
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBase-selective access to highly functionalized heterocycles from multicomponent Ugi adducts†
Beatriz
González-Saiz
a,
Pablo
Pertejo
a,
Pablo
Peña-Calleja
a,
Marcin
Mielczarek
a,
Tomás
Hermosilla
a,
Israel
Carreira-Barral
 a,
Olivia
de Miguel
a,
Francisco
Rodríguez-Vidal
b,
Roberto
Quesada
a and
María
García-Valverde
*a
a,
Olivia
de Miguel
a,
Francisco
Rodríguez-Vidal
b,
Roberto
Quesada
a and
María
García-Valverde
*a
aUniversidad de Burgos, Facultad de Ciencias, Burgos, Castilla y León, Spain
bUniversidad de Burgos, Higher Polytechnic School, Av. Cantabria s/n, Burgos, Spain
First published on 28th September 2022
Abstract
Selection of the appropriate base in Ugi/post-condensation sequences allows the selective syntheses of different ring-size functionalized lactams. The developed methodology uses inexpensive bases under mild and non-sensitive conditions leading to the facile generation of molecular diversity from simple synthons.
1. Introduction
The increasing demand for the development of new and efficient syntheses of N-heterocycles is explained mainly by their importance in the pharmaceutical and fine chemicals industries.1 The synthesis of highly functionalized heterocycles remains a major challenge and continues to drive an intense research effort.2 Different selective methodologies have been described, although many of them present drawbacks such as the need for expensive reagents, specific substrates, harsh or sensitive conditions or long synthetic sequences.3 Furthermore, some substitution patterns have not been described yet despite their promising applications as bioactive compounds or as intermediates in the synthesis of other compounds, mainly due to limitations of synthetic methodologies.4The ring expansion of β-lactams is one of the various approaches to the synthesis of γ-lactam skeletons,5 favored due to the β-lactam ring strain.6 Single-step synthetic methodologies illustrating this reaction can be classified into three main categories: (a) intramolecular transamidation from the proper amino-substituted azetidinone, as 4-(1-aminoalkyl)7 or 3-(2-aminoalkyl)-2-azetidinone derivatives,8 which takes place through the N1–C2 cleavage and is the most common one, (b) the rearrangement favored by an electron-deficient position through the C3–C4 cleavage, as the expansion of 4-(1-haloalkyl)-2-azetidinones to γ-lactams via N-acyliminium intermediates,9 and (c) the rearrangement resulting from an anionic position generated by strong bases, as the benzylic anions generated by LDA in N-benzyl-4-arylazetidinones which rearrange to a γ-lactam through a proposed imine anion intermediate originated in the C4–N1 cleavage.10 The examples within this latter methodology are very scarce,11 probably due to the specificity of the substrates needed as well as the reaction conditions (LDA or n-BuLi in anhydrous THF) employed to achieve the expansion (Scheme 1a).
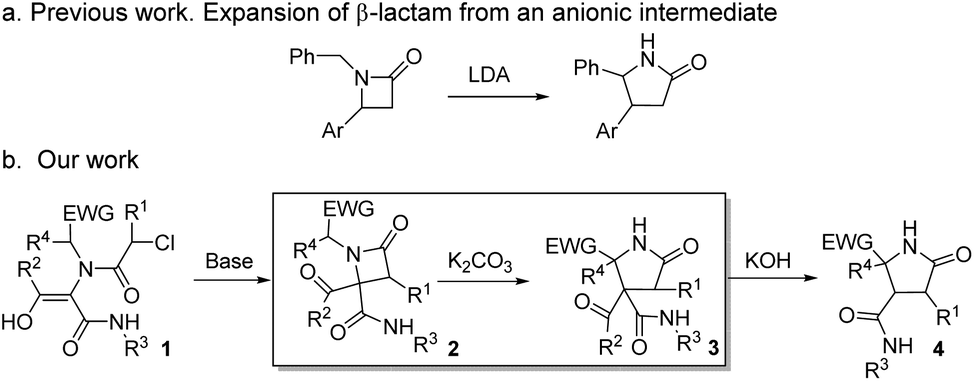 | ||
| Scheme 1 (a) Previous work on ring expansion of β-lactams through an anionic rearrangement and (b) summary of N-heterocycles synthesized from common Ugi adducts. | ||
We envisaged the possibility of expanding the scope of this reaction to other substrates, and we planned to use Ugi/post-condensation sequences, seizing their huge potential to yield structurally complex molecules in expedited syntheses from a reduced number of steps.12 Surprisingly, in addition to the applicability of this strategy to a broad spectrum of substrates, we have found that a rational design of β-lactams, selectively synthesized from Ugi adducts, enables the expansion to highly functionalized γ-lactams using weak bases, under mild and non-moisture-sensitive conditions (box in Scheme 1b). Thus, we have found that variation of the nature of the base employed leads to the selective synthesis of differently functionalized N-heterocyclic systems, some of them with unprecedented functionalization patterns, from common multicomponent Ugi adducts (Scheme 1b).
