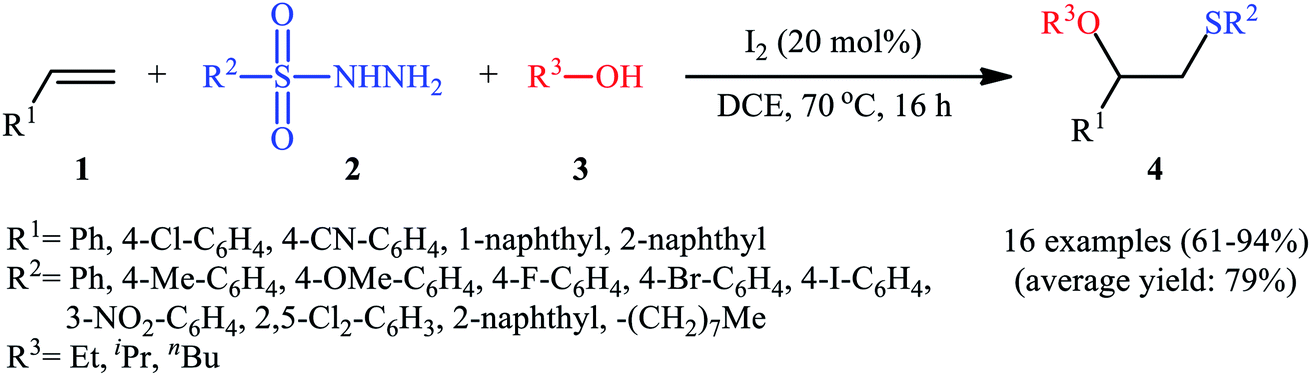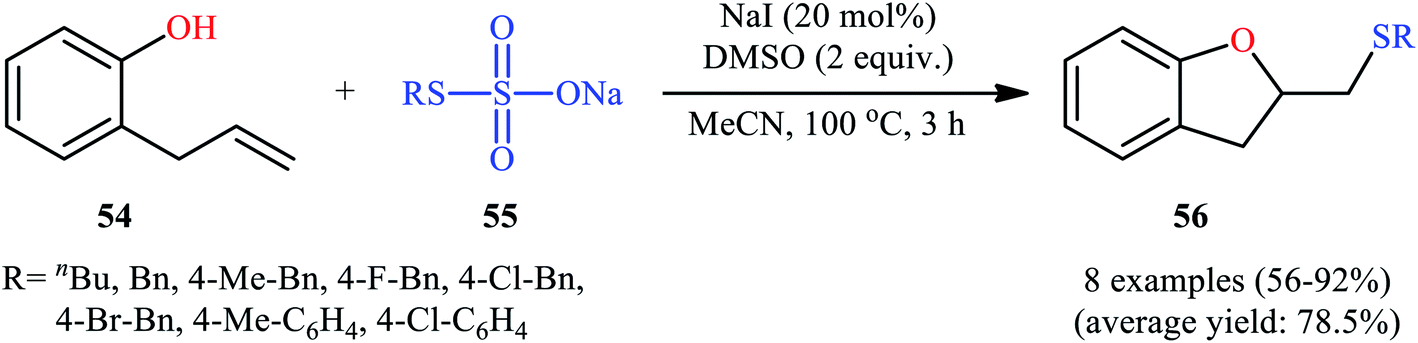DOI:
10.1039/D1RA03980F
(Review Article)
RSC Adv., 2021,
11, 32513-32525
Alkoxysulfenylation of alkenes: development and recent advances
Received
22nd May 2021
, Accepted 24th August 2021
First published on 1st October 2021
Abstract
Among the wide variety of synthetic transformations of inexpensive and abundant feedstock alkenes, vicinal difunctionalization of carbon–carbon double bonds represent one of the most powerful and effective strategies for the introduction of two distinct functional groups into target compounds in a one-pot process. In this context, the direct alkoxysulfenylation of alkenes has emerged as an elegant method to construct valuable β-alkoxy sulfides in an atom- and pot-economic manner utilizing readily accessible starting materials. Here, we review the available literature on this appealing research topic by hoping that it will be beneficial for eliciting further research and thinking in this domain.
 Somayeh Soleimani-Amiri | Somayeh Soleimani-Amiri was born in Tehran, Iran, in 1975. She received her B.S. degree in Pure Chemistry from Shahid Beheshti University, Tehran, Iran, and her M.S. degree in organic chemistry from Alzahra University, Tehran, Iran, in 2002 under the supervision of Prof. S. H. Abdi Oskooie and Prof. M. M. Heravi. She completed his Ph.D. degree in 2009 under the supervision of Prof. M. Z. Kassaee. Now she is working at Karaj Branch, Islamic Azad University as Assistant Professor. Her research interests include Computational Organic Chemistry, Nano Chemistry, Synthesis of Organic Compounds. |
 Roya Ahmadi | Roya Ahmadi began her bachelor's degree in pure chemistry in 1992 at Shahid Beheshti University in Tehran, where she completed her master's degree in inorganic chemistry at Kharazmi University of Tehran (Teacher Training) in 2000 and received her Ph.D. in mineral chemistry from Islamic Azad University, Tehran Science and Research branch in 2007. In the same year, he worked as an assistant professor at Islamic Azad University, Imam Khomeini's Yadegar branch, and in 2014 he was promoted to associate professorship in the field of experimental and computational chemistry over the years, more than 115 articles in chemistry journals and 163 papers in national and international conferences as posters and lectures. Guidance and counseling of more than 75 graduate students, master's and doctoral degrees. |
 Abdolghaffar Ebadi | Abdol Ghaffar Ebadi finished his doctoral degree in Environmental Biotechnology (Algology) from Tajik Academy of Sciences. Now he is a researcher in TAS in Tajikistan and faculty member at the Islamic Azad University of Jouybar in Mazandaran. Dr Ebadi published more than 400 scientific papers in qualified international journals and attended more than 50 international conferences. He has cooperation with many research project teams around world such as China, Malaysia, and Thailand. His interests are Environmental Biotechnology, Biochemistry, Gene pathways in the phytoremediation processes. |
 Esmail Vessally | Esmail Vessally was born in Sharabiyan, Sarab, Iran, in 1973. He received his B.S. degree in pure chemistry from university of Tabriz, Tabriz, Iran, and his M.S. degree in organic chemistry from Tehran university, Tehran, Iran, in 1999 under the supervision of Prof. H. Pirelahi. He completed his Ph.D. degree in 2005 under the supervision of Prof. M. Z. Kassaee. Now he is working at Payame Noor University as Professor in organic chemistry. His research interests include theoretical organic chemistry, new methodologies in organic synthesis and spectral studies of organic compounds. |
1. Introduction
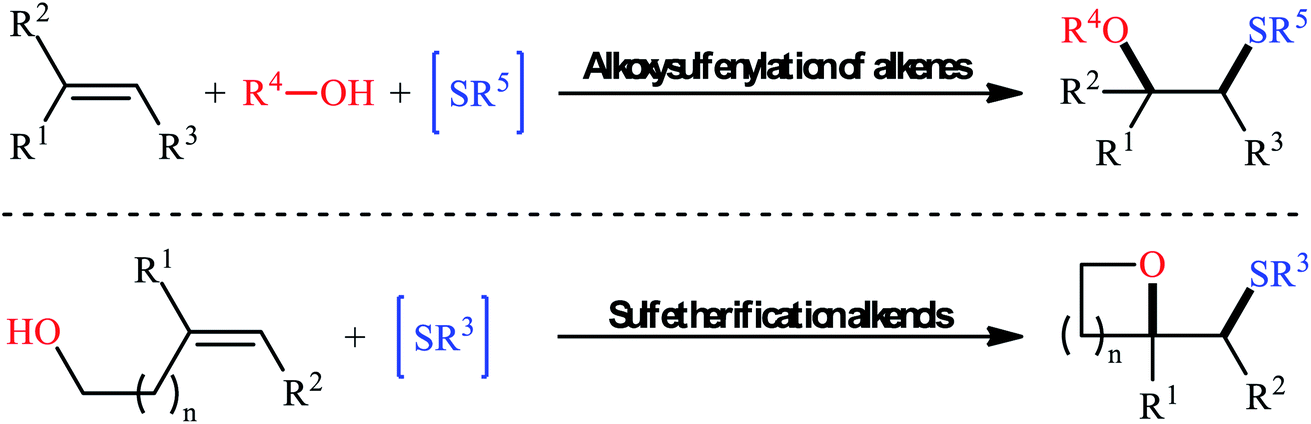
Sulfides (thioethers; R–S–R′) are undoubtedly one of the most important subclasses of organosulfur compounds and play a significant role in the field of drug discovery and development due to their diversified biological activities such as anticancer, antipsychotic, anti-inflammatory, antiretroviral, antimuscarinic, antiarrhythmic, and antiplatelet properties.1,2 Intriguingly, more than thirty FDA-approved drugs contain one or more thioether motifs in their structures.2 It is noteworthy to mention that this privileged moiety is also found in various agrochemicals, mainly in fungicides, herbicides and insecticides.3 In this family of compounds, β-alkoxy sulfides have recently attracted significant interest among chemists not only because of their interesting biological properties but also widespread synthetic applications.4 Traditionally, these compounds are obtained through the reaction of β-alkoxy haloalkoxides with thiophenol sodium salts.5 Another conventional approach is to react β-hydroxy thioethers with haloalkanes under alkaline conditions.6 However, these methods suffer from certain disadvantages, such as limited substrate diversity and complicated multi-step operations. In order to bypass these limitations, the direct alkoxysulfenylation of widely available and inexpensive alkenes with alcohols and various sulfenylating agents (e.g., thiols, disulfides, sulfonyl chlorides, sulfonyl hydrazides, sodium sulfinates) has emerged as a powerful and ideal strategy for the synthesis of the titled compounds which offer numerous advantages over the classical approaches such as simpler starting materials, shorter reaction steps, and higher atom economy. Since a number of remarkable discoveries and developments in this domain have taken place during the past few decades, seems it is an appropriate time to summarize those achievements. In continuation of our interest on organosulfur chemistry7,8 and difunctionalization reactions,9–16 in this review, we intend to highlight the literature reports on the alkoxysulfenylation of alkene substrates from 1981 till today. For clarity, the topic is divided into two major parts. The first section covers methods of intermolecular alkoxysulfenylation reactions, while the second focuses exclusively on the intramolecular reactions (Fig. 1).
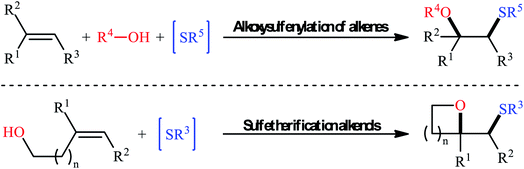 |
| | Fig. 1 Inter- and intramolecular alkoxysulfenylation of C–C double bonds. | |
2. Intermolecular alkoxysulfenylation of C–C double bonds
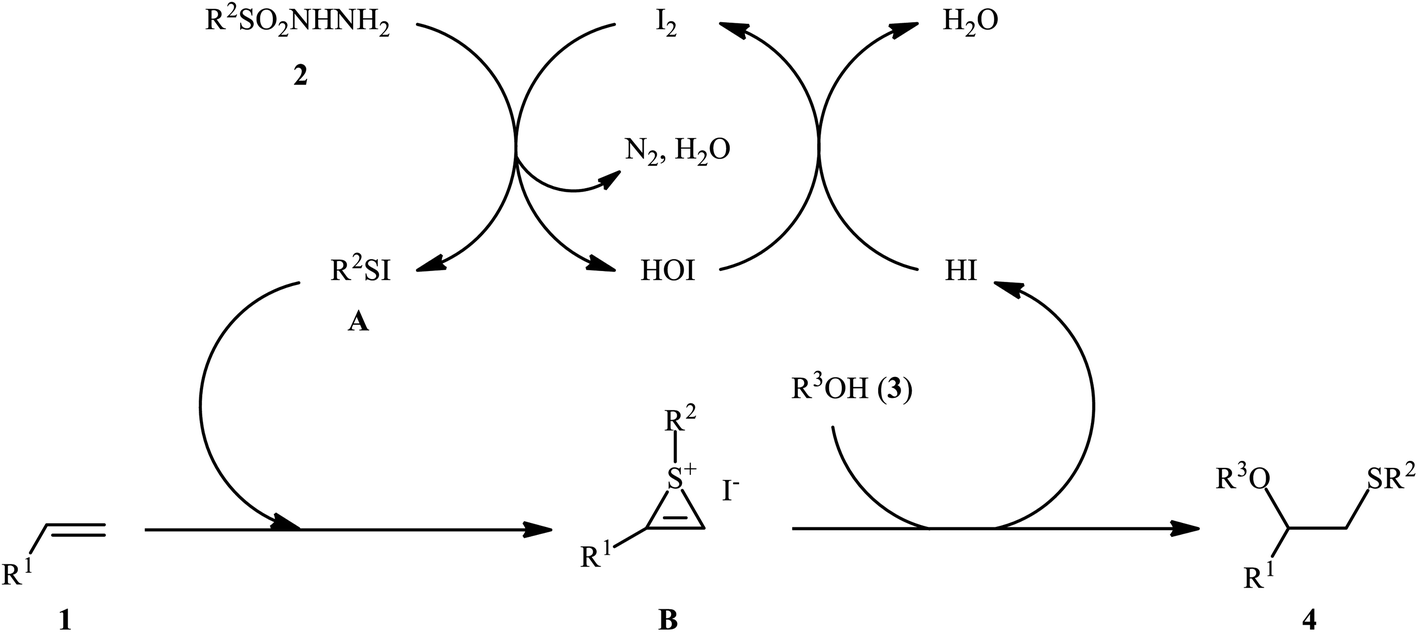
After seminal works by the groups of Ogawa17 and Franck18 on trimethylsilyl trifluoromethanesulfonate (TMSOTf)-catalyzed alkoxylative sulfenylation of a small library of glucal derivatives employing sulfenate esters as both alkoxylating and sulfenylating agents, the first general and practical methodology for intermolecular alkoxysulfenylation of alkenes was published by Tian and co-workers in 2014.19 In this report they showed that the three-component reaction between terminal aromatic alkenes 1, sulfonyl hydrazides 2, and aliphatic alcohols 3 catalyzed by molecular iodine afforded β-alkoxy sulfides 4 in good to excellent yields and outstanding regioselectivity, in which sulfenyl group selectively attached to the terminal carbon atom of the C–C double bond (Scheme 1). However, when aliphatic terminal alkenes were used as the substrates, mixtures of both possible isomers were obtained. Interestingly, while acyclic 1,2-disubstituted alkenes (e.g., stilbene) failed to participate in this reaction, cyclohexene (a cyclic alkene) served as a suitable substrate and the target product was obtained in good yield as a single stereoisomer with trans configuration. Unfortunately, extension of the reaction to 1,1-disubstituted alkenes was not successful. Instead, in these cases, the corresponding vinyl thioethers were obtained in high yields. The following mechanism was proposed by the authors for this difunctionalization reaction: the sulfenyl iodide A formed from sulfonyl hydrazides 2 and I2 is added to alkene 1 to form thiiranium intermediate B, then, the nucleophilic attack of alcohol 3 at the substituted carbon atom of thiiranium ion affords the expected ring-opening products 4 (Scheme 2).
