
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn ultrafast enzyme-free acoustic technique for detaching adhered cells in microchannels†
Alinaghi Salari ab,
Sila Appak-Baskoyac,
Imogen R. Coeacd,
Scott S. H. Tsai*ae and
Michael C. Kolios*af
ab,
Sila Appak-Baskoyac,
Imogen R. Coeacd,
Scott S. H. Tsai*ae and
Michael C. Kolios*af
aInstitute for Biomedical Engineering, Science and Technology (iBEST), Toronto, ON M5B 1T8, Canada
bBiomedical Engineering Graduate Program, Ryerson University, Toronto, ON M5B 2K3, Canada
cDepartment of Chemistry and Biology, Ryerson University, Toronto, ON M5B 2K3, Canada
dMolecular Science Graduate Program, Ryerson University, Toronto, ON M5B2K3, Canada
eDepartment of Mechanical and Industrial Engineering, Ryerson University, Toronto, ON M5B 2K3, Canada. E-mail: scott.tsai@ryerson.ca
fDepartment of Physics, Ryerson University, Toronto, ON M5B 2K3, Canada. E-mail: mkolios@ryerson.ca
First published on 5th October 2021
Abstract
Adherent cultured cells are widely used biological tools for a variety of biochemical and biotechnology applications, including drug screening and gene expression analysis. One critical step in culturing adherent cells is the dissociation of cell monolayers into single-cell suspensions. Different enzymatic and non-enzymatic methods have been proposed for this purpose. Trypsinization, the most common enzymatic method for dislodging adhered cells, can be detrimental to cells, as it can damage cell membranes and ultimately cause cell death. Additionally, all available techniques require a prolonged treatment duration, typically on the order of minutes (5–10 min). Dissociation of cells becomes even more challenging in microfluidic devices, where, due to the nature of low Reynolds number flow and reduced mixing efficiency, multiple washing steps and prolonged trypsinization may be necessary to treat all cells. Here, we report a novel acoustofluidic method for the detachment of cells adhered onto a microchannel surface without exposing the cells to any enzymatic or non-enzymatic chemicals. This method enables a rapid (i.e., on the order of seconds), cost-effective, and easy-to-operate cell detachment strategy, yielding a detachment efficiency of ∼99% and cellular viability similar to that of the conventional trypsinization method. Also, as opposed to biochemical-based techniques (e.g., enzymatic), in our approach, cells are exposed to the dissociating agent (i.e., substrate-mediated acoustic excitation and microstreaming flow) only for as long as they remain attached to the substrate. After dissociation, the effect of acoustic excitation is reduced to microstreaming flow, therefore, minimizing unwanted effects of the dissociating agent on the cell phenotype. Additionally, our results suggest that cell excitation at acoustic powers lower than that required for complete cell detachment can potentially be employed for probing the adhesion strength of cell–substrate attachment. This novel approach can, therefore, be used for a wide range of lab-on-a-chip applications.
Introduction
Cell culture protocols of adherent cells are widely used in many in vitro cell biology studies. Adherent cells are commonly cultured on coated or non-coated polystyrene, polypropylene, or glass substrates. At confluency, or for regular maintenance, the cells typically need to be detached from the substrate. The detachment is traditionally carried out using different enzymes that can cleave the surface proteins involved in forming the focal adhesions, which keep cells attached to the substrate. For this purpose, trypsin, a digestive enzyme discovered in 1876, is commonly used.1 Trypsin is often supplied with the addition of ethylenediaminetetraacetic acid (EDTA), which acts as a metal chelator that removes calcium ions from the cells.2 Although trypsin is widely used in cell culture applications, it has long been known that it can be detrimental to cells due to the cleavage of physiologically important cell surface or extracellular facing glycoproteins, ultimately leading to multiple downstream effects that can result in cell breakdown and death.3–7 Some cell types are particularly susceptible to trypsin-related damage, such as mesenchymal stem cells, where it is important to minimize the application of animal- and human-derived products, including trypsin, to avoid pathogenic transmission.4,8 There is, therefore, much interest in finding alternative enzyme-free methods for dissociating cell monolayers. Current alternatives include buffer treatments and mechanical detachment.9 Enzyme-free cell dissociation buffer treatments can preserve the surface proteins, but they are most effective on lightly adhered cells.10 Mechanical detachment techniques, such as cell scraping,9 and more recently, ultrasound traveling waves, are also enzyme-free.11 However, all these techniques require a treatment duration on the order of minutes (typically 5–10 min).2,11In addition, there is growing interest in microfluidic applications of drug screening12 and organ-on-a-chip13,14 platforms which involve culturing cells inside microchannels. However, trypsinization and other biochemical reactions inside microchannels require multiple washing steps and a complete reagent replacement for the reaction to occur uniformly and thoroughly at the same rate for all cells. With no active mixing strategy, analyte mixing in a low Reynolds number flow regime or in a static fluid in microchannels can be challenging, and trypsinization can be less effective than in macro-scale cell culture flasks and dishes.
