
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChiral 3D DNA origami structures for ordered heterologous arrays†
Md. Sirajul
Islam
a,
Gerrit David
Wilkens
 ab,
Karol
Wolski
c,
Szczepan
Zapotoczny
c and
Jonathan Gardiner
Heddle
*a
ab,
Karol
Wolski
c,
Szczepan
Zapotoczny
c and
Jonathan Gardiner
Heddle
*a
aMalopolska Centre of Biotechnology, Jagiellonian University, Gronostajowa 7A, Kraków 30-387, Poland. E-mail: jonathan.heddle@uj.edu.pl
bSchool of Molecular Medicine, Medical University of Warsaw, Warszawa 02-091, Poland
cFaculty of Chemistry, Jagiellonian University, Gronostajowa 2, Kraków 30-387, Poland
First published on 5th July 2021
Abstract
The DNA origami technique allows the facile design and production of three-dimensional shapes from single template strands of DNA. These can act as functional devices with multiple potential applications but are constrained by practical limitations on size. Multi-functionality could be achieved by connecting together distinct DNA origami modules in an ordered manner. Arraying of non-identical, three-dimensional DNA origamis in an ordered manner is challenging due for example, to a lack of compatible rotational symmetries. Here we show that we can design and build ordered DNA structures using non-identical 3D building blocks by using DNA origami snub-cubes in left-handed and right-handed forms. These can be modified such that one form only binds to the opposite-handed form allowing regular arrays wherein building blocks demonstrate alternating chirality.
Introduction
DNA origami is a versatile means of synthesizing complex molecular architectures with multiple functionalities for wide ranges of applications, including biosensing,1 drug delivery,2–4 nanophotonics and plasmonics,5,6 nanoelectronics7,8 and biomimetics.9,10 DNA origami structures are assembled from a long single-stranded scaffold with the help of a number of DNA oligomer staples through a thermal annealing process. Using this approach higher order structures including one-dimensional (1D), two-dimensional (2D) and three-dimensional (3D) lattices can be achieved by utilising both sticky and blunt ended interactions between discrete and distinct origami modules. These include 1D DNA ribbon structures,11 motif-based 2D lattices12,13 and 3D DNA origami lattices.14 In the case of 1D and 2D repeating structures, two-dimensional DNA origami building blocks such as flat sheets are typically used. This simplifies the design challenge. For example, a 1D array can be made from any number of different flat building blocks as long as each building block has two edges matching perfectly via either base-pairing or mediated by non-DNA linkers15–17 to the building blocks on either side. Similarly, 2D arrays of flat sheets can be made with non-identical, flat building blocks. Such 2D arrays have been produced and modified using a wide variety of experimental approaches.18 Three-dimensional building blocks offer greater capabilities given that they can, for example, act as nanometric cages, carrying cargo. However, forming 1D and 2D arrays from such building blocks is more challenging as they would preferably have matching rotational symmetries at the faces where they join and would be shaped in such a way that they can lie on the same plane. For producing 3D crystalline arrays of three-dimensional building blocks, the challenge is greater still as, for a single type of building block, this can only be achieved using solids that are able to fit inside a cube and have the same symmetry. One solution would be to use convex polyhedra that are identical but of different chirality. Amongst Platonic, Archimedean and Johnson solids there are only seven chiral solids and only the snub-cube has the correct symmetry to allow arranging into a 3D crystal.Chirality has been a focus of some DNA origami work but this has tended to concentrate on using DNA origami structures to change the chiral arrangement of inorganic particles attached to their surface rather than making intrinsically chiral 2D structures from the DNA origami itself. Such arrangements have measurable effects on the plasmonic properties of the resulting structures.19
The original DNA origami concept relied on the parallel packing of DNA double helices.20 An alternative approach uses wireframe designs, a recent innovation based on the scaffolding principle. In this method, the target structure is represented as a polyhedral mesh. The DNA scaffold routing is completed through the target shape and finally staple strand sequences are designed to fold the scaffold into the target shape.21–23 Numerous wireframe structures have been demonstrated and allow increases of both size and complexity of the DNA structures with lower packing density. Advantages over parallel packing include rapid folding and increased stability at low cation concentrations and physiological ion conditions.24 The wireframe approach is particularly useful for formation of convex polyhedra themselves having potential as cargo carrying nanostructures and even as artificial vaccines if their exterior is decorated with antigens.25 Facilitated by in silico modelling tools,21,26–28 many wireframe structures having different shapes and sizes have been designed and reported. However, building arrays of 3D wireframe DNA structures is still difficult compared to parallel packed structures if they are intended to interact to form arrays. Challenges include the necessity for single-stranded DNA scaffolds with custom sequence and length to avoid large unstructured sequences which would interfere with origami–origami interactions.
There have been few examples of well-defined 1D, 2D and 3D arrays of three-dimensional DNA origami nanostructures. Recently, Tian et. al. reported a DNA lattice assembled from anisotropic 3D DNA origami shapes of a regular octahedron and an elongated octahedron, connected by DNA sticky ends at vertices.14 Inspired by this, we designed anisotropic 3D DNA origami snub-cubes able to be connected through sticky ends enabling a face–face interaction which, in principle, can form 1D, 2D or 3D arrays. As proof of principle, we showed that the system is able to form 1D arrays and is also able to interact on a two-dimensional plane. This approach enables us to make chiral 3D DNA origami arrays, where building blocks are connected face-to-face with multiple connections to maximize the number of DNA complementary bonds.
