
Large-scale investigation of the effects of nucleobase sequence on fluorescence excitation and Stokes shifts of DNA-stabilized silver clusters†
Stacy M.
Copp
 *abc and
Anna
Gonzàlez-Rosell
a
*abc and
Anna
Gonzàlez-Rosell
a
aDepartment of Materials Science and Engineering, University of California, Irvine, Irvine, CA 92697-2585, USA. E-mail: stacy.copp@uci.edu
bDepartment of Physics and Astronomy, University of California, Irvine, Irvine, CA 92697-4575, USA
cDepartment of Chemical and Biomolecular Engineering, University of California, Irvine, Irvine, CA 92697-2580, USA
First published on 16th February 2021
Abstract
DNA-stabilized silver clusters (AgN-DNAs) exhibit diverse sequence-programmed fluorescence, making these tunable nanoclusters promising sensors and bioimaging probes. Recent advances in the understanding of AgN-DNA structures and optical properties have largely relied on detailed characterization of single species isolated by chromatography. Because most AgN-DNAs are unstable under chromatography, such studies do not fully capture the diversity of these clusters. As an alternative method, we use high-throughput synthesis and spectroscopy to measure steady state Stokes shifts of hundreds of AgN-DNAs. Steady state Stokes shift is of interest because its magnitude is determined by energy relaxation processes which may be sensitive to specific cluster geometry, attachment to the DNA template, and structural engagement of solvent molecules. We identify 305 AgN-DNA samples with single-peaked emission and excitation spectra, a characteristic of pure solutions and single emitters, which thus likely contain a dominant emissive AgN-DNA species. Steady state Stokes shifts of these samples vary widely, are in agreement with values reported for purified clusters, and are several times larger than for typical organic dyes. We then examine how DNA sequence selects AgN-DNA excitation energies and Stokes shifts, comment on possible mechanisms for energy relaxation processes in AgN-DNAs, and discuss how differences in AgN-DNA structure and DNA conformation may result in the wide distribution of optical properties observed here. These results may aid computational studies seeking to understand the fluorescence process in AgN-DNAs and the relations of this process to AgN-DNA structure.
1. Introduction
Certain DNA oligomers can serve as stabilizing ligands for fluorescent silver clusters1 (AgN-DNAs), whose structures and optical properties are strongly tuned by DNA sequence.2–4 Researchers have collectively reported on AgN-DNAs stabilized by thousands of different DNA strands, representing a diverse palette of fluorescence properties: emission spectra peaked at 450 nm to 1000 nm,5,6 quantum yields from 3% to 93%,7–13 Stokes shifts up to 0.73 eV (5900 cm−1),14 and light-up or color-switching behavior induced by stimuli.15–18 These diverse and sequence-tunable fluorescent AgN-DNAs are promising for applications ranging from sensing19–22 and molecular logic schemes23 to background-free fluorescence microscopy24–26 and nanophotonics.10,27,28 Recent breakthroughs are rapidly advancing our understanding of the structures of certain AgN-DNAs,5,7,14,29–31, largely due to detailed studies of about 20 different AgN-DNA species purified by high performance liquid chromatography (HPLC).32 Purification methods are critical to ensuring interrogation of a single cluster species rather than the heterogenous mixture of fluorescent and nonfluorescent products formed by AgN-DNA synthesis.33 However, because a substantial fraction of AgN-DNAs are unstable under chromatographic separation,3 it remains to be determined whether the purified AgN-DNAs are representative of the entire palette of possible AgN-DNAs. An alternative examination of these clusters which does not select for stability under chromatography may shed light on this issue.Detailed compositions of 14 purified fluorescent AgN-DNAs have been resolved by high resolution mass spectrometry (HR-MS), including numbers of DNA template strands and total silver content (Ntot), which can be separated into neutral and cationic silver content (N0 and N+, respectively).7 (Unpurified AgN-DNAs have also been characterized by HR-MS.8,34–36) For purified samples, total silver content ranges from Ntot = 10 to Ntot = 30 Ag atoms.6,7,29,32 We note that Ntot cannot a priori be assumed to represent cluster size N: recent crystallographic studies find a minority of Ag atoms are attached to the DNA template but unattached to the silver cluster.5,14,31 For other ligand-stabilized noble metal clusters, metal atoms within the cluster core are neutral in character, while metal atoms bonded to the ligands are cationic in character.37 For fluorescent AgN-DNAs analyzed by HR-MS, just 36% to 57% of Ntot are in a neutral state while the remaining silvers are cationic. Nonfluorescent silver clusters have also been found, containing Ntot = 5 to Ntot = 22, with N0 = 2 to N0 = 10.7,29 In both fluorescent and non-fluorescent clusters, a fraction of the cationic silvers, N+, are likely a part of the silver cluster and mediate attachment of the cluster core to the nucleobases, while other Ag+ may be unattached to the DNA template but unattached to the silver cluster itself.
Consistent with other ligand-stabilized metal clusters,37,38 peak excitation wavelength of fluorescent AgN-DNAs scales with the number of neutral silvers, N0, supporting the idea that these silvers form a neutral cluster core.28,29 Compared to monolayer-protected metal clusters, which are largely globular in shape,39 peak excitation wavelengths and extinction coefficients of AgN-DNAs scale more strongly with N0.7,28 Magic number values of N0 for AgN-DNAs29 also differ markedly from globular clusters. These characteristics support a model for HPLC-purified AgN-DNAs whereby DNA imposes a rod-like cluster geometry onto the AgN core.7,8,29,30,40,41 This model was recently verified by the first X-ray crystal structures of purified AgN-DNAs.14,31,42 A prior crystal structure of an Ag8-DNA formed by in situ crystallization is also elongated to a lesser degree,5 agreeing with our prediction that smaller AgN-DNAs have lower aspect ratios.28
While understanding of AgN-DNA structure is rapidly improving for HPLC-purifiable species, less is known about AgN-DNA photophysics. AgN-DNAs luminesce by a fluorescence-like process, with 1–4 ns emission lifetimes and quantum yields >0.1 for most purified AgN-DNAs.7–11 (This contrasts with the ∼μs emission lifetimes and lower quantum yields of many phosphorescent metal clusters.43,44) However, the AgN-DNA luminescence process differs from the simple Jablonski diagram of organic fluorophores.45,46 Excitation and emission spectra of pure AgN-DNAs exhibit single dominant peaks without the vibronic shoulders characteristic of organic fluorophores.3 The broad spectral line widths of AgN-DNAs cooled to below 2 K and the linear scaling of extinction coefficients with N0 could be consistent with an initial collective electronic excitation leading to fluorescence.28,47–49 Ultrafast studies of a purified Ag20-DNA discovered a sub-100 fs relaxation from an initial excited state to a lower energy state from which fluorescence occurs.50 Such ultrafast relaxation, which accounts for most of the Stokes shift in AgN-DNAs,9,50 is also reported for impure AgN-DNAs.51,52 This relaxation is too rapid for typical vibrational relaxation53 but could instead result from dephasing of a collective electronic excitation47 or perhaps unusually rapid vibrational relaxation.
Studies of purified AgN-DNAs also suggest that DNA scaffold conformation and local dielectric environment add diversity and complexity to excited state behavior of AgN-DNAs. Purified AgN-DNAs exhibit solvatochromic behavior that is not described by the simple Onsager-based models typically used for organic fluorophores but instead depends on the specific DNA template, and, potentially, its engagement with local solvent environment.46 Time-resolved and polarization-resolved studies of single AgN-DNAs within a polymer film find that AgN-DNA spectral properties are sensitive to slight variations in local environment.25,41,54 In one case, removing a terminal adenosine from the DNA template of an Ag16-DNA increases Stokes shift while leaving cluster geometry, peak absorbance energy, and quantum yield essentially unchanged.14 Some AgN-DNAs exhibit temperature-dependent excited state relaxation9,14 while others do not.13 Together, these studies paint a complex picture of the range of sequence-dependent excited state behaviors, even for the subset of AgN-DNAs which survive purification.
Because most AgN-DNAs are unstable under HPLC,3 a large-scale investigation is needed to more fully probe the spectral palette of AgN-DNAs and determine whether the few well-studied purified AgN-DNAs are generally representative of these fluorophores. We previously reported high-throughput studies of 103 different AgN-DNA emission spectra, uncovering the magic numbers of AgN-DNAs29 and learning how DNA sequence selects fluorescence emission.55–57 These studies also illustrated the challenges of high-throughput AgN-DNA characterization: ∼25% of 10-base DNA strands can stabilize multiple fluorescent AgN-DNA species with distinct emission peaks.55 Here, we develop a method to rapidly screen steady state fluorescence excitation and emission spectra of AgN-DNAs. Motivated by studies showing that purified AgN-DNAs and single AgN-DNA emitters exhibit single excitation and emission peaks in the visible to near-infrared (NIR) spectrum,28,30,32,47,58 we identify solutions likely to contain one dominant fluorescent AgN-DNA species by using spectral purity as an alternative to harsh purification methods. Performing AgN-DNA synthesis and steady-state fluorimetry identically on 1880 different 10-base DNA oligomers, we find 305 samples which exhibit single excitation and emission peaks. Stokes shifts of these “spectrally pure” samples vary widely, roughly increasing with peak excitation and emission energies, and generally agree with values for purified AgN-DNAs. Stokes shifts are several times larger than for typical organic fluorophores, despite comparable quantum yields of organic fluorophores and AgN-DNAs.7–11,25 Examining the distribution of Stokes shift versus excitation energy, we observe two separate groupings. We hypothesize that these groupings, which appear correlated with the magic cluster sizes of AgN-DNAs,29 arise from structural differences, apart from size only, between smaller, green-emissive AgN-DNAs and larger, red- or NIR-emissive AgN-DNAs, resulting in differences in excited state relaxation. Correlations of DNA sequence with AgN-DNA excitation energy and Stokes shift suggest that the primary role of nucleobase sequence is to select silver cluster size and excitation spectrum; more subtle sequence patterns may vary the magnitude of Stokes shift for clusters of equal size by varying cluster geometry. Finally, we observe 180 AgN-DNA species with single emission peaks but two or more excitation peaks, which may arise from variations in silver cluster geometry, pointing to the diversity of the full palette of possible AgN-DNAs.
