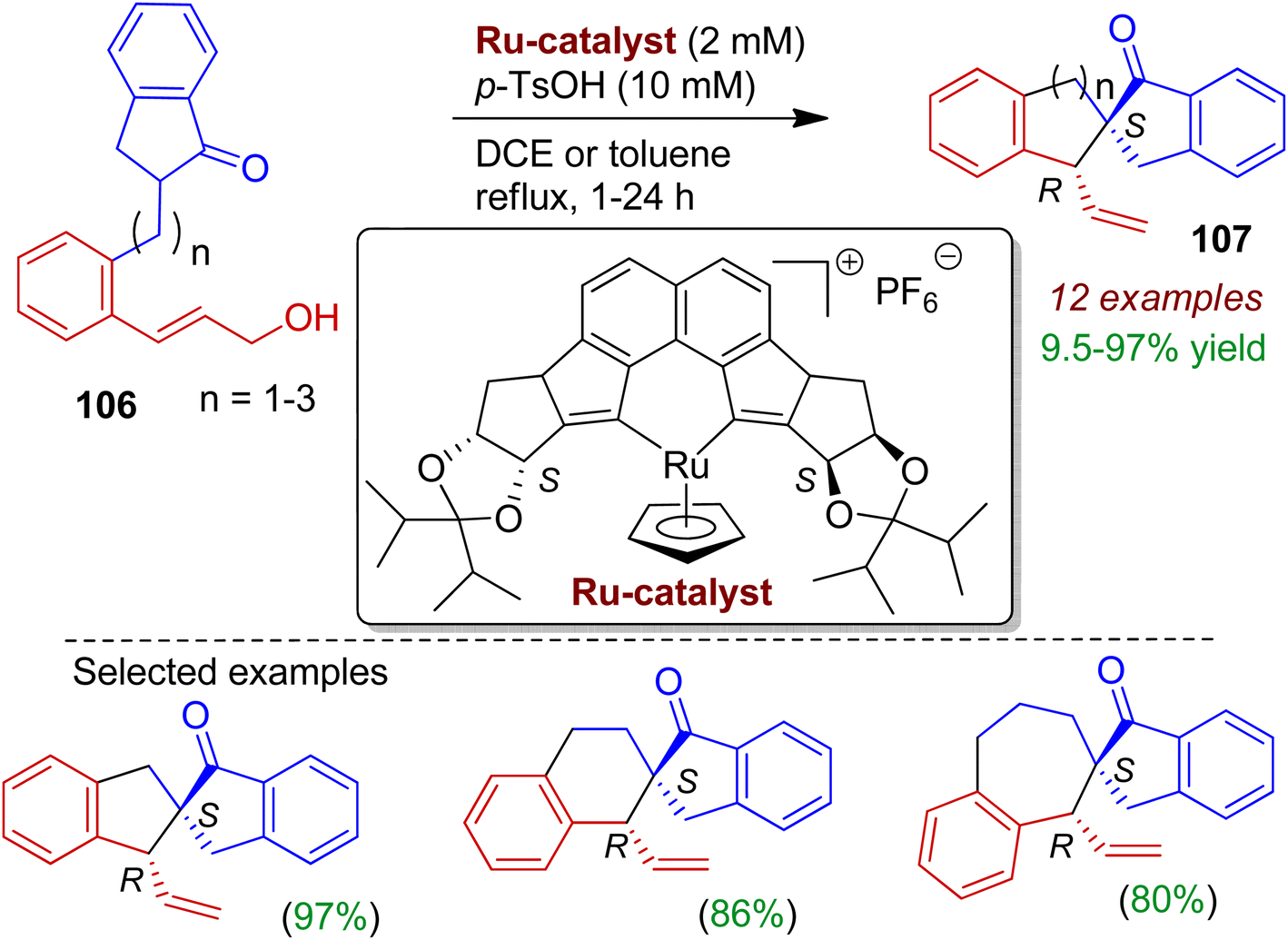
Synthesis of enantiopure spirocycles is important for medicinal chemistry research. Kitamura's group realized stereoselective construction of spiroindanone-carbocycles via an intramolecular Tsuji–Trost strategy.60 The authors applied [Ru(II)Cp((S,S)-Naph-diPIM-dioxo-iPr)]PF6 and sulfonic acid combined catalyst system for the first time to facilitate dehydrative one-pot access to spirocarbocycles from simple racemic ketone-containing allylic alcohols 106 without strochiometric activation of both the CO and OH group (Scheme 28). This enantio- and diastereoselective protocol simultaneously installs the spiro-all-carbon quaternary center at CO β position, resulting corresponding trans isomer 107 among four possible stereoisomers. The selection of sulfonic acid and an E-configured cinnamyl alcohol-type substrate is crucial for high selectivity and reactivity. A double hydrogen bond between OH and SO3H might be important for the activation of allylic alcohol moiety. Notably, synthesis of spiroindanones bearing five-, six-, or seven-membered cyclic systems is possible applying this methodology.
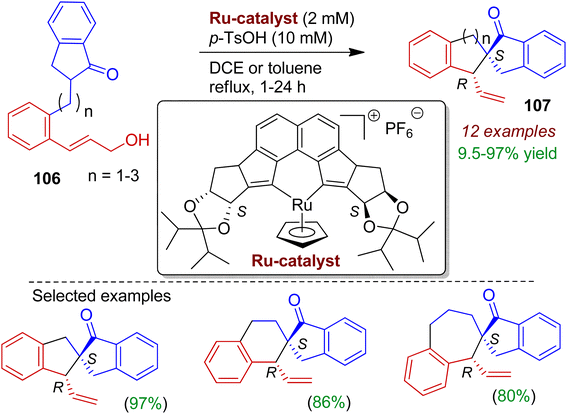 |
| | Scheme 28 Synthesis of enantiopure spiroindanone-carbocycles via intramolecular Tsuji–Trost strategy. | |
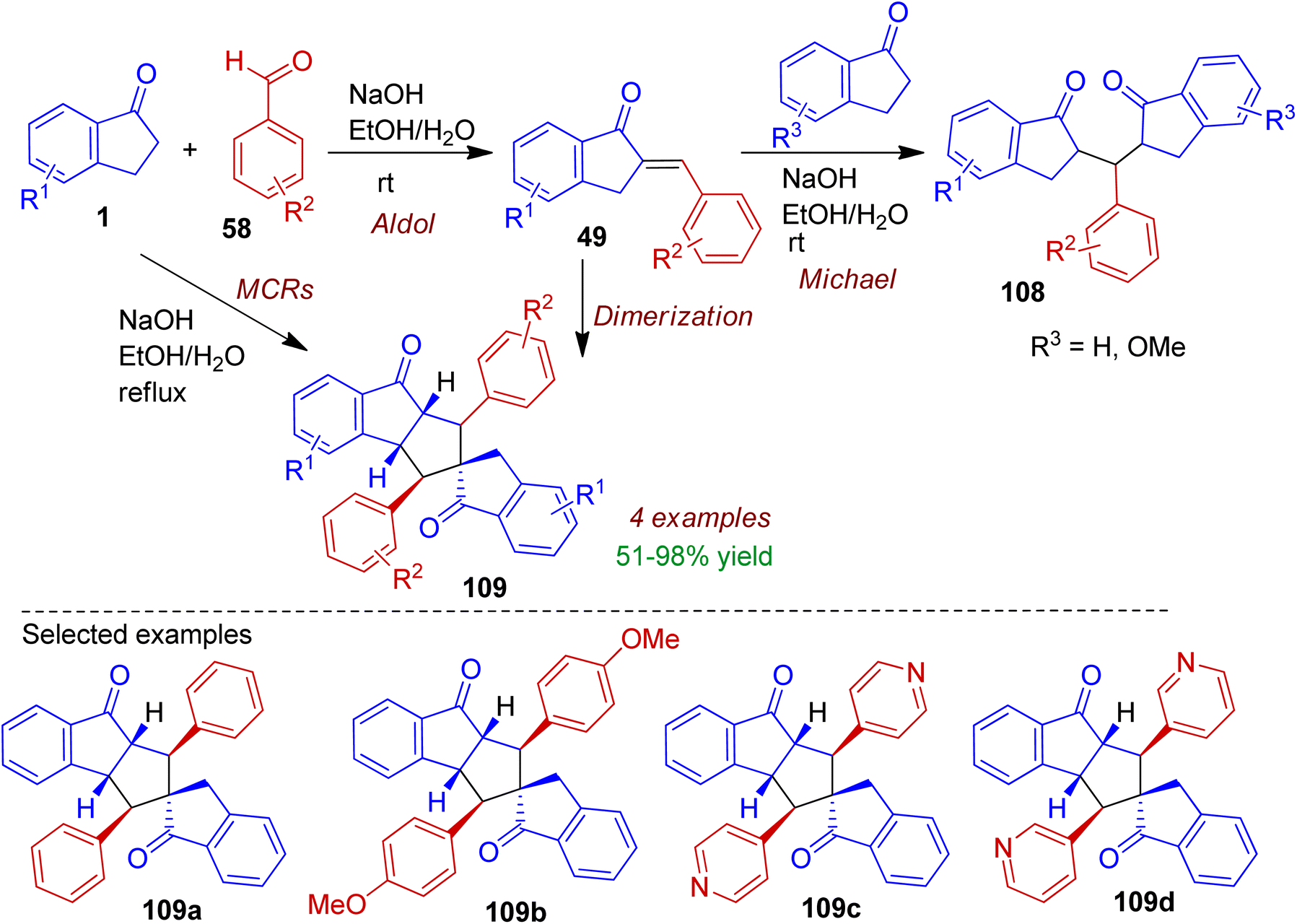
During their research with indanone analogues, Lantano and co-workers studied the influence of reaction temperature and electronic nature of aldehydes under classical Claisen–Schmidt condensation conditions.61 The reaction of 2-arylidene-1-indanones 49 with 1-indanones (R = H, OMe) at room temperature in the presence of aqueous ethanolic NaOH resulted Michael addition product bis-indane-1,5-diketones 108 (Scheme 29). On the other hand, multicomponent reaction between 1-indanone 1 (2 equiv.) and benzaldehydes 58 (2 equiv.) under refluxing conditions accomplished spirocarbocyclic compounds 109a-b in good yields. It is worth mentioned that reaction with 3- and 4-pyridine aldehydes occurred at room temperature affording spiro-products 109c-d in excellent yields (up to 98%). The dimerization of 2-arylidene-1-indanones 49 could also generate corresponding spiro compounds 109.
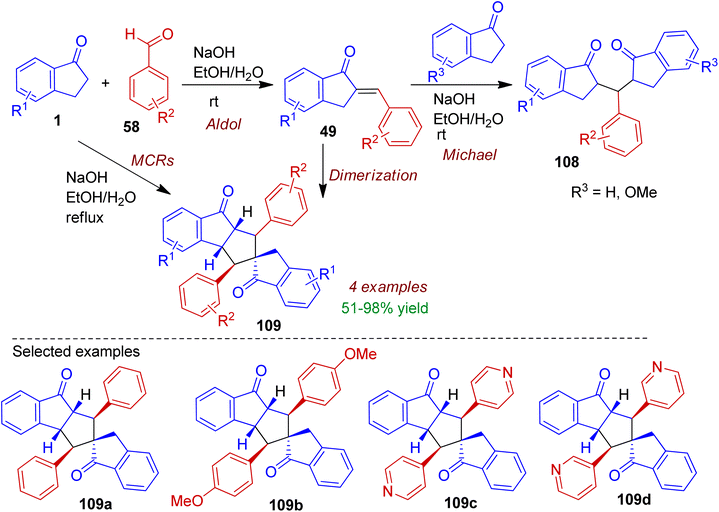 |
| | Scheme 29 Multicomponent synthesis of spirocarbocycles from 1-indanones and aryl aldehydes. | |
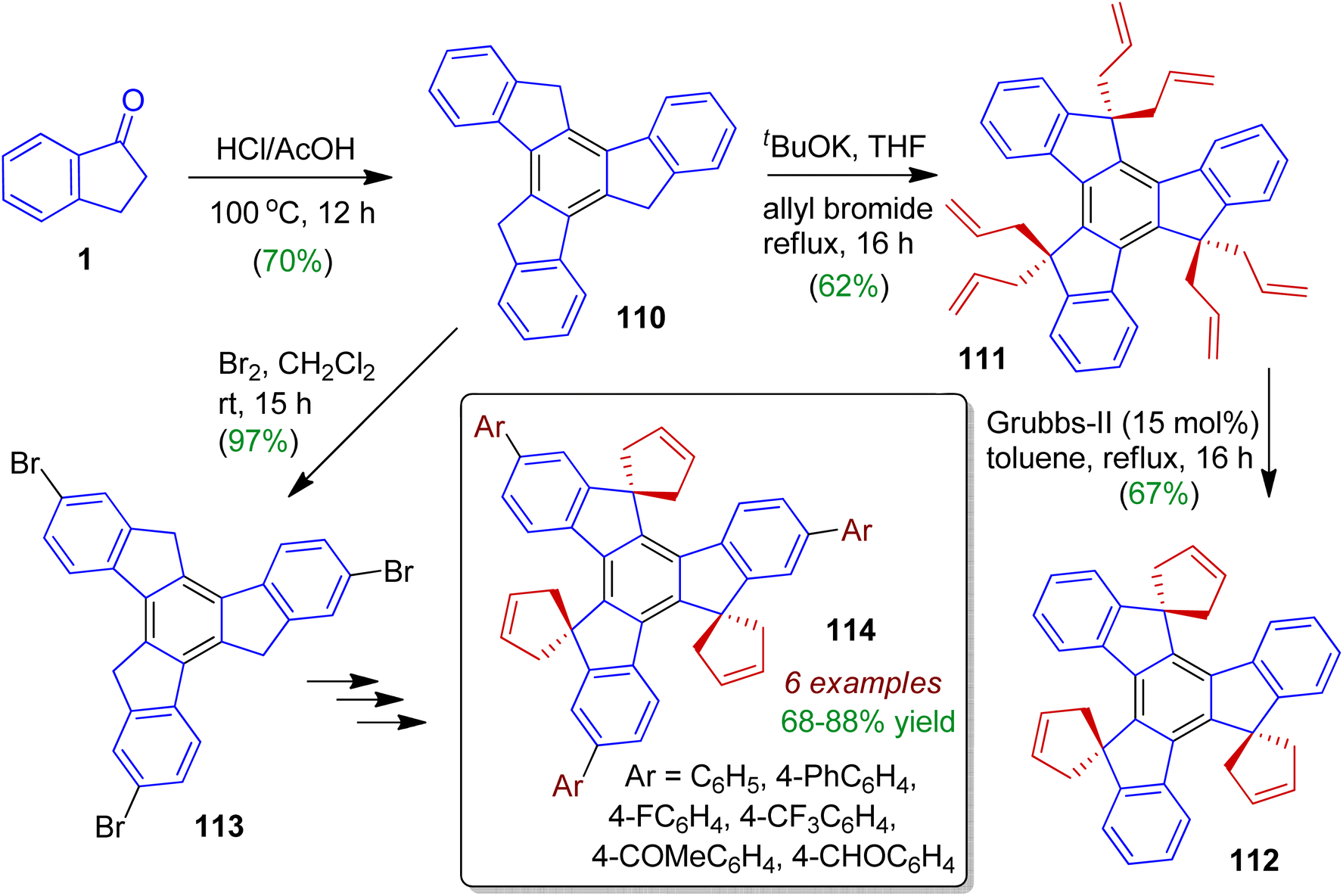
Synthesis as well as investigation of photophysical properties of indanone derived spirotruxenes was carried out by Kotha's group.62 Firstly, cyclotrimerization of 1-indanone 1 was achieved with HCl/AcOH to generate truxene 110 (in 70% yield). Then treatment of tBuOK/allyl bromide in refluxing THF furnished hexallyl derivative 111 in 62% yield (Scheme 30). Subsequently, three-fold ring-closing metathesis (RCM) of 111 was attempted with Grubbs' second-generation catalyst to furnish desired compound 112 (67%). Bromination of truxene 110 may be performed using bromine in dichloromethane to offer 2,7,12-tribromotruxene 113, which was followed by successive steps to deliver various arylated spirotruxenes 114. These arylated substances exhibit fascinating photophysical properties with strong quantum yields (due to enhanced conjugation), and might be considered as C3-symmetric ‘blue-green-light-emitting materials’.
