
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Radical hydroxymethylation of alkyl iodides using formaldehyde as a C1 synthon†
Lewis
Caiger
 a,
Conar
Sinton
a,
Timothée
Constantin
a,
James J.
Douglas
b,
Nadeem S.
Sheikh
c,
Fabio
Juliá
a and
Daniele
Leonori
*a
a,
Conar
Sinton
a,
Timothée
Constantin
a,
James J.
Douglas
b,
Nadeem S.
Sheikh
c,
Fabio
Juliá
a and
Daniele
Leonori
*a
aDepartment of Chemistry, University of Manchester, Manchester M13 9PL, UK. E-mail: daniele.leonori@manchester.ac.uk; Web: https://leonorigroup.com
bEarly Chemical Development, Pharmaceuticals Sciences, R&D, AstraZeneca, Macclesfield, UK
cDepartment of Chemistry, College of Science, King Faisal University, P. O. Box 400, Al-Ahsa 31982, Saudi Arabia
First published on 6th July 2021
Abstract
Radical hydroxymethylation using formaldehyde as a C1 synthon is challenging due to the reversible and endothermic nature of the addition process. Here we report a strategy that couples alkyl iodide building blocks with formaldehyde through the use of photocatalysis and a phosphine additive. Halogen-atom transfer (XAT) from α-aminoalkyl radicals is leveraged to convert the iodide into the corresponding open-shell species, while its following addition to formaldehyde is rendered irreversible by trapping the transient O-radical with PPh3. This event delivers a phosphoranyl radical that re-generates the alkyl radical and provides the hydroxymethylated product.
Introduction
Hydroxymethyl motifs are frequently encountered in the core structure of natural products and drugs and commonly used as handles for further derivatisation (Scheme 1A).1 This structural and practical relevance makes the development of methods able to introduce the “CH2OH” fragment onto advanced building blocks impactful to the discovery and development of high-value materials.2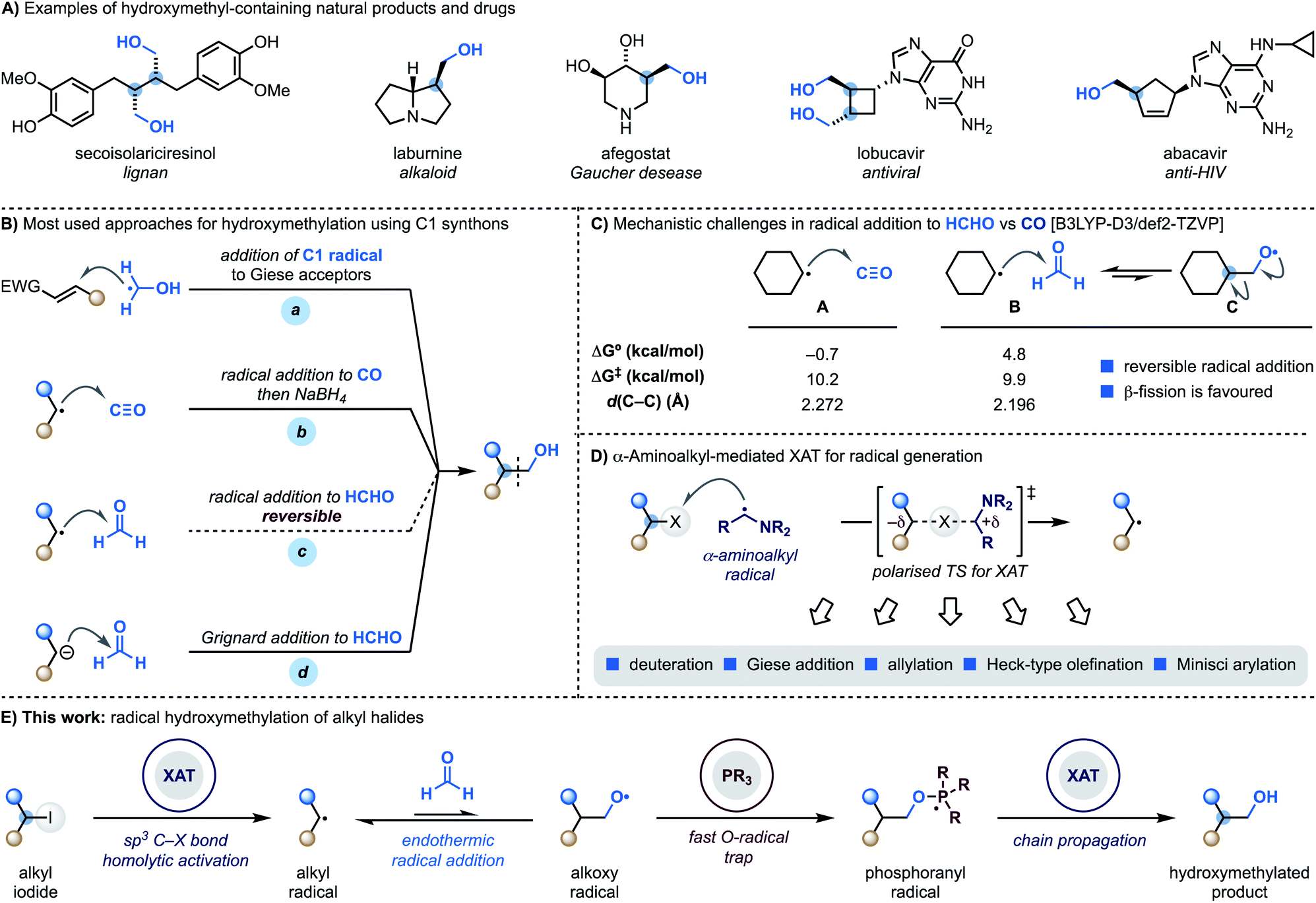 | ||
| Scheme 1 (A) Examples of natural products and therapeutic agents containing the hydroxymethyl motif. (B) Overview of the main methods to install a hydroxymethyl motif using radical chemistry. (C) Radical addition to CO is possible while addition to HCHO is difficult due to reversibility. (D) α-Aminoalkyl radicals enable XAT activation of alkyl iodides and bromides and the corresponding carbon radicals can be engaged in several transformations. (E) This work shows the utilisation of XAT and phosphoranyl radical chemistry for the direct coupling of unactivated alkyl iodides with HCHO. | ||
Retrosynthetically, the most direct approach to achieve programmable small-molecule hydroxymethylation is through the formation of a sp3–sp3 C–C bond with an oxygenated C1 synthon.3 Within the realm of radical chemistry, α-hydroxymethyl radicals, which are easily generated from MeOH via HAT (H-atom transfer), have been successfully applied to the functionalisation of Giese4 and Minisci5 acceptors (Scheme 1B, path a). A significantly less explored avenue is the development of methodologies where C-radicals react with C1 SOMOphiles.6
