
Photochemical reduction of carbon dioxide to formic acid
Robin
Cauwenbergh
 and
Shoubhik
Das
*
and
Shoubhik
Das
*
ORSY Division, Department, of Chemistry, Universiteit Antwerpen, Campus Groenenborger, Groenenborgerlaan 171, 2020 Antwerpen, Belgium. E-mail: Shoubhik.Das@uantwerpen.be
First published on 24th February 2021
Abstract
With the growing awareness of green chemistry, carbon capture and utilization (CCU) has received tremendous attention compared to carbon capture and storage (CCS). Over the past decades, the development of sustainable approaches for converting CO2 to valuable products such as CO, HCOOH, CH3OH, organic carboxylic acids and others has become popular and is of great importance for society. In this review, we particularly focus on the generation of HCOOH using homogeneous catalysts, such as transition metal complexes and organic dyes, under photocatalytic conditions. This review will give an overview of recent reports on this topic to stimulate further research in this appealing and vast growing research area.
Introduction
Carbon dioxide (CO2) emissions into the atmosphere have reached an alarming level of 33.1 Gt in 2020.1,2 Various sectors such as transportation, industry, agriculture, power, etc. have contributed unequivocally to the total amount of global CO2 emissions. Among them, coal- and gas-fired power plants contributed 30% of the global CO2 emission. Therefore, urgent capture and storage of CO2 from different power plants and other sectors is required to avoid further emissions of CO2 into the atmosphere.3 This process, which is well-known as carbon capture and storage (CCS), is effective, however, subsequent utilization of CO2 would be even more attractive as CO2 is a non-toxic molecule.4–6 The aim of this concept is to use CO2 as a C1 source for the sustainable syntheses of hydrocarbon fuels, fine chemicals and pharmaceuticals. The whole procedure is now defined as carbon capture, use and storage (CCUS).7 However, activation of CO2 is highly challenging due to the highest oxidized form of carbon, which is also thermodynamically stable and kinetically inert.8In nature, activation of CO2 has been achieved by several enzymes, such as carbon monoxide dehydrogenases that reduce CO2 to carbon monoxide (CO) and formate dehydrogenases that produce formic acid or generate a methyl group in a tetrahydrofolate-THF mediated process.9 Additionally, CO2 is reduced to sugars using solar energy during photosynthesis.10–12 Indeed, the average intensity of the total solar irradiance is about 1366.1 W m−2, which provides roughly 4.3 × 1020 J of energy per hour, which is more than that consumed globally in 1 year (4.1 × 1020 J)!13,14
Inspired by photosynthesis, visible light-mediated reductions of carbon dioxide have gained increasing attention and CO2 may be converted to CO, formic acid (FA), methanol, methane, etc. using homogeneous catalysts.15–22 Among these products, FA is highly advantageous due to its hydrogen storage capability.23–26 Additionally, FA as a chemical or as a preservative is expected to witness a healthy growth, at an estimated CAGR (Compound Annual Growth Report) of 3.74% during the forecast period of 2019–2024. It should also be noted that 1.137 million metric tons of FA are required per year over the entire world to meet the current demand.27 Therefore, the generation of FA in a sustainable way would be highly advantageous to meet the global demand (Fig. 1).
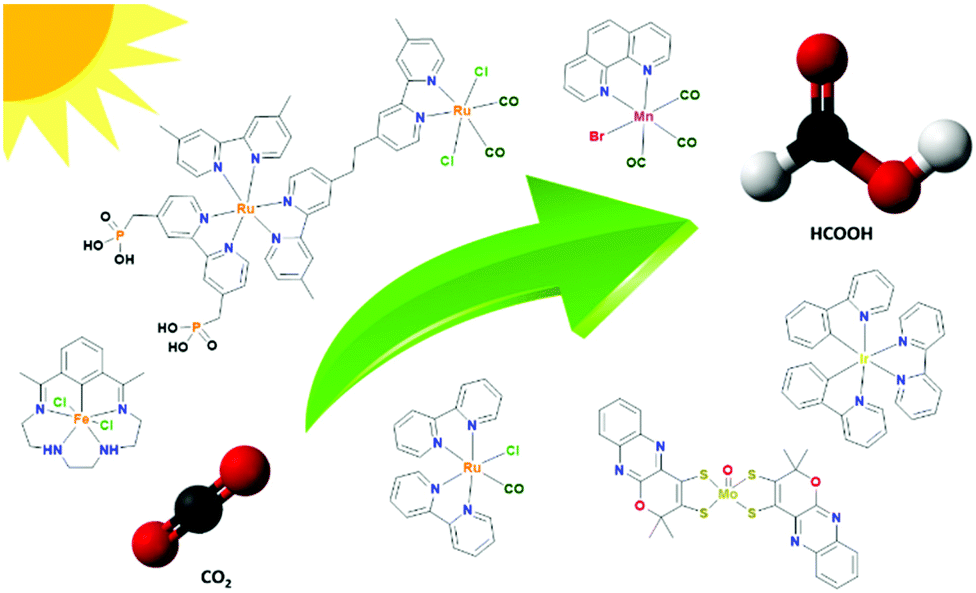 | ||
| Fig. 1 Transformation of CO2 into formic acid using different transition metal catalysts. | ||
The first photocatalytic reduction reaction was established by Fujishima et al. using semiconductor based photocatalysis.28–30 After that, this research field has made incredible progress. Specifically, new transition metal complexes have been synthesized and applied for the reduction of CO2. In some cases, these metal complexes acted as photosensitisers (PSs) or as photocatalysts (PCs) and in some cases, single metal complexes acted both as PSs and PCs in the photocatalytic reductions of CO2. Considering all these, in this review, we would like to give a summary of the reported homogeneous transition metal complexes and organic dyes for the light-induced reduction of CO2 to FA. In this context, it should be noted that many novel reports exist, which make use of MOFs (Molecular organic framework) or others, however, those articles are beyond this review.31–33
Homogeneous CO2 photoreduction
Ruthenium complexes
In 1983, Lehn et al. reported for the first time the photocatalytic conversion of CO2 to CO using a Re(bpy)(CO)3X (X = Cl, Br) catalyst. Although CO was produced in high selectivity, the generation of Re(bpy)(CO)3(HCO2) [bpy = 2,2′-bipyridine] was also observed, which originated from CO2 insertion into the Re–H bond.34 Further to their research, Lehn and co-workers reported for the first time the photocatalytic conversion of CO2 to formate using Ru(bpy)3Cl2 (1) as the photocatalyst in the presence of triethanolamine (TEOA) as the sacrificial oxidant. A turnover number (TONFA) of four was obtained after an irradiation time of 1 hour in the presence of dimethylformamide (DMF) as the solvent. When the solvent was changed to acetonitrile (MeCN), the TONFA was lowered to two. Additionally, when the bipyridine (bpy) ligands were changed to 1,10-phenanthroline (phen) ligands, no formate was observed. Further optimizations led to the highest TONFA of 27 after irradiating the CO2-Ru(bpy)3Cl2-TEOA-DMF mixture for 24 hours.35Lehn et al. continued their research in this area where they initially investigated the effect of CO2 concentration. They found that the generation of formate remained linear over time when CO2 was added regularly, which indicated that CO2 played an important role in the rate determining step. Additionally, they did not find any CO2 pressure effect between 0.5 atm and 2 atm, but at a higher pressure of 7 atm, the formation of formate decreased drastically. The main reason for this was the increased acidity of the reaction mixture, which made TEOA less efficient as an electron donor since it was in its protonated form. Secondly, the effect of water was investigated. They found that without the addition of water no formate was formed, whereas in the presence of water formate was formed. However, with a large amount of water, more CO and H2 were formed at the expense of formate. Thirdly, they reported the increase of formate formation when an excess of bipyridine ligand was added. A reasonable explanation could be a shift in the tris-bpy complex, the PS, to the bis-bpy complex, the PC, ratio. This explanation was based on the observation of new peaks in NMR spectra at around 8.36 ppm, which were most likely coming from RuIIbis-bpy species. Based on these observations, Lehn et al. studied the photoreduction of CO2 to formate in the presence of a RuII bis-bpy or a RuII mono-bpy catalyst and a RuII tris-bpy photosensitizer (Table 1; 2 to 8). The highest yield was obtained using [Ru(bpy)2(CO)(Cl)]+ (2) or [Ru(bpy)2(CO)(H)]+ (3) as the catalyst with a TONFA of 326 or 322, respectively, after 2 hours of irradiation in the presence of Ru(bpy)3 as the photosensitizer. Remarkably, by adding an excess of bpy ligand, the TONFA was dropped from 322 to 245.36
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Cat. | PS | SED | Solvent | Irr. t (h) | Sel. (%) | TONFA | λ (nm) | Ref. |
| Cat. represents catalyst; PS represents photosensitizer; SED represents sacrificial electron donor; Irr. t represents irradiation time; Sel. represents selectivity; TONFA represents turnover number for formic acid generation; λ represents wavenumber of incident light; Ref. represents reference; N.A. represents not applicable. Selectivities and TONs were determined after the corresponding irradiation times unless stated otherwise.a Determined after 2.5 h of irradiation. — = not mentioned. | ||||||||
| 1 | N.A. | TEOA | DMF | 1 | 69a | 4 | >400 | 35 |
| 1 | N.A. | TEOA | DMF | 24 | 69a | 27 | >400 | 35 |
| 1 | N.A. | TEOA | MeCN | 1 | — | 2 | >400 | 35 |
| 1 | N.A. | TEOA | MeCN | 24 | — | 22 | >400 | 35 |
| 2 | 1 | TEOA | DMF | 2 | — | 326 | >400 | 36 |
| 3 | 1 | TEOA | DMF | 2 | — | 322 | >400 | 36 |
| 4 | 1 | TEOA | DMF | 2 | — | 120 | >400 | 36 |
| 5 | 1 | TEOA | DMF | 2 | — | 107 | >400 | 36 |
| 6 | 1 | TEOA | DMF | 2 | — | 51 | >400 | 36 |
| 7 | 1 | TEOA | DMF | 2 | — | 91 | >400 | 36 |
| 8 | 1 | TEOA | DMF | 2 | — | 132 | >400 | 36 |
| 9 | 1 | TEOA | DMF | 2 | — | 40 | >400 | 42 |
| 10 | 1 | TEOA | DMF | 2 | — | 27 | >400 | 42 |
| 11 | 1 | TEOA | DMF | 2 | — | 255 | >400 | 36 |
| 12 | 13 | TEOA | DMA | 23 | 76 | 290 | 436 | 46 |
| 2 | 15 | TEOA | NMP | 5 | 75 | 62 | >400 | 42 |
| 3 | 15 | TEOA | NMP | 5 | 68 | 34 | >400 | 42 |
| 6 | 15 | TEOA | NMP | 5 | 81 | 13 | >400 | 42 |
| 14 | 15 | TEOA | NMP | 5 | 78 | 21 | >400 | 42 |
| 16 | N.A. | TEOA | DMA/H2O | 4 | >99 | 14 | >420 | 48 |
| 17 | 1 | TEOA | DMF | 24 | >99 | 380 | 405 | 49 |
| 18 | 1 | TEOA | DMF | 24 | 83 | 70.5 | 405 | 49 |
| 19 | 23 | BI(OH)H/TEOA | DMA | 24 | 83 | 3296 | >500 | 51 |
| 20 | 23 | BI(OH)H/TEOA | DMA | 24 | 73 | 4593 | >500 | 51 |
| 21 | 23 | BI(OH)H/TEOA | DMA | 24 | 86 | 3792 | >500 | 51 |
| 22 | 23 | BI(OH)H/TEOA | DMA | 24 | 72 | 5634 | >500 | 51 |
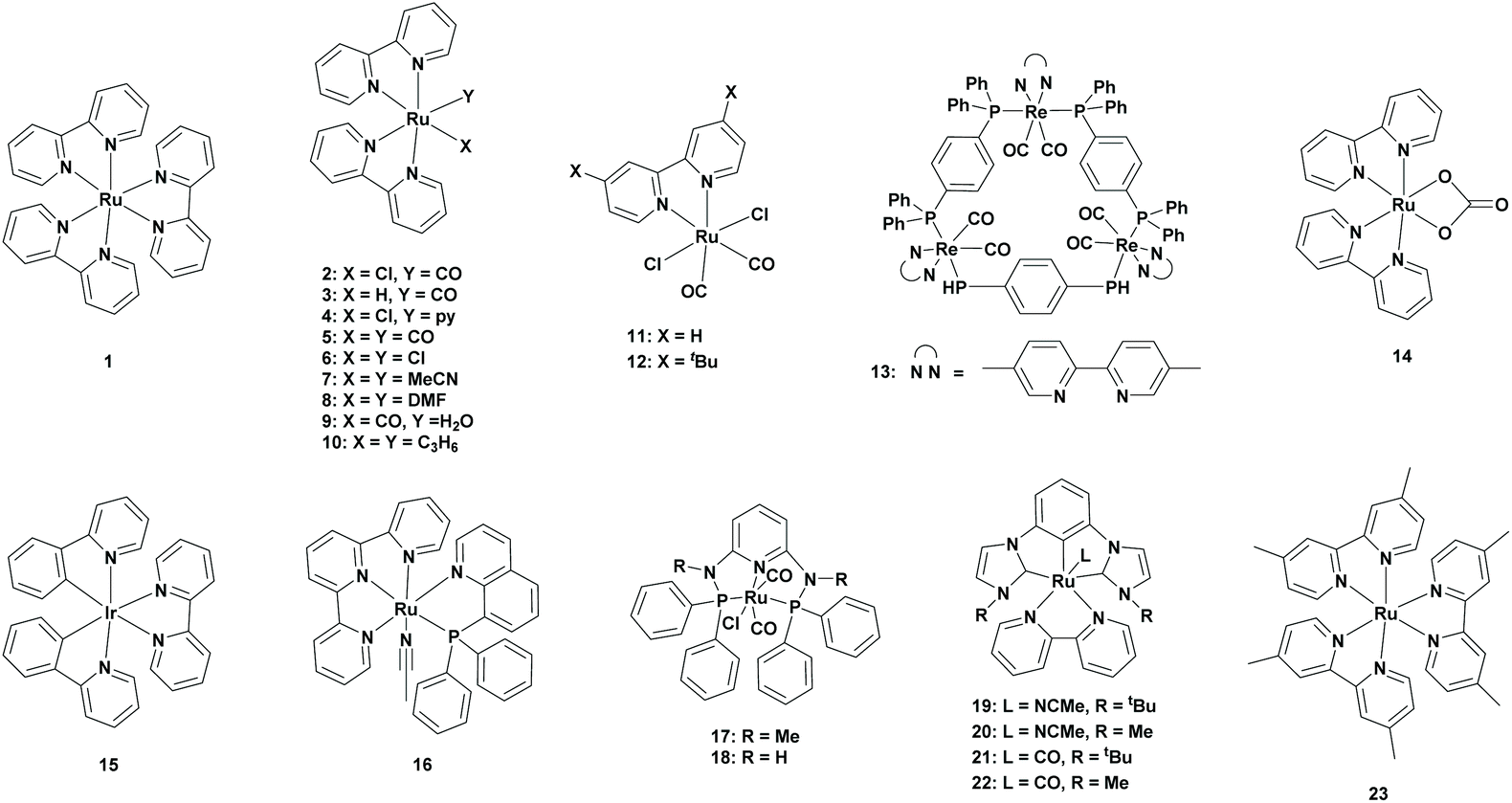