Recently, we reported that exposure of cells to acoustic waves at low and moderate excitation powers (i.e., smaller than ∼20 W) could generate cell-induced microstreaming15 and facilitate the delivery of extracellular materials to the cell.16 The actuation power used in these two works was high enough to generate acoustic microstreaming only, and therefore, most cells retained their attachment to the substrate of the microfluidic channel even during relatively long (e.g., ∼20 min) exposures. However, here, we report that exposure of adhered cells to short (200–500 ms) pulse waves at high excitation powers (∼200 W) can result in cells detaching from the substrate within seconds. As opposed to focused surface acoustic waves which have been shown to locally detach a small number of cells pre-treated with phosphate buffer saline (PBS) from a culture surface,17 in our acoustofluidic technique larger populations of cells can be detached from a microfluidic channel. We use MDA-MB-231 cells as examples of poorly spread cells (i.e., cells with a relatively low spreading area) and human dermal microvascular endothelial cells (HDMECs) as examples of highly spread cells (i.e., cells with a relatively high spreading area).18 At the attachment site, cells form focal adhesion complexes through the linkage between the integrins and the extracellular matrix (i.e., fibronectin in our system).19 As a cell spreads over a surface, the cell-surface adhesion strength increases before it plateaus.20,21 Highly-spread cells (e.g., endothelial cells) generally have a greater adhesion strength compared to highly protrusive cells (e.g., lymphocytes and some cancer cells).18,22,23 The adhesion strength also depends on the surface characteristics of the substrate that the cells are attached to. For example, the spreading area of cells on stiffer substrates is shown to be higher than that on softer ones.24,25 The detached MDA-MB-231 cells show viability of 86–91% and can re-adhere to the substrate surface within hours following the acoustic exposure. We also demonstrate a controlled and gradual detachment of HDMECs from the substrate with high viability (>80%) by exposing the cells to multiple pulse waves. The acoustic technique presented here can be used as a rapid, easy-to-operate, and cost-effective method for detaching or probing the adhesion strength of different adherent cell types in various lab-on-a-chip applications.
Results and discussion
Our methodology for detaching adhered cells from a substrate requires the cells to be exposed to short pulses of acoustic waves. Thin substrates were used to maximize the contribution of Lamb waves to the propagation of sound waves through the substrate.15,26 As a proof-of-concept demonstration and for the ease of culturing, we use a small population of cells (i.e., a few thousand cells in a region of interest (ROI)) inside a polydimethylsiloxane (PDMS) microfluidic channel bonded to a ∼70 μm glass substrate. Once the cells are seeded into a fibronectin-coated microfluidic channel and cultured for at least two days, we expose them to short (in the range of 200–500 ms) pulses of acoustic waves using a piezoelectric (PZT) transducer bonded to the glass surface near the microfluidic channel (see Fig. S1†). The regions of maximum acoustic pressures on the substrate can vary depending on the device geometry, substrate material, and actuation frequency.15,16 We hypothesize that the substrate vibration and the resultant acoustic forces, as well as the microstreaming flow, are responsible for the detachment of the cells. We confirm the vibrations (i.e., flexural waves) of the substrate within the ROI, located inside the microfluidic channel near the PZT, using optical coherence tomography (OCT) (see Fig. S2†).To demonstrate the versatility of our technique, we use an epithelial breast cancer cell line (i.e., MDA-MB-231) as an example of poorly spread cells, i.e., cells with a relatively low spreading area. We then used and an endothelial cell line (i.e., human dermal microvascular endothelial cell (HDMEC)) as an example of highly spread cells, i.e., cells with a relatively high spreading area.18
In our device, when the PZT is actuated at relatively low power (lower than ∼20 W), cells can undergo a stable oscillation, which can give rise to microstreaming flow.15 However, at higher actuation powers, especially above ∼50 W, cells gradually lose their adhesion to the substrate over time (Fig. 1a). We find that a pulse wave of ∼200 W actuation power and ∼200 ms duration is enough to detach MDA-MB-231 cells (Fig. 1b) in less than one second (see Video S1†) with a detachment efficiency of ∼99% (see Note S1†). The acoustically-treated cells that can move freely from their initial (pre-exposure) location are considered as fully detached cells. The detached cells can then be washed and collected for further processing. We also evaluate the re-adhesion capabilities of the acoustically-treated cells to the same glass surface by culturing the detached cells for 48 h inside the same microfluidic channel (Fig. 2a). Our results show that the acoustically-treated cells can re-adhere, spread, and regain their morphology within 16 h post-exposure. In addition, the acoustically-treated cells show viability of 86% using the conventional propidium iodide (PI) staining and 91% using a re-adhesion assay (see Note S2†). The lower viability found from PI staining could be due to the formation of transient pores on the plasma membrane of some of the exposed cells upon the detachment from the substrate.27,28 This can cause PI molecules to be taken up by some cells, which, therefore, can be incorrectly assessed as dead cells.