2. Results and discussion
2.1. Synthesis of azetidinones through Ugi/cyclization sequences
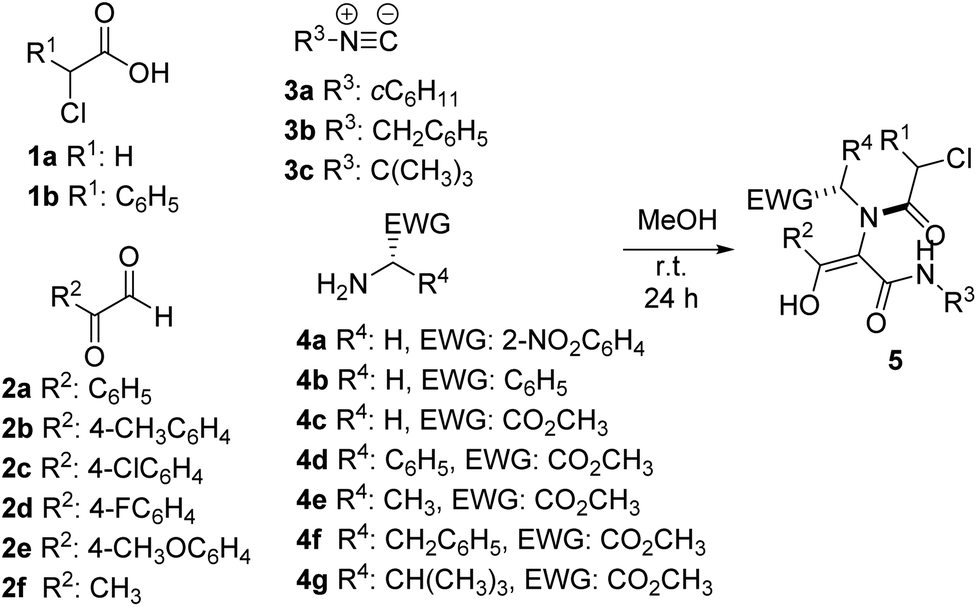
In this way, we prepared different Ugi adducts 5 using 2-nitrobenzylamine 4a, benzylamine 4b and α-amino acid methyl esters 4c–g as amines, along with chloroacetic acids 1a–b, glyoxals 2a–f and different isocyanides 3a–c (Table 1). As we expected, in all cases the enol tautomer was the only one observed, but the spontaneous cyclization never took place.14
|
|
|||||
|---|---|---|---|---|---|
| Entry | 1 (R1) | 2 (R2) | 3 (R3) | 4 (R4, EWG) | 5 (%) |
| a Yield after purification. | |||||
| 1 | 1a (H) | 2a (C6H5) | 3a (cC6H11) | 4a (H, 2-NO2C6H4) | 5a (89) |
| 2 | 1a (H) | 2a (C6H5) | 3b (CH2C6H5) | 4a (H, 2-NO2C6H4) | 5b (67) |
| 3 | 1a (H) | 2a (C6H5) | 3c (C(CH3)3) | 4a (H, 2-NO2C6H4) | 5c (59) |
| 4 | 1a (H) | 2b (4-CH3C6H4) | 3a (cC6H11) | 4a (H, 2-NO2C6H4) | 5d (86) |
| 5 | 1a (H) | 2c (4-ClC6H4) | 3a (cC6H11) | 4a (H, 2-NO2C6H4) | 5e (85) |
| 6 | 1a (H) | 2d (4-FC6H4) | 3a (cC6H11) | 4a (H, 2-NO2C6H4) | 5f (86) |
| 7 | 1a (H) | 2e (4-CH3OC6H4) | 3a (cC6H11) | 4a (H, 2-NO2C6H4) | 5g (54) |
| 8 | 1a (H) | 2f (CH3) | 3a (cC6H11) | 4a (H, 2-NO2C6H4) | 5h (39) |
| 9 | 1b (C6H5) | 2a (C6H5) | 3a (cC6H11) | 4a (H, 2-NO2C6H4) | 5i (93) |
| 10 | 1b (C6H5) | 2d (4-FC6H4) | 3a (cC6H11) | 4a (H, 2-NO2C6H4) | 5j (86) |
| 11 | 1a (H) | 2a (C6H5) | 3a (cC6H11) | 4b (H, C6H5) | 5k (78) |
| 12 | 1a (H) | 2a (C6H5) | 3a (cC6H11) | 4c (H, CO2CH3) | 5l (52) |
| 13 | 1a (H) | 2a (C6H5) | 3a (cC6H11) | 4d (C6H5, CO2CH3) | 5m (70) |
| 14 | 1a (H) | 2a (C6H5) | 3a (cC6H11) | 4e (CH3, CO2CH3) | 5n (65) |
| 15 | 1a (H) | 2a (C6H5) | 3a (cC6H11) | 4f (CH2C6H5, CO2CH3) | 5o (55) |
| 16 | 1a (H) | 2a (C6H5) | 3a (cC6H11) | 4g (CH(CH3)2, CO2CH3) | 5p (78) |
| 17 | 1b (C6H5) | 2a (C6H5) | 3a (cC6H11) | 4c (H, CO2CH3) | 5q (83) |
| 18 | 1b (C6H5) | 2a (C6H5) | 3a (cC6H11) | 4d (C6H5, CO2CH3) | 5r (76) |
| 19 | 1b (C6H5) | 2a (C6H5) | 3a (cC6H11) | 4e (CH3, CO2CH3) | 5s (81) |
| 20 | 1b (C6H5) | 2a (C6H5) | 3a (cC6H11) | 4f (CH2C6H5, CO2CH3) | 5t (82) |
In this way, different bases were employed in order to achieve the selective cyclization of Ugi adduct 5a to the corresponding azetidinone 6a. Initially, we tried triethylamine under different conditions. It turned out that the use of three equivalents of this base in ethanol and ultrasonication for 1 hour afforded azetidinone 6a with a moderate yield (entry 1, Table 2). Then, we tried potassium and sodium carbonates but the obtained yield was low, because the O-alkylation product was obtained along with the desired C-alkylation compound. Despite this result we observed the influence of cation size, as the smaller sodium cation favors C-alkylation15 (entry 2 vs. 4, Table 2). However, when cyclization with lithium carbonate was attempted, the Ugi adduct was recovered (entry 5, Table 2), probably due to the low solubility of this salt in organic solvents.
|
|
|||||
|---|---|---|---|---|---|
| Entry | 5 (R1, R2, R3, X) | Base | Equiv. | 6 (%) | d.r.b |
| a Yield after purification. b Diastereomeric ratio determined by 1H NMR. c In ethanol at room temperature and ultrasonication, 2 h. d In acetone at reflux, 12 h. e In acetonitrile at reflux, 12 h. f O-Alkylation by-product. g Mixture of O-alkylation and a new compound identified as γ-lactam 7a. h The Ugi adduct was recovered. i Relative configuration (3R*,4R*)/(3R*,4S*). j Diastereoisomers were not separated, characterized as a mixture. | |||||
| 1 | 5a (H, C6H5, cC6H11, NO2) | NEt3c | 3 | 6a (58) | — |
| 2 | 5a (H, C6H5, cC6H11, NO2) | K2CO3d | 1.1 | 6a (14)f | — |
| 3 | 5a (H, C6H5, cC6H11, NO2) | K2CO3d | 2 | 6a (—)g | — |
| 4 | 5a (H, C6H5, cC6H11, NO2) | Na2CO3d | 1.1 | 6a (40)f | — |
| 5 | 5a (H, C6H5, cC6H11, NO2) | Li2CO3d | 1.1 | 6a (—)h | — |
