
Preparation of well-defined 2D-lenticular aggregates by self-assembly of PNIPAM-b-PVDF amphiphilic diblock copolymers in solution†
Enrique
Folgado
ab,
Matthias
Mayor
b,
Didier
Cot
b,
Michel
Ramonda
c,
Franck
Godiard
d,
Vincent
Ladmiral
 *b and
Mona
Semsarilar
*a
*b and
Mona
Semsarilar
*a
aInstitut Charles Gerhardt de Montpellier, ICGM, Univ Montpellier, CNRS, ENSCM, Montpellier, France. E-mail: mona.semsarilar@umontpellier.fr
bInstitut Européen des Membranes, IEM, Univ Montpellier, CNRS, ENSCM, Montpellier, France. E-mail: vincent.ladmiral@enscm.fr
cCentre de technologie de Montpellier, CTM, Université de Montpellier, Bât. 5, cc007 Campus Saint Priest, France
dService de Microscopie Electronique, Université Montpellier, Place Eugene Bataillon, 34095 Montpellier Cedex 5, France
First published on 25th November 2020
Abstract
PNIPAM-b-PVDF (poly(N-isopropylacrylamide)-b-poly(vinylidene fluoride)) amphiphilic block copolymers (BCPs) were synthesized via RAFT polymerization from PNIPAM macromolecular chain transfer agents (macro-CTAs). PNIPAM was used as it is one of the few hydrophilic polymers soluble in dimethyl carbonate (DMC) and which can be synthesized using a xanthate CTA with relatively good control. The polymerizations were conducted at 73 °C in DMC using two PNIPAM macro-CTAs of different molar masses and targeting various DPs for the PVDF block. The RAFT polymerization of VDF resulted in relatively well-defined BCPs (Đ ≤ 1.50). These amphiphilic BCPs were able to self-assemble into various morphologies such as spherical, crumpled, lamellar and lenticular 2D aggregates by changing the common solvent or the self-assembly protocol. The size of the aggregates could be controlled by varying the DP of the PVDF block. The polymers were characterized by 1H and 19F NMR, SEC, TGA and DSC, and the assembled structures were studied by TEM, SEM and AFM. The thermosensitive behavior of the 2D lenticular aggregates was also examined.
Introduction
Poly(vinylidene fluoride) (PVDF), despite its remarkable properties such as piezoelectricity, ferroelectricity, chemical inertness, and biocompatibility,1,2 has not received as much attention as other polymers in fields such as macromolecular engineering and polymer self-assembly. This is likely due to the synthesis constraints inherent to the peculiar reactivity of VDF. In addition, since VDF is a gaseous monomer, its polymerization usually requires the use of a high-pressure vessel, not commonly available in every laboratory. However, in recent years, the use of reversible-deactivation radical polymerization techniques to prepare PVDF and PVDF-based BCPs has been investigated with some success.3–6RAFT polymerization and iodine-transfer polymerization of VDF lead to the accumulation of VDF tail-terminated chains (–CF2CF2CH2–X, X = xanthate or iodine).7 These chains are not easily reactivated which hinders the preparation of PVDF-based block copolymers. So far, only PVDF-b-PVAc block copolymers (VAc = vinyl acetate) could be synthesized by chain extension of CF2–CH2–XA-terminated–PVDF (XA = xanthate) prepared by RAFT polymerization.8 Cobalt-mediated radical polymerization was shown to largely mitigate this end-group reactivity issue, and is arguably the best RDRP technique for VDF.4,9 However, it requires the use of air-sensitive organometallic compounds and is thus not easy to implement. Although coupling strategies using click chemistry (CuAAC, or thia-Michael addition for example)10–13 are efficient, they may require the preparation of functional RAFT agents, and often lead to mixtures of block copolymers and homopolymers which are not easy to separate. Perhaps the most straightforward solution is the chain extension of RAFT macromolecular chain transfer agents (macro-CTAs) with PVDF.
To date, only four reports have described the preparation of PVDF-based block copolymers from RAFT macro-CTAs using sequential addition of VDF. Kostov et al. described the synthesis of PVAc-b-P(VDF-co-TFP) block copolymers (TFP = trifluoropropene).14 Girard et al. reported the preparation of PDMA-b-PVDF and PDMA-b-P(VDF-co-PMVE) by chain extension of a PDMA macro-CTA (DMA = dimethylacrylamide, PMVE = perfluorinated methyl vinyl ether).15 Guerre et al. reported the preparation of PEVE-b-PVDF (EVE = ethyl vinyl ether) block copolymers via the sequential combination of cationic RAFT polymerization of vinyl ethers and radical RAFT polymerization of VDF.16 Guerre et al. also reported the polymerization-induced self-assembly (PISA) of PVAc-b-PVDF block copolymers.17 VAc units were then hydrolyzed to prepare PVA-b-PVDF amphiphilic BCPs (VA = vinyl alcohol).18 Only PDMA- and PVA-based PVDF-containing amphiphilic block copolymers have been prepared by sequential addition of VDF.
PVDF is a semi-crystalline fluorinated polymer, which as such is appealing for the preparation of self-assembled block copolymer morphologies through crystallization-driven self-assembly (CDSA) in selective solvents. The self-assembly of non-crystalline (coil–coil) BCPs in solution is well established.19 However, the complexity of the self-assembly process increases when one block of the BCP can crystallize. The formation of semi-crystalline BCP self-assembled structures can be viewed as a two-step process. The structures will first form by minimizing the unfavourable contacts between the solvophobic block and the solvent, and the semi-crystalline block will then start to crystallize in a second step, leading to the final structure. As crystallization takes place in the insoluble core, the initial morphology is either preserved or a morphological transformation into a novel structure is triggered.20 Diverse polymeric architectures have been obtained by the crystallization-driven self-assembly (CDSA) approach. Arno et al. reported recently the preparation of biocompatible and biodegradable 1D cylindrical and 2D platelet micelles of PCL-b-PMMA-b-PDMA (CL = ε-caprolactone, MMA = methyl methacrylate) via CDSA. Interestingly, they were able to control the dimensions and dispersity of the self-assembled nanostructures.21 Li et al. have reported a poly(L-lactide)-based diblock glycopolymer that assembled into 1D cylinders and 2D diamond-shaped platelets.22 Qiu and Gao et al. have reported the preparation of rectangular and hollow structures from polymer blends.23
To date, most reported coil-crystalline self-assembled aggregates with control over their size were prepared via CDSA, requiring heating and aging steps. The most common crystalline blocks in these assembled structures are PEO (poly(ethylene oxide)),24 PCL,21,25 PE (polyethylene),26 and PFS (poly(ferrocenyldimethylsilane)).21,27–30 Only a few studies describe the self-assembly in solution of BCPs containing a fluoropolymer block,10,12,18,31 and even fewer deal with the CDSA behavior of these fluorinated BCPs.10,17
Dimethyl carbonate (DMC) was shown to be the solvent of choice for the RAFT polymerization of VDF as it allows relatively fast polymerization and an acceptable level of transfer reactions.3,6 However, one major limitation of the chain extension with the PVDF approach to prepare amphiphilic PVDF-based BCPs is the rather low solubility of hydrophilic macro-CTAs in DMC.
PNIPAM is hydrophilic and soluble in DMC at the required temperature for VDF polymerization (ca. 73 °C). PNIPAM features a near body lower critical solution temperature (LCST = 32 °C) and biocompatibility, and is thus very appealing for biomedical applications.32–36 So far the reports mentioning both PVDF and PNIPAM only describe polymer blends for the preparation of electrospun fibers or flat membranes, or the grafting of PNIPAM on PVDF membranes.37 To date, the preparation of PNIPAM-b-PVDF block copolymers (BCPs) and the study of their self-assembly in selective solvents have not been reported.
The present study recounts the synthesis via RAFT sequential addition of VDF, characterization and self-assembly in solution of amphiphilic PNIPAM-b-PVDF diblock copolymers. The self-assembly of these BCPs in water led to a wide range of morphologies such as spherical, crumpled-spherical, lamellar and well-defined 2D lenticular aggregates without the need for thermal annealing. The formation and molecular arrangement of these self-assembled structures are discussed, and the thermoresponsive behaviour of the PNIPAM-containing aggregates in water is also examined.
Experimental section
Materials
All reagents were used as received unless otherwise stated. 1,1-Difluoroethylene (vinylidene fluoride, VDF) was supplied by Arkema (Pierre-Bénite, France). O-Ethyl-S-(1-methoxycarbonyl)ethyl dithiocarbonate (CTAXA) was prepared according to the method described by Liu et al.38tert-Amyl peroxy-2-ethylhexanoate (Trigonox 121, purity 95%) was purchased from AkzoNobel (Chalons-en-Champagne, France). Ethanol (EtOH), dimethyl carbonate (DMC), diethyl ether, toluene, laboratory reagent grade hexane (purity >95%) and N-isopropylacrylamide (NIPAM, purity 97%) were purchased from Sigma Aldrich. All deuterated solvents were purchased from Eurisotop. NIPAM was recrystallized twice from hexane/toluene (10/1, v/v).Measurements
Coupling constants and chemical shifts are given in hertz (Hz) and parts per million (ppm), respectively. The experimental conditions for recording 1H and 19F NMR spectra were as follows: flip angle, 30°; acquisition time, 4 s (2 s for 19F NMR); pulse delay, 1 s (2 s for 19F NMR); number of scans, 16; and pulse widths of 9.25 and 11.4 μs for 1H and 19F NMR, respectively.
