
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBasic features of Ne*–HX (X = Cl, Br) chemi-ionization reactions
Stefano Falcinelli *a,
Franco Vecchiocattivia,
Brunetto Giovanni Brunettib,
Marco Parriania,
Giovanni Gigliottia,
Stefano Strangesc and
Fernando Piraniab
*a,
Franco Vecchiocattivia,
Brunetto Giovanni Brunettib,
Marco Parriania,
Giovanni Gigliottia,
Stefano Strangesc and
Fernando Piraniab
aDepartment of Civil and Environmental Engineering, University of Perugia, Via G. Duranti 93, 06125 Perugia, Italy. E-mail: stefano.falcinelli@unipg.it
bDepartment of Chemistry, Biology and Biotechnologies, University of Perugia, Via Elce di Sotto 8, 06123 Perugia, Italy
cDepartment of Chemistry and Drug Technologies, University Sapienza, Rome, 00185, Italy
First published on 8th March 2022
Abstract
Total and partial ionization cross sections for Ne*(3P2,0)–HX (X = Cl, Br) are presented in a comparative way as a function of the collision energy between 0.02–0.5 eV. New mass spectrometric data on Ne*–HBr chemi-ionization are discussed and analyzed with already published data on Ne*–HCl, highlighting similarities and differences of the collisional stereodynamics of the two systems. Basic features of the interaction potentials, driving reactive collisions, suggest that reaction channels, leading to the formation of parent HX+ ions in the ground and excited electronic state and to the formation of associated NeHX+ ions as well as of NeH+ proton transfer species, are selectively opened within angular cones exhibiting different orientation and acceptance.
Introduction
Chemi-ionizations (CHEMI) are elementary gas-phase reactions involving energetic neutral reagents which produce more stable ionic species. They happen in various environments and are very common in nature, as for example in flames, where they are considered as primary steps.1They can be induced by reagent species in an internally excited metastable state: in this case they are commonly known as Penning ionization or collisional autoionization processes. A number of interesting review articles have been published that detail such reactions and to which the reader can refer for a general overview.2 In the case of CHEMI involving electronically excited metastable species (as for example, the rare gas atoms, Rg*, excited in their first electronic level), our group recently proposed a new theoretical approach able to rationalize CHEMI as prototype oxidation processes.3 In such a way, we can schematize such reactions as follows:
| Rg* + X → (Rg⋯X)* | (1) |
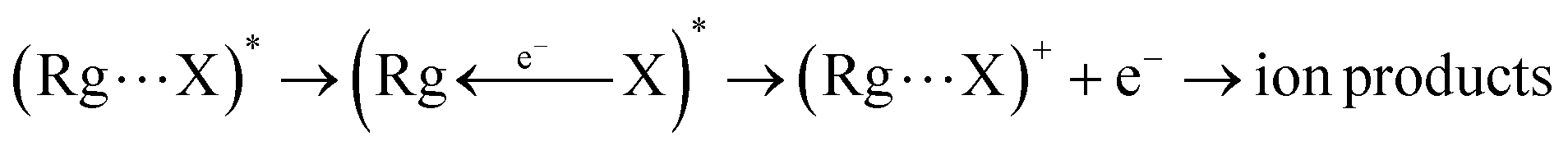 | (2) |
Eqn (1) represents the open-shell excited atom Rg* colliding with X (an atomic or molecular species). Such a collision gives rise to the formation of an electronically excited collision complex (Rg⋯X)* (see eqn (1) and (2) which energetically relaxes through an ionization phenomenon,2 as shown in eqn (2). Here (Rg⋯X)*, showing an electron excited in a high Rydberg state, represents the precursor state of the process, formed by two body collisions, that coincides with the transition state (TS) of CHEMI, which are barrier-less reactions. It must be noted that TS, under thermal conditions, evolves towards final ion products that can be the X+ parent ion, the associated RgX+ ionic aggregate, as well as dissociated or rearranged ions.2
More in detail, at sufficiently high (from thermal up to hyper-thermal) collision energies CHEMI preferentially occur with a direct mechanism, stimulated by “canonical” chemical forces. Such mechanism provides an elementary exothermic oxidation process (schematized by the left-going arrow in eqn (2) representing an electron exchange between the collisional target X and the ionic core of Rg*, which is an high electron affinity open shell species).3 On the other hand, at low (from sub-thermal up to thermal) collision energies and with reagents maintained at large separation distances, the precursor state evolution is basically controlled by the critical balance of weak intermolecular forces, having both physical and chemical nature, that preferentially trigger CHEMI by an indirect mechanism.3 This second mechanism includes virtual photon exchanges between reagents, namely photo-ionization contributions.
In general, CHEMI reactions are of interest in various topics, such as in combustion,4 in chemistry and physics of plasmas,5 planetary atmospheres (where they influence the transmission of radio and satellite signals) and astrochemistry.6 Recently, they have drawn attention from a fundamental point of view to study the nature of quantum resonances that accompany reactive and non-reactive collisions under sub-thermal conditions. In the last years, this very interesting topic has been studied by pioneering and frontier experiments, performed under ultra-cold conditions, using the merged molecular beam technique.7
The ionization processes occurring on HX molecules, where X is a halogen atom, induced by collisions with Rg* have attracted great interest from the scientific community since they are of relevance in the development of excimer lasers. In particular, CHEMI induced by Ne*(3P2,0) occur in the production of the excimer molecule XeCl* generated in Xe + HCl gaseous mixture discharges containing an excess of Ne. A variety of interesting papers have been published on tackling this topic both from a theoretical8 and experimental point of view.9–11
In this paper we report results about Ne*(3P2,0) + HX (where X = Cl and Br) CHEMI, studied by mass spectrometric determinations in a crossed molecular beam apparatus able to measure total and partial ionization cross sections in a thermal collision energy range. The new data obtained for Ne*–HBr system are discussed and analyzed in a comparative way with those of Ne*–HCl, already published,10,11 to enhance similarities and differences in the collisional stereodynamics of the two systems. On such a way, we can rationalize the behaviour of Ne*–HX systems in terms of the main chemical–physical properties of the intermolecular interactions driving the reactive collisions, also considering the Penning ionization electron spectroscopy (PIES) studies previously performed, first, by Hotop and coworkers12,13 and, later, by Ohno and coworkers.14 Our early experiments on CHEMI of HCl showed, among other products, the formation of protonated neon, NeH+. Further experiments, about isotopic effect, have been reported with a study of the CHEMI of DCl. The data have shown a statistical character of the proton transfer process.11
The present investigation extends the study to the HBr CHEMI to achieve a deeper understanding of both these reactions. It will be emphasized that reaction channels, leading to the formation of parent HX+ ions in their ground and excited electronic states, of associated NeHX+ ions, and of NeH+ proton transfer species are selectively opened within angular cones exhibiting different orientation and acceptance.