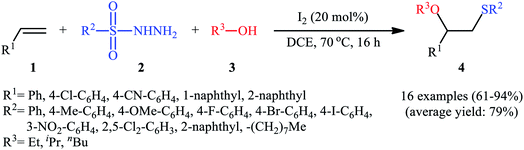 |
| | Scheme 1 Tian's synthesis of β-alkoxy sulfides 4. | |
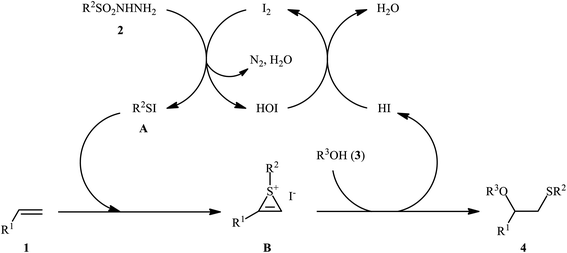 |
| | Scheme 2 I2-catalyzed alkoxylative sulfenylation of alkenes 1 with sulfonyl hydrazides 2 and alcohols 3. | |
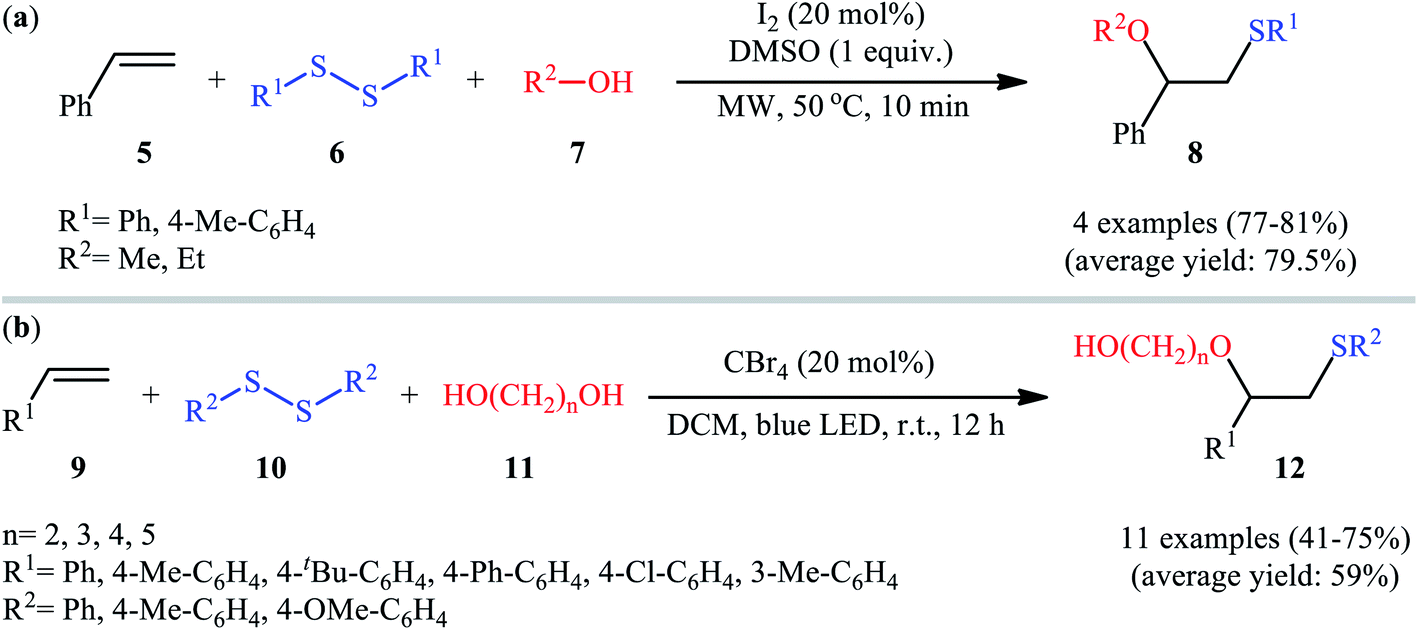
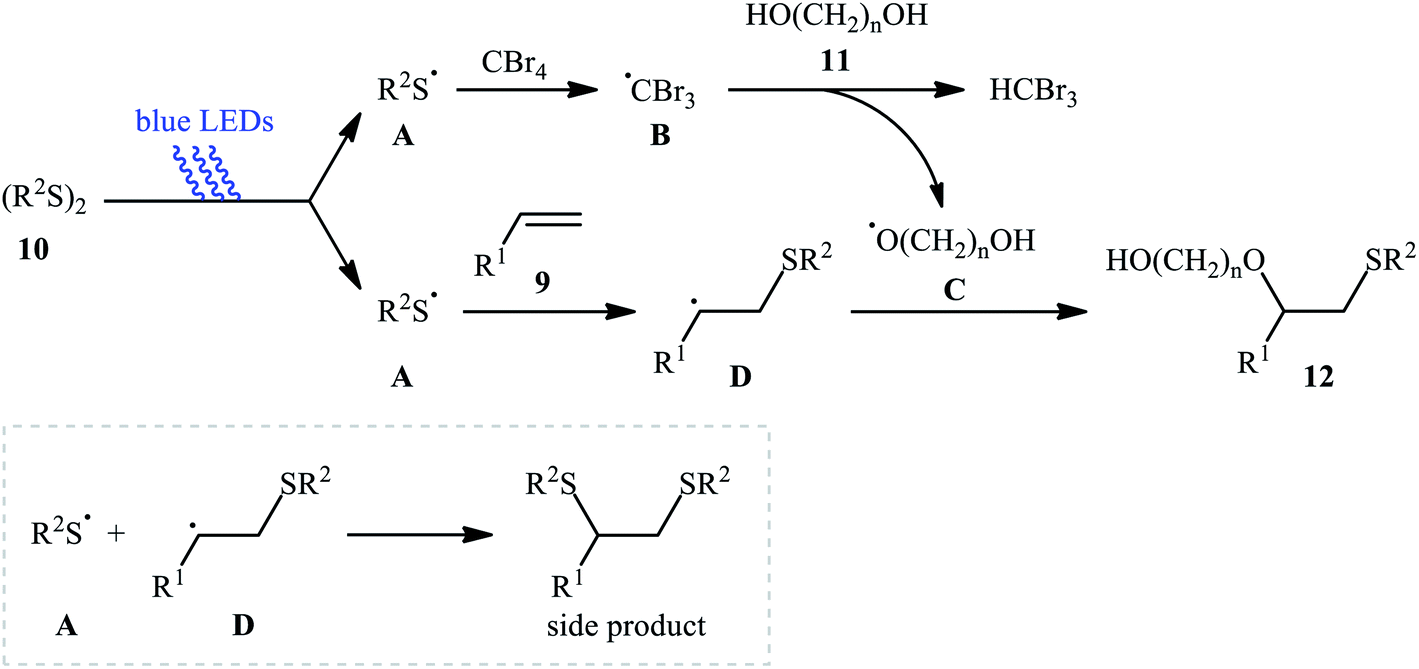
Subsequently, Braga and colleagues demonstrated that the same transformation could be achieved by using disulfides as sulfur sources.20 Thus, in the presence of 20 mol% of I2 as a catalyst and 1.0 equiv. of DMSO as an oxidant under microwave irradiation, alkoxysulfenylation of styrene 5 with a small series of aromatic disulfides 6 and aliphatic alcohols 7 furnished the corresponding β-alkoxy sulfides 8 in high yields and regioselectivities (Scheme 3a). Of note, in this reaction alcohols not only were served as substrates but also as solvents. Intriguingly, when disulfides were replaced with diselenides, the respective β-alkoxy selenide products were obtained in fair to excellent yields. The authors proposed a mechanistic course analogous to that of Tian and co-workers for the alkoxysulfenylation with sulfonyl hydrazides. Recently, Meng and Wang along with their co-workers demonstrated the similar alkoxysulfenylation under visible light irradiation and photocatalyst-free conditions.21 The transformation was performed in DCM at room temperature by using the combination of blue LED and carbon tetrabromide (CBr4). Under these conditions several styrene derivatives 9 carrying various substituents were slowly converted to the corresponding β-alkoxy sulfides 12 in moderate to good yields by treatment with diaryl disulfides 10 and diols 11 (Scheme 3b). The results proved that the lengths of carbon chain in diols had a notable impact on the rate of reaction. Generally, shorter chain diols were found to be more reactive than the longer chain alternatives. The authors attributed this observation to the interaction of the intermolecular hydroxyl groups. It is worth noting that when H2O was used in place of diols under the identical conditions, the hydroxysulfenylated products were obtained in modest to high yields. However, no product was observed when aliphatic alkene or aliphatic disulfide were used as one of the substrates. Furthermore, the process is not viable for gram-scale due to the drastic reduction in the yield (from 69% in the 0.6 mmol scale, to 40% in the 5.0 mmol scale). Several control experiments were implemented to probe the mechanism of this reaction; no reaction was observed in the absence of CBr4 or a light source. Additionally, no reaction could be observed when TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxy) was added to the reaction mixture. On this basis, the authors proposed a plausible mechanistic pathway in which the S–S bond in disulfide 10 undergoes hemolytic cleavage under the irradiation of blue LED to generate thiyl radical A, which after reaction with CBr4 affords a tribromomethyl radical B. Next, abstraction of a hydrogen by the newly formed radical B from diol 11 leads to alkoxyl radical C and HCBr3. Conversely, the addition of another molecule of thiyl radical A to alkene 9 provides carbon-centered radical D. Finally, coupling of alkoxyl radical C with intermediate C gives the desired product 12 (Scheme 4).