 | ||
| Fig. 1 Controlled acoustic actuation can detach the adhered cells. (a) Schematic (not to scale) of the acoustic technique consisting of a PZT transducer attached to a thin glass substrate near a microfluidic channel (not shown), where the cells are adhered. Depending on the acoustic excitation power and the cell adhesion strength, complete cell detachment may require one or numerous acoustic pulse waves. (b) Bright-field and fluorescence images of MDA-MB-231 cells before and after exposure to a ∼200 ms pulse wave when the PZT is driven at ∼200 W input power. The cell detachment occurs immediately after acoustic excitation. Cell nuclei are stained with Hoechst 33342. The scale bar in (b) represents 50 μm and applies to all images. The schematic in (a) is created with Biorender.com. | ||
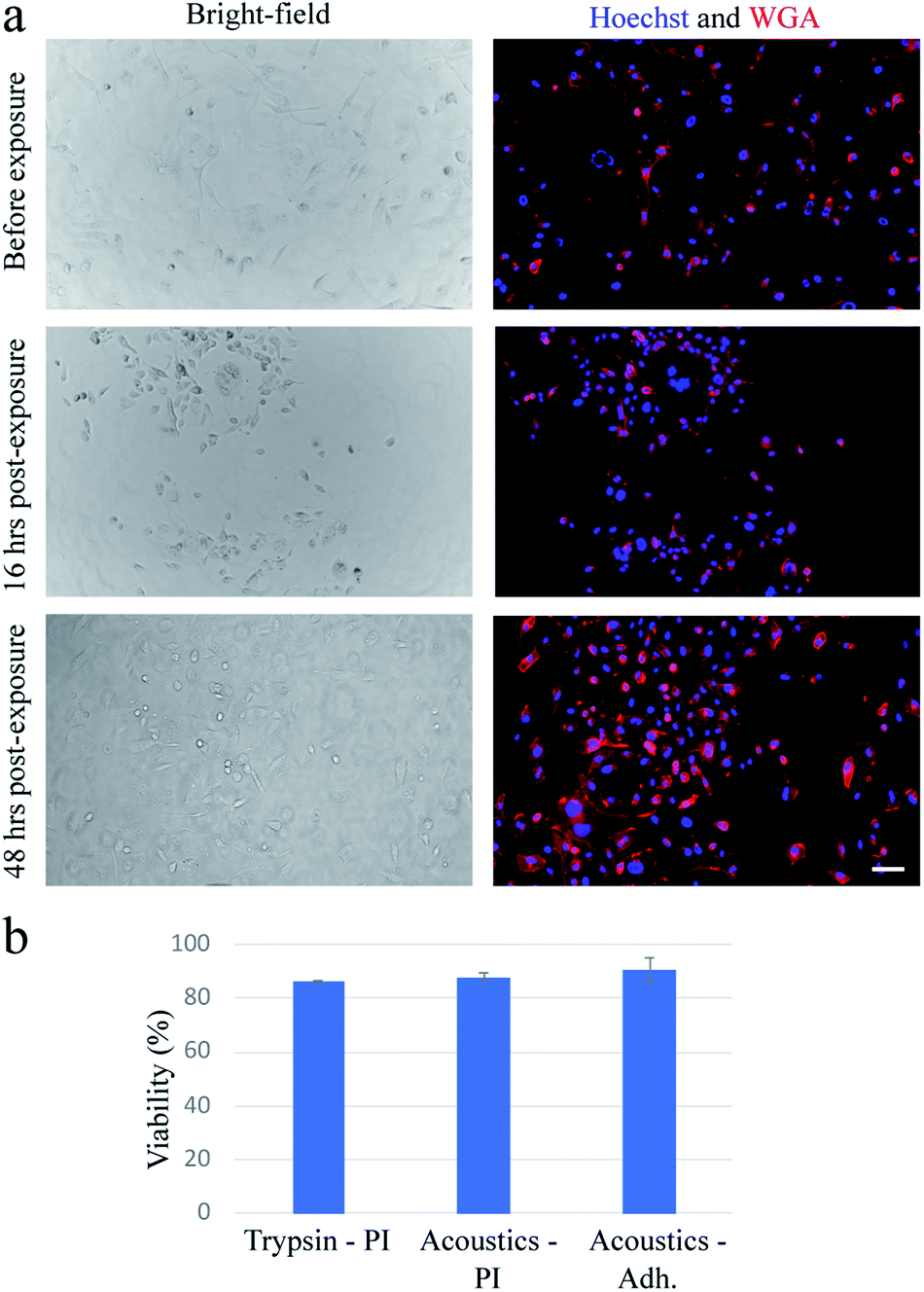 | ||
| Fig. 2 The acoustically-treated MDA-MB-231 cells show equivalent viability to the trypsinized cells. (a) Bright-field and fluorescence images showing the morphology of the MDA-MB-231 cells before exposure, and 16 h and 48 h post-exposure. The acoustically-treated cells can adhere and spread to the glass surface within 16 h post-exposure. (b) Viability results of the acoustically-treated cells against the trypsinized ones. Conventional PI staining is used to determine the number of dead cells to compare cellular viability. The viability is also estimated using a re-adhesion assay at 16 h post-exposure to determine the ratio of the re-adhered cells to the initial number of cells. These results show the acoustically-treated cells can re-adhere and regain their cellular morphology within 