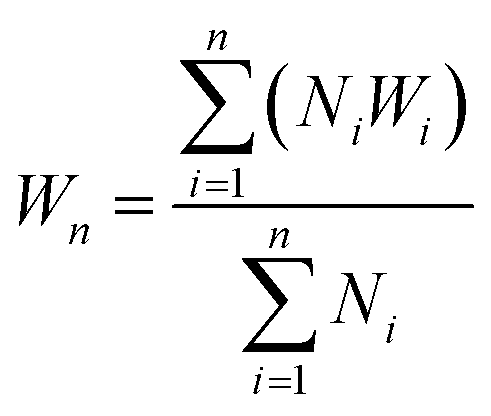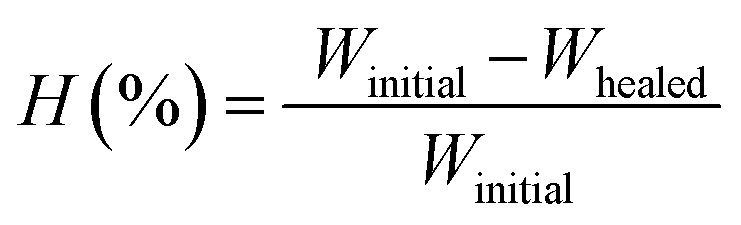DOI:
10.1039/D1RA07425C
(Paper)
RSC Adv., 2021,
11, 39376-39386
Modulation of flexo-rigid balance in photoresponsive thymine grafted copolymers towards designing smart healable coating†
Received
6th October 2021
, Accepted 29th October 2021
First published on 10th December 2021
Abstract
Efficacy and durability of the photovoltaic device mandates its protection against hot, humid weather condition, high energy of UV light and unwanted scratches. Such challenges can be mitigated by smart polymeric coating with inherent properties e.g. hydrophobicity to prevent moisture, optimal viscocity for better processibility and crack-healing. The hydrophobic polymers TP1–TP4 containing pendant photo-crosslinkable thymine moieties are designed that undergo [2 + 2] photocycloaddition upon UVB irradiation and can be dynamically reverted back upon irradiation with UVC light. A judicious control of solvent environment, chain length, functionality% and concentration of the polymers regulate the aspects of photodimerization thereby, rendering intra or inter-chain collapse to form diverse nanostructures. Photodimerization of the thymine moieties renders coil to globule transformation in dilute condition whereas irradiation performed at high macromolecular concentration regime exhibits higher order nanostructures. The photoresponsive chain collapse leads to the formation of rigid crosslinked domains within flexible polymer chains akin to the hard–soft phases of thermoplastic elastomers. Such rigidification of the crosslinked segments endows a tool to photomodulate the glass transition temperature (Tg) that can dynamically revert back upon decrosslinking. Further, the structural modulation of the polymers is explored towards autonomic and nonautonomic self-healing behaviour at ambient conditions. Moreover, the self-healing efficacy can be tuned with the film thickness and it remains unaltered upon using solar simulator or direct sunlight. Overall, such hydrophobic low Tg polymers display photo-regulated self-healing mechanism consisting of both autonomic and non-autonomic self-healing and may find applications in designing smart protective coatings for photovoltaic devices.
1. Introduction
The advent of photovoltaic semiconductor devices as an effective solar energy harvester has revolutionized the global approach to meet the target of total renewable energy production.1 However, the efficiencies of these devices are prone to get downgraded with time upon exposure to hot and humid environmental conditions.2 Moreover, light-induced degradation of semiconductor solar cells with high UV light intensity possess another hurdle for their long life-span.3 Coating of these devices with rigid or flexible organic polymeric materials has gained substantial attention in recent times to preserve the integrity of these devices.4 However, these coatings are susceptible to the formation of cracks or scratches, thereby resulting in exposure to moisture, oxygen etc. In that regard, self-healable coatings from low viscosity processible polymers are of great interest that have the capability of repairing their own damage over time and restore the functional properties of the devices. Moreover, early arrest of the crack propagation circumvents catastrophic device failure and renders rather sustainable and prolonged life span of the devices.5 As compared to the extrinsic self-healing mechanism involving the breakage of microcapsule to release the stored healing agents for single time healing, intrinsic self-healing has the advantages of reduced costs and multiple cycles of healings. Such intrinsic mechanism entails reversible interactions such as hydrogen bonding,6 host–guest interactions,7 metal–ligand complexations,8 ionic interactions,9 π–π stacking interactions,10 dynamic covalent chemistry11 to efficiently mend the damage. Further, control over such interactions by the intervention of external stimuli renders the self-healing processes autonomic or non-autonomic. For example, the thermoplastic elastomer having a segregated hard–soft microphase domain shows a spontaneously autonomic self-healing ability at the ambient condition.12 Among the stimuli-triggered non-autonomic approaches, temperature mediated self-healing was explored through Diels–Alder reactions. However, a high operating temperature especially, for the retro D–A reaction limits its versatility owing to the compromised polymer stability.13 In that regard, the photo-triggered healing process appears to be more advantageous owing to the locally selected area of application, ambient operative temperature and rather inexpensive environment-friendly photoreaction that leads to effective healing of cracks. Light mediated [2 + 2] cyclodimerization in coumarin, anthracene and cinnamate moieties endows excellent dynamic and reversible photoresponsive systems.14,14g Incorporation of these moieties into the polymeric chain is an interesting approach to design photoresponsive self-healing materials for smart coating applications. However, this requires judicious optimization of a number of critical parameters such as low viscosity, ease of processing, good control over the molecular weight, crosslinking ability, flexibility vs. rigidity, architectural control, tacticity in a multiphase, compartmentalized network. A rational design approach to mitigate such multiple structural control and modulate the healing properties is not so far well-explored in literature. In that regard, chain collapse of single chain polymers has emerged as an elegant strategy to design low viscosity polymers to meet most of the above-mentioned parameters. Such facile chain collapse strategy mediated by coumarin photodimerization or encapsulation in cyclodextrin cavity to result concentration dependent nanostructures formation, has been efficiently exploited by our group for self-healing and drug delivery applications.15 The coumarin appended low molecular weight synthetic random copolymers through reversible photodimerization exhibited excellent control over flexibility vs. rigidity to result a smart system with photomodulated interconversion of autonomic and nonautonomic self-healing pathways. However, the generality and the fidelity of the chain collapse strategy towards developing such flexorigid domains has not been studied, more so to find out the material's suitability for coating applications. Thus, we envisaged in designing a photo-crosslinkable polymer as a perfect self-healable coating for photovoltaic cells that would have interesting attributes such as photo-controlled healing response and hydrophobic barrier to prevent moisture seepage. Moreover, photo-responsive thymine moiety in the protective layer would prevent the high intensity UV light, thereby retarding the degradation of solar cells.
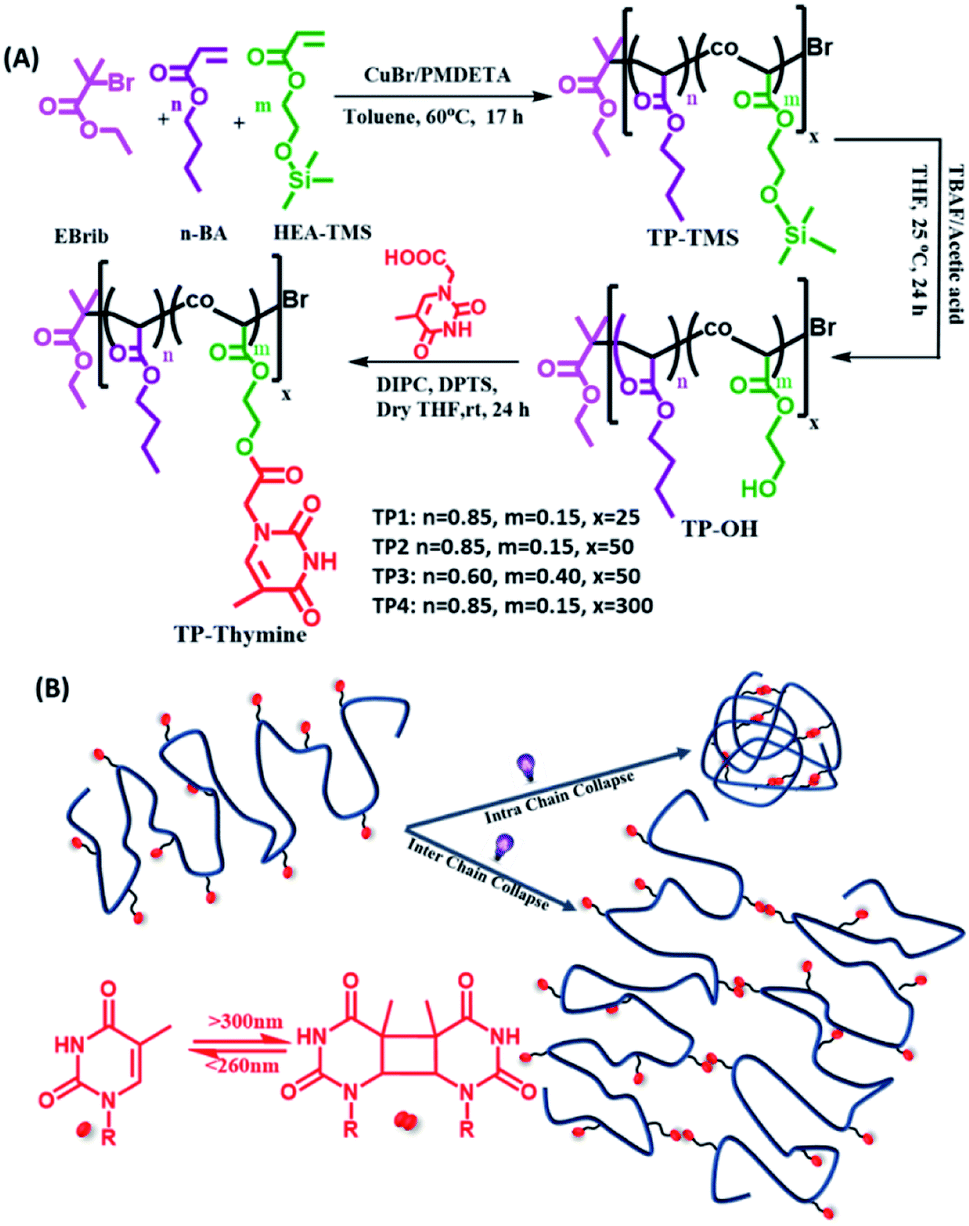
Herein, we designed a new series of single chain random copolymers (TP1–TP4) from butyl acrylate and photoresponsive thymine grafted hydroxyethyl acrylate monomers (Scheme 1). The UVB mediated photodimerization chemistry of the pendant thymine moieties were investigated using UV and NMR spectroscopy to show the formation of cyclobutane adducts that can be dynamically photo-cleaved upon irradiation with UVC light.14d A number of parameters such as solvent environment, chain length, functionality% and concentration of the polymers regulate the efficacy and kinetics of the intra or interchain collapse. This eventually renders a flexorigid compartmentalized network with locally crosslinked compact domains randomly spread in the flexible polymer chains and draws its structural analogy with thermoplastic elastomers. Such strategy for photoresponsive chain collapse is exploited to modulate the thermal property of the material and eventually to control self-healing behaviour by molecular interdiffusion of polymer chain across the cut surfaces. Moreover, the self-healing efficacy could be tuned with the film thickness and it also remains unaltered upon using solar simulator or direct sunlight. Lastly, the hydrophobic nature of the smart polymeric coating surface renders it an ideal candidate for the application of protective coating on the otherwise unstable photovoltaic devices.