Experimental
Total, partial ionization cross sections and branching ratios, defining absolute and relative probability of different reaction channels, have been measured in the Perugia laboratory, from thermal up to hyper-thermal collision energy range, using the apparatus schematized in Fig. 1.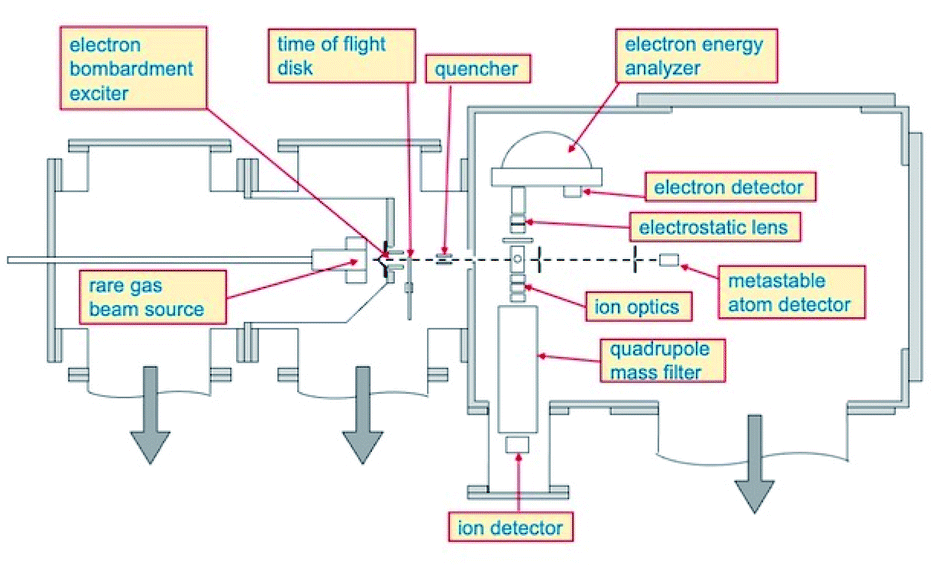 | ||
| Fig. 1 A schematic view of the molecular beam apparatus where the basic devices are indicated. The beams of neutral reagents cross at right angle. The product ions and electrons, are extracted, then analyzed and detected by a quadrupole mass and/or an electron energy analyzer (not used in the present experiment). | ||
Briefly, such apparatus consists of two crossed molecular beams, one of neon metastable atoms and one of target species, that are, in the present case, HCl and HBr molecules. The Ne atoms are excited in their metastable state Ne*(3PJ) by an electron bombardment exciter (at about 150 eV) producing Ne*(3P2) and Ne*(3P0) in a population close to the statistical 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio defined according to the different degeneracy of two atomic states. The metastable atom velocity can be analyzed by a time-of-flight (TOF) technique. The beam of metastable atoms crosses at right angle that of HX molecules emerging from an effusive multi-capillary source. The formed product ions and their relative intensity are obtained by mass spectrometric detection using a quadrupole mass filter. More details about the experimental set up have been already reported.15
:1 ratio defined according to the different degeneracy of two atomic states. The metastable atom velocity can be analyzed by a time-of-flight (TOF) technique. The beam of metastable atoms crosses at right angle that of HX molecules emerging from an effusive multi-capillary source. The formed product ions and their relative intensity are obtained by mass spectrometric detection using a quadrupole mass filter. More details about the experimental set up have been already reported.15
The experimental conditions used to collect the mass spectra as a function of the collision energy were strictly the same for the two investigated systems Ne*–HCl and Ne*–HBr, since CHEMI of HBr was studied after having reproduced the total ionization cross section data for Ne*–HCl (see Fig. 2) obtained previously.10
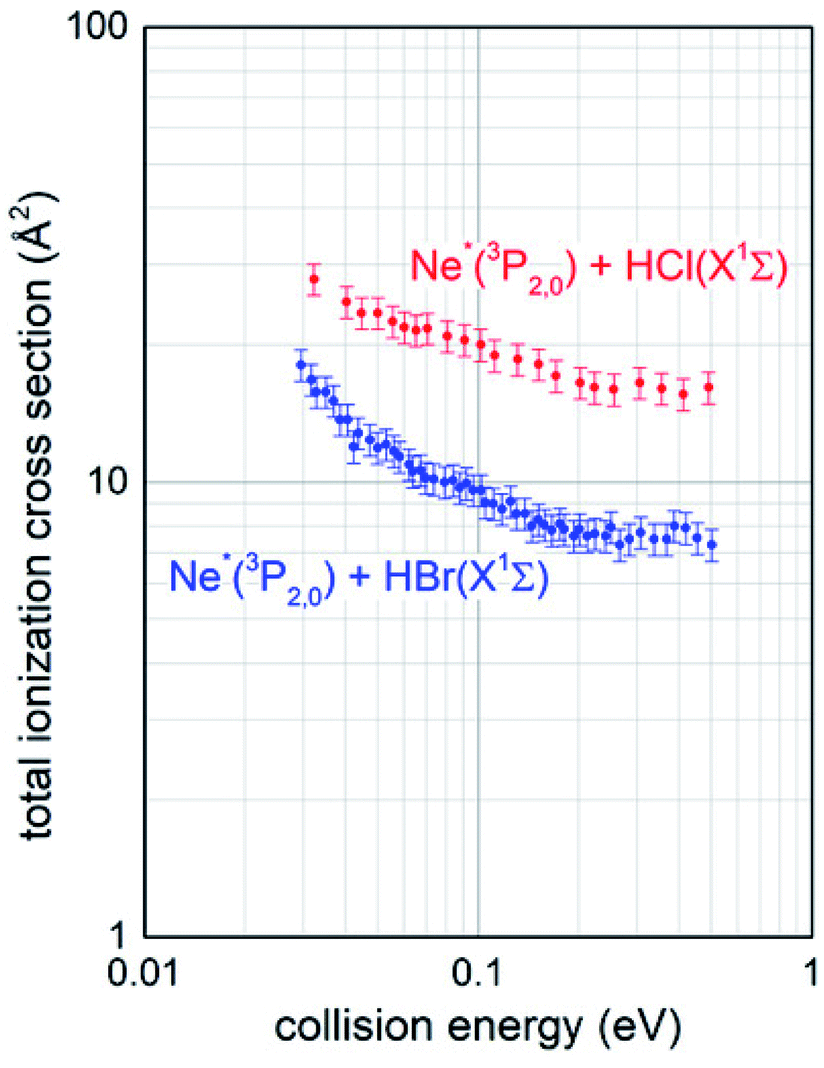 | ||
| Fig. 2 Comparison between total ionization cross sections measured, under the same experimental conditions, for both the ionizations of HCl and HBr molecules by collision with metastable neon atoms, as a function of the collision energy. | ||
High purity hydrogen bromide (min. 99.999%) and hydrogen chloride (min. 99.999%) cylinders, provided by Air Liquide, have been used to generate effusive molecular beams without possible impurities able to affect mass spectrometric determinations.