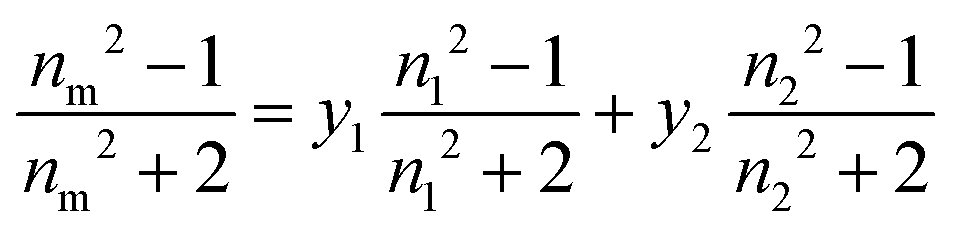 | (1) |
Synthesis
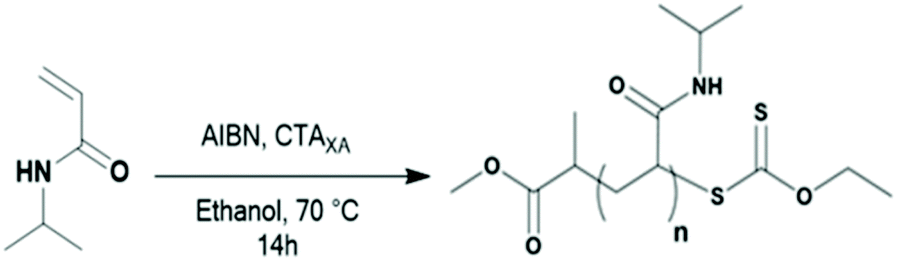 | ||
| Scheme 1 RAFT/MADIX synthesis of the PNIPAM macro-CTA. | ||
| Entry | CTA | M | [M]0/[CTA]0 | Reaction time (h), solvent | Yield (%) | DP(NMR)(R) |
M
n(theo)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) d (g mol−1) d (g mol−1) |
M
n(NMR)(R)e (g mol−1) |
M
n, SECf (g mol−1) |
Đ |
|---|---|---|---|---|---|---|---|---|---|---|
| Reactions conditions: (i) (Entries 1 and 3) NIPAM homopolymerization: [I]/[CTAXA] = 0.1 with I = AIBN and CTAXA = O-ethyl-S-(1-methoxycarbonyl)ethyl dithiocarbonate, T = 70 °C; (ii) (entries 2 and 4–7) chain extension of PNIPAM35-XA:[I]/[CTAXA] = 0.2 with I = Trigonox 121, T = 73 °C.a Determined by 1H NMR using eqn (2).†b Determined gravimetrically.c Determined by 1H NMR using eqn (5).d Calculated using the yield as the conversion and eqn (3)† for PNIPAM and (7) for the BCP.e Calculated from DPNMR using eqn (4)† for PNIPAM and (6) for PVDF.f Determined by SEC (RI detector). |
||||||||||
| 1 | CTAXA | NIPAM | 25 | 14, EtOH | >99 | 25a | 3000 | 3000 | 3200 | 1.30 |
| 2 | PNIPAM25-XA | VDF | 50 | 20, DMC | 60b | 35c | 4900 | 5300 | 6700 | 1.38 |
| 3 | CTAXA | NIPAM | 35 | 14, EtOH | >99 | 35a | 4100 | 4200 | 4400 | 1.19 |
| 4 | PNIPAM35-XA | VDF | 100 | 20, DMC | 60b | 60c | 8000 | 8000 | 7500 | 1.29 |
| 5 | PNIPAM35-XA | VDF | 150 | 20, DMC | 61b | 100c | 10000 |
10600 |
9200 | 1.36 |
| 6 | PNIPAM35-XA | VDF | 200 | 20, DMC | 62b | 150c | 12100 |
13800 |
9800 | 1.43 |
| 7 | PNIPAM35-XA | VDF | 600 | 20, DMC | 61b | 450c | 27600 |
33000 |
25500 |
1.50 |
1H NMR (400 MHz, (CD3)2CO, δ (ppm), Fig. S1†): 0.9–1.28 (m, –NH–CH(CH3)2; –CH(CH3)(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)–), 1.28–1.90 (m, –CH2–CH–(CO)–; CH3–CH2–O–), 1.90–2.50 (m, –CH2–CH–(CO)–), 3.64 (–O–CH3, (R CTA)), 3.95–4.25 (m, –NH–CH(CH3)2), 4.50–4.76 (O–CH2–CH3 (Z CTA)), 6.50–8.00 (m, –NH–CH(CH3)2).
O)–), 1.28–1.90 (m, –CH2–CH–(CO)–; CH3–CH2–O–), 1.90–2.50 (m, –CH2–CH–(CO)–), 3.64 (–O–CH3, (R CTA)), 3.95–4.25 (m, –NH–CH(CH3)2), 4.50–4.76 (O–CH2–CH3 (Z CTA)), 6.50–8.00 (m, –NH–CH(CH3)2).
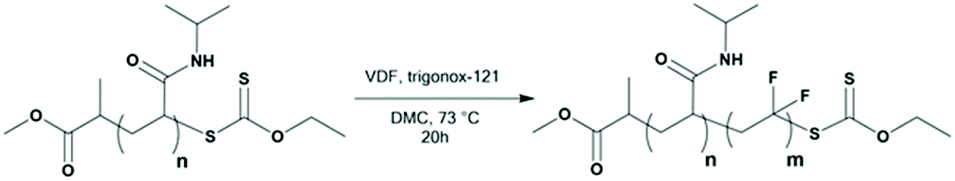 | ||
| Scheme 2 RAFT/MADIX synthesis of PNIPAM-b-PVDF block copolymers using the PNIPAM macro-CTA. | ||
1H NMR (400 MHz, (CD3)2CO, δ (ppm), Fig. S2†): 0.90–1.26 (m, –NH–CH(CH3)2–), 1.28–1.90 (m, –CH2–CH–NIPAM), 1.90–2.50 (m, –CH2–CH–NIPAM), 2.16–2.37 (t, –CF2–CH2–CH2–CF2–, VDF–VDF HH reverse addition), 2.66–3.01 (t, –CF2–CH2–CF2–, VDF–VDF HT regular addition), 3.8 (s, CH3–O–(CO)–(CH3)CH–), 3.95–4.25 (m, –NH–CH(CH3)2), 4.40 (t, CH2–CF2–CF2–CH2–S–), 4.60–4.78 (q, –S(CS)OCH2–CH3, 3JHH = 7.1 Hz), 6.09–6.50 (tt, –CH2–CF2–H, 2JHF = 55.6 Hz, 3JHH = 4.7 Hz), 6.50–8.00 (m, –NH–CH(CH3)2).
19F NMR (376 MHz, (CD3)2CO, δ (ppm), Fig. S3†): −115.64 (–CH2–CF2–CF2–CH2–CH2–, VDF–VDF HH reverse addition), −114.45 (–CH2–CF2–H), −113.36 (–CH2–CF2–CF2–CH2–CH2–, HH reverse addition), −113.09 (CH2–CF2–CF2–CH2–S–), −112.69 (–CH2–CF2–CF2–CH2–S–), −107.40 (–CF2–CH3), −94.81 (–CH2–CH2–CF2–CH2–, TT reverse addition), −93.00 (CH3–O–(CO)–O–CH2–CH2–CF2–, DMC-initiated PVDF), −92.50 (PNIPAM–CH2–CF2–), 92.06 (–CH2–CF2–CH2–CF2H), −91.43 (–CH2–CH2–CF2–CH2–CF2–CH2–CF2–, regular VDF–VDF HT addition), −91.00 (–CH2–CF2–CH2–, regular VDF–VDF HT addition).
The degree of polymerization (DP) of the PNIPAM block was calculated from the 1H NMR spectrum of the purified BCP using the equation provided in the ESI (see S2, eqn (2)†). The DP values were then used for the determination of the experimental Mn (S2, eqn (4)†). The degree of polymerization (DP) of the PVDF block was determined using eqn (5) from the integral of the signals (at 0.9–1.28 ppm) corresponding to the methyl groups (–CH3) of the NIPAM units, used as a reference, and the integral of the signals of the –CH2– group of the normal (HT) VDF additions (at 2.70–3.19 ppm). The signal of the –CH3 group of the NIPAM unit is the only signal visible for the BCP with a DP higher than 150. Regarding the reverse (TT) VDF additions, the average number of monomer additions occurring per chain between two degenerative transfers increases with increasing [VDF]0/[CTA]0. However, the total amount of TT (or HH) VDF additions (intra-chain + chain-end) stabilizes to an identical proportion (ca. 4.1%) for PVDF homopolymerization as previously reported by our group.7 As the signals assigned to these inversions overlap with the signals of the PNIPAM macro-CTA, a 1.041 multiplying factor was employed for the determination of the DP of PVDF.