2. Methods
2.1 Silver cluster synthesis
The 1880 10-base DNA oligomers studied here were subjects of previous studies.29,55–57 A robotic liquid handler was used to perform parallel AgN-DNA synthesis in 384 well microplates (described in ESI† and past work29,56). To summarize, an aqueous solution of AgNO3 and NH4OAc, pH 7, is mixed via pipetting with an aqueous solution of DNA (Integrated DNA Technologies, standard desalting). After 18 minutes, silver-DNA solutions are reduced by a freshly prepared solution of NaBH4 in H2O. Final stoichiometries (20 μM DNA, 10 mM NH4OAc, 100 μM AgNO3, and 50 μM NaBH4) were selected to maximize the number of brightly fluorescent wells and the range of fluorescence colors for 10-base DNA oligomers.55 Well plates were stored in the dark at 4 °C until measurement 7 days after synthesis. Thus, measured products in this study are time-stable to at least one week.2.2 Spectroscopy and spectral analysis
Emission and excitation spectra were collected with a Tecan Infinite M200 PRO, whose software corrects for lamp profile and detector spectral responsivity. First, 280 nm light was used to universally excite all AgN-DNA species.59 Emission spectra were collected from 400 nm to 800 nm (detector sensitivity is significantly limited above ∼800 nm). A custom routine in Igor Pro (Wavemetrics Inc.) identified wells with single dominant fluorescent products, specifically with emission spectra well-fitted to single Gaussians as a function of energy, with peak energy Eem and full width at half maximum < 0.5 eV (threshold chosen by investigating previously reported linewidths of HPLC-purified AgN-DNA solutions7). For samples satisfying these criteria, excitation spectra were collected by monitoring emission at the fitted peak emission wavelength, λem = hc/Eem, while scanning excitation from 230 nm to (λem − 40 nm). The portion of each excitation spectrum above 330 nm (Region 1, Fig. 1) was fitted as a function of energy to a single Gaussian to determine peak excitation energy Eex. The ultraviolet (UV) portion < 306 nm (Region 2, Fig. 1), corresponding to cluster excitation via the DNA,59 was fitted to another single Gaussian. After numerically screening the numbers of peaks and sizes of standard fitting errors to flag outliers, each fit was examined by eye to determine if spectra are truly single Gaussians. For samples whose excitation and emission spectra both exhibit single Gaussian peaks in Region 1, Stokes shift is calculated as SS = Eex − Eem. To compare the relative fluorescence excitation efficiencies in Regions 1 and 2 (Fig. 1), we define the ratio of these peak areas, RUV/vis = (area of Region 2 peak)/(area of Region 1 peak), where each peak area is calculated as the product of fitted peak height and fitted peak full width at half maximum. Data is listed in Table S2.† | ||
| Fig. 1 Prototypical excitation (dashed black line) and emission (solid black line) spectra for an AgN-DNA with single excitation and emission peaks. The emission spectrum is fitted to a single Gaussian (red shading) to determine peak emission energy, Eem. The excitation spectrum is fitted to two single Gaussian peaks, one in the visible-NIR region at fitted peak energy, Eex (green shaded fit in Region 1), and one in the UV (purple shaded fit in Region 2). Signal between 306–330 nm is excluded due to stray light in some regions of the well plate from an imperfection in the plate reader. Stokes shift, SS, is calculated as SS = Eex − Eem. The ratio of excitation peak areas in Regions 1 and 2 is defined as RUV/vis = (area of green Region 2 peak)/(area of purple Region 1 peak). Fig. S7† shows additional excitation spectra of samples excluded from analysis due to multiple peaks in excitation or emission spectra. | ||
3. Results and discussion
AgN-DNAs are well-suited for rapid fluorescence emission spectroscopy due to the universal UV excitation of these clusters via the nucleobases. UV excitation of solutions of a single fluorescent AgN-DNA species (excitation in Region 2, Fig. 1) produces emission spectra of the same line shapes as excitation at the cluster's specific visible-to-NIR excitation peak (green peak in Region 1, Fig. 1).59 We exploit universal UV excitation for high-throughput spectroscopy to identify samples with single-peaked excitation and emission spectra in Region 1 (Fig. 1). We term this characteristic “spectral purity”. HPLC-purified AgN-DNAs with monodisperse sizes are characterized by (i) emission spectra with single Gaussian peaks with < 0.5 eV linewidths in Region 1,7,32 and (ii) excitation spectra with two main peaks: a Gaussian peak corresponding to direct excitation of the silver cluster3 (Fig. 1 Region 1) and a UV peak corresponding to indirect excitation of the cluster via the nucleobases59 (Region 2). Spectral purity in Region 1 can serve as an alternative to harsh chromatographic purification, identifying as-synthesized samples with spectral properties similar to purified AgN-DNA solutions or to single AgN-DNA emitters.47,58 For these spectrally pure AgN-DNA samples, Stokes shift is SS = Eex − Eem, the difference between peak excitation energy Eex in Region 1 and peak emission energy Eem.To identify DNA strands which host single dominant fluorescent species, we scanned UV-excited emission spectra of all 1880 AgN-DNA samples, finding 485 samples with emission spectra well-fitted by a single Gaussian peak of narrow linewidth as a function of energy. Remaining DNA strands either stabilized multiple emissive AgN-DNA species, weakly emissive AgN-DNAs near or below the detector's signal-to-noise ratio, or no detectable fluorescent AgN-DNAs (this study cannot comment on the many AgN-DNAs luminescent above ∼800 nm,6,60 the effective upper limit of our detector). We then collected excitation spectra in Regions 1 and 2 for the 485 DNA strands by monitoring emission signal at each sample's fitted Eem (see Methods). 305 of these exhibit single Gaussian excitation peaks centered at Eex in Region 1. The remaining 180 samples with single peaked emission spectra but poorly resolved or multi-peaked excitation spectra are discussed in Section 3.5; because we cannot accurately assign SS in such cases, these samples are excluded from Fig. 2–6. We do not analyze linewidths here because the described method cannot distinguish single AgN-DNAs species with broad linewidths from multiple AgN-DNA species with very closely spaced energies.