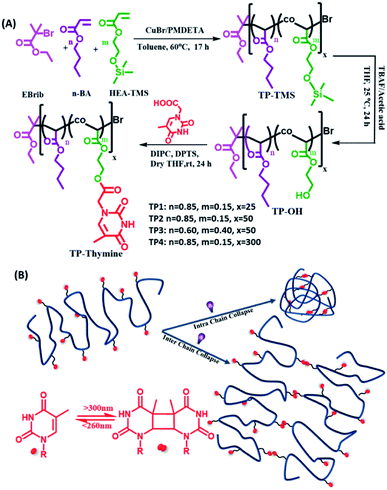 |
| | Scheme 1 (A) Synthetic scheme for the thymine functionalized polymers possessing different functionalities% and degree of polymerizations. (B) Schematic representation of the polymers undergoing homo-functional intra and interchain collapse by means of reversible photodimerization of thymine. | |
2. Experimental section
2.1. Photodimerization study with UV and 1H-NMR
The polymers (TP1–TP4) were taken in DMF at a concentration of 25 mM and diluted with THF, CHCl3, DMF to a concentration of 0.01 mM in mixed solvent system of 0.4 v/v% DMF–THF, DMF–CHCl3. Upon exposure to UVB light (λmax = 320 nm, 1 × 8 Watt UVB lamp) they showed a gradual decrease in the characteristic absorption of the thymine at a wavelength range of 280–350 nm with time. Photodimerization degree was calculated using, PD = (A0 − At)/A0, PD% = (A0 − At)/A0 × 100 where At is the absorbance at time t and A0 is initial absorbance.
The polymer TP1 was taken in CDCl3 (1 mM) and 1H-NMR spectra were recorded upon exposure to UVB light at different time intervals of 0, 3, 6 and 12 h. PD% was calculated according to the formula PD% = {(I0 − It)/I0} × 100 where It = Il/IBA, where Il and IBA are the integral area of the peak corresponding to reactive l proton and non-reactive butyl acrylate (BA) protons. The crosslinked polymer di-TP1 was then irradiated with UVc light at different time intervals of 0, 3, 6 h and PD% was calculated using PD% = {(It − I0)/It} × 100 where It = Il/IBA, with Il and IBA being the integral area of l and BA protons, It is the ratio of the integral area of the said protons at a specific time of UVc irradiation (t).
2.2. Microscopic analysis
10 μL of the diluted polymer samples (TP1–TP4) (c = 10−3, 10−1 and 1 mg mL−1) were drop-casted on silicon wafer followed by washing with the same solvent and dried overnight. AFM height images were recorded by tapping mode on a Bruker Multimode 8 scanning probe microscope with silicon cantilever (Bruker) and analysed using the software NanoScope Analysis 1.5. The size distributions of the nanoparticles were analyzed using image-J software, from the U.S. National Institutes of Health.
15 μL polymer solution at a varied concentration (10−3, 10−1 and 1 mg mL−1) were drop-casted on a 300 mesh carbon-coated copper grid. After ∼5 min, excess solution was blotted using Whatman filter paper. The extra solution was then wicked off from all edges of the grid carefully. Grids having samples were then dried in the desiccator under vacuum for 1 day. TEM images were recorded using JEOL JEM 2100 with a Tungsten filament at an accelerating voltage of 2000 kV.
2.3. Thermal studies by differential scanning calorimetry
The native polymer samples in THF (20 mg mL−1) were drop-casted on the glass substrate and dried properly. The dried sample on the glass sample was exposed to UVB irradiation for 15 h (84 J cm−2) for crosslinking of the polymer. Further, the crosslinked sample was exposed to UVC irradiation for 5 h (15 J cm−2) and decrosslinked. Thermal studies of the polymer samples were investigated on PerkinElmer Differential Scanning Calorimeter DSC 8000 model upon heating the samples from −80 to 150 °C at a heating rate of 10 °C.
2.4. Thickness measurement
20 μL of polymers samples in THF (20 mg mL−1) were drop-casted on coverslip to prepare thin film of the polymers (TP1–TP4) using spin coater Apex Instruments Co. Pvt. Ltd. The thickness of polymeric film on glass slide was measured using spectroscopic ellipsometer from Angstrom Sun Technology Inc. USA. and thickness was calculated using the best-fit parameters in the Cauchy equation.16| | |
n(λ) = A + B/λ2 + C/λ4
| (1) |
2.5. Contact angle measurement
The contact angle of the prepared polymeric film was measured using the sessile drop method in drop shape analyser with model DSA25 from KRUSS GmbH, Germany.
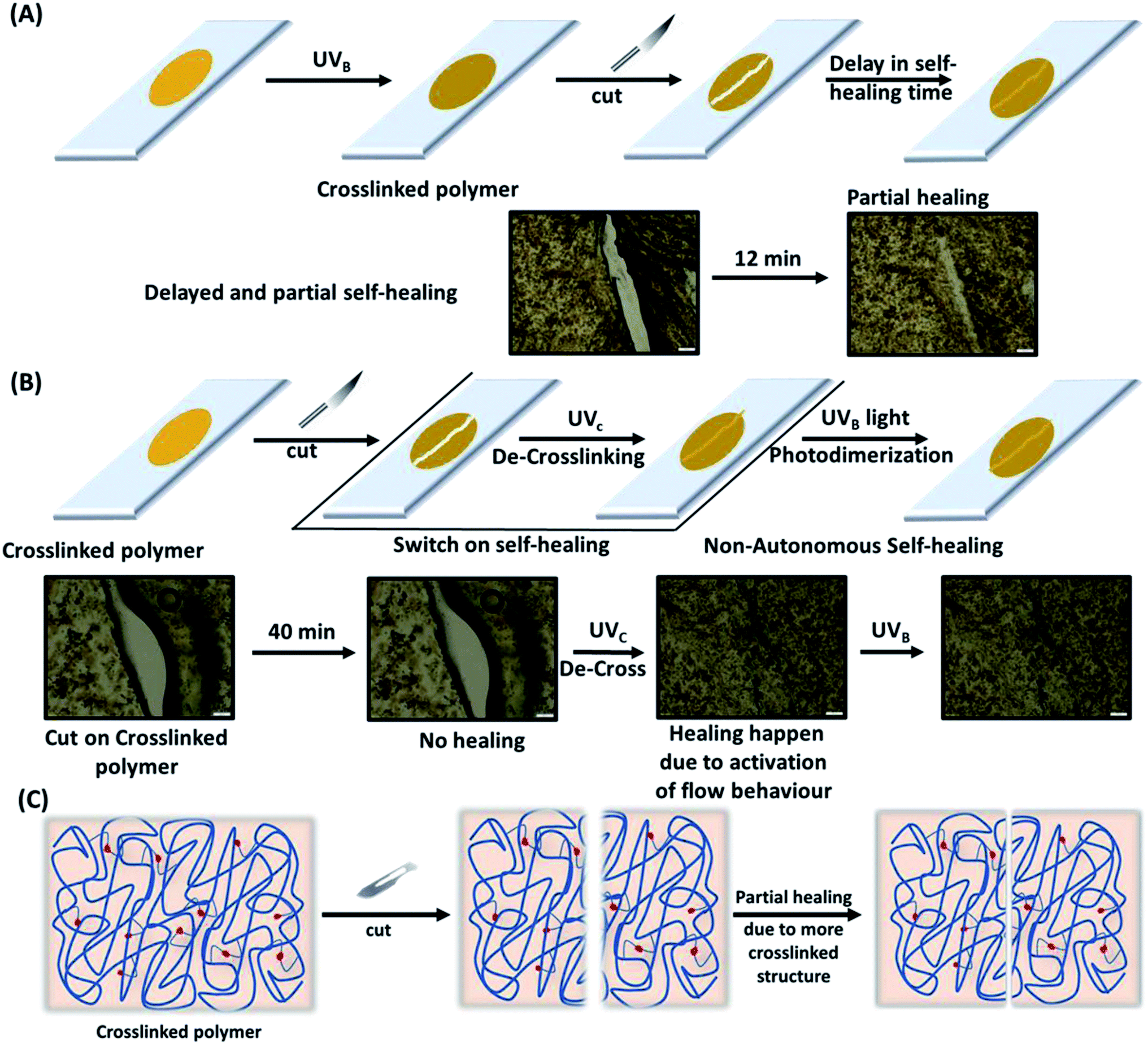
2.6. Self-healing studies
The polymers (20 mg mL−1 in THF) were drop-casted on a glass substrate using spin coater Apex Instruments Co. Pvt. Ltd. and were air-dried to render stable films. A cut was made onto the coated sample using a surgical knife and investigated for autonomic self-healing with the help of an optical microscope Olympus IX73 bright field microscope. The polymeric film was photo-crosslinked using UVB irradiation that resulted in a delay in self-healing. Further, the crosslinked polymer film was exposed to UVC to result de-crosslinking of the polymer with regaining self-healing efficacy in a non-autonomic manner. The percentage of healing was calculated from the ratio of change in the cut width to the initial cut width. The average width of the scratches was determined using image-J software by taking 10 random cut width from different areas of the cut. The average width (Wn), percentage healing efficiency (H%) were calculated17 using eqn (2) and (3), where Ni is the number of widths, Wi width at position n.| |
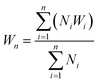 | (2) |
| |
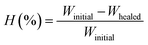 | (3) |
3. Results and discussion
3.1. Synthesis and characterization of photoresponsive copolymers