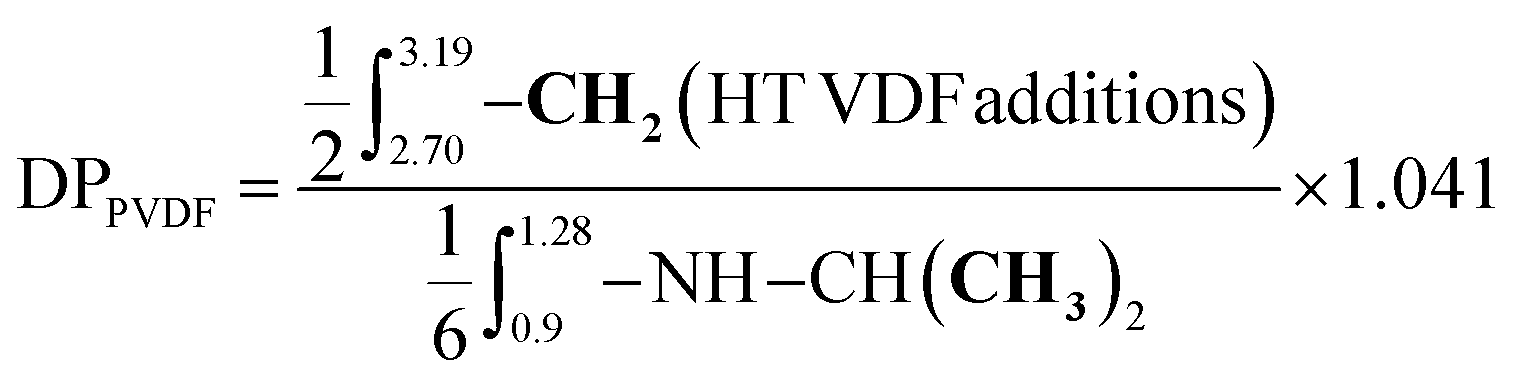 | (5) |
Molar masses were then calculated using eqn (6) (with Mn, PNIPAM-XA calculated using eqn (4) and (2)†):
| Mn, NMR = Mn, PNIPAM-XA + DPPVDF × Mn, VDF | (6) |
Theoretical molar masses were calculated using eqn (6) with yield = conversion and the [VDF]0/[PNIPAM-XA]0 ratios listed in Table 1.
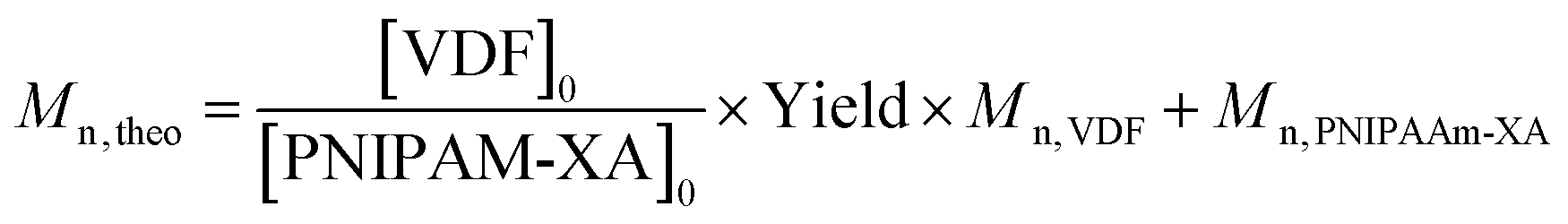 | (7) |
where Mn, VDF = 64.03 g mol−1.
Self-assembly
:water (1:20).
Note: The difference between precipitation and nanoprecipitation is that polymer precipitation refers to a polymer purification technique in which a solution of the polymer is cast dropwise into a non-solvent. Upon contact between the two miscible solvents, the polymer crashes out of the solution while the impurities remain in the solvent phase. In the case of block copolymers, for an efficient precipitation, the different blocks must be insoluble in the non-solvent. In contrast, polymer nanoprecipitation refers to a BCP self-assembly technique in which a polymer solution is cast dropwise into a solvent selective for one of the blocks. The insoluble block forms the core of the self-assembled structure while the soluble block acts as a steric stabilizer of the polymer morphology.
:4, 1:6 and 1:8 solvent:non-solvent ratios.
:1 solvent:non-solvent mixture, and this solution was then heated at 90 °C for 30 min and slowly cooled down to room temperature.
Results and discussion
PNIPAM-XA macro-CTAs were synthesized by RAFT polymerization using CTAXA following protocols described previously by Sistach et al.42 The reactions were stopped when the NIPAM conversion reached at least 99%. After purification by precipitation, 1H NMR spectroscopy of the resulting PNIPAM-XA macro-CTAs was performed to determine their molar masses and degrees of polymerization (DPs). Amphiphilic PVDF-based block copolymers with PVDF of different DPs were prepared from these PNIPAM macro-CTAs (see Table 1) by chain extension with VDF in DMC (a common solvent for PNIPAM and PVDF allowing a relatively high VDF polymerization rate) using Trigonox 121 as the radical initiator.19F NMR spectroscopy of the resulting polymers showed the successful chain extension of PVDF from the PNIPAM macro-CTAs. The presence of the signals of –CF2–CF2–CH2–XA at δ = −113.09 ppm and −112.69 ppm and of –CH2–XA at δ = 4.45 ppm and –CH2–SC(S)–O–CH2– at δ = 4.72 ppm in the 19F NMR and 1H NMR spectra, respectively (Fig. S4 and S3b†), indicates the formation of the diblock copolymers.
As expected, the polymerization of VDF was accompanied by a non-negligible amount of transfer to DMC. The characteristic signals of these transfer reactions can be observed in the 1H NMR spectrum (Fig. S3a and S3b†) as a triplet of triplets at 6.3 ppm corresponding to the –CF2H chain end, and in the 19F NMR spectrum (Fig. S4†) as a doublet of triplet at −114.45 ppm (–CF2H) and a multiplet at −107.3 ppm corresponding to the CF2CH3 chain end. The 1H NMR spectrum also shows a singlet at 5.77 ppm assigned to the DMC–xanthate adduct (CH3O(CO)O–CH2–XA, Fig. S3a†), eliminated by the precipitation step (Fig. S3b†), and a well-defined triplet at 4.35 ppm and a singlet at 3.73 ppm assigned to the –CH2– and –CH3 groups of the DMC moieties of the DMC-initiated PVDF chains, respectively. The RAFT polymerization of VDF is accompanied by a progressive loss of the chain-end functionality (loss of the xanthate group).6,7 This phenomenon has previously been reported for the synthesis of PVAc-b-PVDF BCPs.17 This loss of functionality can be very extensive. Here the remaining molar fraction of the xanthate-functionalized chain end derived from the 19F NMR spectra (Fig. S4 and calculations S5†) was 19.2, 5.4, 2.3 and 1.4% for the PNIPAM35-b-PVDF60, PNIPAM35-b-PVDF100, PNIPAM35-b-PVDF150 and PNIPAM35-b-PVDF450 copolymers respectively.
Despite these transfer reactions and loss of functionality, the chain extension of PNIPAM macro-CTAs with PVDF produced relatively well-defined BCPs with dispersity below 1.50 and monomodal SEC traces without shoulders or significant tailing (Fig. 1). These SEC traces also show a clear shift towards higher molar masses with increasing DP of PVDF.
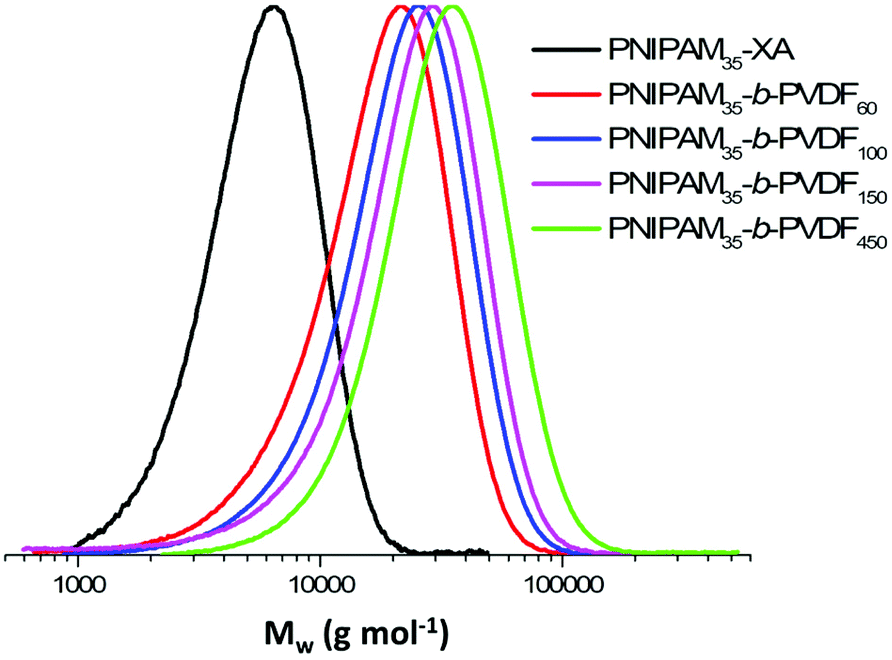 | ||
| Fig. 1 Normalized SEC traces (viscometric detector) of: PNIPAM35-XA (black trace), PNIPAM35-b-PVDF60 (red trace), PNIPAM35-b-PVDF100 (blue trace), PNIPAM35-b-PVDF150 (pink trace) and PNIPAM35-b-PVDF450 (green trace) after purification by precipitation in chilled ether for PNIPAM or cold pentane for the BCPs. | ||
These amphiphilic block copolymers were then used to prepare self-assembled morphologies in different solvents.