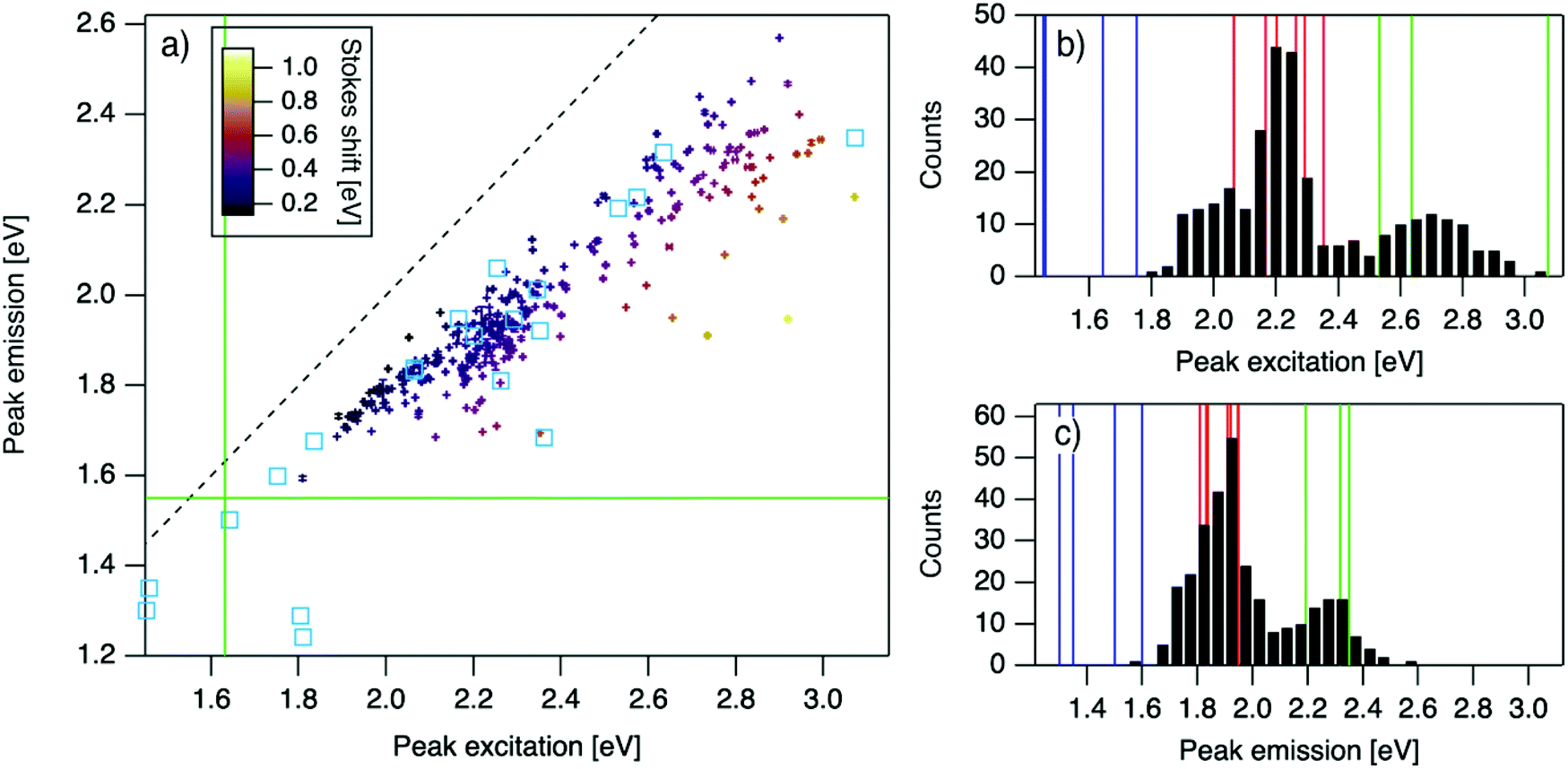 | ||
| Fig. 2 (a) Eemversus Eex for purified AgN-DNAs (cyan squares; data from previous studies,6,7,11,28,29,32) and the spectrally pure AgN-DNAs identified here (data points with error bars; colors correlate to Stokes shift magnitude as indicated by upper left color bar). Error bars represent standard deviations of Gaussian least squares fits. Dashed black line indicates Eem = Eex, (SS = 0). Solid green lines are equivalent to the upper spectral wavelength limits of this study. (b and c) Histograms of (b) Eex and (c) Eem of single-peaked AgN-DNA (black bars). Fourteen colored vertical lines represent (b) Eex and (c) Eem of purified AgN-DNAs with N0 = 4 (green), N0 = 6 (red), and N0 = 10 to 12 (blue), as determined by MS.6,7,28,29 | ||
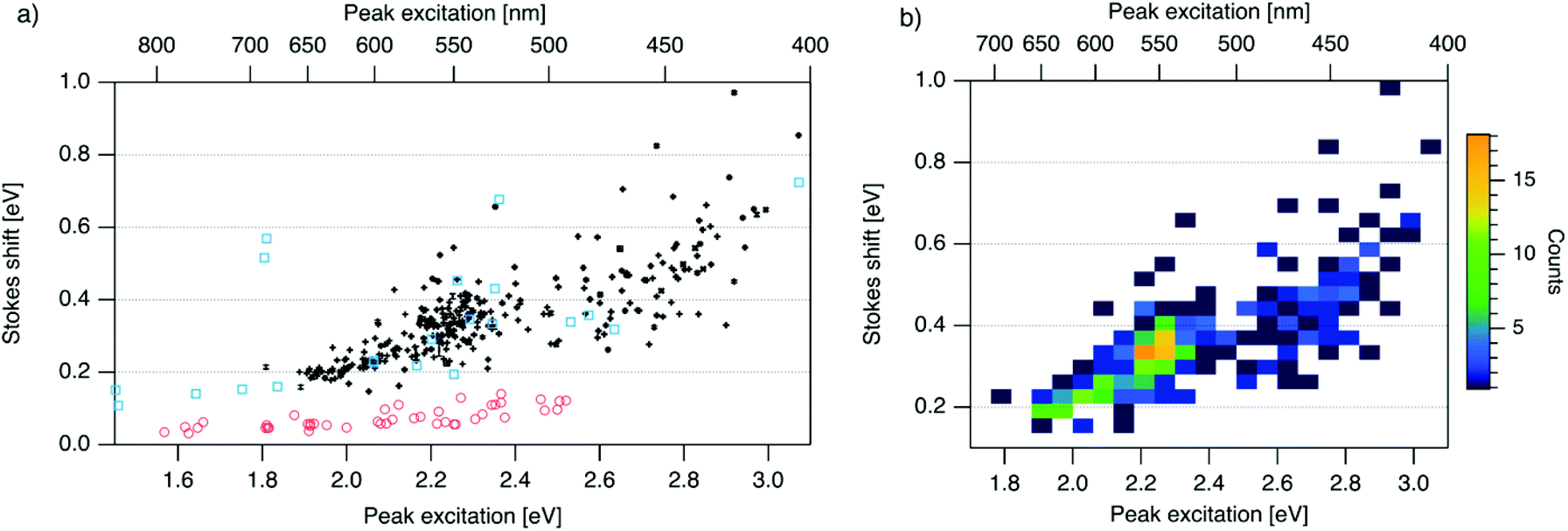 | ||
| Fig. 3 (a) SS versus Eex for spectrally pure solutions of AgN-DNAs (black), previously characterized HPLC-purified AgN-DNAs (cyan squares),6,7,11,28,29,32 and organic fluorophores which are commonly used to label oligonucleotides (red circles).81 Vertical and horizontal error bars of black points represent standard deviations; other markers are larger in size than associated standard deviations. (b) Heatmap of the data for spectrally pure AgN-DNAs from (a) illustrates relative abundance of certain pairs of (Eex, SS) (legend indicates color meaning). | ||
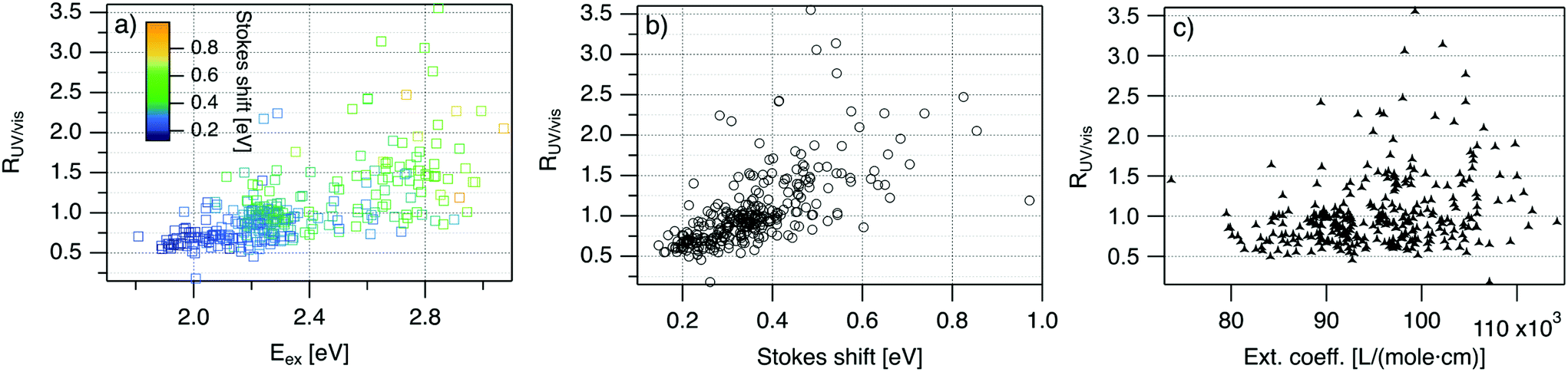 | ||
| Fig. 4 R UV/vis, ratio of areas of UV excitation peak (Region 2) to excitation peak (Region 1), as a function of (a) Eex, (b) SS, and (c) extinction coefficient of the template DNA strand, calculated using the nearest neighbor model.64,65 | ||
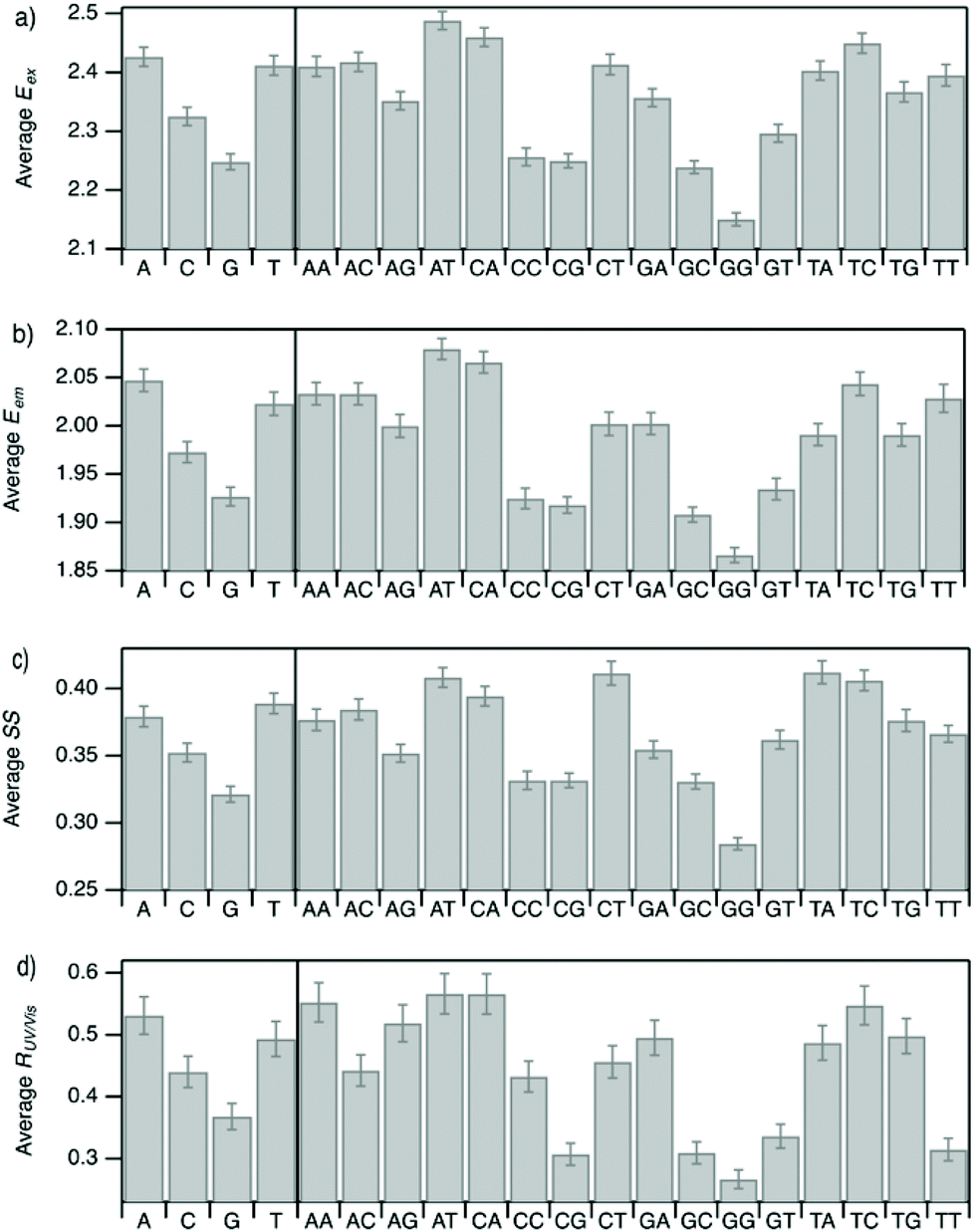 | ||
| Fig. 5 Mean values of (a) Eex, (b) Eem, (c) SS, and (d) RUV/vis for each instance of the four nucleobases and the 16 possible two-base motifs in the set of DNA template sequences for the 305 spectrally pure AgN-DNAs. Error bars represent standard error, as a measure of precision of the mean. | ||
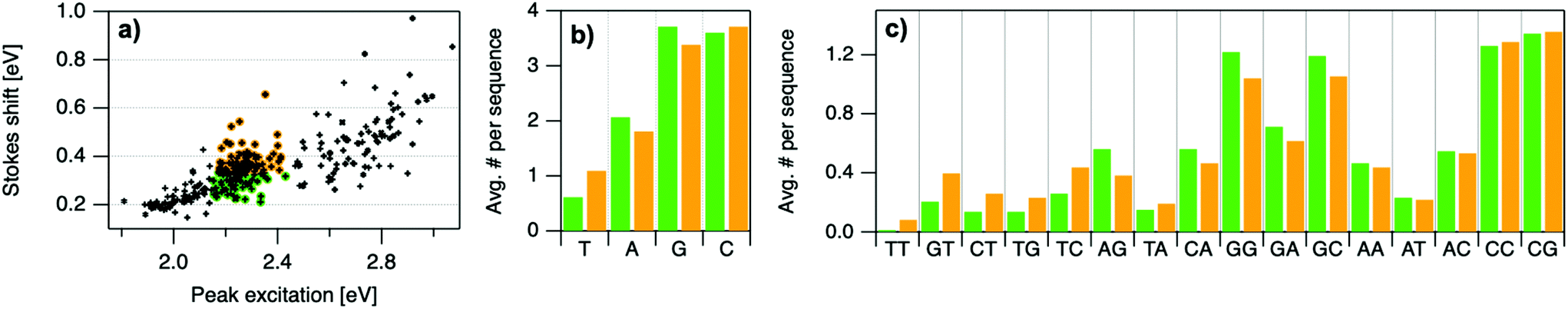 | ||
| Fig. 6 (a) Indication of low SS (green) and high SS (orange) categories, separated at the median value of SS = 0.336 eV. b, (c) Motifs are sorted left to right in order of greatest relative difference between orange and green bars. | ||
3.1 Stokes shifts of spectrally pure AgN-DNAs
For the 305 spectrally pure samples which may be reasonably assigned to a single AgN-DNA species, Eem scales roughly linearly with Eex (Fig. 2a). Such scaling is also characteristic of fluorescent organic dyes.61 The distribution of (Eex, Eem) values for spectrally pure samples are comparable to those reported for purified AgN-DNAs (cyan squares, Fig. 2a).The multimodal histograms of Eex and Eem (Fig. 2b and c) are expected due to the enhanced stabilities of certain AgN-DNAs with “magic numbers” of neutral silver atoms, N0.29 This magic number behavior has been shown to result in a bimodal distribution of Eem in the visible spectrum, which agrees with Fig. 2c. Fig. 2b shows that the distribution of Eex, which has not previously been reported for 102 AgN-DNAs, is also multi-peaked. Comparison of the peaks in Fig. 2b with measured Eex for magic number AgN-DNAs (colored lines, Fig. 2b) suggests that, just as for peak emission, magic sizes of AgN-DNAs lead to enhanced abundances of certain excitation energies due to the strong correlation reported between N0 and Eex.7,28–30 Two of the purified AgN-DNAs with Eex and Eem presented in Fig. 2b and c are stabilized by 10-base DNA