Considering the three most atom-economic and abundant oxygenated C1 synthons – CO2, CO and HCHO – CO is the most used in radical strategies. Indeed, while radical addition to CO2 is endothermic,7 reaction with CO is both kinetically and thermodynamically facile and, as demonstrated by the pioneering work of Ryu, also compatible for application in radical chain propagations.8 This approach however, provides the aldehyde as the product of the radical process and thus requires a subsequent stoichiometric hydride reduction step (Scheme 1B, path b).9
The difficulties in using formaldehyde as a C1 synthon for radical hydroxymethylation (Scheme 1B, path c) come from the reversible nature of the addition process.9a,10 According to our computational studies, the reaction between cyclohexyl radicals and HCHO (B) is kinetically accelerated compared to the reaction with CO (A) but, crucially, endothermic with a later transition state character (as determined by comparison of the respective d(C–C) values) (Scheme 1C).11 This means that the back reaction, involving β-scission of the primary O-radical C, is a fast process which hampers reaction development. Overall, this mechanistic challenge has impacted the practical ways we conduct hydroxymethylation with HCHO, which is generally achieved using Grignard or organolithium reagents that might limit functional group compatibility (Scheme 1B, path d).12
We have recently demonstrated that alkyl radicals can be conveniently accessed from the corresponding halides using halogen-atom transfer (XAT) with α-aminoalkyl radicals (Scheme 1D).13 These open-shell species can be generated from the corresponding amines by SET (single-electron transfer) oxidation14 and deprotonation15 and display an abstracting profile similar to that of tin radicals in the homolytic activation of organic halides. This reactivity mode benefits from a polarised transition state13,16 with significant charge-transfer character and can be used in redox-neutral photoredox manifolds and also as an initiation mechanism in transformation based on radical-chain propagations.13b
We recently became interested in benchmarking XAT reactivity with the aim of enabling radical couplings between alkyl halides and HCHO. Here we demonstrate the realisation of this goal and report the development of a practical approach for direct radical hydroxymethylation (Scheme 1E). The process sequentially exploits XAT to generate an alkyl radical and then harnesses the ability of PPh3 to trap the resulting O-radical. This approach effectively renders the radical addition to HCHO an irreversible process. Crucially, the ensuing phosphoranyl radical is able to sustain a radical chain propagation based on XAT to give the hydroxymethylated product.