Note on solvents: in the following self-assembly study, three solvents were used as good solvents for both the PNIPAM and PVDF segments: acetone, THF and DMF. All of these solvents are good solvents of PNIPAM. Bottino et al.43 investigated the solubility of 450000 g mol−1 PVDF in 46 liquids and concluded that DMF is a solvent of PVDF, while acetone and THF are good swelling agents. The PVDF blocks synthesized here have a much lower molar mass (PVDF with a DP = 450 would have an Mn value of 28800 g mol−1); it is thus not surprising that THF and acetone behave as good solvents especially considering the low concentrations used (2 mg mL−1, whereas Bottino et al. aimed at preparing 100 mg mL−1 solutions). In addition, the self-assembly described and discussed below concerns PNIPAM-b-PVDF block copolymers whose solubility would be enhanced by the PNIPAM block. These copolymers indeed readily dissolved in acetone and in THF, although gentle heating of the solution was necessary for the latter.
The self-assembly of amphiphilic BCPs using rapid solvent exchange usually leads to the formation of colloidal objects via microphase separation. The final structure of these block copolymer colloids is primarily dictated by the volume fraction of the blocks and by the interfacial surface tensions. A feature of the nanoprecipitation process (which consists of the dropwise addition of the BCP solution into a selective solvent under vigorous stirring) is the ability to access kinetically trapped morphologies in nonequilibrium states due to short mixing time. This kinetic trapping is even more pronounced in the case of semicrystalline polymers such as PVDF. In such a case, the PVDF segments often crystallize before the polymer chains can reach the equilibrium morphology during phase separation.
The self-assembly of the PNIPAM-b-PVDF block copolymers was achieved via nanoprecipitation in water from a dilute DMF BCP solution. After stirring for 1 h, the samples were analyzed by DLS and TEM. This approach led to the formation of spherical aggregates of roughly 20 to 60 nm diameter (Fig. 2).
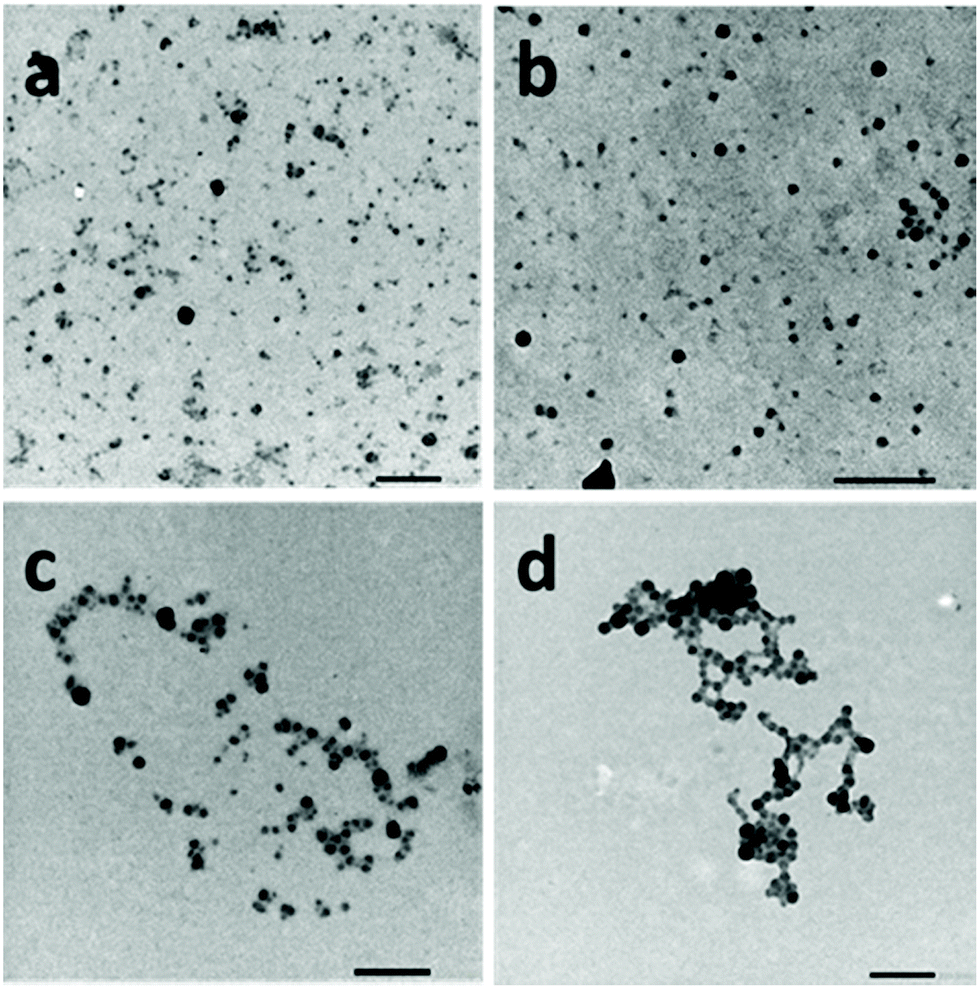 | ||
| Fig. 2 TEM images of spherical aggregates prepared from (a) PNIPAM35-b-PVDF60, (b) PNIPAM35-b-PVDF100, (c) PNIPAM35-b-PVDF150 and (d) PNIPAM35-b-PVDF450. All samples were prepared by nanoprecipitation in water from BCP solutions in DMF at 2 mg mL−1 (final concentration = 0.1 mg mL−1 in DMF:water (1:20)). Scale bars correspond to 200 nm. | ||
The particle sizes measured by TEM were in good agreement with the average hydrodynamic diameters measured by DLS (Fig. S6†) ranging between 30 and 80 nm and increasing with the DP of the core-forming PVDF block.
Solvent switch experiments (slow addition of water into a solution of PNIPAM35-b-PVDFX in DMF, THF or acetone) were also performed using three (1:4, 1:6 and 1:8) solvent:non-solvent ratios.
When DMF was employed as a common solvent and water as a selective solvent for the PNIPAM block, irregular crumpled spherical aggregates were obtained with all the solvent:non-solvent ratios tested (Fig. 3). CDSA self-assembled polymers are thought to favor the formation of aggregates with low interfacial curvature.44 These morphologies may be due to the co-nonsolvency of PNIPAM blocks when the DMF molar fraction changes in the solvent mixture, provoking the collapse of the metastable aggregates before attaining the spherical morphology obtained by nanoprecipitation. A reversible collapse–swelling behavior has already been observed in PNIPAM latexes in water–DMF mixtures.45 When the DMF molar fraction was between 0.02 and 0.25, the latex volume was smaller than that observed in pure water.
 | ||
| Fig. 3 TEM images of crumpled aggregates obtained by the solvent switch of (a and b) PNIPAM25-b-PVDF35 and (b and c) PNIPAM35-b-PVDF450 in DMF at 2 mg mL−1. Final concentration = 0.4 mg mL−1 in the DMF:water (1:4) mixture. | ||
When THF was used as the good solvent for both blocks, solvent switch protocols led to the formation of flat sheet morphologies, presumably 2D bilayer aggregates (Fig. 4).