The copolymer of n-butyl acrylate (BA) and trimethylsilyloxyethyl acrylate (HEA-TMS) were synthesized using atom transfer radical polymerization (ATRP) with different grafting percentage of BA and HEA-TMS and different degrees of polymerization. The TMS group was deprotected to result hydroxy group to which functionalized appended thymine moiety through DIPC/DPTS condensation to form (TP1–TP4) polymers. Thus, all the living polymers had linear polyacrylate backbone backbones with random grafting of n-butyl, hydroxyl ethyl and thymine functionalized hydroxyl ethyl chains. The molar feed ratios of BA and HEA were varied to obtain the polymers with degree of polymerizations 25 (TP1), 50 (TP2, TP3) and 300 (TP4) (Scheme 1). The resulting polymers were characterized by 1H-NMR, UV spectroscopy, elemental analyses and size exclusion chromatography (SEC). 1H NMR spectra of the polymer confirmed the grafting of the thymine characteristic singlet peaks of thymine at chemical shift regions of δ 7.17, 1.92 ppm corresponding to the methylene and methyl protons respectively. The percentage grafting of the thymine group was calculated by end group analysis (the ratio of collective integral for the protons corresponding to the thymine group with that of the terminal methyl proton of butyl acrylate chain) from 1H NMR, UV spectra and elemental analyses (cf. ESI, Fig. S1†). The % of thymine functionality for TP1 and TP2 were similar (∼12%), while TP3 had higher ∼24% of grafting. The longer polymer TP4 showed rather lower ∼8% thymine incorporation. Table 1 summarizes the characteristic details of the polymers including the percentage grafting, average molecular weights (Mn, Mw), PDI values, hydrodynamic size, DDLSH. SEC analysis exhibited good control over the molecular weights and polydispersity indices (PDI) of the polymers in the range of 1.1–1.3. The hydrodynamic sizes of the polymer showed a power law relation, RH ∝ Mwν with a flory exponent value (ν) of 0.56 that closely matches with expanded coil state of the polymer chains (Fig. S2†).18
Table 1 Showing characterization details of the polymers
| Polymer |
Functionalitya (%) |
Mnb (Da) |
Mwb (Da) |
PDIb |
RCALH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c c |
RDLSHd |
| Determined by 1H NMR, UV spectra and elemental analyses. Determined by SEC analysis in THF using polystyrene as standard. Hydrodynamic radii RCALH as calculated according to the formula RCALH (nm) = 1.44 × 10−2Mw0.561.18 Determined by DLS (polymer concentration = 6 mg mL−1 in THF). |
| TP1 |
12 ± 2 |
4729 |
5796 |
1.2 |
1.9 |
20 |
| TP2 |
12 ± 1 |
6759 |
9297 |
1.3 |
2.4 |
28 |
| TP3 |
24 ± 3 |
11624 |
12942 |
1.1 |
2.9 |
22 |
| TP4 |
8 ± 2.5 |
33075 |
35764 |
1.1 |
5.2 |
50 |
3.2. Investigation of dynamic thymine photodimerization
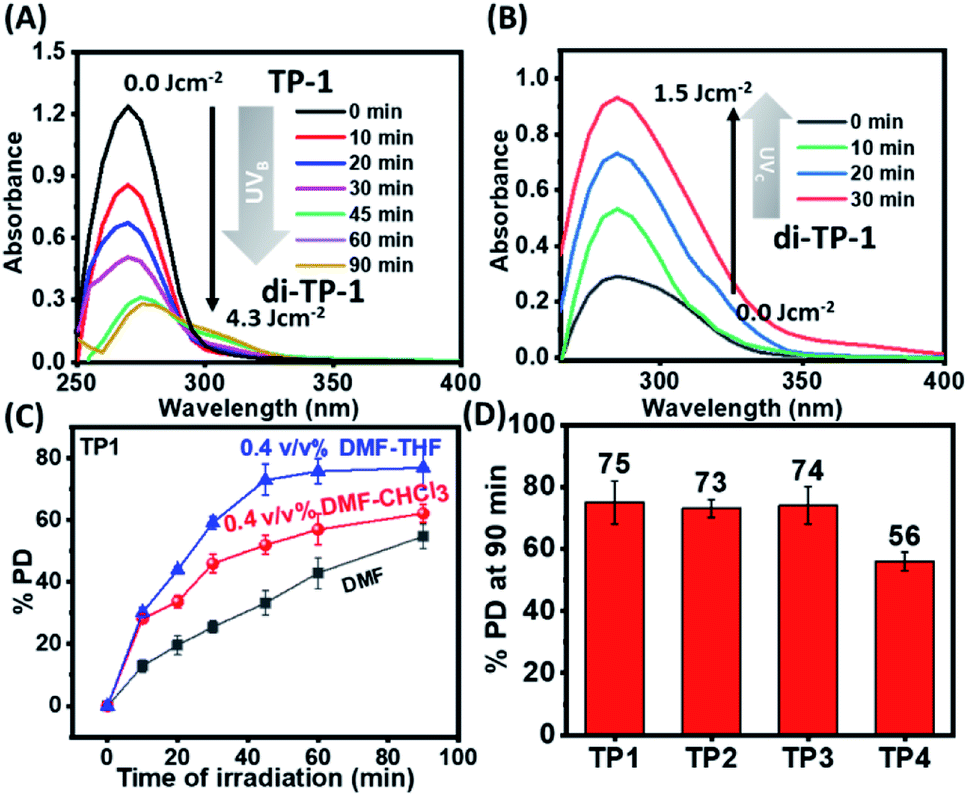
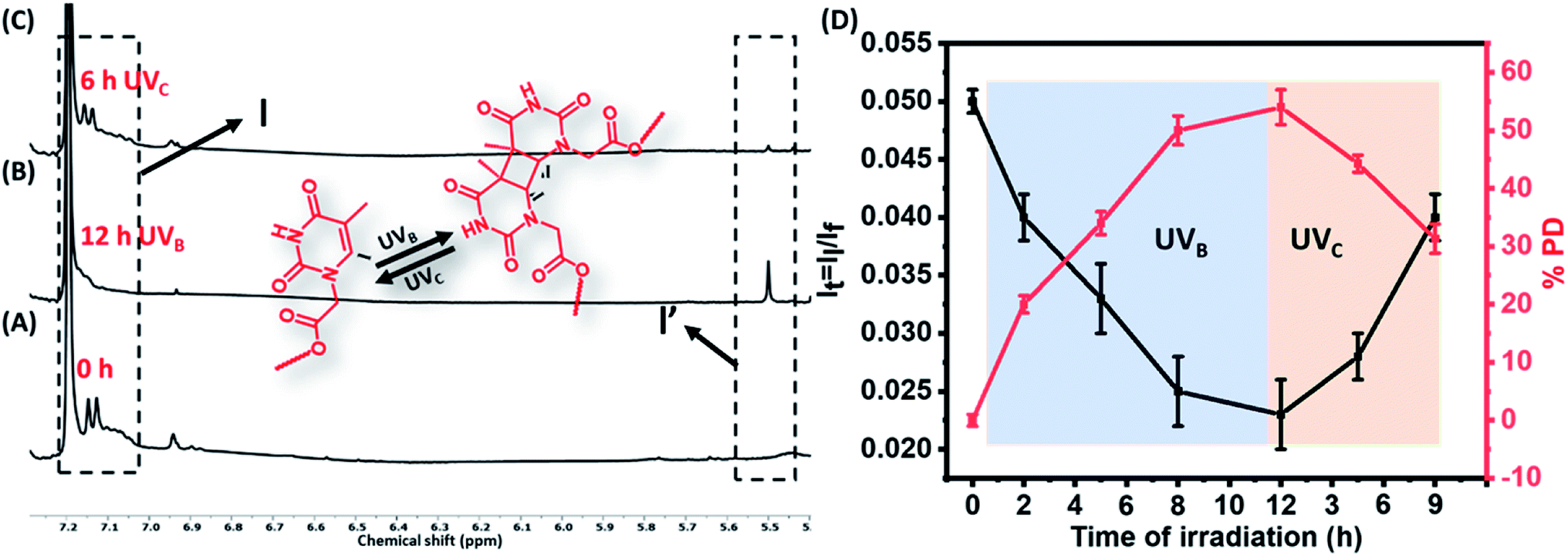
Dilute solution of the polymers (TP1–TP4) (0.01 mM) exhibited a characteristic absorption in the wavelength regime of 270–300 nm that upon UVB irradiation rendered a gradual decrease in absorbance (Fig. 1A & S3†). This suggested the formation of [2π + 2π] cyclobutane adduct between thymine moieties to result di-(TP1–TP4) polymers. Furthermore, the polymers showed retrieval of absorbance upon irradiation of di-(TP1–TP4) with UVC light within 30 min indicating the dynamic nature of photodimerization chemistry (Fig. 1B). In order to avoid the intermolecular hydrogen bonding interactions between the self-complementary groups present in thymine and HEA group side chains,15 the polymers (TP1–TP4) were taken in polar aprotic DMF stock solution. Further, the solution was diluted with THF or CHCl3 to furnish a 0.4% DMF–THF mixed solvent system. The polymer TP1 exhibited faster photodimerization kinetics leading to saturation at higher PD (75%), in 0.4 v/v% DMF–THF and DMF–CHCl3 solvent system as compared to the slower kinetics and low PD% in DMF only solvent (Fig. 1C). This was in corroboration with the compatibility of the Hildebrand solubility parameters (δ) of the solvents with butyl acrylate main chain backbone (17.9 Mpa1/2) that determines solvent polymer interactions.19 Thus, the mixed solvents system with maximum volume percentage of THF (18.5 MPa1/2) or chloroform (18.7 MPa1/2) are good solvents for the polymer owing to high volume of chain interaction and effective exposure of functional groups in expanded coil structures. However, DMF with δ value 24 MPa1/2 results in rather compact and folded polymer chains. Upon comparing the polymers (TP1–TP4), differing in chain length and functionality%, we found that the polymers (TP1–TP3) with smaller chain length showed faster photodimerization (PD = 74–75% in 90 min) with higher second-order rate constants (Fig. 1D and Table S1†), as compared to their longer chain analogues, TP4 (56%). This suggests a direct relation of chain length of the polymer with the polymer chain collapse time in entangled system according to relation (τ ∝ M3) which led to a slower photodimerization rate for TP4 with longer polymer chains.20 Hence, the factors like solvent environment, extent of photoresponsive functionality, the chain length of the polymer moiety played a pivotal role in the kinetics of photodimerization. Further, the percentage of photodimerization and reversal of photodimerization were also analysed by monitoring the polymer solution using 1H-NMR at different time intervals of UVB and UVC irradiations. The intensity of characteristic