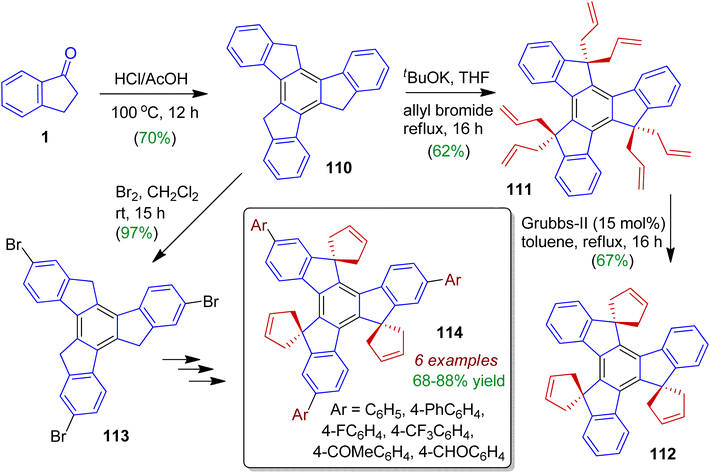 |
| | Scheme 30 Synthesis of various spirotruxenes from 1-indanone via RCM strategy. | |
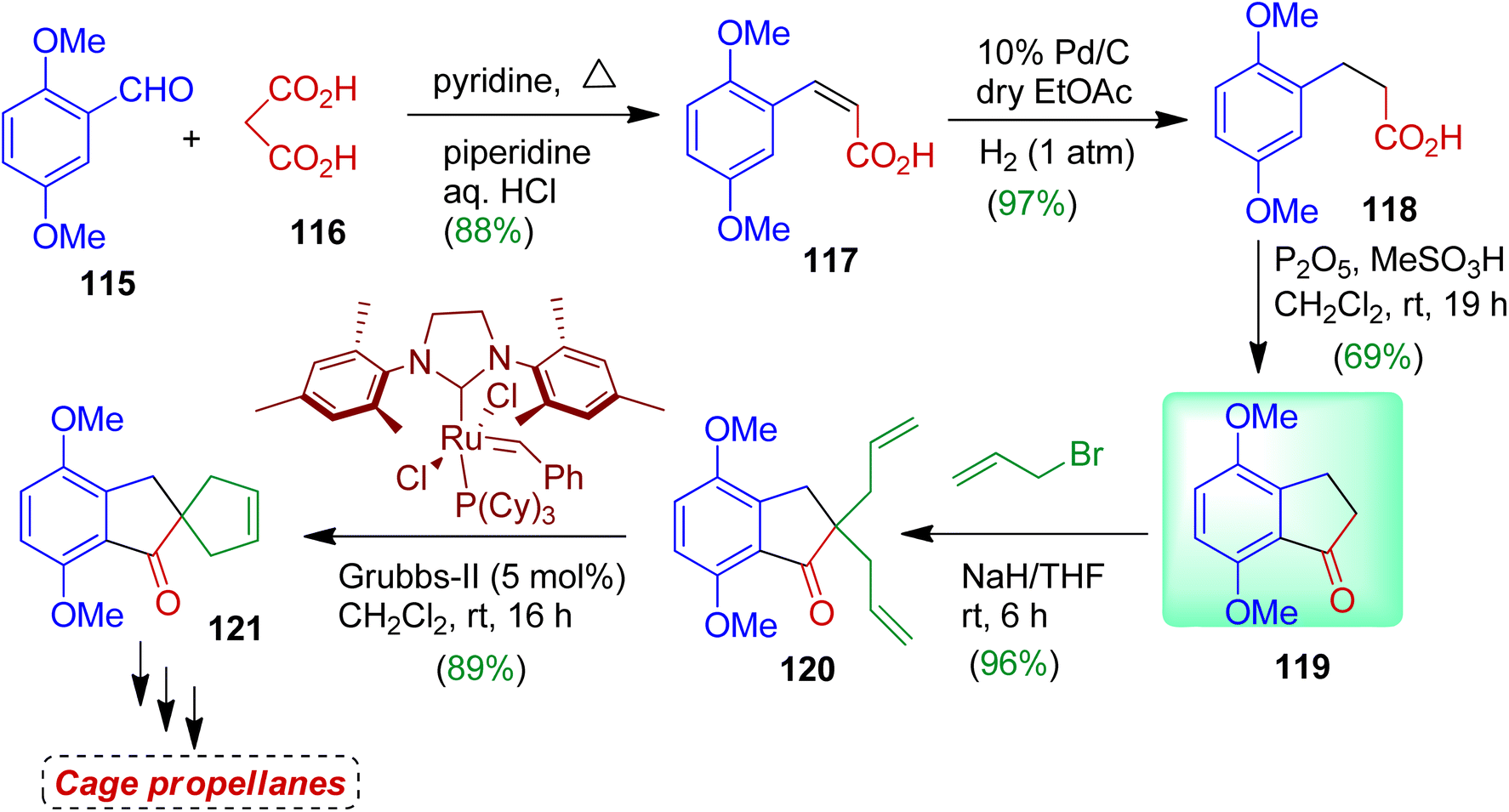
The authors also prepared 1-indanone compound 119 and used as the building block for synthesizing spirocarbocycle as well as cage propellanes.63 The sequence of reactions is outlined in Scheme 31. In the first step, readily available 2,5-dimethoxybenzaldehyde 115 was subjected to react with malonic acid 116 to obtain unsaturated acid derivative 117 (Knoevenagel product) in 88% yield. Then hydrogenation with 10% Pd/C afforded corresponding saturated compound 118 (97% yield). Treatment of 118 with P2O5 and MeSO3H gave 1-indanone derivative 119 which was considered as the key building block for spiro products. Indanone 119 was then treated with allyl bromide in the presence of NaH in THF to furnish diallyl indanone compound 120 (96%). Finally the diallyl derivative 120 was subjected to RCM with Grubbs second generation catalyst resulting ring closure product spiro[4,4]nonane 121 (89%). Compound 121 could be employed as precursor for several cage propellanes and bioactive compounds.64
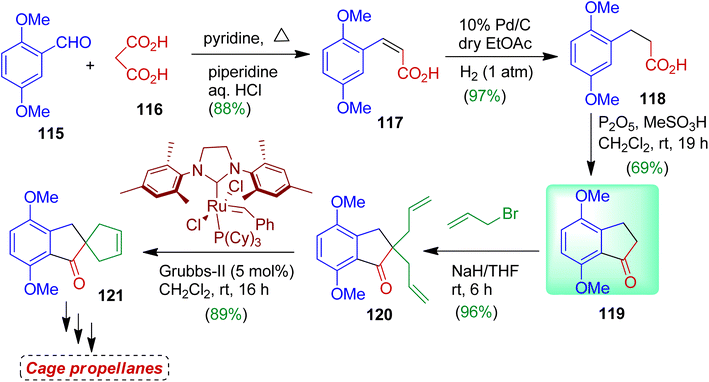 |
| | Scheme 31 Synthesis and application of 1-indanone derivative to access spiro carbocycles. | |
3.2. Spiro heterocycles
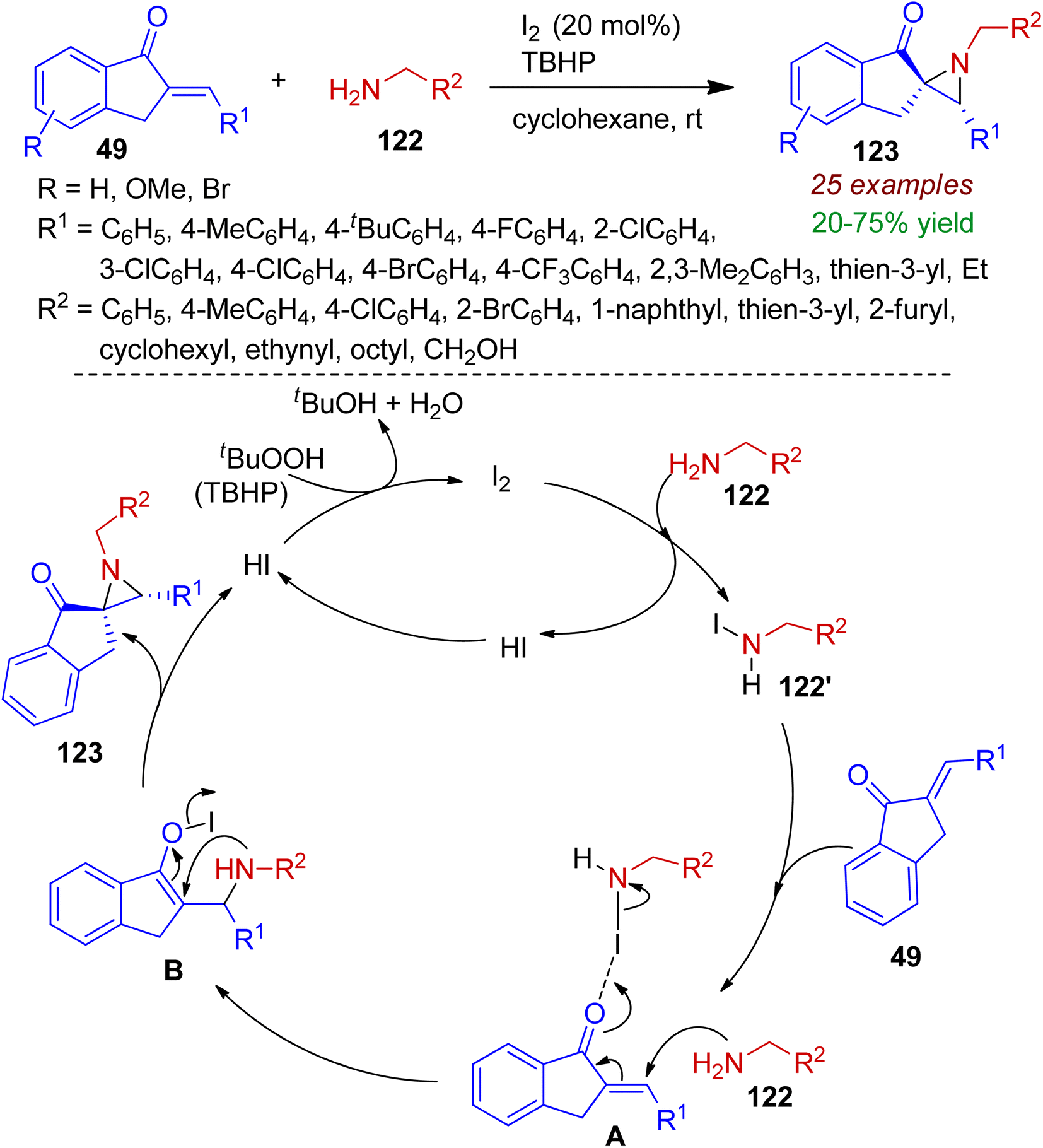
3.2.1. N-containing spiro-heterocycles. Spiro heterocycles are valuable structural motifs for synthetic transformations and considered as emerging drug candidates. Particularly, the synthetic utility of three-membered nitrogen heterocycles (aziridines) can be extended via ring opening reactions due to inherent ring strain.65 Recently, Somappa et al. developed a I2/TBHP mediated method for diastereoselective construction of N-alkylspiroaziridines 123 from primary amines 122 and easily accessible 2-arylated-1-indanones 49.66 Among various types of oxidants TBHP was found to provide the best result in the presence of catalytic amount of I2. Benzyl amines and primary amines bearing aliphatic and heterocyclic core (thiophene, furan) were well tolerated under standard conditions. The proposed mechanism is demonstrated in Scheme 32. Initially primary amine 122 reacted with I2 to generate N-iodoamine species 122′. Afterwards, coordination with N-iodoamine with carbonyl group of indanone 49 assisted the Aza-Michael addition with another amine molecule resulting intermediate A. Intramolecular cyclization of intermediate A delivered the spiroannulated product 123 with regeneration of HI, which is oxidized to I2 by TBHP for next catalytic cycle. Significantly, this transformation involves unprotected primary amines as the nitrogen source.
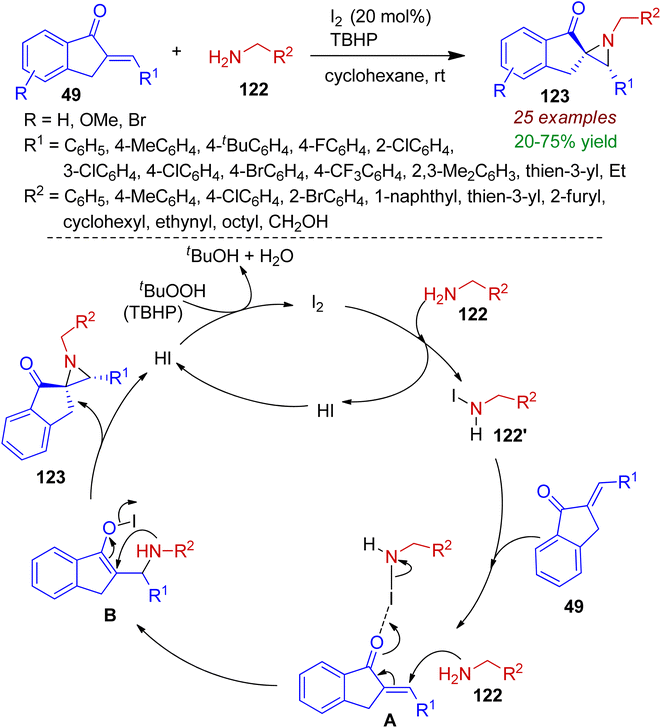 |
| | Scheme 32 I2/TBHP mediated diastereoselective synthesis of N-alkylspiroaziridines from primary amines and 2-arylated-1-indanones. | |
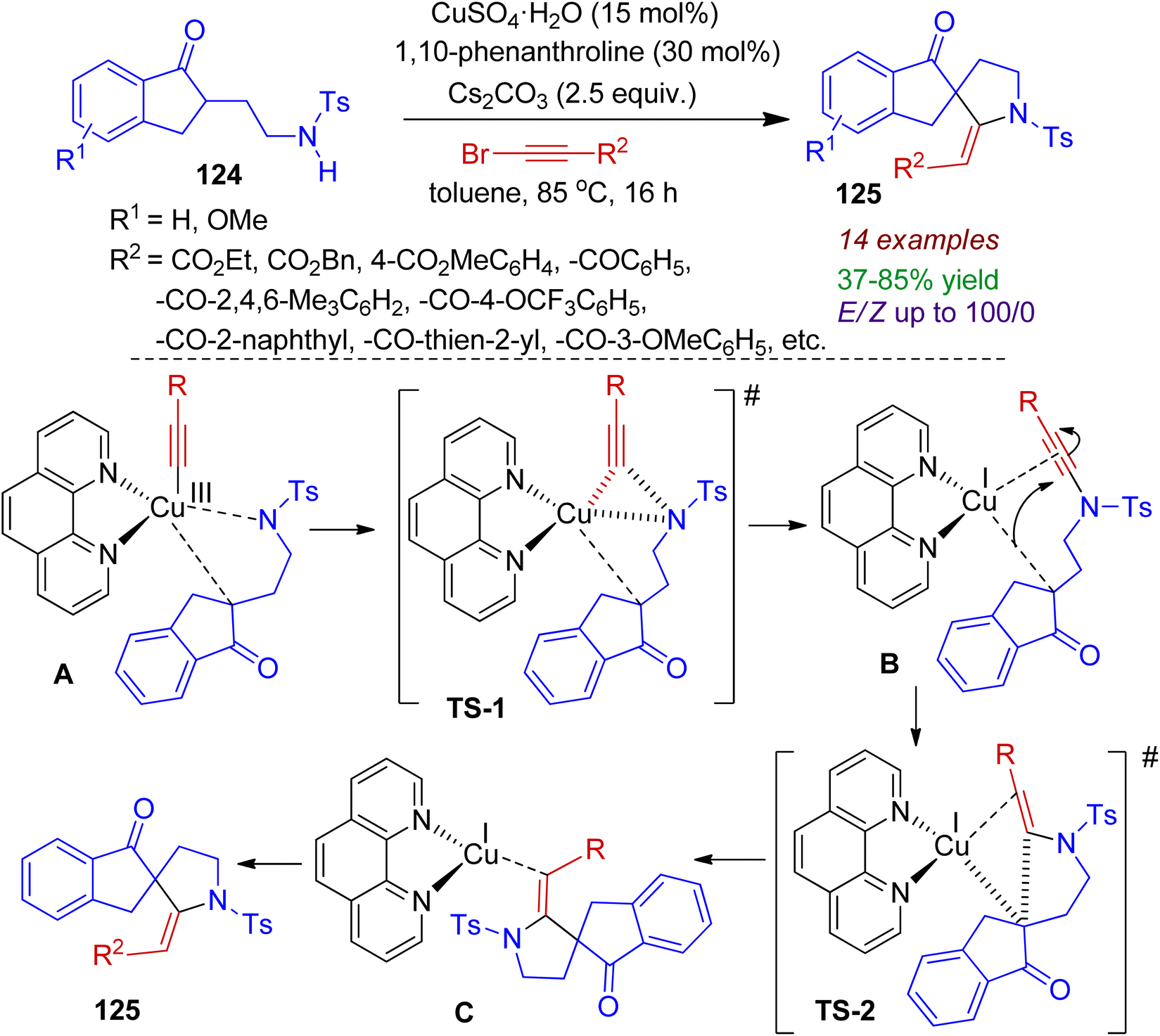
In 2017, spirocyclization of keto-sulfonamides was reported by Miesch and co-workers.67 The copper-catalyzed reaction of keto-sulfonamides 124 with alkynyl bromide could be proceeded in the presence of base Cs2CO3 and ligand 1,10-phenanthroline affording spiroindeno-pyrrolidine compounds 125 in acceptable yields (Scheme 33). The protocol was found to be general for electron-withdrawing groups as substituents affording desired products with E-selectivity (up to E/Z ratio 100/0). Mechanistically, it is conceivable that oxidative addition of in situ formed copper species to the alkynyl bromide generates a Cu(III) alkynylcopper species. The excess base might lead to double deprotonation of the keto sulphonamide which interacts with Cu(III) complex, producing intermediate A. Subsequent reductive elimination (C–N bond formation, intermediate B) and 1,4-addition of the resulting Cu(I) enolate to the alkynoate produces alkenylcopper(I) intermediate C (C–C bond formation). Finally, protonation provides azaspiro compounds 125. The synthesized azaspiro compounds can serve as building blocks for several indole alkaloids.
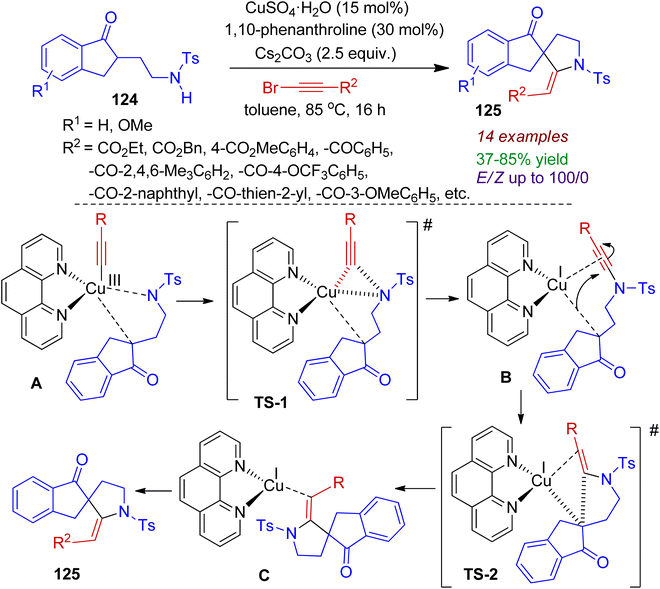 |
| | Scheme 33 Cu-catalyzed reaction of keto-sulfonamides with alkynyl bromide to result azaspiroindanone compounds. | |
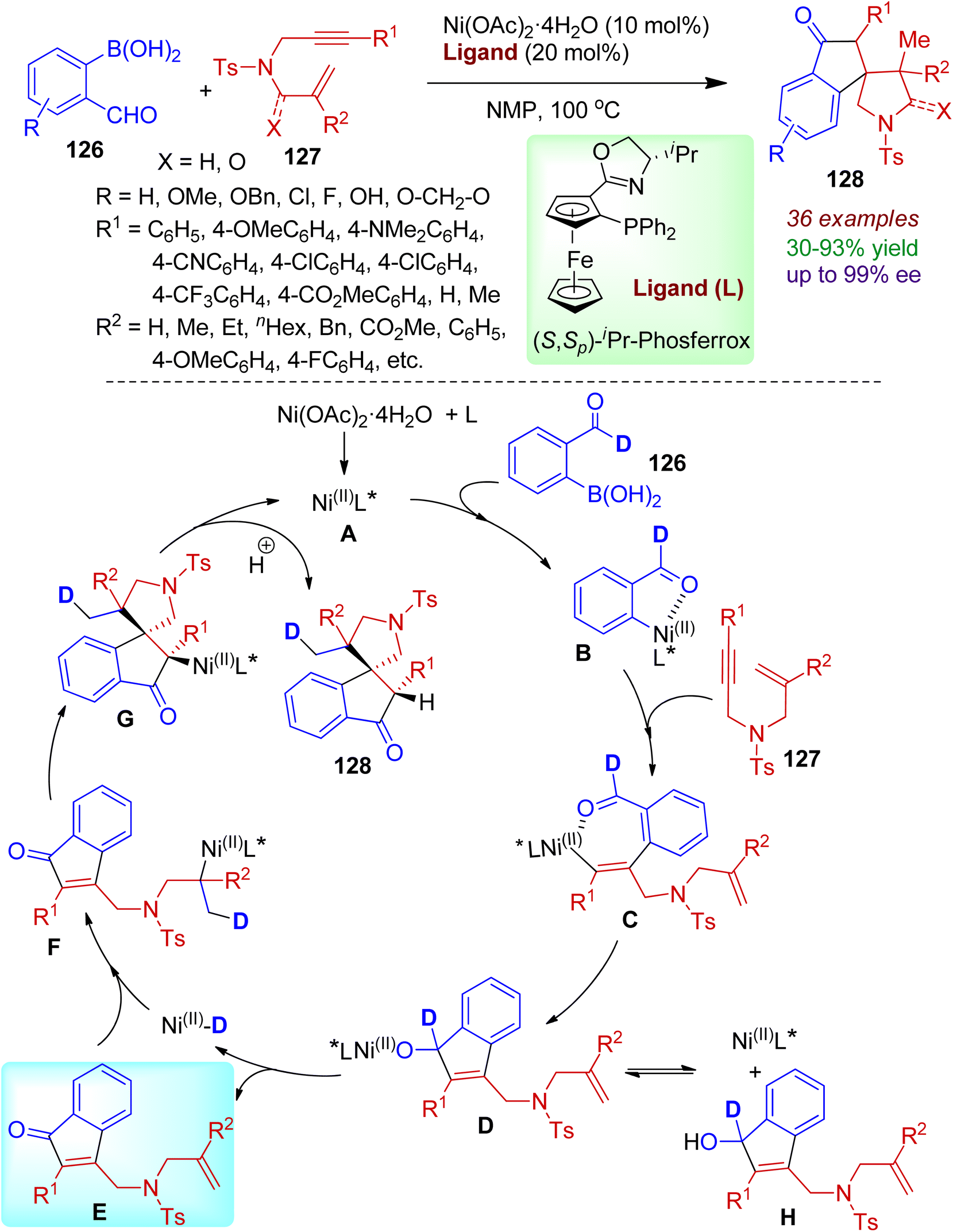
A nickel-catalyzed redox-neutral protocol for one-pot synthesis of spiroindanones from readily accessible o-formylarylboronic acids 126 and 1,6-enynes 127 was reported by Kong's group.68 The reaction comprised in situ formation of indanone species furnishing a library of enantioenriched spiroindanones 128 in good yields with high enantio- and diastereoselectivity (up to 99% ee and >20:1 dr). Phos-type ligands such as, (S,Sp)-iPr-Phosferrox was found to be most effective, and aryl boronic acids containing electron-donating groups (methoxy or benzyloxy) and electron-withdrawing groups (Cl, F) were amenable in this process. Even heteroaromatic boronic acids responded the reaction satisfactorily. However, the reaction failed for substrates containing vinyl moieties instead of aldehydes. The practicality of the Ni-catalyzed asymmetric process was certified by gram-scale synthesis of the desired products. A plausible mechanism of the catalytic conversion is outlined in Scheme 34. Initially, transmetallation of o-formylarylboronic acids 126 with chiral nickel complex A delivered aryl nickel complex B, which possibly be stabilized by coordination between the aldehyde group and the nickel center. In the next step, the seven-membered nickel species C was generated via migratory insertion of the alkyne into the aryl–nickel bond. Intramolecular nucleophilic addition led to alkoxynickel intermediate D. During the process, formation of an appreciable quantity of indenol intermediate H was observed. Subsequent β-H elimination resulted enone E and key Ni(II)–D species. Regioselective 1,2-addition of Ni(II)–D species to the unactivated alkene (intermediate F), followed by spirocyclization gave C(sp3)–nickel intermediate G. Hydrolysis furnished spiroindanone product 128 with regeneration of the nickel catalyst.