Results and discussion
Experimental data
Total ionization cross sections σiontot(E), measured for both Ne* + HCl and Ne* + HBr CHEMI and leading to the global formation (sum) of HX+, NeH+ and NeHX+ final ionic products, are plotted in Fig. 2 as a function of the collision energy E.Data for HCl have been presented and discussed in detail in previous papers,10,11 while those of HBr, measured under the same experimental conditions, are new. It is shown that in the range of probed collision energies, ionization process for HCl is more efficient (cross section of 20 A2 at 0.1 eV collision energy) with respect to HBr (cross section of 10 A2 at the same energy). In addition, the cross sections for HBr show a steeper energy dependence.
Branching ratios (BRs), describing the relative formation probability of selected ionic products, specifically the associate NeHX+ and the proton transfer NeH+ ionic species, respect to the ionization giving the parent ion HX+, have been also obtained and are reported in Fig. 3.
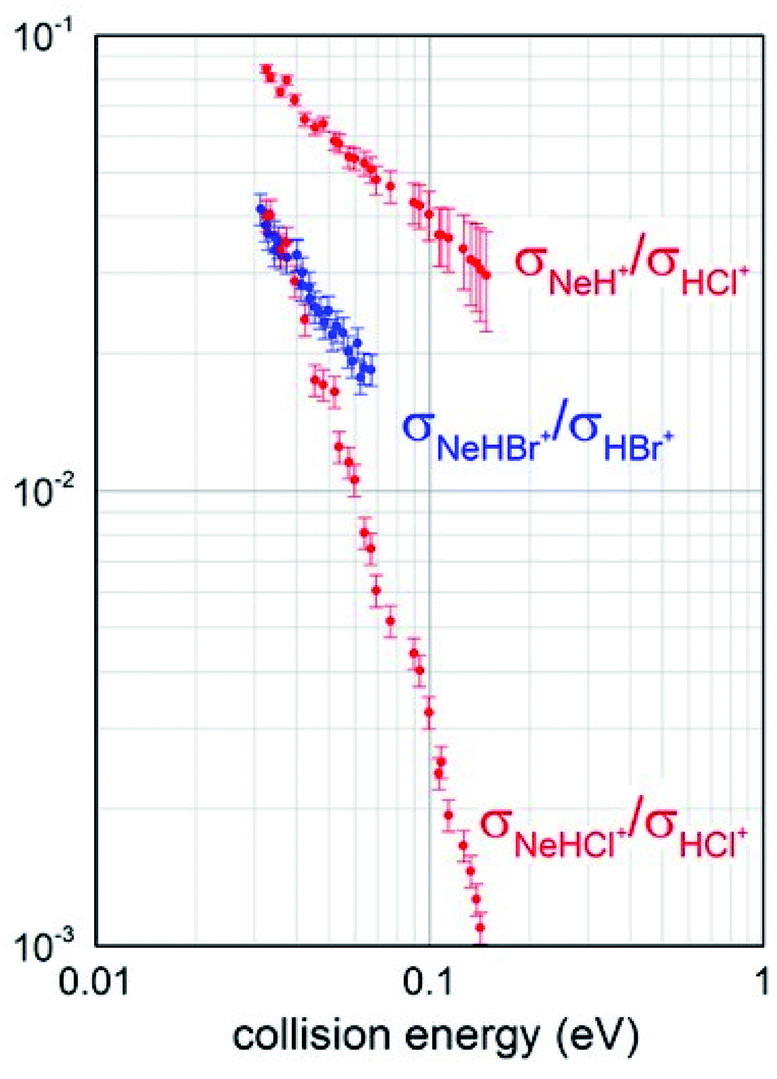 | ||
| Fig. 3 Branching ratio BRs measured for the different channels opened by both Ne* + HCl and Ne* + HBr reactions. | ||
It should be noted that for Ne*–HBr CHEMI a very small formation of NeH+ ion was detected: less than 1% at an average collision energy of 45 meV. For this reason, we have not been able to reliably perform mass spectrometric determinations as a function of collision energy and no branching ratio σNeH+/σHBr+ is available for a comparison with the data reported in Fig. 3. Despite this limitation, such data are useful to carry out an additional and internally consistent comparison between the behaviour of the two systems, and to emphasize the higher efficiency of HCl in leading a proton transfer reaction, with the formation of NeH+.
Obtained BRs also suggest that the relative probability of channels promoting the formation of associate NeHX+ ions from both reactions is similar in the thermal energy range and that of HCl, being measured in an extended collision energy interval because a more efficient total ionization, shows a faster decrease when increasing the collision energy.
The basic features of reagents and of precursor state
In CHEMI of interest for the present study, namely Ne* + HX (X = Cl, Br), it is expected that an initial step of reaction promoted by the well-known harpooning phenomenon, that involves a direct electron transfer between reagents, triggered at the crossing of potential energy surfaces and leading to the formation of the intermediate ion-pair (Ne+HX−), plays a minor role in comparison with CHEMI of the same molecules, promoted by metastable helium atoms. This because in the entrance channel the asymptotic Ne+(2PJ) + HX− ionic state lies at least 5.5 eV above the asymptotic Ne*(3PJ) + HX neutral state, reducing probability/efficiency of possible crossings. This comes from the HX electron affinity, which is estimated to be negative, and from the energy content and the Ne* reagent, that is much lower with respect to that of He*. Therefore, present CHEMI occur with a mechanism rather different from the one promoting harpooning processes.Recently, extending/generalizing some guidelines previously suggested,16 we demonstrated that Ne* + atom3 and Ne* + apolar diatomic molecule17,18 CHEMI occur through two different-complementary mechanisms, whose relative role is modulated by the collision energy. In particular, as stressed above, the direct mechanism, dominant at short separation distances (high collision energy), involves an effective electron transfer between reagents controlled by strong chemical forces. On the other hand, the indirect mechanism (low collision energy), stimulated by a critical balance between weak forces of chemical and physical nature, dominates at large separation distances. Therefore, following the previous general guidelines accounting for the specific nature of reagents and products,3,17,18 important indications on the stereodynamics of present reactions can be obtained.