| 6 | 5a (H, C6H5, cC6H11, NO2) | Cs2CO3/LiCle | 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 :10 |
6a (65)f | — |
| 7 | 5a (H, C6H5, cC6H11, NO2) | Cs2CO3/LiIe | 1:10 |
6a (92) | — |
| 8 | 5a (H, C6H5, cC6H11, NO2) | Cs2CO3/LiIe | 1:2 |
6a (88)f | — |
| 9 | 5b (H, C6H5, CH2C6H5, NO2) | Cs2CO3/LiIe | 1:2 |
6b (79) | — |
| 10 | 5c (H, C6H5, C(CH3)3, NO2) | Cs2CO3/LiIe | 1:2 |
6c (67) | — |
| 11 | 5d (H, 4-CH3C6H4, cC6H11, NO2) | Cs2CO3/LiIe | 1:2 |
6d (73) | — |
| 12 | 5e (H, 4-ClC6H4, cC6H11, NO2) | Cs2CO3/LiIe | 1:2 |
6e (72) | — |
| 13 | 5f (H, 4-FC6H4, cC6H11, NO2) | Cs2CO3/LiIe | 1:2 |
6f (68) | — |
| 14 | 5g (H, 4-CH3OC6H4, cC6H11, NO2) | Cs2CO3/LiIe | 1:2 |
6g (76) | — |
| 15 | 5h (H, CH3, cC6H11, NO2)b | Cs2CO3/LiIe | 1:2 |
6h (90) | — |
| 16 | 5i (C6H5, C6H5, cC6H11, NO2) | Cs2CO3/LiIe | 1:2 |
6i (84) | 24:76i,j |
| 17 | 5i (C6H5, C6H5, cC6H11, NO2) | NEt3c | 3 | 6i (93) | 87:13i,j |
| 18 | 5j (C6H5, 4-FC6H4, cC6H11, NO2) | NEt3c | 3 | 6j (86) | 86:14i,j |
| 19 | 5k (H, C6H5, cC6H11, H) | Cs2CO3/LiIe | 1:2 |
6k (87) | — |
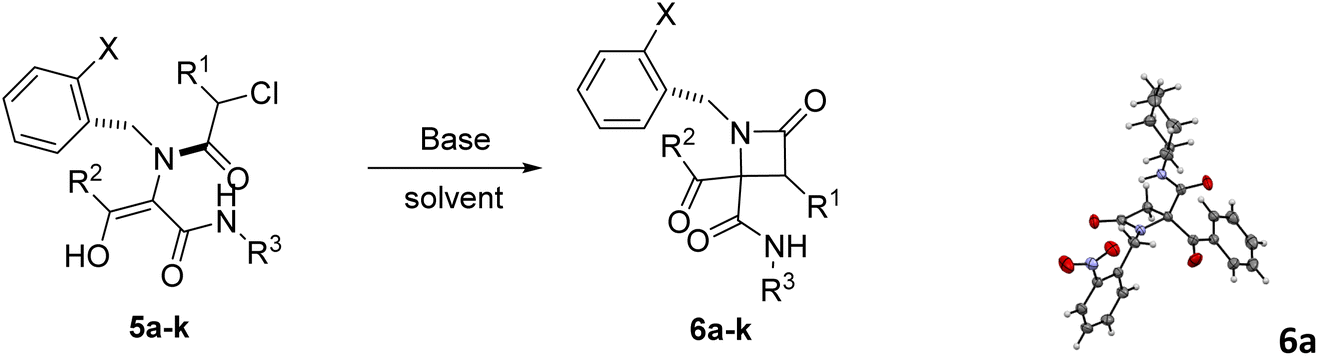
To overcome this limitation, we decided to work with the more soluble cesium carbonate in the presence of an excess of lithium halides to exchange the counter cation. The use of lithium chloride (10 equiv.) improved the C-alkylation yield although the O-alkylation product was still observed (entry 6, Table 2) but, fortunately, the use of lithium iodide yielded β-lactam 6a as a single compound (entry 7, Table 2); furthermore, the amount of this salt may be reduced to 2 equivalents without a significant decrease in chemical yield (entry 8, Table 2), except for the phenylglycine derivative 6m (entries 2 vs. 4, Table 3). These findings could be explained not only by the smaller counter cation, lithium, which binds tightly to oxygen, but also by the Finkelstein transhalogenation reaction.16 Gratefully, this cyclization strategy proved to be general. Thus, simple refluxing of solutions of Ugi adducts 5 with a combination of cesium carbonate and lithium iodide (1.1:2 equiv.) in acetonitrile for 12 hours afforded in almost all cases the corresponding 2-azetidinones 6. However, triethylamine (3 equiv.) in ethanol and ultrasonication for 2 hours proved to be most often the best choice for the cyclization of 2-chloro-2-phenylacetic derivatives (entries 16 vs. 17, Table 2) as a more efficient strategy. Therefore, this last methodology was chosen as the cyclization strategy for the synthesis of 6i–j and 6q–t. In this way, we achieved chemoselective C-alkylation in the cyclization step from Ugi adducts 5, crucial for our purposes in the subsequent synthesis of γ-lactams.
|
|
|||||
|---|---|---|---|---|---|
| Entry | 5 (R1, R2, R3, R4) | Base | Equiv. | 6 (%) | d.r.b |
| a Yield after purification. b Diastereomeric ratio determined by 1H NMR. c In acetonitrile at reflux, 12 h. d In ethanol at room temperature and ultrasonication, 2 h. e In acetone at reflux, 12 h. f Complex reaction mixture. g Mixture of O-alkylation and a new compound identified as a fused pyrrolidinone-imide 8 obtained as the major product. h Mixture of O-alkylation and a new compound identified as γ-lactam 7a. i The Ugi adduct was recovered. j Relative configuration (3R*,4R*)/(3R*,4S*). k Both diastereoisomers were separated and fully characterized. l Only the major diastereoisomer was isolated and fully characterized (n.d.: not detected). | |||||
| 1 | 5l (H, C6H5, cC6H11, H) | Cs2CO3/LiIc | 1:2 |
6l (78) | — |
| 2 | 5m (H, C6H5, cC6H11, C6H5) | Cs2CO3/LiIc | 1:2 |
6m (—)f | — |
| 3 | 5m (H, C6H5, cC6H11, C6H5) | NEt3d | 3 | 6m (—)f | — |
| 4 | 5m (H, C6H5, cC6H11, C6H5) | Cs2CO3/LiIc | 1:10 |
6m (81) | — |
| 5 | 5m (H, C6H5, cC6H11, C6H5) | K2CO3e | 1.1 | 6m (—)g | — |