We designed a 3D wireframe DNA origami snub-cube. The snub-cube is an Archimedean solid with 60 edges, 24 vertices and 38 faces, including 6 squares and 32 equilateral triangles. Snub-cubes occur in both left-handed (L) and right-handed (R) forms and we designed both. We planned to connect the L and R forms of the structure by extending complementary single stranded DNA (ssDNA) on the four corners of each of the six squares that are orthogonal to the surface of the same squares on the L and R snub-cubes in a bottom-up approach for the construction of periodic arrays alternating between L and R.
Taking a step-by-step approach, first we assembled single L and R snub-cubes separately. Next, L and R snub-cubes were constructed with external ssDNA strands with the strands on the R-form being complementary to those on the L-form, with the expectation that these would anneal to form heterodimers. We further modified snub-cubes with external strands having complementary sequences such that continuous L–R–L–R… chains would form. Additional modifications were made such that the alternating pattern would extend in 2 dimensions to form 2D arrays. Our results showed that L and R snub-cubes can be produced and modified such that heterodimers and alternating 1D chains can form. 2D interactions could also be achieved though were somewhat disordered when observed on a mica surface. To our knowledge, this is the first example of two distinct 3D DNA origami structures being combined into regular dimers and 1D chains. Such structures may have utility for example as templates for arranging inorganic molecules for electronic and photonic applications5,7 or position biological macromolecules to study their biomimetic29 functions.
Results and discussion
Structure design
The wireframe snub-cube (Sn) DNA origami structures investigated in this study were designed and constructed in silico in both the L and R forms using an online algorithm framework called “DNA origami Sequence Design Algorithm for User-defined Structures (DAEDALUS)”.16 In the subsequent sections, we designate the L and R forms of the Sn as SnL and SnR respectively. To design the structures, the target geometry of Sn in polygon file format was introduced in the algorithm, where DNA sequences were generated based on the double crossover (DX)-based wireframe motif in which interconnected edges consist of two duplexes joined by means of antiparallel DX (Fig. 1A). In the design algorithm, we defined the length and sequence of the scaffold strand and the staple strands were generated for the Sn structure with 52-bp edge lengths. The wireframe Sn structure was designed to be ∼57 nm in diameter with a scaffold of 6240 bases and 168 staple strands corresponding well with the measured (outer) diameter of ∼58 nm. Note that, as the wireframe contributes a wall thickness equal only to double strand DNA diameter in thickness, the interior is expected to be a large cavity of about 54 nm. Post-design modifications of the appropriate staple strands were accomplished for the design of dimer, 1D chain and 2D structures.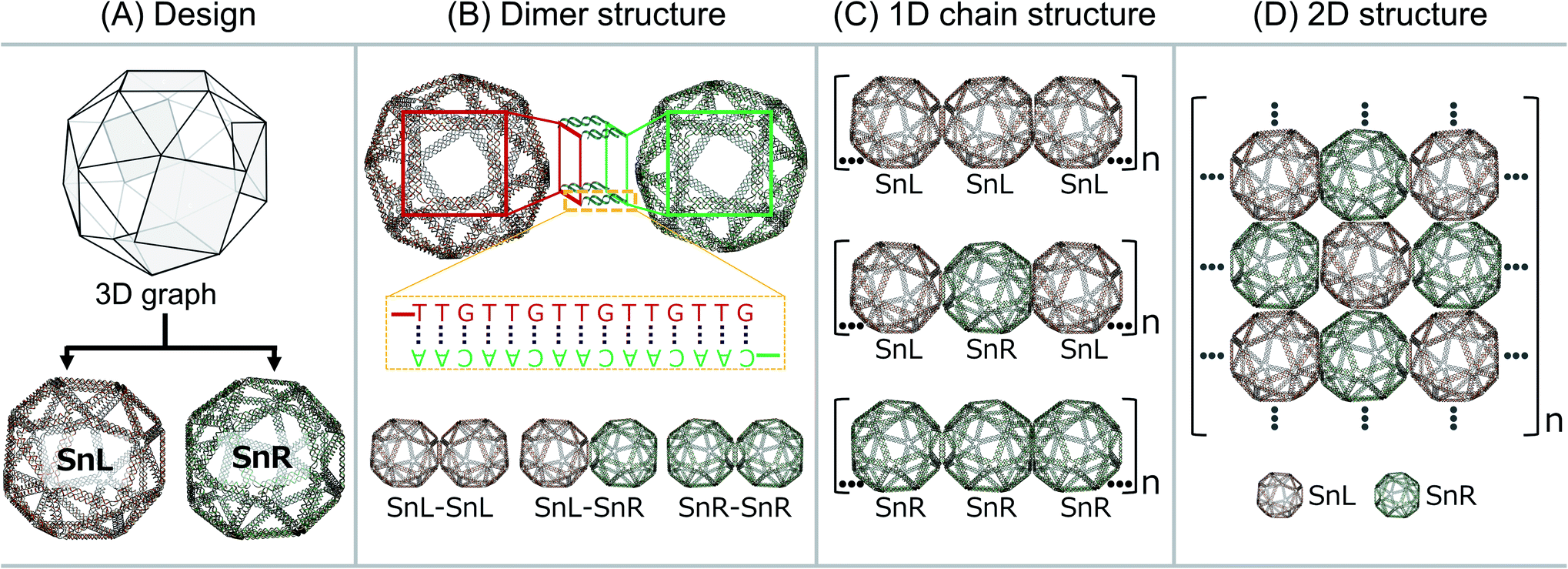 | ||
| Fig. 1 DNA origami designs. (A) Wireframe DNA origami structure of Sn is designed both in the left-handed (SnL) and right-handed (SnR) forms, (B) dimers were designed as SnL–SnL, SnL–SnR or SnR–SnR based on hybridization of complementary DNA sequences extending from both Sns, (C) design of SnL–SnL–SnL, SnL–SnR–SnL or SnR–SnR–SnR 1D chain structures applying the same strategy to connect single units in dimer structures and (D) design of 2D lattice where SnL and SnR structures are connected with unique extension strands. | ||
All the Sn structures including the extensions of complementary staple strands were assembled by mixing custom length scaffolds with an excess of staple strands in an assembly buffer containing 12.5 mM MgCl2. After annealing, the structures were purified using a polyethylene glycol (PEG) precipitation method to remove excess staples (shown in ESI Fig. S1A†). Folded structures were analysed by agarose gel electrophoresis (AGE) and atomic force microscopy (AFM) imaging. The AGE analysis results and AFM image of Sn structures without any staple modification are shown in ESI Fig. S1B and C† respectively. The band shift for the Sn structure compared to the scaffold band was used to confirm the folding of the structure. The gel image also confirms that the excess staples were removed after purification. AFM imaging was employed to visually confirm the overall correct DNA origami structure.