Basic features of HX reagents and of the HX+ products (ionization potential, electron affinity, equilibrium distance, binding energy, electric dipole and quadrupole moment), relevant to rationalize some specific differences observed in CHEMI of HCl and HBr molecules, are summarized in Table 1.
| HCl(X1Σ+) | HBr(X1Σ+) | |
|---|---|---|
| μ/D | 1.103 | 0.827 |
| Q/D Å | 3.8 | 4.3 |
| I/eV | 12.751 | 11.672 |
| EA | <0 | <0 |
| D0/eV | 4.434 | 3.758 |
| Re/Å | 1.275 | 1.414 |
| Asymptotic correlation | H(2S1/2) + Cl(2P3/2) | H(2S1/2) + Br(2P3/2) |
| Energy level/eV | 0 (reference) | 0 (reference) |
| HCl+(X2Π3/2) | HBr+(X2Π3/2) | |
|---|---|---|
| D0/eV | 4.651 | 3.900 |
| Re/Å | 1.315 | 1.448 |
| Asymptotic correlation | H(2S1/2) + Cl+(3P2) | H(2S1/2) + Br+(3P2) |
| Energy level/eV | 12.751 | 11.672 |
| HCl+(A2Σ1/2) | HBr+(A2Σ1/2) | |
|---|---|---|
| D0/eV | 1.760 | 1.775 |
| Re/Å | 1.514 | 1.684 |
| Asymptotic correlation | H+ + Cl(2P3/2) | H(2S1/2) + Br+(1D2) |
| Energy level/eV | 16.272 | 15.296 |
The decreasing trends experimentally observed for σiontot(E) suggest that for Ne*–HX CHEMI reactions the precursor state of most relevant entrance channels correlating with Ne*(3PJ), must be stabilized by pronounced attractive interaction components. In the present cases, a strong attraction can arise from polarization of outer and floppy cloud of 3s external electron on Ne* that stimulates the interaction between the bare Ne+ ionic core with the permanent electric dipole and quadrupole moments of the HX molecule (see Table 1).
Stereodynamics and reaction mechanisms
The symmetry of the molecular orbitals, involved in electron removing and/or electron exchange, discussed in the next subsections, suggests that for both CHEMI, reaction channels leading to the formation of different states of the ionic products must be strongly stereo-selective. Indeed, they are stimulated within specific angular cones, each one exhibiting a well-defined spatial acceptance and space orientation. A representation of angular cones acceptance and orientation is schematized in Fig. 4–6.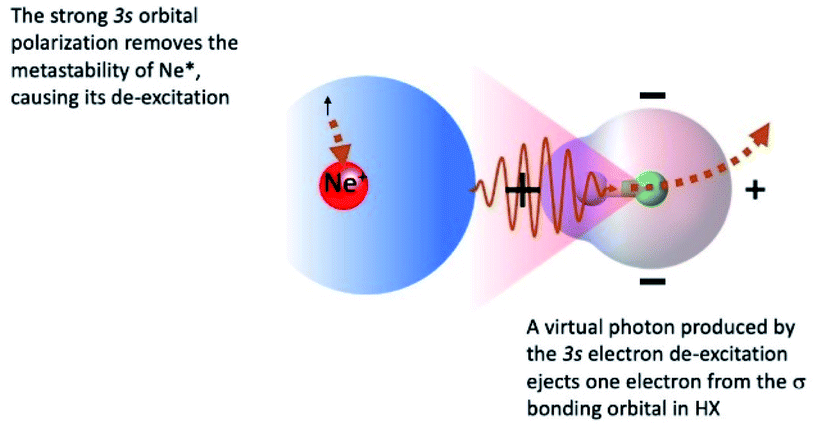 | ||
| Fig. 4 A scheme of atomic and molecular orbitals of reagents in a collinear configuration of the precursor state promoting the formation of HX+ ion in the first excited (A2Σ) electronic state. Most relevant electronic rearrangements are indicated in the figure, where also the shape of acceptance angular cone for the attach on the H side is depicted. As discussed in the text, this approach should stimulate essentially the indirect mechanism, while the role of the direct mechanism is hindered by the 3s floppy cloud polarization of Ne* reagent. | ||
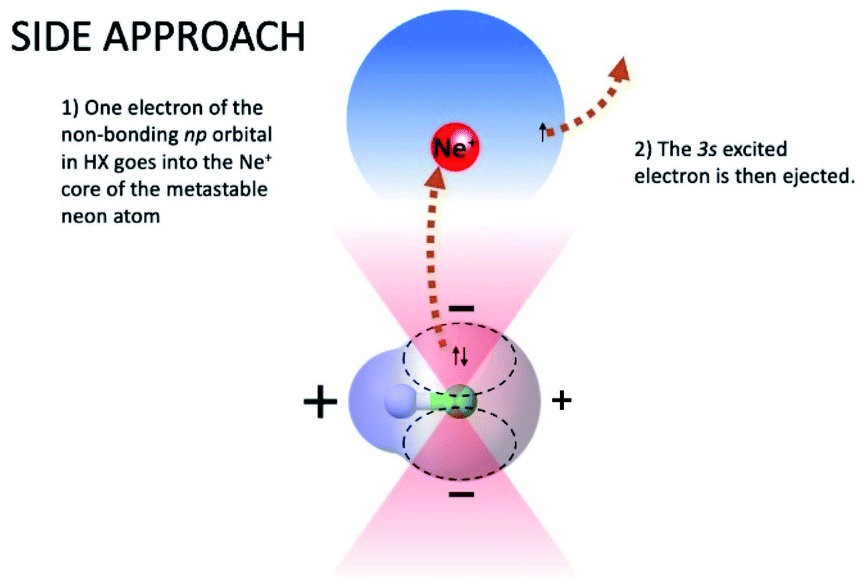 | ||
| Fig. 5 Schematization of atomic and molecular orbitals of reagents and perpendicular configuration of the precursor state promoting the formation of HX+ ion in the ground (X2Π) electronic state. Most relevant electronic rearrangements involved in the direct mechanism are indicated in the figure, where also the shape of acceptance angular cones, where reaction is effectively stimulated, is also depicted. | ||
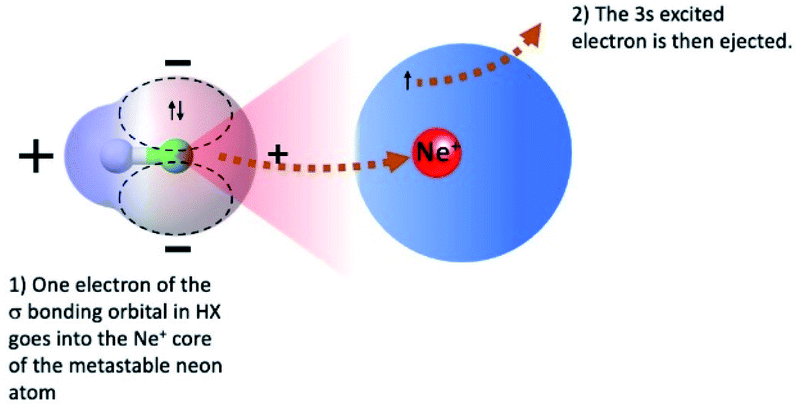 | ||
| Fig. 6 Schematization of atomic and molecular orbitals of reagents and collinear configuration of the precursor state promoting the formation of HX+ ion in the first excited (A2Σ) electronic state. Most relevant electronic rearrangements involved in the direct mechanism are indicated in the figure, where also the shape of acceptance angular cone for the attach on the X side is depicted. | ||
The adopted scheme is applicable to both HCl and HBr reactions. Preliminary, we can analyze the H-end collinear approach of Ne* atom to HX. A strong polarization of 3s floppy atomic cloud, promoted by the positively charged H atom (see Fig. 4) and accompanied by a mixing of atomic orbitals exhibiting different symmetry, can stimulate an indirect reaction mechanism, since the break of optical selection-rules validity promotes a virtual photon exchange between reagents.3,16
Consequently, CHEMI become typical photo-ionization processes. In addition, because of the 3s orbital polarization, any triggering of the direct mechanism is hindered by the reduced overlap integral between atomic orbital, of internal high electron affinity ionic core, and the molecular orbital from which the electron should be extracted.