| 6 | 5n (H, C6H5, cC6H11, CH3) | Cs2CO3/LiIc | 1:2 |
6n (90) | >98:2 |
| 7 | 5n (H, C6H5, cC6H11, CH3) | NEt3d | 3 | 6n (—)h | — |
| 8 | 5o (H, C6H5, cC6H11, CH2C6H5) | Cs2CO3/LiIc | 1:2 |
6o (81) | >98:2 |
| 9 | 5o (H, C6H5, cC6H11, CH2C6H5) | NEt3d | 3 | 6o (—)i | — |
| 10 | 5p (H, C6H5, cC6H11, CH(CH3)2) | Cs2CO3/LiIc | 1:2 |
6p (73) | >98:2 |
| 11 | 5q (C6H5, C6H5, cC6H11, H) | Cs2CO3/LiIc | 1:2 |
6q (71) | 55:45j,k |
| 12 | 5q (C6H5, C6H5, cC6H11, H) | NEt3d | 3 | 6q (77) | 54:46j,k |
| 13 | 5r (C6H5, C6H5, cC6H11, C6H5) | NEt3d | 3 | 6r (45) | 84:16:n.d.:n.d.l |
| 14 | 5r (C6H5, C6H5, cC6H11, C6H5) | K2CO3e | 1.1 | 6r (—)g | — |
| 15 | 5s (C6H5, C6H5, cC6H11, CH3) | Cs2CO3/LiIc | 1:2 |
6s (90) | 53:24:18:11l |
| 16 | 5s (C6H5, C6H5, cC6H11, CH3) | NEt3d | 3 | 6s (93) | 30:26:26:18l |
| 17 | 5t (C6H5, C6H5, cC6H11, CH2C6H5) | NEt3d | 3 | 6t (75) | 58:35:7:n.d.l |
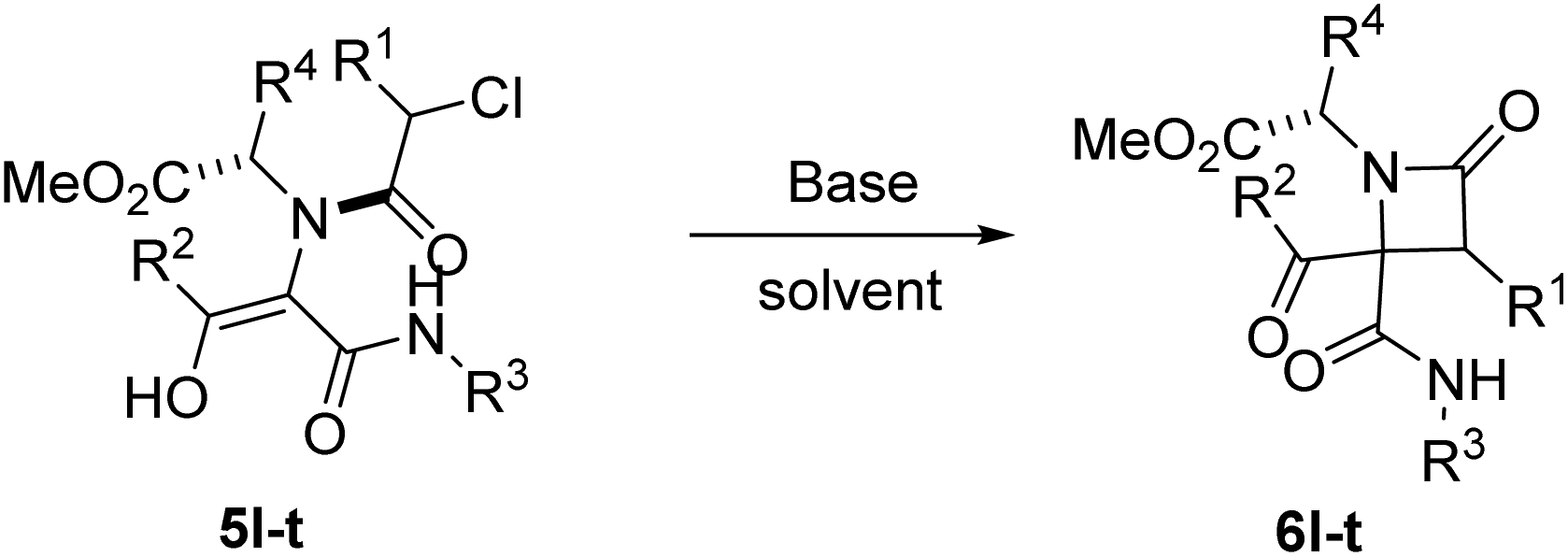
The stereochemical outcome in these reactions was also quite remarkable as the diastereoselectivity observed was fairly good when only one reactant incorporated a stereogenic center, but not when 2-chloro-2-phenylacetic acid (used as a racemate) was combined with α-aminoesters (entries 11–17, Table 3). In addition, the stereochemical results for the 2-chloro-2-phenylacetic acid derivatives were strongly dependent on the cyclization methodology (entry 16 vs. 17, Table 2). Nevertheless, given that the relative configuration of these lactams was not important in the next step, as it will be shown below, the isolation of diastereomers of these β-lactams was not necessary.
2.2. Synthesis of γ-lactams through expansion reactions
During the optimization studies for the synthesis of β-lactams, we gathered some valuable data about the expansion reaction. On one hand, we observed that the treatment of Ugi adduct 5a with two equivalents of potassium carbonate (entry 3, Table 2) afforded γ-lactam 7a as a by-product. On the other hand, the treatment of phenylglycine derivatives 5m and 5r with potassium carbonate (1.1 equiv.) afforded fused pyrrolidinone-imides 8 as the major product (entries 5 and 14, Table 3). We proposed that the syntheses of these γ-lactams probably proceeded through the expansion of the corresponding β-lactam acting as intermediate.|
|
|||
|---|---|---|---|
| Entry | 6 (R1, R2, R3, X) | 7 (%)a | d.r.b |
| a Yield after purification. b Diastereomeric ratio determined by 1H NMR in the reaction mixture. c Relative configuration (4R*,5R*) in the major diastereomer, determined by NOESY and X-ray diffraction experiments. d Yield as mixture of diastereomers. e Relative configuration (3R*,4R*,5S*) and (3R*,4R*,5R*) in the observed diastereomers, determined by NOESY and X-ray diffraction experiments. f Azetidinone 6k was recovered. Treatment of azetidinone 6k with different bases has been further investigated (see Scheme 3). | |||
| 1 | 6a (H, C6H5, cC6H11, NO2) | 7a (90) | >98:2c |
| 2 | 6b (H, C6H5, CH2C6H5, NO2) | 7b (79) | 97:3c |
| 3 | 6c (H, C6H5, C(CH3)3, NO2) | 7c (67) | >98:2c |
| 4 | 6d (H, 4-CH3C6H4, cC6H11, NO2) | 7d (71) | 92:8c |
| 5 | 6e (H, 4-ClC6H4, cC6H11, NO2) | 7e (44) | >98:2c |
| 6 | 6f (H, 4-FC6H4, cC6H11, NO2) | 7f (42) | >98:2c |
| 7 | 6g (H, 4-CH3OC6H4, cC6H11, NO2) | 7g (71) | >98:2c |
| 8 | 6h (H, CH3, cC6H11, NO2) | 7h (76) | >98:2c |
| 9 | 6i (C6H5, C6H5, cC6H11, NO2) | 7i (92)d | 64:36:n.d.:n.d.e |
| 10 | 6j (C6H5, 4-FC6H4, cC6H11, NO2) | 7j (91)d | 57:43:n.d.:n.d.e |
| 11 | 6k (H, C6H5, cC6H11, H) | 7k (—)f | — |
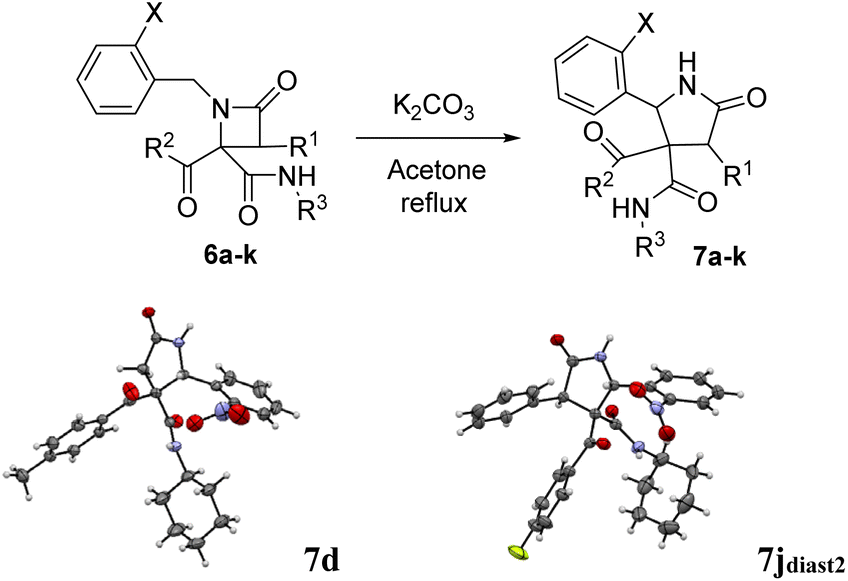