In the same year, Ishida et al. observed that the formate formation was increased drastically with the addition of [Ru(bpy)2(CO)2]2+ (5) to Ru(bpy)3, which resulted in a TONFA of 238 when equimolar amounts of both the complexes were used. In their experiment, [Ru(bpy)3]2+ was used as the photosensitizer, whereas [Ru(bpy)2(CO)2]2+ was used as the catalyst. Additionally, [Ru(phen)3]2+ was also able to reduce CO2 to formate but less efficient than the [Ru(bpy)3]2+ catalyst. This study revealed that a small amount of formate could be formed under the same conditions of Lehn et al. due to the generation of bis(bipyridine)ruthenium species by the dissociation of a bpy ligand from the [Ru(bpy)3]2+ complex. However, when water was added to the reaction mixture, the amount of formate dropped drastically. This can be explained by the decrease of basicity of TEOA which was not suitable as an electron donor due to the conversion to the corresponding protonated form. For this reason, Ishida and co-workers replaced TEOA with 1-benzyl-1,4-dihydronicotinamide (BNAH), since it followed the biological NAD(P)H model in an aqueous medium for CO2 fixation. However, the formate formation was dropped drastically due to the formation of CO under these conditions. The main problem with the CO2-[Ru(bpy)3]2+-[Ru(bpy)2(CO)2]2+-DMF-TEOA system was its stability, where prolonged irradiation induced catalyst decomposition. Later, they proposed a possible mechanism for the reduction of CO2 to FA. They observed that the catalyst released a CO ligand and accepted two photoelectrons from the photosensitizer to form the Ru(bpy˙−)2(CO) intermediate. This intermediate was able to coordinate with a CO2 molecule to form the Ru(bpy)2(CO)(CO2) intermediate. Finally, a proton was added to form [Ru(bpy)2(CO)(COOH)]+, which accepted two electrons to release FA.37 On the other hand, Meyer et al. proposed that Ru(bpy˙−)2(CO) was firstly protonated to Ru(bpy)2(CO)H+, which then gained an electron from the photosensitizer to form Ru(bpy)(bpy˙−)(CO)H+. Here, CO2 was inserted into the Ru–H bond to form Ru(bpy)(bpy˙−)(CO)(OCHO). This complex later gained an electron to release FA.38
In 2012, an important role of DMF in the photocatalytic reduction of CO2 to FA was observed by Vos et al.39 DMF was readily hydrolysed to formate, so the quantification of formate from CO2 became difficult in this medium. Later in 2014, Ishida et al. confirmed this by performing experiments under an argon (Ar) atmosphere, which, surprisingly, formed 25 μmol of formate. This clearly suggested the partial hydrolysis of DMF. For this reason, Ishida et al. proposed dimethylacetamide (DMA) as an alternative solvent for the reduction of CO2 in the presence of [Ru(bpy)3]2+ as the photosensitizer, [Ru(bpy)2(CO)2]2+ as the catalyst and BNAH as the electron donor. Under these reaction conditions, no precipitation was observed from the dimerized catalysts but the reduction of CO2 to CO was dominant.40 Ishida et al. continued their research in this area. After replacing the previous catalyst to trans(Cl)-Ru(bpy)(CO)2Cl2 (Table 1; 11), they observed that the light intensity and the loading of the catalyst affected the CO/HCOO− selectivity. However, mixtures of both the products were still observed and CO remained the major product.41
In 2015, Beller et al. reported iridium-based photosensitizers for the photoreduction of CO2 to FA. N-Methyl-2-pyrrolidone (NMP) was chosen as the solvent due to its similar behaviour to that of DMF, but it has higher stability. At first, they tried different Ru-based catalysts containing bipyridine ligands. All the Ru catalysts (Table 1; 2, 3, 6, 9, 10 and 14) showed reactivity towards the formation of formate, however, Ru-complexes containing associated CO ligands showed higher efficiencies. To observe the structure–activity relationship with the redox properties, they performed cyclic voltammetry of the different catalysts. Ruthenium catalysts without CO ligands showed reduction potentials at lower values, suggesting that the presence of a CO ligand enhanced the reduction process. In their experiment, [Ru(bpy)2(CO)(Cl)]PF6 (2) was the best catalyst, which had a TONFA of 62 for a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 PS to PC ratio. Secondly, a study was also performed on the photosensitizer to catalyst ratio, which clearly showed the increase in formate formation as the concentration of the catalyst was decreased. The maximum reactivity was observed at a 16:1 PS to PC ratio with a TONFA of 419 and a selectivity of 80% for formate formation.42
:1 PS to PC ratio. Secondly, a study was also performed on the photosensitizer to catalyst ratio, which clearly showed the increase in formate formation as the concentration of the catalyst was decreased. The maximum reactivity was observed at a 16:1 PS to PC ratio with a TONFA of 419 and a selectivity of 80% for formate formation.42
Ishitani et al. developed a ring-shaped multinuclear ReI complex with cis,trans-[Re(bpy)(CO)2(P–P)2 (Table 1; 13) as the repeating unit. This complex is known to have excellent photophysical properties and excellent stability in the excited state and is a stronger oxidant in its excited state compared to the corresponding mononuclear Re complex.43–45 In 2016, Ishitani et al. reported the potential of this Re-ring as a photosensitizer in combination with a Ru-based photocatalyst, namely trans(Cl)-Ru(dtbb)(CO)2Cl2 (Table 1; 12) (dtbb = 4,4′-di-tert-butyl-2,2′-bipyridine). A typical experiment included the Ru-based photocatalyst and the trinuclear Re photosensitizer in an equimolar ratio in a CO2-saturated DMA:TEOA (5:1 v/v) solvent mixture. Formate was produced as the major product (TONFA of 290) along with H2 and CO as the minor products (TONH2 of 72 and TONCO of 20). When a stronger electron donor (1,3-dimethyl-2-(o-hydroxyphenyl)-2,3-dihydro-1H-benzo[d]imidazole, BI(OH)H) was added into the solution, no change in TONFA was observed but the selectivity of formate formation was increased. It should be noted that most of the photocatalytic systems used a higher amount of photosensitizer than the catalyst to enhance the electron transfer.46
All the previously described routes included a system where the catalyst and photosensitizer were separate. A major drawback was the requirement of electron transfer from the photosensitizer to the catalyst, and the efficiency was often not optimal and thus was limited in photocatalysis. To overcome this, Masaoka et al. reported in 2018 a function-integrated system, which acted as both the photosensitizer and the catalyst. Based on their earlier electrochemical studies, they chose trans(P,MeCN)-[Ru(tpy)(pqn)(MeCN)]2+ (Table 1; 16) (tpy = 2,2′:6′,2′′-terpyridine, pqn = 8-(diphenylphosphanyl)quinoline) as the catalyst.47 The photocatalytic reduction of CO2 was performed in a DMA/H2O (39:1, v/v) solvent system, which contained 1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzo[d]-imidazole (BIH) as a sacrificial electron donor. In this case, CO was selectively generated. When the reaction mixture was basified using TEOA as a sacrificial electron donor, formate was very selectively generated with a TONFA of 14.48
In 2019, Richeson et al. reported the use of ruthenium PNP pincer complexes as new photocatalysts for the reduction of CO2. CO2 was photoreduced in the presence of [Ru(k3-2,6-(Ph2PNMe)2(NC5H3))(CO)2Cl+]Cl− (Table 1; 17) (1 mM) as the catalyst, [Ru(bpy)3](PF6)2 (1 mM) as the photosensitizer and TEOA as the sacrificial electron donor in DMF. Under these conditions, CO2 was selectively photoreduced to formate (TONFA of 90), where H2 was formed as a by-product. By lowering the concentration of both the catalyst and photosensitizer to 25 μM, the selectivity was increased to 100%, since no H2 was formed, and the TONFA was increased to 380. They also examined the same reaction conditions with [Ru(k3-2,6-(Ph2PNH)2(NC5H3))(CO)2Cl+]Cl− (Table 1; 18) as the catalyst. Similar results were obtained, however a slightly lower efficiency was observed.49
In 2020, Arikawa et al. used CNC pincer complexes since they were known to incorporate CO2.50 In their experiment, CO2 was reduced in the presence of the CNC Ru catalyst [Ru(dmb)3](PF6)2 (23) (dmb = 4,4′-dimethylbipyridine) as a photosensitizer, BI(OH)H as the sacrificial electron donor and DMA/TEOA as the solvent mixture. Four types of ruthenium complexes bearing a CNC ligand were examined: (I) a CO ligand and a CNC ligand which was substituted by a tert-butyl group (II), a CO ligand and a CNC ligand which was substituted by a methyl group (III), a MeCN ligand and a CNC ligand which was substituted by a tert-butyl group (IV), and a MeCN ligand and a CNC ligand which was substituted by a methyl group. Comparison among them revealed that ruthenium complexes bearing a CO ligand (Table 1; 21 and 22) generated more formate compared to ruthenium complexes bearing a MeCN ligand (Table 1; 19 and 20). Furthermore, having a methyl substituent on the CNC yielded a higher TONFA for the formation of formate, however this was at the expense of the selectivity. Among them, [(MeCNC)Ru(bpy)(NCMe)](PF6)2 (20) and [(MeCNC)Ru(bpy)(CO)](PF6)2 (22) were the best catalysts, which obtained TONFA values of 4593 and 5634, respectively. In both cases, a selectivity of 72% was achieved.51
In conclusion, Ru-complexes are the most extensively studied metal centres for the photoreduction of CO2 to formate. As a result, varieties of novel complexes have been synthesized to achieve high reactivity. Although the structure–activity relationship is hard to determine, this diversity can give rise to further investigation of specific ligands on other metals. Additionally, Ru-based catalysts are, until now, the most active and efficient homogeneous photocatalysts for the generation of FA from CO2. However, the selectivity remained a problem, since CO is formed as a by-product.