To produce Sn units capable of forming dimers, one square face was modified such that it would connect to the square face of a second Sn through complementary base pairing. The staple strands of the four corners of the square face were modified with complementary extensions of sequence “5′-TTGTTGTTGTTGTTG” and “5′-CAACAACAACAACAA”. Both were added to both SnL and SnR to produce four Sn structures, named SnL-TTG, SnL-CAA, SnR-TTG and SnR-CAA in the subsequent discussion. The schematic design of the connection strategy to make different dimer structures of Sn i.e. SnL–SnL, SnL–SnR and SnR–SnR is illustrated in Fig. 1B. It is worth noting that two staples pass through each corner junction of the modified square faces. We redesigned these two staples into three and extended one staple at each corner of the square with complementary DNA strands (schematically shown in ESI Fig. S2B† for SnL and S2F for SnR).
We also designed and constructed 1D chain structures using chiral Sns shown schematically in Fig. 1C. The connection strategy was as for dimer structures except that we modified the staple strands of the corners of two opposite square faces on each Sn rather than just one so that each Sn would bind to two others in a linear fashion (shown in Fig. 1C). In this way, we designed three different 1D chains with repeating units of SnL–SnL–SnL, SnL–SnR–SnL and SnR–SnR–SnR.
To produce 2D arrays of Sns, a similar approach was used except two pairs of opposite square faces on each Sn were modified with complementary sequences such that each Sn can attach to four neighbours (Fig. 1D).
The details of the design of the staples at corners of the square faces of SnL and SnR to make dimer, 1D chain and 2D structures is illustrated in ESI Fig. S2.†
Production and validation of DNA–origami Sn dimers
Dimers made from Sn of the same chirality were constructed using Sn origami structures with extensions i.e. SnL-TTG, SnL-CAA, SnR-TTG and SnR-CAA were assembled and purified as described in the methods section. To optimise dimer formation conditions, single origami units were mixed at different concentrations and incubated overnight at different temperatures. First, we assembled the SnL–SnL dimer structures by mixing the SnL-TTG and SnL-CAA single units at concentrations of 10 nM, 20 nM and 30 nM. At each concentration, the dimer structures were incubated at temperatures of 25 °C, 37 °C and 45 °C overnight. Agarose gel analysis (Fig. 2A, top) clearly shows that the dimer structures were formed under all assembly conditions. However, it is obvious that at high concentration i.e. 30 nM for all incubation temperatures, more than one band is present, suggesting the presence of some non-target structures which are less intense at the incubation temperature of 45 °C. We also observed the aggregation of the structures in the wells of the gel at high concentration. This aggregation was reduced at 20 nM and almost no aggregation was observed at 10 nM at the incubation temperature of 45 °C. It is worth mentioning that the incubation temperature 45 °C is higher than the Tm value of the extended complementary DNA sequences (40.7 °C). The dimer structures assembled at different