
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoelectron photofragment coincidence spectroscopy of carboxylates†
J. A. Gibbard and
R. E. Continetti*
and
R. E. Continetti*
Department of Chemistry and Biochemistry, University of California, San Diego, 9500 Gilman Dr, La Jolla, Ca 92093-0340, USA. E-mail: rcontinetti@ucsd.edu
First published on 22nd October 2021
Abstract
Photoelectron–photofragment coincidence (PPC) spectroscopy is a powerful technique for studying the decarboxylation dynamics of carboxyl radicals. Measurement of photoelectron and photofragment kinetic energies in coincidence provides a kinematically complete measure of the dissociative photodetachment (DPD) dynamics of carboxylate anions. PPC spectroscopy studies of methanoate, ethanoate, propanoate, 2-butenoate, benzoate, p-coumarate and the oxalate monoanion are reviewed. All of the systems studied undergo decarboxylation via a two-body DPD channel i.e. 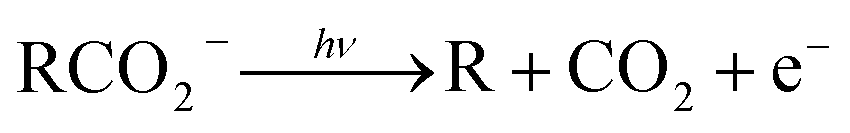 , driven by the thermodynamic stability of CO2. Additionally, decarboxylation is observed via a three-body ionic photodissociation channel for p-coumarate. In some cases photodetachment also results in a stable carboxyl radical (RCO2). The branching ratio for DPD, the threshold detachment energy and the peak of the kinetic energy release spectrum are compared for different carboxylates, as a probe of the character of the potential energy landscape in the Franck–Condon region.
, driven by the thermodynamic stability of CO2. Additionally, decarboxylation is observed via a three-body ionic photodissociation channel for p-coumarate. In some cases photodetachment also results in a stable carboxyl radical (RCO2). The branching ratio for DPD, the threshold detachment energy and the peak of the kinetic energy release spectrum are compared for different carboxylates, as a probe of the character of the potential energy landscape in the Franck–Condon region.
1 Introduction
The facile decarboxylation of a carboxyl radical (RCO2 → R + CO2) is driven by the thermodynamic stability of the CO2 molecule. It is a well known method for the production of radicals in organic chemistry, via the Kolbe electrolysis, the Hunsdiecker reaction and the Barton decarboxylation.1–3 Additionally, studying the dissociation dynamics of carboxyl radicals can probe the potential energy surfaces of many fundamental gas phase reactions of importance in biology, the atmosphere and combustion. One example of this is studying the dissociation dynamics of HCO2 to probe the transition state region of H + CO2 → OH + CO, a fundamental gas-phase reaction that plays a role in combustion and the atmosphere.4,5 Another is probing the competing channels that follow photodetachment of p-coumarate, to investigate the role of the protein environment and the chromophore in the photoactive yellow protein by establishing gas-phase benchmarks for the isolated chromophores.6 Solution-phase decarboxylation has been investigated using experimental and theoretical approaches, and comparison to gas-phase dynamics studies allows the role of solvation to be understood.7,8 The stability of the carboxyl radical, which determines the branching ratio to decarboxylation, depends upon the nature of the R group. Additional factors, such as the presence of stabilizing intermolecular hydrogen bonds in dicarboxylic acids, can also contribute to the stability of the carboxyl radicals in some cases. In this review we will describe the decarboxylation dynamics observed for a number of different carboxylates, with R groups including alkyl chains, aromatic rings and a second carboxylic acid group.Carboxyl radicals can be readily prepared by photodetachment of carboxylate anions 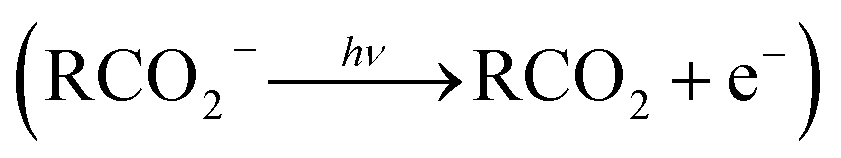 . Photoelectron-photofragment coincidence (PPC) spectroscopy couples photoelectron spectroscopy with translational spectroscopy of the resulting neutrals.9 If, following photodetachment, the neutral is formed on a repulsive region of a neutral potential energy surface, then dissociation into multiple neutral fragments will occur via a process known as dissociative photodetachment (DPD). Alternatively, ionic photodissociation can occur following laser irradiation, producing one anionic fragment and one or more neutral fragments. The dominant DPD channel for carboxylate anions following photodetachment is decarboxylation, driven by the thermodynamic stability of CO2.4–6,10,11 Ionic photodissociation, again thermodynamically driven by the formation of neutral CO2, has also been observed for a handful of carboxylates.6,12 In some cases photodetachment may result in a stable radical with a lifetime longer than the flight time of the neutral fragments (typically ≈10 μs). If dissociation occurs after this, then it cannot be studied using this experimental approach. PPC spectroscopy provides a kinematically complete measure of DPD with the energetics and dynamics determined by the electron affinity (EA) of the carboxyl radical, the peak kinetic energy release (KER) of the fragments and the branching ratio between all possible channels. Therefore, PPC spectroscopy is able to study the decarboxylation dynamics of carboxyl radicals in detail.4–6,10,11
. Photoelectron-photofragment coincidence (PPC) spectroscopy couples photoelectron spectroscopy with translational spectroscopy of the resulting neutrals.9 If, following photodetachment, the neutral is formed on a repulsive region of a neutral potential energy surface, then dissociation into multiple neutral fragments will occur via a process known as dissociative photodetachment (DPD). Alternatively, ionic photodissociation can occur following laser irradiation, producing one anionic fragment and one or more neutral fragments. The dominant DPD channel for carboxylate anions following photodetachment is decarboxylation, driven by the thermodynamic stability of CO2.4–6,10,11 Ionic photodissociation, again thermodynamically driven by the formation of neutral CO2, has also been observed for a handful of carboxylates.6,12 In some cases photodetachment may result in a stable radical with a lifetime longer than the flight time of the neutral fragments (typically ≈10 μs). If dissociation occurs after this, then it cannot be studied using this experimental approach. PPC spectroscopy provides a kinematically complete measure of DPD with the energetics and dynamics determined by the electron affinity (EA) of the carboxyl radical, the peak kinetic energy release (KER) of the fragments and the branching ratio between all possible channels. Therefore, PPC spectroscopy is able to study the decarboxylation dynamics of carboxyl radicals in detail.4–6,10,11
2 Experimental methods
2.1 Techniques for studying decarboxylation
Photoelectron spectroscopy has previously been used to investigate the structure and energetics of both the carboxylate anions and their corresponding carboxyl radicals.13–18 Wang and coworkers have used electrospray ionization (ESI) and photoelectron spectroscopy to study a number of carboxylates, including the aliphatic monoanionic dicarboxylates,13 the dicarboxylate dianions,14,15 the citrate dianion,16 and the linear carboxylates.17 In particular the role of hydrogen bonding has been investigated, and it was shown that the carbon chain backbone would readily distort to accommodate a stabilizing intramolecular interaction within the anion.13 Evidence for intramolecular hydrogen bonding has even been observed in the aliphatic carboxylate anions, between the CO2− group and a C–H bond.17 These wide ranging studies have indicated an EA of ≈3.5 eV for carboxyl radicals and ≈4.5 eV for radicals resulting from photodetachment of dicarboxylate monoanions. As photodetachment ejects an electron in a molecular orbital where the electron density is strongly localized on the O atoms in the CO2− group, similar EAs are observed as the chain length is increased. Small variations are observed from one carboxylate to the next as accommodating the weak intramolecular O−![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) H–C hydrogen bond requires more or less strain. Typically, the EA of a radical formed via the photodetachment of a dicarboxylate monoanion is larger than the EA of an aliphatic carboxyl radical, due to the greater stabilization of the anion as a result of the formation of a stronger intramolecular hydrogen bond, e.g. O−H–O–C in the dicarboxylate monoanion is stronger than O−H–C in the carboxylate. No evidence is seen for intramolecular H bonding in the radicals, as a result of the lower charge density on the O atom. Slow electron velocity map imaging has also been used to record a high resolution photoelectron spectrum of a carboxylate, specifically HCO2−.18
H–C hydrogen bond requires more or less strain. Typically, the EA of a radical formed via the photodetachment of a dicarboxylate monoanion is larger than the EA of an aliphatic carboxyl radical, due to the greater stabilization of the anion as a result of the formation of a stronger intramolecular hydrogen bond, e.g. O−H–O–C in the dicarboxylate monoanion is stronger than O−H–C in the carboxylate. No evidence is seen for intramolecular H bonding in the radicals, as a result of the lower charge density on the O atom. Slow electron velocity map imaging has also been used to record a high resolution photoelectron spectrum of a carboxylate, specifically HCO2−.18
Photoelectron spectroscopy provides no direct insights into dissociation processes however, leading to limited information about the decarboxylation dynamics. Translational spectroscopy of the resulting neutral fragments is required to directly interrogate the neutral fragments and probe decarboxylation directly, but different experimental approaches yield different information. Jouvet and coworkers used cold ion photofragment spectroscopy to study the decarboxylation of benzoate and naphthoate.12 However, no fragment masses or KER spectrum were reported for the DPD channel using this approach. Electrostatic storage rings, absorption spectroscopy, daughter ion mass analysis and time of flight spectroscopy has been used to study the ionic photodissociation of p-coumarate by recording CO2 fragments.19–23 However, this work was undertaken below the photodetachment threshold and decarboxylation via DPD was not directly studied. PPC spectroscopy is the only translational spectroscopy method to measure the fragment mass spectrum and the KER spectrum of neutral fragments for internal-energy-selected carboxyl radicals as determined by the detection of the photoelectron kinetic energy in coincidence.