Cobalt complexes
Ruthenium complexes are the well-studied photocatalysts for the reduction of CO2 to FA. However, recent studies have provided more concerns towards Earth abundant metals. In 1998, Neta et al. reported cobalt porphyrin photocatalysts for the photoreduction of CO2. Reductions of CO2 were performed in acetonitrile containing 5% of triethylamine (TEA). Control experiments clearly proved the formation of CO and formate from the reduction of CO2. When 1 × 10−5 mol L−1 of cobalt tetraphenylporphyrin (Table 2; 24, CoIITPP) was used, about 320–360 × 10−5 mol L−1 of formate was generated under their reaction conditions. However, CO was also produced to a lesser extent, which contributed to a total TONFA of >300 for the CoIITPP catalyst. Derivatives of the CoIITPP catalyst were also examined and the yields of formate using CoT3FPP (Table 2; 25), CoT3CF3PP (Table 2; 26) or CoTM2PyP (Table 2; 27, TM2PyP = tetrakis(N-methyl-2-pyridyl)porphyrin) as the photocatalyst were lowered by a factor of two. Comparatively, the yield was drastically lowered when CoTM3PyP (Table 2; 28, TM3PyP = tetrakis(N-methyl-3-pyridyl)porphyrin) and CoTM4PyP (Table 2; 29, TM4PyP = tetrakis(N-methyl-4-pyridyl)porphyrin) were examined. In all these cases, the porphyrin macrocycle was decomposed which resulted in low efficiency of these photocatalysts.52|
|
|||||||
|---|---|---|---|---|---|---|---|
| Cat. | PS | SED | Solvent | Sel. (%) | TONFA | λ (nm) | Ref. |
| Cat. represents catalyst; PS represents photosensitizer; SED represents sacrificial electron donor; Sel. represents selectivity; TONFA represents turnover number for formic acid generation; λ represents wavenumber of incident light; Ref. represents reference; N.A. represents not applicable. Selectivities and TONs were determined after the corresponding irradiation times unless stated otherwise. — = not mentioned. | |||||||
| 24 | N.A. | TEA | MeCN | >80 | >245 | <320 | 52 |
| 25 | N.A. | TEA | MeCN | — | >120 | <320 | 52 |
| 26 | N.A. | TEA | MeCN | — | >120 | <320 | 52 |
| 27 | N.A. | TEA | MeCN | — | >120 | <320 | 52 |
| 28 | N.A. | TEA | MeCN | — | >120 | <320 | 52 |
| 29 | N.A. | TEA | MeCN | — | >120 | <320 | 52 |
| 30 | 31 | TEA | MeCN | 92 | 110 | 460 | 55 |
| 30 | 31 | BIH/TEA | MeCN | 97 | 386 | 460 | 55 |
| 30 | 31 | BIH/TEOA | MeCN | 76 | 821 | 460 | 55 |
| 30 | 32 | BIH/TEOA | MeCN | 58 | 565 | 460 | 55 |
| 30 | 33 | BIH/TEOA | MeCN | 91 | 493 | 460 | 55 |
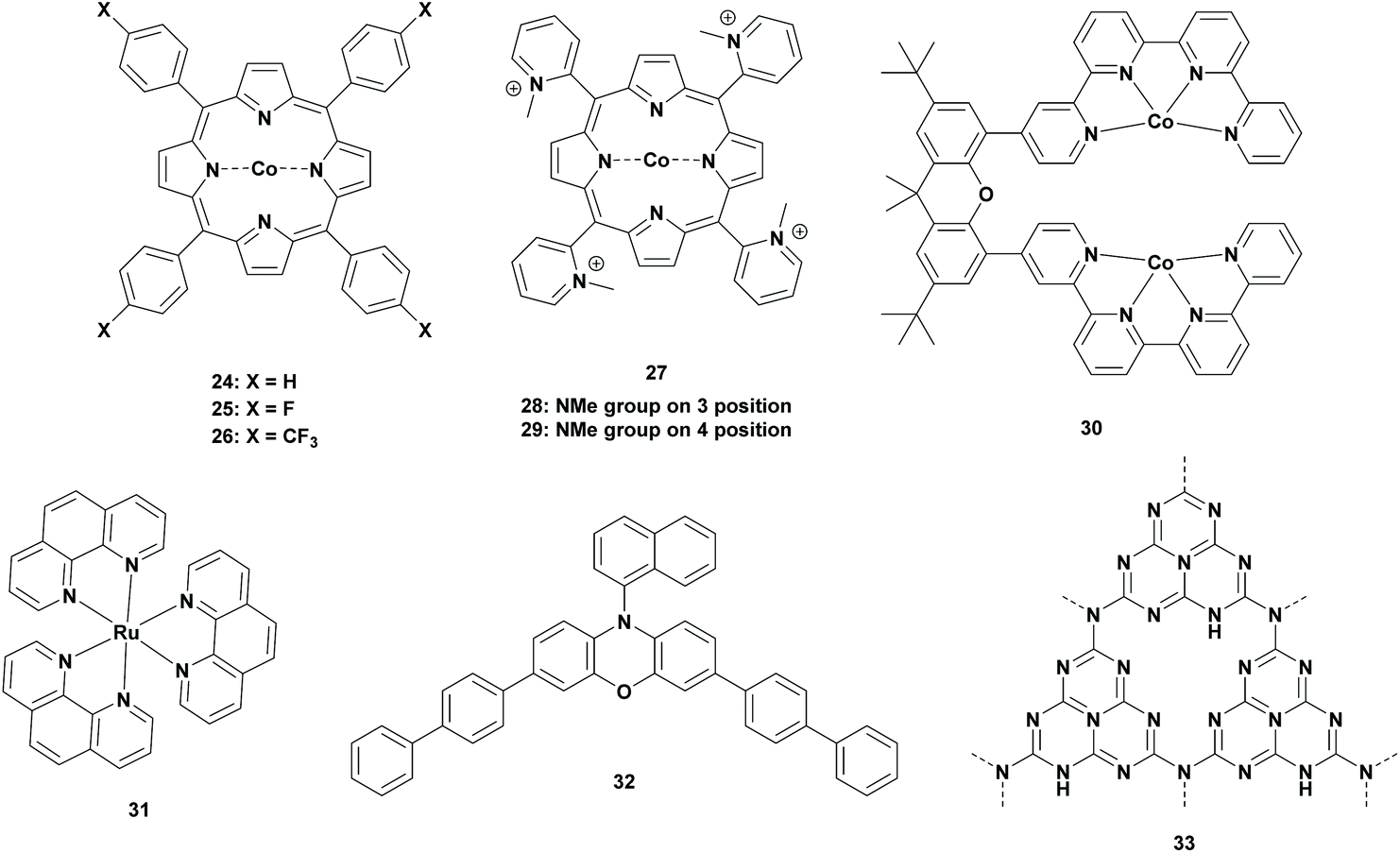
To solve this problem, Neta et al. continued their research where porphyrin complexes were replaced by corrin complexes. It has been shown that Co-corrins are more stable than Co-porphyrins due to the reduced tendency for protonation on the corrin ring.53 Although comparison with their previous studies was not possible since Co-corrins were insoluble in acetonitrile, they performed parallel experiments with CoTPP and hydroxocobalamin (vitamin B12) in acetonitrile/methanol (9:1, v/v) containing 5% TEA for direct comparison. In both of these experiments, CO was produced in the early stage, whereas formate was produced after longer irradiation. In the case of the corrin catalyst, seven times more formate was produced than when using the porphyrin catalyst. In addition, they observed that when 3 mmol L−1 of p-terphenyl (TP) was added as a PS, the yield was increased by a factor of three. This can be explained by the effective photoreduction of TP* by TEA to produce the TP˙− radical anion, which in turn reduced the Co-corrins very efficiently. The main disadvantage of this catalytic system was that Co-corrins could also react with protons to form hydrogen gas, which reduced the corrin macrocycle itself. Although this reduced Co-corrin was able to catalyse the reduction reactions, it bound with CO which lowered the catalytic activity over time.54
Later, in 2019, inspired by the multi-metallic catalytic complexes in nature, Robert et al. used cooperative metal systems for the selective production of formate. To their observation, a binuclear Co complex with a bi-quarterpyridine ligand ([Co2(biqpy)]4+ (Table 2; 30), biqpy = 4,4′′′′-(2,7-di-tert-butyl-9,9-dimethyl-9H-xanthene-4,5-diyl) di-2,2′:6′,2′′:6′′,2′′′-quarter-pyridine) was able to reduce CO2 selectively to formate with a high TONFA. Experiments were carried out using [Co2(biqpy)]4+ as the catalyst and [Ru(phen)3]2+ (Table 2; 31) as the photosensitizer in the presence of a sacrificial reductant in acetonitrile. In the presence of TEA (20 vol%) as the electron donor, formate was generated with 92% selectivity with a TONFA of 110. The selectivity and TONFA were increased to 96.5% and 386, respectively, when BIH was added to the solvent mixture. Moreover, the TONFA was increased even more to 821 when TEA/BIH was replaced by TEOA/BIH. However, the selectivity was dropped to 75.9%. In a second stage, Robert et al. investigated these optimized reaction conditions with cheaper sensitizers such as organic dyes or graphitic carbon nitrides (g-C3N4). In the presence of the phenyl chromophore (Table 2; 32), the TONFA and the selectivity were dropped to 565 and 59%, respectively. For g-C3N4 (Table 2; 33) the selectivity remained almost the same (91%), but the TONFA decreased to 493.55
In conclusion, Co-complexes were the first transition metal complexes used for the photoreduction of CO2 to FA. The main advantage of these complexes is their cheaper price due to high availability in the Earth's crust. Although only a few Co-complexes have been reported, it can be said that they possess moderate to good activities towards the reduction of CO2 to FA. To further enhance the momentum in this direction, ligand diversity should be thoroughly investigated.