Results and discussion
Mechanistic considerations and reaction optimization
In approaching the development of a radical hydroxymethylation reaction using HCHO as a C1 synthon, we initially considered the utilisation of a reductive quenching photoredox cycle (Scheme 2A). In principle, this manifold would enable the generation of the key α-aminoalkyl radical (D → E) for XAT with the alkyl iodide (e.g. 4-iodo-N-Boc-piperidine 1). According to our previous work, this step should be facile and provide radical F along with the iminium iodide G. Addition of F to HCHO is reversible making a potential SET reduction (followed by protonation) of the O-radical H with the reduced photocatalyst challenging. However, our interest was drawn by the fact that this mechanism could potentially benefit from radical-chain reactivity where H undergoes polarity matched HAT (H-atom transfer) with the amine D. Since O-radicals undergo related HAT reactions at fast rates (∼108 M−1 s−1)17 and the amine would be used in excess, this step should provide 2 whilst regenerating the chain-carrying α-aminoalkyl radical E.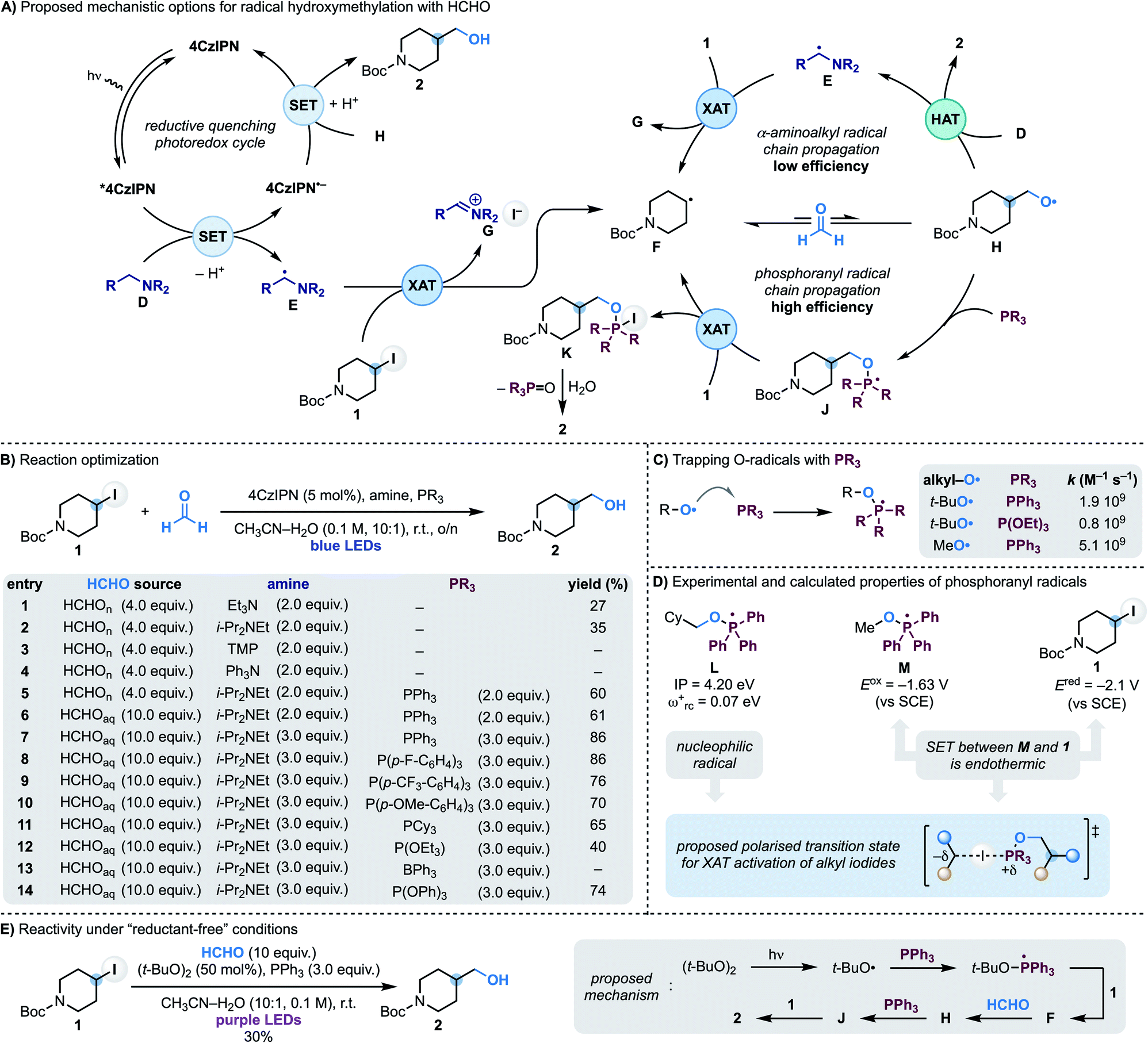 | ||
| Scheme 2 (A) Mechanistic proposal for the coupling of alkyl iodides with HCHO using XAT and phosphoranyl radical chemistry. (B) Optimisation of the radical coupling between iodide 1 and HCHO. (C) Literature rate constants for the reaction of O-radical with P(III) compounds to give phosphoranyl radicals. (D) Experimental and calculated properties of phosphoranyl radicals suggest XAT as the alkyl iodide activation mechanism. (E) “Reductant-free” reaction for hydroxymethylation of alkyl iodides via phosphoranyl radicals. | ||
We started our investigations using 1 as the iodide, 4CzIPN as the photocatalyst and paraformaldehyde in CH3CN–H2O (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) solvent under blue light irradiation at room temperature (Scheme 2B). Pleasingly, using Et3N as the amine, alcohol 2 was formed in moderate yield which was improved by using i-Pr2NEt (entries 1 and 2). In accordance with our mechanistic picture, evaluation of amines that can act as efficient electron donors for *4CzIPN but cannot lead to the formation of an α-aminoalkyl radical (by either oxidation and deprotonation or HAT) led to no product formation [compare entries 3 (TMP = tetramethylpiperidine) and 4 (Ph3N) with 2 (i-Pr2NEt)]. Unfortunately, evaluation of the other reaction parameters11 did not further improve the efficiency of the process which likely underscores the challenges in overcoming the reversibility of the radical addition process (F + HCHO ⇆ H).