2.2 PPC spectroscopy
PPC spectroscopy couples photoelectron spectroscopy with translational spectroscopy to give a kinematically complete picture of DPD. In the case of autodetachment of a fragment anion produced via photodissociation, or a two-photon process of photodissociation and subsequent photodetachment of the anion fragment, a kinematically complete picture of photodissociation can also be gained.25 The structure and energetics of the anion and radical, the fragment masses and KER spectrum of the dissociation products, the branching ratios to different dissociation channels and the distribution of product internal energies are all interrogated. By recording the photoelectron and neutrals from a single dissociation event in coincidence, the nature of dissociative electronic states in the Franck–Condon region can be directly probed. This is important for radicals that exhibit many low-lying electronic states, such as the carboxyl radicals. PPC spectroscopy allows unambiguous assignment of decarboxylation and gives a kinematically complete picture of the dissociation dynamics.As the work detailed in this review has been undertaken over a number of years various PPC spectrometers have been utilized. Fig. 1 is a schematic of the current high beam energy PPC spectrometer for heavy and complex anions.24 Generally, anions are accelerated to a beam energy of several keV before photodetachment. The photoelectrons are imaged on a time and position sensitive detector, recording the electron kinetic energy (eKE) spectrum and the photoelectron angular distribution (PAD). The resulting neutral fragments, whether stable radicals or pairs or triples of neutral photofragments resulting from a dissociation process, are imaged on a time- and position-sensitive crossed-delay-line neutral detector. From this data the fragment mass spectrum and KER spectrum for the neutral products of dissociation can be extracted. As the photoelectrons and neutral fragments are recorded in coincidence a two-dimensional histogram of N(eKE, KER) known as a PPC spectrum can be recorded. The PPC spectrum is unique to PPC spectroscopy, and allows the dissociation to be analyzed in a kinematically complete manner. The total energy (ETOT = eKE + KER) spectrum can also be recorded, which encodes the internal energy distribution of the parent anions and the fragments following dissociation. The parent anions are deflected out of the neutral beam path after the interaction region by an applied electric field, to be monitored in real time on an ion detector.
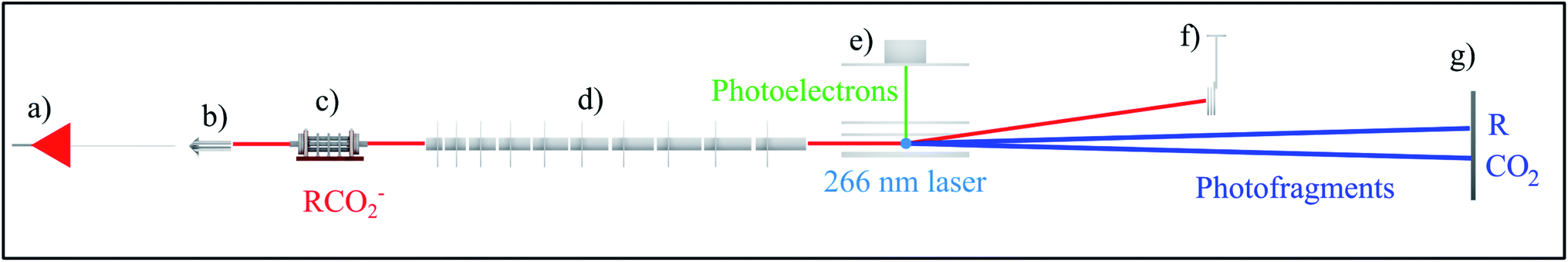 | ||
| Fig. 1 Schematic of the high beam energy PPC spectrometer depicting the (a) ESI source, (b) octopole ion guide, (c) hexapole accumulation trap, (d) LINAC, (e) electron detector, (f) undetached ion deflector and (g) neutral detector.24 The 4.66 eV DPD of RCO2− is also shown. | ||
For most of the systems described in this review the gas phase carboxylate anions were produced by a pulsed electric discharge source, and subsequently accelerated to a beam energy of ≤8 keV.10 The oxalate monoanion, benzoate and p-coumarate were produced using ESI, which is optimal for formation of larger gas phase anions, using the high beam energy PPC spectrometer shown in Fig. 1. For these systems a linear accelerator (LINAC) consisting of ten acceleration stages was used to accelerate the anions to ≤21 keV in order to increase the detection efficiency of light neutral fragments formed by the DPD of heavier parent anions.24 All of the other systems were accelerated using a single acceleration stage. HCO2− was studied using an electrostatic ion beam trap (EIBT) to decouple the source repetition and the data acquisition rate.26 In all cases the harmonics of either a picosecond Nd:YAG laser or a Ti:Sapphire regenerative amplifier with a fundamental output centered at 1.6 eV (775 nm) were used to photodetach the anions.
3 PPC spectroscopy studies of carboxylates
In this review the results of a number of PPC spectroscopy studies on different carboxylates will be discussed. All of the systems studied undergo decarboxylation via DPD of the carboxylate, and in some cases a stable carboxyl radical is also observed. Additionally, decarboxylation via a three-body photodissociation process is seen for p-coumarate. The systems will be discussed in turn from the simplest carboxylate, through increasing alkyl chain length, then aromatic carboxylates, before finally the recent work on the dicarboxylate monoanion is reviewed. Most of this work has been previously published, but propanoate, 2-butenoate and propiolate are described here for the first time. Table 1 details the recorded threshold detachment energy (TDE), peak KER and decarboxylation branching ratio for all the systems studied to date. The TDE is extracted from the onset of photoelectron signal and is used here as there is significant overlap between spectral features attributed to photodetachment to different radical states for many of the carboxylates studied.| Carboxylate | TDE of the carboxyl, eV | KER, eV | DPD branching ratio |
|---|---|---|---|
| Methanoate4,5 (DCO2−) | 3.5 (AEA) | 0.4 | 1 |
| Ethanoate10 (CH3CO2−) | 3.4 (AEA) | 0.7 | 0.9 |
| Propanoate (CH3CH2CO2−) | 3.2 | 0.6 | 1 |
| 2-Butenoate (CH3CH2CH2CO2−) | 3.2 | 0.5 | 1 |
| Benzoate6 (C6H5CO2−) | 3.6 | 0.6 | 0.90 |
| p-Coumarate6 (HOC6H4CHCHCO2−) | 3.9 (carboxylate) | 0.45 | 0.05 (carboxylate) |
| Oxalate monoanion11 (HO2CCO2−) | 4 | 1.1 | 1 |
| Propiolate (HCCCO2−) | 3.8 | 0.45 | 0.04 |
3.1 Aliphatic carboxylates
Over a number of years the role of chain length on the decarboxylation dynamics of the alkyl carboxylates (CH3(CH2)nCO2−, n = 0–2) has been investigated using PPC spectroscopy. DPD dominated in all cases, resulting in CO2 + H(CH2)n + e−, with a peak KER of 0.4–0.7 eV indicating significant repulsion driving dissociation. The TDEs of all of the alkyl carboxylates are between 3.2–3.5 eV, with a small decrease as chain length decreases. These TDEs are similar to those reported in other photoelectron spectroscopy studies.17 Additionally, conjugated 2-butenoate was studied after being generated instead of butanoate in the anion source.In the first study of HCO2− and DCO2− a PPC spectrometer was employed that differed in several ways from the spectrometer shown in Fig. 1.4 Anions were formed by the intersection of a 1 keV electron beam and a supersonic expansion of N2O that had passed over a formic acid solution. The anions were accelerated to 7 keV using a single acceleration stage. The photoelectrons were imaged on two opposing electron detectors, resulting in improved resolution as no electron repellers were required, but only a small fraction of the solid angle was sampled.4 Vibrational structure in the photoelectron spectrum, observed from 2A1 and 2B2 states only, indicated predissociation of the HCO2 radical. State-resolved KER spectra were observed and attributed to bending excitations in the CO2 product, consistent with low rotational excitation in the CO2. Vertical bands in the PPC spectrum indicated that multiple CO2 vibrational states were energetically accessible from each vibrational level in the predissociating DCO2 molecule. The resolved structure was attributed to the near degeneracy of the spacing of the vibrational levels in the 2A1 and 2B2 radical states and the bending mode of the CO2 products. The presence of three low-lying electronic states in the radical, which are nearly degenerate, resulted in a strong pseudo Jahn–Teller coupling that distorted the expected C2v symmetry and made calculations challenging, due to strong vibronic coupling effects. Quantitative vibronic coupling calculations were undertaken on the formoxyl radical by Klein et al.30 and theoretical work has predicted a conical intersections between the 2A1 and 2B2 radical states within the Franck–Condon region.27–29 Therefore it is possible that photodetachment to the 2A1 state results in H + CO2 directly, whereas 2B2 accesses the same vibrational product distribution via the conical intersection. In contrast it has been proposed that the 2A2 state intersects the 2A1 state in a more repulsive region,27,28 resulting in no observed vibrational structure in the photoelectron or PPC spectrum.