aromatic peak of the thymine moieties (δ 6.9–7.3) in the TP1 polymer decreased gradually with increased time of exposure to the irradiation owing to the photodimerization (Fig. 2A–C & S4†). After 12 h of UVB irradiation the PD% values reached 54% (Fig. 2D). The appearance of a new peak δ 5.5 ppm corresponds to cyclobutyl protons owing to thymine photodimerization as corroborated with the literature.21 Further, upon irradiating UVC light, the characteristic signal of cyclobutyl protons gradually diminished, thus indicating photo-reversible formation and cleavage of the [2 + 2] adduct. We performed consecutive irradiations with UVB and UVC light over four cycles to study the photo reversible nature of TP1 (Fig. S3F†). Upon UVB mediated crosslinking the PD% values increased to ∼45–50% and upon UVC mediated decrosslinking it decreased to ∼10–17%. Interestingly, as compared to native TP1 before irradiation, a residual value of PD% after consecutive irradiation indicated partial photo reversible nature of the thymine grafted polymers.
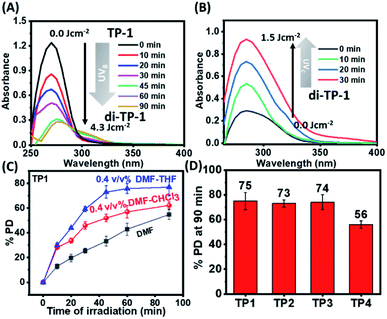 |
| | Fig. 1 Monitoring photodimerization of the polymers by UV spectra. (A) Gradual decrease in UV absorbance with the time of UVB irradiation for the polymer TP1 in 0.4 v/v% DMF–THF and (B) retrieval of absorbance with the time of UVC irradiation for the polymer di-TP1. (C) Graph showing PD% at each time interval of UVB irradiation for the polymer TP1 in different solvent system. (D) % degree of photodimerization for the polymers TP1–TP4 after 90 minutes of UVB irradiation in 0.4 v/v% DMF–THF solvent system (concentration = 0.01 mM, UVB irradiation with 1 × 8 W lamp [λmax = 320 nm], UVC irradiation with 1 × 8 W lamp [λmax = 254 nm]). | |
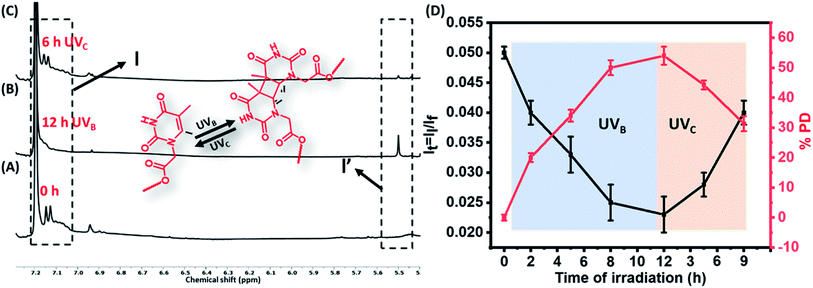 |
| | Fig. 2 Monitoring photodimerization of the polymers 1H NMR. Stacked partial 1H NMR spectra of the polymer TP1 (A) before and (B) after 12 h irradiation of UVB to form di-TP1, a [2π + 2π] cyclobutane adduct. (C) Partial 1H NMR spectra of the polymer TP1 after 6 h irradiation of UVc. (D) Graph showing the change in the integral ratio of the protons of thymine moiety (black line) and photodimerization% degree (red line) with time course of UVB and UVC irradiations. | |
3.3. Investigation of polymer chain collapse
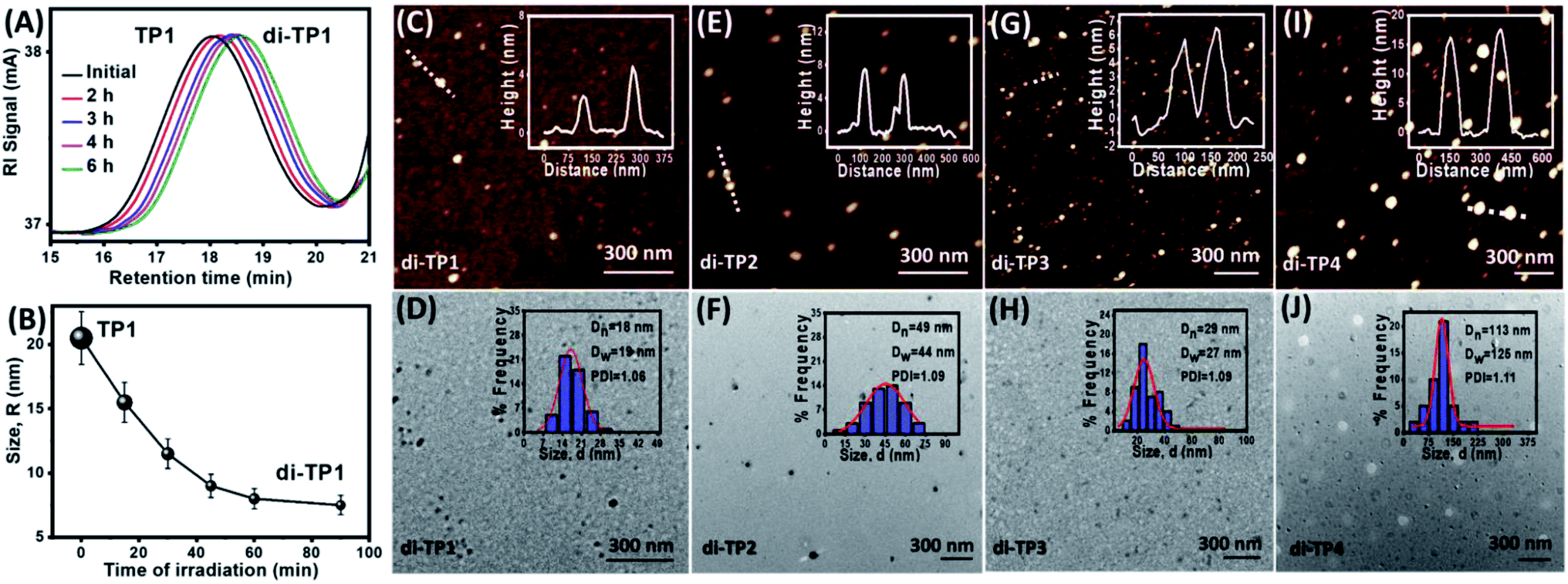
After establishing efficient thymine photodimerization, we further studied the subsequent polymer chain collapse using SEC and DLS techniques. Upon irradiation of TP1, the gradual increase of the retention time in the SEC refractive index signal confirmed a decrease in the size of the polymer (Fig. 3A). This was further corroborated through DLS that exhibited a gradual decrease in the hydrodynamic radii of TP1 upon irradiation suggesting a compaction of the extended coil of the polymer chains (Fig. 3B). Next, the morphology of the nanostructure formation owing to the chain folding was investigated by atomic force microscopy and transmission electron microscopy. Upon irradiation, the polymers exhibited globular nanoparticle like morphologies at dilute concentration (10−3 mg mL−1). Thus, TP1 showed the formation of nanoparticles (∼12 nm) (Fig. 3C, D and S5†) whereas (TP2-TP4) showed comparatively larger nanoparticles (Fig. 3E–J). Among TP2 and TP3 with similar chain length albeit with different functionality% of thymine, TP3 demonstrated higher compaction in size which was in corroboration with the DLS data (Table 2). Such compaction of the expanded coil structures of the polymer chains led to the formation of intrinsically disordered domain. At dilute concentration regime (10−3 mg mL−1) of di-(TP1–TP4), the chain folding led to the formation of hemiellipsoidal nanoparticles in the size range of 10, 22, 15 and 47 nm respectively (Fig. 3C–J). The hemiellipsoidal geometry of the soft particles owing to the surface deposition and drying mediated flattening was corrected for spherical nanoparticle.22 The spherical radii depends on the polymer chain length and functionality% and correlates well with the DLS data. Histogram analyses for the nanoparticles exhibited polydispersity values close to 1 suggesting excellent control over the size of the nanoparticles. Next, the photoresponsive polymers exhibited concentration dependent formation of different nanostructures upon UV exposure (Fig. 4). Thus, irradiation performed at higher polymer concentrations (1 mg mL−1) led to interchain polymer collapse resulting in supramolecular fibers or aggregates. The polymer at lower concentration adopts a globular nanoparticles morphology due to intrachain chain collapse. However, at an intermediate concentration (0.1 mg mL−1) we could observe the onset of transition from intrachain to interchain collapse to render fused nanoparticles and nanofibers. Also, the polymer chain length with higher functionality% on interchain photdimerization led to fiber like or toroidal morphologies of di-TP3. The interchain collapse of flexible polymer with longer chain, TP4 rather exhibited aggregated globules even at a macromolecular high concentration. Thus, the polymer chains (TP1–TP4) in expanded coil could be directed to globular nanoparticles at dilute concentration and higher order structures in high polymer concentration upon tweaking a number of important parameters.