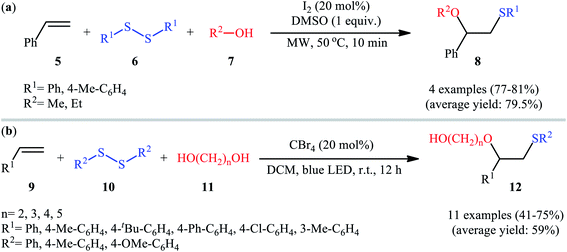 |
| | Scheme 3 (a) I2-catalyzed alkoxysulfenylation of styrene 5 with aromatic disulfides 6 and aliphatic alcohols 7 developed by Braga; (b) visible-light-induced alkoxysulfenylation of alkenes 9 with disulfides 10 and diols 11. | |
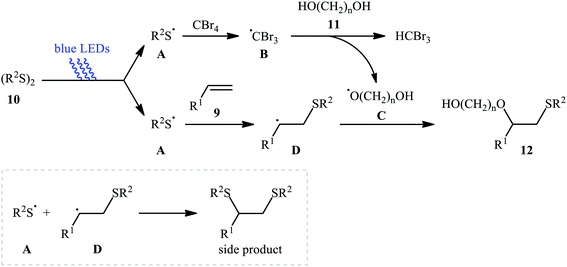 |
| | Scheme 4 Possible mechanism for the formation of β-alkoxy sulfides 12. | |
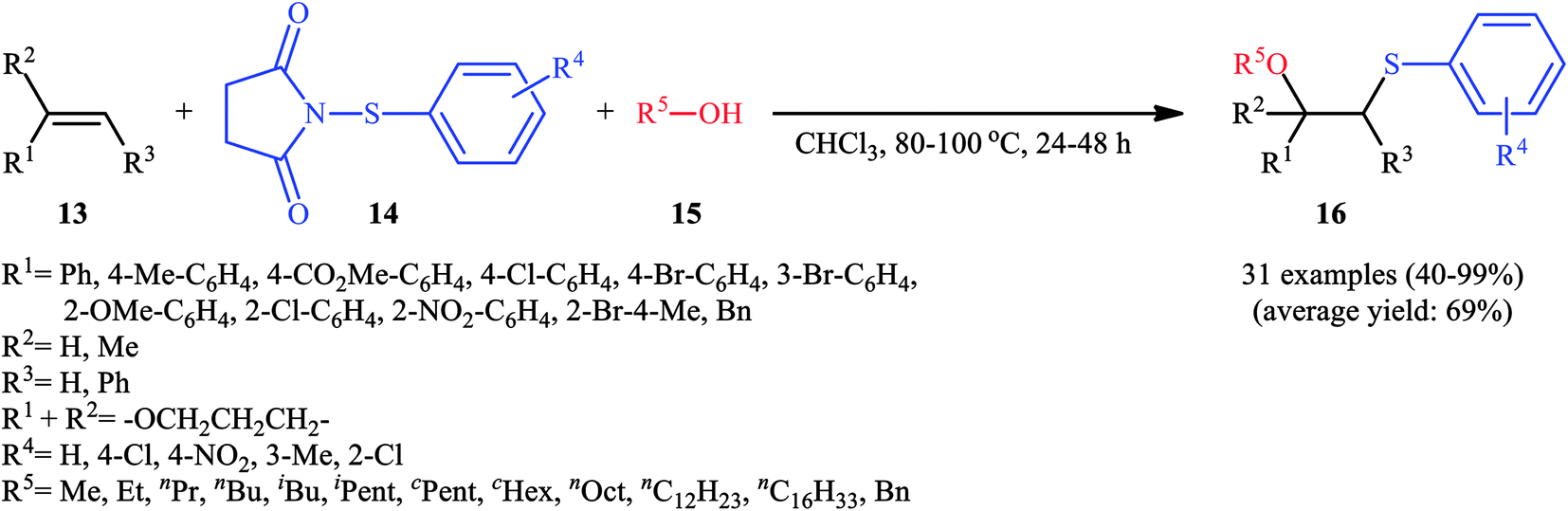
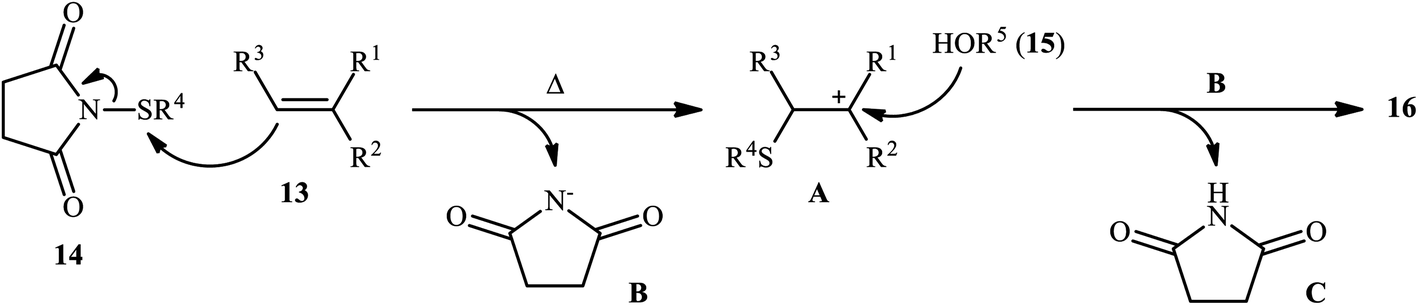
In 2015, Fu's research team disclosed the use of easily available 1-(arylthio)pyrrolidine-2,5-diones as an alternative sulfenylating reagent in oxysulfenylation reactions.22 In this investigation, thirty one β-alkoxy sulfides 16 were synthesized in moderate to quantitative yields by direct reaction of various mono-, 1,1-di-, and 1,2-di-substituted alkenes 13 with functionalized 1-(arylthio)pyrrolidine-2,5-diones 14 and aryl/benzyl alcohols 15 in CHCl3 under catalyst- and additive-free conditions (Scheme 5). Noteworthy, in this transformation, the terminal alkenes were more reactive than the internal ones, and aliphatic alkenes were also tolerated, although the products were obtained in lower yields. Regarding the reaction mechanism, the authors proposed the formation of a carbonium ion intermediate A via the reaction of alkene 13 with 1-(arylthio)pyrrolidine-2,5-dione 14 followed by electrophilic attack of the alcohol 15 on the carbocation center (Scheme 6). Recently, Liang and Zhao applied this chemistry in highly enantiselective synthesis of a library of thiolated 1,3-aminoalcohols using a chiral selenide catalyst.23
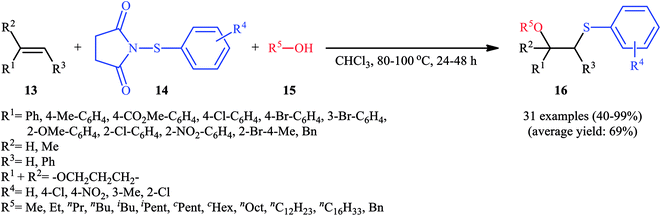 |
| | Scheme 5 Catalyst-free oxysulfenylation of alkenes 13 with 1-(arylthio)pyrrolidine-2,5-diones 14 and alcohols 15. | |
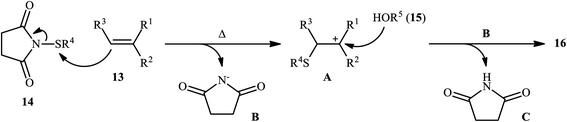 |
| | Scheme 6 The plausible mechanistic pathway for the reaction in Scheme 5. | |
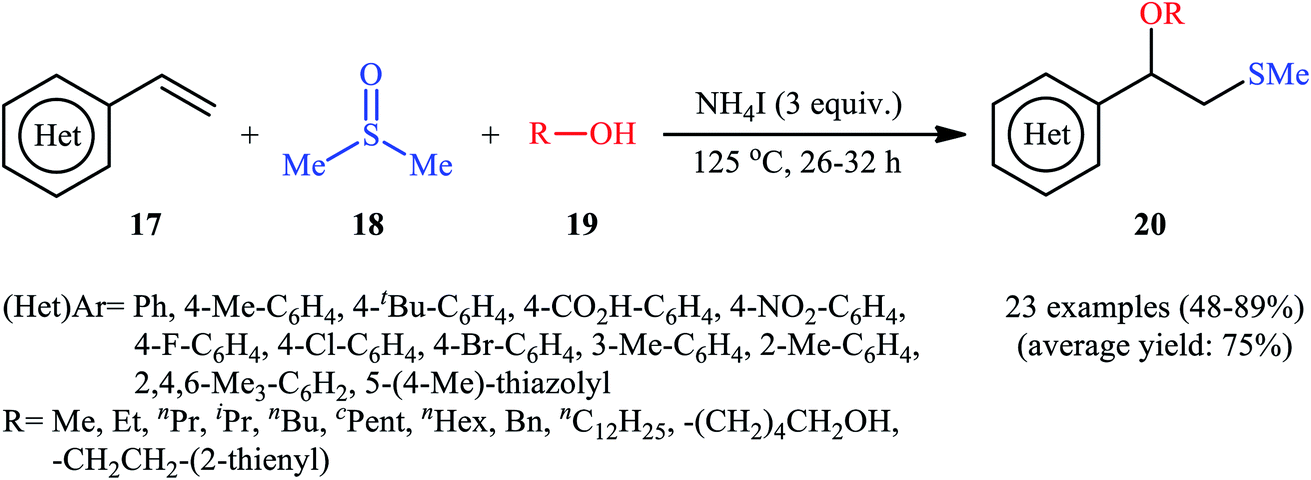
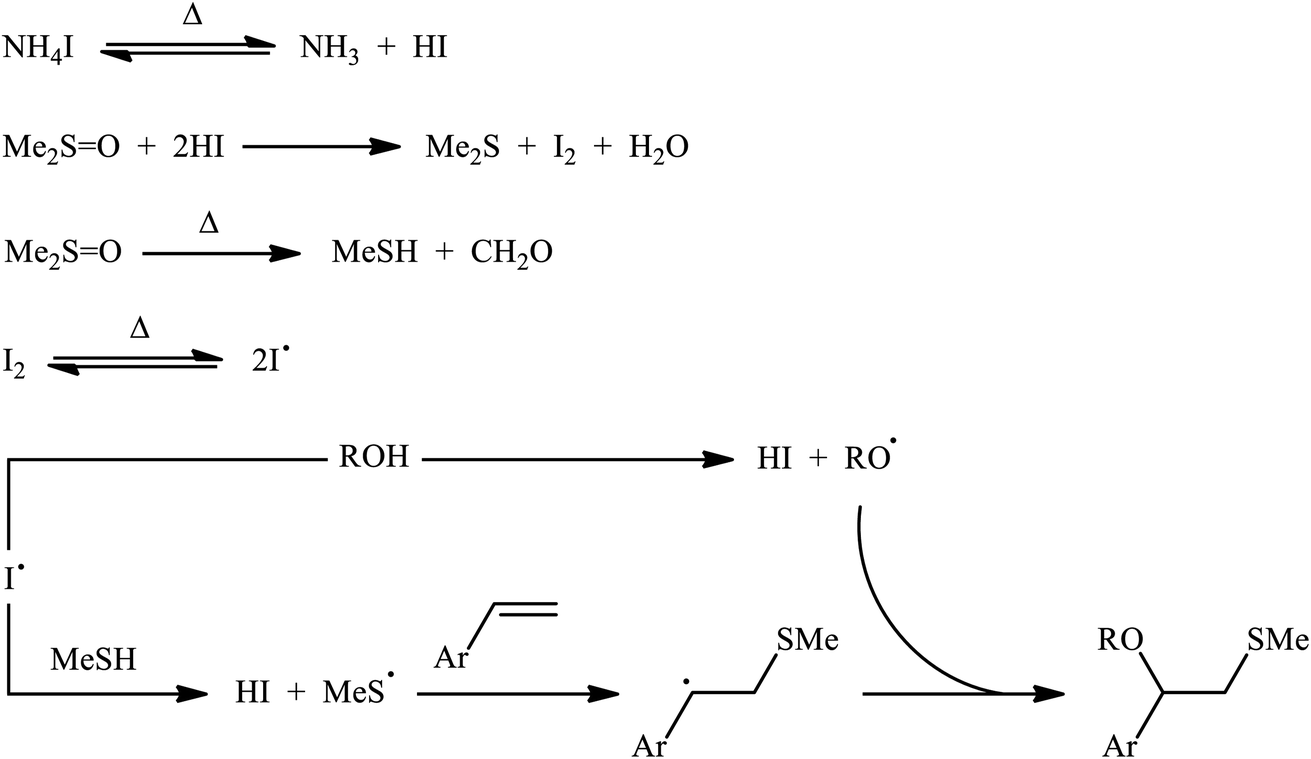
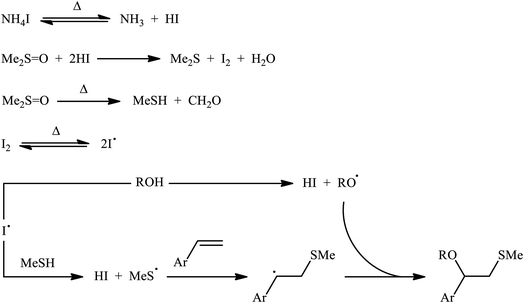
A promising contribution to this field was reported by Yuan, Li, and co-workers in 2015,24 when various terminal aromatic and heteroaromatic alkenes 17 were converted to the corresponding β-alkoxy methyl sulfides 20 through the NH4I-mediated alkoxysulfenylation with dimethyl sulfoxide (DMSO; 18) and alcohols 19 under metal-free conditions. As shown in Scheme 7, this reaction tolerated various primary and secondary alcohols and both electron-rich and electron-poor alkenes, and gave the final products in moderate to high yields and excellent regioselectivities. Two α-substituted styrenes have also been tested and afforded the expected products in good yields. Notably, β-substituted styrenes were also tolerated under the reaction conditions, however, the diastereoselectivity of products was modest at best. The proposed mechanism for this transformation is shown in Scheme 8 which is based on a radical process. First, molecular iodine (I2) and methanethiol (MeSH) are formed from NH4I and DMSO through a series of reactions. Next, I2 is decomposed under the thermal condition to form iodine radical (I˙), which is later reacted with precursor MeSH resulting in a methylthiyl radical (MeSC˙) and concurrently abstracted a hydrogen atom from alcohols to afford alkoxy radical (ROC˙). Subsequently, addition of the methylthiyl radical to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond of alkene leads to an alkyl radical intermediate. Finally, the rapid combination of alkoxy radical and the newly formed carbon-centered radical delivers the desired product. Recently, Liu et al. extended the substrate scope of this reaction to aliphatic (terminal and internal) alkenes by performing the process in the presence of (COCl)2 in MeCN.25 Noteworthy, cycloalkenes selectively afforded trans-adducts under these conditions. Besides alcohols, carboxylic acids also could be applied as suitable O-nucleophiles under the identical conditions.
C double bond of alkene leads to an alkyl radical intermediate. Finally, the rapid combination of alkoxy radical and the newly formed carbon-centered radical delivers the desired product. Recently, Liu et al. extended the substrate scope of this reaction to aliphatic (terminal and internal) alkenes by performing the process in the presence of (COCl)2 in MeCN.25 Noteworthy, cycloalkenes selectively afforded trans-adducts under these conditions. Besides alcohols, carboxylic acids also could be applied as suitable O-nucleophiles under the identical conditions.