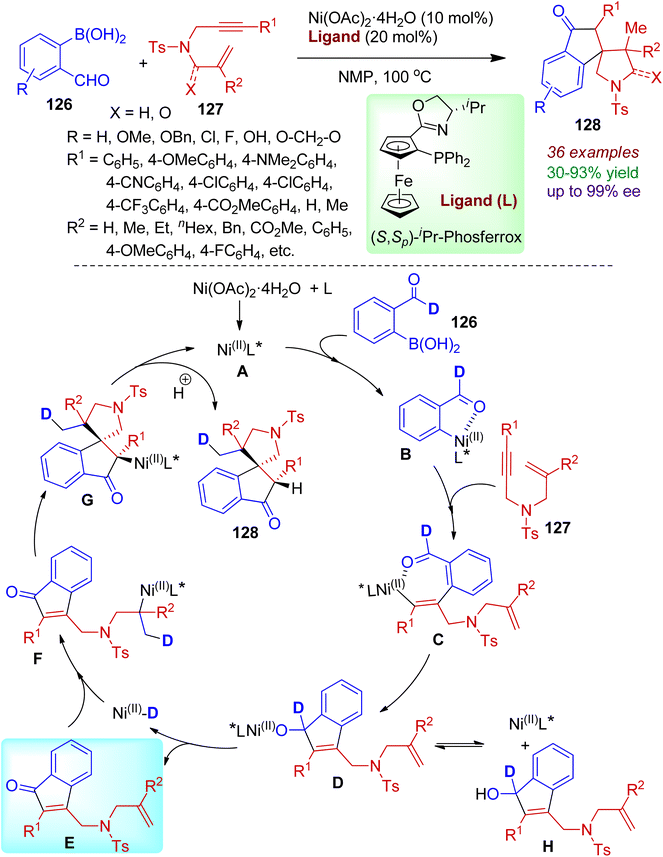 |
| | Scheme 34 Nickel-catalyzed synthesis of spiroindanones from o-formylarylboronic acids and 1,6-enynes. | |
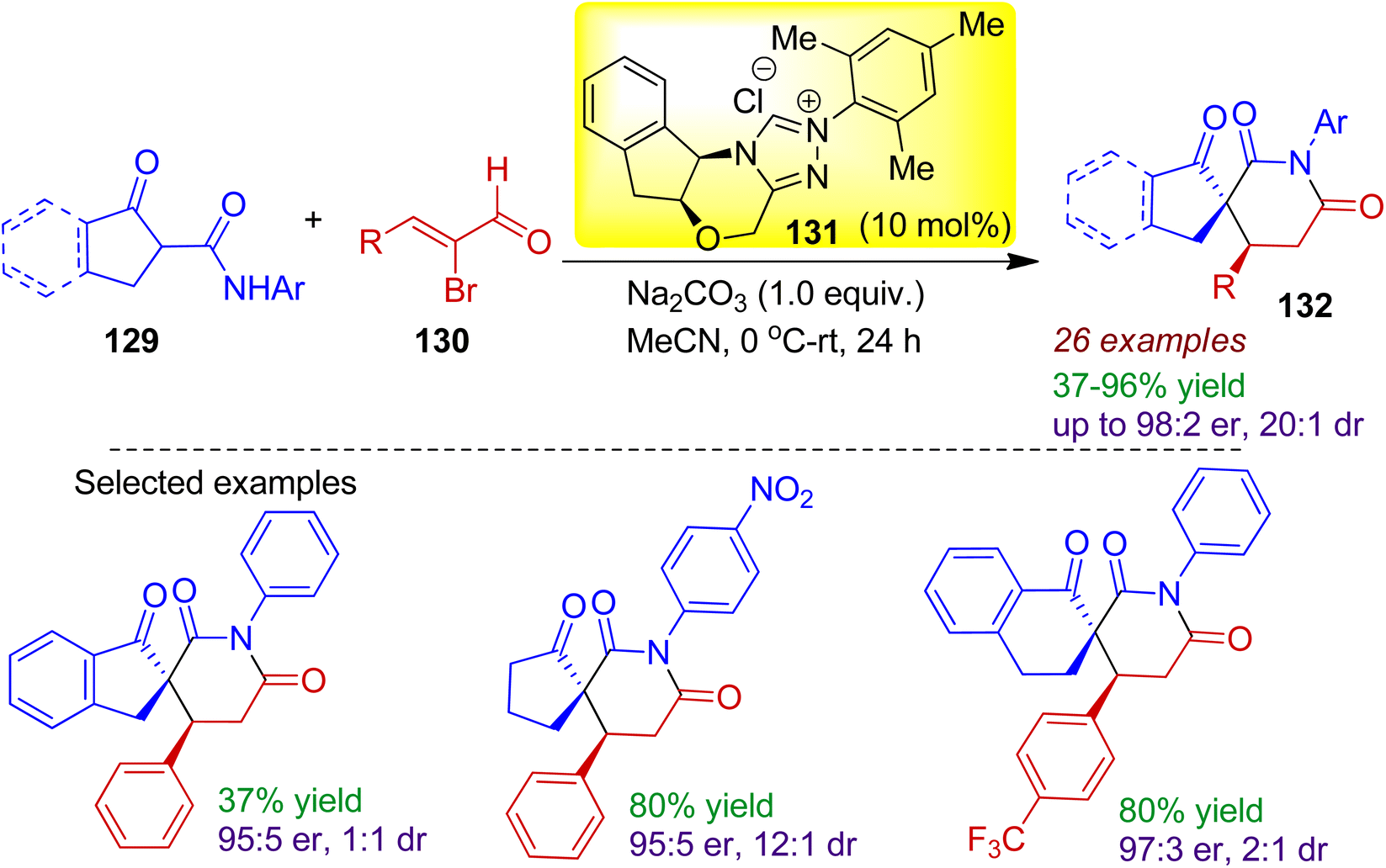
In 2018, Biju's group devised an organocatalytic stereoselective [3+3] spiro-annulation strategy to obtain spiro-glutarimide derivatives 132 containing two contiguous stereocenters including one all-carbon quaternary spirocenter.69 The reaction of cyclic β-ketoamides 129 with enal compounds 130 catalyzed by 10 mol% N-heterocyclic carbene catalyst 131 in acetonitrile solvent efficiently produced spiro-glutarimides 132 (Scheme 35). In addition to indanone derived β-ketoamides, various cyclopentanone and α-tetralone derived β-ketoamides underwent smooth annulation resulting desired compounds with acceptable enantio- and diastereoselectivity. The assembly of the ketoamides with catalytically generated chiral α,β-unsaturated acylazoliums proceeded in a Michael addition-proton transfer-cyclization sequence.
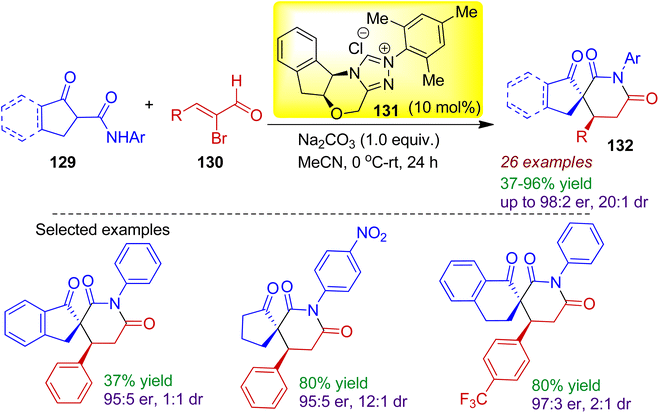 |
| | Scheme 35 Organocatalytic stereoselective synthesis of spiro-glutarimides from cyclic β-ketoamides and enal compounds. | |
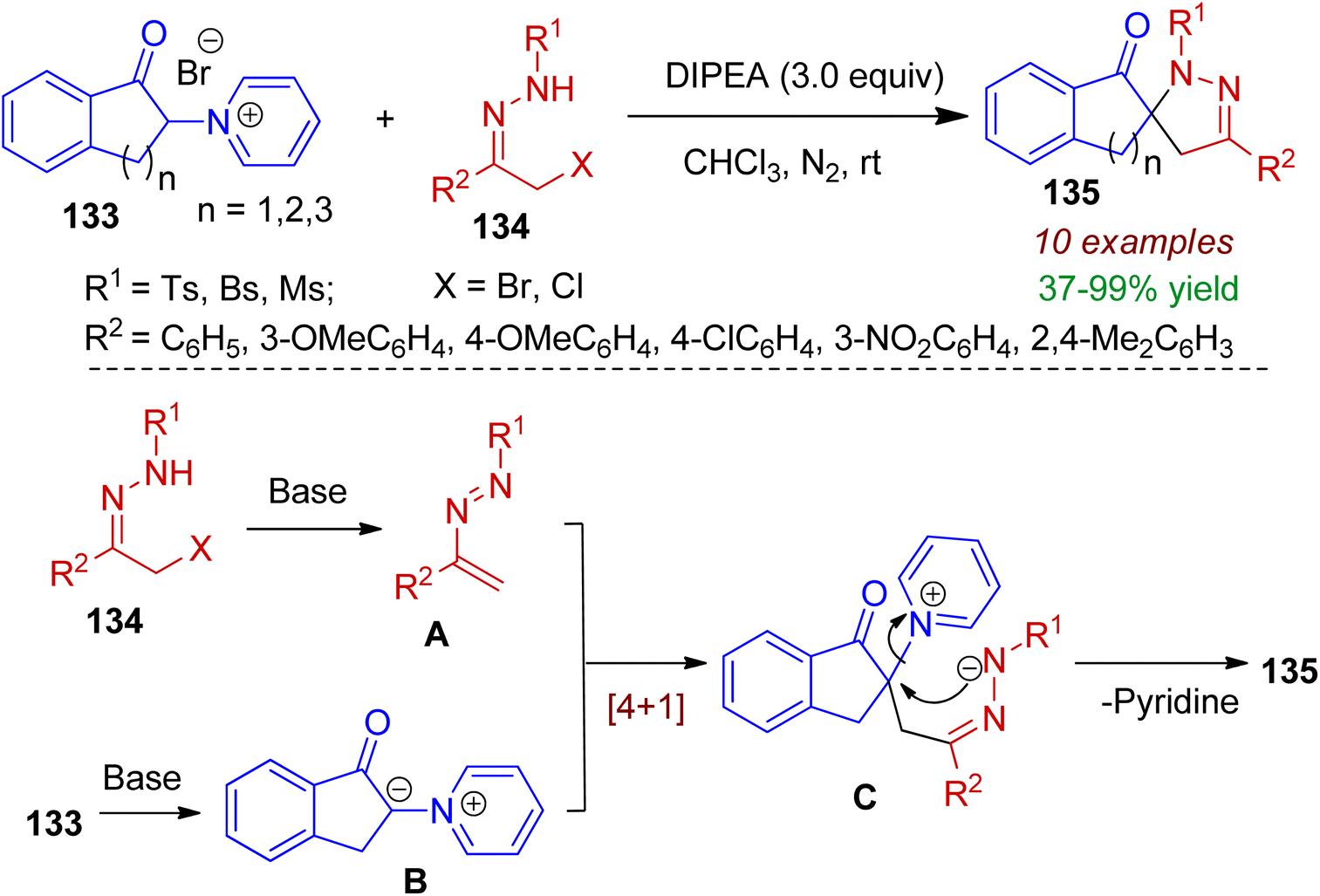
Cyclic pyridinium ylides may also be used as new synthon for spiro-heterocyclic compounds. In 2020, Yuan's group designed indanone-3-pyridinium salts 133 and allowed them to react with α-halo hydrazones 134 for the construction of spiropyrazoline indanones 135 (yield up to 99%) via [4+1] annulation strategy.70 DIPEA was found to be most effective base and chloroform as solvent. Notably, indanone-3-pyridinium ylides acted as C1 synthons in this reaction. Analysis of the X-ray crystallographic data of a single crystal unambiguously confirmed its structure. The probable mechanism is depicted in Scheme 36. The α-halo hydrazone 134 upon base-catalyzed elimination forms azoalkene A which is the key intermediate. Meanwhile, the deprotonation of indanone-3-pyridinium salts furnishes ylide B. Afterwards, conjugate addition of ylide B to intermediate A generates intermediate C. Finally, intramolecular cyclization leads to the formation of spiro-annulated product 135 with elimination of pyridine to complete the [4+1] annulation process.
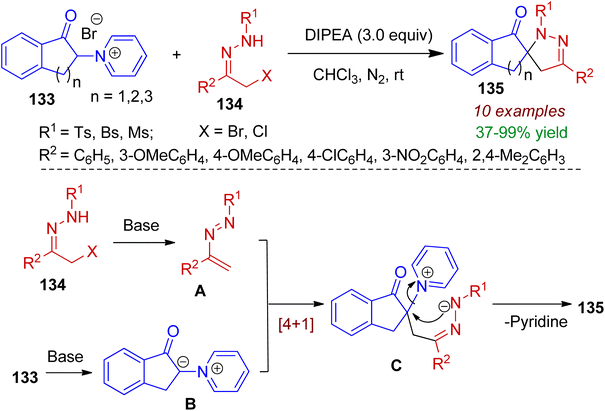 |
| | Scheme 36 Construction of spiropyrazoline indanones via the reaction of indanone-3-pyridinium ylides and α-halo hydrazones. | |
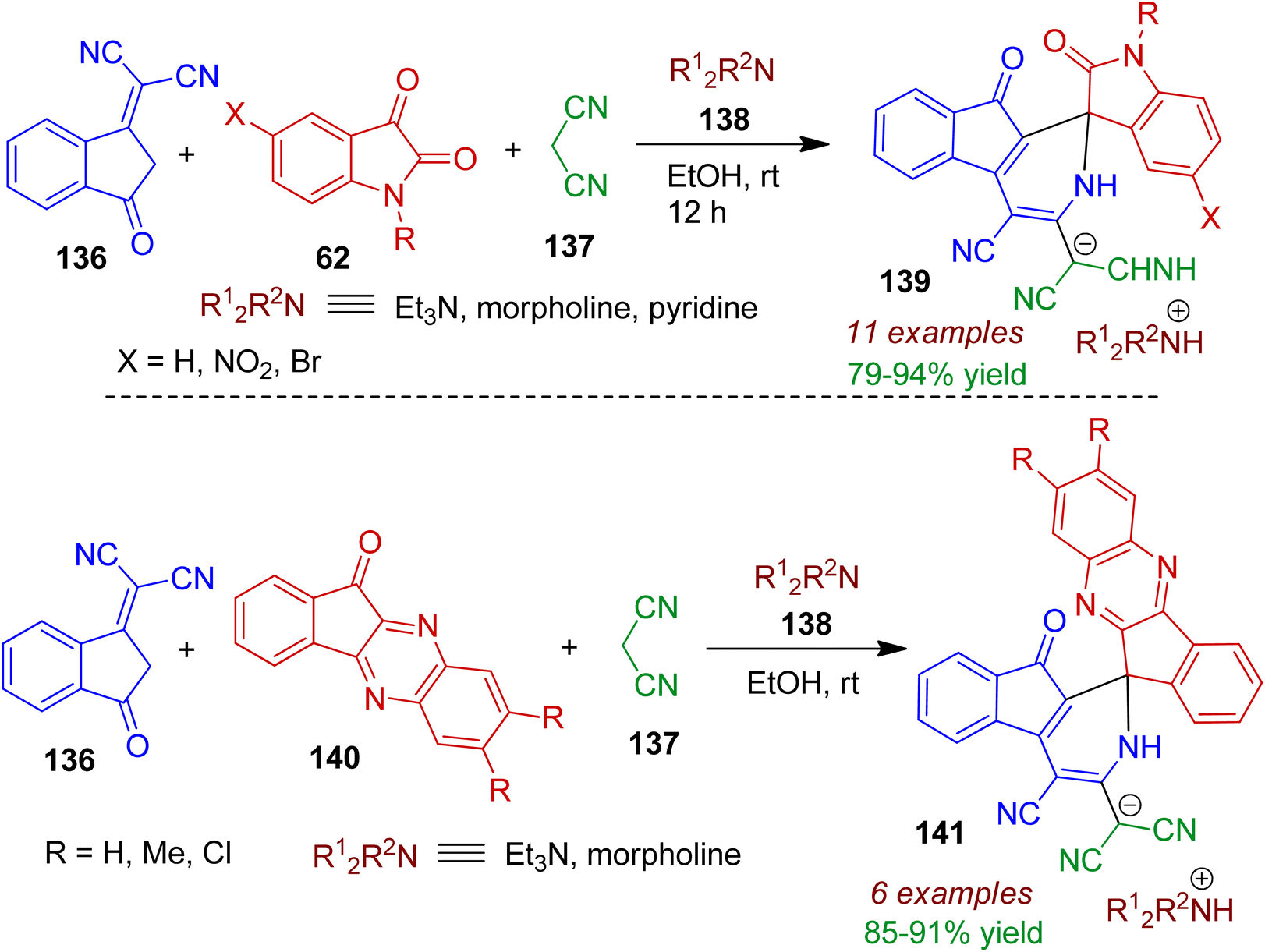
A facile metal-free, green approach towards indenopyridine-spirocyclic systems via multicomponent reaction (MCR) was reported by Shakibaei and Bazgir.71 The four-component reaction of 1,1-dicyanomethylene-3-indanone 136, isatins 62 and malononitrile 137 and amines (morpholine, triethylamine or pyridine) 138 in ethanol regioselectively delivered spiroindenopyridine–oxindole framework 139 at ambient temperature (Scheme 37). The reaction was believed to proceed via tandem knoevenagel/Michael/elimination/[5+1] annulation sequence. The pure products could be isolated simply by evaporation of solvent and washing by cold diethyl ether. As expected, the assembly of indanone 136, malononitrile 137, indenoquinoxalines 140 in the presence of amines 138 under same reaction condition led to the formation of spiroindenopyridine-indenoquinoxaline derivatives 141 in good yields (up to 91%).
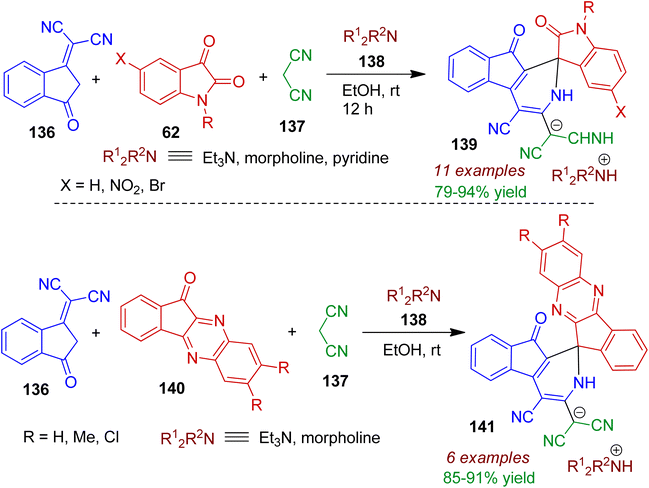 |
| | Scheme 37 Multicomponent synthesis of spiroindenopyridine-oxindoles and spiroindenopyridine-indenoquinoxalines. | |
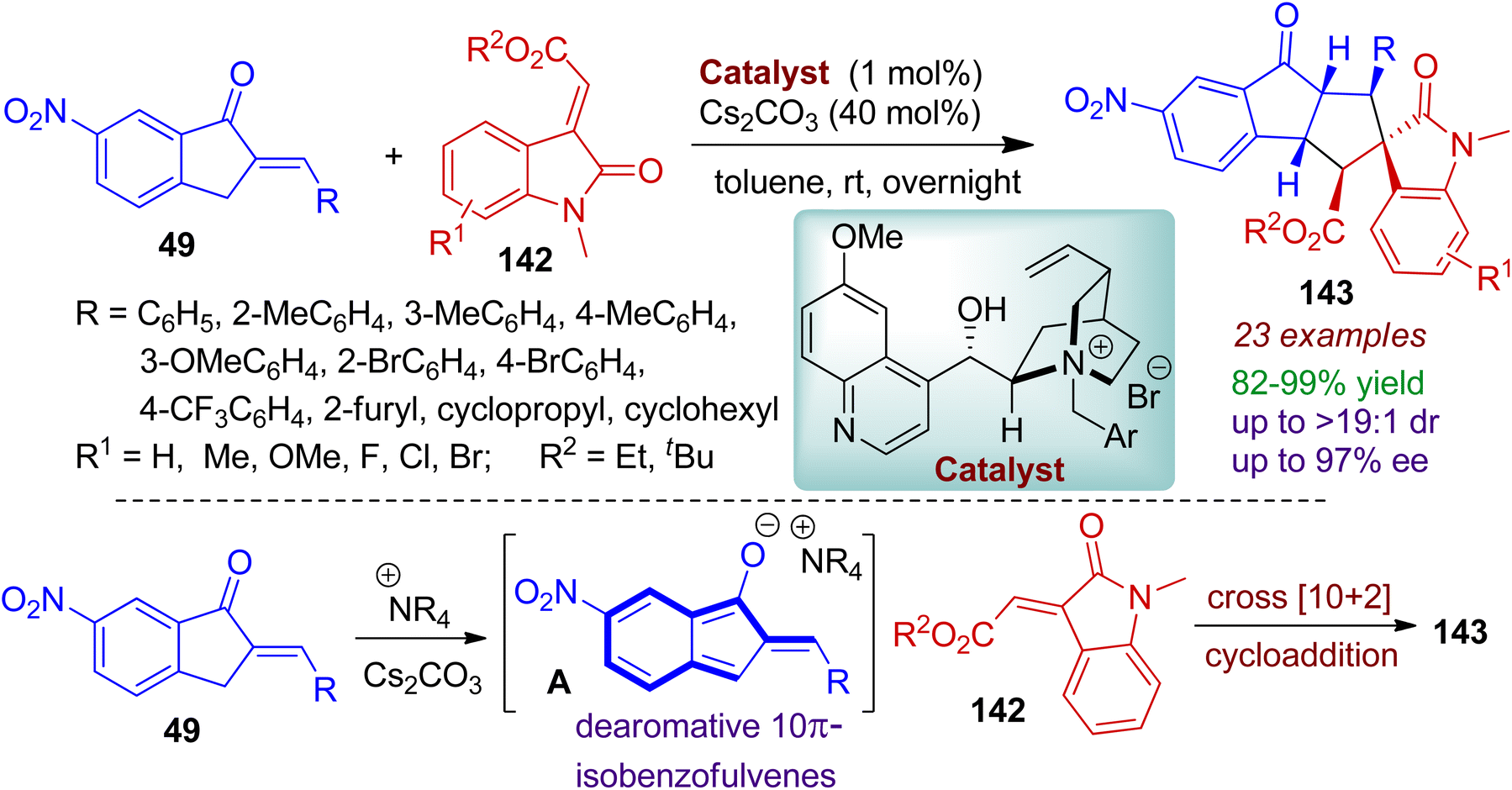
2-Arylidene-1-indanones compounds could be converted to dearomative 1-hydroxyl isobenzofulvene-type species for chemoselective cross formal [10+2] cycloaddition reaction with electron-deficient alkenes 142.72 Excellent diastereo- and enantioselectivity were maintained by employing a novel bulky cinchona-based ammonium salt as catalyst (1 mol%) to afford richly decorated spiro-cyclopentaindeno-indolines 143 (Scheme 38). Significantly, the 2-arylidene-1-indanones 49 possessing aryl groups, cyclopropyl, cyclohexyl, furyl moieties were well tolerated by this process. Considering the acceptor system 142, ethyl as well as sterically hindered tert butyl ester group offered good yields and selectivity. Mechanistically, it is conceivable that under the basic conditions, dearomative 1-hydroxyl isobenzofulvene anion intermediate A was generated from 2-arylidene-1-indanones 49 in the presence of cinchona-derived catalyst. The in situ formed intermediate A, effectively acted as 10π electron system and underwent cross formal [10+2] cycloaddition reaction with electron-deficient alkenes 142 thereby forming the spiro- compound 143 with stereoselectivity.