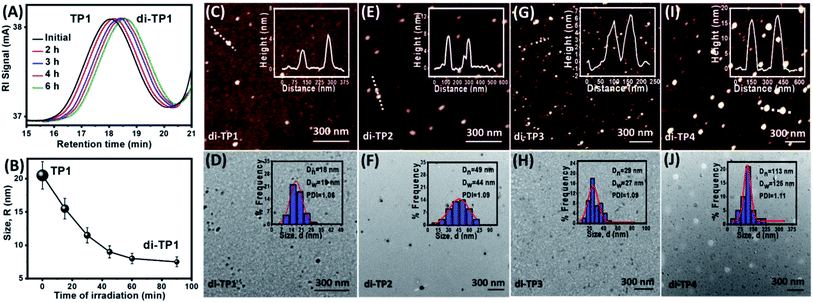 |
| | Fig. 3 (A) SEC analyses showing a delay in retention volume upon UVB irradiation of polymer sample TP1 (irradiation of sample solution (c = 6 mg mL−1 in THF with 1 × 8 W UVB lamp, 6 h) (B) DLS data showing hydrodynamic radii (RH) for the polymer TP1 upon irradiation (irradiation of sample solution, c = 10−3 mg mL−1 in 0.4 v/v% DMF–THF with 1 × 8 W UVB lamp, 8 h). AFM and TEM images of irradiated samples and their corresponding histogram analyses from (C and D) di-TP1, (E and F) di-TP2, (G and H) di-TP3, (I and J) di-TP4. | |
Table 2 Characterization details of the polymers after UV irradiation
| Polymers |
RDLSHa (nm) |
Compaction (%) |
RadiiAFMb (RAFM, nm) |
Radiisphericalc (Rs, nm) |
RadiiTEMd (RTEM, nm) |
| Hydrodynamic radii is determined by the dynamic light scattering studies. Radii determined from the AFM analysis. Spherical radii of nanoparticle absorbed on silicon surface. Radii of nanoparticles determined from TEM analysis. |
| di-TP1 |
7 |
70 |
10 ± 2 |
6 |
9 |
| di-TP2 |
11 |
61 |
22 ± 1 |
13 |
24 |
| di-TP3 |
8 |
64 |
15 ± 2 |
9 |
14 |
| di-TP4 |
24 |
52 |
47 ± 3 |
26 |
55 |
 |
| | Fig. 4 Scheme depicting formation of concentration dependent nanostructures as a result of the crowding of the polymer chains dictating intra vs. interchain collapse. AFM images of the polymers (A) di-TP1, (A) di-TP2, (C) di-TP3, (D) di-TP4 in the concentration regimes of 0.001, 0.1 and 1 mg mL−1 in 0.4 v/v% DMF–THF (left to right) (irradiation with 1 × 8 W UVB lamp, 8 h). | |
3.4. Photo modulation of the thermal properties
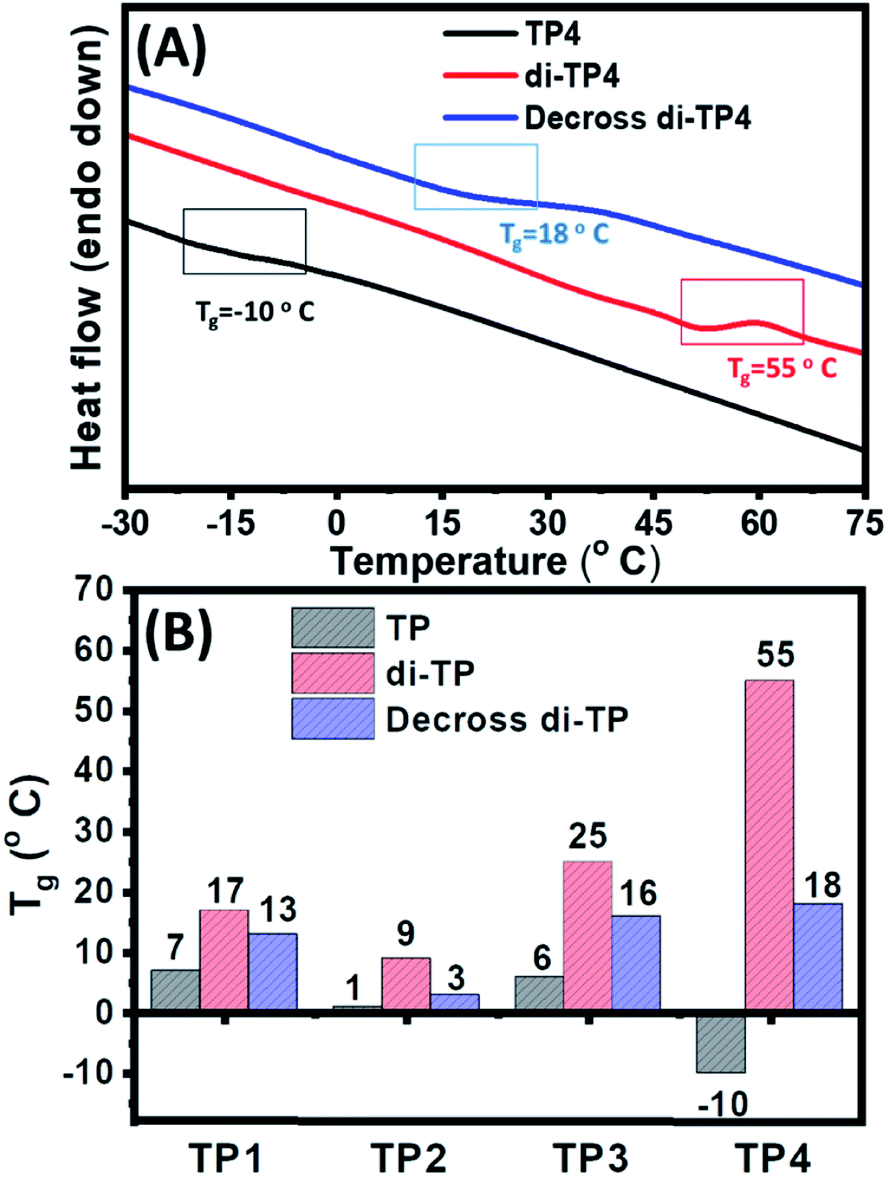
Next, the thermal property of the polymer upon photocrosslinking and decrosslinking was studied using differential scanning calorimetric (DSC) studies in the context of flexible-rigid domain of the copolymer chains. The glass transition temperature (Tg) responsible for a rigid glassy state to a soft rubbery state of polymer, plays a pivotal role in the autonomic self-healing process of the thermoplastic elastomers. Poly butyl acrylate polymer chain shows a typical low value of Tg (−54 °C) owing to the flexibility of the polymeric chain. However, an increase in the Tg value was attributed to the grafting of relatively rigid co-monomers, hydroxyl ethyl acrylate and thymine moieties in the polymers (TP1–TP4) to push the Tg values to a range of −10 °C to 7 °C (Fig. 5 & S6†). This was evident from the highest Tg value (6 °C) of TP3 with 24% of the rigid comonomers, while TP4 (8% rigid co-monomers) exhibited the lowest Tg value (−10 °C) among all the polymers. Photo-irradiation mediated crosslinking of the polymer samples for ∼5 h resulted in an upward shift in the Tg values presumably due to the loss of flexibility and increase in rigidity of the system with concomitant decrease in mobility of the polymer chains.20 This change in the Tg value was found to be highest in the case of di-TP4 (ΔTg = +65 °C) with longest polymer chain (DP 300). It is to be noted that higher amount of thymine crosslinking led to rather high ΔTg for di-TP4 while for the polymers di-(TP1–TP3) factors such as lower DP and rather lower amount of thymine moieties led to the rather moderate change in the Tg value (Table S1 and Fig. S6†). Furthermore, the polymer films also exhibited a reversal of the Tg values upon irradiating the cross-linked polymer samples with UVC light for 3 h indicating the photo-reversible nature of the dynamic covalent bond in the thymine cyclobutane adduct. Such reversible changes in Tg value for the solid film of TP1–TP4 indicated that the photodimerization and reverse cycloaddition along with maximal change of (ΔTg) was observed in TP4 to play an important role in balance of flexo-rigid segments in the chain. This inspired us to investigate the self-healing of the polymer films at ambient condition before and after UV irradiation.