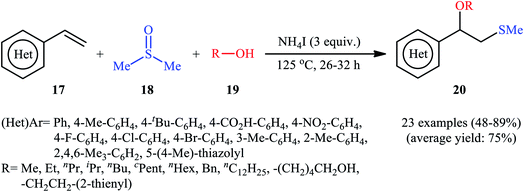 |
| | Scheme 7 Synthesis of β-alkoxy methyl sulfides 20 through the NH4I-mediated three-component coupling alkenes 17, dimethyl sulfoxide 18 and alcohols 19. | |
 |
| | Scheme 8 Possible mechanism for the formation of β-alkoxy methyl sulfides 20. | |
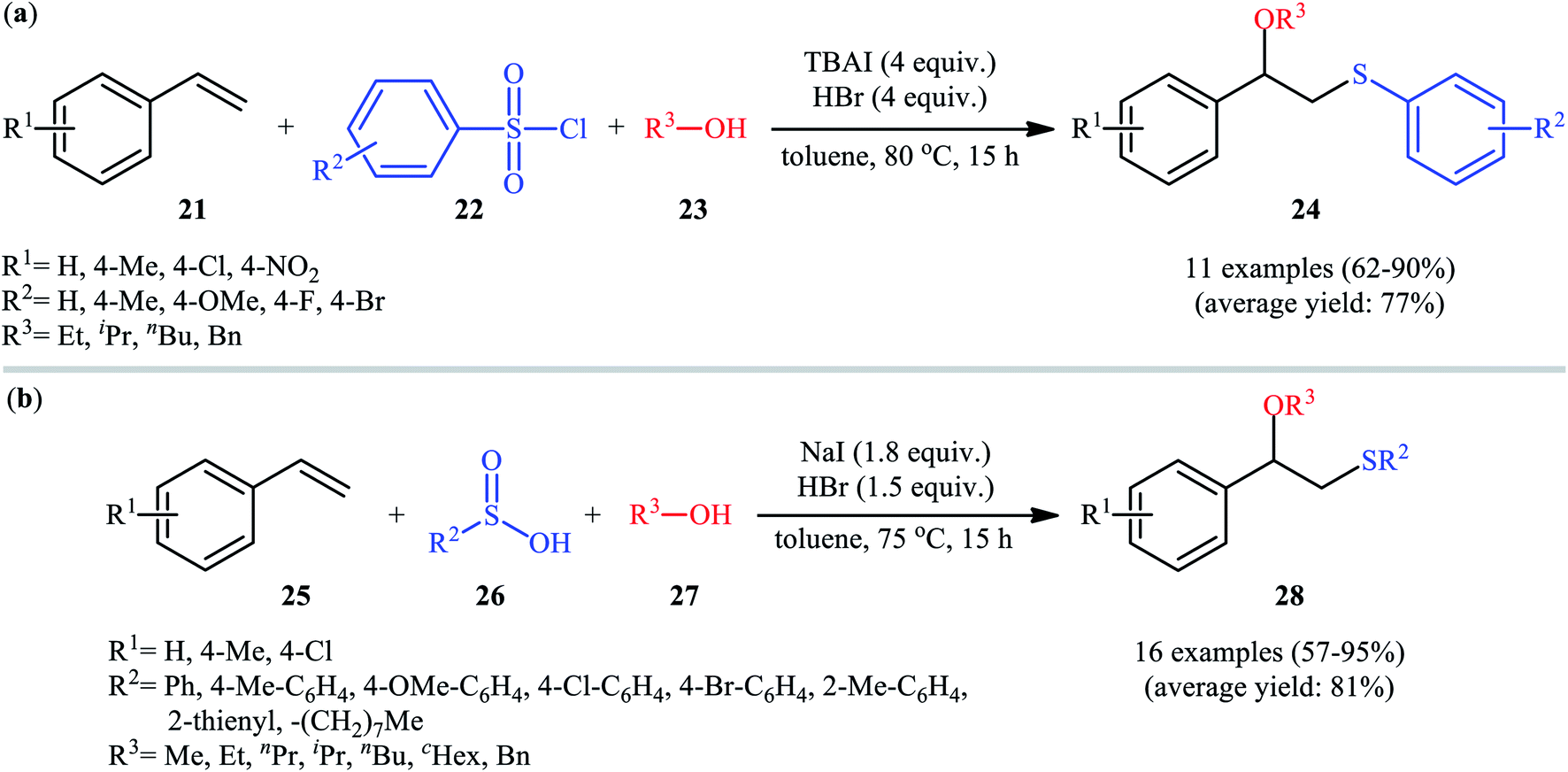
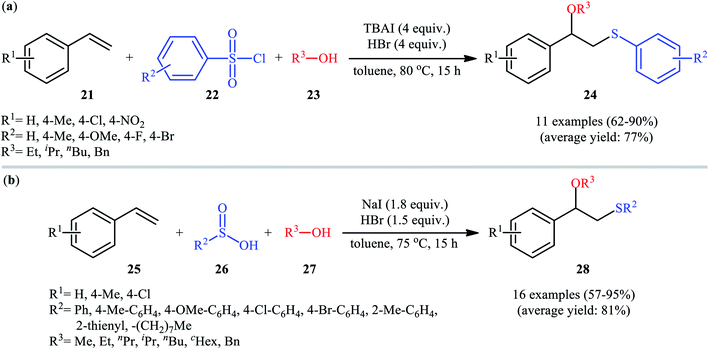
In 2016, Lin and Guo realized the effectiveness of broadly available and bench-stable sulfonyl chlorides as sulfenylating agents in alkoxysulfenylation reactions of C–C double bonds.26 In this study, they showed that treatment of aromatic terminal alkenes 21 with arylsulfonyl chlorides 22 and aliphatic alcohols 23 in the presence of excess amounts of tetrabutylammonium iodide (TBAI) as a promoter and HBr as an acidic additive in toluene furnished the corresponding β-alkoxy sulfides 24 in good to excellent yields, ranging from 62% to 90% (Scheme 9a). Internal aliphatic alkenes were also found to be suitable substrates for this protocol as exemplified by the formation of (2-ethoxycyclohexyl) (p-tolyl)sulfane by using cyclohexene as the substrate. However, just like previous works, aromatic alcohols were inert in this transformation. The mechanism proposed by the authors to explain this difunctionalization reaction is analogous to the one depicted for in Scheme 2. Along this line, the same research group also reported that in the presence of over-stoichiometric amounts of NaI and HBr, styrene derivatives 25 were similarly coupled with various aryl/alkyl-sulfinic acids 26 and alkyl/benzyl-alcohols 27 to obtain β-alkoxy sulfide products 28 with yields of up to 95% (Scheme 9b).27
 |
| | Scheme 9 (a) TBAI-mediated synthesis of β-alkoxy sulfides 24 reported by Lin and Guo; (b) NaI/HBr-mediated three-component alkoxysulfenylation reaction of alkenes 25 with sulfinic acids 26 and alcohols 27. | |
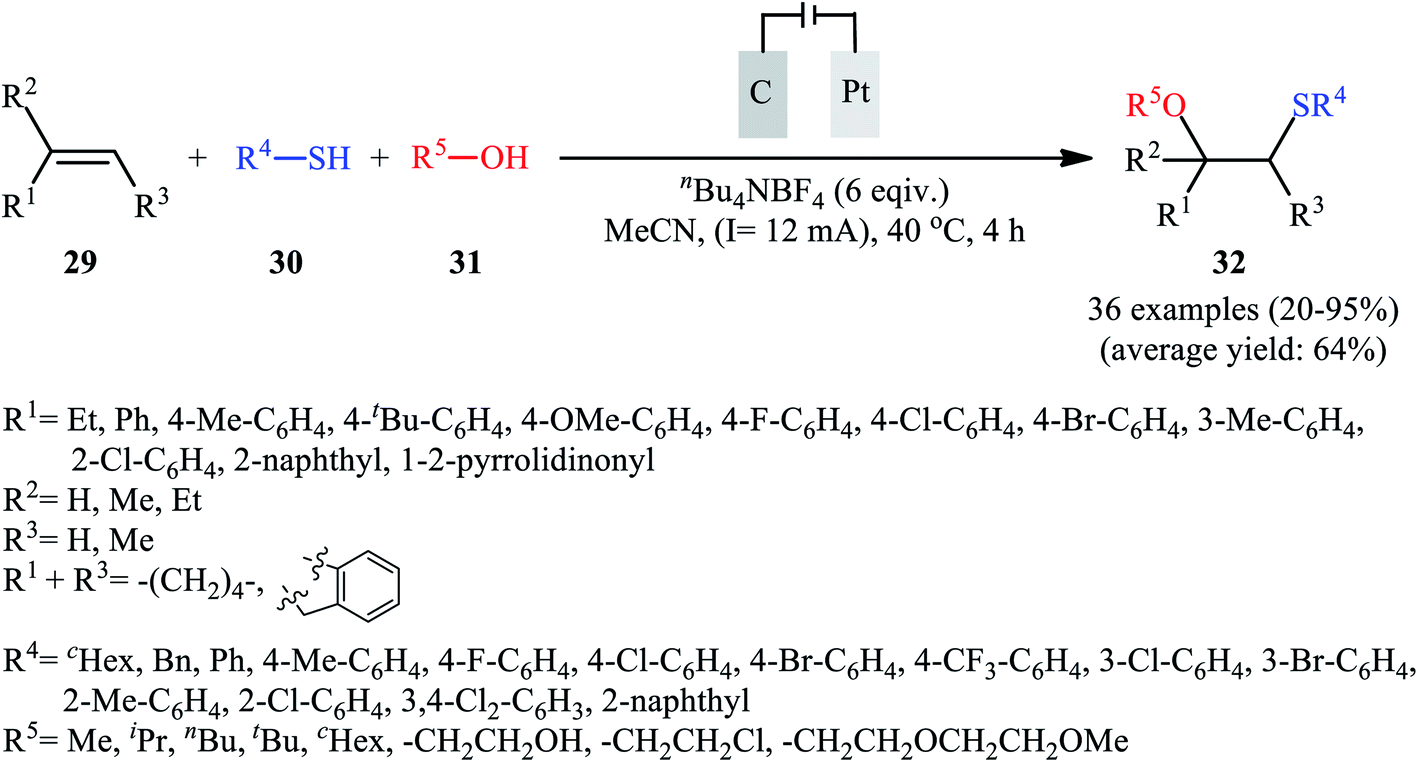
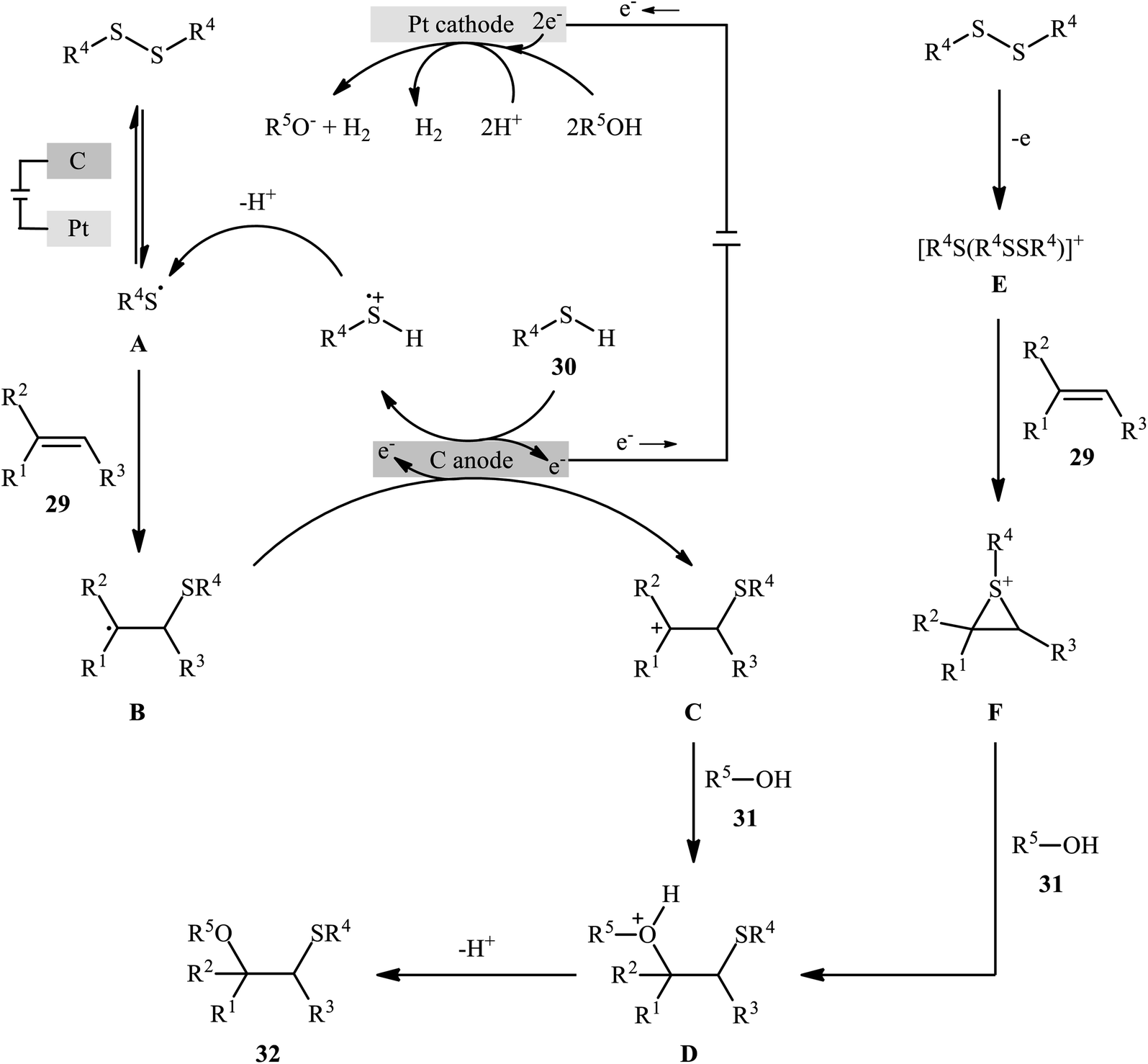
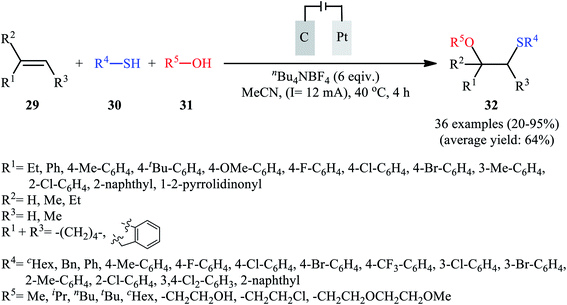
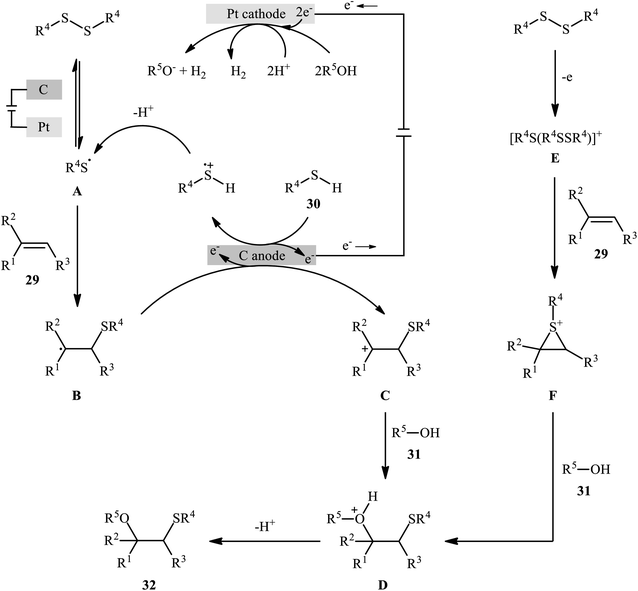
In 2018, Lei and co-workers developed an elegant electrochemical alkoxysulfenylation of alkenes 29 with thiols 30 and alcohols 31 under external oxidant-free conditions which exhibited better substrate scope compared to previously reported examples of this chemistry.28 The reactions were conducted in an undivided cell under a constant current of 12 mA cm−2 with carbon anode and platinum plate cathode employing nBu4NBF4 as the electrolyte and MeCN as the solvent at 40 °C, which afforded the β-alkoxy sulfide products 32 in 20–95% yields (Scheme 10). Noteworthy, other nucleophiles such as amines, acetic acid and water were also developed as the coupling partners in this reaction. Some important information of this oxidative sulfenylation reaction are listed below: (i) although both aromatic and aliphatic thiols were compatible with this reaction, aliphatic thiols afforded significantly poorer yields compared to aromatic ones; (ii) like previous works, the scope of alcohols was restricted to the use of aliphatic and benzylic alcohols; and (iii) the protocol for difunctionalization of aliphatic and internal alkenes was considerably less efficient than aromatic and terminal alkenes. According to the authors proposed mechanism (Scheme 11), this reaction proceeds through a thiiranium ion intermediate.