strands within the 1880 studied in high throughput here. One of these, a red-emissive AgN-DNA with N0 = 6, was identified as spectrally pure in our high-throughput studies. The second, a green-emissive AgN-DNA with N0 = 4, exhibits a small secondary green emission peak (this secondary product may be removed by HPLC) and was therefore excluded from the set of spectrally pure AgN-DNAs.29 HR-MS studies of additional AgN-DNAs may further explain the distribution in Fig. 2b, especially in the 1.9–2.4 eV range where multiple peaks may suggest distinct cluster sizes and/or geometries.
To investigate the energy relaxation process in AgN-DNAs, we examine the distribution of steady-state Stokes shifts, SS, of the 305 spectrally pure AgN-DNAs. Past high-throughput studies of AgN-DNAs focused solely on Eem,29 but it is difficult to discern excited state behavior from Eem = Eex − SS because Eex and SS are determined by distinct processes. AgN-DNA composition and ground state geometry determine Eex (or, rather, a manifold of excitation energies) required to promote the silver cluster to an initial excited state. This excited state then relaxes to a lower-energy excited state from which photoemission occurs. The energy lost between photoexcitation and photoemission determines SS. Thus, to decouple consideration of the initial excitation process from the energetic relaxation process(es), we plot SS as a function of Eex (Fig. 3a). SS grows roughly linearly with Eex, but SS values vary significantly for a given Eex. This variance represents 10–25% of excitation energy at Eex = 2.3 eV (spectral region corresponding to magic number N0 = 6) and 10–30% at Eex = 2.7 eV (corresponding to magic number N0 = 4) (Fig. S1†). While SS variance is lesser for lower values of Eex, we cannot rule out the possibility that this may be caused by reduced spectral sensitivity of the plate reader above 800 nm.
In all spectral regions, AgN-DNAs exhibit SS values which are several times larger than typical organic fluorophores (Table S1†), and SS also grows about four times faster with increasing Eex for Ag-DNAs than for the organic fluorophores (red circles, Fig. 3a, and Fig. S2†). This behavior is remarkable considering the comparably high quantum yields of AgN-DNAs and organic dyes. General agreement between (Eex, SS) values for HPLC-purified AgN-DNAs (cyan squares) and spectrally pure AgN-DNAs suggests that HPLC-purified AgN-DNAs are generally representative of excitation and emission properties of AgN-DNAs produced by the chemical synthesis method here.
We note that black and cyan data points at (Eex, SS) ≈ (2.35 eV, 0.65 eV) represent the same DNA strand, 5′-CACCTAGCGA-3′, which stabilizes an AgN-DNA with exceptionally high SS, whose crystal structure was reported by Cerretani, et al.11,31,42 Second, most of the spectrally pure AgN-DNAs were designed using machine learning methods.55–57 The subset of data for DNA template strands with randomly selected DNA sequences is shown in Fig. S3.† This data generally agrees with the trend in Fig. 3a, supporting that the designed AgN-DNAs do not alter the (Eex, SS) distribution. Fig. S4† displays a histogram of SS for spectrally pure AgN-DNAs with overlaid SS values for purified AgN-DNAs sized by HR-MS.6,7,28,29N0 and N+ values for each of the 14 purified AgN-DNAs are indicated. While SS appears to increase with increasing N+ for AgN-DNAs with N0 = 6, this trend is not preserved at other N0. With few HR-MS data available, more studies are needed to determine how N0 and N+ influence SS.