:1) solvent under blue light irradiation at room temperature (Scheme 2B). Pleasingly, using Et3N as the amine, alcohol 2 was formed in moderate yield which was improved by using i-Pr2NEt (entries 1 and 2). In accordance with our mechanistic picture, evaluation of amines that can act as efficient electron donors for *4CzIPN but cannot lead to the formation of an α-aminoalkyl radical (by either oxidation and deprotonation or HAT) led to no product formation [compare entries 3 (TMP = tetramethylpiperidine) and 4 (Ph3N) with 2 (i-Pr2NEt)]. Unfortunately, evaluation of the other reaction parameters11 did not further improve the efficiency of the process which likely underscores the challenges in overcoming the reversibility of the radical addition process (F + HCHO ⇆ H).
We therefore proposed to overcome the unwanted β-fragmentation by trapping the transient O-radical H with an immediate reaction. Specifically, we were interested by the well-known ability of phosphines to trap O-radicals at diffusion-controlled rates leading to the corresponding phosphoranyl radicals (Scheme 2C).18 These open-shell intermediates can still undergo β-scission across the sp3 C–O bond but the feasibility of this process depends on the nature of the ensuing C-radical, and it is only efficient when tertiary or stabilised (e.g. benzylic) species are generated.19 In our case this fragmentation would lead to a primary alkyl radical, so we were hopeful that J would be long-lived enough to participate in a following reaction. In particular, we anticipated that J might be able to abstract an I-atom from 1 and therefore establish a phosphoranyl radical-based chain propagating system. This step would give the P(V) intermediate K (ref. 20) that could provide the targeted 2 upon hydrolysis. While XAT between phosphoranyl radicals and alkyl halides has not been reported before,21 we speculated that their high nucleophilic character22 should lead to polar effects related to those demonstrated in abstraction reactions with tin, silicon and α-aminoalkyl radicals.13a
This mechanistic proposal was validated by the addition of PPh3 to the reaction mixture, which immediately led to a dramatic increase in the process yield (Scheme 2B, entry 6). Further improvements were made by using formalin in place of paraformaldehyde and increasing the equivalents of both i-Pr2NEt and PPh3. Under the conditions reported in entry 7, 2 was obtained in 86% yield. Other triaryl/trialkyl phosphines were evaluated and were also successful albeit in lower yield (entries 9–12). Phosphites and triaryl boranes are also known to efficiently trap O-radicals18 and were evaluated. While P(OEt)3 resulted in significantly lower yield (entry 12) and BPh3 completely suppressed the reactivity (entry 13), P(OPh)3 gave 2 in 74% yield (entry 14).23 Finally, control experiments demonstrated the reaction required all components as well as continuous blue LEDs irradiation.11
A photoredox initiation based on a reductive quenching cycle is supported by our Stern–Volmer studies whereby the amine quenches *4CzIPN fluorescence with the largest rate constant.11 Obtaining evidence on the XAT reactivity of the phosphoranyl radical J has been more difficult since, despite considerable efforts, we have not be able to locate a transition state for this transformation. Nevertheless, we believe the strong nucleophilic character of phosphoranyl radical [determined by calculating the ionization potential (IP) and electrophilicity index (ωrc+) for L] should provide effective charge-transfer stabilisation in a XAT transition state, just like the chain initiating α-aminoalkyl radicals. Furthermore, its intermediate reductive power (determined by measuring the reduction potential of the phosphonium salt Ph3(MeO)P(OTf) to give M) supports a reaction with alkyl iodide 1 (Ered = −2.09 V vs. SCE) based on XAT over SET (Scheme 2D).11 In addition, we have been able to translate this reactivity under “reductant-free” conditions. As shown in Scheme 2E we have used the photochemical O–O bond homolysis of (t-BuO)2 to generate a t-BuO-containing phosphoranyl radical from which XAT from 1 and a subsequent reaction with HCHO enables formation of 2 (30% yield).11 Overall, we believe this reaction provides supporting evidence for the ability of phosphoranyl radicals to abstract iodine atoms from alkyl residues.
Substrate scope
Having identified optimal conditions for the radical coupling between alkyl iodides and formaldehyde, we evaluated the scope of the process. A series of commercial iodides based on valuable N-heterocyclic systems provided access to C3-functionalised N-Boc-piperidine (3), -azepane (4), -pyrrolidine (5) and -proline (6) in generally high yields. The chemistry was also translated to I-containing N-Boc-azetidine and 4-iodo-(thio)pyran to give alcohols 7–10. Alcohol 9 Represents an example of a chemoselective XAT process as aryl bromides are more difficult to activate using α-aminoalkyl radicals, allowing selective targeting of the sp3 C–I bond. The successful formation of 11 in good yield demonstrates compatibility with carbonyl functionalities that might be problematic when using reactive organometallic intermediates. The chemistry was then applied to the hydroxymethylation of unactivated 4-Ph-cyclohexyl iodide (12) and also used on 2- and 1-iodo-adamantane (13 and 14), thus demonstrating compatibility with tertiary substrates.We also succeeded in employing this chemistry on iodinated N-Boc-protected cyclohexylamine (15) and cyclobutylamine (16) as well as two commercial spirocyclic building blocks (17 and 18) and a bicyclic derivative (19) in high yield.
To showcase the applicability of this methodology we sought to use it to achieve the one-carbon homologation of high-value alcohols using Appel iodination followed by XAT–phosphoranyl radical-mediated hydroxymethylation. As shown in Scheme 3B, the commercial N-heterocycle 20 gave 21 which can lead to the preparation of analogues of the kidney cancer treatment drug tesevatinib. Piperidine 22 provided 23, which is a synthetic intermediate in the manufacture of the orexin antagonist filorexant. The blockbuster cardiac stimulant proxyphylline 24 could also be engaged thus broadening the functional group compatibility and providing access to the 1C-homologated drug analogue 25 in useful yield. Furthermore, subjecting the N-Boc protected alkaloid nortropine 26 and cholesterol 28 to this two-step sequence gave the 1C-homologation products 27 and 29.