 |
| | Scheme 10 Electrochemical alkoxysulfenylation of alkenes 29 with thiols 30 and alcohols 31. | |
 |
| | Scheme 11 A plausible mechanism for the reaction in Scheme 10. | |
3. Intramolecular alkoxysulfenylation of C–C double bonds
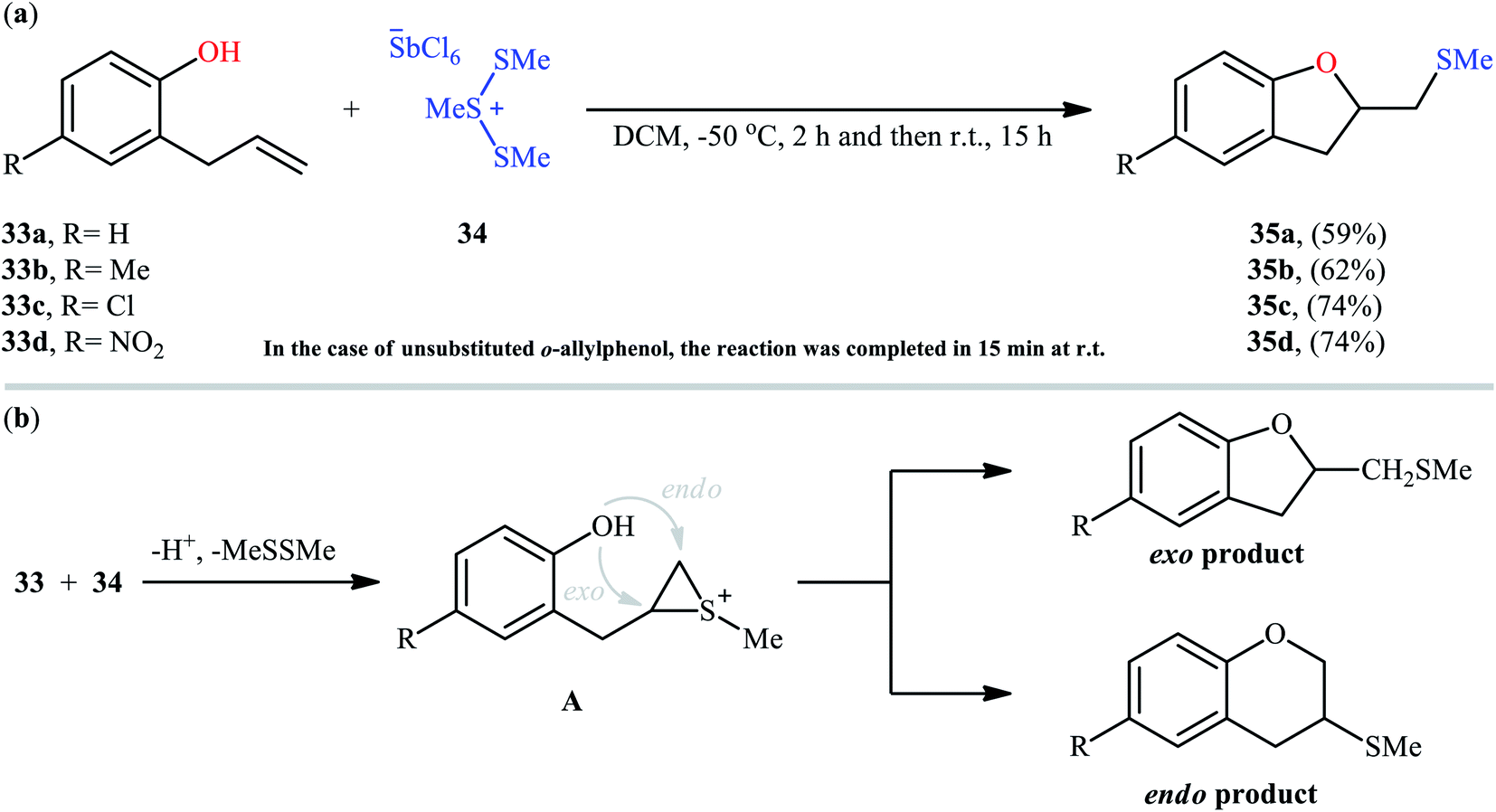
Cyclofunctionalization reactions represent an important strategy for the synthesis of functionalized (hetero)cyclic structures from simple starting materials that would otherwise be difficult to prepare.29 In this family of reactions, sulfetherification of alkenols offers a promising strategy for the selective synthesis of sulfenyl-substituted oxacycles within a single click. One of the earliest reports on the cyclizative sulfenylation of alkenols was published by Capozzi and co-workers in 1981,30 who showed that the treatment of o-allylphenols 33 with methyl(bismethy1thio)sulfonium hexachloroantimonate 34 in the absence of any catalyst or additive in anhydrous DCM, resulted in the formation of methylthio-substituted dihydrobenzofurans 35 in good yields and selectivities (Scheme 12a). The reaction is noteworthy in that both electron-rich and electron-poor o-allylphenols were well tolerated. However, in the case of unsubstituted o-allylphenol, besides the desired 2-((methylthio)methyl)-substituted dihydrobenzofuran, a small amount of 5-(methylthio)-2-((methylthio)methyl)-2,3-dihydrobenzofuran was obtained as a side-product. Intriguingly, the authors nicely controlled the selectivity of this reaction by using different reaction conditions. The formation of dithiolated product was selectively accomplished simply by using a two-fold excess of the sulfonium salt. A reasonable selectivity for the synthesis of the mono-thiolated dihydrobenzofuran 35a was obtained running the reaction at −40 °C. The author also provided the possible mechanism for the formation of benzofuran derivatives 35 (Scheme 12b), in which the thiiranium intermediate A formed via attack of the methylthiolating agent at the double bond of the allylic residue undergoes ring opening through intramolecular nucleophilic attack by the phenolic oxygen at the internal carbon to give regiospecifically the dihydrobenzofuran ring although the possibility of ring closure to a dihydrobenzopyrane ring also exist. Four years later, O'Malley and Cava reported a similar sulfetherification of γ,δ-unsaturated alcohols using dimethyl(methylthio)sulfonium tetrafluoroborate (DMTSF) as the methylsulfenylation reagent and iPr2NEt as a base in anhydrous acetonitrile solvent.31 Notably, γ,δ-unsaturated carboxylic acids could also undergo intramolecular cyclofunctionalization reaction under the identical conditions and give the corresponding lactones in good to high yields. This transformation was later reinvestigated by Okuma and colleagues employing dimethyl(methylthio)sulfonium trifluoromethanesulfonate (DMTST) as the sulfenylation agents and good results were observed.32
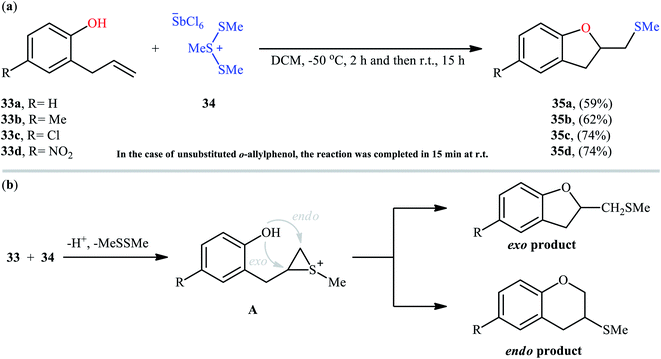 |
| | Scheme 12 (a) Synthesis of 2-((methylthio)methyl)-2,3-dihydrobenzofurans 35 through intramolecular sulfetherification of alkenols 33 with methyl(bismethy1thio)sulfonium hexachloroantimonate 34; (b) the plausible mechanistic pathway for the formation of dihydrobenzofurans 35. | |
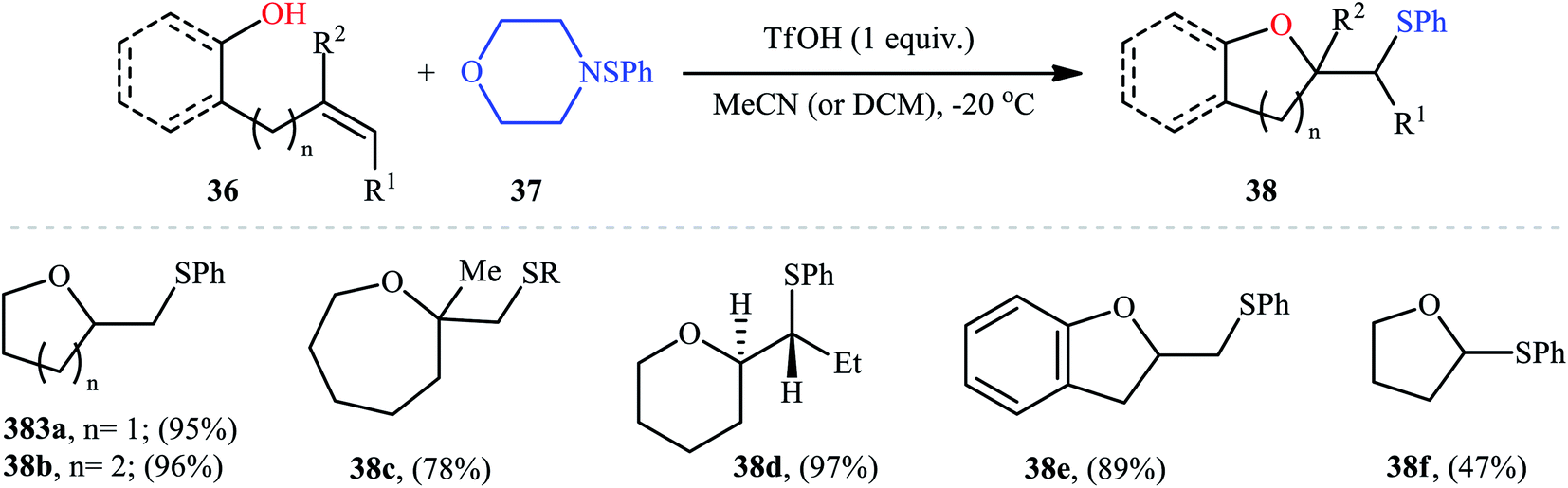