Because many data points overlap in Fig. 3a, a heat map in Fig. 3b quantifies abundance of (Eex, SS) values, showing that data form two distinct groupings below and above Eex = 2.45 eV. These two populations align with Eex values of magic number sizes of HPLC-purified AgN-DNAs: Eex > 2.5 eV corresponds to N0 = 4 neutral Ag atoms, and 2.0 eV < Eex < 2.4 eV corresponds to N0 = 6 (Fig. 2b, vertical colored lines).28,29 It is reasonable to hypothesize that energy relaxation following initial excitation would differ somewhat for distinct sizes of clusters due to differences in energy loss mechanisms. Crystal structures of AgN-DNAs, while few, suggest that a structural difference between N0 = 4 and N0 ≥ 6 AgN-DNAs exists, beyond size only. Huard, et al., found an elongated, planar structure for an Ag8-DNA with 450 nm maximum absorption.5N0 is unknown for this Ag8-DNA, but it is spectrally similar to N0 = 4 AgN-DNAs, and a similar planar structure was suggested by a computational study for a 4-electron cluster.62 Cerretani, et al., reported cylindrical structures for several Ag16-DNAs.14,31,42 These Ag16-DNAs also lack assigned N0 but have Eex and Eem consistent with N0 ≥ 6 AgN-DNAs. Combined with the trend of SS versus Eex of the 305 spectrally pure AgN-DNAs, these few reported crystal structures could support that the silver cluster structure undergoes a planar-to-cylindrical transition between N0 = 4 and N0 = 6. Such a structural transition would change the number of nearest-neighbor bonds formed between Ag atoms in the cluster, possibly resulting in different energy relaxation for these two structures. Future studies are needed to test this hypothesis.
3.2 Comparison of direct and indirect AgN-DNA excitation
In addition to direct excitation of the silver cluster at the visible/NIR excitation peak Eex which depends on cluster structure,3 the same AgN-DNA fluorescence can be excited indirectly via the nucleobases, which absorb most efficiently at 260–280 nm.59 This UV excitation enables rapid emission spectroscopy of AgN-DNA29 but remains poorly understood. For purified fluorescent AgN-DNAs, direct and indirect excitation produce emission spectra with identical shapes and linewidths.59 One study reported ultrafast energy transfer from DNA bases to AgN-DNAs following UV-excitation,63 but the authors are not aware of studies directly comparing lifetimes or quantum yields of direct versus indirect excitation of AgN-DNA fluorescence.To investigate the UV-excited fluorescence process of AgN-DNAs, we compare the excitation efficiencies of indirect and direct excitation. We use the ratio of indirect to direct excitation peak areas, RUV/vis, as a relative metric of excitation efficiency. The absolute excitation peak area, which is determined by a combination of extinction coefficient, fluorescence quantum yield, and chemical yield of AgN-DNA synthesis, cannot be used to compare multiple samples because an AgN-DNA species produced in high synthesis yield but with low extinction coefficient and low quantum yield is indistinguishable from an AgN-DNA species produced in low synthesis yield with high extinction coefficient and high quantum yield. Instead, a relative comparison of excitation peaks in Regions 1 and 2 (Fig. 1) using the metric RUV/vis allows us to decouple chemical yield from other factors determining excitation efficiency.
Fig. 4 displays RUV/vis as a function of Eex, SS, and extinction coefficient. On average, RUV/vis increases with increasing Eex, corresponding to greater UV excitation efficiency of smaller clusters with larger Eex than of larger clusters with lower Eex (Fig. 4a). RUV/vis also generally increases as SS increases (Fig. 4b); this is expected due to the trend of SS versus Eex in Fig. 3, but RUV/vis does trend more strongly with SS than with Eex. RUV/vis does not depend strongly on extinction coefficient of the DNA template strand, which is a function of nucleobase content (Fig. 4c, calculated using the nearest neighbor model.64,65). Thus, trends in Fig. 4 are not only due to how well the DNA template absorbs UV photons but also to the properties of silver clusters themselves.
The correlation of RUV/vis with Eex can be rationalized by two factors. First, AgN-DNA cluster core size, N0, increases as Eex decreases.29 Because visible-NIR extinction coefficients of AgN-DNAs scale linearly with N0,28 visible-NIR excitation peak area will increase as Eex decreases, causing RUV/vis to decrease as Eex decreases. Second, UV excitation may be more efficient for smaller, greener AgN-DNAs than for larger, redder clusters due to increased overlap of small clusters’ energy levels with the nucleobase energy levels. More detailed studies, such as time-resolved infrared spectroscopy to monitor nucleobase excitations,66 could investigate whether one or both of these factors determine the behavior in Fig. 4a.
The correlation of RUV/vis with SS is, in large part, due to the linear correlation of SS with Eex (Fig. 3a). We speculate that the slightly stronger correlation of RUV/vis with SS than with Eex may be caused by stronger coupling of smaller, greener clusters with the DNA, resulting in greater energy relaxation relative to larger clusters. Significant spread of RUV/vis values for a single SS or Eex value may arise from variations in cluster attachment to the DNA, e.g., the strength of coupling of the cluster to the nucleobases. Further studies are needed to determine the origin of this spread.
3.3 Role of base sequence in selecting AgN-DNA optical properties
Improved understanding of the sequence-dependence of AgN-DNAs can enable rational design of fluorophores with custom properties. We previously applied data mining and machine learning to a data library of 103 DNA sequences to uncover nucleobase patterns (“motifs”) correlated with the value of Eem.56 When combined with machine learning, these motifs are effective building blocks for designing new AgN-DNAs with desired values of Eem.56,57 To also investigate how DNA sequence selects Eex, SS, and RUV/vis, we analyze correlations of DNA base motifs to values of Eex, Eem, SS, and RUV/vis. (305 spectrally pure AgN-DNAs is rather few for machine learning, so we examine statistical correlations of base patterns with AgN-DNA optical properties.)Fig. 5 displays mean values of Eex, Eem, SS, and RUV/vis for single nucleobases (A, C, G, T) and two-base motifs in the 305 spectrally pure AgN-DNA template sequences (means are weighted by number of occurrences of each base pattern in a given DNA sequence). Fig. S5† also displays means for three-base motifs. Correlations of DNA sequence with Eem (Fig. 5b) agree with previous findings56,57 that A-rich motifs are correlated with greener emission (higher energies) and consecutive G's are strongly correlated with redder emission (lower energies). This agreement is expected because the 305 spectrally pure AgN-DNAs were included in those previous studies. The association of smaller, greener clusters with A-rich motifs and larger, redder clusters with G-rich motifs has been suggested to arise from varying affinities of silver cations for different nucleobases.67
We observe that relative trends of mean Eex, Eem, SS, and RUV/vis values in Fig. 5 are similar to one another. (Because Eem is a linear combination of Eex and SS, this similarity is expected for Eem.) In Fig. 5a, c, and d, nucleobases A and T are associated with higher values of Eex, SS, and RUV/vis, while G is associated with lower values of each of these parameters. Trends for two-base motifs are also similar among all parameters, with some distinct differences for RUV/vis. This trend is expected given roughly linear correlations of SS and RUV/vis with Eex (Fig. 2a, 3a and 4a) and shows that DNA sequence patterns which select for the excitation energy Eex from ground state to initial unrelaxed excited state (Franck Condon state) are also predictive of the magnitude of SS, the energy lost during relaxation following excitation. A lack of obviously distinct sequence patterns which encode SS and Eex could support that energy relaxation is due to a delocalized relaxation across the entire cluster and/or DNA template, as opposed to specific relaxation of one or a few excited nucleobases. Because Eex, SS, and RUV/vis do not appear to be separately tuned by distinct DNA base motifs to a significant degree, designing an AgN-DNA with custom values for each of these optical properties is likely to be a significant challenge, although three-base motifs may enable such prediction (Fig. S5†), which is the subject of ongoing work.
To elucidate if subtler sequence features determine SS irrespective of Eex, we consider the most abundant peak in Fig. 2b: 2.15 eV < Eex < 2.45 eV. HPLC-purified AgN-DNAs in this spectral range have N0 = 6 neutral Ag atoms. The 145 DNA sequences in this peak are separated into two categories above and below their median SS value (Fig. 6a). Average numbers of nucleobase, 2-base, and 3-base patterns are calculated for both categories. Fig. 6b and c display average numbers of bases and 2-base motifs in sequences correlated to higher (orange) and lower (green) SS categories, with base patterns ordered left-to-right in order of the relative difference between average occurrence in the two classes, which is a metric of predictiveness of SS value for each base pattern (3-base patterns in Fig. S6†). For this specific spectral window, inclusion of T bases generally increases SS, while A and G bases tend to reduce SS. Interestingly, A-rich motifs were correlated to higher SS values when considering the entire 305 spectrally pure AgN-DNAs, but their role for selecting SS for a given color window may be more nuanced. C is less predictive of SS, and behavior of 2-base motifs containing C's is generally predicted by A, G, and T content.