Manganese complexes
In 2014, Ishitani et al. replaced the precious metal complexes with a manganese complex. The research was based on the report of Deronzier et al., where fac-Mn(bpy)(CO)3Br (Table 3; 34) was successfully used as an electrocatalyst for the reduction of CO2 to CO.56 Ishitani et al. performed the photocatalytic reduction of CO2 using this catalyst (0.05 mM) in the presence of [Ru(dmb)3]2+ (Table 1; 23) as the photosensitizer and BNAH (0.1 M) as a reductant in a 4:1 (v/v) DMF:TEOA solvent mixture under a CO2 atmosphere. Remarkably, 29.8 μmol of formate was formed (TONFA of 149) with a selectivity of 85% after 12 h. When the reaction mixture was changed to a 4:1 (v/v) ratio of DMA:TEOA, the TONFA dropped to 98 with similar selectivity. In a MeCN:TEOA solvent mixture, not only the TONFA was dropped to 78, but the selectivity was also dropped and a higher amount of CO was formed. Changing the photosensitizer to [Ru(bpy)3]2+ increased the TONFA slightly to 157, without changing the selectivity. UV-VIS studies revealed new absorption peaks at λmax = 630 nm and 820 nm after the start of the irradiation. The changes in the IR spectrum supported the formation of the [Mn(bpy)(CO)3]2 dimer. This could be explained by the selective photoexcitation of [Ru(dmb)3]2+, which was reductively quenched by BNAH to give [Ru(dmb)2(dmb˙−)]+. From this state, an electron transfer process proceeded to the Mn-photocatalyst to release Br− for the rapid formation of the Mn dimer. Within 30 minutes, this dimer disappeared, which enhanced the formation of formate and the formation of CO diminished. The active catalyst for the reduction of CO2 to formate was not clarified but it was proposed to be the five-coordinated manganese radical species [Mn(bpy)(CO)3]˙, which resulted from homolytic dimer cleavage.57
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Cat. | PS | SED | Solv. | Irr. t (h) | Sel. (%) | TONFA | λ (nm) | Ref. |
| Cat. represents catalyst; PS represents photosensitizer; SED represents sacrificial electron donor; Irr. t represents irradiation time; Sel. represents selectivity; TONFA represents turnover number for formic acid generation; λ represents wavenumber of incident light; Ref. represents reference. Selectivities and TONs were determined after the corresponding irradiation times unless stated otherwise. | ||||||||
| 34 | 23 | BNAH/TEOA | DMF | 12 | 85 | 149 | 480 | 57 |
| 34 | 23 | BNAH/TEOA | DMA | 12 | 81 | 98 | 480 | 57 |
| 34 | 23 | BNAH/TEOA | MeCN | 12 | 58 | 78 | 480 | 57 |
| 34 | 1 | BNAH/TEOA | DMF | 12 | 89 | 157 | 480 | 57 |
| 34 | 1 | BNAH/TEOA | MeCN | 16 | 24 | 15 | 480 | 59 |
| 34 | 1 | TEOA | DMF | 16 | 87 | 39 | 480 | 59 |
| 34 | 42 | BIH/TEOA | DMA | 2 | 75 | 157 | 436 | 62 |
| 35 | 23 | BNAH/TEOA | DMF | 15 | 94 | 127 | 470 | 58 |
| 35 | 23 | BNAH/TEOA | MeCN | 15 | 29 | 9 | 470 | 58 |
| 36 | 13 | TEA | DMA | 15 | 73 | 85 | 436 | 46 |
| 37 | 42 | BIH/TEOA | DMA | 2 | 28 | 65 | 436 | 62 |
| 37 | 42 | BIH/TEOA | DMA | 24 | <24 | 310 | 436 | 62 |
| 38 | 1 | BNAH/TEOA | MeCN | 16 | 87 | 52 | 480 | 59 |
| 39 | 1 | BNAH/TEOA | MeCN | 16 | 84 | 48 | 480 | 59 |
| 38 | 1 | BNAH/TEOA | DMF | 16 | 51 | 22 | 480 | 59 |
| 40 | 41 | BIH/TEOA | MeCN/H2O | 3 | 14 | 19 | ∼500 | 60 |
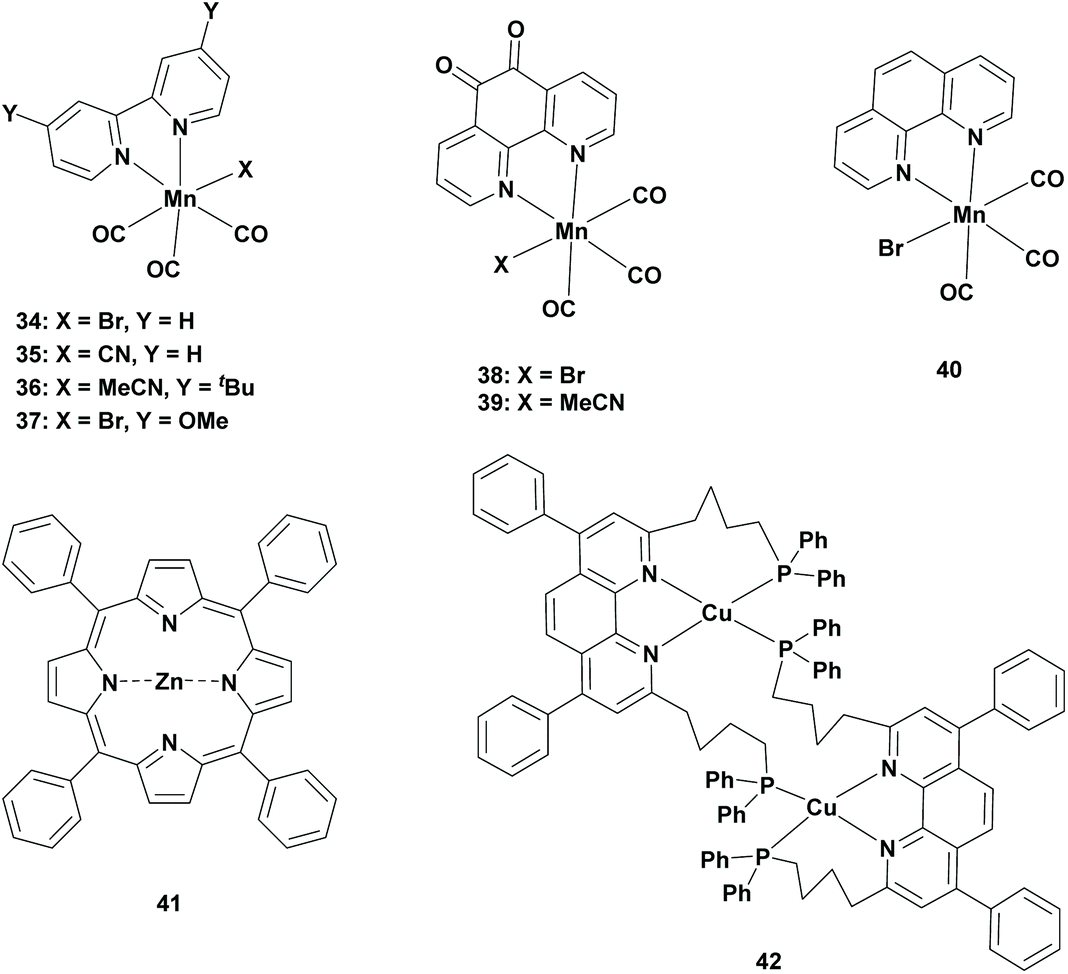
Later, Kubiak et al. have substituted the Br− ligand with the pseudohalogen cyanide (CN−) ligand. Electrocatalytic behaviour was proven to reduce CO2 to CO to a large extent with a faradaic yield close to 100%. The same study revealed that the CN− ligand did not readily dissociate from the Mn-based electrocatalyst. As a result, the dimer was not obtained as an intermediate for the formation of the active catalyst [Mn(bpy)(CO)3]˙ radical. Instead, the slightly different active catalyst [Mn(bpy)(CO)3]− anion was generated by a disproportionation reaction. This triggered the research for using this electrocatalyst as a photocatalyst to observe the selectivity pattern in the photoreduction of CO2. Typical experiments were conducted using fac-Mn(bpy)(CO)3(CN) (Table 3; 35) as the photocatalyst, [Ru(dmb)3]2+ as a photosensitizer and BNAH as a sacrificial reductant in a 4:1 (v/v) DMF:TEOA solvent mixture under a CO2 atmosphere. A maximum TONFA of 127 was obtained using 0.10 mM of the photocatalyst, 1.00 mM of the photosensitizer and 0.1 M of the reductant after the irradiation of 15 h. Remarkably, when the solvent mixture was changed to 4:1 (v/v) MeCN:TEOA, a decrease of formate was observed (TONFA of 9), whereas CO became the major reduction product (TONCO of 21). It was noted that fac-Mn(bpy)(CO)3(CN) underwent a disproportionation reaction under photocatalytic conditions. First, the photosensitizer was photoexcited and was reduced by BNAH to form [Ru(dmb)3]˙+. This species transferred an electron to the Mn-based photocatalyst to form a [Mn(CN)(bpy)−(CO)3]˙− radical anion. Two equivalents of this radical anion underwent a disproportionation reaction to yield the active photocatalyst [Mn(bpy)(CO)3]− and fac-Mn(bpy)(CO)3(CN) was regenerated. Later, it has been found that the [Mn(CN)(bpy)−(CO)3]˙− intermediate was not stable in MeCN, which provided a possible clarification for the low formation of formate in MeCN.58
The photocatalytic reduction of CO2 using Mn-based photocatalysts was best performed in DMF as the solvent. However, DMF is a toxic solvent with a high boiling point, thus making it hard to remove from the reaction mixture. Additionally, it has been proven that hydrolysis of DMF results in the formation of FA, making it difficult for the accurate quantification of the product by the photoreduction of CO2.39 Therefore, Chardon-Noblat et al. searched for new Mn-based photocatalysts in alternative solvents. They proposed Mn tricarbonyl complexes, which contained a 1,10-phenanthroline-5,6-dione ligand (Table 3; Mn(phen-dione)(CO)3Br, 38 and Mn(phen-dione)(CO)3MeCN, 39). Photocatalytic experiments were performed using a Mn-based photocatalyst (0.1 mM), Ru(bpy)3Cl2 as the photosensitizer (0.1 mM) and BNAH (0.1 M) as the sacrificial reductant in a 4:1 (v/v) MeCN:TEOA solvent mixture (Table 3). Both of these Mn catalysts generated formate as the major reduction product with a TONFA of 52 and 48, respectively. When the solvent mixture was changed to DMF:TEOA, formate generation decreased to a TONFA of 22 and the selectivity decreased as well (TONCO of 21). Additionally, a reference Mn photocatalyst ([Mn(bpy)(CO)3Br]) was examined under these reaction conditions which provided CO as the major product (TONCO of 47, TONFA of 15) in the MeCN:TEOA solvent mixture whereas formate was produced in the DMF:TEOA mixture (TONFA of 39). Notably, previous photoreductions using Mn-based photocatalysts in MeCN provided CO as the major reduction product. The difference in selectivity could be explained by in situ UV-VIS studies, which revealed a different intermediate from the usual Mn dimer, although these intermediates were too unstable for further characterization.59
As discussed earlier, Ishitani et al. have developed a ring-shaped multinuclear ReI complex with cis,trans-[Re(bpy)(CO)2(P–P)2] (Table 1; 13) as the repeating unit, which exhibited excellent photochemical properties. This Re-complex was used as a photosensitizer using fac-[Mn(dtbb)(CO)3(CH3CN)]+ (Table 3; 36) as the photocatalyst in an equimolar ratio in a CO2-saturated DMA:TEOA (5:1 v/v) solvent mixture. Formate was produced as the main reduction product with a TONFA of 85, while CO was formed as a minor product with a TONCO of 32. The addition of a strong sacrificial reductant such as BIH did not improve the reaction yield, which stated that the Re photosensitizer was excellent in exciting the photocatalyst itself.46
Ruthenium and to a lesser extent rhenium complexes have been widely used as photosensitizers in the reduction of CO2, since the Mn-based photocatalyst was not able to absorb visible light irradiation itself. Owing to the higher price of Ru-based photosensitizers, Bian et al. reported a Mn-based photosensitizer. Their experiments were conducted using fac-[Mn(phen)(CO)3Br] (Table 3; 40) (0.5 mM) as the precursor for the photocatalyst, ZnTPP (Table 3; 41) as the photosensitizer and TEA as a sacrificial reductant in a 20:1 (v/v) MeCN:H2O solvent mixture under a CO2 atmosphere. Several photocatalyst to photosensitizer ratios were examined, where a ratio of 4:1 provided the highest formation of formate (TONFA of 19). However, CO was the major reduction product (TONCO of 119).60
Later, Ishitani et al. substituted the precious metal photosensitizer with a copper photosensitizer. Previous studies revealed CuPS complexes as an excellent photosensitizer for the reduction of CO2.61 As a result, Ishitani et al. performed experiments using a Mn-based photocatalyst (0.05 mM), CuPS (Table 3; 42) as the photosensitizer (0.25 mM) and BIH (10 mM) as the sacrificial reductant in a 4:1 (v/v) DMA:TEOA solvent mixture under a CO2 atmosphere. In the case of Mn(bpy)(CO)3Br, formate was the major product with a TONFA of 157. However, CO was also generated (TONCO of 50). In the case of Mn(4-OMe-bpy)(CO)3Br (Table 3; 37), CO was the main product (TONCO of 164) and formate was produced to a lesser extent (TONFA of 65). After 2 hours of irradiation, BIH was completely consumed, and hence the reaction stopped after 2 h. To see the effect of the amount of BIH, the reaction was repeated using 0.1 M of BIH in the Mn(4-OMe-bpy)(CO)3Br system. After 24 h, the TONFA of formate was increased to 310 and simultaneously the TONFA of CO was increased to 1004, which indicated that consumption of BIH stopped the reaction process. Although the reaction using Mn(bpy)(CO)3Br was not performed with prolonged irradiation times with a larger amount of BIH, it is safe to say that the TONFA would also increase. Furthermore, the increased reaction time showed that CuPS was a very stable and efficient redox photosensitizer for the photoreduction of CO2.62
In summary, like the Co-complexes, the main advantage is the use of an Earth-abundant metal instead of a precious metal. A high TONCO can be obtained, but the TONFA still remains a problem. Additionally, ligand diversity should be thoroughly investigated to achieve high selectivity.