On the contrary, the reactions leading to the formation of HX+ ions in the ground (X2Π) and in the first excited (A2Σ) electronic state are selectively triggered, by both the direct and indirect mechanism of the reaction, within different angular cones, consistent with those depicted in Fig. 5 and 6. In particular, as confirmed by the detailed experimental investigation of other systems,3,17,18 the indirect mechanism, controlled by the critical balance of weak interaction components of physical and chemical nature, dominates at large separation distances, while the direct mechanism stimulated by chemical forces, like the charge transfer, emerges at intermediate and short separation distances. Moreover, the formation of the ionic products in their ground state involves a precursor state having a perpendicular geometry, while the one promoting the formation of ionic products in their first excited electronic state preferentially assumes a collinear configuration. It is important to note that, as recently demonstrated for Ne* + N2 reactions,18 the perpendicular approach is the proper one giving associated NeN2+ ion formation, since, under such configuration, pre-dissociation effects due to the formation of vibration excited molecular ions are ineffective.
In addition, the removing of one electron from a non-bonding orbital, leading to the formation of HX+ in the ground electronic state (X2Π) (see Fig. 5), maintains the binding energy HX+ ion practically at the same value of neutral HX state. However, the removing of one electron from a bonding orbital, leading to the formation of HX+ in the first excited electronic state (A2Σ) (see Fig. 6–8), strongly reduces the binding energy value respect to that of neutral HX state and favors the dissociation of the parent ion HX+.
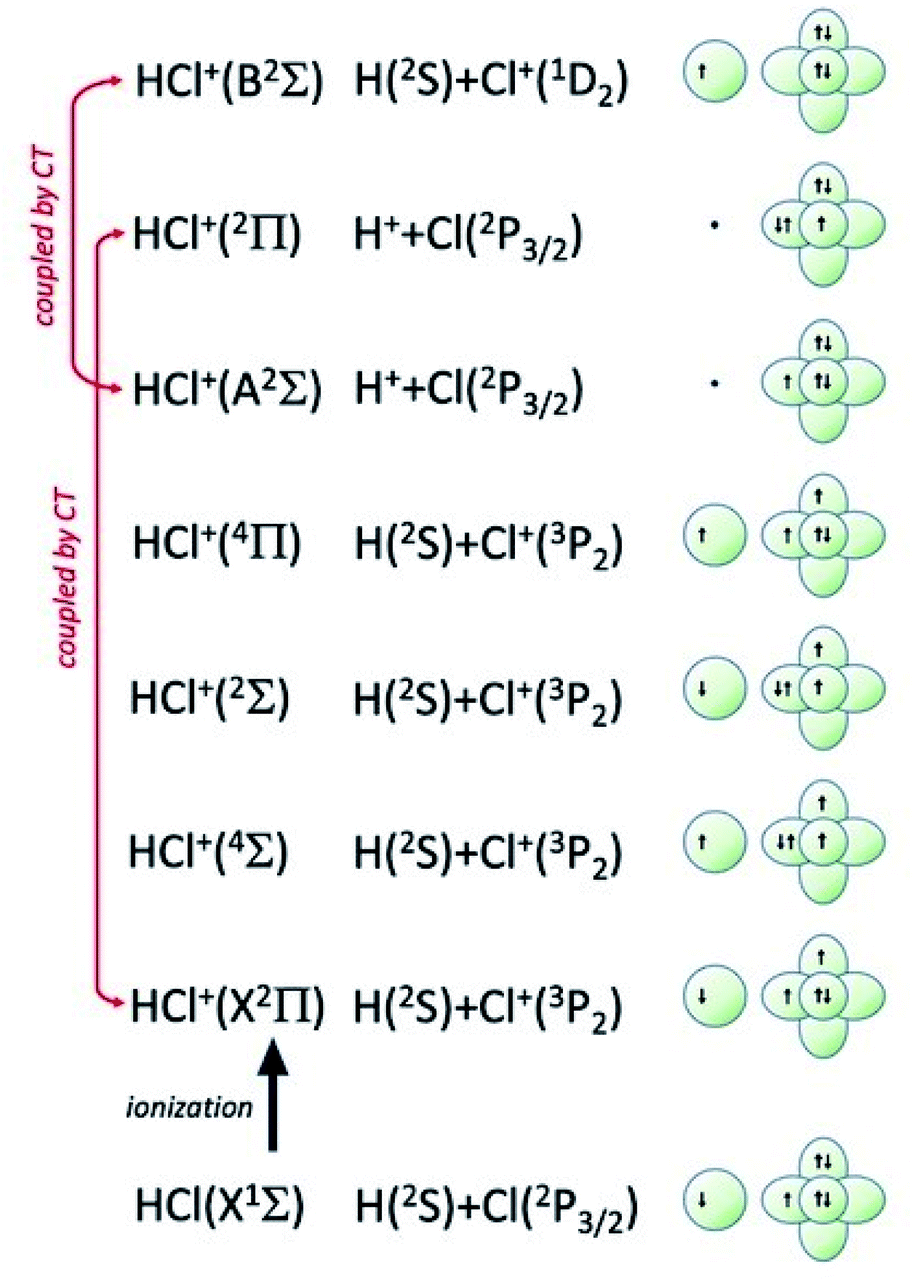 | ||