The differences in SS behavior of AgN-DNAs stabilized by A- and G-rich strands as compared to T-rich strands are somewhat unexpected if one assumes that strength of silver-nucleobase bonds should control SS magnitude. Thymines are not expected to bind the silver cluster and adenines are expected to bind rather weakly, as compared to strong affinities of cytosine and guanine for silver.67,68 The few crystal structures available for AgN-DNAs show that A can coordinate the silver cluster,69 although a separate study of a DNA-Ag+ complex finds that adenines protrude outward from Ag+-mediated DNA duplexes.5,14,31 T is similarly observed to protrude away from silver clusters.70 While solved AgN-DNA structures are scant and may not be representative of general behavior, we note that differences in SS of AgN-DNAs in Fig. 6 do not appear to be explained simply by total silver-binding power of the DNA template, due to the similar SS values correlated with A- and G-rich motifs. The relaxation dynamics of a more tightly bound cluster would be expected to differ from a weakly bound cluster. Since G likely binds to the cluster more strongly than A, this simple model is unlikely.
Other possible mechanisms for finer variations in SS behavior include cluster structural variations and slight differences in vibrational modes of nucleic acids, which are measured in the range of 0.19 eV to 0.21 eV.14,31 Thymine has the highest frequency modes of the set of natural nucleobases in DNA,71 which could increase SS of T-containing AgN-DNAs if energy relaxation occurs via vibrational modes (discussed below). A and G are both purines with bulkier sizes than T, which may affect energy relaxation of the excited cluster. It is unclear whether the vibrational modes of free nucleobases are representative of silver-bound nucleobases, and more crystal structures must be solved to understand how structure contributes to SS and how the four nucleobases coordinate silver clusters.
3.4 Possible origins of the Stokes shift in AgN-DNAs
By the Franck-Condon principle, SS is determined by processes which follow photoexcitation.71 Possible mechanisms for energy relaxation processes in AgN-DNAs include electronic dephasing, vibrational relaxation, and charge transfer. Measured vibrational modes of nucleic acids, 0.19 eV to 0.21 eV,72 roughly correspond to the lower limit of SS values observed (Fig. 3a), which could support an energetic relaxation process via excitation of one or more vibrational modes in the nucleic acids of the surrounding DNA ligand. However, vibrational modes of nucleic acids bound to silver cations may differ from free nucleic acids. Calculations by Weerawardene and Aikens for thiolate-protected gold clusters found that changes in cluster geometry after excitation, i.e. nuclear rearrangement of Au atoms, lead to electronic structure changes that produce a Stokes shifts, and the magnitude of these geometric changes are correlated to the magnitude of SS.53 Calculations of thiolate-protected Ag clusters also find geometric relaxation following excitation with a magnitude that is smaller for larger clusters, leading to inverse correlation of cluster size and SS73–75 which matches what is observed here for AgN-DNAs.The extremely rapid 10–100 fs energy relaxation observed immediately following AgN-DNA excitation45 may be too fast to be caused solely by geometric relaxation of cluster and/or DNA ligand structure or by charge transfer.50–52 However, ab initio calculations of small atomic chains of Ag atoms found ∼100 fs plasmon dephasing due to certain molecular vibrations in 1-dimensional atomic Ag chains.71 Thus, the unusual ultrafast energy relaxation process in AgN-DNAs may be of mixed character, involving both electronic dephasing and geometric relaxation. A recent study used ultrafast time-resolved infrared spectroscopy to monitor vibrational modes of DNA nucleobases following excitation of two green-emissive AgN-DNAs at their visible excitation wavelengths. Upon excitation, vibrational modes of certain nucleobases were found to bleach.76 Because isolated nucleobases require higher energy (UV) excitation to undergo bleaching, this suggests an intimate and intriguing connection between the silver cluster and its nucleobase ligands. Future studies are needed to understand how electronic dephasing, vibrational relaxation, and/or charge transfer contribute to the origins of Stokes shift in AgN-DNAs. Such studies could shed light on why AgN-DNAs retain high quantum yields and SS values several times larger than organic fluorophores, even into the NIR.66
3.5 Multipeaked excitation spectra: heterogeneous samples or fundamentally different AgN-DNA structures?
Of the 485 AgN-DNA samples which exhibited single-peaked emission spectra, 305 of these also exhibited single-peaked excitation spectra (e.g.Fig. 1) while the remaining 180 samples exhibited excitation spectra with poorly resolved peaks, asymmetric peaks or, for 88 samples, multiple peaks > 350 nm. Fig. S7† illustrates representative examples of these excitation spectra. The most common secondary peak observed was in the near UV between the nucleobase absorption band and the dominant Region 1 peak (Fig. 1); this peak appeared in 63 samples. While it is most convenient to simply dismiss these samples as heterogeneous mixtures of different clusters indiscernible by emission spectra only (which is reasonable for poorly resolved or asymmetric peaks), we cannot rule out the possibility that some multipeaked samples may represent single AgN-DNA species with spectral features which fundamentally differ from the well-studied HPLC-stable AgN-DNAs with single Gaussian excitation peaks in Region 1 (Fig. 1). As described in Methods, all measurements were performed one week after AgN-DNA synthesis to capture only time-stable products. AgN-DNAs evolve in the hours to few days after synthesis but generally stabilize well before one week.13 Thus, it is unlikely that AgN-DNAs undergo significant evolution in size/shape during collection of emission spectra and excitation spectra, which would could cause the appearance of multiple excitation peaks. Another possible cause of AgN-DNAs with single-peaked emission spectra but multipeaked excitation spectra is when a single DNA template strand forms two different AgN-DNA species with equal Eem, one with low SS and one with high SS. The probability of this occurring in such a large fraction of samples is highly unlikely. Finally, larger silver nanoparticles display size-dependent surface plasmon resonances,77 but excitation of surface plasmons in nanoparticle impurities should not induce fluorescence in separate AgN-DNAs that coexist in solution.A final possibility is that certain AgN-DNAs inherently have multipeaked excitation spectra, with a dominant visible-NIR peak and a secondary peak, typically between 3 eV and 3.5 eV. Secondary excitation peaks at 3–3.5 eV have been observed in a few HPLC-purified AgN-DNAs.78 A particularly stable and bright AgN-DNA which emits at 670 nm has a primary excitation peak at 600 nm and a secondary excitation peak at 400 nm.28 Secondary excitation peaks at energies >Eex are consistent with fluorescence arising from an initial collective excitation in AgN-DNAs.10 In this model, the dominant visible-NIR excitation peak corresponds to a longitudinal collective electronic oscillation along the silver cluster rod. In this rod, a transverse collective mode is also expected. Computational studies find that this mode is damped in straight atomic Ag chains,28 but simulations of atomic Ag chains with chiral twist find that as chirality increases, transverse excitation peaks become more prominent.79 We hypothesize that some AgN-DNAs with increased curvatures host more prominent transverse excitation modes which also lead to AgN-DNA fluorescence. AgN-DNAs with higher curvature may not have been observed in large numbers due to lower stabilities under HPLC as compared to straighter AgN-DNAs.
4. Conclusions
We present the first high-throughput study probing Stokes shifts of AgN-DNAs across the diverse spectral range of these nanoclusters. By using spectral purity to identify 305 samples containing a single fluorescent AgN-DNA species, we found that Stokes shifts for these samples vary widely and are several times larger than organic fluorophores, despite the molecular-like high quantum yields of AgN-DNAs.30,80 The distribution of peak excitation energies and Stokes shifts suggests that there may exist distinctions between excited state relaxation processes in AgN-DNAs with N0 = 4 and N0 ≥ 6 neutral Ag atoms. By comparing to crystal structures available for AgN-DNAs,5,14,31,42 we propose that AgN-DNA cluster structure transitions from planar to cylindrical between N0 = 4 and N0 = 6, resulting in different magnitudes of energy relaxation following photoexcitation. Future studies are needed to test this hypothesis.We also found strong correlations between certain DNA sequence patterns and peak excitation energy. Because Stokes shift scales strongly with excitation energy, sequence patterns predictive only of Stokes shift but not of excitation energy are more subtle. This suggests that the primary role of DNA sequence is to select the silver cluster's structure, which plays the dominant role in determining the energy relaxation process following photoexcitation. Finer variations of cluster geometry and/or the cluster's interaction with its DNA template may cause the significant spread of Stokes shifts observed for AgN-DNAs with equal excitation energies. Finally, we observe a significant minority of AgN-DNAs with single-peaked emission spectra yet multiple peaks in excitation spectra, particularly in the near UV. The prevalence of these samples, and the near UV energies of their peaks, could correspond to transverse collective excitation modes of clusters with greater chirality, as predicted by ab initio calculations.5,14,31 We hope that the extensive data set presented here will enable computational studies of fluorescence excitation and emission processes in AgN-DNAs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We dedicate this paper to the essential workers whose heroism and dedication continue to save countless lives during the COVID-19 pandemic. This work was supported by NSF-DMR-1309410 and NSF-CBET-2025790. A.G.R. acknowledges a Balsells Graduate Fellowship. S.M.C. acknowledges use of the Biological Nanostructures Laboratory within the California NanoSystems Institute, supported by the University of California, Santa Barbara and the University of California, Office of the President. The pipetting robot was acquired with support from NIH-NEI 5-R24-EY14799. We are grateful to Elisabeth Gwinn for helpful discussions and to Steven Swasey for synthesis and characterization of some samples studied here.Notes and references
- J. T. Petty, J. Zheng, N.,V. Hud and R. M. Dickson, DNA-templated Ag nanocluster formation, J. Am. Chem. Soc., 2004, 126, 5207–5212 CrossRef CAS.