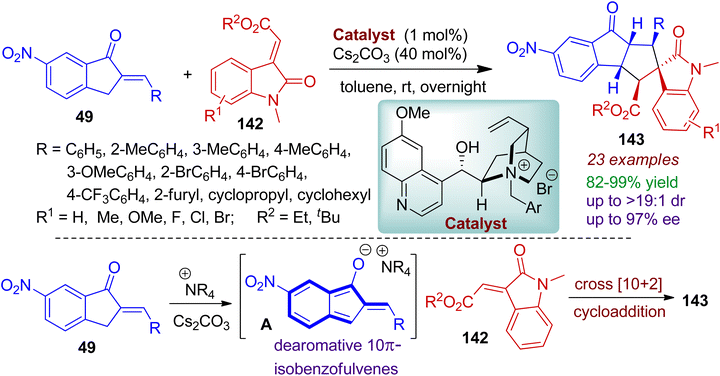 |
| | Scheme 38 Cross formal [10+2] cycloaddition of 1-hydroxyl isobenzofulvene species with electron-deficient alkenes to form spiro-cyclopentaindeno-indolines. | |
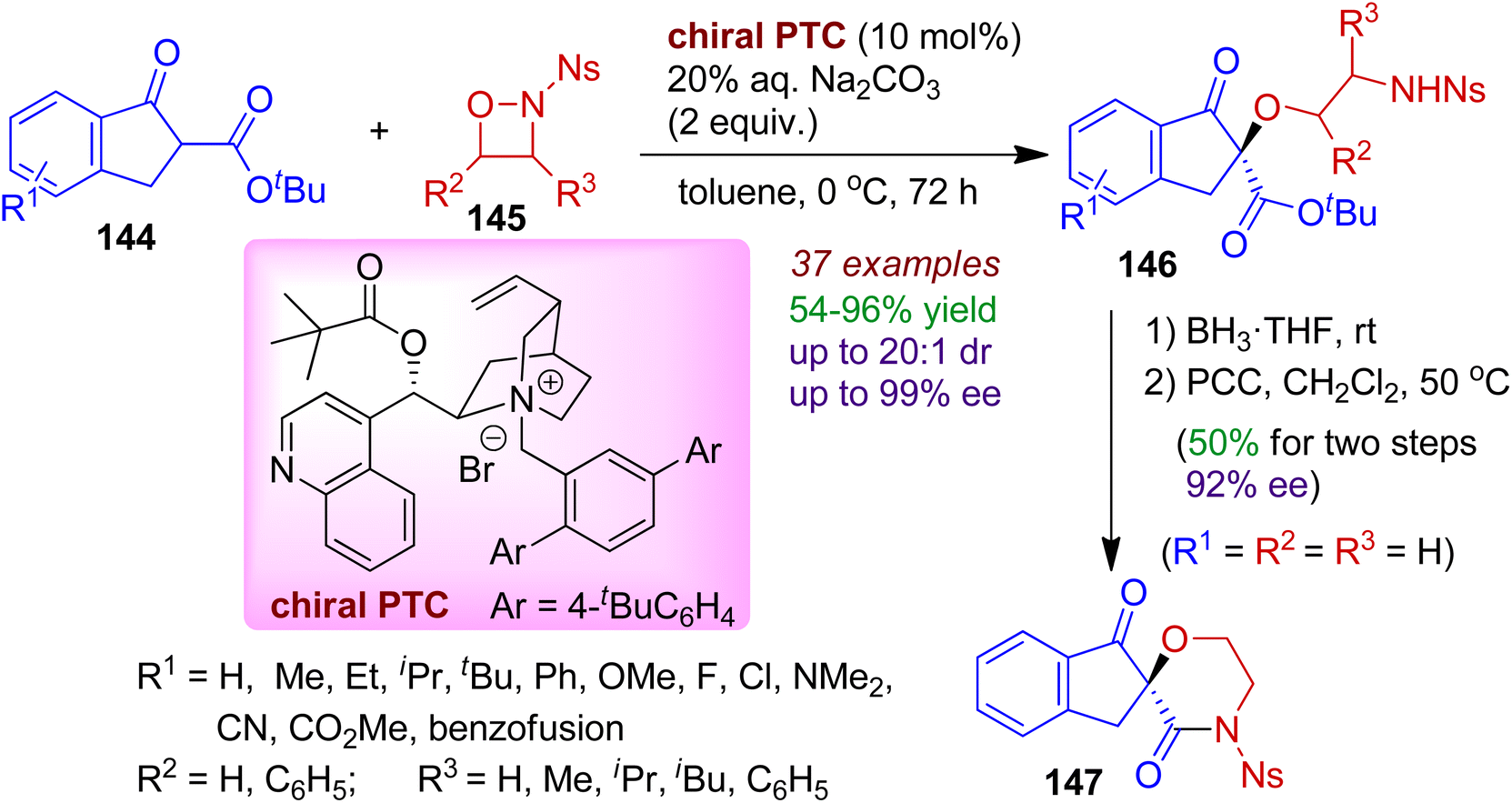
A highly enantio- and diastereoselective ring opening reaction of 1,2-oxazetidines 145 with indanone carboxylates 144 in the presence of a chiral phase transfer catalyst (PTC) was reported by Hu's group.73 Exploiting the unique electrophilic oxygen reactivity of highly functionalized N-nosyl 1,2-oxazetidines, a library of N,O-containing chiral ether 146 bearing a quaternary carbon center was obtained in good yields and stereoselectivities (up to 97% ee and 20:1 dr) under mild conditions. This is an interesting example of catalytic asymmetric umpolung reaction involving indanone system. Significantly, the products could be effectively converted into biologically important spiroindeno-morpholine derivatives 147 via two steps (Scheme 39).
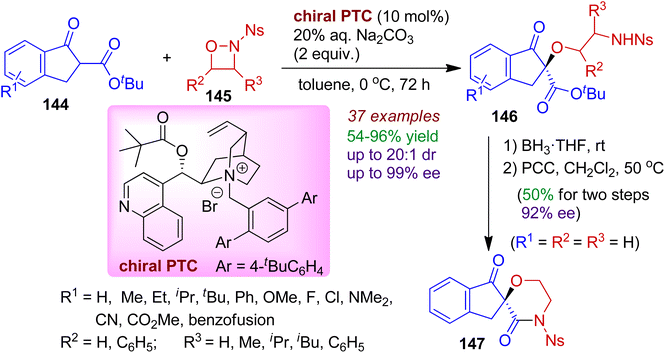 |
| | Scheme 39 Asymmetric synthesis of spiroindeno-morpholines via the reaction of 1,2-oxazetidines and indanone carboxylates. | |
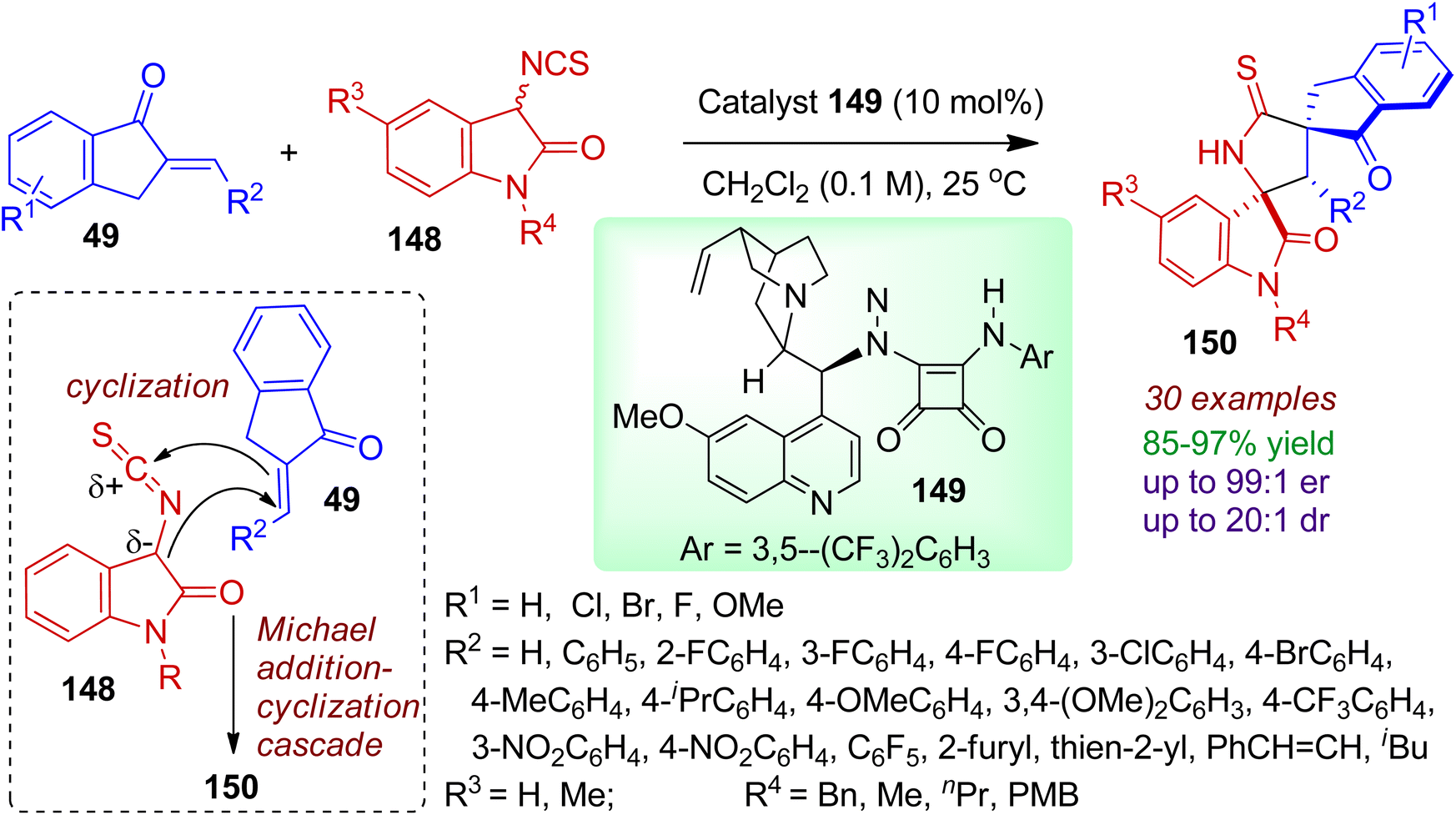
3.2.2. N-Containing bispiro-heterocycles. Synthesis of bispiro skeleton with stereochemical diversity is a challenging task in organic chemistry. Kayal and Mukherjee synthesized enantioenriched 3,2′-pyrrolidinyl bispirooxindole derivatives 150 from readily available 3-isothiocyanato oxindoles 148 and 2-arylidene-indanones 49 catalyzed by a quinine-derived tertiary amino-squaramide 149 (Scheme 40).74 The bispiro-oxindole indanone compounds 150 were produced via Michael addition/cyclization cascade in excellent yields with high enantioselectivity and diastereoselectivity (up to 99:1 er and 20:1 dr). Various arylidene-indanones with diverse steric and electronic character were compatible under ambient conditions. Notably, heteroarylidene-indanones were also well tolerated to form desired products in good selectivity. The protocol was equally efficient for different 3-isothiocyanato oxindoles containing varied N-substituents (Bn, Me, nPr, PMB). The absolute configuration was determined by single crystal XRD analysis.
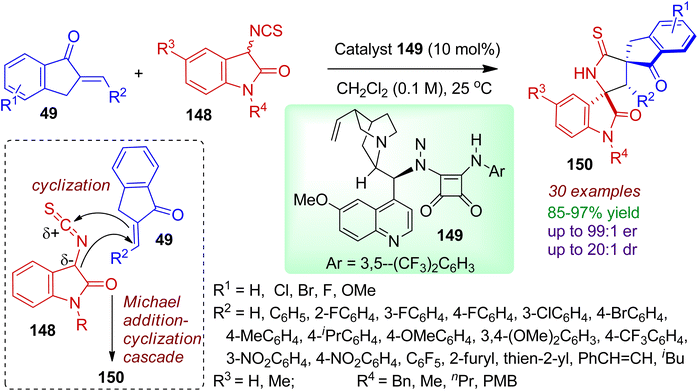 |
| | Scheme 40 Catalytic enantioselective cyclization of 3-isothiocyanato oxindoles and 2-arylidene-indanones towards 3,2′-pyrrolidinyl bispirooxindoles. | |
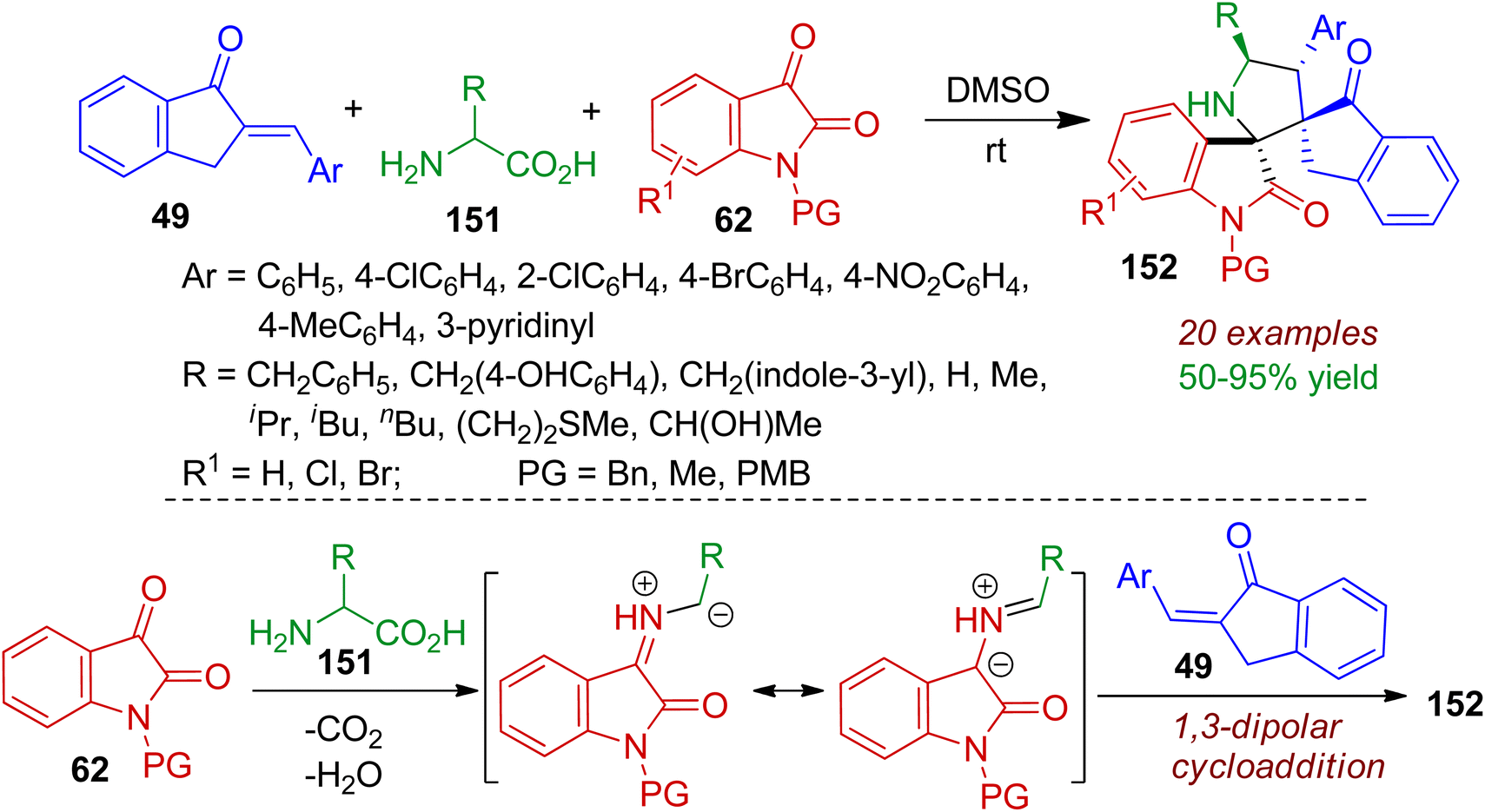
Construction of pyrrolidine–bispirooxindole frameworks bearing sterically congested two vicinal spiro-centers was realized via a three-component reaction between 2-arylidene-indanones 49, isatins 62, primary amino acids 151.75 The 1,3-dipolar cycloaddition reaction accomplished pyrrolidine–bispirooxindoles 152 in highly regio- and diastereoselective manner. In most of the cases, single diastereoisomer were obtained in good yields (up to 95%) at room temperature in DMSO solvent. Significantly, primary amino acids 151 acted as amine component (via decarboxylation) for the in situ generation of azomethine ylides from isatin (Scheme 41). The azomethine ylides underwent 1,3-dipolar cycloaddition reaction with 2-arylidene-indanones 49 offering bispiro skeleton 152. Relative configurations of the products were determined by single crystal X-ray diffraction analysis. The practicality of the reaction was certified by gram-scale synthesis. Molecular docking studies revealed that the some products can act as inhibitors of the epidermal growth factor receptor (EGFR).