| Fig. 7 Symmetry of the ground state of HCl molecule and sequence of molecular states accessible to HCl+ molecular ion. The asymptotic correlation with the states of atomic partners involved in the molecular bond is also represented. The energies of most relevant atomic and molecular states are given in absolute scale in Fig. 9. | ||
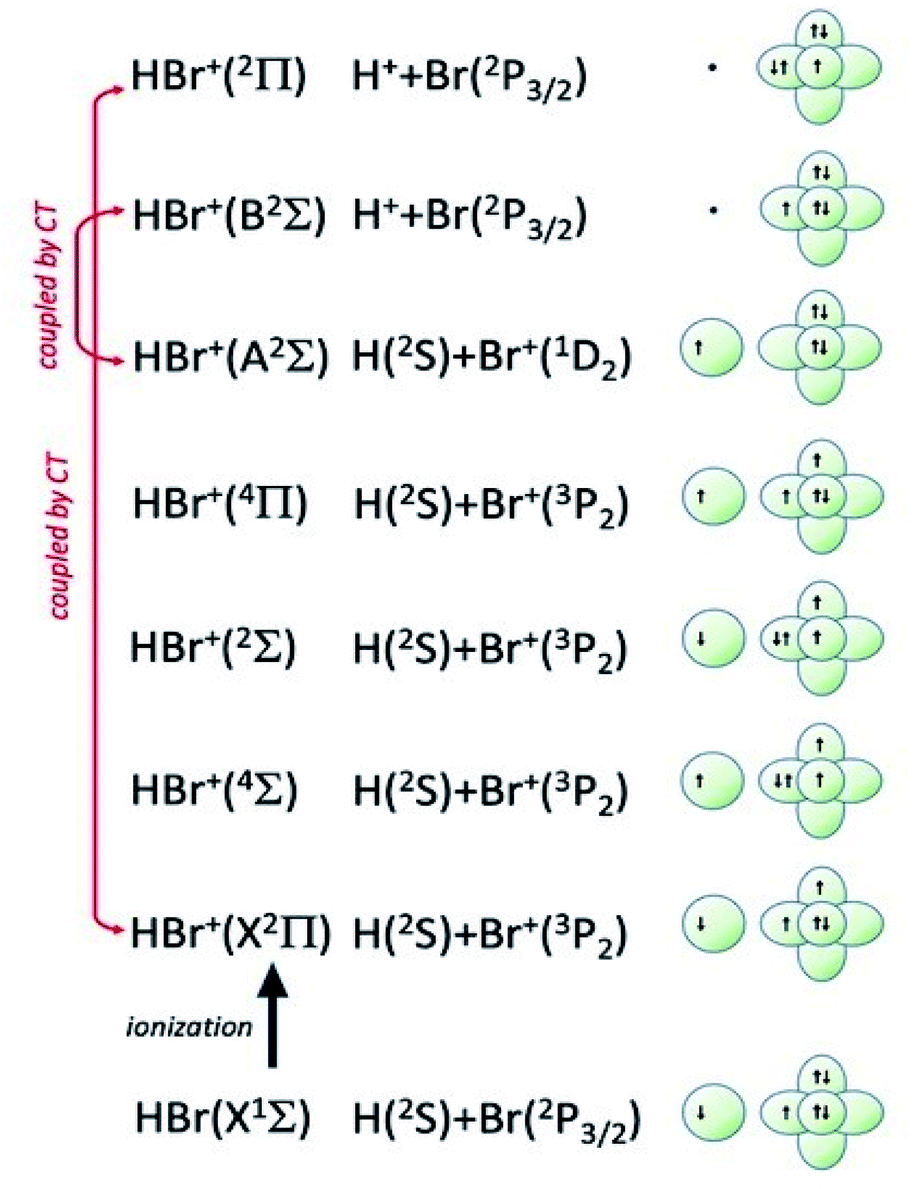 | ||
| Fig. 8 Symmetry of the ground state of HBr molecule and sequence of molecular states accessible to HBr+ molecular ion. The asymptotic correlation with the states of atomic partners involved in the molecular bond is also represented. The energies of most relevant atomic and molecular states are given in absolute scale in Fig. 9. | ||
The lower efficient CHEMI with HBr, compared to that of HCl, can be therefore explained by the combination of three different factors:
(a) The HBr molecule is less polar with respect to HCl (see dipole moments in Table 1) and, consequently, polarization effects on Ne* that trigger the indirect mechanism of the reaction, are attenuated.
(b) The molecular orbitals of HBr are more diffuse respect to those of HCl and this character tends to reduce the overlap integral between orbital exchanging the electron.
(c) Because of the molecule polar character change, the role of the so called “σ hole”, localized along HX bond length on the external X part, tends to increase its effect in the case of X = Br, reducing the reaction efficiency along the corresponding angular cone.
Symmetry and energy of molecular orbitals involved
Additional selectivity of the active reaction channels is suggested by basic differences in molecular orbitals involved. It is of interest to note for both CHEMI that the ground state of the ionic HX+ product is stabilized by the configuration interaction with an excited state of symmetry 2Π, asymptotically correlating with H+ + X(2P3/2).Some important details on the correlations between molecular and asymptotic states are reported in Fig. 7 and 8, while the diagram of the main energetic levels is given in Fig. 9.