- E. G. Gwinn, P. O'Neill, A. J. Guerrero, D. Bouwmeester and D. K. Fygenson, Sequence-Dependent Fluorescence of DNA-Hosted Silver Nanoclusters, Adv. Mater., 2008, 20, 279–283 CrossRef CAS.
- E. G. Gwinn, D. Schultz, S. M. Copp and S. M. Swasey, DNA-Protected Silver Clusters for Nanophotonics, Nanomaterials, 2015, 5, 180–207 CrossRef.
- Y. Chen, M. L. Phipps, J. H. Werner, S. Chakraborty and J. S. Martinez, DNA Templated Metal Nanoclusters: From Emergent Properties to Unique Applications, Acc. Chem. Res., 2018, 51, 2756–2763 CrossRef CAS.
- D. J. E. Huard, et al., Atomic Structure of a Fluorescent Ag8 Cluster Templated by a Multistranded DNA Scaffold, J. Am. Chem. Soc., 2019, 141, 11465–11470 CrossRef CAS.
- S. M. Swasey, et al., High throughput near infrared screening discovers DNA-templated silver clusters with peak fluorescence beyond 950 nm, Nanoscale, 2018, 10, 19701–19705 RSC.
- D. Schultz, et al., Evidence for rod-shaped DNA-stabilized silver nanocluster emitters, Adv. Mater., 2013, 25, 2797–2803 CrossRef CAS.
- J. T. Petty, M. Ganguly, I. J. Rankine, D. M. Chevrier and P. Zhang, A DNA-Encapsulated and Fluorescent Ag 106+ Cluster with a Distinct Metal-Like Core, J. Phys. Chem. C, 2017, 121, 14936–14945 CrossRef CAS.
- C. Cerretani, M. R. Carro-Temboury, S. Krause, S. A. Bogh and T. Vosch, Temperature dependent excited state relaxation of a red emitting DNA-templated silver nanocluster, Chem. Commun., 2017, 53, 12556–12559 RSC.
- S. A. Bogh, C. Cerretani, L. Kacenauskaite, M. R. Carro-Temboury and T. Vosch, Excited-State Relaxation and Förster Resonance Energy Transfer in an Organic Fluorophore/Silver Nanocluster Dyad, ACS Omega, 2017, 2, 4657–4664 CrossRef CAS.
- S. A. Bogh, et al., Unusually large Stokes shift for a near-infrared emitting DNA-stabilized silver nanocluster, Methods Appl. Fluoresc., 2018, 6, 024004 CrossRef.
- P. R. O'Neill, L. R. Velazquez, D. G. Dunn, E. G. Gwinn and D. K. Fygenson, Hairpins with Poly-C Loops Stabilize Four Types of Fluorescent Ag n, DNA, 2009, 4229–4233 Search PubMed.
- V. A. Neacşu, et al., Unusually large fluorescence quantum yield for a near-infrared emitting DNA-stabilized silver nanocluster, Chem. Commun., 2020, 56(47), 6384–6387 RSC.
- C. Cerretani, J. Kondo and T. Vosch, Removal of the A10 adenosine in a DNA-stabilized Ag16 nanocluster, RSC Adv., 2020, 10, 23854–23860 RSC.
- H.-C. Yeh, et al., A fluorescence light-up Ag nanocluster probe that discriminates single-nucleotide variants by emission color, J. Am. Chem. Soc., 2012, 134, 11550–11558 CrossRef CAS.
- M. Ganguly, C. Bradsher, P. Goodwin and J. T. Petty, DNA-Directed Fluorescence Switching of Silver Clusters, J. Phys. Chem. C, 2015, 119, 27829–27837 CrossRef CAS.
- C. Cerretani and T. Vosch, Switchable Dual-Emissive DNA-Stabilized Silver Nanoclusters, ACS Omega, 2019, 4, 7895–7902 CrossRef CAS.
- L. E. Yourston and A. V. Krasnoslobodtsev, Micro RNA Sensing with Green Emitting Silver Nanoclusters, Molecules, 2020, 25, 3026 CrossRef CAS.
- S. W. Yang and T. Vosch, Rapid detection of microRNA by a silver nanocluster DNA probe, Anal. Chem., 2011, 83, 6935–6939 CrossRef CAS.
- J. Liu, DNA-stabilized, fluorescent, metal nanoclusters for biosensor development, TrAC, Trends Anal. Chem., 2014, 58, 99–111 CrossRef CAS.
- Y.-A. Chen, et al., NanoCluster Beacons Enable Detection of a Single N(6)-Methyladenine, J. Am. Chem. Soc., 2015, 137, 10476–10479 CrossRef CAS.
- J. T. Del Bonis-O'Donnell, D. Vong, S. Pennathur and D. K. Fygenson, A universal design for a DNA probe providing ratiometric fluorescence detection by generation of silver nanoclusters, Nanoscale, 2016, 8, 14489–14496 RSC.
- Z. Huang, Y. Tao, F. Pu, J. Ren and X. Qu, Versatile logic devices based on programmable DNA-regulated silver-nanocluster signal transducers, Chemistry, 2012, 18, 6663–6669 CrossRef CAS.
- B. C. Fleischer, J. T. Petty, J.-C. Hsiang and R. M. Dickson, Optically Activated Delayed Fluorescence, J. Phys. Chem. Lett., 2017, 8, 3536–3543 CrossRef CAS.
- S. Krause, M. R. Carro-Temboury, C. Cerretani and T. Vosch, Probing heterogeneity of NIR induced secondary fluorescence from DNA-stabilized silver nanoclusters at the single molecule level, Phys. Chem. Chem. Phys., 2018, 20, 16316–16319 RSC.
- S. Krause, C. Cerretani and T. Vosch, Disentangling optically activated delayed fluorescence and upconversion fluorescence in DNA stabilized silver nanoclusters, Chem. Sci., 2019, 10, 5326–5331 RSC.
- D. Schultz, et al., Dual-color nanoscale assemblies of structurally stable, few-atom silver clusters, as reported by fluorescence resonance energy transfer, ACS Nano, 2013, 7, 9798–9807 CrossRef CAS.
- S. M. Copp, D. Schultz, S. M. Swasey, A. Faris and E. G. Gwinn, Cluster Plasmonics: Dielectric and Shape Effects on DNA-Stabilized Silver Clusters, Nano Lett., 2016, 16, 3594–3599 CrossRef CAS.
- S. M. Copp, et al., Magic Numbers in DNA-Stabilized Fluorescent Silver Clusters Lead to Magic Colors, J. Phys. Chem. Lett., 2014, 5, 959–963 CrossRef CAS.
- S. M. Swasey, et al., Chiral Electronic Transitions in Fluorescent Silver Clusters Stabilized by DNA, ACS Nano, 2014, 8, 6883–6892 CrossRef CAS.
- C. Cerretani, H. Kanazawa, T. Vosch and J. Kondo, Crystal structure of a NIR-Emitting DNA-Stabilized Ag16 Nanocluster, Angew. Chem., Int. Ed., 2019, 58, 17153–17157 CrossRef CAS.
- D. Schultz and E. G. Gwinn, Silver atom and strand numbers in fluorescent and dark Ag:DNAs, Chem. Commun., 2012, 48, 5748–5750 RSC.
- M. R. Carro-Temboury, V. Paolucci, E. N. Hooley, L. Latterini and T. Vosch, Probing DNA-stabilized fluorescent silver nanocluster spectral heterogeneity by time-correlated single photon counting, Analyst, 2016, 141, 123–130 RSC.
- K. Koszinowski and K. A. Ballweg, Highly Charged Ag 64+ Core in a DNA-Encapsulated Silver Nanocluster, Chem. – Eur. J., 2010, 16, 3285–3290 CrossRef CAS.
- J. T. Petty, M. Ganguly, I. J. Rankine, D. M. Chevrier and P. Zhang, A DNA-Encapsulated and Fluorescent Ag106+Cluster with a Distinct Metal-Like Core, J. Phys. Chem. C, 2017, 121, 14936–14945 CrossRef CAS.
- J. T. Petty, et al., Repeated and Folded DNA Sequences and Their Modular Ag106+Cluster, J. Phys. Chem. C, 2018, 122, 4670–4680 CrossRef CAS.
- H. Häkkinen, Atomic and electronic structure of gold clusters: understanding flakes, cages and superatoms from simple concepts, Chem. Soc. Rev., 2008, 37, 1847–1859 RSC.
- M. Walter, et al., A unified view of ligand-protected gold clusters as superatom complexes, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9157–9162 CrossRef CAS.
- I. Chakraborty and T. Pradeep, Atomically Precise Clusters of Noble Metals: Emerging Link between Atoms and Nanoparticles, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS.
- N. Markešević, S. S. R. Oemrawsingh, D. Schultz, E. G. Gwinn and D. Bouwmeester, Polarization Resolved Measurements of Individual DNA-Stabilized Silver Clusters, Adv. Opt. Mater., 2014, 2, 765–770 CrossRef.
- E. N. Hooley, M. R. Carro-Temboury and T. Vosch, Probing the Absorption and Emission Transition Dipole Moment of DNA Stabilized Silver Nanoclusters, J. Phys. Chem. A, 2017, 121, 963–968 CrossRef CAS.