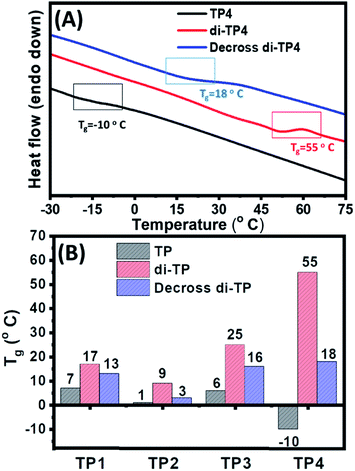 |
| | Fig. 5 (A) Representative DSC traces showing effect of photodimerization on the glass transition temperature of the polymer films (20 mg mL−1) of native TP4, crosslinked di-TP4 and decrosslinked di-TP4. (B) The glass transition temperatures for TP1–TP4 polymers in native, crosslinked and decrosslinked forms. | |
3.5. Self-healing behaviours of the polymeric films
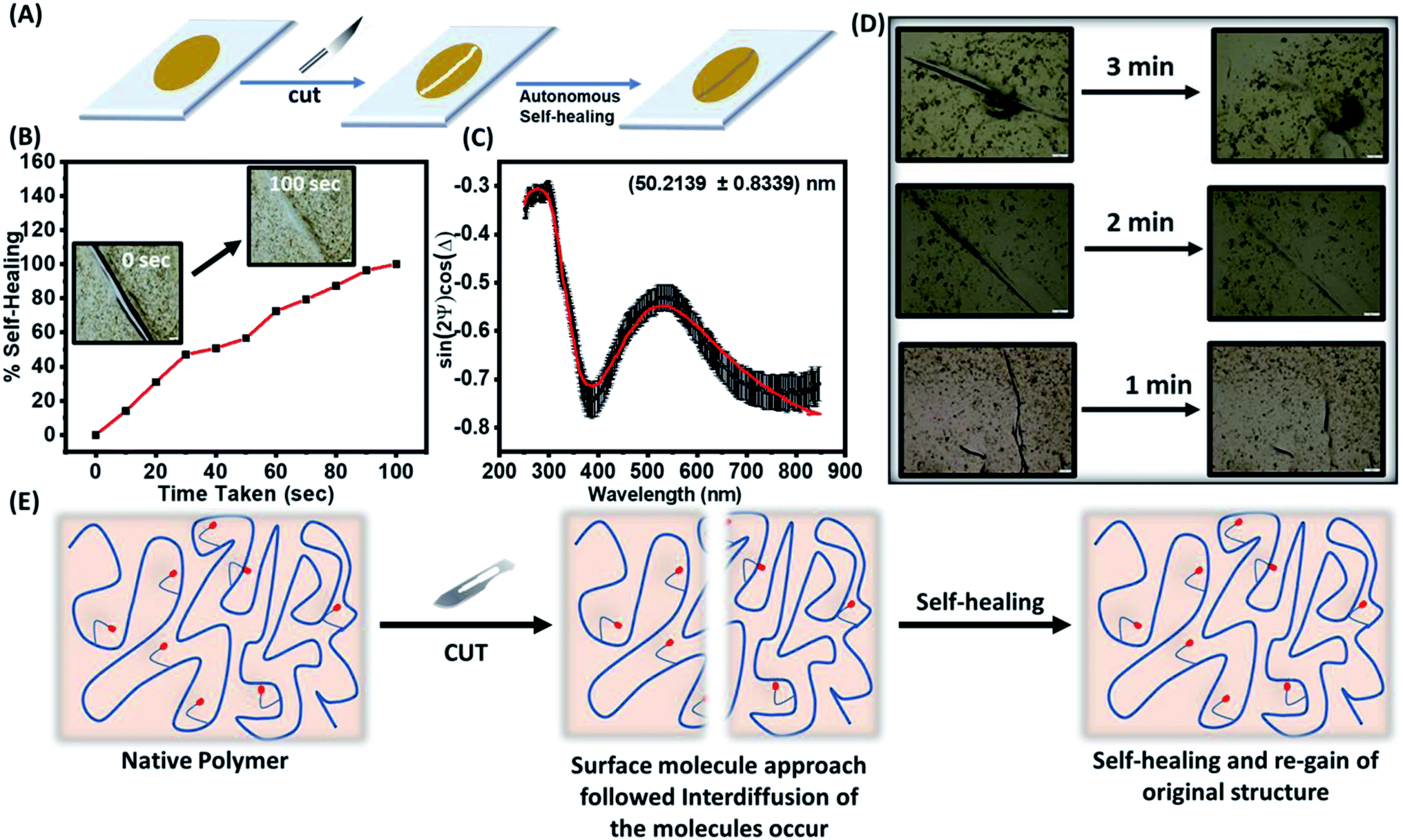
3.5.1 Autonomic self-healing. The transparent polymeric film of TP1–TP4 on a glass surface were prepared following two approaches: firstly, the polymers (TP1–TP4) were drop-casted on a glass plate and dried to render stable films. Secondly, thin film of polymers with different thickness on a glass substrate was made using the spin-coating to get a uniform polymeric layer. A surgical blade was used to make a micron level cut on the surface of the film that was investigated for healing at room temperature under the bright-field optical microscope (Fig. 6A). Polymer chain mobility at the temperature greater than the Tg values promotes the enthalpy driven interfacial diffusion of the chains across the cross-sectional cut surface leading to the healing of the cracks as shown in self-healing mechanism in (Fig. 6E). Thus, a polymer with low Tg value owing to increased molecular diffusion across the interface could have a higher polymer–polymer interface for autonomic crack healing even without the intervention of external environment condition.23 Upon studying different polymeric films towards autonomic and intrinsic self-healing behaviour without any external healing agent at ambient temperature, we found that TP1–TP3 film exhibited slower autonomic self-healing or no effective healing (Fig. S7†) owing to containing higher percentage of rigid comonomers in the polymer backbone. However, optical microscopic images of the native TP4 film with time exhibited a speedy and effective self-healing behaviour (Fig. 6B). Thus, the polymer TP4 having low Tg, high ΔTg values upon photo-crosslinking ability emerged as an ideal candidate for investigating further healing behaviour.
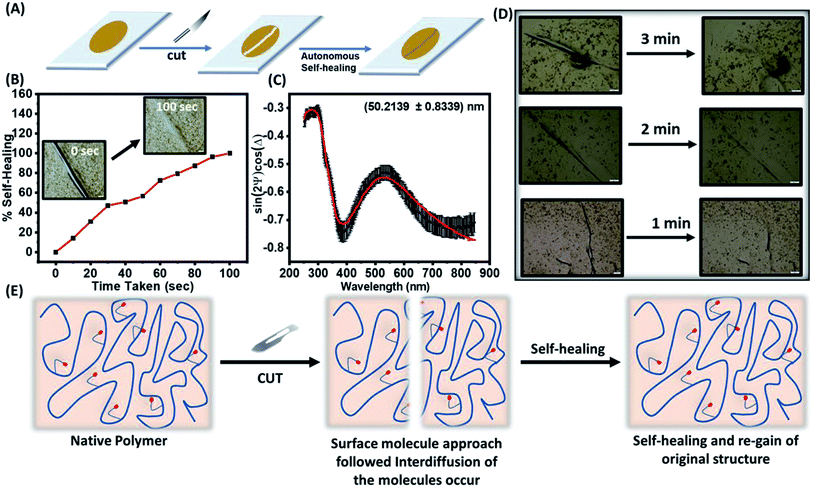 |
| | Fig. 6 (A) Schematic diagram to depict autonomic self-healing pathway after introduction of a crack. (B) A cut of average width 62 μm on a film of TP4 showing complete autonomic healing captured at different time intervals. (C) Ellipsometric parameters vs. wavelengths for a single polymeric layer upon fitting to Cauchy equation rendered ∼50 nm thickness of the film. (D) Thickness dependent autonomic self-healing of TP4 films with the thickness of 50, 115 and 183 nm. (E) Mechanism of self-healing via interdiffusion of the polymer chains across the cut surfaces. | |
3.5.2. Effect of thickness on self-healing properties. Next, the effect of film thickness over the autonomic self-healing behaviour was studied (Fig. 6C). The polymeric films spin-coated on the glass surface were subjected the thickness measurement using ellipsometer. Fitting in the Cauchy equation that accounts for the dispersion formula to calculate the thickness of the film, rendered us a reasonable value of the thickness of the polymeric films (Table 3 & S3†). We noted that the healing efficiency slightly enhanced upon increase in the thickness of the films from 50 nm to 115 to 183 nm against a same size of the cut (Fig. 6D). The increased mobility of polymeric chain with increased cross-sectional area resulted in improved self-healing at a faster rate (Fig. 6E).24
Table 3 The thickness dependent healing efficiency
| Polymer |
Thickness (nm) |
Goodness of fit |
Healing time |
Healing efficiency (%) |
| TP4 |
50 ± 0.84 |
0.9910 |
3 min |
95 |
| 115 ± 4.74 |
0.9613 |
2 min |
92 |
| 184 ± 5.3 |
0.9645 |
1 min |
94 |
3.5.3. Non-autonomic self-healing. Furthermore, a cut in the photo-crosslinked di-TP4 polymer films exhibited a delay in the healing process (up to 12–15 min) (Fig. S8†). This is due to the photo-crosslinking of the thymine moiety present in the polymeric chain that led to an increase in rigidity in the system with eventual decrease in mobility of the polymeric chain to render a delay in autonomic mechanism of healing (Fig. 7A). Next, we envisaged on the non-autonomic self-healing behaviour of the polymeric film. A cut was introduced on the photo-crosslinked sample that does not exhibit self-healing even after 40 min. This upon irradiation with UVC light, led to decrosslinking with concomitant increase in the mobility of the polymer chain, thereby switching the healing process on. Further, the polymer was photo-crosslinked with UVB light to switch off the self-healing process. Thus, formation of the cracks and photo modulated self-healing could be repeated in cycle to promote longer durability of the coating.