Iron, molybdenum, iridium and rhenium complexes
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Cat. | PS | SED | Solv. | Irr. t (h) | Sel. (%) | TONFA | λ (nm) | Ref. |
| Cat. represents catalyst; PS represents photosensitizer; SED represents sacrificial electron donor; Irr. t represents irradiation time; Sel. represents selectivity; TONFA represents turnover number for formic acid generation; λ represents wavelength of the incoming radiation; Ref. represents reference; N.A. represents not applicable. Selectivities and TONs were determined after the corresponding irradiation times unless stated otherwise. | ||||||||
| 43 | 44 | TEA | MeCN | 20 | >99 | 5 | 460 | 63 |
| 46 | 1 | BIH/TEOA | MeCN | 15 | 33 | 31 | 400 | 65 |
| 47 | 1 | BIH/TEOA | MeCN | 15 | 39 | 83 | 400 | 65 |
| 48 | 1 | BIH/TEOA | MeCN | 15 | 10 | 80 | 400 | 65 |
| 50 | N.A. | TEOA | MeCN | 24 | 90 | 20 | >410 | 67 |
| 51 | N.A. | BIH | DMA/H2O | 145 | 87 | 2080 | >400 | 69 |
| 53 | N.A. | TEOA | DMA | 24 | >99 | 10 | 405 | 71 |
| 53 | 1 | TEOA | DMA | 24 | 88 | 2750 | 405 | 71 |
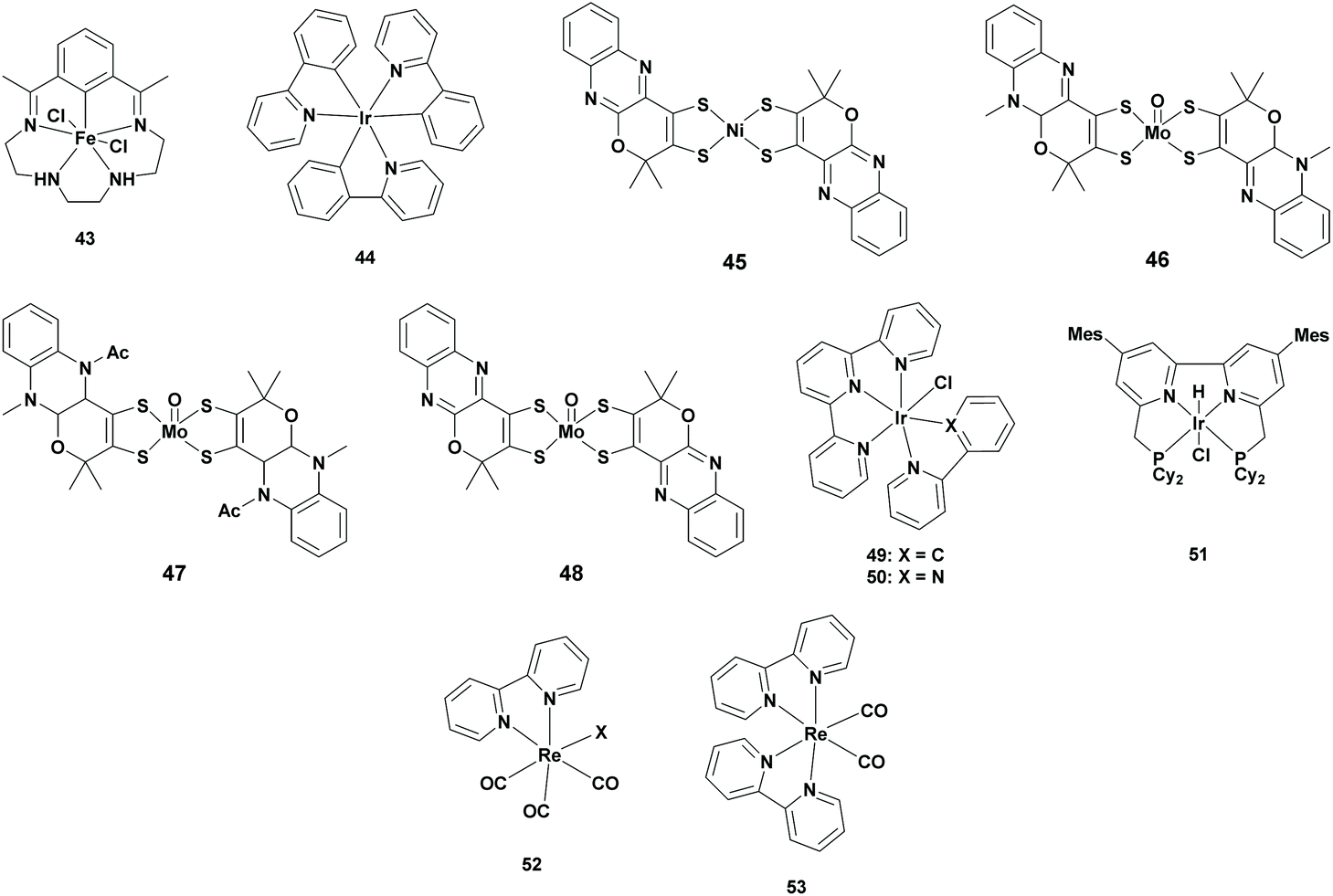 :10 ratio. BIH was added as the sacrificial reductant and a 5:1 (v/v) MeCN:TEOA solvent mixture was used. All of these three complexes reduced CO2 to formate with a TONFA of 31, 83 and 80 using ([Mo(O)(H-qpdt)2]2−, [Mo(O)(2H-qpdt)2]− and [Mo(O)(qpdt)2]2− respectively, however all of them also produced an equal or larger amount of H2.65
:10 ratio. BIH was added as the sacrificial reductant and a 5:1 (v/v) MeCN:TEOA solvent mixture was used. All of these three complexes reduced CO2 to formate with a TONFA of 31, 83 and 80 using ([Mo(O)(H-qpdt)2]2−, [Mo(O)(2H-qpdt)2]− and [Mo(O)(qpdt)2]2− respectively, however all of them also produced an equal or larger amount of H2.65
Most photocatalytic systems made use of both a photosensitizer and a catalyst for the conversion of CO2 to FA. However, using a metal complex which exhibits both of these properties gives the advantage of lowering the activation energy and controlling the selectivity. For this reason, many systems were reported using metal complexes possessing both these properties. However, in most of the cases, CO was formed as the major product.68 In 2020, Saito et al. reported the use of a tetradentate PNNP-type Ir complex Mes-IrPCy2 (Table 4; 51) as a photosensitizer and catalyst for the conversion of CO2 to FA. The key aspects for designing this catalyst were based on molecular engineering: (i) use of bulky PNNP ligands to prevent catalyst degradation and for controlling the selectivity and (ii) use of bipyridyl ligands with CH2P groups, which can act as additional proton donors. The catalyst was examined in a 9:1 (v/v) DMA:H2O solution containing BIH as the sacrificial reductant under a CO2 atmosphere. Formate was selectively (87%) produced with a TONFA of 2080, whereas CO was also generated with a TONCO of 470.69
In conclusion, Ir-based complexes are highly suitable for the photocatalytic reduction of CO2 to FA. Moreover, reported Ir-based catalytic systems do not require an additional PS since the catalyst itself can adsorb the incident light very efficiently. Here, formate was formed with high selectivity and with a higher TONFA. Despite these great advantages, the main disadvantage of Ir-based metal complexes is their higher price.
:1 (v/v) DMA:TEOA solvent mixture. Remarkably, almost no CO was detected, whereas formate was selectively formed (TONFA of 10). In the second experiment, [Ru(bpy)3]2+ (1 mM) was added as a photosensitizer to 0.01 mM of the catalyst in a 4:1 (v/v) DMA:TEOA solvent mixture. As expected, the reactivity of the photocatalytic system increased (TONFA of 2750), where at the same time the selectivity slightly dropped (88%).71
In conclusion, switching from Earth abundant metal catalysts to Ir- and Re-based photocatalysts ensured higher TONs (TONFA > 2000). Compared to the Rh-based catalysts, the activity was lower, but the selectivity increased to almost 90%. However, it will be excellent progress if this high selectivity can be achieved by Earth abundant metal complexes.