A second PPC spectroscopy study was subsequently undertaken,5 utilizing a modified PPC spectrometer which had implemented an EIBT, to decouple the source repetition and the data acquisition rates.26 This second study was undertaken at a photon energy of 4.27 eV, closer to the photodetachment threshold of DCO2−. The PPC spectrum was gated by selecting specific eKE values, which were associated with each predissociated vibrational state in the radical. This resulted in KER spectra which exhibited clear vibrational progressions in the CO2 bending modes. Quantum dynamics calculations were undertaken in conjunction with this study and predicted less vibrational excitation in the CO2 bending mode than was observed in the experimental spectrum.31 The discrepancy between the predicted and measured distribution of vibrational energy suggested inaccuracies in the neutral potential energy surface utilized in the quantum dynamics calculations, particularly in the lower total energy region probed by the predissociating formyloxyl radical, demonstrating the need for more accurate potential energy surface calculations.
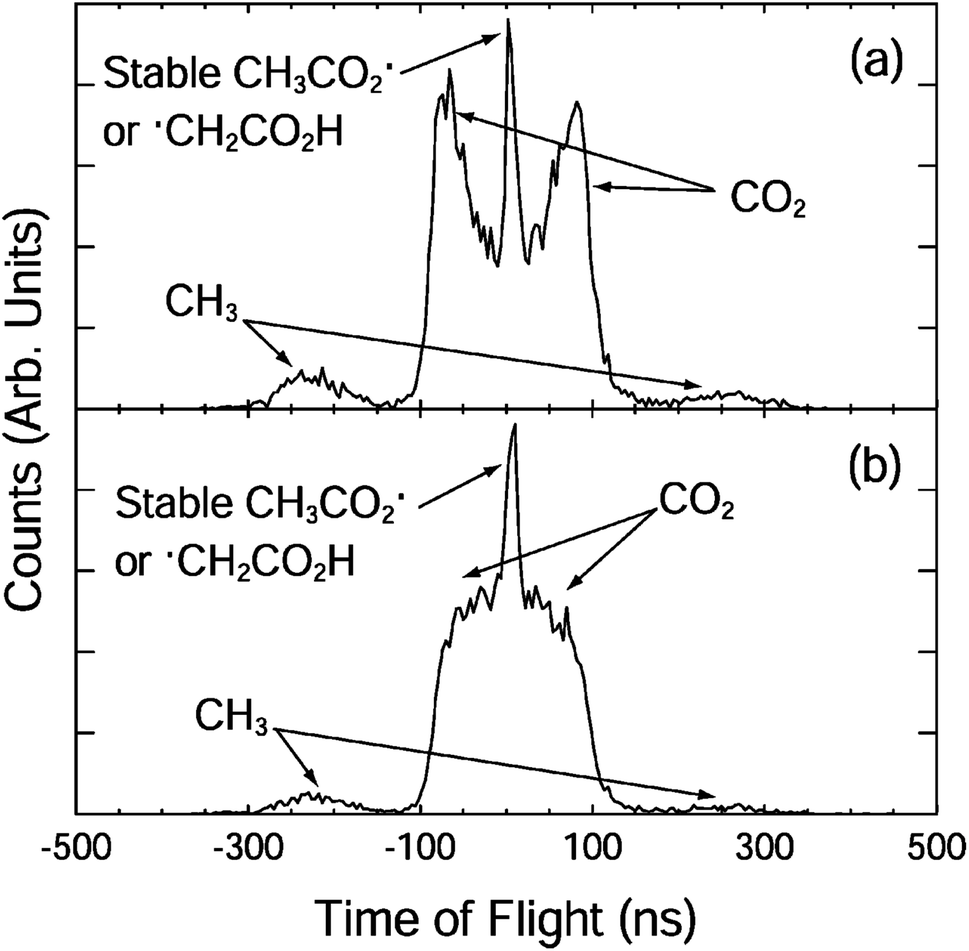 | ||
| Fig. 2 The fragment TOF spectra for CH3CO2− at 3.5 eV with the electric vector of the laser (a) parallel and (b) perpendicular to the direction of ion beam.10 Reprinted with permission from Journal of Physical Chemistry A, 2004, 108, 9962. Copyright 2004 American Chemical Society. | ||
The photoelectron spectrum suggested the presence of several low-lying electronic states and an upper limit to the EA of 3.47 ± 0.01 eV. A more accurate EA of 3.25 ± 0.01 eV was subsequently reported by Wang and coworkers using high-resolution photoelectron spectroscopy of cryogenically cooled ethanoate.34 The 3.5 eV photoelectron spectrum showed two nearly degenerate states of CH3CO2, and the 4.8 eV spectrum showed an additional two peaks at 3.68 eV and 3.88 eV above the CH3CO2− ground state. The electronic structure of CH3CO2 is difficult to calculate due to the presence of multiple low-lying electronic states and symmetry breaking from C2v to Cs. Theoretical studies have indicated the presence of two low-lying minima and the two transition states.29 The O–C–O bond angle is calculated to be 130° in the anion and 121° in the radical.35 The reported stable and dissociative photoelectron spectra were identical providing evidence for a sequential DPD process. In effect photodetachment did not result in a neutral on a bound or repulsive potential energy surface, but instead on some intermediate state, which then rapidly evolved to either dissociation or stable products. No evidence was seen for an internal energy dependence on the fate of the radical. An anisotropic photofragment distribution was observed, consistent with a one-photon photofragment angular distribution characterized by β = 1.2, indicative of prompt dissociation on a timescale shorter than molecular rotation. The KER spectrum recorded at 4.82 eV peaked at 0.8 eV, whereas the spectrum recorded at lower photon energies peaked at 0.6 eV, but both spectra had the same shape on the low KER side, indicative of the involvement of a higher-lying, more repulsive, dissociative electronic state at the higher photon energy. From the PPC spectra recorded at the lower photon energy a KEmax = 0.93 eV was extracted, a value smaller than expected from calculations, suggesting that the products were vibrationally excited. Excitation of specific modes was not observed in the PPC spectrum due to the numerous energetically accessible rotational and vibrational modes in both products. However, it is logical that DPD would result in excitation of the bending mode in CO2 and the out-of-plane umbrella CH3 mode. The experimental KEmax indicated the presence of a total of three or four quanta of vibrational excitation in the products. Measurements on the deuterated analogue (CD3CO2−) indicated a larger partitioning of energy to internal modes following DPD and higher vibrational energy in the deuterated products, especially as CD3 has smaller vibrational quanta than CH3.
Propanoate (mass 73 a.m.u.) was produced in the discharge source, as assigned by TOF mass spectrometry. PPC spectroscopy of propanoate (CH3CH2CO2−) indicated that decarboxylation dominated, with all of the anions photodetached at 258 nm undergoing DPD, and resulting in CH3CH2 + CO2 + e−. Fig. 3a shows the 4.8 eV photoelectron spectrum of propanoate. The experimental adiabatic TDE for propanoyoxyl was determined to be ≈3.20 ± 0.20 eV in this PPC spectroscopy study, similar to the 300 K photoelectron spectrum reported by Wang and coworkers.17 The spectra exhibited some evidence for macro-scale structure (spacing 0.15 ± 0.25 eV), which may be attributable to vibrational excitation or the presence of the low-lying electronic states expected for the aliphatic carboxyl radicals seen in the formyloxyl and acetyloxyl radical cases. The KER spectrum of propanoate resulting in CO2 + CH3CH2 + e− is shown in Fig. 3b. It consists of a single structureless peak with maximum intensity at ≈0.6 eV. The peak KER was slightly lower than for ethanoate discussed in Section 3.1.2.