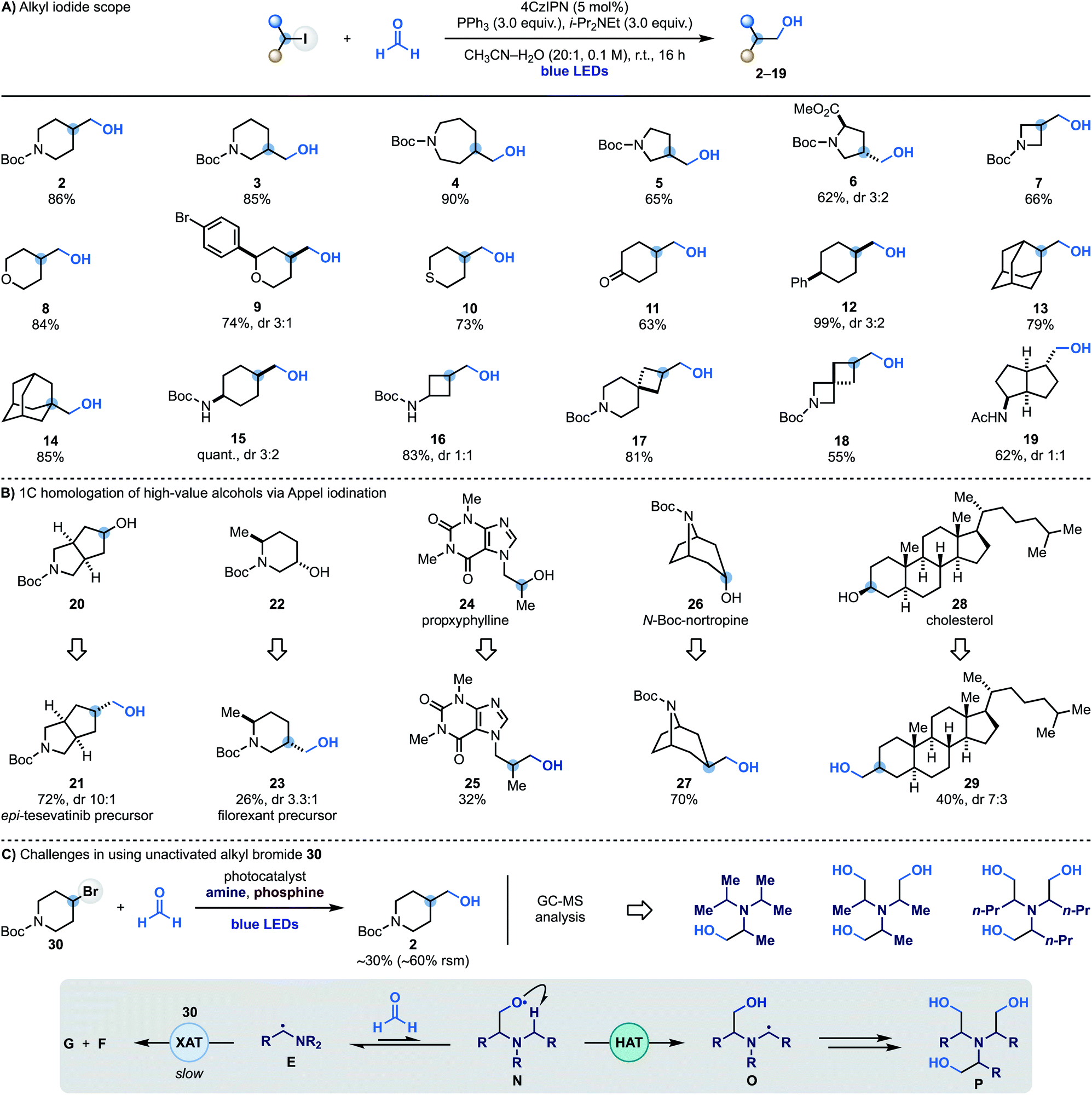 | ||
| Scheme 3 (A) Alkyl iodide scope. (B) Application of the hydroxymethylation strategy in the 1C homologation of high-value alcohols via Appel iodination followed by radical coupling with HCHO. (C) Difficulties in extending the hydroxymethylation reactivity to unactivated alkyl bromide 30. rsm is remaining starting material. | ||
While α-aminoalkyl radicals can be successfully used for the homolytic activation of both alkyl iodides and bromides,13a this hydroxymethylation strategy is at the moment synthetically useful only for the iodides. In the case of the bromides, as XAT is slower (kXAT < 105 M−1 s−1 for Cy–Br and the Et3N α-aminoalkyl radical),13a other unwanted reactivities become competitive which may hamper product formation. Indeed, when we attempted the reaction with 4-bromo-N-Boc-piperidine 30 using various combinations of photocatalysts, amines and oxidants, we obtained 2 in up to 30% yield, recovered 30 in >60% yield and identified by mass spectrometry analysis several by-products resulting from the hydroxymethylation of the amine reagent. We believe that in these cases where XAT is slower, the nucleophilic α-aminoalkyl radical E can be trapped by HCHO leading to O-radical N. This species can either react with PPh3, or, in the case of linear trialkyl amines (e.g. Et3N), undergo intramolecular and polarity matched HAT from the other α-N-positions (O). This radical translocation process ultimately leads to the accumulation of polar poly-hydroxymethylated derivatives (P).11