16 h post-exposure while maintaining high viability. A detachment efficiency of ∼99% is obtained from the acoustically-treated cells. The plasma membrane and nucleus of the cells are stained with wheat germ agglutinin (WGA) and Hoechst 33342, respectively. The scale bar in (a) represents 50 μm and applies to all images. The error bars represent the standard deviation of at least two independent experiments. | ||
The temperature in the ROI inside the microfluidic channel near the PZT and on the PZT surface both are measured using an infrared camera (see Fig. S4†). These measurements show that once a pulse is applied to the PZT, the maximum temperature in the ROI does not surpass ∼33 °C. This increase in temperature lasts only a few seconds. Even though the role of temperature on cell detachment in our system cannot be ruled out completely, we hypothesize that this role is minimal, if not negligible, due to the relatively small temperature rise and short duration of the exposure.
To confirm that these findings were applicable for cells with a high spreading area, we repeat the experiments with HDMECs, which are primary endothelial cells. We find that multiple pulse waves, with actuation amplitude similar to those used for the exposure of the MDA-MB-231 cells, are required to detach HDMECs from the substrate fully. As highlighted by dashed lines on the fluorescence images in Fig. 3, upon the actuation of each pulse wave, the cells lose a portion of their projected area. As a result, the average changes in the projected area of the cells gradually decrease as five pulse waves actuate the cells during a ∼1 min exposure.