Metal-free catalysts or photosensitizers
Homogeneous photoreduction of CO2 is extensively studied using metal complexes. However, photoreduction of CO2 using metal-free photocatalysts has been reported in 1990 by Yanagida et al. Earlier, they reported that p-terphenyl (Table 5; 54) was an effective catalyst for the photoreduction of acetaldehyde to ethanol in the presence of TEA as an electron donor, which induced the interest in using p-terphenyl as a catalyst for the photoreduction of CO2.72 Although previous reports stated that photochemical reactions using CO2 and hydrocarbons generated the carboxylated hydrocarbon, Yanagida et al. performed research on the photoreduction of CO2 in the presence of p-terphenyl.73 To their delight, a CO2-saturated DMF:TEA mixture in the presence of p-terphenyl was irradiated for 15 hours, which resulted in the formation of formate (TONFA of 3) and a trace amount of CO. The experiment was repeated with different solvents, where MeCN and THF (tetrahydrofuran) yielded a trace amount of reduction products. It was also found that methanol and ethanol yielded no reduction products. In the second stage, they examined other terphenyls for the photoreduction of CO2. Both o-terphenyl (Table 5; 55) and m-terphenyl (Table 5; 56) showed less reactivity compared to p-terphenyl. Additionally the insoluble polymer PPP (Table 5; 57, poly(p-phenylene)) was examined, which resulted in the formation of formate with low efficiency.74
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Cat. | PS | SED | Solv. | Irr. t (h) | Sel. (%) | TONFA | λ (nm) | Ref. |
| Cat. represents catalyst; PS represents photosensitizer; SED represents sacrificial electron donor; Irr. t represents irradiation time; Sel. represents selectivity; TONFA represents turnover number for formic acid generation; λ represents wavelength of the incoming radiation; Ref. represents reference; N.A. represents not applicable. Selectivities and TONs were determined after the corresponding irradiation times unless stated otherwise. — = not mentioned.a Phen ligand in stead of bpy ligand.b KCl was used as an electrolyte. | ||||||||
| N.A. | 54 | TEA | DMF | 15 | 86 | 3 | >290 | 74 |
| N.A. | 54 | TEA | MeCN | 15 | 95 | 1 | >290 | 74 |
| N.A. | 54 | TEA | THF | 15 | 77 | <1 | >290 | 74 |
| N.A. | 54 | TEA | MeOH | 15 | N.A. | 0 | >290 | 74 |
| N.A. | 54 | TEA | EtOH | 15 | N.A. | 0 | >290 | 74 |
| N.A. | 55 | TEA | DMF | 15 | 79 | <1 | >290 | 74 |
| N.A. | 56 | TEA | DMF | 15 | 78 | <1 | >290 | 74 |
| N.A. | 57 | TEA | DMF | 15 | 11 | <1 | >290 | 74 |
| 58 | 54 | TEA | DMF/MeOH | 1 | N.A. | 0 | >290 | 76 |
| 58 | 54 | TEA | MeCN/MeOH | 1 | 30 | 1 | >290 | 76 |
| 59 | 54 | TEA | MeCN/MeOH | 1 | N.A. | 0 | >290 | 76 |
| 58 | 54 | TEA | MeCN/H2O | 1 | 64 | <1 | >290 | 76 |
| 58 | 54 | TEOA | MeCN/MeOH | 1 | 38 | 3 | >290 | 22 |
| 58 | 54 | TIPOA | MeCN/MeOH | 1 | 21 | >3 | >290 | 22 |
| 58 | 67 | TEA | MeCN/MeOH | 3 | 92 | — | >290 | 80 |
| 58 | 68 | TEA | MeCN/MeOH | 3 | 94 | — | >290 | 80 |
| Py | 1 | Vit. C. | Aq. Py | 6 | >99 | 11 | ∼470 | 81 |
| Py | 1 | Vit C. | Aq. Py | 6 | >99 | 76b | ∼470 | 81 |
| 69 | 71 | TEOA | EtOH/H2O | 10 | >99 | 14000 |
420 | 85 |
| 70 | 71 | TEOA | EtOH/H2O | 10 | >99 | 13100 |
420 | 85 |
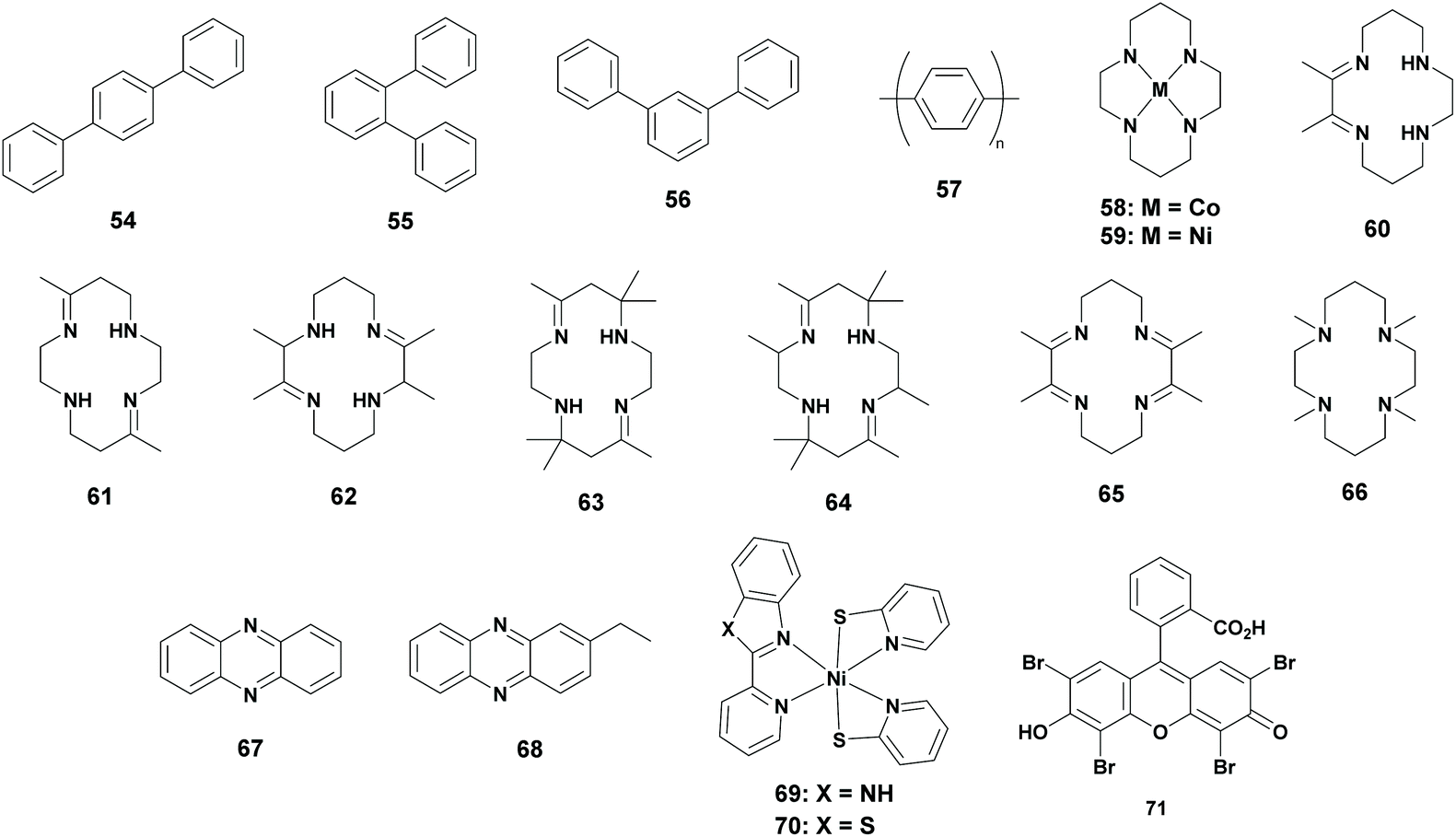
Based on a report where metal cyclam complexes were used as an electron mediator, Yanagida and co-workers continued their research.75 They investigated the photoreduction of CO2 by p-terphenyl in the presence of Co3+-cyclam (Table 5; 58) or Ni2+-cyclam (Table 5; 59) for investigating the electron mediation properties. It has to be mentioned that methanol had to be added to the mixture in order to solubilize the cyclam complex. They found that no formate was formed in the DMF/MeOH solvent mixture when the Co3+-cyclam complex was used, whereas CO was selectively formed. Switching the solvent mixture to MeCN/MeOH, formate was formed to some extent, whereas CO remained the major product (1:2 ratio). Unfortunately, the Ni2+-cyclam complex was unable to reduce CO2 photochemically to neither formate nor CO. When the reaction was performed in an MeCN:H2O mixture, formate was formed as the major product, however, the activity of the system was dropped.76
Lehn et al. reported that the tertiary amine electron donor influenced the photoreduction of CO2, hence further research was performed by Yanagida and co-workers.77 It was found that when β-hydroxylated tertiary amines, such as TEOA and TIPOA (tri-2-propanolamine) were used, the activity increased more than two fold. Additionally, the structural effects of the 14-membered tetraazamacrocyles (Table 5; 60 to 66) were also examined. Partially unsaturated cyclams with two or four C-methyl groups provided comparable activities, whereas more extensively C-methylated partially unsaturated cyclams decreased the activity drastically. This was explained by the steric hindrance of the methyl groups which reduced the accessibility of CO2 on the metal centre and also reduced the ligand exchange of the Cl atom with TEA or TEOA. CO2 reduction was lowered when a completely reduced cyclam was used whereas hydrogen production dominated. This result implied that the absence of N–H groups reduced the CO2 binding probability. N-Methylated cyclams did not show any activity in the photoreduction of CO2, which was explained by the steric hindrance for CO2 binding.22 Since the results of these photoreductions were comparable or worse, they are not given in the table.
On the other hand, photoreaction systems using phenazine (derivatives) as a photosensitizer were reported.78,79 Yanagida et al. continued their research with the use of phenazine (Table 5; 67) instead of p-terphenyl as a photosensitizer. A typical experiment included the Co-cyclam complex as the catalyst, phenazine as the photosensitizer and TEA as the sacrificial reductant in a 1:10 (v/v) MeCN:MeOH solvent mixture under a CO2 atmosphere. Remarkably, they observed the selective formation of formate with a trace amount of CO and H2. Phenazine was gradually degraded in the photocatalytic system. GCMS and NMR analyses revealed that 2-ethylphenazine (Table 5; 68) was formed, along with other unidentified products. Later, it was found that 2-ethylphenazine was as effective as phenazine for the photoreduction of CO2 to formate.80
Previous examples only made use of a metal-free photosensitizer. It took until 2013 before a metal-free catalyst was reported for the photoreduction of CO2 to FA, although the main goal was to obtain methanol. This research was based on the electrocatalytic reduction of CO2 to methanol using a pyridine (Py) catalyst. Only Pt or Pd electrodes were active in the electrocatalytic system, since they shift the reduction potential to more positive values compared to Hg electrodes. MacDonnell and co-workers investigated this protocol in terms of photochemistry by replacing the electrodes with a homogeneous photosensitizer and by adding a sacrificial reducing agent. Their photochemical experiments contained pyridine as the photocatalyst, [Ru(phen)3]2+ as the photosensitizer and ascorbic acid as the sacrificial donor. Although formate was the main product, the system was optimized according to the formation of methanol, since it was the desired product. In a 1 to 1 ratio of cat/PS, formate was produced with a TONFA of 11, whereas methanol was not formed. By decreasing the ratio of cat/PS to 1:200, the formation of methanol was increased (TONMeOH of 0.15), whereas the formation of HCOOH decreased (Table 5). In electrochemical systems an electrolyte is added and therefore, MacDonnell and co-workers investigated the addition of KCl to the mixture. This immensely increased the formation of formate to a TON of 76 and doubled the formation of MeOH. Other salts (LiCl, NaCl, RbCl, CsCl and MgCl2) had little to no effect on the system. It is important to mention that the pH of the mixture had to be 5.0, which implied that both pyridine and the protonated pyridinium salt were important for the catalytic activity. Moreover, in an acidic environment ascorbic acid was protonated which was a less prominent electron donor than the ascorbate molecule.81
In biological systems, CO dehydrogenase is responsible for the reduction of CO2 to CO. It was reported that the excited state of CO dehydrogenase possesses an unsaturated NiII species containing three S ligands.82 However, these Nickel thiolate complexes have only been examined for H2 production.83,84 In 2020, Jinheung et al. reported the first use of new mononuclear N/S ligated NiII complexes using eosin Y (71) as the photosensitizer and TEOA as the sacrificial reductant in an EtOH/H2O solvent mixture. Interestingly, complexes Ni(pbi)(pyS)2 (Table 5; 69, pbi = 2-(2-pyridyl)-benzimidazole; pyS = pyridine-2-thiolate) and Ni(pbt)(pyS)2 (Table 5; 70, pbt = 2-(2-pyridyl)benzothiazole) photoreduced CO2 to formate in a selective way, with a TONFA of 14000 and 13100, respectively.85
In general, the use of organic molecules instead of metal-based compounds increased the sustainability of these reactions. Moreover, organic molecules are often cheaper compared to the transition metal complexes. However, when an organic compound was used as the photocatalyst, a very low TONFA was observed. Moreover, UV-light was needed to excite these organocatalysts. Therefore, from the reported values it can be assumed that the organic compounds are not highly efficient at reducing CO2 to FA. On the contrary, when an organic compound was used as a photosensitizer, a very high TONFA is reported. Moreover, the use of eosin Y as the photosensitizer yielded FA in a very selective way and visible light can be used. This generated a very interesting avenue for further research to develop novel organic dyes as photosensitizers.