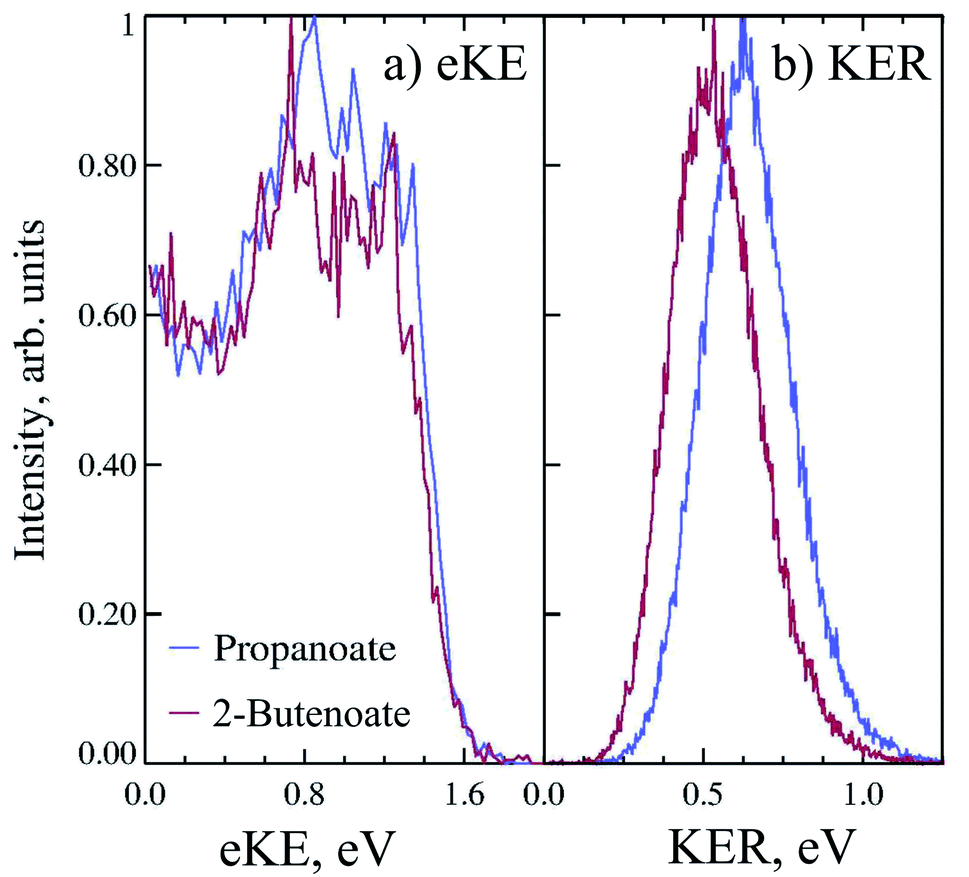 | ||
| Fig. 3 The 11 keV (a) eKE and (b) KER spectra of propanoate (blue) and 2-butenoate (red) at 4.8 eV. | ||
The PPC spectrum of 8 keV propanoate is shown in Fig. 4a, resulting in CH3CH2 + CO2 + e−. A single, vertical intense feature is observed with a diagonal cutoff at high total energy, indicative of a DPD channel. The total kinetic energy partitioned to the photoelectrons and neutral fragments (KEmax) can be extracted from the experimental spectrum by drawing a diagonal line at the limit of the most intense portion to the PPC spectrum, KEmax = 2.1 eV. This is an experimental measure of the energy difference between the anion ground state and the dissociation asymptote i.e. E(RCO2− → R + CO2) = hν − KEmax. The majority of photodetachment events have a much lower total kinetic energy than the KEmax, indicating substantial internal excitation of the products. No stable propanoyloxyl radicals were observed. The fragment mass spectrum is shown in the Fig. S1 (ESI†), and shows two clear peaks at 29 a.m.u. and 44 a.m.u. The propanoyoxyl radicals were unstable on the 10 μs time of flight from the interaction region to the detector, fragmenting to ethyl radical (CH3CH2, 29 a.m.u.) and carbon dioxide (CO2, mass 44 a.m.u.). It is somewhat unexpected that this larger alkyloxyl radical is completely dissociative, given that the smaller acetyloxyl radical in Section 3.1.2 exhibited both dissociative (90%) and stable (10%) channels. One possible explanation is that the longer alkyl chain may stabilize the alkyl radical (R) produced via DPD more effectively than the carboxyl radical (RCO2) produced via photodetachment. Fig. S2† is the total energy spectrum of propanoate at 4.8 eV, where ETOT = eKE + KER. The KEmax = 2.1 eV, extracted from the PPC spectrum, is also shown on the ETOT spectrum.
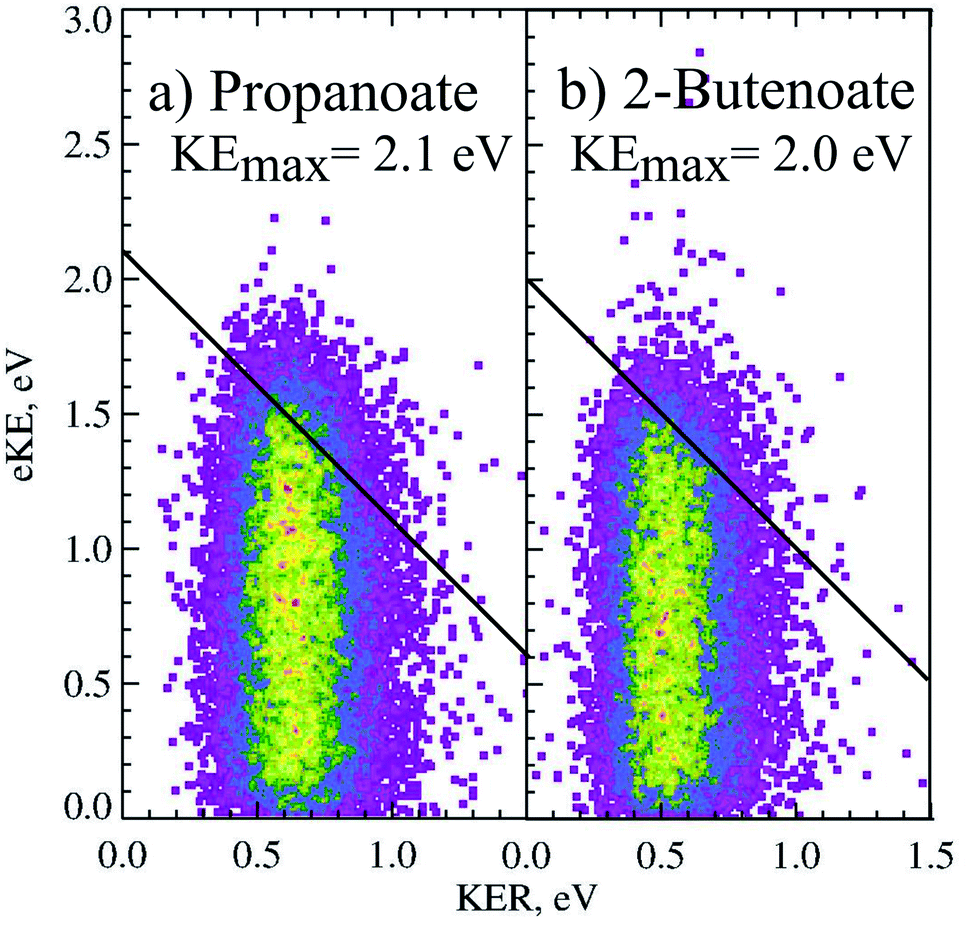 | ||
| Fig. 4 The PPC spectrum of 11 keV (a) propanoate and (b) 2-butenoate at 258 nm. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) CHCO2−). PPC spectroscopy of 2-butenoate was undertaken at the same time and using the same spectrometer as propanoate. Butanoic acid (CH3CH2CH2CO2H) was used to produce the molecular beam however, TOF mass spectrometry indicated a parent anion mass of 85 a.m.u. rather than the expected 87 a.m.u., attributable to the formation of CH3CH
CHCO2−). PPC spectroscopy of 2-butenoate was undertaken at the same time and using the same spectrometer as propanoate. Butanoic acid (CH3CH2CH2CO2H) was used to produce the molecular beam however, TOF mass spectrometry indicated a parent anion mass of 85 a.m.u. rather than the expected 87 a.m.u., attributable to the formation of CH3CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCO2−, rather than CH3CH2CH2CO2−. This may be a result of the greater anion stability afforded by the conjugated double bond and carbonyl group, driving H2 loss in the discharge anion source. Fig. 3 is the photoelectron spectrum of CH3CHCHCO2− recorded at 4.8 eV. The measured TDE was ≈3.20 ± 0.20 eV, which is lower than the only previously reported estimate of the EA (3.49 eV) extracted from the measurement of proton transfer equilibria.36 Additionally, there was some evidence for macro-scale structure in the photoelectron spectrum, that may be attributable to unresolved vibrational excitation or the presence of multiple low lying electronic states. This structure is similar to the macro-scale structure observed in the propanoate spectrum and discussed in Section 3.1.3.
CHCO2−, rather than CH3CH2CH2CO2−. This may be a result of the greater anion stability afforded by the conjugated double bond and carbonyl group, driving H2 loss in the discharge anion source. Fig. 3 is the photoelectron spectrum of CH3CHCHCO2− recorded at 4.8 eV. The measured TDE was ≈3.20 ± 0.20 eV, which is lower than the only previously reported estimate of the EA (3.49 eV) extracted from the measurement of proton transfer equilibria.36 Additionally, there was some evidence for macro-scale structure in the photoelectron spectrum, that may be attributable to unresolved vibrational excitation or the presence of multiple low lying electronic states. This structure is similar to the macro-scale structure observed in the propanoate spectrum and discussed in Section 3.1.3.DPD resulting in CH3CHCH + CO2 + e− (mass 41 a.m.u and 44 a.m.u) was the only observed channel, with no stable CH3CHCHCO2 being observed. The fragment mass spectrum is shown in the ESI (ESI, Fig. S1†). A single peak is observed with maximum intensity near 42.5 a.m.u. which shows clear evidence for the mass 41 and 44 a.m.u., rather than 43 and 44 a.m.u., and supports the attribution of the dominant anion to mass 85 following proton abstraction and H2 elimination from butanoic acid, i.e. CH3CHCHCO2−. Fig. 3b is the KER spectrum of CH3CHCHCO2− recorded at 4.8 eV for DPD resulting in CH3CHCH + CO2 + e−. As shown in Fig. 3 the peak of the KER spectrum is ≈0.5 eV, indicating a slight reduction in peak KER as the carboxylate chain length is increased from ethanoate to propanoate and finally 2-butenoate. The PPC spectrum of 8 keV CH3CHCHCO2− is shown in Fig. 4b. The spectrum is similar in structure to that of propanoate, with an intense vertical feature, and a high energy diagonal cutoff. This spectrum is indicative of a one-photon two-body DPD channel, where the products have significant vibrational excitation. An experimental KEmax = 2.0 eV can be extracted by drawing a diagonal line at the limit of the most intense portion of the PPC spectrum. Fig. S2† is the total energy spectrum of 2-butenoate at 4.8 eV and also shows the KEmax. The ETOT spectrum is shifted by ≈0.1 eV compared to propanoate. One explanation is that the difference is attributable to the change in the peak KER, indicating that the potential energy surface is sightly less repulsive in the Franck–Condon region. Alternatively, it could be a result of the greater number of degrees of freedom in 2-butenoate compared to propanoate, and therefore the increased density of rovibrational states. It is interesting to note the similarities between the data for propanoate and 2-butenaote, considering one is an alkyl carboxylic acid and the other is an α,β-unsaturated carboxylic acid.