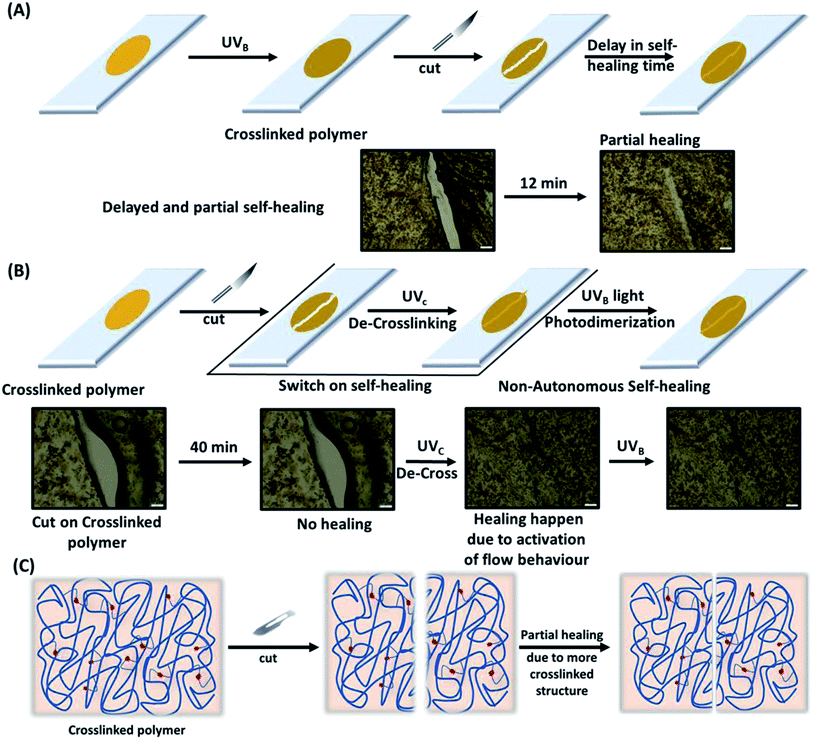 |
| | Fig. 7 Self-healing pathway with corresponding optical microscopy images depicting the autonomic and non-autonomic mechanisms of the healing. (A) Photo-crosslinked film of polymer di-TP4 exhibits delayed and inefficient autonomic healing of a cut on the film. (B) Non-autonomic self-healing of the cut on the crosslinked polymer film of di-TP4 as mediated by UVC light and further photo-fixed with the help of UVB light. (C) Schematic mechanism showing intervention of rigidity in the crosslinked polymer network leading to reduction in the healing efficacy. | |
3.5.4. Effect of artificial and natural sunlight on self-healing properties. After establishing single wavelength mediated switching on–off healing process, we intended to investigate the healing ability of the film under simulated solar spectrum conditions. This is critical as in natural conditions both the forward and reverse photoreactions happens simultaneously, thereby establishing possible dynamic photostationary states. Thus, we investigated the effect of artificial and natural sunlight on the self-healing property for both native and cross-linked TP4 samples (Fig. 8A and B). The polymeric film of thickness 183 nm was irradiated with oreal solar simulator (100 mW cm−2) to create artificial sunlight and with natural sunlight (max 83 mW cm−2) in two different experiments. This could establish the efficacy of in situ dynamic photo-healable coating in presence of a standard solar spectra range that is more ideal than single wavelength lamps. However, it is noteworthy that the maximum radiation intensity of the solar spectra lies at 500 nm in the visible range and less on the UV side. Thus, working wavelength of the photovoltaic device (∼500 nm) and coating materials (250–320 nm) could be complementary to each other. Moreover, the coating materials might prevent the excess UV light that could potentially damage the photovoltaic cells with hot electron generation. For native TP4 the efficacy of healing behaviour remained comparable in the presence of both natural sunlight and artificial sunlight with crack of getting healed in 2 min. Interestingly, the crosslinked di-TP4 polymer also exhibited self-healing albeit in 15 min, that is in line with the previous self-healing experiments with single wavelength. Thus, in natural sunlight and artificial sunlight, we anticipate that both autonomic and non-autonomic self-healing mechanisms work in tandem. Moreover, the working principle of photovoltaic device was not affected by the coating of our polymeric materials that work on the UV region to make it an ideal candidate for coating application.
 |
| | Fig. 8 Self-healing of native and crosslinked polymeric films, (A) TP4 and (B) di-TP4 (thickness 183 nm) as seen from the optical microscopy images in presence of natural sunlight and artificial sunlight using solar simulator. | |
3.6. Surface hydrophobicity
Next, the surface hydrophobicity was measured using static contact angle measurement. For the polymeric film to be envisaged as hydrophobic, its static contact angle should be greater than equal to 90°.25 As compared to the control glass substrate (∼35°), TP1–TP4 native polymers film exhibited contact angles ∼90° suggesting hydrophobic nature of the polymeric films (Fig. 9 & S9†). The increase in thickness of the polymer film resulted in gradual increase in the hydrophobicity of the film. Moreover, TP4 of thickness 183 nm showed a contact angle ∼94° that increased to 97.5° with photo-crosslinking of the film. Such control of hydrophobicity with film thickness and photo-crosslinking may potentially be interesting to prevent permeation of oxygen and moisture through the coating. This is especially important to protect the next generation photo-voltaic cells that are susceptible to the efficiency loss by the moisture mediated degradation.
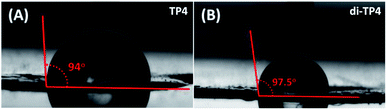 |
| | Fig. 9 The static contact angle of the polymeric film TP4 (thickness 183 nm) as measured using the sessile drop method. (A) Measured contact angles of the control (B) native TP4, (B) crosslinked di-TP4 and (C) decrosslinked TP4. | |
4. Conclusions
To summarize, we designed a series of smart photoresponsive polymer system with pendant thymine moiety using controlled free radical polymerization. The polymers exhibited thymine [2 + 2] cyclodimerization mediated crosslinking and decrosslinking that were monitored through UV and 1H NMR. The polymers stayed in extended coil state in good solvent condition and could be subjected to photodimerization mediated chain collapse depending on a number of factors e.g. concentration, chain length, molecular weight and functionality% of photoactive thymine moieties. At dilute concentration regime condition intrachain collapse rendered extended coil to globule transformation, while interchain crosslinking at high macromolecular concentration regime led to polymer chain compaction to furnish higher order nanostructures. Such formation of rigid photocrosslinked domains within flexible polymer chains possess a structural analogy to the hard–soft phase of thermoplastic elastomer. The rigidification of the crosslinked segments endowed a tool to photo-modulate the glass transition temperatures (Tg) that could be dynamically reverted back upon decrosslinking. Further, the structural modulation of the polymers was explored towards autonomic and non-autonomic self-healing behaviour at ambient conditions. Moreover, the self-healing efficacy could be tuned with the film thickness and it also remained unaltered upon using solar simulator or direct sunlight indicating both autonomic and non-autonomic self-healing mechanism working in tandem. Finally, the contact angle for the polymer surfaces exhibited a value ∼90° that can be efficiently tuned with film thickness and photo-crosslinking. Such hydrophobic nature of surface coating may be ideal barrier for oxygen and moisture to permeate through. Overall, this approach using photo-regulated self-healing system with low Tg hydrophobic polymers displays both autonomic and non-autonomic self-healing mechanism and may find its application in designing smart coatings of the photovoltaic device.
Conflicts of interest
The authors declare no competing financial interests.
Acknowledgements
The authors are thankful to the Institute of Nano Science and Technology (INST), Mohali, for providing infrastructure and characterization facilities. A. Pal thanks the Department of Science & Technology (DST, SERB project ECR/2016/000401 & CRG/2020/004251) for financial support. A. Singh thanks UGC for the SRF fellowship (SR no. 2121310082). C. Miglani, J. P. Joseph and D. Gupta acknowledge the INST fellowship.
Notes and references
- R. Kiran, P. Leev, A. R. Aleksandra, A. Kirill, L. Valerii and S. Elena, J. Eng. Appl. Sci., 2016, 14, 481 CrossRef.
-
(a) J. Gaume, P. W. W. Chung, A. Rivaton, S. Thérias and J. L. Gardette, RSC Adv., 2011, 1, 1471 RSC;
(b) P. R. Elowe, M. A. Stempki, S. J. Rozeveld and M. W. DeGroot, Chem. Mater., 2011, 23, 3915 CrossRef CAS;
(c) S. Banerjee, R. Tripathy, D. Cozzens, T. Nagy, S. Keki, M. Zsuga and R. Faust, ACS Appl. Mater. Interfaces, 2015, 7, 2064 CrossRef CAS PubMed.
-
(a) J. Lindroosa and H. Savin, Sol. Energy Mater. Sol. Cells, 2016, 147, 115 CrossRef;
(b) H. Hashigami, Y. Itakura and T. Saitoh, Int. J. Appl. Phys., 2003, 93, 4240 CrossRef CAS;
(c) K. Bothe, R. Hezel and J. Schmidt, Appl. Phys. Lett., 2003, 83, 1125 CrossRef CAS.
-
(a) T. Krajewski and K. Hardikar, US pat., 12/824,646, 2011 Search PubMed;
(b) J. Ackermann and F. Schwager, US pat., CA2806311A1, 2012 Search PubMed.
-
(a) J. Li, J. Liang, L. Li, F. Ren, W. Hu, J. Li, S. Qi and Q. Pei, ACS Nano, 2014, 8, 12874 CrossRef CAS;
(b) J. P. Joseph, A. Singh and A. Pal, in Smart polymer nanocomposites-energy harvesting, self-healing and shape memory applications, ed. D. Ponnamma, K. K. Sadasivuni, J. J. Cabibihan and M. Al-Maadeed, Springer, AG, 2017, p. 181 Search PubMed;
(c) S. Roy, A. Baral and A. Banerjee, Chem.–Eur. J., 2013, 19, 14950 CrossRef CAS;
(d) V. K. Thakur and M. R. Kessler, Polymer, 2015, 69, 369 CrossRef CAS.