Hydroxymethylation of other alkyl radical precursors
Since phosphoranyl radicals are relatively good reductants (Eox = −1.63 V vs. SCE, see Scheme 2D), we wondered if this strategy could be extended to the hydroxymethylation of other alkyl radical precursors based on electrophores easier to reduce than alkyl iodides. We were particularly interested by the use of Katritzky's pyridinium salts24 and imidazole thiocarbonyls25 as these species would enable the overall hydroxymethylation of amines and alcohols. As shown in Scheme 4, we succeeded in engaging pyridinium 31 (Ered = −0.94 V vs. SCE) in this reactivity using either photoredox or EDA (electron donor–acceptor)26 conditions. In the first case an oxidative quenching photoredox cycle using Ir(ppy)3 photocatalyst was used as the initiation mechanism for alkyl radical F generation. In the latter case, the supramolecular association of 31 and 4-Me-Hantzsch ester (4-Me-HE) generated an EDA complex2632 that underwent photoinduced SET upon irradiation of its charge-transfer band with blue light.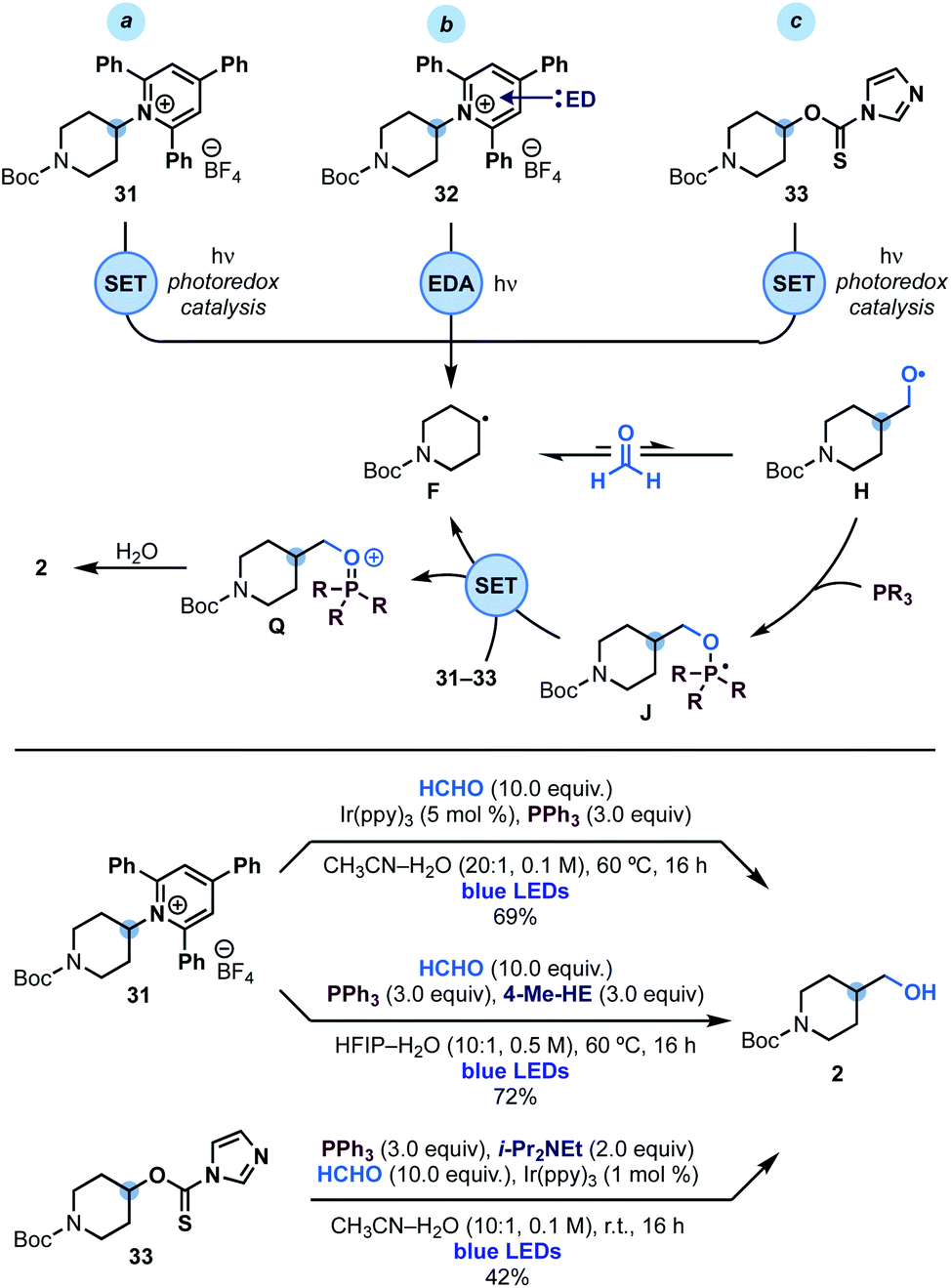 | ||
| Scheme 4 Mechanistic analysis and reaction conditions for the hydroxymethylation of Katritzky's pyridinium 31 and xanthate 33. | ||
Extension of this chemistry to thiocarbamate 33 proved more challenging due to the tendency of these species to undergo radical O → S rearrangement.27 Nevertheless, optimisation of the reaction parameters led to conditions for its implementation in useful yield using oxidative quenching of the photocatalyst Ir(ppy)3 as the initiation step.