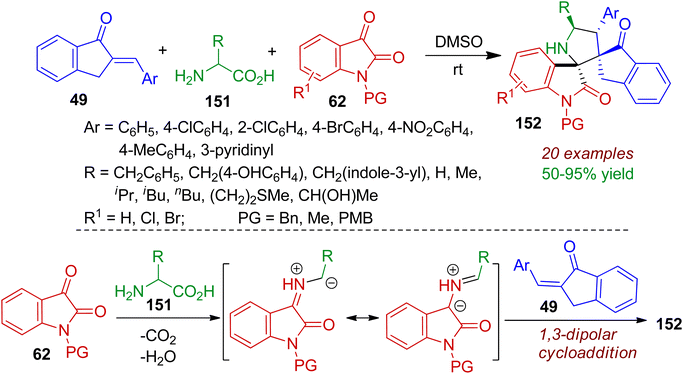 |
| | Scheme 41 Formation of bispiro compounds via three-component reaction between 2-arylidene-indanones, isatins and primary amino acids. | |
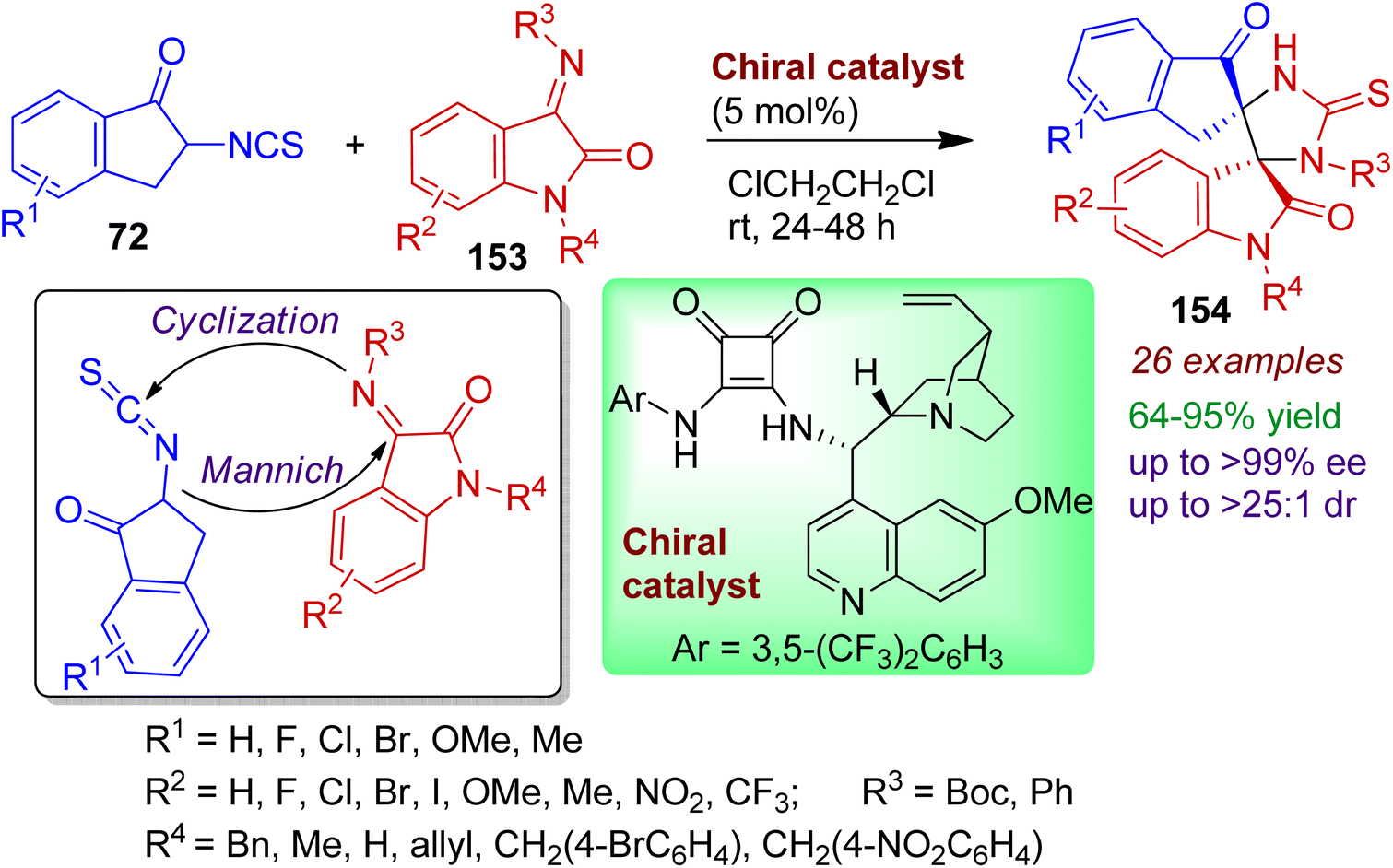
An efficient protocol for the stereoselective construction of bispiro indanon-thioimidazolidine-oxindoles bearing two adjacent spiro-quaternary stereocenters has been reported by Zhao and Du.76 The cinchona alkaloid derived squaramide-based chiral organocatalyst promotes reaction between 2-isothiocyanato-1-indanones 72 and isatinimines 153 to deliver desired thioimidazolidine-oxindole bispiro system 154 with excellent enantio- and diatereoselectivity (up to >99% ee, >25:1 dr). The reaction takes place via initial Mannich reaction followed by cyclization sequence (Scheme 42). Indanones and isatnimines containing electron-withdrawing (F, Cl, Br) and electron-donating substituents (Me, OMe) responds well under standard conditions. The synthesized bispiro products can easily be transformed into various bioactive compounds.
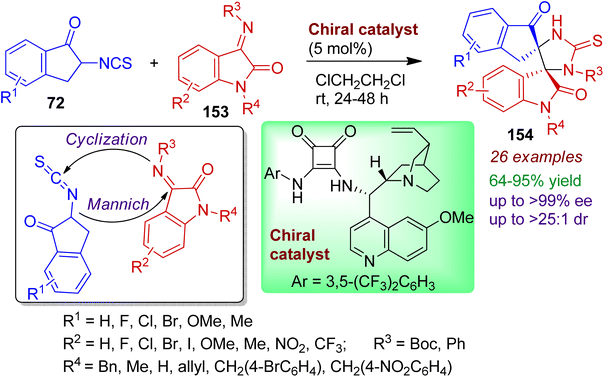 |
| | Scheme 42 Access to bispiro indanone-thioimidazolidine-oxindoles from 2-isothiocyanato-1-indanones and isatinimines. | |
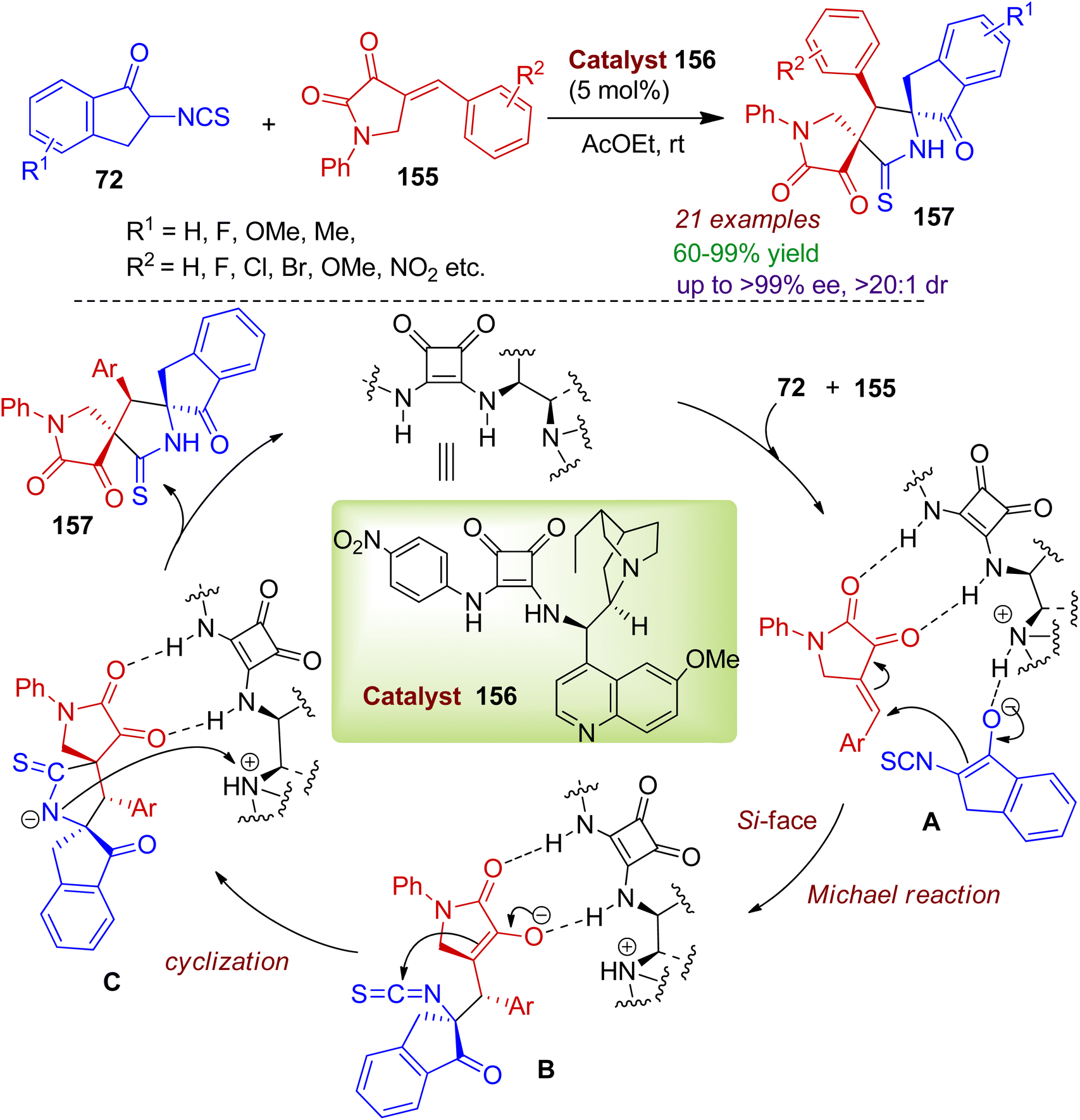
The authors also synthesized spiropyrrolidone indanone scaffolds using squaramide catalyst 156 using readily available 4-arylmethylidene-2,3-dioxopyrrolidines 155 and 2-isothiocyanato-1-indanones 72.77 This method provides mild access to bispirocyclic compounds 157 containing three contiguous stereocenters in excellent yields (up to 99%) and enantio-/diatereoselectivity (up to 99% ee, >20:1 dr). A plausible mechanism for the squaramide-triggered cycloaddition reaction is outlined in Scheme 43. Initially, the isothiocyanato-1-indanone 72 is enolized via quinidine amine in the Michael addition process. Meanwhile, dioxopyrrolidine 155 is activated by the catalyst through hydrogen bonding. The enol ion attacks the double bond of 155 from the Si-face via intermediate A. Subsequently, the newly formed anion on the α-carbon center of 155 attacks the carbon atom of the isothiocyanato group in the intramolecular cyclization fashion involving intermediate B. The desired bispiro skeleton 157 is constructed through protonation of intermediate C along with the regeneration of the catalyst for the next cycle.
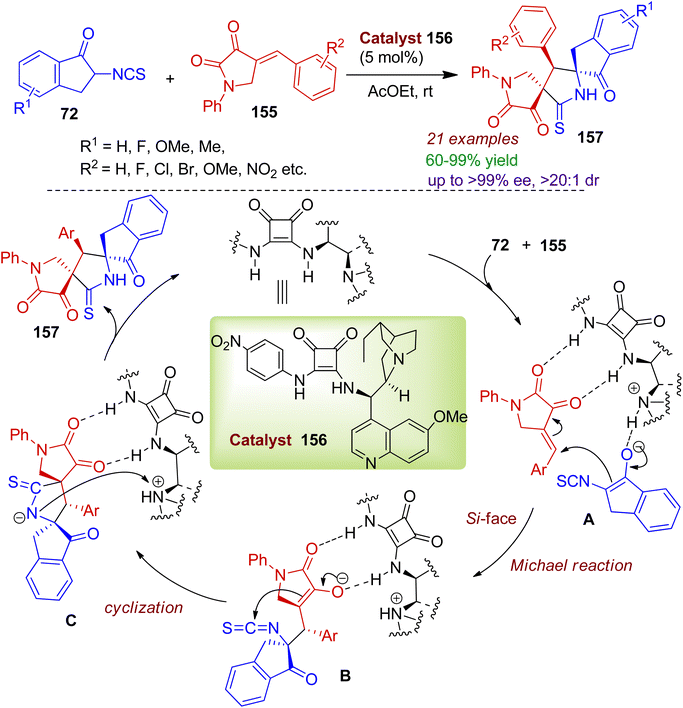 |
| | Scheme 43 Synthesis of bispiropyrrolidone indanones from 2,3-dioxopyrrolidines and 2-isothiocyanato-1-indanones. | |
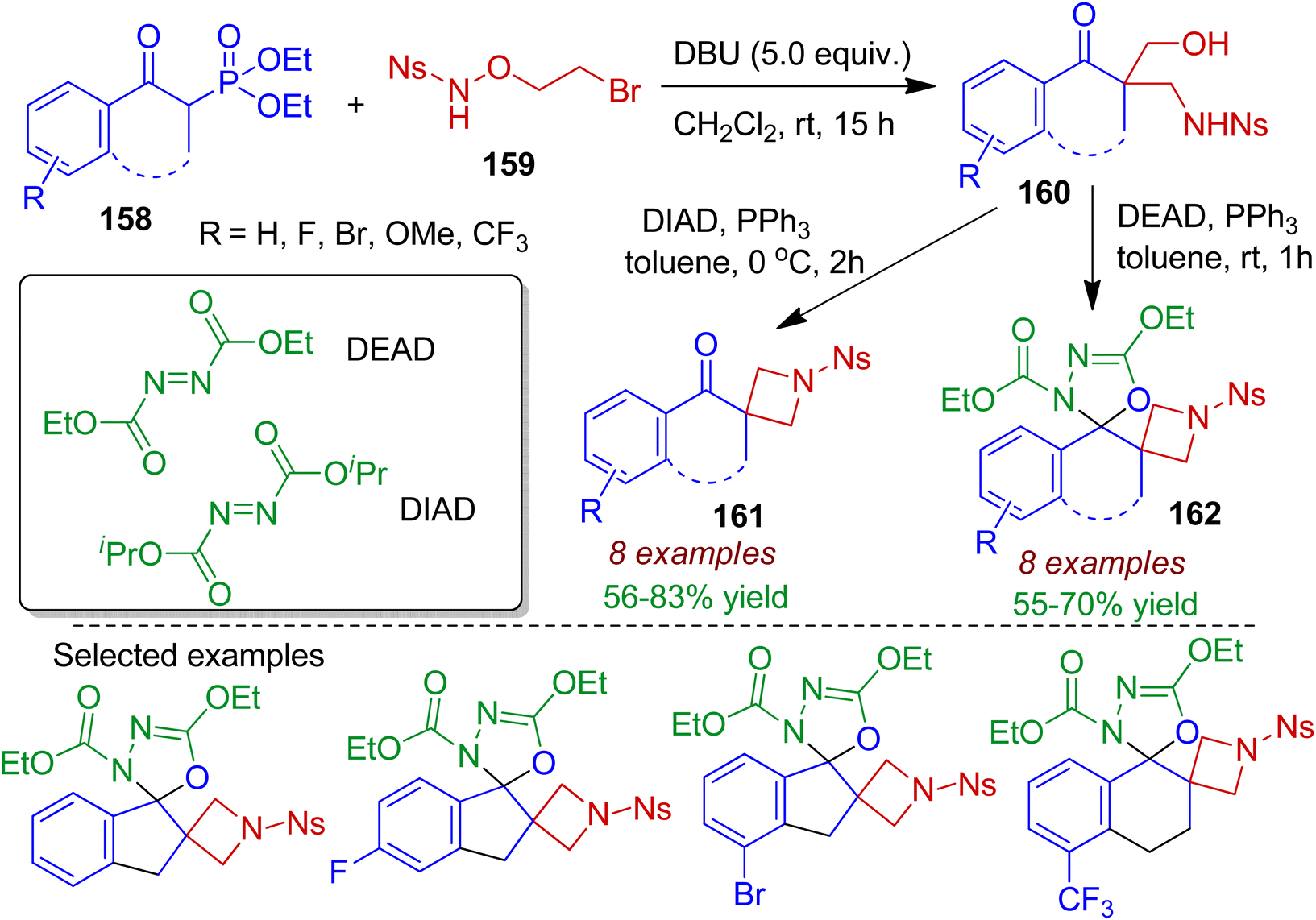
A tandem hydroxymethylation and aminomethylation reaction of cyclic β-keto phosphonates 158 with N-nosyl-O-(2-bromoethyl)hydroxylamine 159 in the presence of DBU was devised by Hu et al.78 The reaction led to the formation of 1,3-aminoalcohols 160 at room temperature through sequential Horner–Wadsworth–Emmons/Michael addition/aldol reactions. The resultant 1,3-aminoalcohols 160 could readily be transformed into biologically relevant spirocyclic azitidines 161 and azitidine-oxadiazoline bispirocycles 162 via Mitsunobu cyclization reaction (Scheme 44). It is worth mentioned that synthesis of bispirocyclic core bearing two vicinal azetidine and oxadiazoline rings otherwise difficult to achieve.
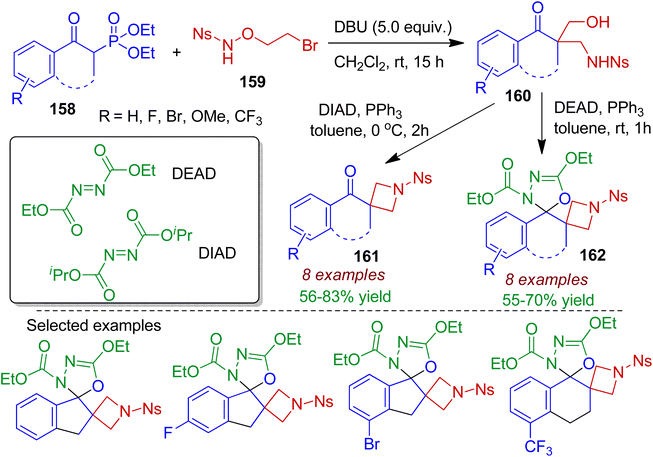 |
| | Scheme 44 Synthesis of spirocyclic azitidines starting from cyclic β-keto phosphonates and N-nosyl-O-(2-bromoethyl)hydroxylamine. | |
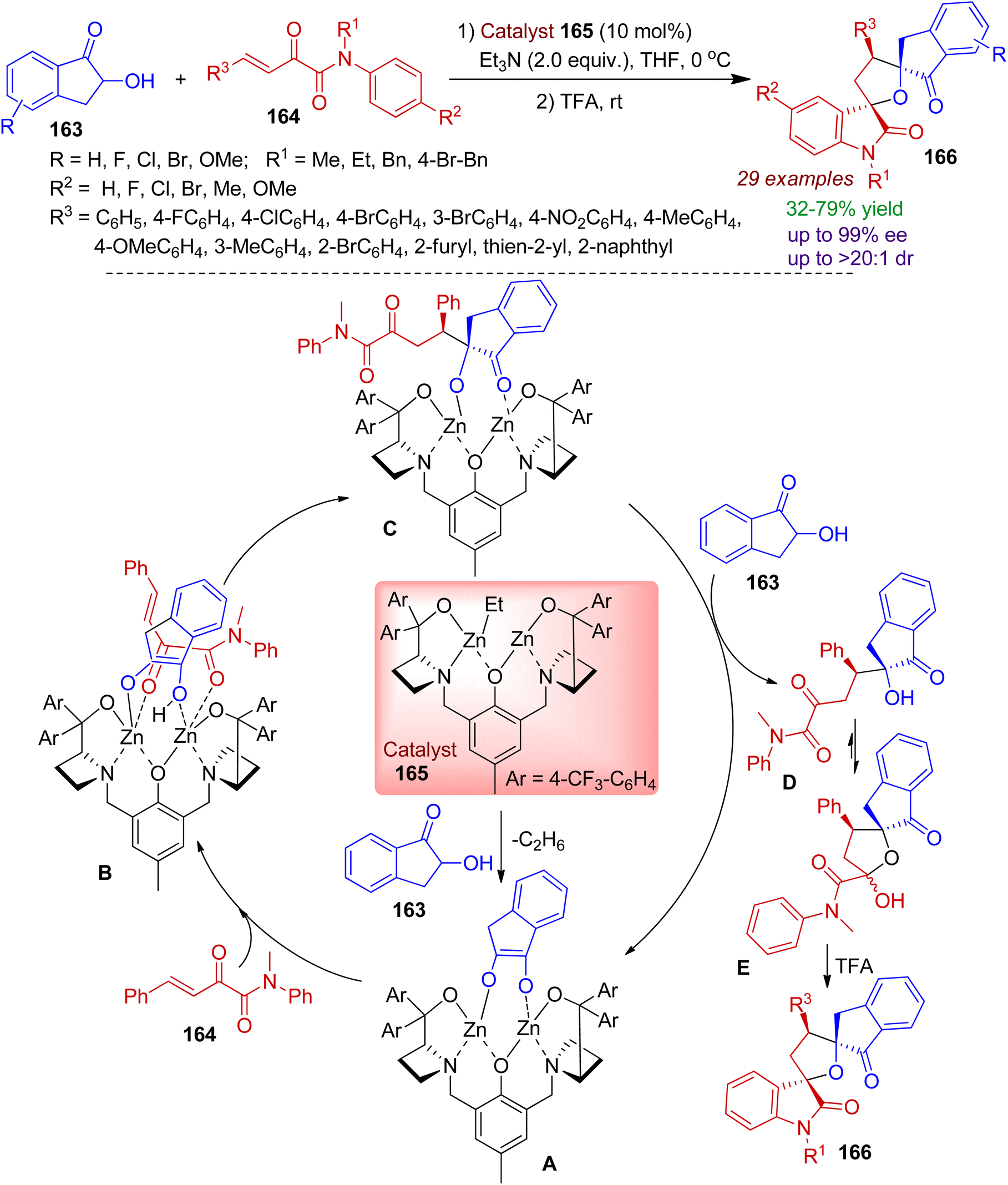
Recently, 2-hydroxy-1-indanone scaffold has been exploited by researchers to access diverse spiro heterocyclic frameworks. A facile synthesis of bispirotetrahydrofuran oxindoles 166 by cooperative bimetallic-catalyst is realized by Chang, Wang and co-workers via the reaction of 2-hydroxy-1-indanone 163 and β,γ-unsaturated α-ketoamide 164.79 The strategy involves cascade Michael/hemiketalization/Friedel–Crafts reaction sequence. The reaction is highly step-economic and can run on a gram scale with minimum catalyst loading. A plausible mechanism for the catalytic conversion is proposed in Scheme 45. Coordination and deprotonation of nucleophilic 2-hydroxy-1-indanone 163 with dinuclear zinc–ligand catalyst 165 gives intermediate A. Afterwards, electrophilic β,γ-unsaturated α-ketoamide 164 is activated by zinc–oxygen coordination to afford complex B from the sterically less hindered site. Subsequently, B undergoes Michael addition reaction to generate intermediate C. Next, proton transfer with another 2-hydroxy-1-indanone produces the Michael addition product D which is converted into its hemiketal form E. Finally, intramolecular Friedel–Crafts alkylation leads to bispiro indanone compound 166.