The possibility of synthesizing new, complex, and highly functionalized γ-lactams through a new and simple methodology prompted us to explore the potential of this reaction. Thankfully, all azetidin-2-ones 6 treated with carbonate afforded expansion products, although the results depended on the nature of the electron-withdrawing group (EWG) on the N-substituent of the β-lactam (Tables 4 and 5). Thus, for 2-nitrobenzylamine derivatives, the treatment of azetidin-2-ones 6a–k with potassium carbonate (1.1 equiv.) in boiling acetone for 12 hours afforded the corresponding 4-acyl-4-alkylaminocarbamoyl-5-(2-nitrophenyl)pyrrolidin-2-ones 7a–k (Table 4). Moreover, these reactions proceeded with high diastereoselectivities. Hence, for 2-chloroacetic acid derivatives 7a–h the observed diastereoselectivities were excellent (entries 1–8, Table 4), while for 2-chloro-2-phenylacetic acid derivatives 7i–j only two of the four possible diastereomers were observed. We also tried the expansion on lactam 6k, derived from benzylamine, a non-activated amine, in order to explore the influence of acidity in the benzylic position. As it was expected, azetidinone 6k was recovered after the treatment with potassium carbonate (entry 11, Table 4). However, in an attempt to achieve expansion from this lactam we tried different bases and conditions, as it will be discussed later.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | 6 (R1, R2, R3, R4) | Carbonate | Equiv. | t (h) | 7 (%)a | 8 (%)a | 9 (%)a |
|
a Yield after purification.
b Synthesis carried out from the Ugi adduct.
c Mixture of O-alkylation and expansion products.
d After 12 h most of the azetidine was recovered.
e After 72 h some azetidine was recovered.
f Two diastereomers were observed (d.r. 68:32).
g Yield referred to the major diastereomer after purification.
h Complex reaction mixture.
i A single diastereomer was observed.
|
|||||||
| 1 | 6l (H, C6H5, cC6H11, H) | Cs2CO3 | 1.2 | 1 | — | — | 9l (73) |
| 2 | 6l (H, C6H5, cC6H11, H) | K2CO3 | 1.2 | 12 | 7l (18) | — | 9l (58) |
| 3 | 6m (H, C6H5, cC6H11, C6H5) | Cs2CO3b,c | 2.2 | 1 | — | — | 9m (65) |
| 4 | 6m (H, C6H5, cC6H11, C6H5) | K2CO3b,c | 1.1 | 12 | — | 8m (83) | — |
| 5 | 6m (H, C6H5, cC6H11, C6H5) | Cs2CO3 | 1.2 | 1 | — | 8m (30) | 9m (52) |
| 6 | 6m (H, C6H5, cC6H11, C6H5) | K2CO3 | 1.2 | 12 | — | 8m (68) | 9m (12) |
| 7 | 6n (H, C6H5, cC6H11, CH3) | Cs2CO3 | 1.2 | 1 | — | — | 9n (75) |
| 8 | 6n (H, C6H5, cC6H11, CH3) | K2CO3 | 1.2 | 12 | 7n (58) | 8n (9) | 9n (10) |
| 9 | 6o (H, C6H5, cC6H11, CH2C6H5) | Cs2CO3 | 1.2 | 1 | — | — | 9o (57) |
| 10 | 6o (H, C6H5, cC6H11, CH2C6H5) | K2CO3 | 1.2 | 12 | 7o (38) | 8o (36) | 9o (10) |
| 11 | 6p (H, C6H5, cC6H11, CH(CH3)2) | Cs2CO3 | 1.2 | 1 | — | — | 9p (65) |
| 12 | 6p (H, C6H5, cC6H11, CH(CH3)2) | K2CO3 | 1.2 | 12d | — | 8p (5) | — |
| 13 | 6p (H, C6H5, cC6H11, CH(CH3)2) | K2CO3 | 1.2 | 72 | — | 8p (78) | 9p (8) |
| 14 | 6q (C6H5, C6H5, cC6H11, H) | Cs2CO3 | 1.2 | 72e | 7q (32)g | — | — |
| 15 | 6q (C6H5, C6H5, cC6H11, H) | Cs2CO3 | 2.0 | 12 | —h | — | — |
| 16 | 6r (C6H5, C6H5, cC6H11, C6H5) | K2CO3b,c | 1.2 | 5 | — | 8r (70) | — |
| 17 | 6r (C6H5, C6H5, cC6H11, C6H5) | Cs2CO3 | 1.2 | 1 | — | 8r (72) | — |
| 18 | 6s (C6H5, C6H5, cC6H11, CH3) | K2CO3 | 1.2 | 12 | 7s (75) | — | — |
| 19 | 6s (C6H5, C6H5, cC6H11, CH3) | Cs2CO3 | 1.2 | 1 | 7s (80) | — | — |
| 20 | 6t (C6H5, C6H5, cC6H11, CH2C6H5) | Cs2CO3 | 1.2 | 1 | 7t (67) | — | — |
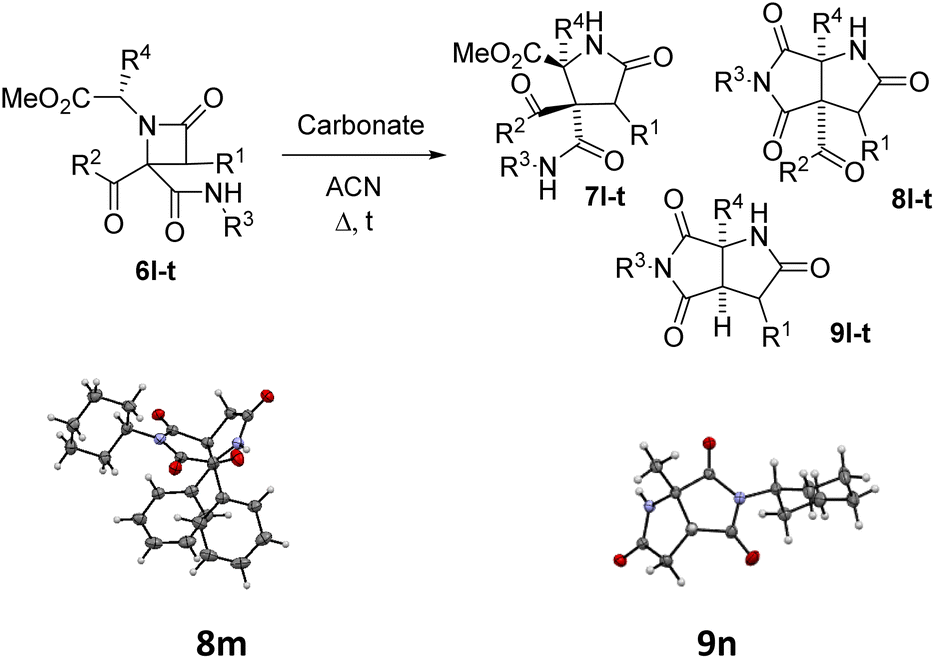
Thus, the chemical results for the aminoester series were controlled mainly by the substituents size, firstly by the C3 substitution on the γ-lactam (R1) (entries 1–13 vs. 14–20, Table 5) and secondly by the C5 substitution (R4) (entries 8, 10 and 13, Table 5), which is explained by the steric hindrance exerted by the different substituents, and additionally by the nature of the carbonate employed. This shows the importance of the counter cation on the stereoselectivity of enolate reactions,17 which is reflected on the chemical result (entries 7, 9 vs. 8, 10, Table 5). These results are remarkable taking into account that ring expansion is promoted by a weak base in a wet solvent under an air atmosphere, in contrast to the strong bases and dry atmosphere needed for the ring expansion previously reported;10,11 moreover, high diastereoselectivities are observed in these syntheses.