concentrations and incubation temperatures were imaged under AFM (Fig. 2A, bottom). To estimate dimer yield we carried out particle counting from AFM results (Fig. 2B, ESI Fig. S3–S11 and Table S1†). The results showed that the percentage of dimer structures is much higher than that of the single, trimer and tetramer structures at all assembly concentrations. There was also a temperature dependence with the yield of correctly formed dimer structures increasing with increasing temperature over the range tested, consistent with AGE results (Fig. 2A). Next, we assembled SnR–SnR dimer structures from SnR-TTG and SnR-CAA at concentrations of 10 nM, 20 nM and 30 nM and incubated at 25 °C, 37 °C and 45 °C for each concentration. The agarose gel image (Fig. 2C, bottom) shows that the assembly of SnR–SnR dimer structures follows the same trends as observed for SnL–SnL dimers. We found a single band for dimer structures for 10 nM at an incubation temperature of 45 °C. This sample was used in AFM imaging (Fig. 2C, right). Comparison graphs were made from analysis of the structures imaged under AFM (ESI Fig. S12, S13 and Table S2†). As shown in Fig. 2D, the percentage (92%) of the dimer structures for SnR–SnR (ESI Fig. S13†) is significantly higher than that of the single and tetramer structures, which is in good agreement with the results for SnL–SnL (Fig. 2B).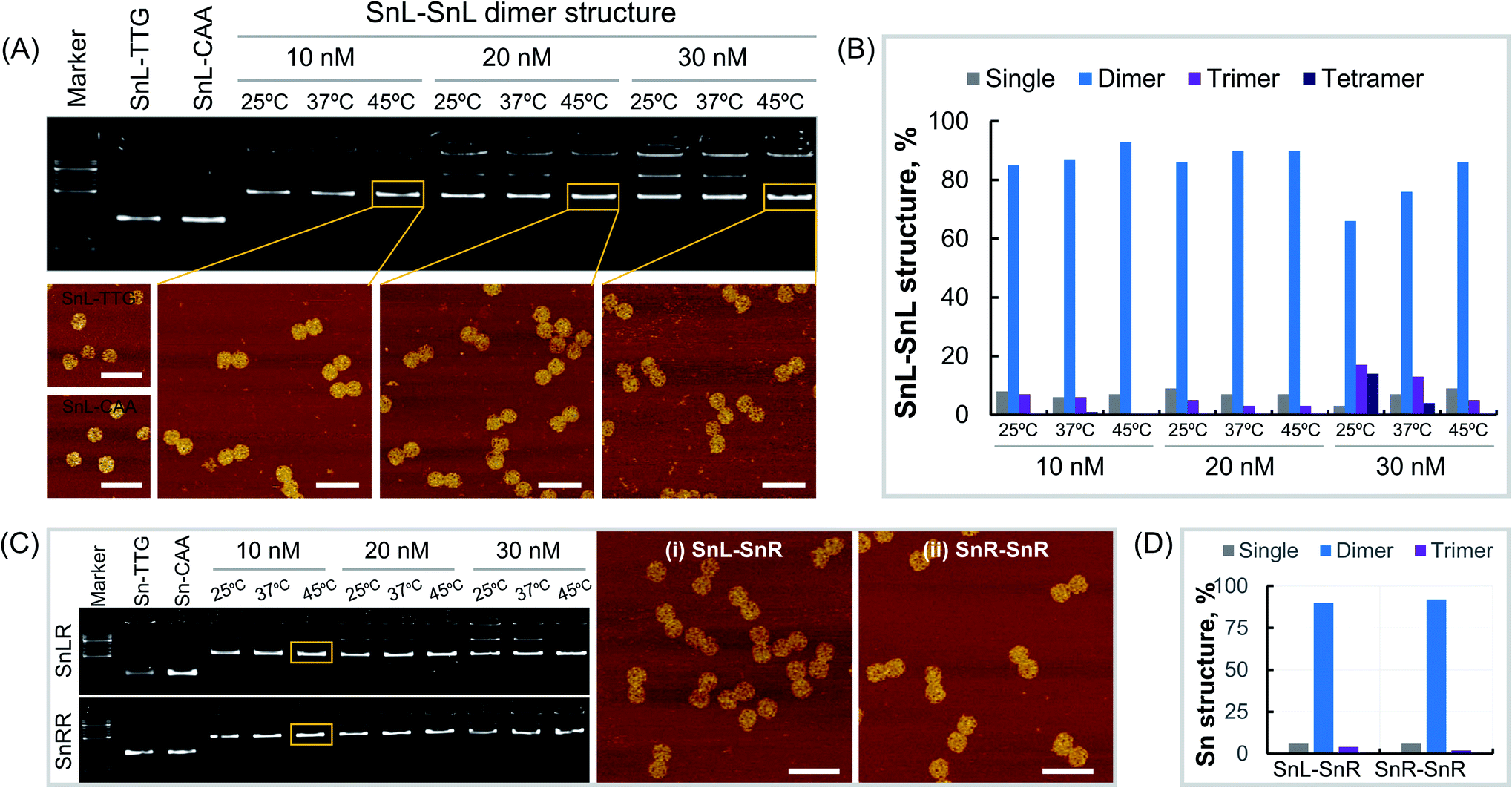 | ||
| Fig. 2 Production of Sn DNA origami dimers. (A) Agarose gel of SnL monomers and SnL–SnL dimers assembled at different concentrations and incubation temperatures (top) and AFM images for corresponding monomer and dimer structures (bottom), (B) effect of SnL monomer concentration on SnL–SnL dimer yields, (C) agarose gel of SnL–SnR and SnR–SnR structures assembled at different monomer concentrations and incubation temperatures (left) and AFM images for SnL–SnR (middle) and SnR–SnR (right) structures, (D) yield of SnL–SnR and SnR–SnR structures formed at 10 nM and incubation temperature 45 °C. AFM scale bars represent 200 nm. | ||
We next made mixed chiral dimers consisting of SnL and SnR by mixing the SnL-TTG and SnR-CAA single units. The assembly concentrations and incubation temperatures were the same as for non-chiral dimers. We observed similar results, and found highest assembly yield at 10 nM, 45 °C (Fig. 2C top). AFM imaging of resulting dimers (Fig. 2C, middle panel) confirmed their structure. We also estimated the dimer yield by counting the particles from AFM images (ESI Fig. S12 and Table S2†), which is 90% for dimer structures, significantly higher than the percentage of the single and tetramer structures.
To validate the mixed chirality of the SnL–SnR dimers, we further modified a staple strand at the corner of a square face in SnL-TTG and SnL-CAA with biotin for binding of streptavidin (SA) (Fig. 3A). Using the modified Sns, dimers SnL–SnL, SnL–SnR and SnR–SnR were assembled and imaged with AFM after addition of SA samples. The results (Fig. 3, ESI Fig. S15–S17 and Table S3†) clearly showed that SnL–SnL and SnL–SnR dimer structures bind two SA and one SA respectively, whereas the SnR–SnR dimer, having no biotinylated staples, binds essentially no SA.