Supramolecular complexes
Ishitani et al. found in 2005 a new route for the photoreduction of CO2 to CO using a supramolecular photocatalyst, which possessed both a photosensitizer unit and a photocatalyst unit. The RuRe supramolecular catalysts were already studied for the photoreduction of CO2 to FA or CO.86–88 However, studies using aqueous solutions have not been published. Ishitani et al. stated that the use of water as a solvent and an electron donor would be the ultimate goal, since it is non-toxic and abundantly present on the Earth. Therefore, they continued the research on the photoreduction of CO2 with the use of supramolecular complexes in aqueous solutions. Typically, experiments were carried out using BNAH or BIH. However, these were not water soluble, so other electron donors were also examined and only sodium ascorbate was found to be efficient in this system. Their experiments contained RuReCl (Table 6; 76) as both the photocatalyst and photosensitizer and sodium ascorbate as the sacrificial reductant in water under a CO2 atmosphere. Since RuRe supramolecular complexes form CO in organic solvents, it was remarkable that formate was produced with a selectivity of 83% and a TONFA of 25. For comparison, this experiment was repeated with the photocatalyst and photosensitizer mononuclear complexes, which only produced a small amount of formate (TONFA of 2). The low efficiency of this photocatalytic system was explained by the fast back electron transfer (BET) from the one electron reduced species (OERS) of the photosensitizer unit to the oxidized ascorbate. Furthermore, they investigated the influence on the formation of formate when a mononuclear photosensitizer was added to the mixture. This clearly accelerated the reaction, probably due to the possibility of a new electron supply route to the photocatalyst unit in the RuReCl supramolecular complex.89|
|
|||||||
|---|---|---|---|---|---|---|---|
| Cat. + PS | SED | Solv. | Irr. t (h) | Sel. (%) | TONFA | λ (nm) | Ref. |
| Cat. represents catalyst; PS represents photosensitizer; SED represents sacrificial electron donor; Irr. t represents irradiation time; Sel. represents selectivity; TONFA represents turnover number for formic acid generation; λ represents wavelength of the incoming radiation; Ref. represents reference; N.A. represents not applicable. Selectivities and TONs were determined after the corresponding irradiation times unless stated otherwise. | |||||||
| 72 | BNAH/TEOA | DMF | 20 | 88 | 315 | >500 | 90 |
| 73 | BNAH/TEOA | DMF | 20 | 50 | 353 | >500 | 90 |
| 74 | BNAH/TEOA | DMF | 20 | 38 | 234 | >500 | 90 |
| 75 | BNAH/TEOA | DMF | 20 | 89 | 562 | >500 | 90 |
| 75 | BNAH/TEOA | DMF | 20 | 90 | 671 | >500 | 90 |
| 76 | Vit C. | H2O | 24 | 83 | 25 | >500 | 89 |
| 75 | BIH/TEOA | DMF | 20 | 72 | 641 | >500 | 91 |
| 75 | BI(OH)H/TEOA | DMF | 20 | 87 | 2766 | >500 | 91 |
| 77 | BI(OH)H | DMA | 20 | 8 | 11 | 480 | 92 |
| 78 | BI(OH)H | DMA | 20 | 93 | 181 | 480 | 92 |
| 79 | BIH/TEOA | DMA | 6 | 77 | 98 | ∼550 | 93 |
| 80 | BIH/TEOA | DMA | 6 | 65 | 33 | ∼550 | 93 |
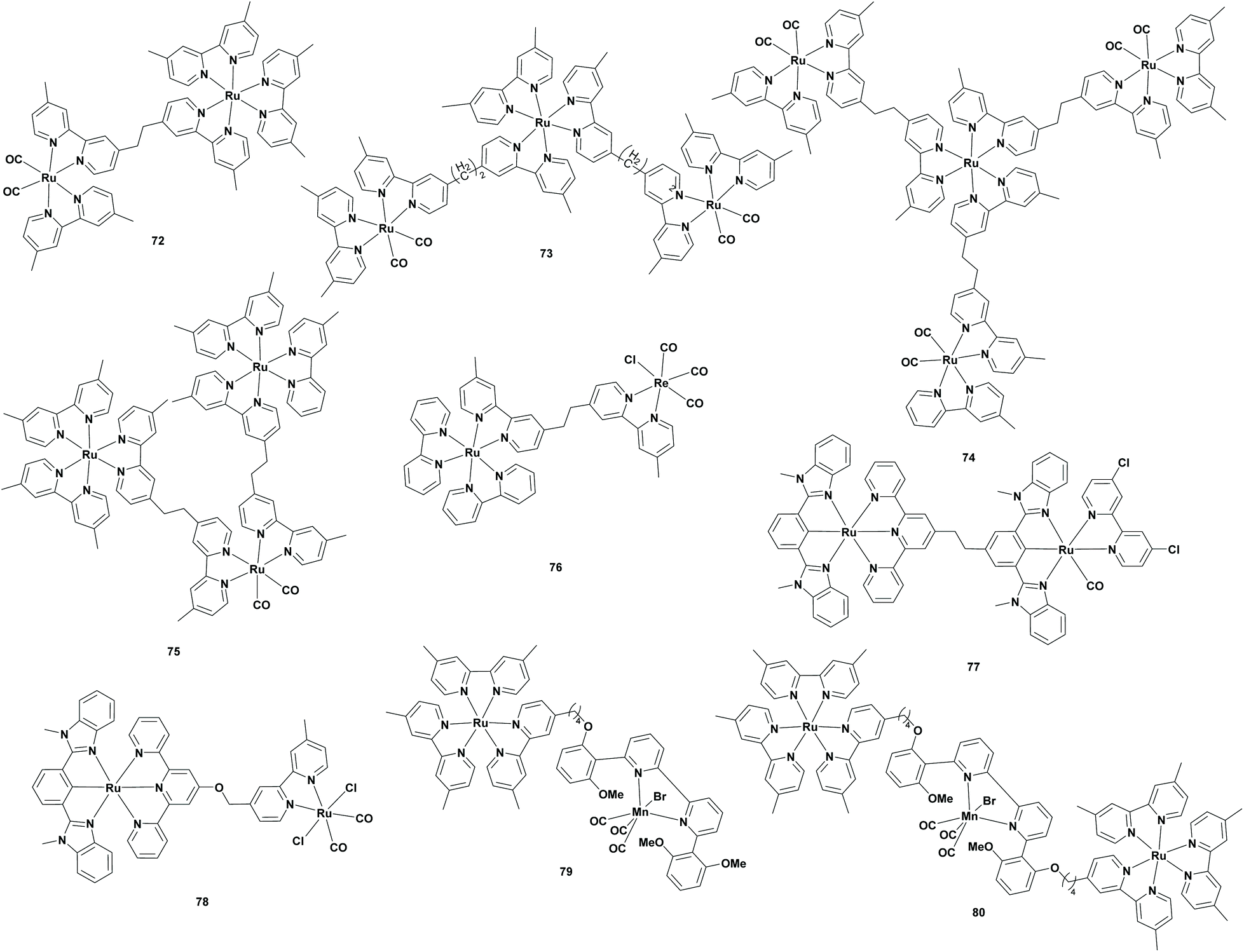
Ishitani et al. applied the supramolecular complex concept to the [Ru(bpy)3]2+ (Table 1; 1) photosensitizer and the [Ru(bpy)2(CO2)]2+ (Table 1; 5) catalyst. Compared to mononuclear Ru complexes, supramolecular complexes required BNAH, since TEOA cannot reduce CO2 under photocatalytic conditions. However, TEOA still played an important role as a base for hydrogen abstraction from the one-electron oxidation product of BNAH (BNAH˙+). Additionally, formate was formed under basic conditions, so TEOA basified the mixture for selective formate generation. Firstly, a supramolecular structure containing one unit of both the photosensitizer and catalyst (Table 6; 72) was investigated. Furthermore, they compared the supramolecular system to the system where the photosensitizer and catalyst were separated and concluded that in the case of the supramolecular system more formate was generated (26.9 μmol compared to 5.3 μmol). Moreover, no precipitate was formed using the supramolecular system. Later, they changed the ratio of the photosensitizer to catalyst in the supramolecular system. When more catalyst was present compared to the photosensitizer (Table 6; 73 and 74), the yield of formate was dropped and the CO yield was increased. For example, a supramolecular structure consisting of one unit of the photosensitizer and three units of the catalyst (Table 6; 74) yielded 5.3 μmol of formate and 8.1 μmol of CO. In contrast, when more photosensitizer compared to the catalyst was present, the formate yield was increased as seen in the experiment with a supramolecular structure consisting of two units of photosensitizer and one unit of catalyst (Table 6; 75) (30.4 μmol and a TONFA of 562). In the second stage of the experiment, they tried to improve the reductive quenching of the excited state of the supramolecular system for improved catalyst stability and more efficient formate formation. For this purpose, 1-(4-methoxybenzyl)1,4-dihydronicotinamide (MeO-BNAH) was used as the sacrificial electron donor, where the stability was drastically improved and the formate formation was increased to 36.8 μmol (TONFA of 671).90
Ishitani et al. were not satisfied with their reported system, since the quantum yield for formate formation was relatively low (ΦHCOOH = 0.061). Moreover, studies revealed that BNAH was inefficient in quenching the excited state of the Ru photosensitizer unit, that a relatively fast BET occurs from the reduced photosensitizer unit to the oxidized BNAH+ and that BNA dimers accumulated by deprotonation of BNAH˙+ molecules followed by coupling processes.88,90 Therefore, they replaced BNAH with a benzimidazoline derivative, BIH, which was able to quench the excited state of the photosensitizer unit more efficiently. Furthermore, it was more effective than BNAH in decreasing the BET. In the first experiment, the DMF:TEOA solvent mixture containing the Ru2Ru(CO) supramolecular complex (Table 6; 75) and BIH as the sacrificial reducing agent was irradiated under a CO2 atmosphere. Here, both the formate and CO were produced with a TONFA of 641 and a TONCO of 237, respectively. It should be noted that by switching BNAH to BIH, the TONFA and the ΦHCOOH increased, whereas the selectivity dropped. In the second stage, Ishitani et al. replaced BIH with another benzimidazoline derivative BI(OH)H, since this compound was able to supply two electrons and two protons to eliminate the proton shortage in the reaction mixture. Using BI(OH)H instead of BIH under the same conditions, formate was produced with higher efficiency (TONFA of 2766) and selectivity (87%).91
Due to the hybridization of supramolecular complexes to semiconducting heterogeneous materials, Ishitani and co-workers developed a new type of supramolecular complex, consisting of a Ru catalyst unit and a Ru photosensitizer unit with two different tridentate ligands. In their first experiment, a DMA:H2O solution containing RuRu(Cl)(bpy)(CO) (Table 6; 77) as the supramolecular complex and BI(OH)H as the sacrificial reductant was irradiated under a CO2 atmosphere. In this case, CO was formed as the main product (TONCO of 134) with a minor amount of FA (TONFA of 11). In contrast, when RuRu(CO)2Cl2 (Table 6; 78) was used as the supramolecular complex under the same conditions, FA was the main product (TONFA of 181). It is noteworthy that the water was the proton source instead of TEOA, which could be the possible reason for the lower TONs.92
With the intention of replacing the precious Ru catalyst unit of the supramolecular complex, Ishitani et al. investigated the use of a Mn catalyst unit for supramolecular CO2 reduction. This proposal was supported by the Earth abundant Mn, its catalytic behaviour (see the Manganese section) and the fact that mononuclear Mn photocatalysts formed dimers, which decomposed under visible light irradiation. The incorporation of a photosensitizer should suppress the dimerization. In the first reaction, a DMA:TEOA solvent mixture containing the Ru–Mn supramolecular complex (Table 6; 79) and BIH as a sacrificial reductant was irradiated under a CO2 atmosphere. Here, formate was produced as the major product (TONFA of 98) and CO as the minor product (TONCO of 29). When the mononuclear complexes were added instead of the supramolecular complex, the production rate of formate slowed down. When the Ru2–Mn (Table 6; 80) supramolecular complex was used, the formate formation slowed down even further (TONFA of 33).93
It can be concluded that the supramolecular complexes are viable alternatives for the photoreduction of CO2 to FA. One of the main advantages is that these complexes possess a PS and the active catalytic centre in the same molecule and for that reason, a straightforward electron transfer occurs from the PS unit to the PC unit. However, synthesis of these complexes is quite challenging and tedious.