In 1987, Brownbridge introduced the use of N-(phenylthio)morpholine as an efficient phenylthiolating agent for intramolecular sulfenyl etherification of alkenols under metal-free conditions.33 Thus, a library of thiophenol-functionalized five-, six-, and seven-membered cyclic ethers 38 were successfully synthesized by treatment of the corresponding unsaturated alcohols 36 with N-(phenylthio)morpholine 37 in the presence of a stoichiometric amount of triflic acid (TfOH) as a promoter in a dry solvent at −20 °C (Scheme 13). However, cyclization to give four-membered ring systems did not take place by this method. Interestingly, when a cis-disubstituted alkenol (5-octen-1-ol) subjected to this reaction condition, a single regio- and stereo-isomer 38d was formed, indicated trans-addition without any alkene isomerization. Shortly afterwards, the author found that the regio- and stereo-selectivity of this cyclofunctionalization reaction is highly dependent on the substitution patterns of the employed alkenols.34 Benzenesulfenyl chloride was also found to be useful for the cyclization, though few examples are available.35–37
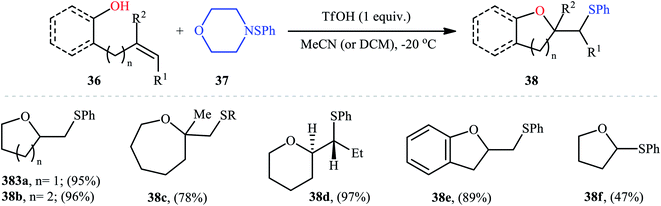 |
| | Scheme 13 Brownbridge's synthesis of thiol-substituted cyclic ethers 38. | |
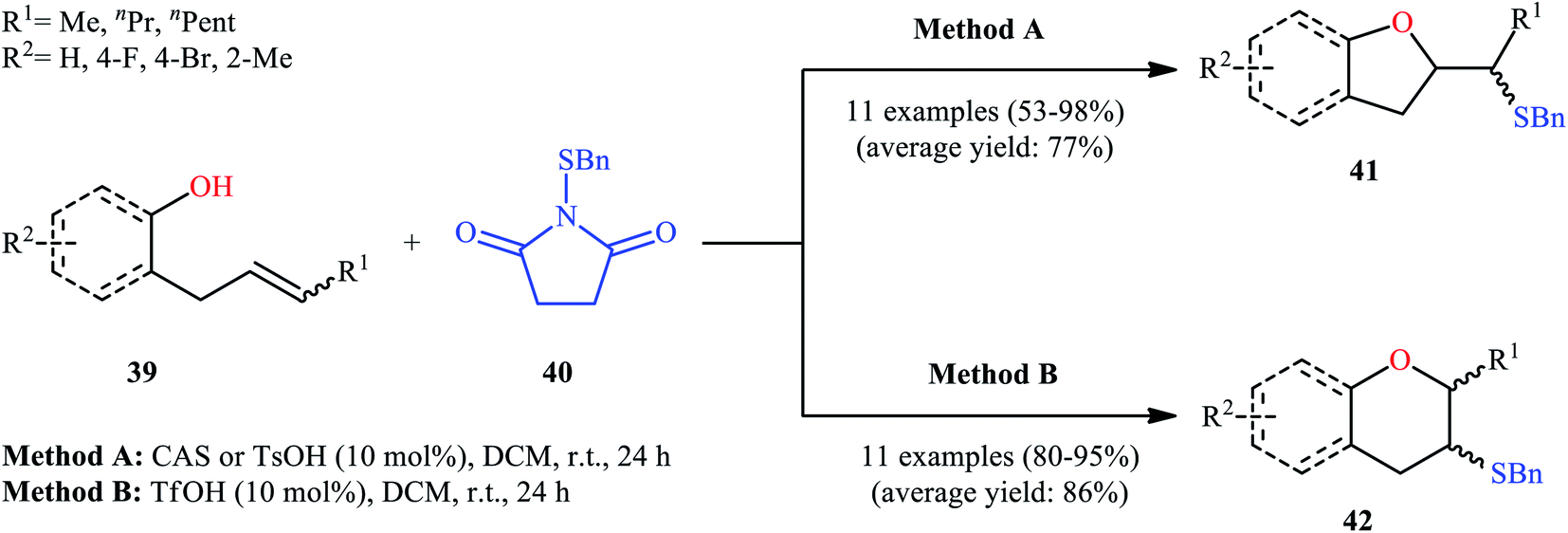
Drawing inspiration from these works, in 2011, Shi's group demonstrated an interesting acid-catalyzed regio- and stereo-selective sulfetherification of alkenols 39 with the use of N-(benzylthio)succinimide 40, thus providing either the corresponding tetrahydrofurans 41 and tetrahydropyrans 42 depending upon the acid catalyst used (Scheme 14).38 Studies indicated the formation of 5-exo cyclization products with camphorsulfonic acid (CSA) or tosylic acid (TsOH) and 6-endo cyclization products in the presence of triflic acid (TfOH). The results proved that cis-alkenes gave higher 5-exo selectivity as compared to trans-alkenes. The authors ascribed this observation by the steric effect during the cyclization. Studies also showed that when isolated 5-exo products were treated with a catalytic amount of TfOH at room temperature, 6-endo products were formed in high yields. These results clearly indicated that the cyclization route to tetrahydrofurans 41 was under kinetic control, while the formation of tetrahydropyrans 42 was thermodynamically favored.
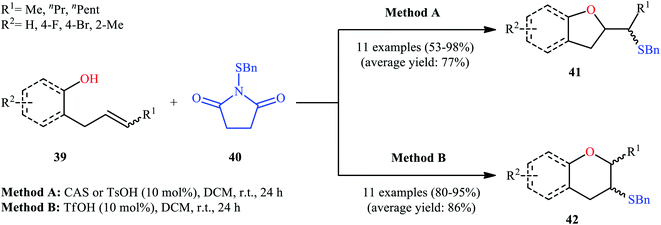 |
| | Scheme 14 Acid-dependent regioselective sulfetherification of alkenols 39 in the presence of N-(benzylthio)succinimide 40 reported by Shi. | |
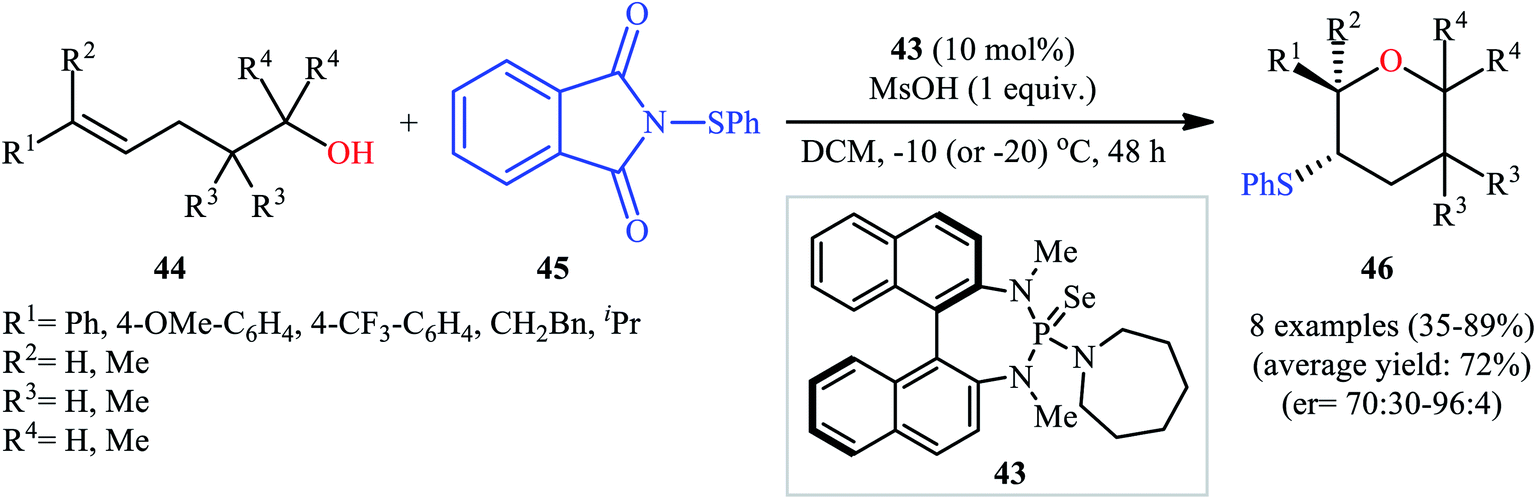
Concurrently, Denmark and colleagues developed a robust asymmetric sulfetherification of various alkenols using a chiral BINAM-based selenophosphoramide catalyst 43 and a Brønsted acid (methanesulfonic acid, MsOH) additive.39 A library of conjugated and trans nonconjugated alkenes 44 reacted efficiently with N-phenylsulfenyl-phthalimide 45 in the presence of 10 mol% of 43 to afford sulfenylated tetrahydropyrans 46 in modest to excellent yields and outstanding enantioselectivities along with a small amount of sulfenylated tetrahydrofuran side-products (Scheme 15). Notably, when unsubstituted, cis nonconjugated and geminally disubstituted alkenes were subjected to the same reaction conditions, sulfenylated tetrahydrofurans were formed as the main reaction products, thus suggesting that the regioselectivity of this transformation is strongly dependent on the substitution patterns of the employed alkenols. Besides alkenols, unsaturated carboxylic acids could also be elegantly used in this cyclofunctionalization reaction. Moreover, several successful examples of the intermolecular sulfetherification of simple alkenes with methanol also reported under this reaction condition.