The strong vertical structure of the PPC spectra for 2-butenoate and propanoate are unusual for DPD processes, as a fixed KER is usually associated with a photodissociation process. Typically PPC spectra for DPD processes are characterized by strong diagonal structure, which is also observed here in the diagonal high energy cutoff. Similar vertical structure is seen in the PPC spectra of methanoate and ethanoate,4,5,10 suggesting that it is a distinctive characteristic of the DPD dynamics of alkyl carboxylates. The strong vertical structure indicates that the KER is constant, regardless of the eKE or the ETOT. Additionally, there is a total energy difference of 1.5 eV between events near the KEmax and those at low eKE, suggesting that following photodetachment the radical is formed in a wide range of electronic and vibrational states that all dissociate with high KER. An excited radical has a lifetime longer than the vibrational period, and ultimately evolves to a repulsive transition state structure, that dissociates with a well defined KER. Therefore the strong vertical structure of the PPC spectra indicates the presence of non-adiabatic dynamics in the decarboxylation of alkyl carboxylates.
3.2 Aromatic carboxylates
Photodetachment of aromatic carboxylates can result in carboxyl radicals that are resonance stabilized, resulting in a lower branching ratio to DPD. Photoelectron spectroscopy, electrostatic ion storage rings and cold ion photofragment spectroscopy have been used to study the aromatic carboxylates previously,12,20,37,38 but none of these experiments have been able to study the decarboxylation in a kinematically complete manner. The dissociation dynamics of benzoate and p-coumarate have recently been investigated using high beam energy PPC spectroscopy, using the apparatus shown in Fig. 1.6 Photodetachment and DPD are shown to occur for both of these anions, resulting in a mixture of stable carboxyl radicals and the products of decarboxylation. The ground state radicals are longer lived than the flight time between the interaction region and the neutral detector (10 μs). However, calculations indicate that the radical ground state is slightly higher in energy or the same energy as the DPD asymptote, with a number of low-lying dissociative excited states, indicating that the aromatic carboxyl ground state is metastable in character. This contrasts to the alkyl carboxylates where multiple dissociative low-lying electronic states are observed at similar energies.4,5,10 DPD is shown to occur via multiple excited states, and requires vibrational excitation in the first excited radical state to undergo decarboxylation in both cases. However, very different branching ratios between DPD and photodetachment are observed and an additional three-body photodissociation channel is seen for p-coumarate.Only one dissociation channel was observed for benzoate at 4.66 eV, assigned to a one-photon, two-body DPD channel that resulted exclusively in C6H5 + CO2 + e− via the A or B radical states. The PPC spectrum of benzoate is shown in Fig. 5a and the calculated maximum kinetic energy of photoelectrons and fragments (KEmax = 1.11 eV) is plotted as a solid black line. Two broad regions are observed in the PPC spectrum; one high intensity feature at eKE <0.5 eV, and a lower intensity feature at higher eKE. These features are not resolved but it is probable that they correspond to DPD via the B or A state of the radical respectively. The KEmax (dashed line) is on the high energy side of the B state feature, suggesting that the B state radicals undergo DPD to produce vibrationally excited products. Conversely the KEmax is to the low energy side of the A state feature, indicating that these parent anions are vibrationally excited. The high beam energy PPC spectrometer is characterized by a vibrational temperature of 298 K, such that a small proportion of the anions have significant vibrational excitation. It is this high energy tail of the vibrational Boltzmann distribution that undergoes DPD via the A state, and results in a higher overall ETOT. Additionally, the energy resolution of a PPC spectrum is 12%, such that only those events with ETOT ≥ 1.24 eV can be definitively attributed to vibrationally excited parent anions. Similarities are observed between the photodetachment dynamics of benzoate and the alkyl carboxylates, in particular ethanoate. However, benzoate has a slightly higher EA and a slightly lower branching ratio to DPD, suggesting that the aromatic ring stabilizes both the aromatic carboxylate and carboxyl radical compared to the alkyl analogues.
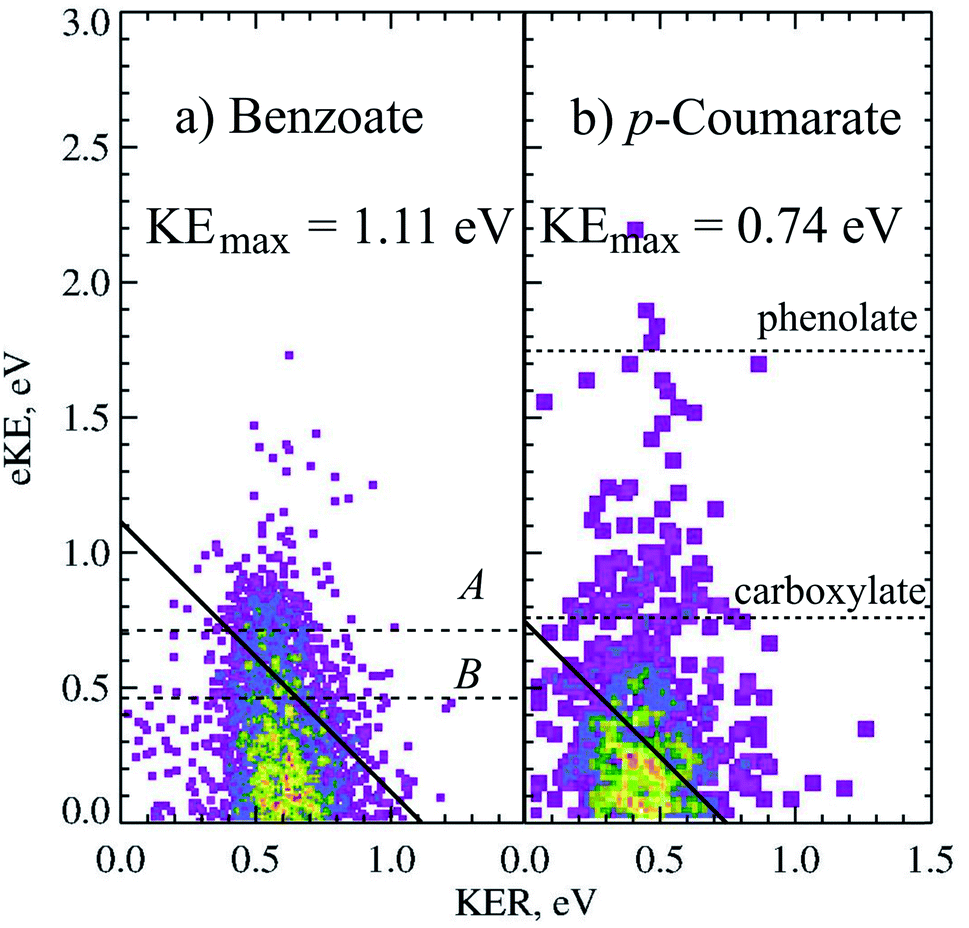 | ||
| Fig. 5 The PPC spectrum for decarboxylation of 11 keV benzoate and p-coumarate recorded at 4.66 eV. The calculated KEmax are depicted by the solid diagonal lines. The ADEs for photodetachment of benzoate to the A and B radical states and VDEs of the carboxylate or phenolate p-CA− to the corresponding radicals are shown in the dashed horizontal lines. Reproduced from ref. 6 with permission from the PCCP Owner Societies. | ||
Clear evidence was seen in the stable photoelectron spectra for the formation of carboxyl or alkoxyl radicals with a lifetime longer than the flight time from the interaction region to the neutral detector, following photodetachment of both tautomers. The phenolate form is the most stable in the gas phase and has a VDE of 2.7 eV, observed in the photoelectron spectrum as the feature centered at eKE = 1.7 eV in the stable photoelectron spectrum (one photoelectron in coincidence with neutral fragment i.e. one stable radical). A broad feature centered at the expected VDE of the carboxylate form of p-coumarate (3.9 eV) was also recorded in the stable photoelectron spectrum, attributed to the photodetachment of the carboxylate anion to form the stable carboxyl radical. The stable photoelectron spectrum dominated the total photoelectron spectrum and indicated the presence of significant amounts of both tautomers in the anion beam. Therefore it was clear that the carboxyl form of p-coumarate is the most stable carboxyl radical studied to date, as a result of resonance stabilization via the conjugated π system.
The dissociative photoelectron spectrum (one photoelectron recorded in coincidence with two neutral fragments) was dominated by a low eKE feature and of much lower intensity than the stable photoelectron spectrum. The fragment mass spectrum indicated a DPD process resulting in CO2 + OHC6H4CHCH + e−. As decarboxylation is a signature of carboxyl dissociation, the observation of CO2 in the fragment mass spectrum confirms both the presence of the carboxylate tautomer in the anion beam and that dissociation is occurring from the carboxylate tautomer. The KER spectrum of p-coumarate peaked at 0.45 eV, indicating a less repulsive dissociative potential energy surface than observed for other carboxyl radicals or a greater partitioning of energy into the internal degrees of freedom of the products. The PPC spectrum showed a single feature where the most intense portion of the spectrum was to the low energy side of the calculated KEmax. This is also shown in the PPC spectrum of the decarboxylation of 11 keV p-coumarate at 4.66 eV (Fig. 5). Similar to the decarboxylation of benzoate, the only other aromatic carboxylate studied to date, the PPC spectrum extends beyond the calculated KEmax (solid line). Again, this may indicate the presence of two or more dissociative excited states, where radicals in the lowest lying excited state only undergo DPD when vibrationally excited, whereas radicals in higher energy excited states undergo decarboxylation readily to produce vibrationally excited products. The extension of events beyond the KEmax, after accounting for the 12% resolution of the PPC spectrum (ETOT >0.83 eV), may also suggest that some high frequency modes in p-coumarate are ineffectively cooled in the trap. The branching ratio to DPD in this system is reduced to 5% as a result of the increased stability of the radical, due to stabilization by conjugation and resonance. This is an overall branching ratio for both tautomers, so that the true carboxylate branching ratio to DPD is nearer 10%, assuming an equal amount of phenolate and carboxylate in the anion beam.