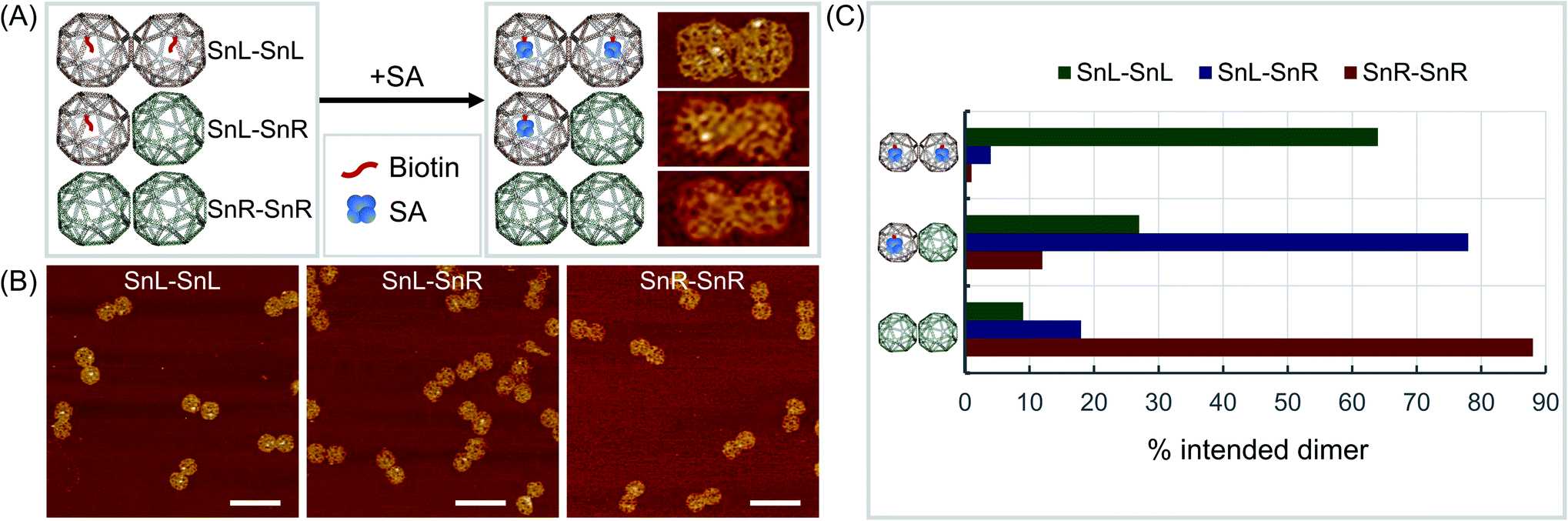 | ||
| Fig. 3 Validation of chirality. (A) Schematic diagrams (left) and AFM results after reaction with biotin (right) showing the different produced Sn dimers, biotin modifications and SA attachment where SA is visible in AFM images as white dots. (B) Wider field AFM images of the 3 forms of Sn dimers after reaction with biotin for bound SA with dimer structures of SnL–SnL (left), SnL–SnR (middle) and SnR–SnR (right). (C) Quantitation of intended dimers formed. AFM scale bars represent 200 nm. | ||
Production and validation of DNA–origami Sn 1D chains
Next, we designed and constructed a 1D chain of Sn origamis (Fig. 1C) where the SnL and SnR were arranged to make chains of repeating SnL–SnL–SnL, SnL–SnR–SnL and SnR–SnR–SnR structures. The modified single units of Sn with complementary DNA extensions assembled and purified following the procedure described in the methods section. We constructed the SnL–SnL–SnL chain structure by mixing SnL-TTG and SnL-CAA single units, where SnR-TTG and SnR-CAA were combined to make the SnR–SnR–SnR chain structure. The chain structure in the chiral form i.e. SnL–SnR–SnL was constructed from the SnL-TTG and SnR-CAA single units. In all of the chain structures, the modified single units were mixed at the concentration 10 nM and incubated overnight at 45 °C. AFM imaging showed that SnL–SnL–SnL, SnL–SnR–SnL and SnR–SnR–SnR chain structures all assembled and formed 1D chains (Fig. 4 middle panel, ESI Fig. S14†). All chains were bent, consistent with the design given that the modified squares in a Sn are not precisely opposite. | ||
| Fig. 4 1D chain structures and visualisation of alternating chirality of Sn origami. Schematic designs (top) and corresponding AFM images of chains without SA (middle panel) and with SA (bottom panel) of (A) SnL–SnL–SnL chains, (B) SnL–SnR–SnL alternating chains and (C) SnR–SnR–SnR chains. AFM scale bars represent 200 nm. | ||
To show the expected sequence of L- and R-building blocks in the chain, a similar approach as for dimer structures, using biotinylation and SA labeling was used with the biotinylated staple being housed on the SnL unit (Fig. 4, bottom panel and ESI Fig. S18†). The results clearly show that all building blocks in the SnL–SnL–SnL chain are capable of binding SA while only alternating blocks are able to do so in the SnL–SnR–SnL chain while the SnR–SnR–SnR chain is incapable of binding SA.