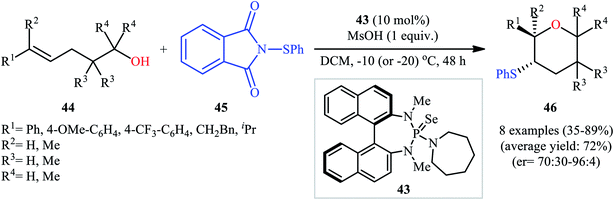 |
| | Scheme 15 Denmark's synthesis of chiral sulfenylated tetrahydropyrans 45. | |
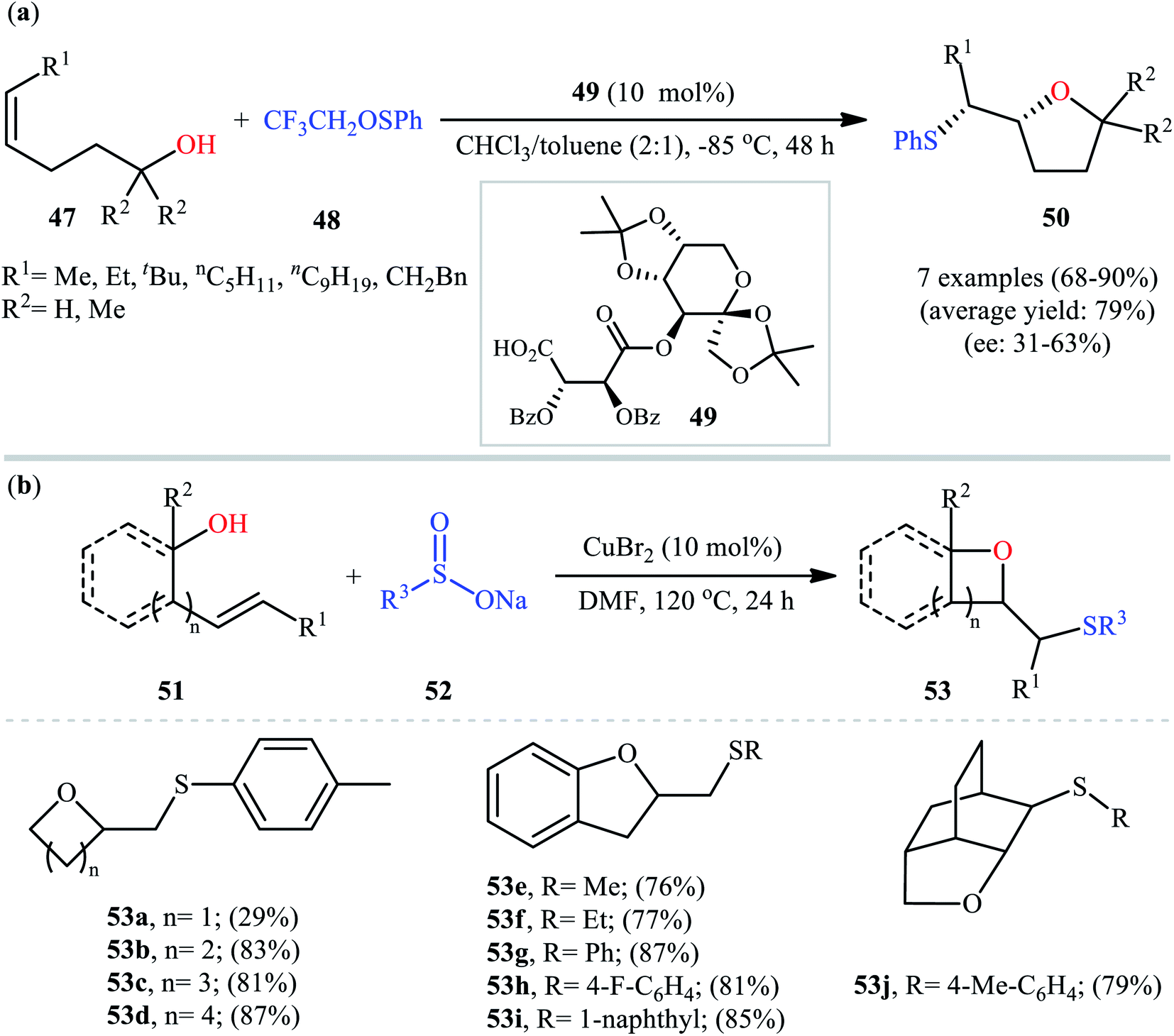
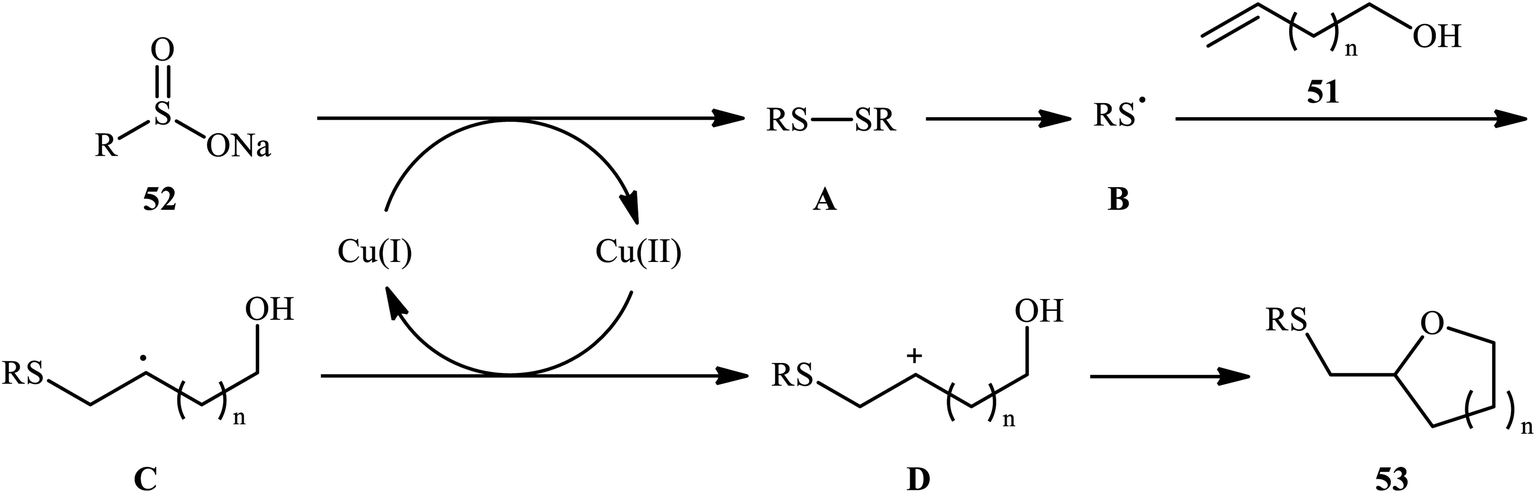
In a related investigation, Shi and co-workers reacted 5-substituted-pent-4-en-1-ols 47 with trifluoroethyl benzenesulfenate 48 in the presence of chiral tartaric acid-based catalyst 49 to selectively provide enantioenriched tetrahydrofurans 50 in good to excellent yields (68–90%) and low to good enantioselectivities (31–63% ee) (Scheme 16a).40 Although the reaction showed good tolerance to a series of cis-alkenols, trans-alkenols completely failed to participate in this transformation. Subsequently, Wu and Jiang along with their co-workers demonstrated the synthesis of a diverse range of sulfenylated four-, five-, six-, and seven-membered cyclic ether derivatives 53 via CuBr2-catalyzed sulfetherification of the corresponding alkenols 51 employing sodium sulfinates 52 as sulfenylating agents.41 The reaction was run in DMF at 120 °C, tolerated various aliphatic, benzylic, and (hetero)aromatic sodium sulfinates, and provided the desired sulfenylated O-heterocycles in good to excellent yield. However, the only example of strained four-membered ring product was obtained in medium yield. Some reported examples are shown in Scheme 16b. It should be mentioned that other simple copper salts such as CuCl, CuBr, CuI, and CuCl2 were also found to catalyze this sulfenylative cyclization reaction, albeit with reduced efficiencies. On the basis of a series of preliminary control experiments, it was suggested that this sulfetherification reaction starts with the formation of disulfide intermediate A via Cu-catalyzed dimerization of sodium sulfinate 52, which then undergoes radical cracking to give the sulfur radical B. Subsequently, this radical adds to the double bond of alkene 51 to form the α-sulfenyl-alkyl radical C that, after a single-electron oxidation converts to the carbocation D. Finally, the internal nucleophilic trapping of the newly formed intermediate D affords the desired product 53 (Scheme 17).
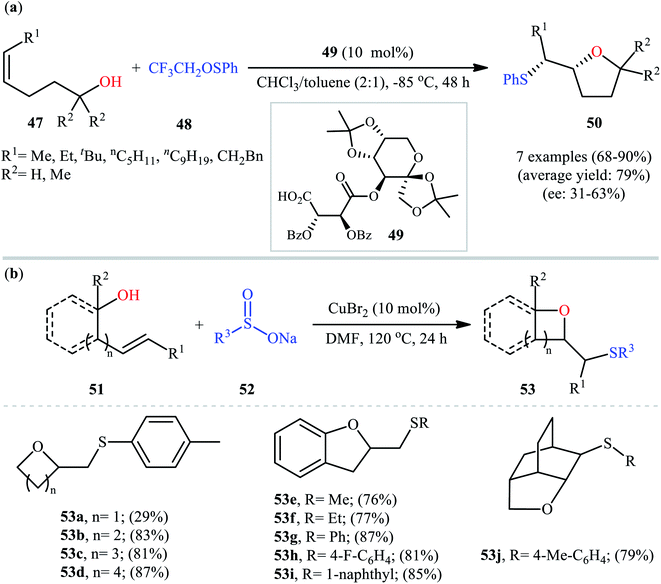 |
| | Scheme 16 (a) Shi's synthesis of enantioenriched tetrahydrofurans 50; (b) selected examples of the Cu(II)-catalyzed sulfenyl etherification of alkenols 51 with sodium sulfinates 52 reported by Wu and Jiang. | |
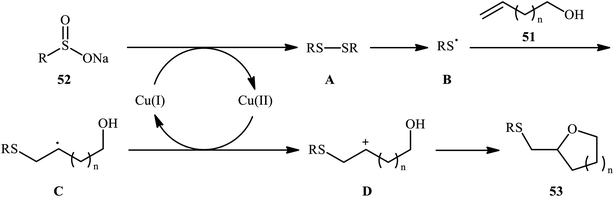 |
| | Scheme 17 Mechanism of the Cu(II)-catalyzed sulfetherification of alkenols 51 with sodium sulfinates 52. | |
In 2017, Zhang, Yan, and Lin studied the possibility of synthesizing sulfenylated 2,3-dihydrobenzofurans through the intramolecular sulfetherification of 2-allylphenol 54 with environmentally friendly thiosulfates 55.42 To determine the best conditions, they meticulously probed the activities of different iodine-based catalysts (e.g., I2, NaI, TBAI, NH4I) and oxidants (e.g., H2O2, DMSO, TBHP, Cu(OAc)2, K2S2O8) in the sulfenyl etherification of 2-allylphenol with sodium S-benzyl sulfothioate in MeCN, as a model reaction. The optimal system was recognized with the used of 20 mol% NaI and 2 equiv. of DMSO at 100 °C. Under the optimized conditions, 2-allylphenol 54 was reacted with various aromatic, aliphatic, and benzylic thiosulfates 55 to provide the corresponding 2-(thiomethyl)-2,3-dihydrobenzofurans 56 in good to excellent yields within 3 h (Scheme 18). The authors have also examined pent-4-en-1-ol in place of 2-allylphenol to give the respective tetrahydrofuran in good yield. Furthermore, three component reaction between alkenes, acetic acid, and thiosulfates also successfully implemented under the identical conditions to provide the desired β-acetoxy sulfides in good yields.
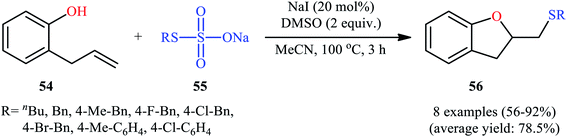 |
| | Scheme 18 NaI-catalyzed sulfetherification of 2-allylphenol 54 with thiosulfates 55. | |
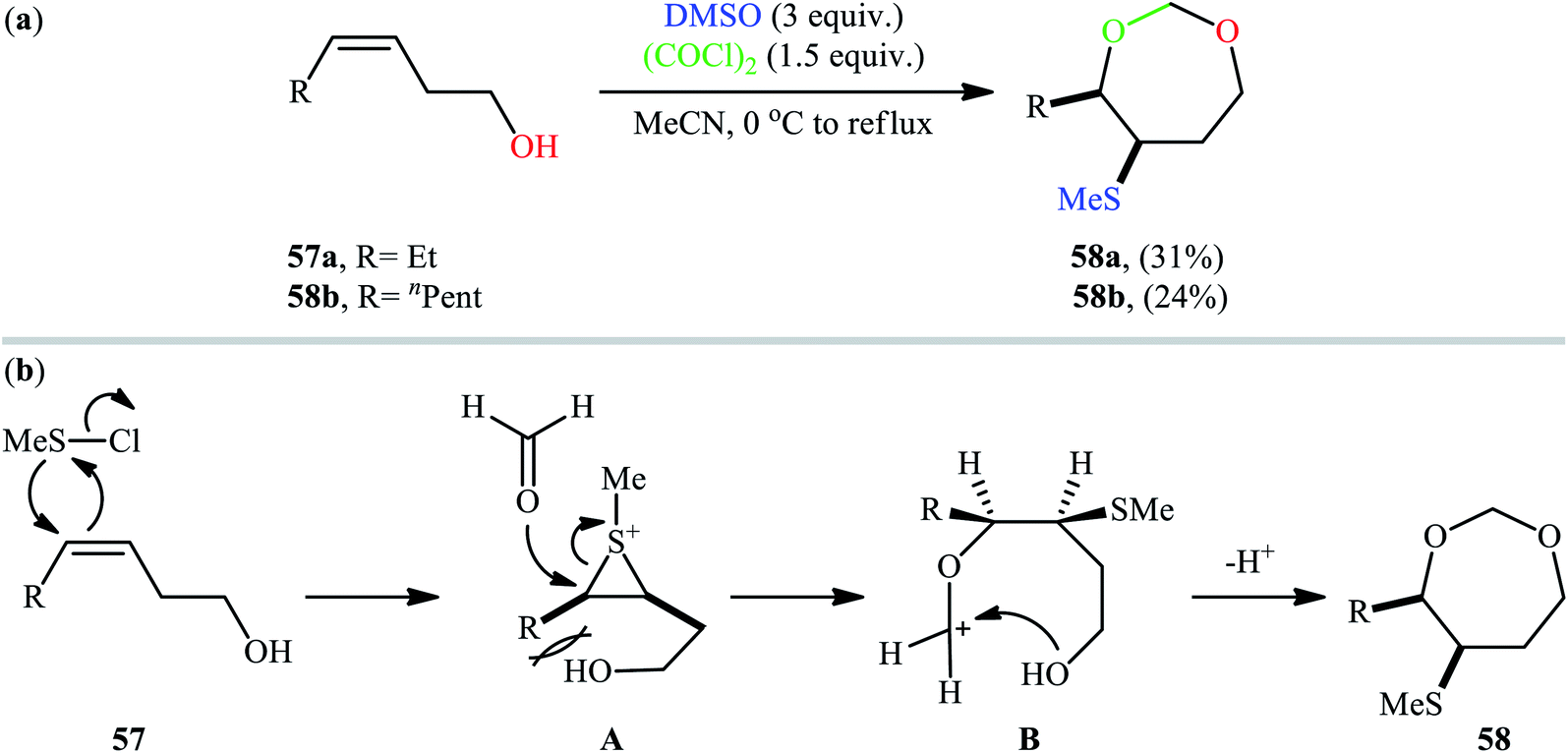
Along this line, recently, Gao et al. found that a combination of dimethyl sulfoxide (DMSO) and oxalyl chloride [(COCl)2] is a suitable reagent for sulfenyletherification of unsaturated alcohols under catalyst- and additive-free conditions.43 Methanesulfenyl chloride (MeSCl) is assumed to be the compound responsible for the sulfenyletherification, which is generated by the reaction of DMSO and (COCl)2. Various unsaturated alcohols including 3, 4, and 5-alkenols were all compatible by this reaction, thus indicating its broad applicability. However, cis-3-alkenols 57 cannot be converted to cyclic ethers due to the effect of steric bulk in the intermediates. In these cases, instead of cyclic ethers, seven-membered cyclic acetals 58 with cis configurations were generated as the sole reaction products (Scheme 19a). According to the authors proposed mechanism (Scheme 19b), this transformation began with the addition of methanechloride to the double bond of cis-3-alkenol 57 to achieve cis-thiiranium ion intermediate A. Then, the oxygen of carbonyl group of formaldehyde, generated in situ during the formation of methanesulfenyl chloride, was attached at the less substituted carbon atom of thiiranium ion to form carbocation intermediate B. Finally, intramolecular cyclization of intermediate B led to the observed seven-membered cyclic acetal 58.