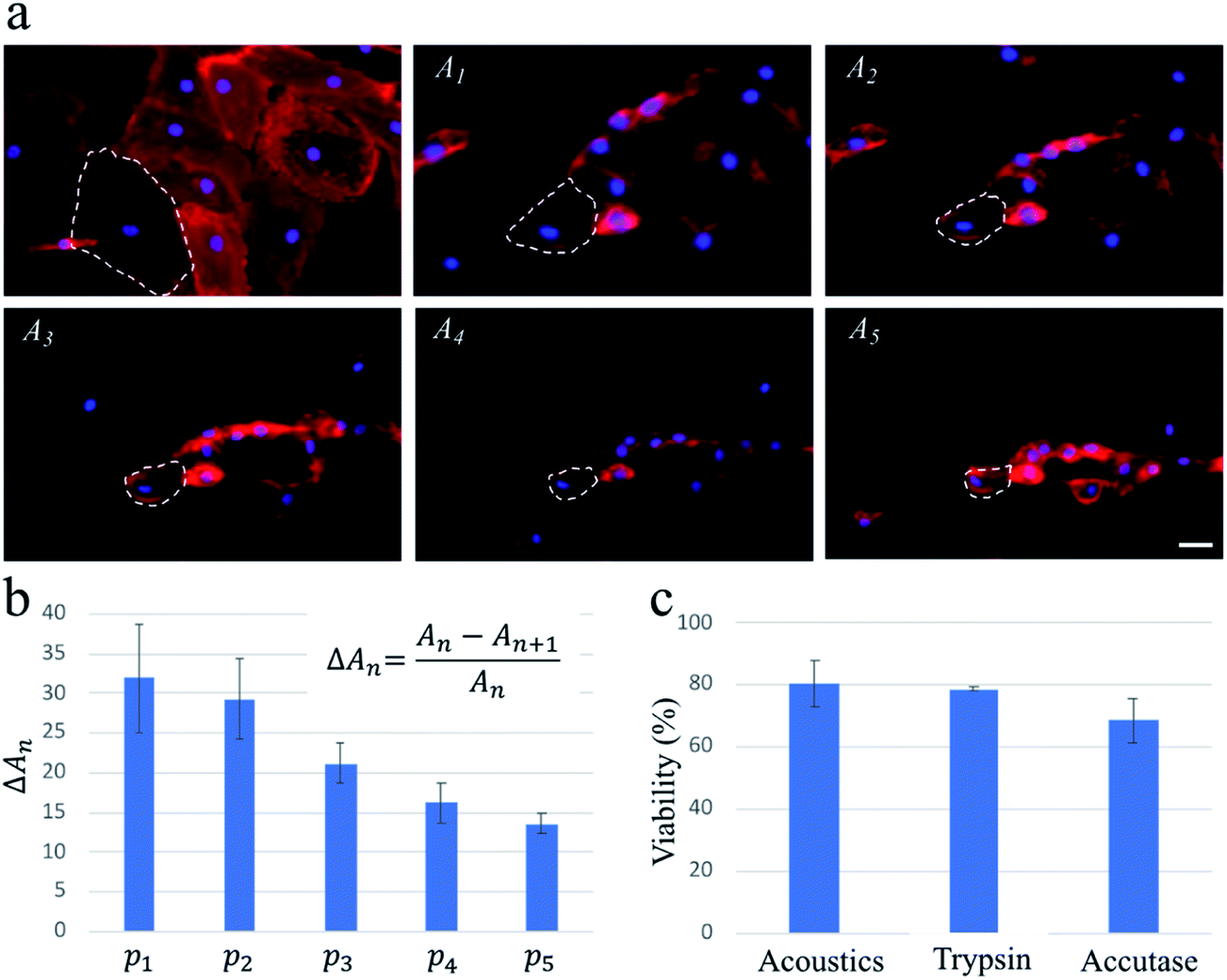 | ||
| Fig. 3 The acoustic treatment with five consecutive pulse waves (i.e., p1 to p5) enables a highly controlled approach with high viability for gradually detaching HDMECs. (a) The red areas named A1 to A5 are the projected area of the fluorescently labelled HDMECs captured right before actuating pulses p1 to p5, respectively. For all pulse waves, the PZT is actuated at ∼200 W input power for ∼500 ms, and the total duration of the experiment is ∼1 min. The plasma membrane and the nucleus are stained with WGA and Hoechst, respectively, to better visualize the projected area of the cells. To illustrate the effect of consecutive pulses, the shrinking area of one cell is outlined by dashed lines in all images. (b) The graph shows the average reduction in the projected area of ∼30 cells. Upon the actuation of each pulse wave, the cells lose some of their projected areas. (c) Viability results of acoustically-treated cells and cells treated with trypsin and accutase. Conventional PI and Hoechst staining is used to compare the cellular viability of the three methods. The scale bar in (a) represents 25 μm and applies to all images. The error bars in (b and c) represent the standard deviation of at least two independent experiments. | ||
The capillary endothelial cells have been shown to be greater in size compared to the MDA-MB-231 cells.29,30 Also, fibronectin used in our system for enhancing the cell binding onto the channel surface can contribute to HDMECs adhesion being stronger, due to their larger spreading area, than that of MDA-MB-231 cells (see Fig. 2a and 3a for a rough comparison between their spreading areas). These factors could be the reasons why multiple pulse waves are needed for gradually detaching HDMECs, as opposed to MDA-MB-231 cells which can be fully detached after applying only one pulse wave of the same power. This important finding suggests a potential application of our acoustic methodology for probing cell–substrate adhesive forces in a controlled and high-throughput manner compared to conventional techniques such as atomic force microscopy.31
In addition to the drawbacks of trypsinization mentioned above, the optimal concentration and incubation time needed for an effective treatment, as well as the cellular response, can vary for different cell lines. Primary cells (e.g., stem cells) are more resistant to the activity of trypsin, mostly due to the expression of extracellular matrix proteins that are not digested by trypsin. However, due to the nature of sound wave excitation and propagation in our system, upon cell detachment, the substrate-mediated acoustic energy experienced by the cells drops significantly, even if the acoustic excitation continues. Therefore, to account for the variability in adhesion strength in cells, the acoustic excitation can continue until all cells are detached, as a longer acoustic excitation cannot damage the already detached cells. This is a unique characteristic of our system compared to the chemical-based (e.g., enzymatic) cell treatment methods where the entire cell population is incubated with the dissociating agent for a particular time.