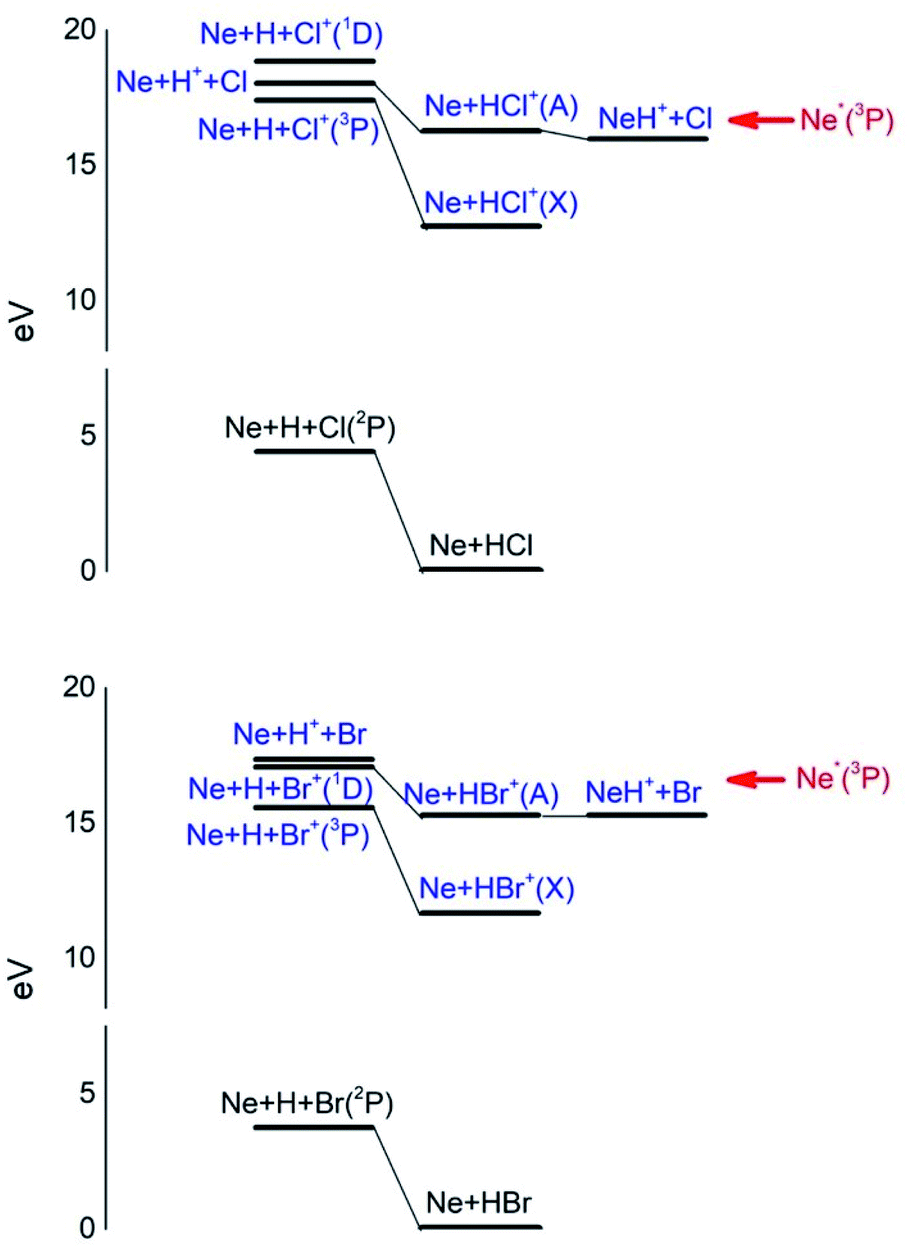 | ||
| Fig. 9 Energy levels of neutrals and ionic species involved in CHEMI of HCl and HBr with metastable Ne*(3P) atoms. See also Fig. 7 and 8 for the symmetry of the states. | ||
Fig. 7 also suggests that for CHEMI of HCl, the excited state HCl+(A2Σ) asymptotically correlates with H+ + Cl (2P3/2) (18.032 eV) and is stabilized by the configuration interaction with the state of B2Σ symmetry, asymptotically correlating with H(2S) + Cl+(1D2) (18.847 eV) (see cartoon in Fig. 1 and 7 of ref. 12).
The nascent proton existing in HCl+(A2Σ) structure favors the possibility of a proton transfer with the appreciable formation of NeH+, stimulated also by the approach along the cones parallel to the HX bond, as confirmed by the experimental findings of Perugia laboratory (see Fig. 3). It has been also found11 that this reaction channel is exothermic of about 0.5 eV and exhibits a small barrier in the exit channel. Moreover, the short average rotational period of HCl+ (of the magnitude order of 6 × 10−13 s at room temperature), tends to favor, especially under long collision time (low collision energy conditions), an efficient synchronization between the collision time and the rearrangement time requested by the system to assume the most proper configurations giving proton transfer. In contrast, in CHEMI of HBr, the excited state HBr+(A2Σ) asymptotically correlates with H(2S) + Br+(1D2) (17.071 eV) and is stabilized by the configuration interaction with the state of the same symmetry (B2Σ), asymptotically correlating with H+ + Br(2P3/2) (17.356 eV) (see cartoon in Fig. 1 and 8 of ref. 13). Therefore, the presence of a nascent Br+ ion into the HBr+(A2Σ) structure hinders the possibility of proton transfer.
Moreover, the change in the sequence of molecular states of HBr+ respect to those in HCl+ has as consequence that although NeH+ + Br rearrangement state is confined at about the same thermodynamic level of Ne + HBr+(A2Σ) ionic product, its formation is hindered by an energy barrier, expected to be higher than that controlling the rearrangement leading to NeH+ + Cl, and by the higher rearrangement time requested. This indication is supported by the lack of appreciable NeH+ detection in the reaction with HBr (see Fig. 3). Finally, Fig. 9 compares the energy levels of NeH+ + X with those of Ne + HX+ (A2Σ) ionic products, showing the most favorable thermodynamic evolution of proton transfer for HCl CHEMI respect to those of HBr.
Conclusions
This paper reports new experimental results for Ne* + HBr CHEMI processes. They are compared with results previously obtained, under the same conditions, for Ne* + HCl reactions. This comparison has been exploited to emphasize similarities and differences in the stereodynamics of both processes. The present analysis, that considers the role of the fundamental interaction components combined with energy and symmetry of HOMO and LUMO orbitals of HX molecules and HX+ molecular ions, suggests that the stereodynamics of opened reaction channels is confined along angular cones showing different orientation and acceptance.Moreover, proton transfer rearrangements, occurring in exit channels of reactions, are more efficient for Ne* + HCl respect to Ne* + HBr CHEMI. The present study suggests that the different behaviour of the two cases can be associated to the different charge distribution on HX+ ionic products, to the various energy barriers and to the diverse time required for the rearrangement.
The reactions discussed in this study have been experimentally investigated using beams of Ne*(3PJ) atoms with a statistical population of J = 2, 0 fine levels. Generalizing the detailed behaviour recently emphasized for Ne*(3PJ) + N2 CHEMI,18 it can be expected that under the collision energy conditions here adopted, essentially confined in the thermal range, the atoms in the two different spin orbit levels J behave similarly because a compensation between direct and indirect mechanism features. It has been demonstrated18 that, under hyper-thermal conditions, J = 0 atoms become more reactive since promoting a more efficient direct mechanism.
Finally, present results, combined with those recently obtained in experiments performed under state resolved conditions on other systems,20 provide additional-basic details on the stereodynamics of CHEMI involving molecules.