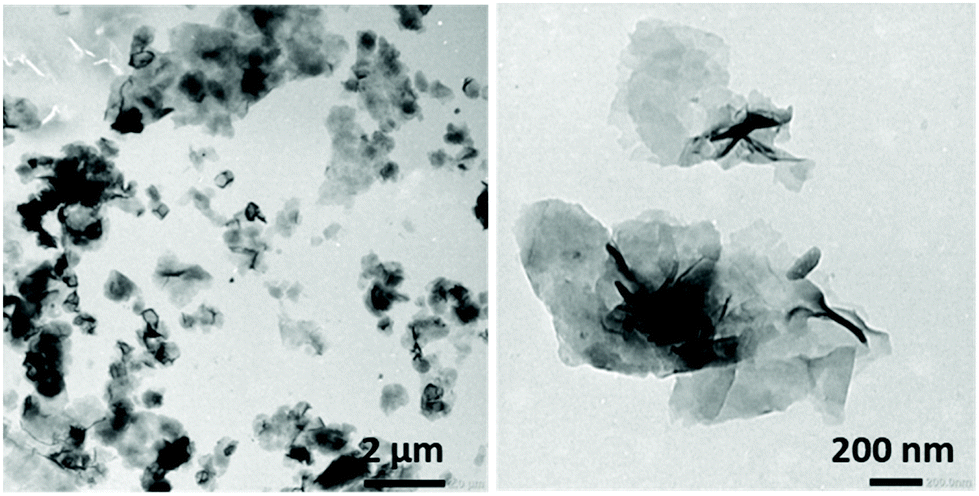 | ||
| Fig. 4 TEM images of the bilayer aggregates obtained by the solvent switch of PNIPAM35-b-PVDF450. Initial polymer concentration in THF = 2 mg mL−1. Final concentration = 0.4 mg mL−1 in the THF:water (1:4) mixture. The water addition rate was 4 mL h−1. | ||
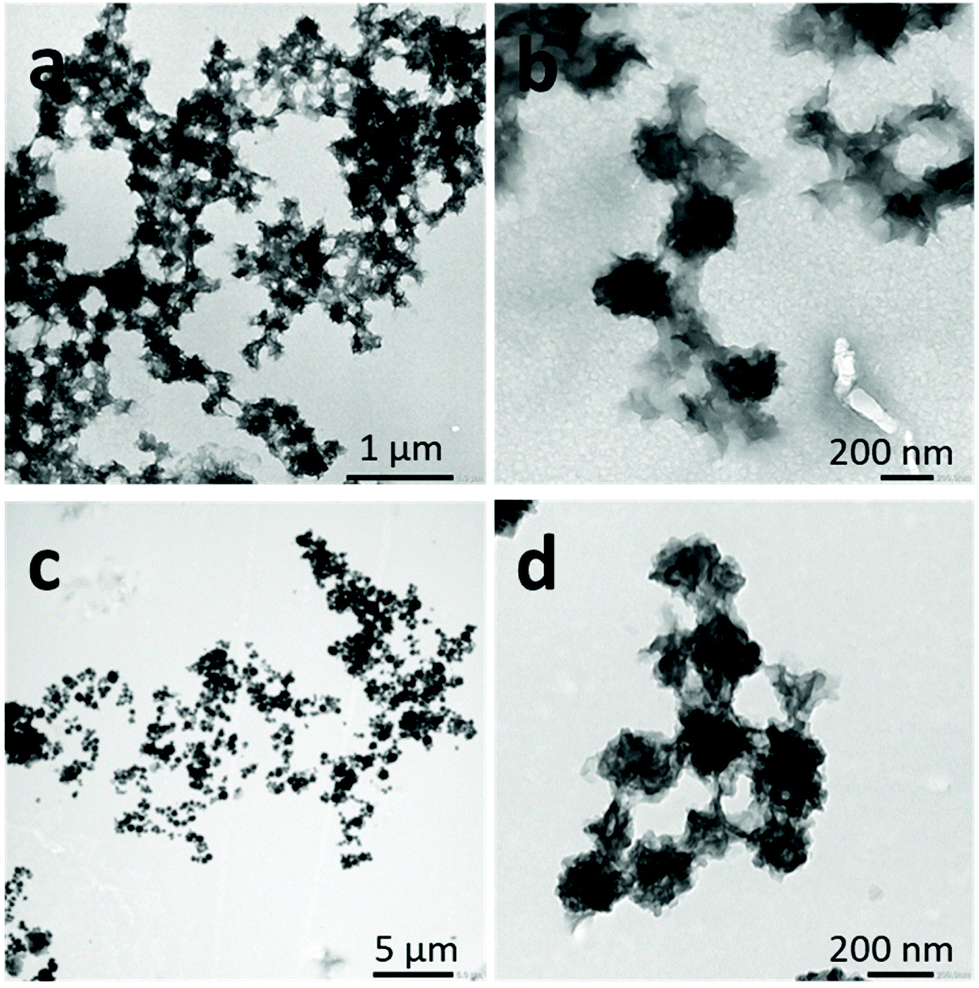
When acetone was used as the good solvent, well-defined flat 2D lenticular morphologies with relatively low dispersity and good dimensional control were obtained (Fig. 5–7).
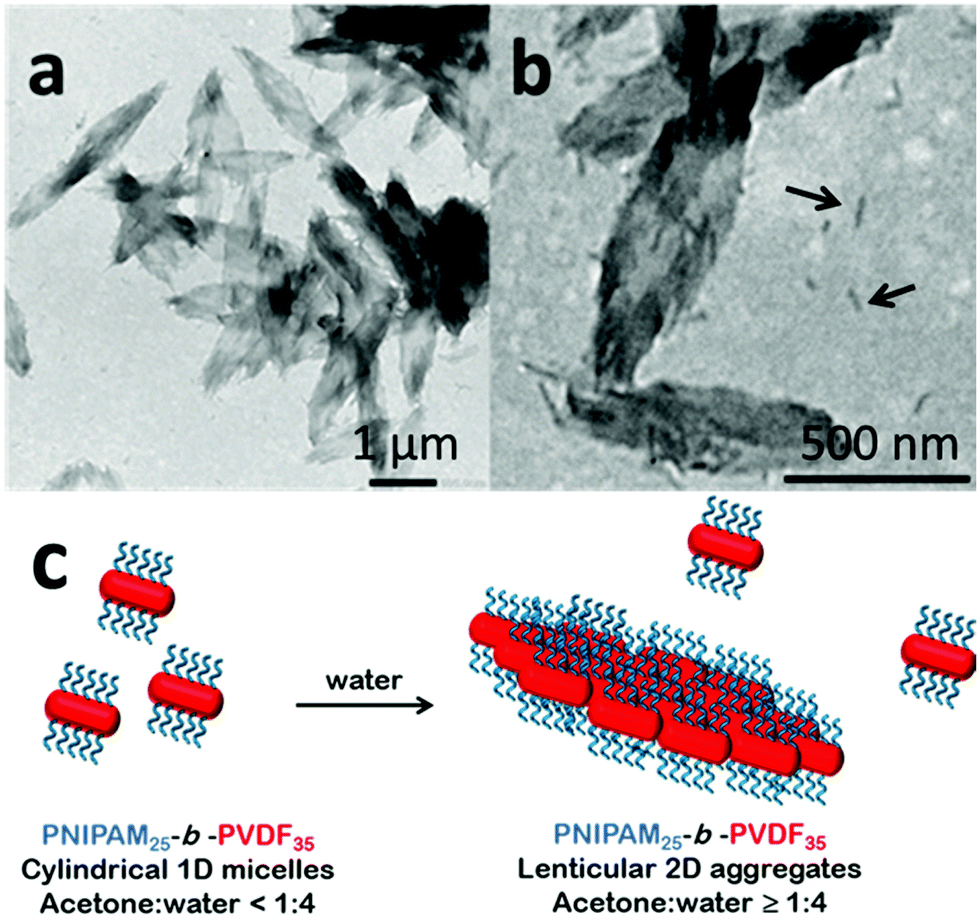 | ||
| Fig. 5 TEM images of 2D lenticular micelles obtained by self-assembly via the solvent switch protocol of PNIPAM25-b-PVDF35 (a and b). Initial BCP concentration in acetone = 2 mg mL−1. Final concentration = 0.4 mg mL−1 in the acetone:water (1:4) mixture. The non-solvent addition rate was 4 mL h−1. The arrows indicate the isolated 1D micelles. Schematic representation of the proposed mechanism of the formation of PNIPAM25-b-PVDF35 lenticular aggregates from short 1D rod-like micelles (c). | ||
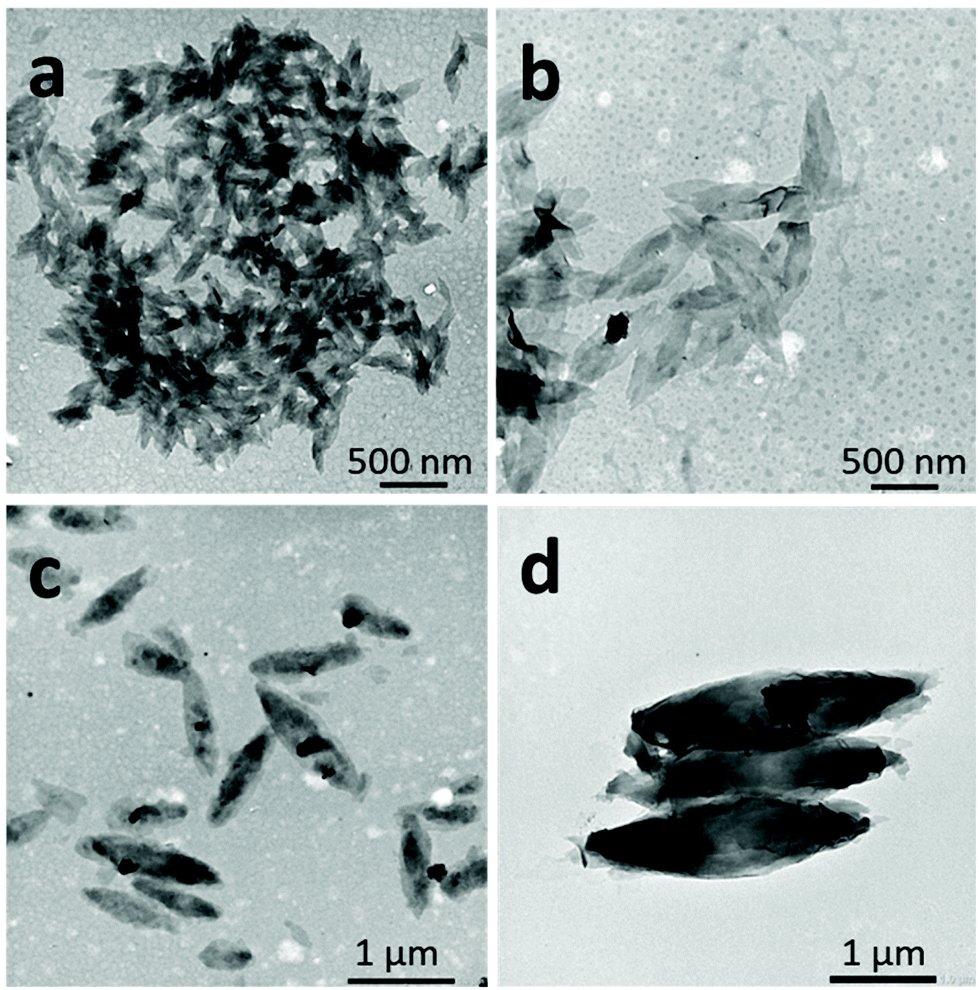 | ||
| Fig. 6 TEM images of representative self-assembled 2D lenticular morphologies prepared from (a) PNIPAM35-b-PVDF60, (b) PNIPAM35-b-PVDF100, (c) PNIPAM35-b-PVDF150 and (d) PNIPAM35-b-PVDF450. All samples were prepared via the solvent switch in acetone:water mixtures. Initial polymer concentration in acetone = 2 mg mL−1. Final concentration = 0.4 mg mL−1 in the acetone:water (1:4) mixture. Water addition rate = 4 mL h−1. | ||
 | ||
| Fig. 7 SEM images of 2D lenticular objects prepared from: PNIPAM25-b-PVDF35 (a and b) and PNIPAM35-b-PVDF450 (c and d). | ||
In the case of the PNIPAM25-b-PVDF35 BCP, these lenticular objects had an average length and width of 600 nm and 250 nm, respectively, and ragged edges that seemed to be formed by aggregation of smaller 1D rod-like aggregates (Fig. 5b and S7†). Crystallization-induced aggregation of micelles has already been observed for PB-b-PEO BCPs.46 These short 1D micelles have an average length and width of 98 and 19 nm, respectively.