In summary, we have demonstrated a novel enzyme-free methodology for dissociating adhered cells using acoustofluidics. Our technique can significantly reduce the time needed for cell treatment while maintaining high cellular viability. Depending on the adhesion strength of the cells, one or multiple pulse waves of different powers and durations can be introduced in order to detach different cell types. Compared to the biochemical4,5,32,33 and the more recent ultrasound11,34 techniques, which require up to several minutes (typically 5–10 min) for an effective dissociation of an entire cell population, our technique shortens the treatment duration by two orders of magnitude to seconds. As opposed to enzymatic techniques, cells are exposed to the dissociating agent only for as long as they remain attached to the substrate. Finally, our technique can potentially be used to probe the cell–substrate adhesion strength in a label-free and high-throughput manner, which will be explored in the future.
Methods
Device fabrication
We used standard photolithography and soft lithography35 techniques to fabricate the PDMS microfluidic channel (145 μm × 2 mm × 3 cm). Briefly, a PDMS mixture (with the base to crosslinker ratio at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was poured on the silicon wafer. After curing at 70 °C overnight, the PDMS channel was cut, and the inlets were punched using a biopsy punch. The PDMS channel was then bonded to a borosilicate glass substrate (Agar Scientific, UK) with a thickness of ∼70 μm after the two surfaces are functionalized using plasma treatment. A PZT transducer (diameter: 27 mm, 7BB-27-4L0, Mouser Electronics, USA) was then bonded on the glass substrate near the microfluidic channel using the PDMS mixture. Actuation pulse waves at 96 kHz were first generated at 300 m Vpp using a function generator (33521A, Agilent, USA) and then amplified using an amplifier (2200L, E&I, USA). Substrate vibrations were characterized using an optical coherence tomography system (Swept-Source OCT, VEGA series, Thorlabs, USA). Temperature measurements were conducted using an infrared camera (Thermovision A40, FLIR, USA).
:1) was poured on the silicon wafer. After curing at 70 °C overnight, the PDMS channel was cut, and the inlets were punched using a biopsy punch. The PDMS channel was then bonded to a borosilicate glass substrate (Agar Scientific, UK) with a thickness of ∼70 μm after the two surfaces are functionalized using plasma treatment. A PZT transducer (diameter: 27 mm, 7BB-27-4L0, Mouser Electronics, USA) was then bonded on the glass substrate near the microfluidic channel using the PDMS mixture. Actuation pulse waves at 96 kHz were first generated at 300 m Vpp using a function generator (33521A, Agilent, USA) and then amplified using an amplifier (2200L, E&I, USA). Substrate vibrations were characterized using an optical coherence tomography system (Swept-Source OCT, VEGA series, Thorlabs, USA). Temperature measurements were conducted using an infrared camera (Thermovision A40, FLIR, USA).