Evidence was also seen for a second decarboxylation channel; three-body ionic photodissociation resulting in CO2 + HCC− + C6H5OH. At the lower laser powers used in this study, the most likely detected products of a three-body photodissociation channel were a pair of neutral fragments. In this case the undetected anionic fragment was closest to the center-of-mass velocity, with the CO2 and C6H5OH recoiling away from the parent anion trajectory by the KER. This allowed a fragment mass spectrum to be recorded, but a KER spectrum was not extracted as the neutral fragments are not of equal mass. The fragment mass spectrum for p-coumarate indicated the presence of coincident CO2 + C6H5OH confirming the presence of this channel. The occurrence of this photodissociation channel is driven by the thermodynamic stability of CO2 and phenol.
3.3 Dicarboxylic acids
Dicarboxylic acids have two carboxylic groups, resulting in a wider range of possible dissociation channels than mono-carboxylates, including the potential for a double decarboxylation. Intramolecular hydrogen bonds between the two carboxylic acid groups stabilize the dicarboxylic acid monoanions relative to the neutral, resulting in larger EAs than for the monocarboxylic acid radicals.13 Photoelectron spectroscopy has been used to study the monoanions and dianions of these species, and much work has focused on the geometry adopted by the anion and radicals to accommodate these hydrogen bonds.13–16,39,40 Recent work using the high beam energy PPC spectrometer24 has focused on the dissociation dynamics of the oxalate monoanion,11 the conjugate base of the shortest dicarboxylic acid. This was the first study that detected the neutral fragments, as well as the photoelectrons, and therefore directly probed the decarboxylation dynamics of a dicarboxylic acid.The KER for the decarboxylation of the oxalate monoanion was large, peaking at 1.1 eV indicating a large degree of repulsion driving dissociation. The PPC spectrum, shown in Fig. 6, consisted of a single structureless feature, which was localized on the low energy side of the calculated maximum kinetic energy (KEmax). The O–C–O bond angle in C2O4H− was calculated to be 122.3°, which is significantly smaller than the analogous bond angle in other carboxylates (typically ≈130°) and the C–C bond length in C2O4H− was longer than in C2O4H. Both the smaller O–C–O bond angle and the longer C–C bond in the anion support accommodation of the intramolecular H bond. Therefore upon photodetachment, when the stabilizing H bonding interaction is weakened, there is a significant amount of strain in the radical molecular framework which drives the repulsive dissociation and leads to the high energy KER spectrum recorded. The vertical transition resulted in a radical with the strained anion geometry, within the Born–Oppenheimer approximation, but no stabilization from a strong intermolecular H bond. Calculations also suggested that a three-body DPD channel producing CO2 + CO2 + H + e− was energetically accessible, but despite high beam energy studies on the deuterated analogue, no H/D atoms were observed. A preference for trans-HOCO was expected due to geometric constraints, but it was not possible to conclusively assign due to the experimental resolution of the PPC spectrum. No evidence for a stable oxalate radical or an ionic photodissociation channel was observed. Clear differences between the dynamics of the monoanions of carboxylates and dicarboxylates following photodetachment were observed in this study.
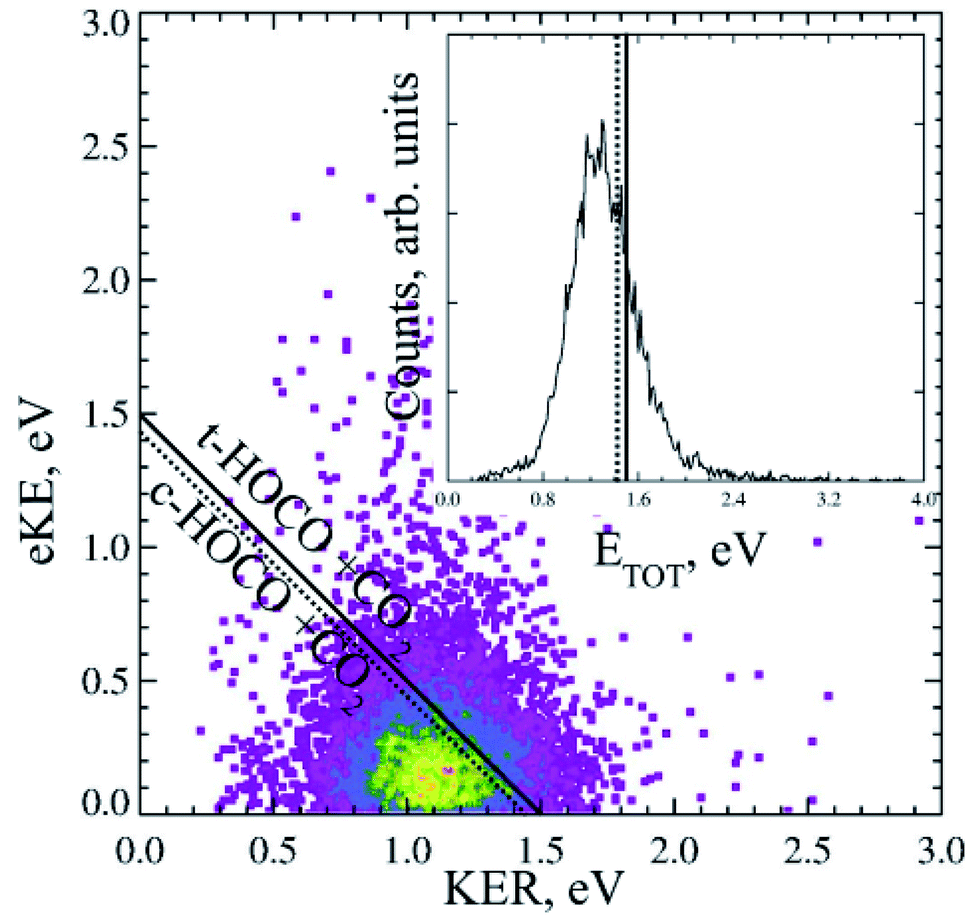 | ||
| Fig. 6 The PPC and ETOT spectrum for decarboxylation of the 21 keV oxalate monoanion, recorded at 4.66 eV. Reproduced from ref. 11 with permission from the PCCP Owner Societies. | ||
3.4 Other systems
There has been another relevant PPC spectroscopy study that will not be discussed in detail here, as it will be the focus of a future publication. The decarboxylation of the propiolate anion (HC = CCO2−) was studied using a PPC spectrometer with an EIBT.26 The predominant channel was photodetachment resulting in a stable radical, with only 4% undergoing DPD to form HCC + CO2 + e−.41 The peak KER, EA and branching ratio to DPD are included in Table 1 and Fig. 7. This ion source has been shown to inefficiently cool high frequency modes, explaining the presence of events with ETOT larger than the calculated KEmax. It is interesting to note the similarities between the data recorded for propiolate and p-coumarate, where similar branching ratios to DPD channels are observed and the ETOT spectra extend to higher energies than expected from the calculated KEmax. In both cases this can be attributed to only the most vibrationally excited anions in the anion beam undergoing DPD following photodetachment. Additionally, despite the two studies utilizing different anion sources, it is possible that high frequency modes in both the propiolate and p-coumarate are less efficiency cooled than the vibrational modes in other carboxylates, leading to the presence of some vibrationally hot ions in the molecular beam. Current work underway in the laboratory is a high beam energy PPC spectroscopy study of the aromatic carboxylate, phenylacetate. Preliminary results suggest clear similarities between the photodetachment dynamics of the phenylacetate and the longer alkyl carboxylates, in particular propanoate and 2-butenoate, and benzoate.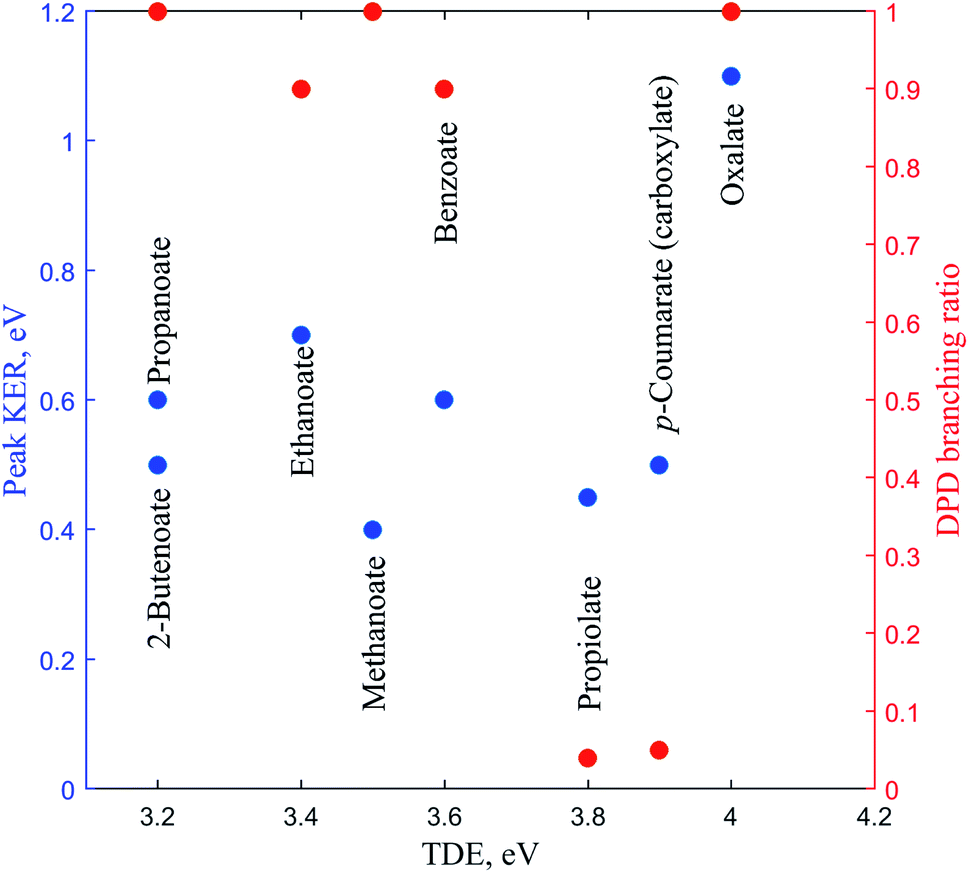 | ||
| Fig. 7 The TDE, peak KER and the DPD branching ratio of the carboxylates as recorded from PPC spectroscopy studies. | ||
4 Discussion
Table 1 and Fig. 7 summarizes the findings of the numerous PPC spectroscopy studies investigating the decarboxylation dynamics of the carboxylates reviewed here. Similar to the previous photoelectron spectroscopy studies by Wang and coworkers, the TDEs reported here for the carboxyl radicals are near 3.5 eV. The highest TDE was found for the oxalate mononanion, which is also in keeping with the previously recorded EAs for radicals formed via photodetachment of dicarboxylic acid monoanions, which are typically 1 eV higher than for carboxylates due to the stabilization of the anion by intramolecular hydrogen bonds.13 For all of the anions the dominant dissociation channel is DPD e.g.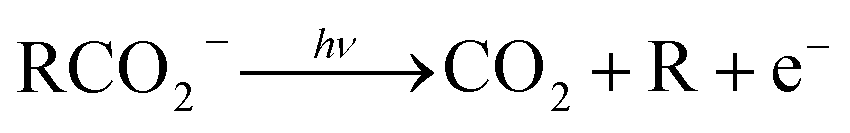 resulting in decarboxylation of the corresponding carboxyl radical following photodetachment. In addition, decarboxylation via a three-body ionic photodissociation channel is observed for p-coumarate, driven by the thermodynamic stability of the closed-shell CO2 and phenol products. Additionally, no three-body DPD processes are observed, either as a direct three-body DPD process or where the loss of CO2 leaves an unstable radical which fragments further upon the timescale of the flight time between the interaction region and the neutral detector.
resulting in decarboxylation of the corresponding carboxyl radical following photodetachment. In addition, decarboxylation via a three-body ionic photodissociation channel is observed for p-coumarate, driven by the thermodynamic stability of the closed-shell CO2 and phenol products. Additionally, no three-body DPD processes are observed, either as a direct three-body DPD process or where the loss of CO2 leaves an unstable radical which fragments further upon the timescale of the flight time between the interaction region and the neutral detector.