In contrast, the PNIPAM-b-PVDF BCP prepared from the PNIPAM35 macro-CTA self-assembled into lenticular objects with much smoother-looking edges with average lengths of 200, 600, 1000 and 2300 nm and average widths of 90, 250, 400 and 850 nm for PVDF DPs of 60, 100, 150 and 450, respectively (Fig. 6).
The length and width of the lenticular 2D micelles showed a linear dependence on the degree of polymerization of PVDF up to 150 (Fig. S7†). The loss of linearity observed for the PVDF450 BCP may be due to the poor accuracy of the DP value calculated from weak NMR signals. The real DP of this PVDF block might be closer to 300 than the calculated 450.
Due to their relatively large size (in the micrometer range), these morphologies were not colloidally stable and sedimented over time (ca. 14 h). However, they were easily redispersed by simple shaking.
The shape and size of these morphologies were not affected by the concentration of the initial BCP solutions over the concentration range studied (Fig. S9†). In addition, once the self-assembled structures were formed in an acetone:water mixture (1:4), they did not evolve within the 2-week observation time; they thus seem to be kinetically trapped. Increasing the water content of the self-assembly media or decreasing the acetone content (by evaporation) of the solvent mixture only affected the aggregation rate. However, the lenticular shape of these assemblies was strongly affected by the ratio of the length of the hydrophobic and hydrophilic blocks. The morphologies were rather patchy with sharp edges for PNIPAM25-b-PVDF35 (Fig. 5), while better-defined objects were found for PNIPAM35-b-PVDFn BCPs with n > 60 (Fig. 6). Better-defined lenticular objects were obtained as higher DPs of PVDF were targeted.
The PVDF fraction of these BCPs was crystalline as determined from DSC thermograms (Fig. S10†). However, the PVDF crystallinity could not be observed on these self-assembled structures by electron diffraction during TEM analysis. This was ascribed to the thinness of these lenticular objects, undergoing fast amorphisation under the electron beam.
Nevertheless, the XRD diffraction pattern (Fig. 8) obtained from freeze-dried 2D aggregates formed via self-assembly of a 2 mg mL−1 solution of PNIPAM35-b-PVDF100 BCPs in acetone:water (1:4) suggests that the PVDF phase of these aggregates was indeed crystalline and in the α-form with characteristic 2θ peaks at 17.6 (100), 18.5 (020), 19.8 (110), 26.5 (021), 36.1 (200) and 39 (002).47 The α-crystal phase is hexagonal, with aligned polymer chains anti-parallel to each other, in the conformation of trans-gauche–trans-gauche′ (TGTG′). The α-form is the most common and spontaneously forming crystalline form of PVDF.48,49
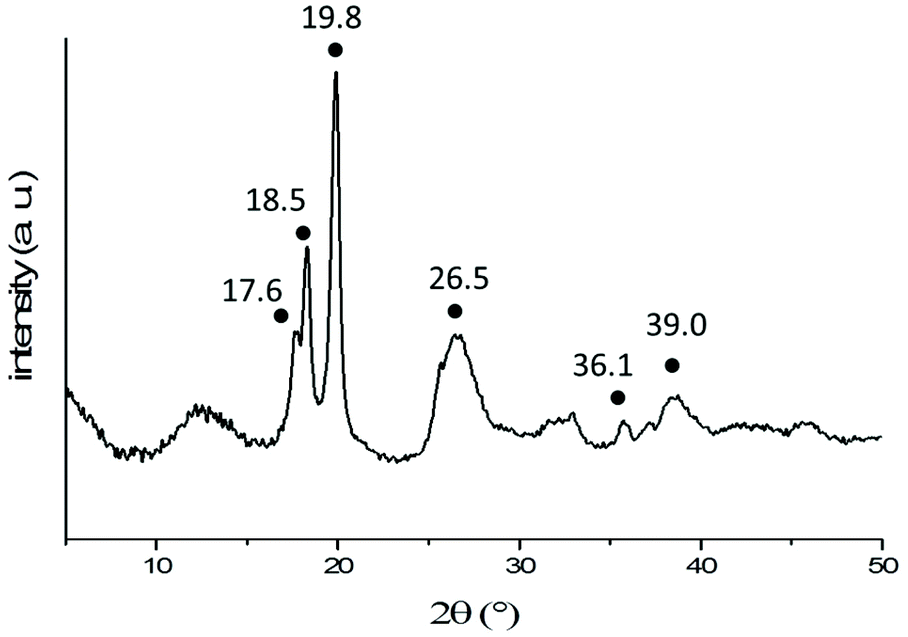 | ||
| Fig. 8 X-Ray diffraction pattern of freeze-dried 2D lenticular aggregates obtained from PNIPAM35-b-PVDF100 BCPs. The black dots indicate the characteristic peaks of the α-phase of PVDF. | ||
SEM images of the lenticular objects (Fig. 7) suggest that they can fold and twist to an extent, and are often stacked on top of each other, making their characterization quite difficult. AFM images (Fig. 9) confirmed the stacking of the objects but also revealed the increase of their thickness with the increase of the DP of the PVDF block.
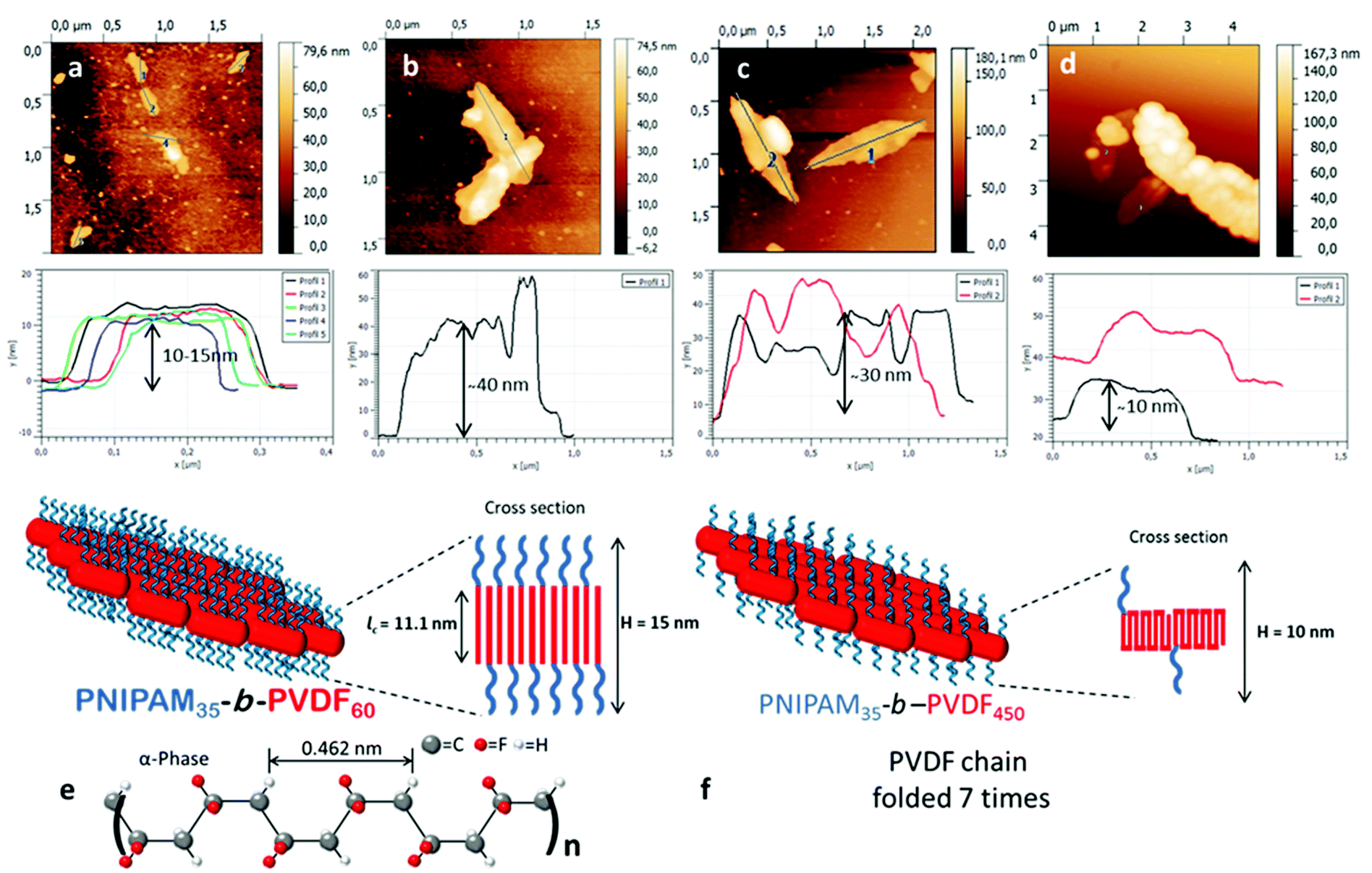 | ||
| Fig. 9 AFM images of 2D lenticular aggregates prepared from: (a) PNIPAM35-b-PVDF60, (b) PNIPAM35-b-PVDF100, (c) PNIPAM35-b-PVDF150, and (d) PNIPAM35-b-PVDF450. (e and f) Schematic representation and suggested molecular arrangement of the 2D lenticular aggregates formed by the assembly of PNIPAM35-b-PVDF60 and PNIPAM35-b-PVDF450 respectively. | ||
The presence of short cylindrical 1D micelles (Fig. 6b) suggests that the lenticular morphologies may form by aggregation of these smaller structures. In addition, the thickness of the lenticular aggregates seems to be related to the length and folding of the PVDF segments. As the degree of polymerization of the PVDF core-forming block increased, the thickness of the aggregates increased.