Cell preparation
A fibronectin (Sigma, USA) solution at a concentration of 100 μg ml−1 was used to functionalize the glass surface of the microfluidic channel and to facilitate cell adhesion to the substrate. The cells were then seeded into the microfluidic channel and were incubated at 37 °C for at least two days before the experiments were conducted. The cell culture medium used for culturing MDA-MB-231 cells is composed of RPMI 1640 (Wisent Bioproducts, Canada) supplemented with 10% (v/v) fetal bovine serum (Wisent Bioproducts, Canada) and 1% (v/v) PenStrep (Wisent Bioproducts, Canada). For the enzymatic detachment experiments, MDA-MB-231 cells were incubated with 0.25% trypsin–EDTA (Wisent Bioproducts, Canada) for 4 min. To enzymatically dissociate HDMECs, we incubated them with trypsin and accutase (Gibco, Thermofisher, USA) for 15 and 10 min, respectively. We used the endothelium growth medium (PromoCell, Germany) for culturing HDMECs. For the cellular membrane staining, WGA Alexa Fluor 633 Conjugate (Thermofisher, USA) was used at a dilution ratio of 1:100 in the cell culture medium, and nuclear staining was carried out with Hoechst 33342 (Thermofisher, USA) at a concentration of 0.1 μg ml−1. For the PI staining viability test, PI at a final concentration of 0.02 mg ml−1 was used. The cells were incubated at 37 °C for 15 min before fluorescence images were taken.
Author contributions
A. S. and S. A. B. wrote the manuscript. A. S. designed, fabricated, and tested the devices, conducted all experiments, and collected and analyzed data. S. A. B. provided discussion and optimized protocols for staining. I. R. C., S. S. H. T., and M. C. K. contributed to experimental design, provided equipment, discussion, and supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Dr Coe acknowledges support from the Canadian Natural Sciences and Engineering Research Council (NSERC) Discovery Grant Program (No. RGPIN-2017-05193). Dr Tsai is thankful for funding from the NSERC Strategic Projects Grant program (No. STGP 506318). Dr Kolios acknowledges support from the NSERC Discovery Grant Program (No. RGPIN-2017-06496). Equipment funding is from the Canada Foundation for Innovation (CFI, projects # 30994, 36687 & 36442), the Ontario Research Fund (ORF), and Ryerson University.References
- W. Kühne, FEBS Lett., 1976, 62, E8–E12 CrossRef.
- T. Tokiwa, T. Hoshika, M. Shiraishi and J. Sato, Acta Med. Okayama, 1979, 33, 1–4 CAS.
- L. J. Anghileri and A. Dermietzel, Oncology, 1976, 33, 17–23 CrossRef CAS PubMed.
- B. C. Heng, C. M. Cowan and S. Basu, Biol. Proced. Online, 2009, 11, 161–169 CrossRef CAS PubMed.
- K. H. Sit, K. P. Wong and B. H. Bay, J. Tissue Cult. Methods, 1991, 13, 257–259 CrossRef.
- U. S. Ryan, M. Mortara and C. Whitaker, Tissue Cell, 1980, 12, 619–635 CrossRef CAS PubMed.
- H. L. Huang, H. W. Hsing, T. C. Lai, Y. W. Chen, T. R. Lee, H. T. Chan, P. C. Lyu, C. L. Wu, Y. C. Lu, S. T. Lin, C. W. Lin, C. H. Lai, H. T. Chang, H. C. Chou and H. L. Chan, J. Biomed. Sci., 2010, 17, 1–10 CrossRef PubMed.
- N. Meuleman, T. Tondreau, A. Delforge, M. Dejeneffe, M. Massy, M. Libertalis, D. Bron and L. Lagneaux, Eur. J. Haematol., 2006, 76, 309–316 CrossRef PubMed.
- M. Mutin, F. George, G. Lesaule and J. Sampol, Endothelium : Journal of Endothelial Cell Research, 1996, 4, 289–295 CrossRef CAS.
- S. Mahabadi, F. H. Labeed and M. P. Hughes, Electrophoresis, 2015, 36, 1493–1498 CrossRef CAS PubMed.
- Y. Kurashina, C. Imashiro, M. Hirano, T. Kuribara, K. Totani, K. Ohnuma, J. Friend and K. Takemura, Commun. Biol., 2019, 2, 1–11 CrossRef CAS PubMed.