The branching ratio to DPD is a measure of the stability of the carboxyl radical relative to the dissociation asymptote, whereas the EA and peak KER probe the potential energy landscape of the carboxylate and carboxyl radical. Methanoate, ethanoate and propanoate are alkyl carboxylates characterized by a large branching ratio to DPD, substantial peak KER and an EA that drops with increasing chain length. 2-Butenoate is characterized by a similar peak KER, EA and branching ratio to DPD as the alky carboxylates, in particular propanoate, despite the presence of a conjugated CC double bond. Benzoate is an aromatic carboxylate with extended conjugation of the π system, which has similar peak KER and DPD branching ratios, but a higher EA, than the alkyl carboxylates. The only other aromatic carboxylate studied to date, p-coumarate, has a lower peak KER and much lower branching ratio to DPD than the alkyl carboxylates or benzoate, but the carboxylate isomer has a higher EA. Propiolate is characterized by a similar peak KER, EA and DPD branching ratio to p-coumarate. The oxalate monoanion, the only dicarboxylic acid anion studied to date, has the largest peak KER, the largest branching ratio to DPD and the largest EA of any carboxylate studied so far. The oxalate monoanion has a strained geometry with a smaller O–C–O bond angle than other carboxylates (130° for CH3CO2−, 129.2° for C6H5CO2−, and 122.3° for C2O4H−)10,11,37 in order to accommodate an intramolecular H bond. Upon photodetachment the H bond is substantially weakened, leading to strain in the molecular framework, and resulting in a larger peak KER than for other carboxylates. From Fig. 7 it is clear that the relationship between EA and the branching ratio to DPD is bimodal, with DPD dominating for all the carboxylates, except propiolate and p-coumarate. Similar values of peak KER are seen for all of the carboxylates studied, regardless of EA, except for the oxalate monoanion where a large amount of repulsion drives dissociation.
Many of the carboxyl radicals investigated here exhibit multiple energetically accessible excited states at the photon energies used. The simplest carboxyl radical HCO2 has three dissociative, low-lying electronic states in order of increasing energy 2A1, 2B2 and 2A2. Benzoate also exhibits three low-lying states of the same symmetry. However the ground state is 2B2 state and is metastable in character, where the 2A1 becomes the first excited state. Therefore as the complexity of the R group increases the relative ordering of the carboxyl radical states may change. For the aromatic carboxylates the ground state is stabilized significantly, becoming metastable in character. More complex radicals or those with different symmetries often have additional low-lying electronic states which are dissociative in character. Ethanoate, benzoate, propiolate and p-coumarate all result in stable carboxyl radicals with a lifetime longer than the flight time between the interaction region and the detector (≈10 μs). The C2O4H radical was predicted to have an energetically accessible bound radical, but it was not observed experimentally.11 This may be due to poor Franck–Condon overlap as the calculations indicated a shorter C–C bond length than the anion. All of the carboxyl radicals exhibit decarboxylation, indicating the presence of at least one energetically accessible dissociative potential energy surface for each system. In some cases, including methanoate, ethanoate, benzoate and p-coumarate, DPD is shown to occur from multiple excited states at the photon energies studied. No evidence is seen for excited, bound radical states.
One of the most interesting aspects of these PPC spectroscopy studies investigating the decarboxylation of carboxyl radicals following photodetachment is the distribution of vibrational excitation in the parent anions and products. Various PPC spectrometers, with different ion sources, have been used to study the decarboxylation of the carboxylates, which may result in a variety of parent anion vibrational temperatures. For the high beam energy PPC spectrometer shown in Fig. 1, which has a buffer-gas-cooled ion trap, a vibrational temperature of 298 K has been reported.24 This is consistent with some vibrational excitation in the parent anions, particularly for large systems with many normal modes. Some evidence is seen in the p-coumarate measurements, for ineffective cooling of high-frequency normal modes in the buffer-gas-cooled accumulation ion trap. The kHz pulsed discharge source used in previous PPC spectrometers has been shown to result in a non-Boltzmann distribution of anion vibrational temperatures, where high frequency modes are ineffectively cooled, as is evident in the propiolate measurements.25,42 The PPC spectroscopy studies reviewed here demonstrate that decarboxylation often results in vibrational excitation in the radical and the products, as in many of the photoelectron spectra resolved and unresolved vibrational features are observed. In the case of methanoate a vibrationally resolved PPC spectrum is recorded, with clear features attributed to excitation of the CO2 modes. The resolved structure in this case is a result of the near degeneracy of the vibrational energy levels of the DCO2 electronic states and the CO2 modes. Benzoate and p-coumarate undergo DPD from multiple excited states. Decarboxylation from the lowest-lying excited state occurs only for the aromatic carboxyls with the most vibrational excitation, whereas the higher energy excited state radicals undergo dissociation readily without vibrational excitation and result in vibrationally excited products. Previous calculations on benzoate have indicated that photodetachment is expected to result in excitation of the carboxyl group bending mode.37 The PPC and ETOT spectra are broad for the aromatic carboxylates indicating vibrationally excited products as well as vibrationally excited parent anions. Calculations for many different carboxylates have indicated a substantial change in O–C–O bond angle upon photodetachment, suggesting excitation of the carboxyl bending mode in the radicals and the CO2 bend in the products.10,11,37
In the future the hexapole accumulation trap on the high beam energy PPC spectrometer will be cryogenically cooled. This will allow the decarboxylation dynamics of vibrationally cold carboxylates to be studied in a kinematically complete manner, with the aim to probe the effect of vibrational excitation on the decarboxylation dynamics and reduce spectral congestion.25,43 These studies will also be extended to other biologically and atmospherically relevant carboxylates such as citrate and pyruvate. The implementation of the LINAC on the high beam energy PPC spectrometer has increased the mass difference between the parent anion and the neutral fragment that can be successfully studied by PPC spectroscopy, by increasing the detectability of the light fragments. This allows for the DPD of more complex carboxylates to be investigated where the mass of the carboxylate is far larger than the mass of CO2. In addition the dissociation dynamics of dicarboxylate dianions will be studied, where multiple plausible decarboxylation pathways, including double decarboxylation, exist.