Production and validation of DNA–origami Sn 2D arrays
To assemble a chiral 2D lattice, the assembled SnL and SnR structures with the unique extension strands were mixed at concentration of 10 nM and incubated overnight at 43 °C, that is above the Tm of the extension complementary strands to avoid aggregation. The design of the Sn for 2D lattice formation is illustrated schematically in Fig. 5A. Fig. 5B shows AFM results of trials where 3D SnL and SnR origamis were mixed into 2D lattices. Although the structures are not assembled in a perfectly aligned manner, it is obvious from the AFM image that the SnL and SnR structures are connected and expanded only in the X and Y direction. As a control experiment, we mixed SnL and SnR structures without staple modifications. It is evident from AFM images in Fig. 5C that no 2D lattice is formed. The irregularity of the 2D array is likely due to difficulties in depositing/assembling on the mica surface and may require extensive probing of conditions to ensure ordered surface deposition as well as potential redesign to make origami shapes with higher rigidity.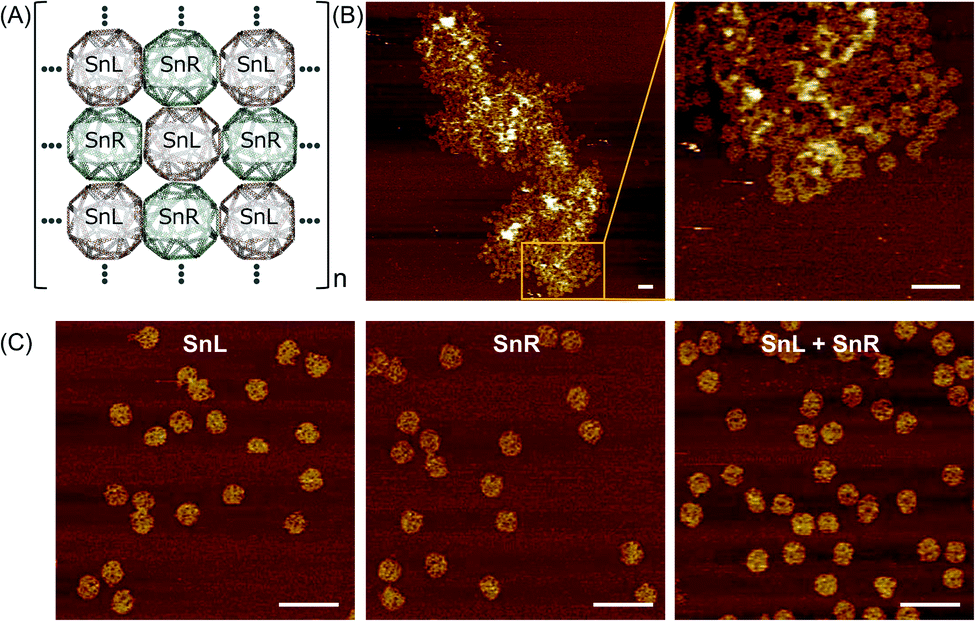 | ||
| Fig. 5 2D arrays of Sn origami. (A) Schematic illustration of the array, (B) AFM images of the 2D structure (left) and a magnified image (right) and (C) AFM images of SnL (left) and SnR (middle) alone without staple modifications and after mixing together SnL + SnR (right), showing no interaction in absence of connecting staples. AFM scale bars represent 200 nm. | ||
Conclusions
Overall, these results show that, as predicted, DNA origami snub-cubes allow close attachment of two different 3-dimensional, convex, polyhedral structures in an alternating fashion. This enables an ABAB… type arrangement where the constituent building blocks are able to remain aligned in the same plane. Our attempts to form a 2D array using the snub-cube designs clearly show interaction between the origamis but when deposited onto a surface from solution, do not result in a truly regular alternating array meaning that improved deposition and design are likely necessary.These results provide a promising basis for extension of the technique into three dimensions where the chiral properties of the snub-cube should allow a perfect crystal to be formed from two three-dimensional building blocks of different chirality (L and R snub-cubes). The current designs will likely need to be improved to facilitate this due to the general intrinsic flexibility of wireframe DNA origami structures with duplex edges,30 which likely disfavours crystal formation. This might be circumvented by converting the edge design to a 6 helix bundle cross section that has been shown to display dramatically increased structural stiffness compared to DNA duplexes,31 with 6 helix bundles having a persistence length in the micrometre scale.32 Also, in previous work, crystals have been made using two octahedral 3D wireframe structures having 6 helix bundle edges,14 an approach that may also help to improve 2D array formation.
Methods
Structure design
The Sn structures including the SnL and SnR were designed using the DAEDALUS package, which is an algorithm framework to design wireframe DNA origami structures. The algorithm generates the staple DNA sequences from the input 3D shapes in the polygon file format. The structure design was computed with a custom-scaffold sequence and a defined minimal edge length of 52 bp. The sequences of the custom scaffold and staple strands for SnL and SnR are shown in ESI Tables S4–S6.† Post-design modifications of the staple strands with complementary sequences for the construction of dimer, 1D chain and 2D structures were accomplished manually and sequences are shown in ESI Tables S7–S10.†Plasmid mutagenesis to produce custom scaffold
To produce pScaf_6240, we deleted 1824 bases from pScaf_8064.4 (a kind gift of Shawn Douglas, Addgene plasmid #111520)33 using an inverse PCR reaction with following primers: pScaf_6240_for: TAATCAGGATCAATGGATTCAGTGTG and pScaf_6240_rev: CTGAGCACCTGCACGACG. Amplification was carried out in a 50 μL reaction in 1× HF buffer (NEB) using 200 nM primer, 500 nM dNTPs and 0.5 μL Phusion polymerase with an initial denaturation step at 98 °C for 30 s followed by 30 cycles at 98 °C for 10 s, 67 °C for 10 s and 72 °C for 125 s and a final incubation for 5 min at 72 °C. The PCR product was purified from template by treatment with 1 μL Dpn I (Roche) for 60 min at 37 °C followed by heat inactivation for 20 min at 80 °C. Next, 17 μL of the PCR product was phosphorylated by treatment with 1 μL polynucleotide kinase (PKN, Thermo Fisher Scientific) and addition of 2 μL 10× T4 ligase buffer (NEB) for 20 min at 37 °C, followed by heat inactivation at 75 °C for 10 min. 1 μL T4 ligase was added and incubated overnight at 16 °C. Resulting plasmid was transformed into chemo competent E. coli Dh5α. The DNA sequence of the pScaf_6240 plasmid was confirmed by Sanger sequencing.Scaffold amplification and purification