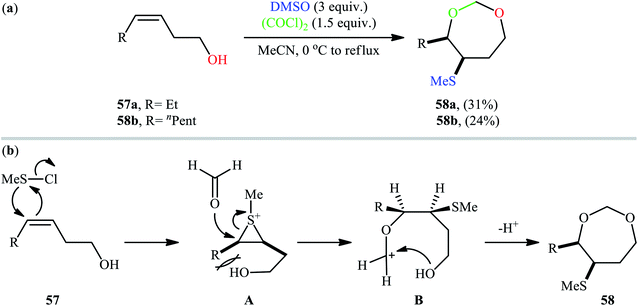 |
| | Scheme 19 (a) Gao's synthesis of seven-membered cyclic acetals 58; (b) plausible reaction mechanism for the formation of seven-membered cyclic acetals 58. | |
4. Conclusion
Vicinal difunctionalization of alkenes is an attractive and efficient way to incorporation of two new adjacent functional groups onto a carbon–carbon bond, producing complicated molecular scaffolds in one step. In this family of reactions, the direct alkoxysulfenylation of alkenes with alcohols and appropriate sulfenylating agents has been attracted considerable attention as an ideal strategy for the preparation of β-alkoxy sulfide derivatives, which serve as versatile building blocks in chemical synthesis. As illustrated, not only intermolecular but also intramolecular version of this transformation using alkenols as both the C–C double bond and hydroxy sources have been extensively investigated. Although several common sulfenylating agents have been successfully applied in this difunctionalization reaction, the scope of alcohols is mainly restricted to the aliphatic alcohols. Additionally, the scope of alkenes is largely limited to styrene derivatives. Therefore, of course, further investigations and improvements are still needed that this research field matures.
Conflicts of interest
There are no conflicts to declare.
References
-
(a) M. Feng, B. Tang, S. H. Liang and X. Jiang, Curr. Top. Med. Chem., 2016, 16, 1200–1216 CrossRef CAS PubMed;
(b) X. Lin, Z. Li, B. Liang, H. Zhai, W. Cai, J. Nan and A. Wang, Water res, 2019, 162, 236–245 CrossRef CAS PubMed;
(c) X. Li, Sci. Total environ., 2021, 765, 144497 CrossRef CAS PubMed;
(d) X. Wang, et al., Green Chem., 2020, 22, 6157–6169 RSC;
(e) H. Wang, J. Cui, Y. Zhao, Z. Li and J. Wang, Green Chem., 2021, 23, 405–411 RSC;
(f) Z. Li, et al., Angew. Chem., Int. Ed., 2021, 60, 3928–3933 CrossRef CAS PubMed;
(g) P. Dong, T. Zhang, H. Xiang, X. Xu, Y. Lv, Y. Wang and C. Lu, J. Mater. Chem. B, 2021, 9, 958–968 RSC;
(h) Z. Wang, T. Zhang, L. Pi, H. Xiang, P. Dong, C. Lu and T. Jin, Biointerfaces, 2021, 198, 111480 CrossRef PubMed.
-
(a) K. A. Scott and J. T. Njardarson, Top. Curr. Chem., 2018, 376, 5 CrossRef PubMed;
(b) L. Zhang, et al., J. Environ. Sci., 2020, 91, 212–221 CrossRef PubMed;
(c) X. Wang, P. Gao, Y. Liu, H. Li and F. Lu, Curr. Bioinform., 2020, 15, 493–502 CrossRef;
(d) Q. Wang, et al., Bioresour. Technol., 2021 DOI:10.1016/j.biortech.2021.125120;
(e) S. Sun, L. Xu, Q. Zou, G. Wang and J. Gorodkin, Bioinformatics, 2021, 37, 1319–1321 CrossRef PubMed.
-
(a) P. Devendar and G. F. Yang, Top. Curr. Chem., 2017, 375, 82 CrossRef PubMed;
(b) M. Niu, Y. Lin and Q. Zou, Plant Mol. Biol., 2021, 105, 483–495 CrossRef PubMed;
(c) H. Wang, et al., ACS Appl. Mater. Interfaces, 2021, 13, 25918–25925 CrossRef PubMed;
(d) Q. Pan, et al., Angew. Chem., Int. Ed., 2021, 60, 11835–11840 CrossRef CAS PubMed.
-
(a) L. P. Yang and S. J. Keam, Drugs, 2009, 69, 919–942 CrossRef CAS PubMed;
(b) C. M. Rayner, in Organosulfur Chemistry: Synthetic Aspects, ed. P. Page, Academic Press, London, 1995, p. 89 Search PubMed.
- W. E. Doering and K. C. Schreiber, J. Am. Chem. Soc., 1955, 77, 514–520 CrossRef CAS.
- T. Takeda, H. Furukawa, M. Fujimori, K. Suzuki and T. Fujiwara, Bull. Chem. Soc. Jpn., 1863, 57, 1863–1869 CrossRef.
- Selected reviews:
(a) F. A. H. Nasab, L. Z. Fekri, A. Monfared, A. Hosseinian and E. Vessally, RSC Adv., 2018, 8, 18456–18469 RSC;
(b) A. Hosseinian, S. Ahmadi, F. A. H. Nasab, R. Mohammadi and E. Vessally, Top. Curr. Chem., 2018, 376, 1–32 CrossRef;
(c) M. Hamzehloo, A. Hosseinian, S. Ebrahimiasl, A. Monfared and E. Vessally, J. Fluorine Chem., 2019, 224, 52–60 CrossRef CAS.
- Z. Liu, A. Ebadi, M. Toughani, N. Mert and E. Vessally, RSC Adv., 2020, 10, 37299–37313 RSC.
- Selected reviews:
(a) A. Bakhtiary, M. R. P. Heravi, A. Hassanpour, I. Amini and E. Vessally, RSC Adv., 2020, 11, 470–483 RSC;
(b) M. R. J. Sarvestani, N. Mert and E. Vessally, J. Chem. Lett., 2020, 1, 93–102 Search PubMed;
(c) L. Sreerama, E. Vessally and F. Behmagham, J. Chem. Lett., 2020, 1, 9–18 Search PubMed;
(d) L. Sreerama, E. Vessally and F. Behmagham, J. Chem. Lett., 2020, 1, 25–31 Search PubMed;
(e) E. A. Mahmood, B. Azizi and S. Majedi, Chem. Rev. Lett, 2020, 3, 2–8 CAS;
(f) N. Salehi and B. Azizi, J. Chem. Lett., 2021, 2, 2–8 Search PubMed;
(g) D. Tayde and M. Lande, Chem. Rev. Lett., 2021, 4, 30–36 CAS;
(h) M. Daghagheleh, M. Vali, Z. Rahmani, S. Sarhandi and E. Vessally, Chem. Rev. Lett., 2018, 1, 23–30 Search PubMed.
- B. Azizi, M. R. P. Heravi, Z. Hossaini, A. Ebadi and E. Vessally, RSC Adv., 2021, 11, 13138–13151 RSC.
- X. Wang, W. Ping, A. G. Ebadi, S. Majedi, Z. Hossaini and M. Toughani, J. CO2 Util., 2021, 50, 101592, DOI:10.1016/j.jcou.2021.101592.
- X. Ma, Z. Kexin, W. Yonggang, A. G. Ebadi and M. Toughani, Iran. J. Chem. Chem. Eng., 2021 DOI:10.30492/IJCCE.2021.529010.4694.
- A. Hassanpour, E. Ghavidelaghdam, A. G. Ebadi, M. R. P. Heravi and E. Vessally, RSC Adv., 2021, 11, 22305–22316 RSC.
- Z. Hossaini, E. A. Mahmood, M. R. P. Heravi, A. G. Ebadi and E. Vessally, RSC Adv., 2021, 11, 21651–21665 RSC.
- S. Ahmadi, A. Hosseinian, P. D. Kheirollahi Nezhad, A. Monfared and E. Vessally, Iran. J. Chem. Chem. Eng., 2019, 38, 1–19 CAS.
- E. Vessally, S. Mohammadi, M. Abdoli, A. Hosseinian and P. Ojaghloo, Iran. J. Chem. Chem. Eng., 2020, 39, 11–19 CAS.
- Y. Ito and T. Ogawa, Tetrahedron Lett., 1987, 28, 2723–2726 CrossRef CAS.
- S. Ramesh and R. W. Franck, J. Chem. Soc., Chem. Commun., 1989, 960–962 RSC.
- F. L. Yang, F. X. Wang, T. T. Wang, Y. J. Wang and S. K. Tian, Chem. Commun., 2014, 50, 2111–2113 RSC.
- A. A. Vieira, J. B. Azeredo, M. Godoi, C. Santi, E. N. da Silva Junior and A. L. Braga, J. Org. Chem., 2015, 80, 2120–2127 CrossRef CAS.
- H. Ruan, L. G. Meng, L. Zhu and L. Wang, Adv. Synth. Catal., 2019, 361, 3217–3222 CrossRef CAS.
- J. Yu, C. Gao, Z. Song, H. Yang and H. Fu, Org. Biomol. Chem., 2015, 13, 4846–4850 RSC.
- Y. Liang and X. Zhao, ACS Catal., 2019, 9, 6896–6902 CrossRef CAS.
- X. Gao, X. Pan, J. Gao, H. Jiang, G. Yuan and Y. Li, Org. Lett., 2015, 17, 1038–1041 CrossRef CAS PubMed.
- Y. Liu, Y. Gao, Z. Wang, J. Dong, R. Ding, B. Sun and H. Tian, Synth. Commun., 2019, 49, 2662–2670 CrossRef CAS.
- D. Wang, R. Zhang, S. Lin, Z. Yan and S. Guo, Synlett, 2016, 27, 2003–2008 CrossRef CAS.
- D. Wang, R. Zhang, W. Ning, Z. Yan and S. Lin, Org. Biomol. Chem., 2016, 14, 5136–5140 RSC.
- Y. Yuan, Y. Chen, S. Tang, Z. Huang and A. Lei, Sci. Adv., 2018, 4, eaat5312 CrossRef CAS PubMed.
-
(a) H. T. Huang, T. C Lacy, B. Błachut, G. X. Ortiz Jr and Q. Wang, Org. Lett., 2013, 15, 1818–1821 CrossRef CAS PubMed;
(b) N. Liu, H. Y. Wang, W. Zhang, Z. H. Jia, I. A. Guzei, H. D. Xu and W. Tang, Chirality, 2013, 25, 805–809 CrossRef CAS PubMed;
(c) X. F. Xia, S. L. Zhu, J. B. Liu, D. Wang and Y. M. Liang, J. Org. Chem., 2016, 81, 12482–12488 CrossRef CAS PubMed;
(d) W. Wei, L. Liao, T. Qin and X. Zhao, Org. Lett., 2019, 21, 7846–7850 CrossRef CAS PubMed;
(e) W. Huang, C. Xu, J. Yu and M. Wang, J. Org. Chem., 2021, 86, 1987–1999 CrossRef CAS PubMed.
- G. Capozzi, V. Lucchini, F. Marcuzzi and G. Modena, J. Chem. Soc. Perkin Trans. 1, 1981, 3106–3110 RSC.
- G. J. O'Malley and M. P. Cava, Tetrahedron Lett., 1985, 26, 6159–6162 CrossRef.
- K. Okuma, T. Yasuda, I. Takeshita, K. Shioji and Y. Yokomori, Tetrahedron, 2007, 63, 8250–8254 CrossRef CAS.
- P. Brownbridge, J. Chem. Soc., Chem. Commun., 1987, 1280–1281 RSC.
- P. L. López-Tudanca, K. Jones and P. Brownbridge, Tetrahedron Lett., 1991, 32, 2261–2264 CrossRef.
- S. M. Tuladhar and A. G. Fallis, Can. J. Chem., 1987, 65, 1833–1837 CrossRef CAS.
- S. M. Tuladhar and A. G. Fallis, Tetrahedron Lett., 1987, 28, 523–526 CrossRef CAS.
- K. Hegemann, B. Wibbeling and G. Haufe, J. Prakt. Chem. Chem., 1998, 340, 455–458 CrossRef CAS.
- H. Wang, D. Huang, D. Cheng, L. Li and Y. Shi, Org. Lett., 2011, 13, 1650–1653 CrossRef CAS PubMed.
- S. E. Denmark, D. J. Kornfilt and T. Vogler, J. Am. Chem. Soc., 2011, 133, 15308–15311 CrossRef CAS PubMed.
- H. Guan, H. Wang, D. Huang and Y. Shi, Tetrahedron, 2012, 68, 2728–2735 CrossRef CAS.
- Y. Gao, Y. Gao, X. Tang, J. Peng, M. Hu, W. Wu and H. Jiang, Org. Lett., 2016, 18, 1158–1161 CrossRef CAS PubMed.
- R. Zhang, Z. Yan and S. Lin, Synlett, 2018, 29, 336–339 CrossRef CAS.
- Y. Gao, S. Cheng, T. Zhang, R. Ding, Y. Liu, B. Sun and H. Tian, Synth. Commun., 2018, 48, 2773–2781 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b,
Roya Ahmadi
c,
Alibek Issakhovde,
Abdol Ghaffar Ebadi
f and
Esmail Vessally
*b,
Roya Ahmadi
c,
Alibek Issakhovde,
Abdol Ghaffar Ebadi
f and
Esmail Vessally