Previous photoelectron spectroscopy studies of carboxylates have demonstrated that photodetachment occurs via the removal of an electron localized within the CO2− group.17 Furthermore it has previously been demonstrated that the chain length for alkyl carboxylates has a minimal effect upon the EA. It is clear that this trend continues when studying the dissociation dynamics of the carboxylates, as the results of many of the carboxylate PPC spectroscopy studies are very similar, in particular for ethanoate, methanoate, propanoate, 2-butenoate and benzoate. Typically, following photodetachment of a carboxylate, decarboxylation occurs characterized by a TDE of 3.2–3.6 eV, a DPD branching ratio ≈1, a peak KER of 0.4–0.7 eV. Only those carboxylates with additional carboxylate groups or extended π systems within the R group, such as p-coumarate or the oxalate monoanion, exhibit substantially different dissociation dynamics. In effect, the dissociation dynamics of a carboxyl radical are dominated by the carboxylate functional group, with only a minimal effect from the R group, unless the R group strongly stabilizes the carboxylate or carboxyl.
5 Conclusions
PPC spectroscopy allows the decarboxylation dynamics of the carboxylates to be investigated in a kinematically complete manner. All of the systems studied undergo decarboxylation, but the branching ratio to DPD provides a measure of the stability of the carboxyl radical. Conjugated or aromatic R groups are shown to increase the stability of the radical, and increase the preference for photodetachment rather than DPD. In the cases where DPD dominates large peak KERs are recorded, consistent with substantial repulsion in the transition state driving dissociation. The largest peak KER is reported for the oxalate monoanion, where there is substantial strain in the radical upon photodetachment, as a result of the weakening of the intramolecular hydrogen bond. PPC spectroscopy probes the potential energy landscape of both the carboxylate anion and the carboxyl radical, providing new insights into decarboxylation processes of importance in biology and atmospheric chemistry.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors would like to acknowledge the contributions of the many group members, past and present, who have studied the decarboxylation dynamics of carboxylates using PPC spectroscopy. The high beam energy PPC spectrometer was developed via support from the NSF Division of Chemistry under grant CHE-1464548 and grant CHE-1955449.References
- H. Kolbe, Justus Liebigs Ann. Chem., 1849, 69, 257–294 CrossRef.
- D. H. R. Barton, H. A. Dowlatshahi, W. B. Motherwell and D. Villemin, J. Chem. Soc., Chem. Commun., 1980, 732–733 RSC.
- R. G. Johnson and R. K. Ingham, Chem. Rev., 1956, 56, 219–269 CrossRef CAS.
- T. G. Clements and R. E. Continetti, J. Chem. Phys., 2001, 115, 5345–5348 CrossRef CAS.
- A. W. Ray, B. B. Shen, B. L. Poad and R. E. Continetti, Chem. Phys. Lett., 2014, 592, 30–35 CrossRef CAS.
- J. Gibbard, E. Castracane, A. I. Krylov and R. Continetti, Phys. Chem. Chem. Phys., 2021, 23, 18414–18424 RSC.
- X.-Q. Hu, Z.-K. Liu, Y.-X. Hou and Y. Gao, iScience, 2020, 23, 101266 CrossRef CAS PubMed.
- R. W. Molt, A. M. Lecher, T. Clark, R. J. Bartlett and N. G. J. Richards, J. Am. Chem. Soc., 2015, 137, 3248–3252 CrossRef CAS PubMed.
- R. E. Continetti, Int. Rev. Phys. Chem., 1998, 17, 227–260 Search PubMed.
- Z. Lu and R. E. Continetti, J. Phys. Chem. A, 2004, 108, 9962–9969 CrossRef CAS.
- J. A. Gibbard, E. Castracane, A. J. Shin and R. E. Continetti, Phys. Chem. Chem. Phys., 2020, 22, 1427–1436 RSC.
- G. A. Pino, R. A. Jara-Toro, J. P. Aranguren-Abrate, C. Dedonder-Lardeux and C. Jouvet, Phys. Chem. Chem. Phys., 2019, 21, 1797–1804 RSC.
- H.-K. Woo, X.-B. Wang, K.-C. Lau and L.-S. Wang, J. Phys. Chem. A, 2006, 110, 7801–7805 CrossRef CAS PubMed.
- X.-B. Wang, X. Yang, J. B. Nicholas and L.-S. Wang, Science, 2001, 294, 1322–1325 CrossRef CAS PubMed.
- X.-P. Xing, X.-B. Wang and L.-S. Wang, Phys. Rev. Lett., 2008, 101, 083003 CrossRef PubMed.
- X.-B. Wang, C.-F. Ding and L.-S. Wang, Phys. Rev. Lett., 1998, 81, 3351–3354 CrossRef CAS.
- X.-B. Wang, H.-K. Woo, B. Kiran and L.-S. Wang, Angew. Chem., Int. Ed., 2005, 44, 4968–4972 CrossRef CAS PubMed.
- E. Garand, K. Klein, J. F. Stanton, J. Zhou, T. I. Yacovitch and D. M. Neumark, J. Phys. Chem. A, 2010, 114, 1374–1383 CrossRef CAS PubMed.
- I. B. Nielsen, S. Boyé-Péronne, M. O. A. El Ghazaly, M. B. Kristensen, S. Brøndsted Nielsen and L. H. Andersen, Biophys. J., 2005, 89, 2597–2604 CrossRef CAS PubMed.
- L. Lammich, J. Rajput and L. H. Andersen, Phys. Rev. E, 2008, 78, 051916 CrossRef PubMed.
- T. Rocha-Rinza, O. Christiansen, J. Rajput, A. Gopalan, D. B. Rahbek, L. H. Andersen, A. V. Bochenkova, A. A. Granovsky, K. B. Bravaya, A. V. Nemukhin, K. L. Christiansen and M. B. Nielsen, J. Phys. Chem. A, 2009, 113, 9442–9449 CrossRef CAS PubMed.
- T. Rocha-Rinza, O. Christiansen, D. B. Rahbek, B. Klærke, L. H. Andersen, K. Lincke and M. B. Nielsen, Chem.–Eur. J., 2010, 16, 11977–11984 CrossRef CAS PubMed.
- L. H. Andersen, A. V. Bochenkova, J. Houmøller, H. V. Kiefer, E. Lattouf and M. H. Stockett, Phys. Chem. Chem. Phys., 2016, 18, 9909–9913 RSC.
- J. A. Gibbard, A. J. Shin, E. Castracane and R. E. Continetti, Rev. Sci. Instrum., 2018, 89, 123304 CrossRef CAS PubMed.
- B. B. Shen, Y. Benitez, K. G. Lunny and R. E. Continetti, J. Chem. Phys., 2017, 147, 094307 CrossRef PubMed.
- C. J. Johnson, B. B. Shen, B. L. J. Poad and R. E. Continetti, Rev. Sci. Instrum., 2011, 82, 105105 CrossRef PubMed.
- D. Feller, E. S. Huyser, W. T. Borden and E. R. Davidson, J. Am. Chem. Soc., 1983, 105, 1459–1466 CrossRef CAS.
- S. D. Peyerimhoff, P. S. Skell, D. D. May and R. J. Buenker, J. Am. Chem. Soc., 1982, 104, 4515–4520 CrossRef CAS.
- A. Rauk, D. Yu and D. A. Armstrong, J. Am. Chem. Soc., 1994, 116, 8222–8228 CrossRef CAS.
- K. Klein, E. Garand, T. Ichino, D. M. Neumark, J. Gauss and J. F. Stanton, Theor. Chem. Acc., 2011, 129, 527 Search PubMed.
- J. Ma and H. Guo, Chem. Phys. Lett., 2011, 511, 193–195 CrossRef CAS.
- Z. Zhou, X. Cheng, X. Zhou and H. Fu, Chem. Phys. Lett., 2002, 353, 281–289 CrossRef CAS.
- E. A. Lissi, G. Massiff and A. E. Villa, J. Chem. Soc., Faraday Trans. 1, 1973, 69, 346–351 RSC.
- X.-B. Wang, H.-K. Woo, L.-S. Wang, B. Minofar and P. Jungwirth, J. Phys. Chem. A, 2006, 110, 5047–5050 CrossRef CAS PubMed.
- D. Yu, A. Rauk and D. A. Armstrong, J. Chem. Soc., Perkin Trans. 2, 1994, 2207–2215 RSC.
- G. Caldwell, R. Renneboog and P. Kebarle, Can. J. Chem., 1989, 67, 611–618 CrossRef CAS.
- H.-K. Woo, X.-B. Wang, B. Kiran and L.-S. Wang, J. Phys. Chem. A, 2005, 109, 11395–11400 CrossRef CAS PubMed.
- C. R. S. Mooney, M. A. Parkes, A. Iskra and H. H. Fielding, Angew. Chem., Int. Ed., 2015, 54, 5646–5649 CrossRef CAS PubMed.
- X.-B. Wang, X. Yang, J. B. Nicholas and L.-S. Wang, J. Chem. Phys., 2003, 119, 3631–3640 CrossRef CAS.
- C.-F. Ding, X.-B. Wang and L.-S. Wang, J. Phys. Chem. A, 1998, 102, 8633–8636 CrossRef CAS.
- A. Ray, S. M. Rabidoux, T. Ichino, A. L. Cooksy, J. F. and R. E. Continetti, in preparation, 2021.
- Z. Lu, Q. Hu, J. E. Oakman and R. E. Continetti, J. Chem. Phys., 2007, 126, 194305 CrossRef PubMed.
- B. B. Shen, K. G. Lunny, Y. Benitez and R. E. Continetti, Front. Chem., 2019, 7, 295 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra06340e |
| This journal is © The Royal Society of Chemistry 2021 |