- C. Cerretani, J. Kondo and T. Vosch, Mutation of position 5 as a crystal engineering tool for a NIR-Emitting DNA-Stabilized Ag16 Nanocluster, CrystEngComm, 2020, 46, 8136–8141 RSC.
- S. Chakraborty, et al., A Hybrid DNA-Templated Gold Nanocluster for Enhanced Enzymatic Reduction of Oxygen, J. Am. Chem. Soc., 2015, 137, 11678–11687 CrossRef CAS.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Atomically Precise Colloidal Metal Nanoclusters and Nanoparticles: Fundamentals and Opportunities, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, in Principles of Fluorescence Spectroscopy, Springer Science & Business Media, 2006. DOI:10.1007/978-0-387-46312-4.
- S. M. Copp, A. Faris, S. M. Swasey and E. G. Gwinn, Heterogeneous Solvatochromism of Fluorescent DNA-Stabilized Silver Clusters Precludes Use of Simple Onsager-Based Stokes Shift Models, J. Phys. Chem. Lett., 2016, 7, 698–703 CrossRef CAS.
- S. S. R. Oemrawsingh, N. Markešević, E. G. Gwinn, E. R. Eliel and D. Bouwmeester, Spectral Properties of Individual DNA-Hosted Silver Nanoclusters at Low Temperatures, J. Phys. Chem. C, 2012, 116, 25568–25575 CrossRef CAS.
- S. Bernadotte, F. Evers and C. R. Jacob, Plasmons in Molecules, J. Phys. Chem. C, 2013, 117, 1863–1878 CrossRef CAS.
- C. M. Krauter, S. Bernadotte, C. R. Jacob, M. Pernpointner and A. Dreuw, Identification of Plasmons in Molecules with Scaled Ab Initio Approaches, J. Phys. Chem. C, 2015, 119, 24564–24573 CrossRef CAS.
- E. Thyrhaug, et al., Ultrafast coherence transfer in DNA-templated silver nanoclusters, Nat. Commun., 2017, 8, 15577 CrossRef CAS.
- S. H. Yau, et al., Bright two-photon emission and ultra-fast relaxation dynamics in a DNA-templated nanocluster investigated by ultra-fast spectroscopy, Nanoscale, 2012, 4, 4247–4254 RSC.
- S. A. Patel, et al., Electron transfer-induced blinking in Ag nanodot fluorescence, J. Phys. Chem. C. Nanomater. Interfaces, 2009, 113, 20264–20270 CrossRef CAS.
- D. Brinks, et al., Ultrafast dynamics of single molecules, Chem. Soc. Rev., 2014, 43, 2476–2491 RSC.
- E. N. Hooley, V. Paolucci, Z. Liao, M. R. Carro Temboury and T. Vosch, Single-Molecule Characterization of Near-Infrared-Emitting Silver Nanoclusters, Adv. Opt. Mater., 2015, 3, 1109–1115 CrossRef CAS.
- S. M. Copp, P. Bogdanov, M. Debord, A. Singh and E. Gwinn, Base Motif Recognition and Design of DNA Templates for Fluorescent Silver Clusters by Machine Learning, Adv. Mater., 2014, 26, 5839–5845 CrossRef CAS.
- S. M. Copp, et al., Fluorescence Color by Data-Driven Design of Genomic Silver Clusters, ACS Nano, 2018, 12, 8240–8247 CrossRef CAS.
- S. M. Copp, S. M. Swasey, A. Gorovits, P. Bogdanov and E. G. Gwinn, General Approach for Machine Learning-Aided Design of DNA-Stabilized Silver Clusters, Chem. Mater., 2020, 32, 430–437 CrossRef CAS.
- T. Vosch, et al., Strongly emissive individual DNA-encapsulated Ag nanoclusters as single-molecule fluorophores, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 12616–12621 CrossRef CAS.
- P. R. O'Neill, E. G. Gwinn and D. K. Fygenson, UV Excitation of DNA Stabilized Ag Cluster Fluorescence via the DNA Bases, J. Phys. Chem. C, 2011, 115, 24061–24066 CrossRef.
- S. M. Swasey, et al., Adaptation of a visible wavelength fluorescence microplate reader for discovery of near-infrared fluorescent probes, Rev. Sci. Instrum., 2018, 89, 095111 CrossRef.
- B. Valeur and M. N. Berberan-Santos, Molecular Fluorescence: Principles and Applications, Wiley-VCH Verlag & Co., KGaA, 2012 Search PubMed.
- X. Chen, M. Boero and O. Lopez-Acevedo, Atomic structure and origin of chirality of DNA-stabilized silver clusters, Phys. Rev. Mater., 2020, 4, 065601 CrossRef CAS.
- I. L. Volkov, Z. V. Reveguk, P. Y. Serdobintsev, R. R. Ramazanov and A. I. Kononov, DNA as UV light-harvesting antenna, Nucleic Acids Res., 2018, 46, 3543–3551 CrossRef CAS.
- M. M. Warshaw and I. Tinoco, Optical properties of sixteen dinucleoside phosphates, J. Mol. Biol., 1966, 20, 29–38 CrossRef CAS.
- M. J. Cavaluzzi and P. N. Borer, Revised UV extinction coefficients for nucleoside-5′-monophosphates and unpaired DNA and RNA, Nucleic Acids Res., 2004, 32, e13 CrossRef.
- Y. Zhang, C. He, J. T. Petty and B. Kohler, Time-Resolved Vibrational Fingerprints for Two Silver Cluster-DNA Fluorophores, J. Phys. Chem. Lett., 2020, 8958–8963, DOI:10.1021/acs.jpclett.0c02486.
- S. M. Swasey, L. E. Leal, O. Lopez-Acevedo, J. Pavlovich and E. G. Gwinn, Silver(I) as DNA glue: Ag(+)-mediated guanine pairing revealed by removing Watson-Crick constraints, Sci. Rep., 2015, 5, 10163 CrossRef CAS.
- S. M. Swasey and E. G. Gwinn, Silver-mediated base pairings: Towards dynamic DNA nanostructures with enhanced chemical and thermal stability, New J. Phys., 2016, 18, 045008 CrossRef.
- S. M. Swasey, F. Rosu, S. M. Copp, V. Gabelica and E. G. Gwinn, Parallel Guanine Duplex and Cytosine Duplex DNA with Uninterrupted Spines of AgI-Mediated Base Pairs, J. Phys. Chem. Lett., 2018, 9, 6605–6610 CrossRef CAS.
- J. Kondo, et al., A metallo-DNA nanowire with uninterrupted one-dimensional silver array, Nat. Chem., 2017, 9, 956–960 CrossRef CAS.
- C. S. Peng, K. C. Jones and A. Tokmakoff, Anharmonic vibrational modes of nucleic acid bases revealed by 2D IR spectroscopy, J. Am. Chem. Soc., 2011, 133, 15650–15660 CrossRef CAS.
- L. Gell, et al., Tuning Structural and Optical Properties of Thiolate-Protected Silver Clusters by Formation of a Silver Core with Confined Electrons, J. Phys. Chem. C, 2013, 117, 14824–14831 CrossRef CAS.
- K. L. D. M. Weerawardene and C. M. Aikens, Theoretical Insights into the Origin of Photoluminescence of Au25(SR)18- Nanoparticles, J. Am. Chem. Soc., 2016, 138, 11202–11210 CrossRef CAS.
- K. L. D. M. Weerawardene, E. B. Guidez and C. M. Aikens, Photoluminescence Origin of Au 38 (SR) 24 and Au 22 (SR) 18 Nanoparticles: A Theoretical Perspective, J. Phys. Chem. C, 2017, 121, 15416–15423 CrossRef CAS.
- K. L. D. M. Weerawardene and C. M. Aikens, Origin of Photoluminescence of Ag 25 (SR) 18 – Nanoparticles: Ligand and Doping Effect, J. Phys. Chem. C, 2018, 122, 2440–2447 CrossRef CAS.
- G. Donati, D. B. Lingerfelt, C. M. Aikens and X. Li, Molecular Vibration Induced Plasmon Decay, J. Phys. Chem. C, 2017, 121, 15368–15374 CrossRef CAS.
- J. J. Mock, M. Barbic, D. R. Smith, D. A. Schultz and S. Schultz, Shape effects in plasmon resonance of individual colloidal silver nanoparticles, J. Chem. Phys., 2002, 116, 6755–6759 CrossRef CAS.
- V. Amendola, O. M. Bakr and F. Stellacci, A study of the surface plasmon resonance of silver nanoparticles by the discrete dipole approximation method: Effect of shape, size, structure, and assembly, Plasmonics, 2010, 5, 85–97 CrossRef CAS.
- J. Yan and S. Gao, Plasmon resonances in linear atomic chains: Free-electron behavior and anisotropic screening of d electrons, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 235413 CrossRef.
- N.,V. Karimova and C. M. Aikens, Time-Dependent Density Functional Theory Investigation of the Electronic Structure and Chiroptical Properties of Curved and Helical Silver Nanowires, J. Phys. Chem. A, 2015, 119, 8163–8173 CrossRef CAS.
- Integrated DNA Technologies Fluorophores Modifications. Available at: http://www.idtdna.com/site/Catalog/Modifications/Dyes (accessed: 23rd August 2018).
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S7, Tables S1 and S2 and experimental details. See DOI: 10.1039/d0nr08300c |
| This journal is © The Royal Society of Chemistry 2021 |