Author contributions
Conceptualization, S. F., F. V. and F. P.; methodology, validation and formal analysis, S. F., F. V., B. G. B. and F. P.; experimental investigation, S. F., B. G. B., M. P., G. G., and S. S.; writing – original draft preparation, S. F., F. V., B. G. B. and F. P.; writing-review and editing, S. F., F. V., B. G. B., M. R., G. G., S. S., and F. P.; all authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support from Italian MIUR and University of Perugia (Italy) is acknowledged within the program “Dipartimenti di Eccellenza 2018–2022”.References
- H. F. Calcote, Electrical properties of flames, Symp. Combust. Flame Explos. Phenom., 1948, 3(1), 245–253 CrossRef CAS; T. M. Sugden, Annu. Rev. Phys. Chem., 1962, 13(1), 369–390 CrossRef.
- P. E. Siska, Rev. Mod. Phys., 1993, 65, 337–412 CrossRef CAS; B. Brunetti and F. Vecchiocattivi, Current Topic on Ion Chemistry and Physics, ed. C. Y. Ng, T. Baer and I. Powis, John Wiley &Sons Ltd, New York, 1993, pp. 359–445 Search PubMed.
- S. Falcinelli, F. Vecchiocattivi and F. Pirani, Commun. Chem., 2020, 3, 64 CrossRef CAS; S. Falcinelli, J. M. Farrar, F. Vecchiocattivi and F. Pirani, Acc. Chem. Res., 2020, 53(10), 2248–2260 CrossRef PubMed.
- T. M. Sugden, Annu. Rev. Phys. Chem., 1962, 13(1), 369–390 CrossRef CAS.
- A. A. Mihajlov, L. M. Ignjatović, V. A. Srećković and M. S. Dimitrijević, Astrophys. J., Suppl. Ser., 2011, 193(2), 1–7 Search PubMed.
- S. Falcinelli, F. Pirani and F. Vecchiocattivi, Atmosphere, 2015, 6, 299–317 CrossRef.
- A. B. Henson, S. Gersten, Y. Shagam, J. Narevicius and E. Narevicius, Science, 2012, 338, 234–238 CrossRef CAS PubMed; S. D. S. Gordon, J. Zou, S. Tanteri, J. Jankunas and A. Osterwalder, Phys. Rev. Lett, 2017, 119, 053001 CrossRef PubMed; S. D. S. Gordon, J. J. Omiste, J. Zou, S. Tanteri, P. Brumer and A. Osterwalder, Nat. Chem., 2018, 10, 1190–1195 CrossRef PubMed; P. Paliwal, N. Deb, D. M. Reich, A. van der Avoid, C. P. Koch and E. Narevicius, Nat. Chem., 2021, 13, 94–98 CrossRef PubMed.
- M. Bettendorff, S. D. Peyerimhoff and R. J. Buenker, Chem. Phys., 1982, 66, 261 CrossRef CAS; H.-J. Werner, P. Rosmus, W. Schätzl and W. Meyer, J. Chem. Phys, 1984, 80, 831 CrossRef; D. A. Chapman, K. Balasubramanian and S. H. Sin, Phys. Rev.A, 1988, 38, 6098 CrossRef PubMed; P. Candori, S. Falcinelli, F. Pirani, F. Tarantelli and F. Vecchiocattivi, Chem. Phys. Lett., 2007, 436, 322–326 CrossRef.
- H. L. Snyder, B. T. Smith and R. M. Martin, Chem. Phys. Lett., 1983, 94, 90 CrossRef CAS; M. S. de Vries, G. W. Tyndall and R. M. Martin, J. Chem. Phys, 1984, 80, 1366 CrossRef; M. Tsuji, J. P. Maier, H. Obase and Y. Nishimura, Chem. Phys., 1986, 110, 17–26 CrossRef; H. Obase, M. Tsuji and Y. Nishimura, J. Chem. Phys., 1987, 87, 2695–2699 CrossRef; W. Simon, A. J. Yencha, M.-W. Ruf and H. Hotop, Z. Phys. D, 1988, 8, 71 CrossRef; I. Tokue, H. Tanaka and K. Yamasaki, J. Phys. Chem. A, 2002, 106, 6068–6074 CrossRef.
- A. Aguilar Navarro, B. Brunetti, S. Falcinelli, M. Gonzalez and F. Vecchiocattivi, J. Chem. Phys., 1992, 96(1), 433–439 CrossRef.
- B. Brunetti, R. Cambi, S. Falcinelli, J. M. Farrar and F. Vecchiocattivi, J. Phys. Chem., 1993, 97(46), 11877–11882 CrossRef CAS.
- A. J. Yencha, J. Ganz, M.-W. Ruf and H. Hotop, Z. Phys. D: At., Mol. Clusters, 1989, 14, 57–76 CrossRef CAS.
- A. J. Yencha, M.-W. Ruf and H. Hotop, Z. Phys. D: At., Mol. Clusters, 1991, 21, 113–130 CrossRef CAS.
- K. Imura, N. Kishimoto and K. Ohno, J. Phys. Chem. A, 2002, 106, 3759–3765 CrossRef CAS.
- F. Biondini, B. G. Brunetti, P. Candori, F. De Angelis, S. Falcinelli, F. Tarantelli, F. Pirani and F. Vecchiocattivi, J. Chem. Phys., 2005, 122(16), 164308 CrossRef CAS; S. Falcinelli, M. Rosi, S. Cavalli, F. Pirani and F. Vecchiocattivi, Chem. Eur. J., 2016, 22(35), 12518–12526 CrossRef.
- W. H. Miller and J. Chem, Phys, 1970, 52, 3563–3572 Search PubMed; W. H. Miller and H. Morgner, J. Chem. Phys., 1977, 67, 4923–4930 CrossRef CAS; R. W. Gregor and P. E. Siska, J. Chem. Phys., 1981, 74, 1078–1092 CrossRef.
- S. Falcinelli, F. Vecchiocattivi, J. M. Farrar, B. G. Brunetti, S. Cavalli and F. Pirani, Chem. Phys. Lett., 2021, 778, 138813 CrossRef CAS.
- S. Falcinelli, F. Vecchiocattivi and F. Pirani, Sci. Rep., 2021, 11, 19105 CrossRef CAS.
- NIST, Computational Chemistry Comparison and Benchmark DataBase, 2021. A. A. Radzig and B. M. Smirnov, Reference Data on Atoms, Molecules and Ions, Springer-Verlag, Berlin Heidelberg, 1985 Search PubMed.
- L. Ploenes, P. Straňák, H. Gao, J. Küpper and S. Willitsch, Mol. Phys., 2021, 119, e1965234 CrossRef.
| This journal is © The Royal Society of Chemistry 2022 |