For scaffold production, we transformed E. coli XL1-blue with the M13KO7 based helper phage plasmid HP17_KO7 (a kind gift of Hendrik Dietz, Addgene plasmid #120346).34 The resulting helper strain was made chemically competent again and transformed with pScaf_6240. Scaffold production was carried out as described previously33 with the exception that 6 mL lysis buffer were used per 100 mL culture volume.Assembly of monomeric DNA–origami snub-cubes
Staple strands were purchased from Integrated DNA Technologies. The scaffold strand was prepared as described above. The assembly reactions were prepared with a custom scaffold strand (12 nM) and staple strands (60 nM for each strands) in 1× TAE/Mg2+ buffer (Tris-base 5 mM, EDTA 1 mM and MgCl2 12.5 mM) in 50 μL. The annealing was performed in a PCR thermal cycler as follows: 95 °C for 5 min, 80 °C to 75 °C at a rate of −1 °C per 5 min, 75 °C to 30 °C at a rate of −1 °C per 15 min and 30 °C to 25 °C at a rate of −1 °C per 10 min. After folding, the assembled structures were purified by PEG precipitation method to remove excess staples from the folding reactions: the folded structures were mixed with PEG buffer (5 mM Tris, pH 8.0, 15% PEG-8000, NaCl 500 mM, MgCl2 20 mM) at a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 and incubated overnight. Then, the mixture was centrifuged at 10000 rpm for 10 min at room temperature. The supernatant was removed, and the pellet was dissolved in 20 μL 1× TAE/Mg2+ buffer.
:2 and incubated overnight. Then, the mixture was centrifuged at 10000 rpm for 10 min at room temperature. The supernatant was removed, and the pellet was dissolved in 20 μL 1× TAE/Mg2+ buffer.
Assembly of dimers, 1D chains and 2D lattices
Sn dimers, 1D chains and 2D lattices were assembled from the monomeric Sn structures with complementary DNA extension strands, assembled and purified according to the method described above. For the assembly of all structures, the monomeric Sn with complementary extension strands were mixed at the ratio of 1:1 and incubated at different temperatures overnight.
Preparation of SA bound dimer and 1D chains
To bind the SA, 3′ end of a staple strand (5′-CGA AGC ACT CAT TTT TGG GAA CTG GAG TTA TCC CTA TTT TTT CCT GAA GTA C) at the corner of square 5 of SnL was modified with BiotinTEG and was purchased from Sigma-Aldrich. The monomeric Sn for dimers and 1D chains were assembled with staple strand with biotin and purified according to the method described above. The assembled dimers and 1D chains from the 10 nM monomeric Sn were mixed with SA at a ratio of 1:3 and incubated 1 h at room temperature. Then, the excess SA was removed using the Microspin S-400 HR column (GE Healthcare Life Sciences) following the manufacturer's protocol.
AFM imaging
The assembled monomer, dimer, 1D chain and 2D structures were imaged via AFM using a Bruker Dimension Icon microscope with a Bruker ScanAsyst-Fluid+ probe in the PeakForce QNM® mode. ScanAsyst-Fluid+ probes (Bruker) with nominal spring constant equal to 0.7 N m−1 and sharpened tip (nominal radius equal to 2 nm) were used in all the measurements. For imaging, 1.5 μL samples were deposited on freshly cleaved mica. After 1 min incubation, 20 μL of 1× TAE/12.5 mM Mg2+ buffer was added followed by immediate addition of 1.5 μL of 100 mM NiCl2 solution and the sample on mica was incubated further 1 min. Then, 140 μL of 1× TAE/Mg2+ buffer was added to the sample and imaged under AFM.Author contributions
M. S. I. designed DNA origami snub-cube structures, and carried out DNA origami assembly, purification and AFM imaging and contributed to pScaf_6240 cloning and scaffold production. G. D. W. cloned pScaf_6240 and carried out scaffold production and some DNA origami assembly and purification. K. W. and S. Z. carried out additional AFM analysis. J. G. H. acquired funding, conceived the study, supervised experimental work and wrote the manuscript together with all authors.Conflicts of interest
There are no conflicts of interest.Acknowledgements
This work was funded by the TEAM Programme of the Foundation for Polish Science co-financed by the European Union under the European Regional Development Fund (TEAM/2016-3/23) awarded to J. G. H. The authors thank Bernard Piette and Agnieszka Kowalczyk for helpful discussions.References
- S. Wang, Z. Zhou, N. Ma, S. Yang, K. Li, C. Teng, Y. Ke and Y. Tian, Sensors, 2020, 20, 6899 CrossRef CAS PubMed.
- Q. Zhang, Q. Jiang, N. Li, L. Dai, Q. Liu, L. Song, J. Wang, Y. Li, J. Tian and B. Ding, ACS Nano, 2014, 8, 6633–6643 CrossRef CAS PubMed.
- Q. Chi, Z. Yang, K. Xu, C. Wang and H. Liang, Front. Pharmacol., 2020, 10, 1585 CrossRef PubMed.
- D. Balakrishnan, G. D. Wilkens and J. G. Heddle, Nanomedicine, 2019, 14, 911–925 CrossRef CAS PubMed.