-
(a) B. K. Ahn, D. W. Lee, J. N. Israelachvili and J. H. Waite, Nat. Mater., 2014, 13, 867 CrossRef CAS;
(b) C. Wang, H. Wu, Z. Chen, M. T. McDowell, Y. Cui and Z. Bao, Nat. Mater., 2013, 5, 1042 CrossRef CAS;
(c) A. Faghihnejad, K. E. Feldman, J. Yu, M. V. Tirrell, J. N. Israelachvili, C. J. Hawker, E. J. Kramer and H. Zeng, Adv. Funct. Mater., 2014, 24, 2322 CrossRef CAS.
-
(a) M. Nakahata, Y. Takashima, H. Yamaguchi and A. Harada, Nat. Commun., 2011, 2, 1 Search PubMed;
(b) T. W. Chuo, T. C. Wei and Y. L. Liu, J. Polym. Sci., Part A-1: Polym. Chem., 2013, 51, 3395 CrossRef CAS;
(c) X. Yang, H. Yu, L. Wang, R. Tong, M. Akram, Y. Chen and X. Zhai, Soft Matter, 2015, 11, 1242 RSC.
-
(a) N. H. Andersen, M. J. Harrington, H. Birkedal, B. P. Lee, P. B. Messersmith, K. Y. C. Lee and J. H. Waite, Proc. Natl. Acad. Sci., 2011, 108, 2651 CrossRef PubMed;
(b) S. Bode, L. Zedler, F. H. Schacher, B. Dietzek, M. Schmitt, J. Popp, M. D. Hager and U. S. Schubert, Adv. Mater., 2013, 25, 1634 CrossRef CAS PubMed;
(c) B. Sharma, A. Singh, T. K. Sharma, N. Sardana and A. Pal, New J. Chem., 2018, 42, 6427 RSC.
-
(a) M. A. Rahman, G. Spagnoli, A. M. Grande and L. D. Landro, Macromol. Mater. Eng., 2013, 298, 1350 CrossRef CAS;
(b) H. Gong, Y. Gao, S. Jiang and F. Sun, ACS Appl. Mater. Interfaces, 2018, 10, 26694 CrossRef CAS PubMed;
(c) A. Singh, J. P. Joseph, D. Gupta, I. Sarkar and A. Pal, Chem. Commun., 2018, 54, 10730 RSC;
(d) N. Gupta, A. Singh, N. Dey, S. Chattopadhyay, J. P. Joseph, D. Gupta, M. Ganguli and A. Pal, Chem. Mater., 2021, 33, 589 CrossRef CAS.
-
(a) S. Burattini, H. M. Colquhoun, B. W. Greenland and W. Hayes, Faraday Discuss., 2009, 143, 251 RSC;
(b) L. Li, B. Yan, J. Yang, L. Chen and H. Zeng, Adv. Mater., 2015, 27, 1294 CrossRef CAS PubMed;
(c) L. Li, B. Yan, J. Q. Yang, W. J. Huang, L. Y. Chen and H. B. Zeng, ACS Appl. Mater. Interfaces, 2017, 9, 9221 CrossRef CAS.
-
(a) H. Otsuka, S. Nagano, Y. Kobashi, T. Maeda and A. Takahara, Chem. Commun., 2010, 46, 1150 RSC;
(b) N. Roy, E. Buhlerb and J. M. Lehn, Polym. Int., 2014, 63, 1400 CrossRef CAS;
(c) P. Casuso, I. Odriozola, A. P. Vicente, I. Loinaz, G. Cabañero, H. Grande and D. Dupin, Biomacromolecules, 2015, 16, 3552 CrossRef CAS PubMed.
-
(a) H. Kautz, D. J. M. van Beek, R. P. Sijbesma and E. W. Meijer, Macromolecules, 2006, 39, 4265 CrossRef CAS;
(b) H. P. Wang, Y. C. Yuan, M. Z. Rong and M. Q. Zhang, Macromolecules, 2010, 43, 595 CrossRef CAS;
(c) Y. Chen, A. M. Kushner, G. A. Williams and Z. Guan, Nat. Chem., 2012, 4, 447 Search PubMed.
-
(a) B. J. Adzima, C. J. Kloxin and C. N. Bowman, Adv. Mater., 2009, 22, 2784 CrossRef PubMed;
(b) Z. Wei, J. H. Yang, X. J Du, F. Xu, M. Zrinyi, Y. Osada and Y. M. Chen, Macromol. Rapid Commun., 2013, 34, 1464 CrossRef CAS PubMed.
-
(a) Y. Zheng, M. Micic, S. V. Mello, M. Mabrouki, F. M. Andreopoulos, V. Konka and R. M. Leblanc, Macromolecules, 2002, 35, 5228 CrossRef CAS;
(b) S. Dai, P. Ravi and K. C. Tam, Soft Matter, 2009, 5, 2513 RSC;
(c) J. He, L. Tremblay, S. Lacelle and Y. Zhao, Soft Matter, 2011, 7, 2380 RSC;
(d) G. Kaur, P. Johnston and K. Saito, Polym. Chem., 2014, 5, 2171 RSC;
(e) P. Froimowicz, H. Frey and K. Landfester, Macromol. Rapid Commun., 2011, 32, 468 CrossRef CAS PubMed;
(f) J. P. Joseph, A. Singh, D. Gupta, C. Miglani and A. Pal, ACS Appl. Mater. Interfaces, 2019, 11, 28213 CrossRef CAS;
(g) A. Singh, J. P. Joseph, D. Gupta, C. Miglani and N. A. Mavlankar, Nanoscale, 2021, 13, 13401 RSC.
-
(a) J. P. Joseph, C. Miglani, A. Singh, D. Gupta and A. Pal, Soft Matter, 2020, 16, 2506 RSC;
(b) J. P. Joseph, C. Miglani, A. Bhatt, D. Ray, A. Singh, D. Gupta, M. E. Ali, V. K. Aswal and A. Pal, Polym. Chem., 2021, 12, 1002 RSC.
-
(a) L. Cauchy, Bull. des sc. Math., 1830, 14, 9 Search PubMed;
(b) R. A. Synowicki, G. K. Pribil, J. N. Hilfiker and K. Edwards, AIP Conf. Proc., 2005, 788, 324 CrossRef CAS;
(c) A. Franquet, J. D. Laet, T. Schram, H. Terryn, V. Subramanian, W. J. van Ooij and J. Vereecken, Thin Solid Films, 2001, 384, 37 CrossRef CAS.
-
(a) J. Dahlke, J. Kimmig, M. Abend, S. Zechel, J. Vitz, U. S. Schubert and M. D. Hager, NPG Asia Mater., 2020, 12, 13 CrossRef CAS;
(b) B. J. Briscoe, P. D. Evans, E. Pellilo and S. K. Sinha, Wear, 1996, 200, 137 CrossRef CAS.
- J. A. Pomposo, I. P. Baena, F. L. Verso, A. J. Moreno, A. Arbe and J. Colmenero, ACS Macro Lett., 2014, 3, 767 CrossRef CAS.
-
(a) C. M. Hansen, J. Paint Technol., 1967, 39, 511 CAS;
(b) C. M. Hansen, Solubility Parameters'’ in Paint Testing Manual, Manual 17, ed. J. V. Koleske, American Society for Testing and Materials, 1995, p. 383 Search PubMed.
-
(a) W. W. Graessley, J. Chem. Phys., 1967, 47, 1942 CrossRef CAS;
(b) Z. Huang, C. Lu, Y. Cao, B. Dong, G. Xu, C. Ji, K. Zhao and L. Yan, Nanoscale, 2016, 8, 1024 RSC;
(c) J. R. Hollis, F. Fleischli, A. A. Jahnke, N. Stingelin, D. S. Seferos and J. S. Kim, J. Phys. Chem. C, 2017, 121, 2088 CrossRef;
(d) Y. Yang, P. Chen, Y. Cao, Z. Huang, G. Zhu, Z. Xu, X. Dai, S. Chen, B. Miao and L. Yan, Langmuir, 2018, 34, 9477 CrossRef CAS PubMed;
(e) Y. Maki, Polym. J., 2014, 46, 641 CrossRef CAS.
-
(a) T. Murata, S. Iwai and E. Ohtsuka, Nucleic Acids Res., 1990, 18, 7279 CrossRef CAS;
(b) D. Elad, I. Rosenthal and S. Sasson, J. Chem. Soc. C, 1971, 2053 RSC;
(c) M. Barbatti, ChemPhysChem, 2014, 15, 3342 CrossRef CAS PubMed.
-
(a) V. A. Ivanov, W. Paul and K. Binder, J. Chem. Phys., 1998, 109, 5659 CrossRef CAS;
(b) H. Noguchi and K. Yoshikawa, J. Chem. Phys., 1998, 109, 5070 CrossRef CAS;
(c) Y. A. Kuznetsov and E. G. Timoshenko, J. Chem. Phys., 1999, 111, 3744 CrossRef CAS;
(d) D. Hu, J. Yu, K. Wong, B. Bagchi and P. J. Rossky, Nature, 2000, 405, 1030 CrossRef CAS PubMed.
-
(a) R. Wool and K. M. O’connor, J. Appl. Phys., 1981, 52, 5953 CrossRef CAS;
(b) K. Jud and H. H. Kausch, Polym. Bull., 1967, 1, 697 Search PubMed;
(c) Y. H. Kim and R. P. Wool, Macromolecules, 1983, 16, 1115 CrossRef CAS;
(d) D. Davydovich and D. M. W. Urban, Nat. Commun., 2020, 11, 5743 CrossRef CAS.
-
(a) Y. M. Malinskii, V. V. Prokopenko and V. A. Kargin, Polym. Mech., 1970, 6, 969 CrossRef;
(b) B. A. Beiermann, M. W. Keller and N. R. Sottos, Smart Mater. Struct., 2009, 18, 085001 CrossRef.
-
(a) Y. Li, J. Q. Pham, K. P. Johnston and P. F. Green, Langmuir, 2007, 23, 9785 CrossRef CAS;
(b) K. Y. Law and K. Yee, J. Phys. Chem. Lett., 2014, 5, 686 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra07425c |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*