In all three cases, we believe that upon alkyl radical F generation, reversible reaction with HCHO and fast trapping of H with PPh3, leads to phosphoranyl radical J that could sustain a chain propagation based on SET (instead of XAT) as the alkyl radical re-generation step. The feasibility of this step is supported by the matching redox potentials for SET between M (see Scheme 2D) and those of 31 and 33.
Conclusions
We have demonstrated that the unfavorable addition of carbon radicals to HCHO can be overcome by trapping the resulting O-radical with Ph3P in a fast and irreversible reaction. This leads to the generation of a phosphoranyl radical that can sustain chain propagations based on either XAT or SET. This provides versatility to the hydroxymethylation protocol that can be applied to alkyl iodides and Katritzky's salts in high yield and also thiocarbamates.Data availability
The data that support the findings of this study are available in the ESI and from the corresponding author upon reasonable request.Author contributions
FJ and DL designed the project. LC, CS, TC and FJ performed all the synthetic experiments. JJD performed some mechanistic experiments. NSS performed the computational studies. All authors analysed the results and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
DL thanks EPSRC for a Fellowship (EP/P004997/1), and the European Research Council for a research grant (758427). LC thanks the Integrated Catalysis (iCAT) CDT (EPSRC grant: EP/S023755/1) for a PhD Studentship. CS thanks AstraZeneca for a PhD Studentship. NSS acknowledges the Deanship of Scientific Research at the King Faisal University for the financial support under the annual research grants.Notes and references
- (a) R. Richa and R. P. Sinha, EXCLI J., 2014, 13, 592–610 Search PubMed; (b) M. C. White and J. Zhao, J. Am. Chem. Soc., 2018, 140, 13988–14009 CrossRef CAS PubMed; (c) S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451–3479 CrossRef CAS PubMed; (d) D. G. Brown and J. Boström, J. Med. Chem., 2016, 59, 4443–4458 CrossRef CAS.
- D. C. Blakemore, L. Castro, I. Churcher, D. C. Rees, A. W. Thomas, D. M. Wilson and A. Wood, Nat. Chem., 2018, 10, 383–394 CrossRef CAS.
- (a) I. Ryu, N. Sonoda and D. P. Curran, Chem. Rev., 1996, 96, 177–194 CrossRef CAS; (b) G. Yuan, C. Qi, W. Wu and H. Jiang, Current Opinion in Green and Sustainable Chemistry, 2017, 3, 22–27 CrossRef; (c) Y. Shi, B.-W. Pan, Y. Zhou, J. Zhou, Y.-L. Liu and F. Zhou, Org. Biomol. Chem., 2020, 18, 8597–8619 RSC; (d) T. E. Hurst, J. A. Deichert, L. Kapeniak, R. Lee, J. Harris, P. G. Jessop and V. Snieckus, Org. Lett., 2019, 21, 3882–3885 CrossRef CAS PubMed.
- B. Fraser-Reid, N. L. Holder and M. B. Yunker, J. Chem. Soc., Chem. Commun., 1972, 1286–1287 RSC.
- (a) A. Citterio, A. Gentile, F. Minisci, M. Serravalle and S. Ventura, Tetrahedron, 1985, 41, 617–620 CrossRef CAS; (b) C. A. Huff, R. D. Cohen, K. D. Dykstra, E. Streckfuss, D. A. DiRocco and S. W. Krska, J. Org. Chem., 2016, 81, 6980–6987 CrossRef CAS PubMed; (c) F. Rammal, D. Gao, S. Boujnah, A. A. Hussein, J. Lalevée, A.-C. Gaumont, F. Morlet-Savary and S. Lakhdar, ACS Catal., 2020, 10, 13710–13717 CrossRef CAS.
- (a) S. Kim, I. Y. Lee, J.-Y. Yoon and D. H. Oh, J. Am. Chem. Soc., 1996, 118, 5138–5139 CrossRef CAS; (b) S. Kim and S. Y. Jon, Tetrahedron Lett., 1998, 39, 7317–7320 CrossRef CAS; (c) M. Shee, S. S. Shah and N. D. P. Singh, Chem. Commun., 2020, 56, 4240–4243 RSC; (d) D. J. Hart and F. L. Seely, J. Am. Chem. Soc., 1988, 110, 1631–1633 CrossRef CAS; (e) H. Miyabe, R. Shibata, C. Ushiro and T. Naito, Tetrahedron Lett., 1998, 39, 631–634 CrossRef CAS.
- Z. Fan, Z. Zhang and C. Xi, ChemSusChem, 2020, 13, 6201–6218 CAS.