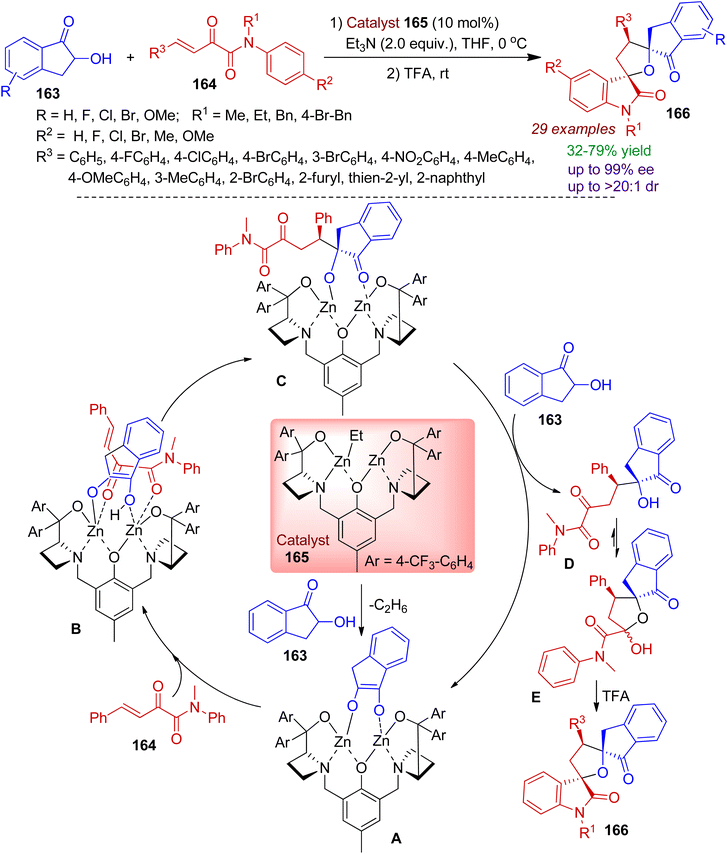 |
| | Scheme 45 Formation of chiral bispirotetrahydrofuran oxindoles via the reaction of 2-hydroxy-1-indanone and β,γ-unsaturated α-ketoamides. | |
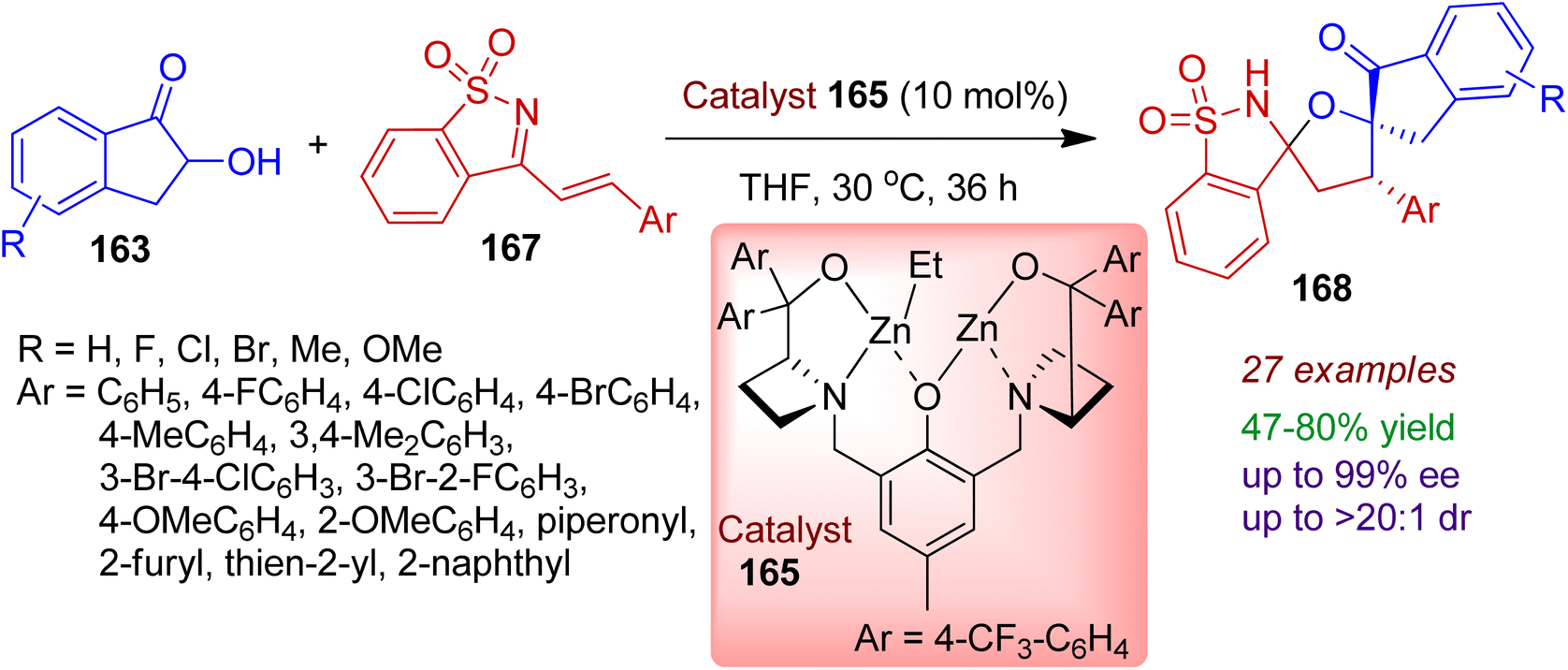
Very recently, an efficient synthesis of bispirocyclic saccharine system is realized involving asymmetric [3+2] spiroannulation reaction of saccharine-derived cyclic azadienes 167 with 2-hydroxy-1-indanones 163.80 The bimetallic cooperative catalytic conversion was attributed via Michael/O-Mannich cascade process resulting highly stereoselective formation of bispirocyclic compounds 168 in which indanone, tetrahydrofuran, and saccharine moieties are embedded (Scheme 46). 2-Hydroxy-1-indanones bearing electron-donating groups (Me, OMe) and electron-withdrawing groups (F, Cl, Br) underwent the [3+2] spiroannulation reaction with excellent enantioselectivity (up to 99% ee). A wide range of saccharine derivatives with varied aryl substitution including heterocycles (thienyl, furyl, piperonyl) was fruitful under the standard conditions. This strategy could be run on a gram scale without significant loss of stereoselectivities.
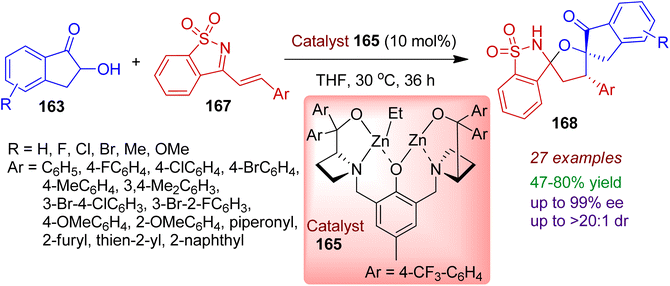 |
| | Scheme 46 Construction of bispirocyclic saccharine scaffolds via reaction of cyclic azadienes and 2-hydroxy-1-indanones. | |
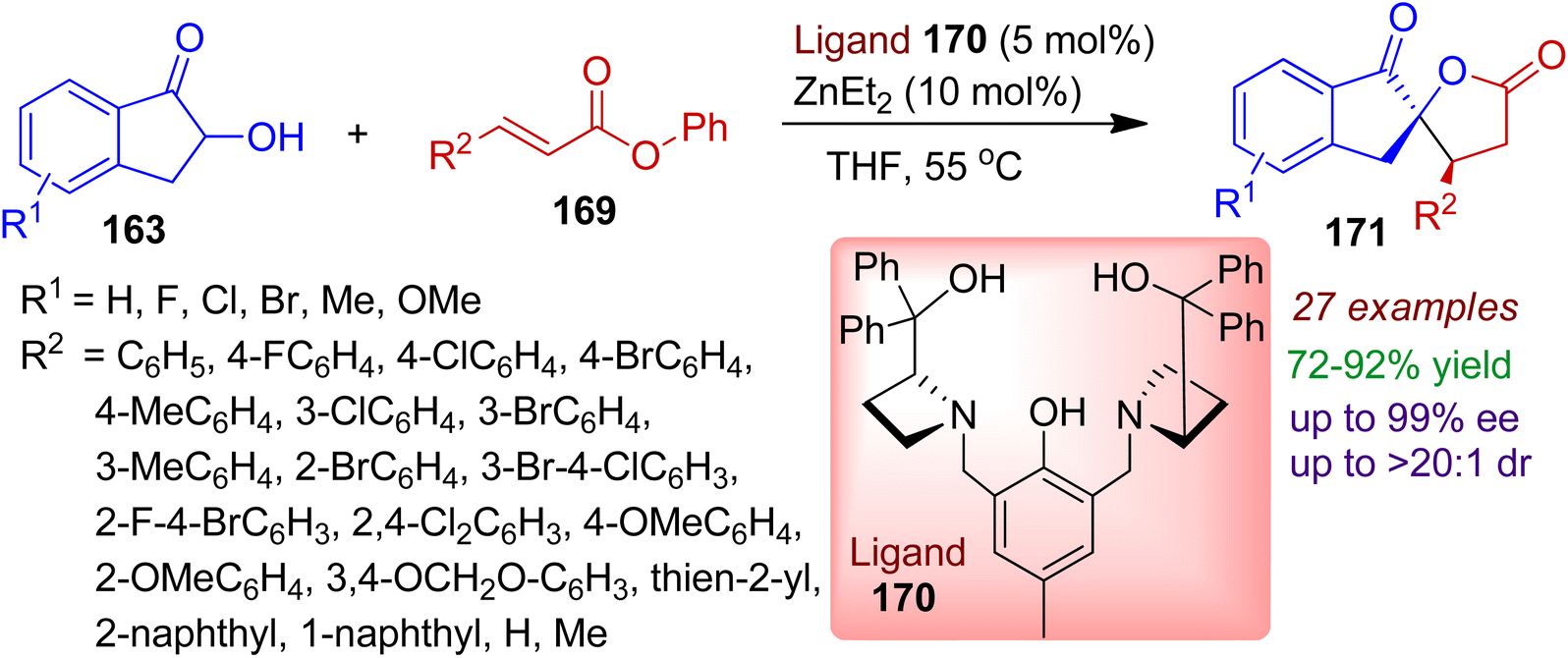
3.2.3. O-containing spiro-heterocycles. Stereoselective construction of spirolactones has attracted significant attention due to their great medicinal value as well as diverse synthetic applications. In 2019, Wang et al. developed a facile chiral dinuclear zinc-AzePhenol complex catalytic reaction employing 2-hydroxy-1-indanones 163 and α,β-unsaturated esters 169 (Scheme 47).81 The use of 5 mol% of ligand 170 and 10 mol% of Et2Zn was found to be effective for cyclization in THF medium. The one-step cascade Michael addition/transesterification reaction well accomplished wide range of spiro[indanone-5,2′-γ-butyrolactones] 171 with contiguous stereocenters in excellent yields and stereoselectivities (up to >99% ee, >20:1 dr). α,β-Unsaturated esters bearing aromatic (aryl, naphthyl, piperonyl, thienyl) as well as nonaromatic moieties smoothly delivered corresponding products. The electronic natures and positions of substituents had very little influence on the stereoselectivities. The absolute configuration of the synthesized products was established by X-crystal structure.
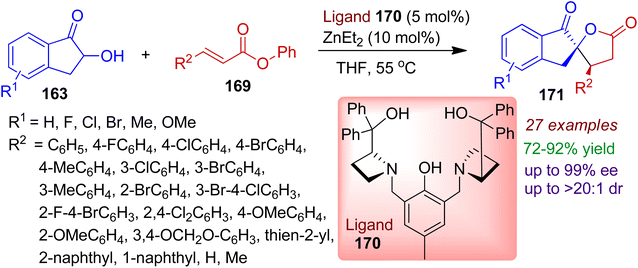 |
| | Scheme 47 Dinuclear zinc-AzePhenol complex catalyzed synthesis of spiro[indanone-5,2′-γ-butyrolactones] from 2-hydroxy-1-indanones and α,β-unsaturated esters. | |
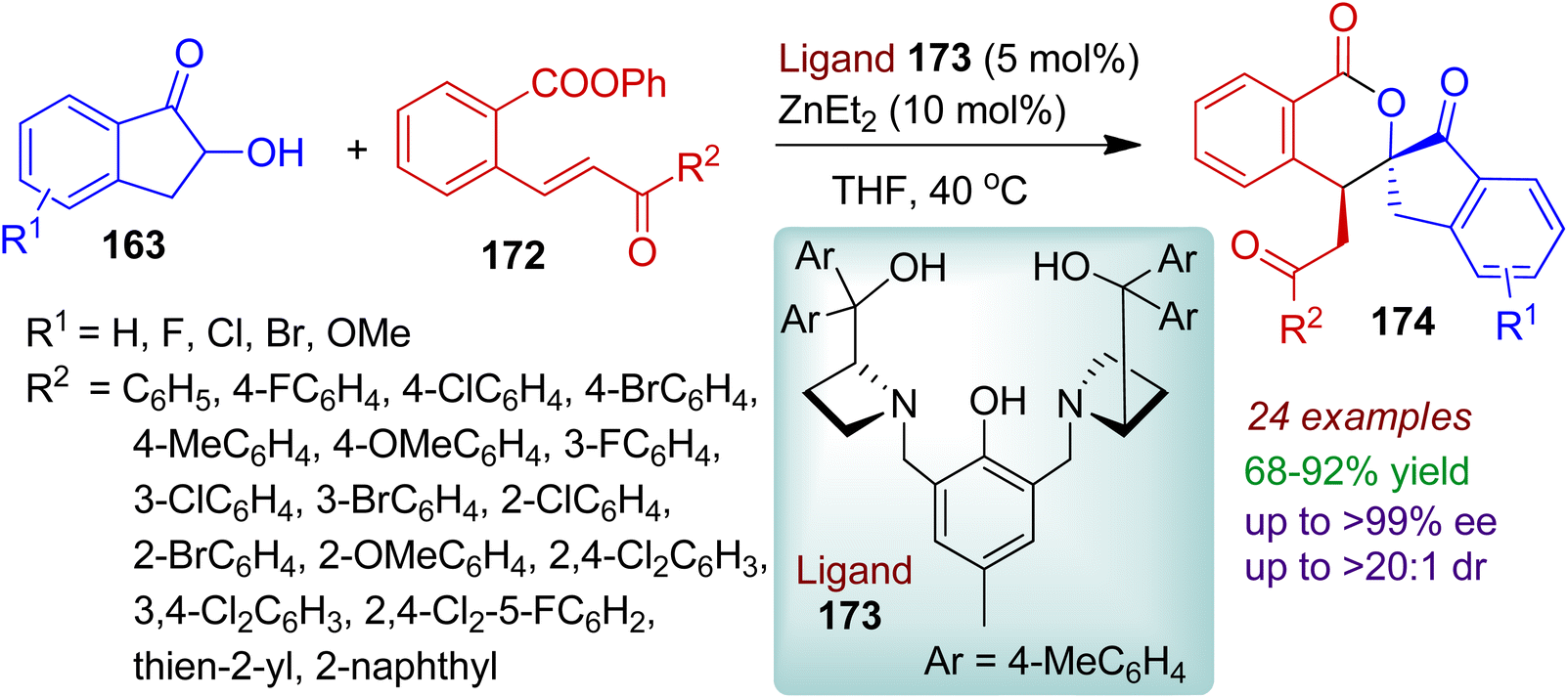
The authors also successfully carried out similar type of asymmetric Michael addition/transesterification tandem reaction between 2-hydroxy-1-indanones 163 and ortho-ester chalcones 172 using a dinuclear zinc catalyst (ligand 173 and Et2Zn).82 A library of chiral spiro[indanone-2,3′-isochromane-1-one] derivatives 174 possessing two adjacent chiral centers was obtained up to >99% ee, >20:1 dr (Scheme 48). Notably, the ortho-ester chalcones 172 bearing mono, di, and trisubstituted aromatic rings (R2 = aromatics) were proved to be suitable substrates for this conversion. However, the reaction failed for ortho-ester chalcones with aliphatic substituents (R2 = Me). The practicality of the reaction was manifested by the gram-scale synthesis of the desired spiro products without affecting stereoselectivities.
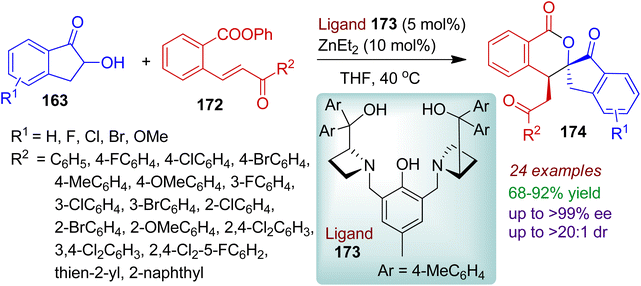 |
| | Scheme 48 Dinuclear zinc catalyzed synthesis of spiro[indanone-2,3′-isochromane-1-one] derivatives from 2-hydroxy-1-indanones and ortho-ester chalcones. | |
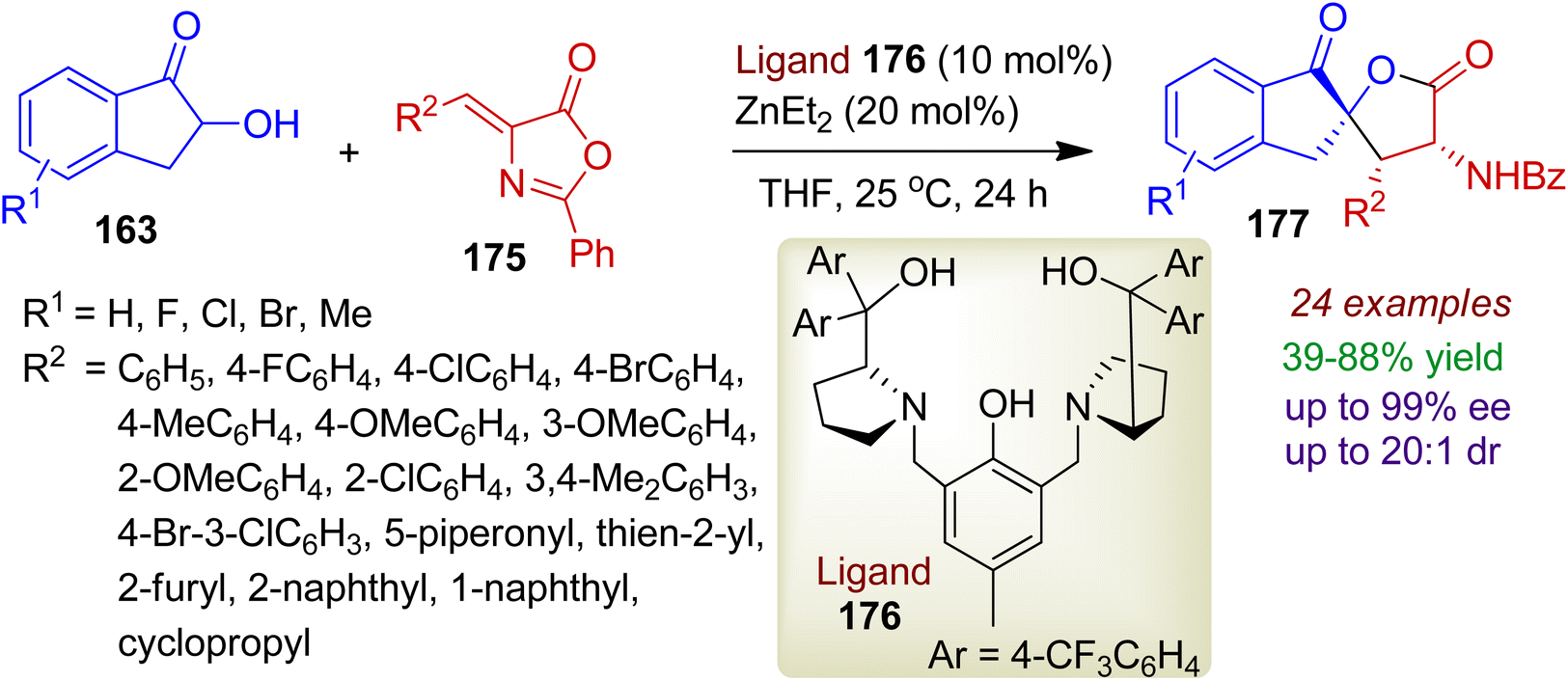
Alkylidene azlactones may also be used for the preparation of spirolactone derivatives. An efficient enantioselective annulation of 2-hydroxy-1-indanones 163 and alkylidene azlactones 175 was realized with chiral dinuclear zinc catalyst (ligand 176 and Et2Zn).83 The reaction enabled the formation of polyfunctional spirocyclic α-amino-γ-butyrolactones 177 bearing three stereocenters via [3+2] cyclization (Scheme 49). A wide range of ortho-, meta-, para- and multisubstituted phenyl azlactone substrates 175 could be applied to afford α-amino-γ-butyrolactones 177 in good yields (up to 88%) and stereoselectivities (up to 99% ee, 20:1 dr). However, corresponding aliphatic substitution (R2 = cyclopropyl) led to decreased yield (70%) and enantioselectivity (73% ee). The methodology was also amenable for α-hydroxyacetophenones and 3-hydroxychroman-4-ones.