2.3. Mechanism proposal for the expansion reaction
In order to understand the mechanism of expansion reactions, it is important to emphasize two significant points in their stereochemical outcome: (1) the results were independent of the diastereomeric purity of the β-lactam employed, which simplifies the experimental work to a great extent, and (2) the use of enantiomerically pure α-aminoesters leads to γ-lactams as racemates. In this way, an anionic rearrangement can be proposed starting from the deprotonation of the acidic position in the N-substituent of the azetidinone favoring its opening, which would destroy the chiral centers on C4 and the N-substituent on azetidinone 6, affording an intermediate containing an imine and an enolate, which would react intramolecularly yielding the corresponding pyrrolidinone (Scheme 2).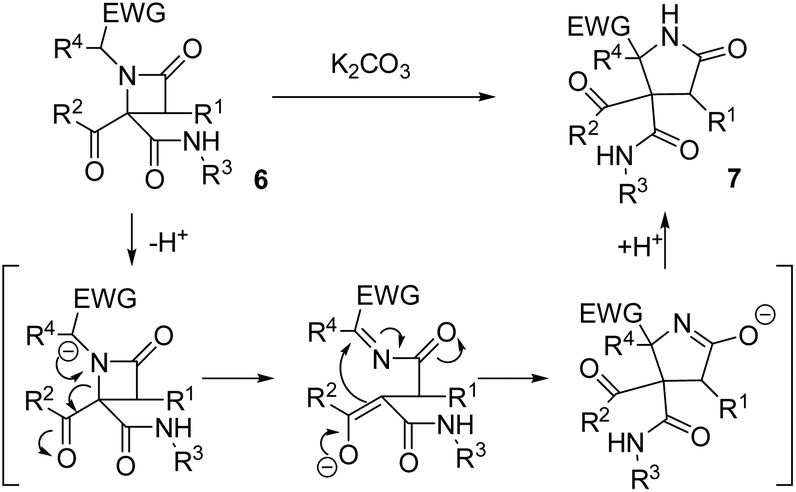 | ||
| Scheme 2 Proposed mechanism for the anionic rearrangement from azetidinones 6 to pyrrolidinones 7. | ||
2.4. Synthesis of debenzoylated γ-lactams
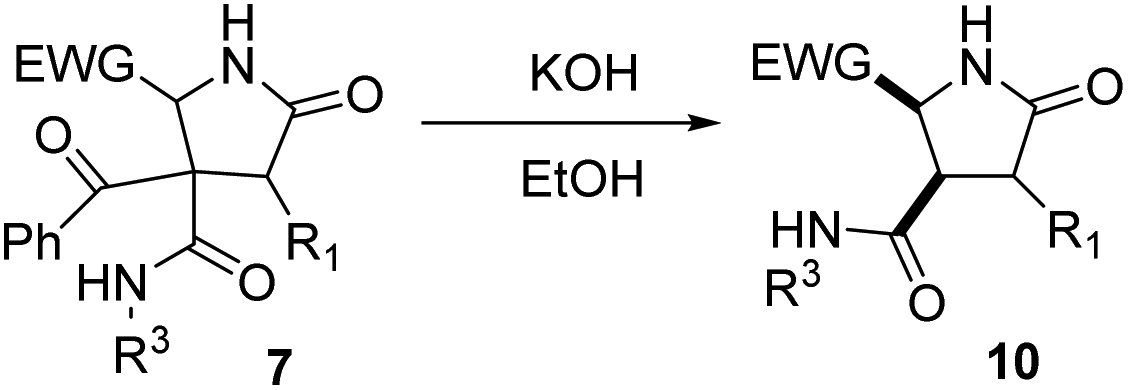
As γ-lactams fused with an imide ring 8 were debenzoylated under some of the conditions tried (entries 1–3, 5–11 and 13, Table 5), we tried the debenzoylation for the different γ-lactams synthesized, in order to obtain a new family of pyrrolidine-2-ones. Thus, the treatment of pyrrolidin-2-ones 7a–c with a catalytic amount of potassium hydroxide in ethanol at room temperature afforded the corresponding deacylated pyrrolidin-2-one 10 as the result of a retro-Claisen reaction; moreover, only the cis diastereoisomer was observed by 1H NMR in the reaction mixture (entries 1–3, Table 6). However, when we tried debenzoylation on lactams derived from 2-chloro-2-phenylacetic acid and/or α-aminoester derivatives, it was unsuccessful, probably due to steric hindrance for the former and saponification of the ester group for the latter (entries 4 and 5, Table 6).|
|
|||
|---|---|---|---|
| Entry | 4 (R1, R3, EWG) | 10 (%)a | d.r.b,c |
| a Yield after purification. b Diastereomeric ratio determined by 1H NMR. c Relative configuration (4R*,5S*) in the major diastereomer, determined by NOESY experiments. d The γ-lactam was recovered. e Complex reaction mixture. | |||
| 1 | 7a (H, cC6H11, 2-NO2C6H4) | 10a (76) | >98:2 |
| 2 | 7b (H, CH2C6H5, 2-NO2C6H4) | 10b (58) | >98:2 |
| 3 | 7c (H, C(CH3)3, 2-NO2C6H4) | 10c (72) | >98:2 |
| 4 | 7i (C6H5, cC6H11, 2-NO2C6H4) | 10i (—)d | — |
| 5 | 7l (H, cC6H11, CO2CH3) | 10l (—)e | — |
2.5. Study of the expansion reaction on azetidine 6k, derived from a non-activated benzyl amine
Finally, we studied the expansion on azetidinone 6k, derived from a non-activated amine. As noted above, the azetidinone was recovered when it was treated with potassium carbonate (entry 11, Table 4). This proves the key role of acidity of the α-CH substituent on the nitrogen, so we tested stronger bases. When we tried potassium hydroxide debenzoylated azetidinone 11 was the only product observed (entry 2, Table 7). Therefore, we tried LDA in dry THF, which afforded γ-lactam 7k but in low yield, with the result depending on the amount of base employed. Thus, when 1.2 equivalents were used, γ-lactam 7k was obtained together with debenzoylated azetidinone 11 (entry 3, Table 7) because of the presence of traces of water in the medium. In order to reduce the formation of this compound we increased the amount of LDA employed (3.0 equiv.). In this way, the formation of azetidinone 11 was not observed, but a new compound identified as succinimide 12 was generated (entry 4, Table 7). These results highlight the importance of the nature of the N-substituent on the azetidine system.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Base | Solvent | Equiv. | 11 (%) | 7k (%) | 12 (%) |