- (a) I. Ryu, K. Kusano, A. Ogawa, N. Kambe and N. Sonoda, J. Am. Chem. Soc., 1990, 112, 1295–1297 CrossRef CAS; (b) I. Ryu, K. Kusano, H. Yamazaki and N. Sonoda, J. Org. Chem., 1991, 56, 5003–5005 CrossRef CAS.
- (a) V. Gupta and D. Kahne, Tetrahedron Lett., 1993, 34, 591–594 CrossRef CAS; (b) H. Matsubara, S. Yasuda, H. Sugiyama, I. Ryu, Y. Fujii and K. Kita, Tetrahedron, 2002, 58, 4071–4076 CrossRef CAS; (c) S. Kobayashi, T. Kawamoto, S. Uehara, T. Fukuyama and I. Ryu, Org. Lett., 2010, 12, 1548–1551 CrossRef CAS PubMed; (d) S. Kobayashi, T. Kinoshita, T. Kawamoto, M. Wada, H. Kuroda, A. Masuyama and I. Ryu, J. Org. Chem., 2011, 76, 7096–7103 CrossRef CAS PubMed; (e) T. Kawamoto, T. Fukuyama and I. Ryu, J. Am. Chem. Soc., 2012, 134, 875–877 CrossRef CAS PubMed; (f) T. Kawamoto, T. Okada, D. P. Curran and I. Ryu, Org. Lett., 2013, 15, 2144–2147 CrossRef CAS PubMed.
- M. Oyama, J. Org. Chem., 1965, 30, 2429–2432 CrossRef CAS.
- See ESI† for more information.
- C. Bernardon, J. Organomet. Chem., 1989, 367, 11–17 CrossRef CAS.
- (a) T. Constantin, M. Zanini, A. Regni, N. S. Sheikh, F. Juliá and D. Leonori, Science, 2020, 367, 1021–1026 CrossRef CAS PubMed; (b) T. Constantin, F. Juliá, N. S. Sheikh and D. Leonori, Chem. Sci., 2020, 11, 12822–12828 RSC , for a Cu/peroxide mediated approach for α-aminoalkyl radical generation and XAT on activated alkyl halides, see; (c) R. K. Neff, Y.-L. Su, S. Liu, M. Rosado, X. Zhang and M. P. Doyle, J. Am. Chem. Soc., 2019, 141, 16643–16650 CrossRef CAS PubMed.
- P. J. DeLaive, T. K. Foreman, C. Giannotti and D. G. Whitten, J. Am. Chem. Soc., 1980, 102, 5627–5631 CrossRef CAS.
- (a) S. F. Nelsen and J. T. Ippoliti, J. Am. Chem. Soc., 1986, 108, 4879–4881 CrossRef CAS; (b) J. P. Dinnocenzo and T. E. Banach, J. Am. Chem. Soc., 1989, 111, 8646–8653 CrossRef CAS.
- J. Lalevée, X. Allonas and J. P. Fouassier, Chem. Phys. Lett., 2008, 454, 415–418 CrossRef.
- D. Griller, J. A. Howard, P. R. Marriott and J. C. Scaiano, J. Am. Chem. Soc., 1981, 103, 619–623 CrossRef CAS.
- (a) D. Griller, K. U. Ingold, L. K. Patterson, J. C. Scaiano and R. D. Small, J. Am. Chem. Soc., 1979, 101, 3780–3785 CrossRef CAS; (b) A. Inial, F. Morlet-Savary, J. Lalevée, A.-C. Gaumont and S. Lakhdar, Org. Lett., 2020, 22, 4404–4407 CrossRef CAS PubMed.
- (a) W. G. Bentrude, E. R. Hansen, W. A. Khan and P. E. Rogers, J. Am. Chem. Soc., 1972, 94, 2867–2868 CrossRef CAS; (b) M. Bietti, A. Calcagni and M. Salamone, J. Org. Chem., 2010, 75, 4514–4520 CrossRef CAS PubMed.
- Intermediate K has been depicted as an iodophosphorane but it could also exist as the corresponding phosphonium iodide.
- For mechanistic studies on XAT with diethoxyphosphonyl radicals, see: (a) M. Anpo, R. Sutcliffe and K. U. Ingold, J. Am. Chem. Soc., 1983, 105, 3580–3583 CrossRef CAS; (b) S. Kim, C. H. Cho and C. J. Lim, J. Am. Chem. Soc., 2003, 125, 9574–9575 CrossRef CAS PubMed.
- W. G. Bentrude, Acc. Chem. Res., 1982, 15, 117–125 CrossRef CAS.
- The lower yield observed in the case of P(OEt)3 with respect to P(OPh)3 might be due a β-fragmentation of the corresponding phosphoranyl radical.
- A. R. Katritzky and S. S. Thind, J. Chem. Soc., Perkin Trans. 1, 1980, 1895–1900 RSC.
- D. H. R. Barton and S. W. McCombie, J. Chem. Soc., Perkin Trans. 1, 1975, 1574–1585 RSC.
- C. G. S. Lima, T. de M. Lima, M. Duarte, I. D. Jurberg and M. W. Paixão, ACS Catal., 2016, 6, 1389–1407 CrossRef CAS.
- L. Chenneberg, A. Baralle, M. Daniel, L. Fensterbank, J.-P. Goddard and C. Ollivier, Adv. Synth. Catal., 2014, 356, 2756–2762 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc03083c |
| This journal is © The Royal Society of Chemistry 2021 |