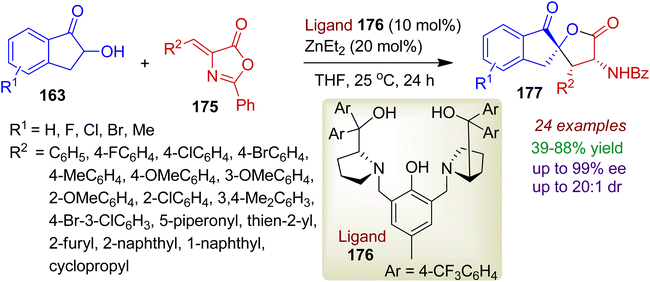 |
| | Scheme 49 Dinuclear zinc catalyzed synthesis of spirocyclic α-amino-γ-butyrolactones from 2-hydroxy-1-indanones and alkylidene azlactones. | |
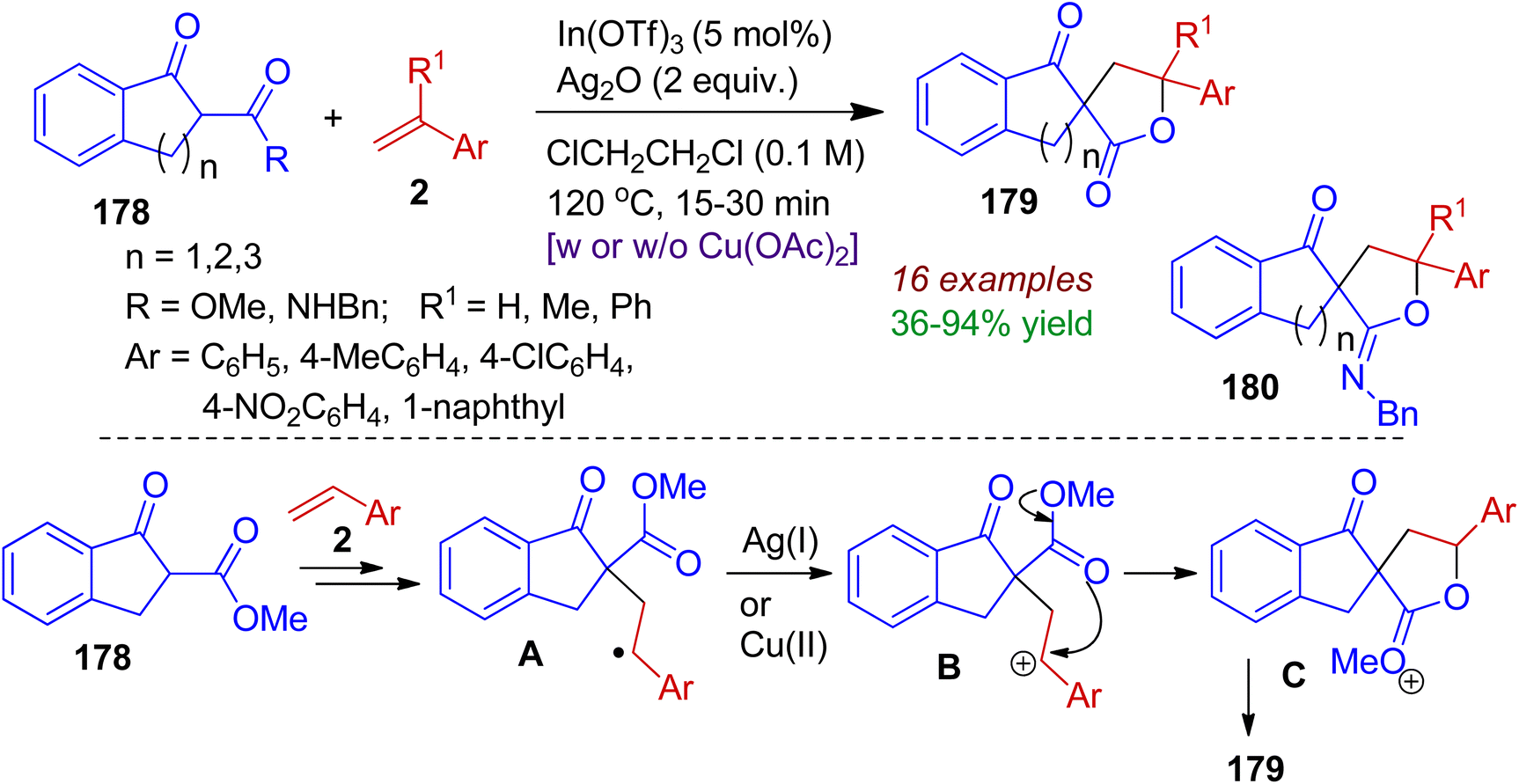
During their synthetic programme, Ko and Youn realized a cooperative indium(III)/silver(I)-catalyzed synthesis of spirolactones 179 and spiroiminolactones 180 starting from readily available 2-substituted 1-indanones 178 and substituted styrenes 2.84 This method involves sequential formation of C–C and C–O (or C–N) bonds with good substrate scope within short time (15–30 min). Mechanistically, it is conceivable that (Scheme 50) the reaction of indanones 178 with styrene 2 in the presence of indium catalyst generates radical intermediate A. Then, intermediate A is oxidized to the benzylic carbocation B by Ag(I) alone or with the help of co-oxidant Cu(OAc)2. Subsequent intramolecular cyclization (lactonization) furnishes spirolactone species C, which after hydrolysis produces spirolactone product 179.
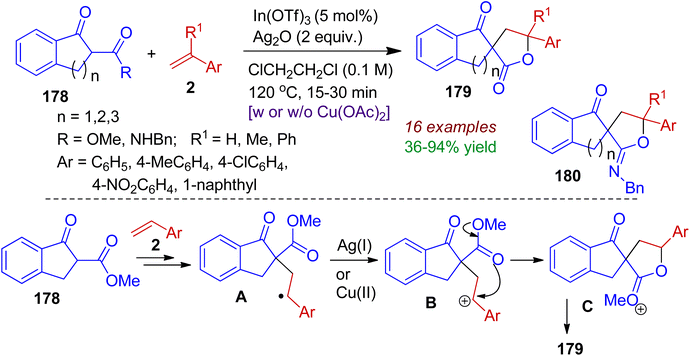 |
| | Scheme 50 Indium(III)/silver(I)-catalyzed synthesis of spirolactones/spiroiminolactones from 2-substituted 1-indanones and styrene. | |
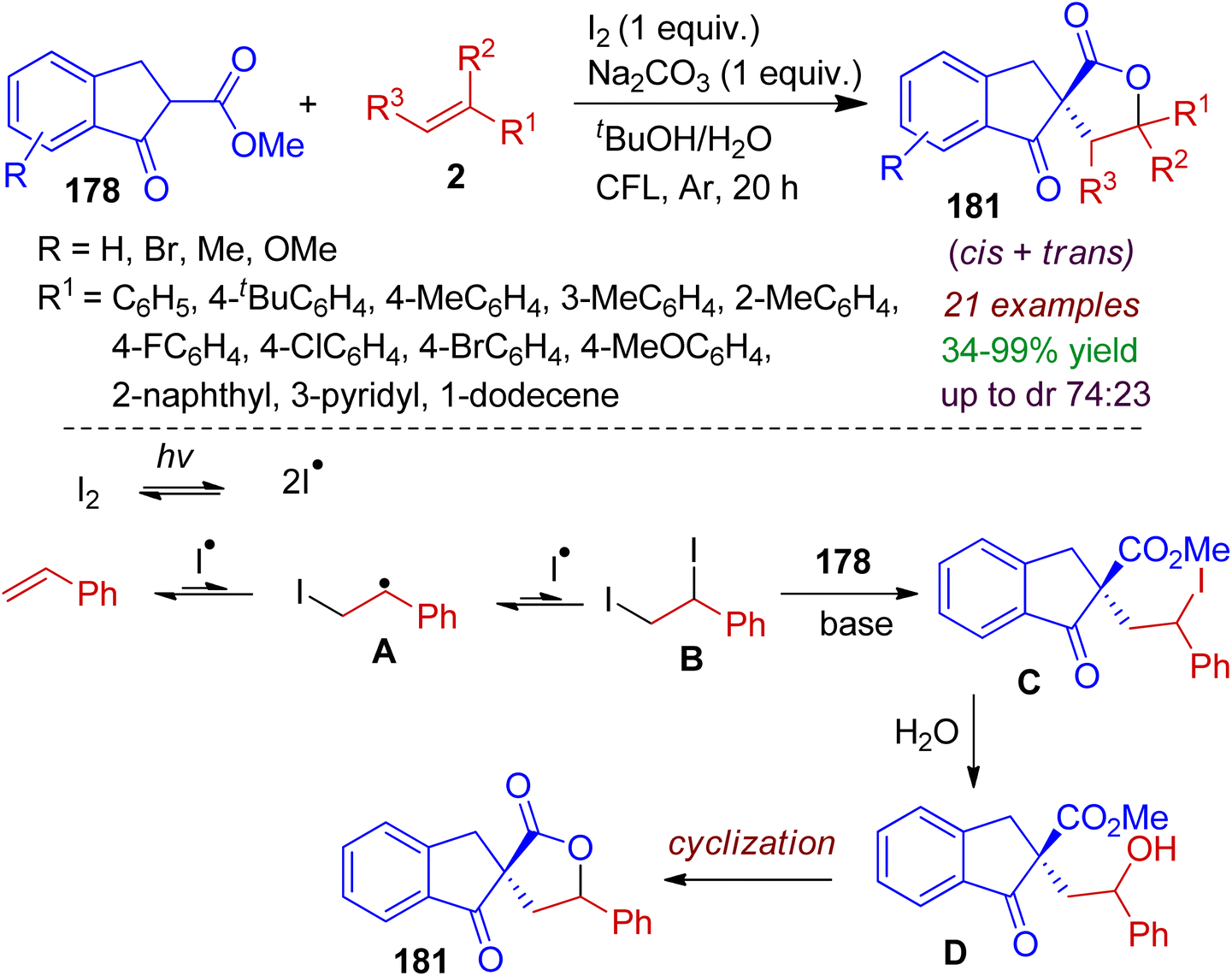
Finally, we discuss an interesting photooxidative intermolecular spirolactonization reaction as reported by Maejima, Yamaguchi and Itoh.85 The visible light/molecular iodine-mediated reaction between 1-indanone derivative 178 (acts as β-keto ester) and olefins 2 proceeded to afford indeno spirolactones 181 in a single step. Iodine radical generation from molecular iodine triggered by visible light was the key step in this process. A plausible mechanism is outlined in Scheme 51. Initially, iodine radicals are generated via activation of molecular iodine by visible light. Afterwards, iodine radicals are added to the olefin to form radical intermediate A, which is added to another iodine radical, thereby producing vic-diiodide intermediate B. Subsequently, the diiodide B reacted with β-keto ester 178 in the presence of base to obtain compound C. Hydrolysis of C gives corresponding hydroxyl intermediate D, which after intramolecular cyclization forms desired spirolactone 181. The reaction is cost effective, atom economic, without using transition-metal catalyst and environmentally benign.
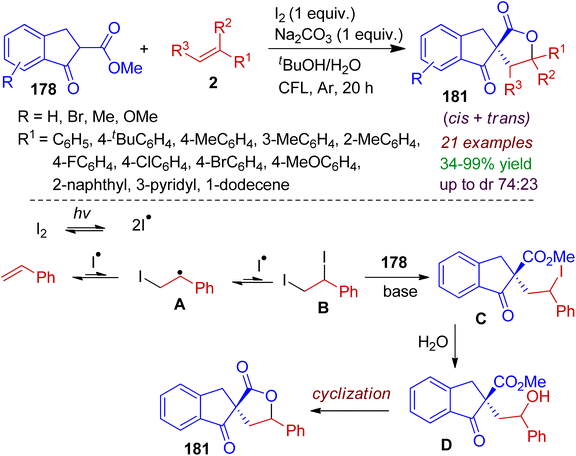 |
| | Scheme 51 Photooxidative reaction between substituted 1-indanones and olefins to afford spirolactones. | |
4. Conclusion
Indanone scaffolds comprise key structural components of numerous bioactive natural products. Because of their versatile reactivity they are efficiently employed in various organic transformations. In the last few years, a number of novel methodologies have been adopted to access diverse annulated scaffolds involving 1-indanones. This review emphasizes recent (2016–2022) applications of 1-indanones for the construction of various fused- and spiro carbo-/heterocyclic compounds.
In the fused carbocycle section we discuss synthesis of benzocycloheptenones, fluorenones, dibenzo-azulenes, indanone fused cyclopentanes, and photodimerized products which are difficult to prepare otherwise.86 In the next part, formation of fused N- and O-containing heterocycles, such as indeno-indoles, indeno-pyridines, indenoquinoxalines, cyclopenta-isoquinolines, indeno[1,2-b]chromenes, indeno[1,2-c]furans, etc. is highlighted. The spirocarbocyclic sector consists of cyclopropane/cyclopentane/cyclohexane-spiroindanone compounds. Construction of various N-containing spiro heterocycles viz. spiroindeno-aziridines, spiroindeno-pyrrolidines, spiro-glutarimides, spiroindeno-pyrazoles, spiroindeno-pyridine-oxindoles, spiro-cyclopentaindeno-indolines, spiroindeno-morpholines as well as interesting bispirocyclic skeletons is demonstrated accordingly. Finally, methods of synthesis of several spiroindeno-γ-butyrolactones, spiroindeno-isochromanes are unveiled in the O-containing spiro-heterocycles section.
Interestingly, a large number of reactions described in this review are associated with stereoselective formation of annulated scaffolds. Moreover, mechanistic aspects of representative reactions have been illustrated for better understanding of the reaction pathways. Some of the reactions offer biologically relevant compounds as well as natural products, viz., plecarpenene/plecarpenone, swinhoeisterol A, cephanolides A–D, diptoindonesin G and atlanticone C. We believe that the results presented in this article will attract the attention of organic and medicinal chemistry researchers for future developments of cyclization methods involving indanone analogues.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
The technical support of Rishi Bankim Chandra College for Women, Naihati, INDIA is gratefully acknowledged. The authors would like to thank the authors of the papers collected here for their hard work that made this review possible. We are grateful to the reviewers for their constructive suggestions.
References
- S.-H. Kim, S. H. Kwon, S.-H. Park, J. K. Lee, H.-S. Bang, S.-J. Nam, H. C. Kwon, J. Shin and D.-C. Oh, Tripartin, a Histone Demethylase Inhibitor from a Bacterium Associated with a Dung Beetle Larva, Org. Lett., 2013, 15, 1834–1837, DOI:10.1021/ol4004417.
- X. Luo, C. Li, P. Luo, X. Lin, H. Ma, N. P. Seeram, C. Song, J. Xu and Q. Gu, Pterosin Sesquiterpenoids from Pteris cretica as Hypolipidemic Agents via Activating Liver X Receptors, J. Nat. Prod., 2016, 79, 3014–3021, DOI:10.1021/acs.jnatprod.6b00558.
- G. Assante, R. Locci, L. Camarda and G. Nasini, Screening of the genus Cercospora for secondary metabolites, Phytochemistry, 1977, 16, 243–247, DOI:10.1016/S0031-9422(00)86794-1.
- S. Das, Recent applications of 1,3-indanedione in organic transformations for the construction of fused- and spiro scaffolds, Tetrahedron, 2022, 122, 132954, DOI:10.1016/j.tet.2022.132954.
- S. Das, Annulations involving 2-arylidene-1,3-indanediones: stereoselective synthesis of spiro- and fused scaffolds, New J. Chem., 2020, 44, 17148–17176, 10.1039/d0nj03968.
- S. Das, Recent applications of ninhydrin in multicomponent reactions, RSC Adv., 2020, 10, 18875–18906, 10.1039/d0ra02930k.
- S. Sutthivaiyakit, W. Mongkolvisut, S. Prabpai and P. Kongsaree, Diterpenes, Sesquiterpenes, and a Sesquiterpene-Coumarin Conjugate from Jatropha integerrima, J. Nat. Prod., 2009, 72, 2024–2027, DOI:10.1021/np900342b.
- M. J. D. Peña, J. M. B. Ares, I. G. Collado and R. H. Galán, Biologically active diterpenes containing a gem-dimethylcyclopropane subunit: an intriguing source of PKC modulators, Nat. Prod. Rep., 2014, 31, 940–952, 10.1039/c4np00008k.
- K. E. Wilson, N. N. Tsou, Z. Guan, C. L. Ruby, F. Pelaez, J. Gorrochategui, F. Vincente and H. R. Onishi, Isolation and structure elucidation of coleophomone A and B, novel inhibitors of bacterial cell wall transglycosylase, Tetrahedron Lett., 2000, 41, 8705–8709, DOI:10.1016/S0040-4039(00)01532-X.
- K. C. Nicolaou, T. Montagnon and G. Vassilikogiannakis, Total synthesis of celeophomone D, Chem. Commun., 2002, 2478–2479, 10.1039/b208236p.
- J. A. Wendt, P. J. Gauvreau and R. D. Bach, Synthesis of (.+−.)-Fredericamycin A, J. Am. Chem. Soc., 1994, 116, 9921–9926, DOI:10.1021/ja00101a013.
- S. Kotha and A. Fatma, Synthetic Approaches to the Anticancer Agent Fredericamycin A, Asian J. Org. Chem., 2021, 10, 129–148, DOI:10.1002/ajoc.202000484.
- M. A. Ernst-Russell, C. L. L. Chai, J. H. Wardlaw and J. A. Elix, Euplectin and Coneuplectin, New Naphthopyrones from the Lichen Flavoparmelia euplecta, J. Nat. Prod., 2000, 63, 129–131, DOI:10.1021/np9903245.
- H. L. Holland, D. B. MacLean, R. G. A. Rodrigo and R. F. H. Manske, A new synthesis of spirobenzylisoquinolines. Analogues of sibiricine and corydaine, Tetrahedron Lett., 1975, 16, 4323–4326, DOI:10.1016/S0040-4039(00)91114-6.
- G. Blaskó, N. Murugesan, A. J. Freyer, D. J. Gula, B. Şener and M. Shamma, The indenobenzapine-spirobenzylisoquinoline rearrangement; stereocontrolled syntheses of (±)-raddeanine and (±)-yenhusomine, Tetrahedron Lett., 1981, 22, 3139–3142, DOI:10.1016/S0040-4039(01)81847-5.
- X. Wang, T. Yang, X. Cheng and Q. Shen, Enantioselective Electrophilic Trifluoromethylthiolation of β-Ketoesters: A Case of Reactivity and Selectivity Bias for Organocatalysis, Angew. Chem., Int. Ed., 2013, 52, 12860–12864, DOI:10.1002/anie.201305075.
- Q.-H. Deng, T. Bleith, H. Wadepohl and L. H. Gade, Enantioselective Iron-Catalyzed Azidation of β-Keto Esters and Oxindoles, J. Am. Chem. Soc., 2013, 135, 5356–5359, DOI:10.1021/ja402082p.
- M. Majdecki, P. Grodek and J. Jurczak, Stereoselective α-Chlorination of β-Keto Esters in the Presence of Hybrid Amide-Based Cinchona Alkaloids as Catalysts, J. Org. Chem., 2021, 86, 995–1001, DOI:10.1021/acs.joc.0c02486.
- S. Dai, F. Zhao, Q. Zhang, T.-K. Lau, T. Li, K. Liu, Q. Ling, C. Wang, X. Lu, W. You and X. Zhan, Fused Nonacyclic Electron Acceptors for Efficient Polymer Solar Cells, J. Am. Chem. Soc., 2017, 139, 1336–1343, DOI:10.1021/jacs.6b12755.
- Q. Liao, Q. Kang, Y. Yang, C. An, B. Xu and J. Hou, Tailoring and Modifying an Organic Electron Acceptor toward the Cathode Interlayer for Highly Efficient Organic Solar Cells, Adv. Mater., 2019, 32, 1906557, DOI:10.1002/adma.201906557.
- L. A. Estrada, J. E. Yarnell and D. C. Neckers, Revisiting Fluorenone Photophysics via Dipolar Fluorenone Derivatives, J. Phys. Chem. A, 2011, 115, 6366–6375, DOI:10.1021/jp200507j.
- K.-C. Tang, M.-J. Chang, T.-Y. Lin, H.-A. Pan, T.-C. Fang, K.-Y. Chen, W.-Y. Hung, Y.-H. Hsu and P.-T. Chou, Fine Tuning the Energetics of Excited-State Intramolecular Proton Transfer (ESIPT): White Light Generation in A Single ESIPT System, J. Am. Chem. Soc., 2011, 133, 17738–17745, DOI:10.1021/ja2062693.
- Y. Cui, H. Ren, J. Yu, Z. Wang and G. Qian, An indanone-based alkoxysilane dye with second order nonlinear optical properties, Dyes Pigm., 2009, 81, 53–57, DOI:10.1016/j.dyepig.2008.09.002.