|
a Reflux, 12 h. Azetidinone 6k was recovered.
b Ultrasonication, 30 min.
c Room temperature, 5 h.
d Diastereomeric ratio determined by 1H NMR as 1:1.
e Diastereoisomers were not separated.
|
||||||
| 1 | K2CO3a | Acetone | 1.1 | — | — | — |
| 2 | KOHb | EtOH | Cat. | 94 | — | — |
| 3 | LDAc | THF | 1.2 | 21 | 23d,e | — |
| 4 | LDAc | THF | 3.0 | — | 27d,e | 14 |
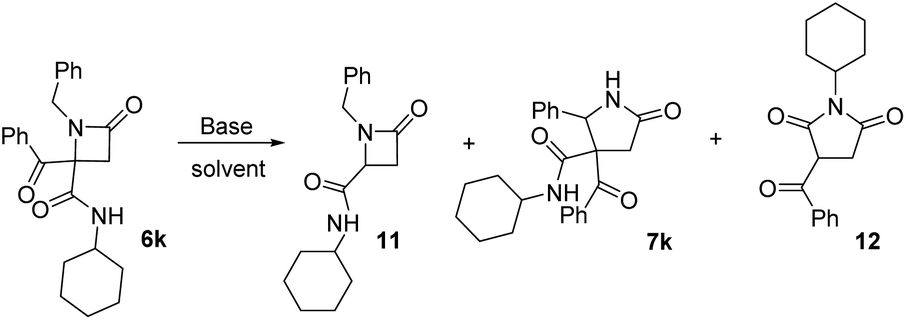
This succinimide was probably generated through the competing formation of a C-anion on the azetidine ring. Thus, the new C-anion would favor the ring opening to a diamide anion, which after an intramolecular cyclization to a five-member ring followed by an intramolecular Cannizaro-type reaction, would afford imide 12 (Scheme 3).
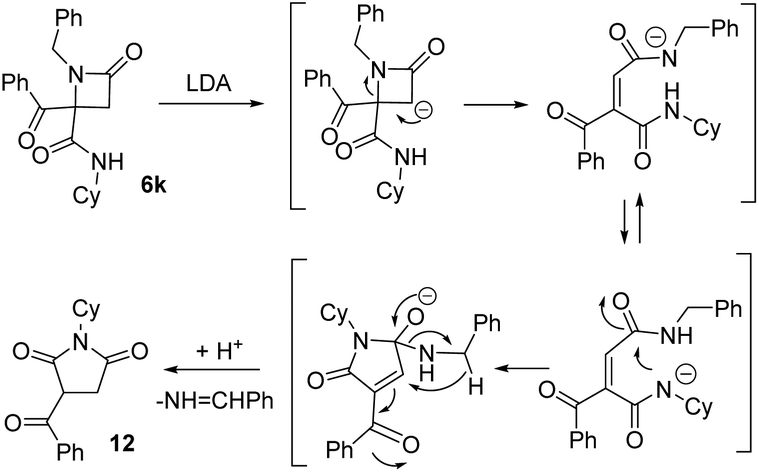 | ||
| Scheme 3 Results for the treatment of azetidinone 6l with base and proposed mechanism for the synthesis of succinimide 12 (Cy = cyclohexyl). | ||
3. Conclusions
These results show the versatility of Ugi adducts in the synthesis of highly functionalized N-heterocycles through post-condensation reactions. Different ring-size lactams with different substituents have been synthesized using simple protocols, in many cases with a high diastereoselectivity. Moreover, the rational design of Ugi adducts allows the synthesis of β-lactams which rearrange to γ-lactams using simple, economical and low moisture-sensitive bases, in an air atmosphere and in a robust manner. Interestingly, the substitution patterns achieved in some structures have not been reported before.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding from Consejería de Educación de la Junta de Castilla y León and European Regional Development Fund (ERDF) (project BU075G19 and project BU067P20) and MCIN/AEI/10.13039/501100011033 [grant PID2020-117610RB-I00] is gratefully acknowledged. B. G.-S. and I. C.-B. thank Consejería de Educación de la Junta de Castilla y León, European Social Fund (ESF) and ERDF for their predoctoral (B. G.-S) and postdoctoral (I. C.-B.) contracts.References
- (a) Heterocycles in Life and Society, ed. A. F. Pozharskii, A. T. Soldatenkov and A. R. Katrizky, John Wiley & Sons, Ltd, 2nd edn, 2011, pp. 209–244 Search PubMed; (b) C. Lamberth, Pest Manage. Sci., 2013, 69, 1106–1114 CrossRef CAS; (c) E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- (a) A. Lei, J. P. Waldkirch, M. He and X. Zhang, Angew. Chem., Int. Ed., 2002, 41, 4526–4529 CrossRef CAS; (b) P. Y. Ng, Y. Tang, W. M. Knosp, H. S. Stadler and J. T. Shaw, Angew. Chem., Int. Ed., 2007, 46, 5352–5355 CrossRef CAS; (c) J. Yu, F. Shi and L.-Z. Gong, Acc. Chem. Res., 2011, 44, 1156–1171 CrossRef CAS; (d) R. Narayan, M. Potowski, Z. J. Jia, A. P. Antonchick and H. Waldmann, Acc. Chem. Res., 2014, 47, 1296–1310 CrossRef CAS; (e) A. McNally, B. Haffemayer, B. S. L. Collins and M. J. Gaunt, Nature, 2014, 510, 129–133 CrossRef CAS; (f) N. Fuentes, W. Kong, L. Fernández-Sánchez, E. Merino and C. Nevado, J. Am. Chem. Soc., 2015, 137, 964–973 CrossRef CAS PubMed; (g) A. P. Taylor, R. P. Robinson, Y. M. Fobian, D. C. Blakemore, L. H. Jones and O. Fadeyi, Org. Biomol. Chem., 2016, 14, 6611–6637 RSC; (h) A. Geranurimi and W. D. Lubell, Org. Lett., 2018, 20, 6126–6129 CrossRef CAS PubMed; (i) A. Tyszka-Gumkowska and J. Jurczak, Org. Chem. Front., 2021, 8, 5888–5894 RSC; (j) L.-S. Wu, Y. Ding, Y.-Q. Han and B.-F. Shi, Org. Lett., 2021, 23, 2048–2051 CrossRef CAS PubMed.