Since the PVDF in these aggregates is crystallized in the α-form, the number of foldings of the PVDF chains can be estimated. In the α-form crystal, 0.462 nm is the dimension of 2.5 VDF units in that specific conformation.50 Crystallized PVDF chains of DP 60, 100, 150 and 450 would thus extend over 11.1, 18.5, 27.7 and 83.2 nm, respectively, in the α-form. The thicknesses of the lenticular objects derived from the AFM topographic images shown in Fig. 9 are in agreement with these calculations for PVDF60, PVDF100 and PVDF150. Fig. 9a, corresponding to PNIPAM35-b-PVDF60, shows a thickness of 10–15 nm. Fig. 9b and Fig. S11,† corresponding to PNIPAM35-b-PVDF100, shows a thickness of about 40 nm. This value is roughly twice the calculated length of PVDF100 (18.5 nm). This is because the AFM images in Fig. 9b show two aggregates stacked on top of each other. The thickness measured for PNIPAM35-b-PVDF150 in Fig. 9c is 30 nm which also is in agreement with the corresponding calculated value of 27.7 nm. In the case of the PNIPAM35-b-PVDF450 BCP, the calculated thickness of 83.2 nm does not match the thickness of 10 nm measured in Fig. 9d. This discrepancy may be explained by considering that in this case the crystallized PVDF chains are folded rather than extended. Indeed, the calculated and measured values can be reconciled if the PVDF450 chains were folded 7 times (or 5 times if the real DP is actually 300 as mentioned above). This explanation is also consistent with the observation of larger aggregates for a higher PVDF degree of polymerization.
As PVDF chains fold more, the repulsion between the PNIPAM hydrophilic chains decreases, allowing the formation of larger but thinner 2D aggregates.
Since the degree of crystallinity can be modified via temperature annealing, a heating and cooling treatment was applied to the crumpled structures (Fig. 3) obtained by self-assembly from the DMF solution. In addition, at higher temperature, the solubility of PVDF in a DMF/water mixture increases. A 5 mg mL−1 PNIPAM25-b-PVDF35 BCP solution in DMF was self-assembled by adding water to reach a 1:1 solvent:non-solvent ratio; then this solution was heated at 90 °C for 30 min and slowly cooled down to room temperature. Fig. S12† shows that this temperature treatment led to a mixture of ill-defined aggregates and spindle-shaped morphologies with lengths ranging from 300 nm to 1 μm. The crumpled sphere to spindle morphology transition is thought to proceed via temperature-induced crystallization-driven self-assembly (TI-CDSA) due to the degradation of the solvent quality for the crystallisable block caused by the slow cooling.20
To sum up, five different morphologies were observed by the self-assembly of these PNIPAM-b-PVDF BCPs as presented in Table 2.
| BCP | NP | SS | SS | SS | SS + TI-CDSA |
|---|---|---|---|---|---|
| DMF:H2O |
DMF:H2O |
THF:H2O |
Acetone:H2O |
DMF:H2O |
|
| NP, SS and TI-CDSA stand for nanoprecipitation, solvent switch and temperature-induced crystallization-driven self-assembly protocols respectively.a Sharp edges. In all PNIPAM35-b-PVDFm systems examined, the size of the nanoaggregates increased with increasing DP of the PVDF block. The initial concentration for nanoprecipitation and solvent switch protocols was 2 mg mL−1 (5 mg mL−1 in the case of the sample used for TI-CDSA). The rate of selective solvent addition was 4 mL h−1 for the solvent switch protocol and the solvent was added dropwise during nanoprecipitation. The TI-CDSA sample was placed for 30 min in an oil bath at 90 °C and slowly cooled after that time. | |||||
| PNIPAM25-b-PVDF35 | Spherical | Crumpled spherical | Flat sheet | Lenticulara | Spindle |
| PNIPAM35-b-PVDF60 | Spherical | — | — | Lenticular | — |
| PNIPAM35-b-PVDF100 | Spherical | — | — | Lenticular | — |
| PNIPAM35-b-PVDF150 | Spherical | — | — | Lenticular | — |
| PNIPAM35-b-PVDF450 | Spherical | Crumpled spherical | Flat sheet | Lenticular | — |
Without surprise, the solvents used and the self-assembly protocol played a crucial role in the resulting BCP structures. The same DMF/water binary mixture of solvents afforded three distinct morphologies depending on the self-assembly process used. Spherical nanoparticles were obtained when nanoprecipitation was used while a solvent switch protocol led to the formation of crumpled spheres. These crumpled spheres evolved into spindle-like aggregates when a TI-CDSA protocol was applied. The solvent switch protocol produced 2D lamellar aggregates in the case of THF/water and well-defined lenticular shaped aggregates of increasing length and width with increasing DP of the PVDF block in an acetone/water binary mixture.
Thermoresponse of PNIPAM-b-PVDF lenticular nanoparticles
The thermoresponsiveness of the PNIPAM-b-PVDF lenticular aggregates was also investigated. The usual turbidity test could not be used due to the poor colloidal stability of the BCP aggregates even at room temperature. Since the LCST of PNIPAM is affected by the presence of organic solvents, acetone was completely removed from the PNIPAM-b-PVDF BCP suspensions under reduced pressure using a rotary evaporator at room temperature to prepare an aqueous suspension of the self-assembled morphologies. TEM analysis of this suspension (Fig. S13†) showed that the size and shape of the assemblies were not affected by the removal of acetone.The samples were heated at 50 °C under gentle stirring for a period of 30 min and then cooled down to room temperature.
Fig. 10 shows the effect of temperature on the aqueous PNIPAM-b-PVDF BCP morphologies. The lenticular objects partly lost their well-defined shape, crumpled to an extent and also broke into smaller flat sheet aggregates. The change of conformation of the PNIPAM blocks provoked by the decrease of the PNIPAM solubility in water at T > LCSTPNIPAM is thought to be responsible for the aggregates to irreversibly break or evolve into the observed crumpled morphologies.
 | ||
| Fig. 10 TEM images of the representative samples of 2D lenticular aggregates prepared from: (a and b) PNIPAM35-b-PVDF150 BCPs and (c and d) PNIPAM35-b-PVDF450 BCPs in pure water after being heated at 45 °C for 30 min. | ||
Conclusions
A series of relatively well-defined PNIPAM-b-PVDF semicrystalline amphiphilic diblock copolymers were successfully synthesized by RAFT polymerization using PNIPAM macro-CTAs. These diblock copolymers were fully characterized by 1H and 19F NMR spectroscopy, TGA, DSC and SEC. Despite the loss of the xanthate functionality observed when DPs of PVDF higher than 50 were targeted, BCPs with relatively low dispersity (Đ < 1.50) were achieved. Due to their amphiphilic nature, these BCPs had the ability to self-assemble in aqueous solutions. The self-assembled structures were characterized by TEM, SEM and AFM microscopies. The final structures were strongly affected by the choice of common solvents and the self-assembly protocol used. Five types of morphologies were obtained depending on the common solvent and the self-assembly protocol. Surprisingly, self-assembly from acetone solutions led to the formation of well-defined 2D lenticular aggregates whose dimensions increased with the degree of polymerization of PVDF, reaching a length of 2.3 μm for the longest PVDF synthesized. These aggregates were morphologically stable over time and accessible even at higher concentrations. A formation pathway and a possible molecular arrangement of these 2D lenticular aggregates were proposed.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Arkema for providing VDF, and the Institut Carnot Chimie Balard Cirimat, the LabEx CheMISyst (ANR-10-LABX-05-01), IEM and ICGM for funding the PhD of EF.Notes and references
- T. Soulestin, V. Ladmiral, F. Domingues, D. Santos and B. Améduri, Prog. Polym. Sci., 2017, 72, 16–60 CrossRef CAS.