- J. Ryssy, A. K. Natarajan, J. Wang, A. J. Lehtonen, M. K. Nguyen, R. Klajn and A. Kuzyk, Angew. Chem., Int. Ed., 2021, 60, 5859–5863 CrossRef CAS PubMed.
- B. Shen, M. A. Kostiainen and V. Linko, Langmuir, 2018, 34, 14911–14920 CrossRef CAS PubMed.
- Y. Geng, A. C. Pearson, E. P. Gates, B. Uprety, R. C. Davis, J. N. Harb and A. T. Woolley, Langmuir, 2013, 29, 3482–3490 CrossRef CAS PubMed.
- X. Dai, Q. Li, A. Aldalbahi, L. Wang, C. Fan and X. Liu, Nano Lett., 2020, 20, 5604–5615 CrossRef CAS PubMed.
- K. Göpfrich, C.-Y. Li, M. Ricci, S. P. Bhamidimarri, J. Yoo, B. Gyenes, A. Ohmann, M. Winterhalter, A. Aksimentiev and U. F. Keyser, ACS Nano, 2016, 10, 8207–8214 CrossRef PubMed.
- H. Shen, Y. Wang, J. Wang, Z. Li and Q. Yuan, ACS Appl. Mater. Interfaces, 2018, 11, 13859–13873 CrossRef PubMed.
- Z. Li, M. Liu, L. Wang, J. Nangreave, H. Yan and Y. Liu, J. Am. Chem. Soc., 2010, 132, 13545–13552 CrossRef CAS PubMed.
- W. Liu, H. Zhong, R. Wang and N. C. Seeman, Angew. Chem., Int. Ed., 2011, 50, 264–267 CrossRef CAS PubMed.
- S. Woo and P. W. Rothemund, Nat. Commun., 2014, 5, 1–11 Search PubMed.
- M. Ji, J. Liu, L. Dai, L. Wang and Y. Tian, J. Am. Chem. Soc., 2020, 142, 21336–21343 CrossRef CAS PubMed.
- W. Pfeifer, P. Lill, C. Gatsogiannis and B. Saccà, ACS Nano, 2018, 12, 44–55 CrossRef CAS PubMed.
- S. Loescher and A. Walther, Angew. Chem., Int. Ed. Engl., 2020, 59, 5515–5520 CrossRef CAS PubMed.
- J. Ye, S. Helmi, J. Teske and R. Seidel, Nano Lett., 2019, 19, 2707–2714 CrossRef CAS PubMed.
- J. M. Parikka, K. Sokołowska, N. Markešević and J. J. Toppari, Molecules, 2021, 26, 1502 CrossRef CAS PubMed.
- M. Wang, J. Dong, C. Zhou, H. Xie, W. Ni, S. Wang, H. Jin and Q. Wang, ACS Nano, 2019, 13, 13702–13708 CrossRef CAS PubMed.
- P. W. K. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS PubMed.
- R. Veneziano, S. Ratanalert, K. Zhang, F. Zhang, H. Yan, W. Chiu and M. Bathe, Science, 2016, 352, 1534 CrossRef CAS PubMed.
- E. Benson, A. Mohammed, J. Gardell, S. Masich, E. Czeizler, P. Orponen and B. Högberg, Nature, 2015, 523, 441–444 CrossRef CAS PubMed.
- F. Zhang, S. Jiang, S. Wu, Y. Li, C. Mao, Y. Liu and H. Yan, Nat. Nanotechnol., 2015, 10, 779 CrossRef CAS PubMed.
- P. Piskunen, S. Nummelin, B. Shen, M. A. Kostiainen and V. Linko, Molecules, 2020, 25, 1823 CrossRef CAS PubMed.
- R. Veneziano, T. J. Moyer, M. B. Stone, E.-C. Wamhoff, B. J. Read, S. Mukherjee, T. R. Shepherd, J. Das, W. R. Schief and D. J. Irvine, Nat. Nanotechnol., 2020, 15, 716–723 CrossRef CAS PubMed.
- H. Jun, F. Zhang, T. Shepherd, S. Ratanalert, X. Qi, H. Yan and M. Bathe, Sci. Adv., 2019, 5, eaav0655 CrossRef PubMed.
- H. Jun, T. R. Shepherd, K. Zhang, W. P. Bricker, S. Li, W. Chiu and M. Bathe, ACS Nano, 2019, 13, 2083–2093 CAS.
- H. Jun, X. Wang, W. P. Bricker and M. Bathe, Nat. Commun., 2019, 10, 1–9 CrossRef CAS PubMed.
- V. Linko, M. Eerikäinen and M. A. Kostiainen, Chem. Commun., 2015, 51, 5351–5354 RSC.
- E. Benson, A. Mohammed, D. Rayneau-Kirkhope, A. Gådin, P. Orponen and B. Högberg, ACS Nano, 2018, 12, 9291–9299 CrossRef CAS PubMed.
- D. J. Kauert, T. Kurth, T. Liedl and R. Seidel, Nano Lett., 2011, 11, 5558–5563 CrossRef CAS PubMed.
- T. Liedl, B. Högberg, J. Tytell, D. E. Ingber and W. M. Shih, Nat. Nanotechnol., 2010, 5, 520–524 CrossRef CAS PubMed.
- P. M. Nafisi, T. Aksel and S. M. Douglas, Synth. Biol., 2018, 3, ysy015 CrossRef CAS PubMed.
- F. Praetorius, B. Kick, K. L. Behler, M. N. Honemann, D. Weuster-Botz and H. Dietz, Nature, 2017, 552, 84–87 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1na00385b |
| This journal is © The Royal Society of Chemistry 2021 |