- X. Su, E. W. K. Young, H. A. S. Underkofler, T. J. Kamp, C. T. January and D. J. Beebe, J. Biomol. Screening, 2011, 16, 101–111 CrossRef CAS PubMed.
- Q. Wu, J. Liu, X. Wang, L. Feng, J. Wu, X. Zhu, W. Wen and X. Gong, Biomed. Eng. Online, 2020, 19, 1–19 CrossRef PubMed.
- M. Tehranirokh, A. Z. Kouzani, P. S. Francis and J. R. Kanwar, Biomicrofluidics, 2013, 7, 051502 CrossRef PubMed.
- A. Salari, S. Appak-Baskoy, M. Ezzo, B. Hinz, M. C. Kolios and S. S. H. Tsai, Small, 2019, 16, 1903788 CrossRef PubMed.
- A. Salari, S. Appak-Baskoy, I. R. Coe, J. Abousawan, C. N. Antonescu, S. S. H. Tsai and M. C. Kolios, Lab Chip, 2021, 21, 1788–1797 RSC.
- T. Inui, J. Mei, C. Imashiro, Y. Kurashina, J. Friend and K. Takemura, Lab Chip, 2021, 21, 1299–1306 RSC.
- J. T. Parsons, A. R. Horwitz and M. A. Schwartz, Nat. Rev. Mol. Cell Biol., 2010, 11, 633–643 CrossRef CAS PubMed.
- A. A. Khalili and M. R. Ahmad, Int. J. Mol. Sci., 2015, 16, 18149–18184 CrossRef CAS PubMed.
- K. K. Elineni and N. D. Gallant, Biophys. J., 2011, 101, 2903–2911 CrossRef CAS PubMed.
- C. A. Reinhart-King, M. Dembo and D. A. Hammer, Biophys. J., 2005, 89, 676–689 CrossRef CAS PubMed.
- S. F. Kemeny, S. Cicalese, D. S. Figueroa and A. M. Clyne, J. Cell. Physiol., 2013, 228, 1727–1736 CrossRef CAS PubMed.
- A. Fuhrmann, A. Banisadr, P. Beri, T. D. Tlsty and A. J. Engler, Biophys. J., 2017, 112, 736–745 CrossRef CAS PubMed.
- S. J. Han, K. S. Bielawski, L. H. Ting, M. L. Rodriguez and N. J. Sniadecki, Biophys. J., 2012, 103, 640–648 CrossRef CAS PubMed.
- C. M. Lo, H. B. Wang, M. Dembo and Y. L. Wang, Biophys. J., 2000, 79, 144–152 CrossRef CAS PubMed.
- K. Worden, Strain, 2001, 37, 167–172 CrossRef.
- M. Fechheimer, J. F. Boylan, S. Parker, J. E. Sisken, G. L. Patel and S. G. Zimmer, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 8463–8467 CrossRef CAS PubMed.
- P. L. Mcneil, Methods Cell Biol., 1988, 29, 153–173 CrossRef.
- S. Connolly, K. McGourty and D. Newport, Sci. Rep., 2020, 10, 1–13 CrossRef PubMed.
- R. H. Adamson, Microvasc. Res., 1993, 46, 77–88 CrossRef CAS PubMed.
- P. H. Puech, A. Taubenberger, F. Ulrich, M. Krieg, D. J. Muller and C. P. Heisenberg, J. Cell Sci., 2005, 118, 4199–4206 CrossRef CAS PubMed.
- M. A. Brown, C. S. Wallace, C. C. Anamelechi, E. Clermont, W. M. Reichert and G. A. Truskey, Biomaterials, 2007, 28, 3928–3935 CrossRef CAS PubMed.
- T. Ikeda, K. Ichikawa, H. Shigeto, T. Ishida, R. Hirota, H. Funabashi and A. Kuroda, Biosci. Biotechnol. Biochem., 2019, 83, 2272–2275 CrossRef CAS PubMed.
- H. Tauchi, C. Imashiro, T. Kuribara, G. Fujii, Y. Kurashina, K. Totani and K. Takemura, Biotechnol. Bioprocess Eng., 2019, 24, 536–543 CrossRef CAS.
- Y. Xia and G. M. Whitesides, Annu. Rev. Mater. Sci., 1998, 28, 153–184 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra04875a |
| This journal is © The Royal Society of Chemistry 2021 |