- Y. Yu, H. Sun, H. Orbay, F. Chen, C. G. England, W. Cai and X. Wang, Nano Energy, 2016, 27, 275–281 CrossRef CAS.
- A. D. Asandei, O. I. Adebolu and C. P. Simpson, J. Am. Chem. Soc., 2012, 134, 6080–6083 CrossRef CAS.
- B. M. Ameduri, R. Poli, V. Ladmiral, S. Banerjee, A. Debuigne and C. Detrembleur, Angew. Chem., Int. Ed., 2018, 57, 2934–2937 CrossRef.
- D. Valade, C. Boyer, B. Ameduri and B. Boutevin, Macromolecules, 2006, 39, 8639–8651 CrossRef CAS.
- M. Guerre, B. Campagne, O. Gimello, K. Parra, B. Ameduri and V. Ladmiral, Macromolecules, 2015, 48, 7810–7822 CrossRef CAS.
- M. Guerre, S. M. W. Rahaman, B. Améduri, R. Poli and V. Ladmiral, Macromolecules, 2016, 49, 5386–5396 CrossRef CAS.
- M. Guerre, S. M. Wahidur Rahaman, B. Améduri, R. Poli and V. Ladmiral, Polym. Chem., 2016, 7, 6918–6933 RSC.
- P. G. Falireas, V. Ladmiral, A. Debuigne, C. Detrembleur, R. Poli and B. Ameduri, Macromolecules, 2019, 52, 1266–1276 CrossRef.
- M. Guerre, M. Semsarilar, C. Totée, G. Silly, B. Améduri and V. Ladmiral, Polym. Chem., 2017, 5203–5211 RSC.
- M. Guerre, B. Ameduri and V. Ladmiral, Polym. Chem., 2016, 7, 441–450 RSC.
- G. Lopez, M. Guerre, J. Schmidt, Y. Talmon, V. Ladmiral, J.-P. Habas and B. Améduri, Polym. Chem., 2016, 7, 402–409 RSC.
- E. Folgado, M. Guerre, A. Da Costa, A. Ferri, A. Addad, V. Ladmiral and M. Semsarilar, Polym. Chem., 2020, 11, 401–410 RSC.
- G. Kostov, F. Boschet, J. Buller, L. Badache, S. Brandsadter and B. Ameduri, Macromolecules, 2011, 44, 1841–1855 CrossRef CAS.
- E. Girard, J. D. Marty, B. Ameduri and M. Destarac, ACS Macro Lett., 2012, 1, 270–274 CrossRef CAS.
- M. Guerre, M. Uchiyama, E. Folgado, M. Semsarilar, B. Améduri, K. Satoh, M. Kamigaito and V. Ladmiral, ACS Macro Lett., 2017, 6, 393–398 CrossRef CAS.
- M. Guerre, M. Semsarilar, F. Godiard, B. Améduri and V. Ladmiral, Polym. Chem., 2017, 8, 1477–1487 RSC.
- M. Guerre, J. Schmidt, Y. Talmon, B. Améduri and V. Ladmiral, Polym. Chem., 2017, 8, 1125–1128 RSC.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- J. J. Crassous, P. Schurtenberger, M. Ballauff and A. M. Mihut, Polymer, 2015, 62, A1–A13 CrossRef CAS.
- M. C. Arno, M. Inam, Z. Coe, G. Cambridge, L. J. Macdougall, R. Keogh, A. P. Dove and R. K. O'Reilly, J. Am. Chem. Soc., 2017, 139, 16980–16985 CrossRef CAS.
- Z. Li, Y. Zhang, L. Wu, W. Yu, T. R. Wilks, A. P. Dove, H. M. Ding, R. K. O'Reilly, G. Chen and M. Jiang, ACS Macro Lett., 2019, 8, 596–602 CrossRef CAS.
- H. Qiu, Y. Gao, C. E. Boott, O. E. C. Gould, R. L. Harniman, M. J. Miles, S. E. D. Webb, M. A. Winnik and I. Manners, Science, 2016, 352, 697–702 CrossRef CAS.
- M. S. Hsiao, J. X. Zheng, S. Leng, R. M. Van Horn, R. P. Quirk, E. L. Thomas, H. L. Chen, B. S. Hsiao, L. Rong, B. Lotz and S. Z. D. Cheng, Macromolecules, 2008, 41, 8114–8123 CrossRef CAS.
- H. Qi, T. Zhou, S. Mei, X. Chen and C. Y. Li, ACS Macro Lett., 2016, 5, 651–655 CrossRef CAS.
- L. Yin and M. A. Hillmyer, Macromolecules, 2011, 44, 3021–3028 CrossRef CAS.
- Q. Zhang, Y. He, A. M. Oliver, S. Pearce, R. L. Harniman, G. R. Whittell, Y. Liu, S. Du, J. Leng and I. Manners, Polym. Chem., 2019, 10, 3973–3982 RSC.
- G. Guerin, P. Rupar, G. Molev, I. Manners, H. Jinnai and M. A. Winnik, Macromolecules, 2016, 49, 7004–7014 CrossRef CAS.
- S. F. M. Yusoff, M. S. Hsiao, F. H. Schacher, M. A. Winnik and I. Manners, Macromolecules, 2012, 45, 3883–3891 CrossRef.
- H. Zhou, Y. Lu, Q. Yu, I. Manners and M. A. Winnik, ACS Macro Lett., 2018, 7, 166–171 CrossRef CAS.
- Y. Wu, L. Chen, X. Sun, J. Xu, G. Gu and J. Qian, J. Saudi Chem. Soc., 2017, 21, 713–719 CrossRef CAS.
- C. Lv, Z. Zhang, J. Gao, J. Xue, J. Li, J. Nie, J. Xu and B. Du, Macromolecules, 2018, 51, 10136–10149 CrossRef CAS.
- T. Zeng, D. Yang, H. Li and Y. Gao, Polym. Chem., 2018, 9, 627–636 RSC.
- V. F. Korolovych, A. Erwin, A. Stryutsky, H. Lee, W. T. Heller, V. V. Shevchenko, L. A. Bulavin and V. V. Tsukruk, Macromolecules, 2018, 51, 4923–4937 CrossRef CAS.
- M. Cao, H. Nie, Y. Hou, G. Han and W. Zhang, Polym. Chem., 2019, 10, 403–411 RSC.
- P. Zhao, M. Ni, C. Chen, Z. Zhou, X. Li, C. Li, Y. Xie and J. Fei, Nanoscale, 2019, 11, 7394–7403 RSC.
- P. N. Porous, Y. Li, L. Chu, J. Zhu, H. Wang, S. Xia and W. Chen, Ind. Eng. Chem. Res., 2004, 43, 2643–2649 CrossRef.
- X. Liu, O. Coutelier, S. Harrisson, T. Tassaing, J. Marty and M. Destarac, ACS Macro Lett., 2015, 4, 89–93 CrossRef CAS.
- J. M. Bernal-garcía, A. Guzmán-lópez, A. Cabrales-torres, A. Estrada-baltazar and G. A. Iglesias-silva, J. Chem. Eng. Data, 2008, 53, 1024–1027 CrossRef.
- C. Yang, Y. Sun, Y. He and P. Ma, J. Chem. Eng. Data, 2008, 53, 293–297 CrossRef CAS.
- Acetone-Water Mixture Viscosity: Datasheet from “Dortmund Data Bank (DDB) – Thermophysical Properties Edition 2014”, ed. J. Gmehling, SpringerMaterials, 2014, https://materials.springer.com/thermophysical/docs/vism_c4c174 Search PubMed.
- S. Sistach, M. Beija, V. Rahal, A. Brûlet, J. D. Marty, M. Destarac and C. Mingotaud, Chem. Mater., 2010, 22, 3712–3724 CrossRef CAS.
- A. Bottino, G. Capannelli, S. Munari and A. Turturro, J. Polym. Sci., Part B: Polym. Phys., 1988, 26, 785–794 CrossRef CAS.
- M. Inam, G. Cambridge, A. Pitto-Barry, Z. P. L. Laker, N. R. Wilson, R. T. Mathers, A. P. Dove and R. K. O'Reilly, Chem. Sci., 2017, 8, 4223–4230 RSC.
- P. W. Zhu and D. H. Napper, Chem. Phys. Lett., 1996, 256, 51–56 CrossRef CAS.
- A. M. Mihut, J. J. Crassous, H. Schmalz, M. Drechsler and M. Ballauff, Soft Matter, 2012, 8, 3163–3173 RSC.
- W. Li, Q. Meng, Y. Zheng, Z. Zhang, W. Xia and Z. Xu, Appl. Phys. Lett., 2010, 96, 192905 CrossRef.
- P. Martins, A. C. Lopes and S. Lanceros-Mendez, Prog. Polym. Sci., 2014, 39, 683–706 CrossRef CAS.
- B. E. El Mohajir and N. Heymans, Polymer, 2001, 42, 7017–7023 CrossRef CAS.
- L. Yu and P. Cebe, J. Polym. Sci., Part B: Polym. Phys., 2009, 47, 2520–2532 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0py01193b |
| This journal is © The Royal Society of Chemistry 2021 |