- For the synthesis of β-lactams see: (a) C. R. Pitts and T. Lectka, Chem. Rev., 2014, 114, 7930–7953 CrossRef CAS; (b) S. Hosseyni and A. Jarrahpour, Org. Biomol. Chem., 2018, 16, 6840–6852 RSC For the synthesis of γ-lactams see: (c) F. Rivas and T. Ling, Org. Prep. Proced. Int., 2016, 48, 254–295 CrossRef CAS; (d) J. Caruano, G. G. Muccioli and R. Robiette, Org. Biomol. Chem., 2016, 14, 10134–10156 RSC.
- (a) S. Dandapani and L. A. Marcaurelle, Nat. Chem. Biol., 2010, 6, 861–863 CrossRef CAS; (b) A. Barker, J. G. Kettle, T. Nowak and J. E. Pease, Drug Discovery Today, 2013, 18, 298–304 CrossRef CAS PubMed.
- S. Comesse, M. Sanselme and A. Daïch, J. Org. Chem., 2008, 73, 5566–5569 CrossRef CAS.
- B. Alcaide, P. Almendros and C. Aragoncillo, Chem. Rev., 2007, 107, 4437–4492 CrossRef CAS.
- (a) B. Alcaide, P. Almendros, C. Aragoncillo, R. Callejo and M. P. Ruiz, J. Org. Chem., 2013, 78, 10154–10165 CrossRef CAS PubMed; (b) B. Alcaide, P. Almendros, R. Martín-Montero and M. P. Ruiz, Adv. Synth. Catal., 2016, 358, 1469–1477 CrossRef CAS.
- (a) S. Kano, T. Ebata and S. Shibuya, Chem. Pharm. Bull., 1979, 27, 2450–2455 CrossRef CAS; (b) T. J. Fleck, W. W. McWhorter Jr., R. N. DeKam and B. A. Pearlman, J. Org. Chem., 2003, 68, 9612–9617 CrossRef CAS PubMed.
- (a) W. Van Brabandt and N. De Kimpe, J. Org. Chem., 2005, 70, 3369–3374 CrossRef CAS PubMed; (b) W. Van Brabandt, R. Van Landeghem and N. De Kimpe, Org. Lett., 2006, 8, 1105–1108 CrossRef CAS; (c) S. Dekeukeleire, M. D'Hooghe and N. De Kimpe, J. Org. Chem., 2009, 74, 1644–1649 CrossRef CAS; (d) M. D′hooghe, S. Dekeukeleire, E. Leemans and N. De Kimpe, Pure Appl. Chem., 2010, 82, 1749–1759 Search PubMed.
- T. Durst, R. Van Den Elzen and M. J. LeBelle, J. Am. Chem. Soc., 1972, 94, 9261–9263 CrossRef CAS.
- (a) J. Escalante and M. A. Gonzalez-Tototzin, Tetrahedron: Asymmetry, 2003, 14, 981–985 CrossRef CAS; (b) T. Sakai, K. Yamada and K. Tomioka, Chem. – Asian J., 2008, 3, 1486–1493 CrossRef CAS PubMed; (c) M.-J. Lee and C. Ahn, Bull. Korean Chem. Soc., 2016, 37, 580–583 CrossRef CAS.
- (a) Z. Xu, F. De Moliner, A. P. Cappelli and C. Hulme, Angew. Chem., Int. Ed., 2012, 51, 8037–8040 CrossRef CAS; (b) U. K. Sharma, N. Sharma, D. D. Vachhani and E. V. Van der Eycken, Chem. Soc. Rev., 2015, 44, 1836–1860 RSC; (c) Y. He, Z. Li, K. Robeyns, L. Van Meervelt and E. V. Van der Eycken, Angew. Chem., Int. Ed., 2018, 57, 272–276 CrossRef CAS; (d) S. Wang, Y. Tao, J. Wang, Y. Tao and X. Wang, Chem. Sci., 2019, 10, 1531–1538 RSC; (e) K. Singh, B. K. Malviya, P. K. Jaiswal, V. P. Verma, S. S. Chimni and S. Sharma, Org. Lett., 2019, 21, 6726–6730 CrossRef CAS PubMed.
- X.-H. Zeng, H.-M. Wang, Y.-M. Yan, L. Wu and M.-W. Ding, Tetrahedron, 2014, 70, 3647–3652 CrossRef CAS.
- P. Pertejo, A. Sancho-Medina, T. Hermosilla, B. González-Saiz, J. Gómez-Ayuso, R. Quesada, D. Moreno, I. Carreira-Barral and M. García-Valverde, Molecules, 2021, 26, 919 CrossRef CAS PubMed.
- L. M. Jackman and B. C. Lange, Tetrahedron, 1977, 33, 2737–2769 CrossRef CAS.
- H. Finkelstein, Ber. Dtsch. Chem. Ges., 1910, 43, 1528–1532 CrossRef CAS.
- Y. Hu, R. L. Bishop, A. Luxenburger, S. Dong and L. A. Paquette, Org. Lett., 2006, 8, 2735–2737 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2193426, 2193428–2193430 and 2193435. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2gc02896d |
| This journal is © The Royal Society of Chemistry 2022 |