- K. Kolmakov, C. Wurm, M. V. Sednev, M. L. Bossi, V. N. Belov and S. W. Hell, Masked red-emitting carbopyronine dyes with photosensitive 2-diazo-1-indanone caging group, Photochem. Photobiol. Sci., 2012, 11, 522–532, 10.1039/C1PP05321C.
- E. Benedetti, L. S. Kocsis and K. M. Brummond, Synthesis and Photophysical Properties of a Series of Cyclopenta[b]naphthalene Solvato-chromic Fluorophores, J. Am. Chem. Soc., 2012, 134, 12418–12421, DOI:10.1021/ja3055029.
- L. Fang, X.-Y. Zhang, Q. Yuan, D.-D. Li, Q.-C. Jiao, Y.-S. Yang and H.-L. Zhu, A novel indanone-derivated fluorescence sensor for Cysteine detection and biological imaging, Dyes Pigm., 2020, 175, 108122, DOI:10.1016/j.dyepig.2019.108122.
- C. R. Monzón-González, R. Corona-Sánchez, W. E. V. Narvaáez, T. Rocha-Rinza, M. E. Sánchez-Vergara, R. A. Toscano and C. Álvarez-Toledano, Synthesis and photophysical properties of conformationally restricted difluoroboron β-diketonate complexes of 1-indanone derivatives, Tetrahedron, 2020, 76, 131457, DOI:10.1016/j.tet.2020.131457.
- S. Das and A. Dutta, Recent advances in transition-metal-catalyzed annulations for the construction of a 1-indanone core, New J. Chem., 2021, 45, 4545–4568, 10.1039/D0NJ06318E.
- S. Das, Transition-Metal-Free Synthesis of 1-Indanone Skeleton: A Brief Update, ChemistrySelect, 2021, 6, 4761–4781, DOI:10.1002/slct.202101460.
- T. Chanda and M. S. Singh, Developments toward the synthesis and application of 3-hydroxyindanones, Org. Biomol. Chem., 2016, 14, 8895–8910, 10.1039/C6OB01648K.
- Y. Xia, S. Ochi and G. Dong, Two-Carbon Ring Expansion of 1-Indanones via Insertion of Ethylene into Carbon–Carbon Bonds, J. Am. Chem. Soc., 2019, 141, 13038–13042, DOI:10.1021/jacs.9b07445.
- R. Zhang, Y. Xia and G. Dong, Intermolecular [5+2] Annulation between 1-Indanones and Internal Alkynes by rhodium-Catalyzed C-C Activation, Angew. Chem., Int. Ed., 2021, 60, 20476–20482, DOI:10.1002/ange.202106007.
- Z. Xing, B. Fang, S. Luo, X. Xie and X. Wang, Generation of Fuesed seven-Membered Polycyclic Systems via Ring Expansion and Application to the Total Synthesis of Sesquiterpenoids, Org. Lett., 2022, 24, 4034–4039, DOI:10.1021/acs.orglett.2c01401.
- P. R. Throve, M. M. Guru and B. Maji, Manganese-Catalyzed Divergent Markovnikov Addition and [2+2+2] Cycloaddition of 2-Carbonyl Indanone with Terminal Alkyne, J. Org. Chem., 2019, 84, 8185–8193, DOI:10.1021/acs.joc.9b01173.
- K. Patel, U. K. Mishra, D. Mukhopadhyay and S. S. V. Ramasastry, Beyond the Corey-Chaykovsky reaction: synthesis of Unusual Cyclopropanoids via Desymmetrization and Thereof, Chem.–Asian J., 2019, 14, 4568–4571, DOI:10.1002/asia.201901108.
- T. Mita, M. Uchiyama and Y. Sato, Catalytic Intramolecular Coupling of Ketoalkenes by Allylic C(sp3)–H Bond Cleavage: Synthesis of Five- and Six-Membered Carbocyclic Compounds, Adv. Synth. Catal., 2020, 362, 1275–1280, DOI:10.1002/adsc.201901533.
- M. E. Jung and G. Piizzi, gem-Disubstituent Effect: Theporetical basis and Synthetic Applications, Chem. Rev., 2005, 105, 1735–1766, DOI:10.1021/cr940337h.
- N. Uemura, H. Ishikawa, N. Tamura, Y. Yoshida, T. Mino, Y. Kasashima and M. Sakamoto, Stereoselective Photodimerization of 3-Arylindenones in Solution and in the Solid State, J. Org. Chem., 2018, 83, 2256–2262, DOI:10.1021/acs.joc.7b03147.
- N.-N. Zhou, S.-S. Ning, L.-Q. Li, J. Y. Zhang, M.-J. Fan, D.-S. Yang and H.-T. Zhu, Brønsted Acid-Catalyzed One-Pot Tandem Annulation/[5+2]-Cycloaddition of o-Propargyl Alcohol Benzaldehydes with Alkynes: Regioselective and Stereoselective Synthesis of Dibenzo[a,f]azulen-12-ones, Org. Chem. Front., 2020, 7, 2664–2669, 10.1039/D0QO00522C.
- Z.-J. Shen, H.-N. Shi, W.-J. Hao, S.-J. Tu and B. Jiang, Visible-light photocatalytic bicyclization of β-alkynyl propenones for accessing diastereoenriched syn-fluoren-9-ones, Chem. Commun., 2018, 54, 11542–11545, 10.1039/c8cc06086j.
- J. Zhang, H. Jiang and S. Zhu, Cascade One-pot Synthesis of Indanone-Fused Cyclopentanes from the Reaction of Donor-Acceptor Cyclopropanes and Enynals via a Sequential Hydrolysis/Knoevenagel Condensation/[3+2] Cycloaddition, Adv. Synth. Catal., 2017, 359, 2924–2930, DOI:10.1002/adsc.201700345.
- T. Kurzawa, K. Harms and U. Koert, Stereoselective Synthesis of the Benzodihydropentalene Core of the Fijiolides, Org. Lett., 2018, 20, 1388–1391, DOI:10.1021/acs.orglett.8b00163.
- A. Takano, Y. Zhao, T. Ohyoshi and H. Kigoshi, Synthetic studies toward swinhoeisterol A, a novel steroid with an unusual carbon skeleton, Tetrahedron Lett., 2019, 60, 591–593, DOI:10.1016/j.tetlet.2019.01.027.
- M. Kashyap, S. Kandekar, A. T. Baviskar, D. Das, R. Preet, P. Mohapatra, S. R. Satapathy, S. Siddharth, S. K. Guchhait, C. N. Kundu and U. C. Banerjee, Indenoindolone derivatives as topoisomeraseII-inhibiting anticancer agents, Bioorg. Med. Chem. Lett., 2013, 23, 934–938, DOI:10.1016/j.bmcl.2012.12.063.
- A. G. K. Reddy and G. Satyanarayana, A Facile, Superacid-Promoted Sequential Domino One-Pot Dual C–C Bond Formation and Fischer Indole Synthesis: Rapid Access to 10-Phenyl-5,10-dihydroindeno[1,2-b]indoles, Synthesis, 2015, 1269–1279, DOI:10.1055/s-0034-1380287.
- Z. Wang, M. Wang, S. Xu, M. Gu, Z. Meng, C. Li, P. Cai and L. Rong, Efficient and facile synthesis of 5-arylindeno[2′,1′:5,6]pyrido[2,3-d]pyrimidine-2,4(3H)-dione and 7-arylbenzo[h]pyrimido[4,5-b]quinoline-8,10(5H,9H)-dione under mild conditions, Synth. Commun., 2016, 46, 1887–1892, DOI:10.1080/00397911.2016.1234621.
- H. Nagarajaiah, A. K. Mishra and J. N. Moorthy, Mechanochemical Solid-State Synthesis of 2-Aminothiazoles, Quinoxalines and Bezoylbenzofurans from Ketones by One-Pot Sequential Acid- and Base-Mediated reactions, Org. Biomol. Chem., 2016, 14, 4129–4135, 10.1039/C6OB00351F.
- T. M. Kadayat, S. Park, A. Shrestha, H. Jo, S.-Y. Hwang, P. Katila, R. Shrestha, M. R. Nepal, K. Noh, S. K. Kim, W.-S. Koh, K. S. Kim, Y. H. Jeon, T. C. Jeong, Y. Kwon and E.-S. Lee, Discovery and Biological Evaluations of Halogenated 2,4-Diphenyl Indeno[1,2-b]pyridinol Derivatives as Potent Topoisomerase IIα-Targeted Chemotherapeutic Agents for Breast Cancer, J. Med. Chem., 2019, 62, 8194–8234, DOI:10.1021/acs.jmedchem.9b00970.
- J.-Q. Zhao, S. Zhou, H.-L. Qian, Z.-H. Wang, Y.-P. Zhang, Y. You and W.-C. Yuan, Higher-order [10 + 2] cycloaddition of 2-alkylidene-1-indanones enables the dearomatization of 3-nitroindoles: access to polycyclic cyclopenta[b]indoline derivatives, Org. Chem. Front., 2022, 9, 3322–3327, 10.1039/d2qo00289b.
- S. K. Sarkar, O. Osisioma, W. L. Karney, M. Abe and A. D. Gudmundsdottir, Using Molecular Architecture to Control the Reactivity of a Triplet Vinylnitrene, J. Am. Chem. Soc., 2016, 138, 14905–14914, DOI:10.1021/jacs.6b05746.
- D. F. Vargas, E. L. Larghi and T. S. Kaufman, Concise Synthesis of the ABC-Ring System of the Azafluoranthene, Tropoisoquinoline and Proaporphine Alkaloids: An Olefin Hydroacylation/Pomeranz–Fritsch Cyclization Approach, Synthesis, 2019, 51, 2030–2038, DOI:10.1055/s-0037-1611711.
- X.-Q. Hou, Y. Lin and D.-M. Du, Organocatalytic domino annulation of in situ generated tert-butyl 2-hydroxybenzylidenecarbamates with 2-isothiocyanato-1-indanones for synthesis of bridged and fused ring heterocycles, Org. Chem. Front., 2021, 8, 4183–4187, 10.1039/d1qo00626f.
- X. Liu, K. Wang, Y. Liu and C. Li, Divergent Asymmetric Reactions of ortho-Quinone Methides with α-Thiocyanato Indanones for the Synthesis of Spiro- and Fusedindanones, Chem.–Eur. J., 2021, 27, 735–739, DOI:10.1002/chem.202003647.
- B. Li, N. Shen, Y. Yang, X. Zhang and X. Fan, Tunable Synthesis of Indeno[1,2-c]furans and 3-Benzoylindenones via FeCl3-Catalyzed Carbene/Alkyne Metathesis Reaction of o-Alkynylbenzoyl Diazoacetates, Org. Lett., 2021, 23, 388–393, DOI:10.1021/acs.orglett.0c03882.
- M. Haider, G. Sennari, A. Eggert and R. Sarpong, Total Synthesis of the Cephalotaxus Norditerpenoids (±)-Cephanolides A–D, J. Am. Chem. Soc., 2021, 143, 2710–2715, DOI:10.1021/jacs.1c00293.
- D. K. Singh and I. Kim, Skeletal Reorganization: Synthesis of Diptoindonesin G from Pauciflorol F, J. Org. Chem., 2018, 83, 1667–1672, DOI:10.1021/acs.joc.7b03089.
- J. Proessdorf, A. Zech, C. Jandl and T. Bach, Concise Total Synthesis of (+)-Atlanticone C, Synlett, 2020, 31, 1598–1602, DOI:10.1055/s-0040-1707215.
- S. P. Midya, E. Gopi, N. Satam and I. N. N. Namboothiri, Synthesis of fused cyanopyrroles and spirocyclopropanes via addition of N-ylides to chalconimines, Org. Biomol. Chem., 2017, 15, 3616–3627, 10.1039/c7ob00529f.
- J.-N. Wang, S.-Q. Chen, Z.-W. Liu and X.-Y. Shi, Divergent Syntheses of Spiroindanones and 2-Substituted 1-Indanones by Ruthenium-Catalyzed Tandem Coupling and Cyclization of Aromatic Acids with α,β-Unsaturated Ketones, J. Org. Chem., 2019, 84, 1348–1362, DOI:10.1021/acs.joc.8b02820.
- Y. Suzuki, N. Vatmurge, S. Tanaka and M. Kitamura, Enantio- and Diastereoselective Dehydrative ''One-Step'' Construction of Spirocarbocycles via a Ru/H+-Catalyzed Tsuji-Trost Approach, Chem.–Asian J., 2017, 123, 633–637, DOI:10.1002/asia.201700013.
- B. Lantaño, J. M. Aguirre, E. V. Drago, M. Bollini, D. J. De la Faba and J. D. Mufato, Synthesis of benzylidenecycloalkan-1-ones and 1,5-diketones under Claisen–Schmidt reaction: Influence of the temperature and electronic nature of arylaldehydes, Synth. Commun., 2017, 47, 2202–2214, DOI:10.1080/00397911.2017.1367819.
- S. Kotha, R. Ali, N. R. Panguluri, A. Datta and K. K. Kannaujiya, Synthesis and photophysical properties of star-shaped
blue green emitting π-conjugated spirotruxenes, Tetrahedron Lett., 2018, 59, 4080–4085, DOI:10.1016/j.tetlet.2018.10.005.
- S. Kotha and S. R. Cheekatla, Synthesis and Acid Catalyzed Rearrangement of Cage Propellanes, ChemistrySelect, 2019, 4, 13440–13445, DOI:10.1002/slct.201903441.
- S. Kotha, S. R. Cheekatla and A. Fatma, Synthetic Approach to the ABCD Ring System of Anticancer Agent Fredericamycin A via Claisen Rearrangement and Ring-Closing Metathesis as Key Steps, ACS Omega, 2019, 4, 17109–17116, DOI:10.1021/acsomega.9b01178.
- S. Sabir, G. Kumar, V. P. Verma and J. L. Jat, Aziridine Ring Opening: An Overview of Sustainable Methods, ChemistrySelect, 2018, 3, 3702–3711, DOI:10.1002/slct.201800170.
- K. T. Ashitha, P. P. Vinaya, A. Krishna, D. C. Vincent, R. Jalaja, S. Varughese and S. B. Somappa, I2/TBHP Mediated Diastereoselective Synthesis of Spiroaziridines, Org. Biomol. Chem., 2020, 18, 1588–1593, 10.1039/C9OB02711D.
- F. Beltran, I. Fabre, I. Ciofini and L. Miesch, Direct Spirocyclization from Keto-sulfonamides: An Approach to Azaspiro Compounds, Org. Lett., 2017, 19, 5042–5045, DOI:10.1021/acs.orglett.7b02216.
- Z. Ding, Y. Wang, W. Liu, Y. Chen and W. Kong, Diastereo- and Enantioselective Construction of Spirocycles by Nickel-Catalyzed Cascade Borrowing Hydrogen Cyclization, J. Am. Chem. Soc., 2021, 143, 53–59, DOI:10.1021/jacs.0c10055.
- S. Mondal, A. Ghosh, S. Mukherjee and A. T. Biju, N-Heterocyclic Carbene-Catalyzed Enantioselective Synthesis of Spiro-glutarimides via α,β-Unsaturated Acylazoliums, Org. Lett., 2018, 20, 4499–4503, DOI:10.1021/acs.orglett.8b01799.
- B.-X. Quan, J.-R. Zhuo, J.-Q. Zhao, M.-L. Zhang, M.-Q. Zhou, X.-M. Zhang and W.-C. Yuan, [4+1] Annulation reaction of cyclic pyridinium ylides with in situ generated azoalkenes for the construction of spirocyclic skeletons, Org. Biomol. Chem., 2020, 18, 1886–1891, 10.1039/C9OB02733E.
- G. I. Shakibaei and A. Bazgir, A highly efficient one-pot synthesis of indenopyridine-fused spirocyclic systems, RSC Adv., 2016, 6, 22306–22311, 10.1039/C6RA00869K.
- Y. Yang, Y. Jiang, W. Du and Y.-C. Chen, Asymmetric Cross [10+2] Cycloadditions of 2-Alkylidene-1-indanones and Activated Alkenes under Phase-Transfer Catalysis, Chem.–Eur. J., 2020, 26, 1754–1758, DOI:10.1002/chem.201904930.
- B. Wu, J. Yang, M. Gao and L. Hu, Ring-Strain-Enabled Catalytic Asymmetric Umpolung C–O Bond-Forming Reactions of 1,2-Oxazetidines for the Synthesis of Functionalized Chiral Ethers, Org. Lett., 2020, 22, 5561–5566, DOI:10.1021/acs.orglett.0c01916.
- S. Kayal and S. Mukherjee, Catalytic enantioselective cascade Michael/cyclization reaction of 3-isothiocyanato oxindoles with exocyclic α,β-unsaturated ketones en route to 3,2’-pyrrolidinyl bispirooxindoles, Org. Biomol. Chem., 2016, 14, 10175–10179, 10.1039/c6ob02187e.
- K. Dutta, M. Kumar, S. K. Ghosh, A. Das and R. Chowdhury, Diastereoselective synthesis of diverse 3,2′-pyrrolidinyl bispirooxindoles via 1,3-dipolar cycloaddition reactions of azomethine ylides with exocyclic α,β-unsaturated ketones and in silico studies for prediction of bioactivity, Synth. Commun., 2019, 49, 444–455, DOI:10.1080/00397911.2018.1560472.
- B.-L. Zhao and D.-M. Du, Catalytic Asymmetric Mannich/Cyclization of 2-Isothiocyanato-1-indanones: An Approach to the Synthesis of Bispirocyclic Indanone-Thioimidazolidine-Oxindoles, Org. Lett., 2018, 20, 3797–3800, DOI:10.1021/acs.orglett.8b01389.
- X.-Q. Hou, J.-B. Wen, L. Yan and D.-M. Du, Squaramide-catalysed asymmetric Michael addition/cyclization cascade reaction of 4-arylmethylidene-2,3-dioxopyrrolidines with 2-isothiocyanato-1-indanones, Org. Biomol. Chem., 2021, 19, 7181–7185, 10.1039/d1ob01223a.
- B. Wu, H. Chen, M. Gao, X. Gong and L. Hu, Synthesis of 1,3-Aminoalcohols and Spirocyclic Azetidines via Tandem Hydroxymethylation and Aminomethylation Reaction of β-Keto
Phosphonates with N-Nosyl-O-(2-bromoethyl)hydroxylamine, Org. Lett., 2021, 23, 4152–4157, DOI:10.1021/acs.orglett.1c01091.
- M.-M. Liu, X.-C. Yang, Y.-Z. Hua, J.-B. Chang and M.-C. Wang, Synthesis of Chiral Bispirotetrahydrofuran Oxindoles by Cooperative Bimetallic-Catalyzed Asymmetric Cascade Reaction, Org. Lett., 2019, 21, 2111–2115, DOI:10.1021/acs.orglett.9b00386.
- S.-H. Chen, Y.-H. Miao, G.-J. Mei, Y.-Z. Hua, S.-K. Jia and M.-C. Wang, Dinuclear zinc catalyzed asymmetric [3+2] spiroannulation for the synthesis of diverse bispirocyclic saccharines, Org. Chem. Front., 2022, 9, 5010–5015, 10.1039/d2qo01039a.
- M.-M. Liu, X.-C. Yang, Y. Z. Hua, J.-B. Chang and M.-C. Wang, Dinuclear Zinc-Catalyzed Asymmetric Tandem Reaction of α-Hydroxy-1-indanone: Access to Spiro[1-indanone-5,2′-γ-butyrolactones], Org. Lett., 2019, 21, 7089–7093, DOI:10.1021/acs.orglett.9b02658.
- X.-C. Yang, M. Xu, J.-B. Wang, M.-M. Liu, F. Mathey, Y.-Z. Hua and M. C. Wang, Enantioselective Synthesis of Indanone Spiro-Isochromanone Derivatives via Dinuclear Zinc-Catalyzed Michael/Transesterification Tandem Reaction, Org. Biomol. Chem., 2020, 18, 3917–3926, 10.1039/D0OB00541J.
- W.-P. Yang, S.-K. Jia, T.-T. Liu, Y.-Z. Hua and M.-C. Wang, Dinuclear zinc-catalyzed asymmetric [3 + 2] cyclization reaction for direct assembly of chiral α-amino-γ-butyrolactones bearing three stereocenters, Org. Chem. Front., 2021, 8, 6998–7003, 10.1039/d1qo01338f.
- T. Y. Ko and S. W. Youn, Cooperative Indium(III)/Silver(I) System for Oxidative Coupling/Annulation of 1,3-Dicarbonyls and Styrenes: Construction of Five-Membered Heterocycles, Adv. Synth. Catal., 2016, 358, 1934–1941, DOI:10.1002/adsc.201600280.
- S. Maejima, E. Yamaguchi and A. Itoh, Visible Light/Molecular-Iodine-Mediated Intermolecular Spirolactonization Reaction of Olefins with Cyclic Ketones, J. Org. Chem., 2019, 84, 9519–9531, DOI:10.1021/acs.joc.9b01081.
- H. Cao, F. Chen, C. Su and L. Yu, Construction of Carbocycles from Methylenecyclopropanes, Adv. Synth. Catal., 2020, 362, 438–461, DOI:10.1002/adsc.201900615.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a and
Arpita Dutta
b
*a and
Arpita Dutta
b