Partially homogeneous CO2 photoreduction
Metal-based semiconductors
From an economic point of view, heterogenization of the homogeneous catalyst is interesting, since the catalyst can easily be recovered from the reaction mixture and can be recycled. Furthermore, semiconductor-based heterogeneous photocatalysts possess superior oxidation ability. It wasn't until 2010 that Sato et al. linked a Ru complex to a semiconductor for the selective reduction of CO2 to FA. In this combination, the semiconductor was photoexcited, which induced electron transfer from its conduction band to the catalyst in a Z-scheme system. However, the system was not solely photocatalytic since an electric current was required. They chose [Ru(bpy)2(CO)2]2+ (Table 1; 5), [Ru(dcbpy)(bpy)(CO)2]2+ (Table 7; 82) and [Ru(dcbpy)2(CO)2]2+ (Table 7; 81) (dcbpy = 4,4′-dicarboxy-2,2′-bipyridine) as the catalysts and N-doped Ta2O5 as the semiconductor-based photocatalyst. The [Ru-(dcbpy)2]/N-Ta2O5 exhibited the highest TONFA of 89 for the formation of FA per metal complex with more than 75% selectivity. It was observed that the amount of FA formation was increased linearly upon increasing the amount of the photocatalyst. Using [Ru-(dcbpy)(bpy)CO]/N-Ta2O5, the TONFA decreased to around 30, whereas using a non-linked catalyst and semiconductor mixture, the TONFA decreased to less than 10. It was shown that the carboxylic acid group improved the electron transfer from the semiconductor to the catalyst by direct linkage, however, the exact reason was not clear.94|
|
|||||||
|---|---|---|---|---|---|---|---|
| Cat. | SC | Solv. | Irr. t (h) | Sel. (%) | TONFA | λ (nm) | Ref. |
| Cat. represents catalyst; SC represents semiconductor; Solv. represents solvent; Irr. t represents irradiation time; Sel. represents selectivity; TONFA represents turnover number for formic acid generation; λ represents wavelength of the incoming radiation; Ref. represents reference; N.A. represents not applicable. Selectivities and TONs were determined after the corresponding irradiation times unless stated otherwise. — = not mentioned.a TiO2 photoanode was used.b SrTiO3 photoanode was used.c TEOA as sacrificial electron donor was used.d Aqueous solution.e Two-cell configuration.f One-cell configuration. | |||||||
| 81 | N-Ta2O5a | MeCNc | 8 | >75 | 89 | >410 | 94 |
| 82 | N-Ta2O5a | MeCNc | 8 | >75 | >30 | >410 | 94 |
| 83 | N-Ta2O5a | MeCNc | 60 | <50 | 118 | >410 | 95 |
| 84 | InPa | H2O | N.G. | — | — | >400 | 20 |
| 84 + 85 | InPa | H2O | N.G. | — | — | >400 | 20 |
| 84 + 85 | InPa | NaHCO3d | N.G. | — | — | >400 | 20 |
| 84 + 85 | InPb | NaHCO3d | N.G. | — | 18e | >400 | 97 |
| 84 + 85 | InPb | NaHCO3d | N.G. | — | <12f | >400 | 97 |
| 86 | TaON | MeOH | 9 | 70 | 41 | >400 | 21 |
| 86 | CaTaO2N | DMAc | 15 | >99 | 9 | >400 | 98 |
| 86 | Ag-CaTaO2N | DMAc | 15 | >99 | 32 | >400 | 98 |
| 86 | Pb2Ti2O5.4F1.2 | MeCNc | 15 | >99 | 26 | >480 | 102 |
| 86 | GaN:ZnO |
Na2CO3d | 15 | 78 | 359 | 365 | 101 |
| 86 | TiO2:Ta/N |
Na2CO3d | 15 | 67 | 18 | 365 | 101 |
| 86 | Li2LaTa2O6N | MeCNc | 15 | 97 | 50 | >400 | 103 |
| 86 | Ag-Li2LaTa2O6N | MeCNc | 15 | 99 | 110 | >400 | 103 |
| 86 | Y2Ta2O5N2 | DMAc | 24 | >99 | 5 | >400 | 100 |
| 86 | Ag-Y2Ta2O5N2 | DMAc | 24 | >99 | 18 | >400 | 100 |
| 86 | Ta3N5 | MeCNc | 15 | 93 | — | >480 | 107 |
| 86 | Ag-Ta3N5 | MeCNc | 15 | 98 | — | >480 | 107 |
| 86 | TbTaOxNy | MeCNc | 15 | >99 | 19 | >400 | 106 |
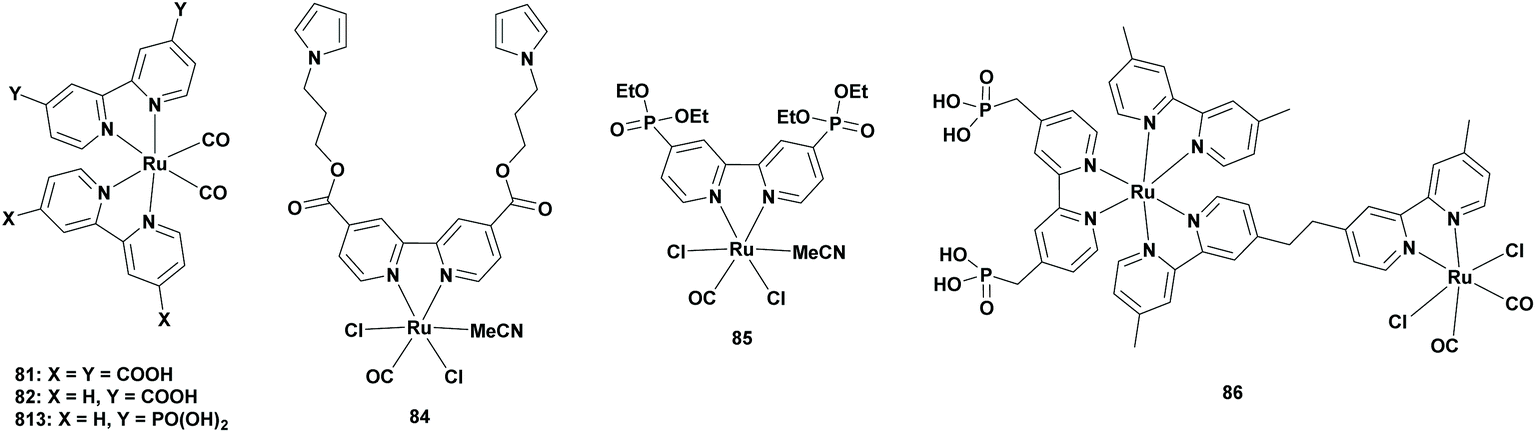
One year later, Suzuki et al. started to find better anchoring groups to enhance the electron transfer, which should exhibit better photoredox activity. Hybrid catalysts with these anchoring groups were typically synthesized by the adsorption of metal complexes onto a semiconductor. In the quest for optimal anchoring groups, each of the metal complexes was synthesized individually. However, with the polar anchoring groups, purification problems of the hybrid material started to arise. To solve this, they reported an alternative approach via direct assembly of the metal complex with ligand-functionalized semiconductors. Here, the phosphonate group was observed to improve the photocatalytic ability of the hybrid catalyst. With the catalyst bearing this anchoring group (Table 7; 83), FA was formed with a TONFA of 118, but CO was also produced with a TONCO of 67.95
In 2011, Sato et al. transferred their protocol to an aqueous solution, so that carbon-based fuels could be produced from CO2 and H2O. Since the previously reported [Ru-(dcbpy)2]/N-Ta2O5 could not maintain its linkage in aqueous solutions, they selected a wide range of different photocatalysts and/or semiconductors such as N-Ta2O5, gallium phosphide (GaP) and indium phosphide (InP). Among these hybrid catalysts, the ruthenium-based complexes linked to InP yielded the highest quantity of formate, with in particular the Ru-(dppc)(bpy) complex (Table 7; 84, dppc = 4,4′-di(1H-pyrrolyl-3-propyl carbonate)) being linked to InP. When a hybrid catalyst was made using two different Ru-complexes (Table 7; 84 and Ru-(dpe)(bpy), 85, dpe = 4,4′-diphosphate ethyl-2,2′-bipyridine) linked to InP, the FA formation increased even more. This was explained by the dpe ligand which acted as an anchor to form a tight linkage with the surface of InP to accelerate the electron transfer. Later, the reactivity of the hybrid catalyst was determined in the presence of electrolyte solutions such as NaHCO3, Na3PO4 and Na2SO4, since it could increase the photocurrent due to its higher electrical conductivity compared to water. In all of these electrolytes, the production of FA increased, parallel to the drastic increase of the production of hydrogen gas.20
Most of the semiconducting material-based photocatalysts produce a tremendous amount of hydrogen gas as the by-product. Therefore, to gain better selectivity towards the formation of FA, the search for new heterogenized photocatalysts was continued. Moreover, the photoreduction of CO2 to FA in the presence of a hybrid catalyst was very complex and was performed in a two-compartment cell, which was divided by a proton exchange membrane, since the produced FA could be re-oxidized by the anode current. Furthermore, these photocatalysts could not oxidize water themselves to utilize it as an electron donor. For this reason, TiO2 was often used as a photocathode. In 2013, Arai et al. proposed the use of a SrTiO3 photoanode since it had a similar bandgap and had already been reported for the oxidation of water.96 The catalyst was a RuCP polymer linked to a zinc-doped indium phosphide (Zn-InP) semiconductor (Table 7). Using this catalyst in a two-cell set-up, formate was formed with a TONFA of 20. For comparison, they performed the reaction with TiO2 as the photoanode and found that the formation of formate was less than a fourth compared to when the SrTiO3 photoanode was used. In addition, Arai et al. established a one-cell reactor, where the formation of formate dropped by more than 30%. The increase in formate formation using SrTiO3 was explained by its more negative energy of the conduction band minimum (ECBM), which enhanced electron transfer from the photoanode to the photocathode.97
Later in 2013, Ishitani et al. reported the first hybrid photocatalyst which was able to reduce CO2 and oxidize methanol at the same time. They chose methanol as the electron donor, since it formed formaldehyde, a widely known bulk chemical. As the heterogeneous unit, they chose a supramolecular complex consisting of [Ru(dmb)2(MPA-dmb)]2+ (MPA = methylene phosphonic acid) as the photosensitizer unit and [Ru(CH2-dmb)(CO)2Cl2] as the catalyst unit (Table 7; 86, RuRu′), whereas tantalum oxynitride (TaON) with 1 wt% metallic Ag was chosen as the semiconductor. Using this hybrid structure in methanol, formaldehyde was produced to a great extent with FA as the main reduction product from CO2 (TONFA of 41).21
It should be noted that hybrid systems for the reduction of CO2 to FA never exceeded the selectivity of more than 80%. Therefore, new hybrid systems were desirable for improving selectivity. The photocatalytic activity of the Z-scheme was highly dependent on the potential of the conduction band of the semiconductor, since electron transfer from the semiconductor to the photosensitizer unit plays a crucial role. Therefore, Ishitani et al. continued their research by investigating perovskites as new semiconductors. Using CaTaO2N as the perovskite semiconductor with RuRu′ (Table 7; 86) as the catalyst, FA was produced with high selectivity (>99%) with a TONFA of 9. When Ag (1 wt%) was loaded on the semiconductor, the TONFA was increased to 32 with the same selectivity. The deposited Ag was explained to trap electrons to mediate charge transfer from the semiconductor to the photosensitizer unit of the RuRu′ complex.98 Additionally, based on the report of Li et al., Ishitani et al. also investigated an yttrium–tantalum oxynitride (YTON) as a semiconductor since it was proven to possess photocatalytic activity.99 However, when the YTON semiconductor was linked to the RuRu′ catalyst to reduce CO2, a lower TONFA of 5 was achieved but with similar selectivity.100
Although improvements were made for the catalytic activity and selectivity of the hybrid catalysts for the CO2 reduction to FA, these systems required strong sacrificial reductants. The main goal of using semiconductors was the use of weak reductants such as water. Therefore, Ishitani et al. further investigated the photocatalytic reactivity of GaN:ZnO, a metal oxynitride solid solution between GaN and ZnO, and TiO2:Ta/N, rutile titania dioxide co-doped with tantalum and nitrogen, since they are well known to oxidize water. In this way, investigation of interfacial electron transfer could be done, since they possessed different conduction band potentials. The photocatalytic activities of both of the semiconductors were investigated when RuRu′ was linked to it (Table 7). Both of them provided FA as the main product, where TiO2:Ta/N had lower activity compared to GaN:ZnO due to the inefficient light absorption and this observation was explained by the energy of the conduction band. Back electron transfer from the photosensitizing unit of the RuRu′ to the semiconductor was suppressed when the conduction band was elevated in energy. GaN:ZnO possesses a conduction band with negative potential, which exhibited higher reactivity for the photoreduction of CO2.101 This research triggered the interest for the use of mixed anions such as oxyfluorides, oxynitrides and oxysulfides in the photocatalysis discipline, since their valence band can be adjusted to a more negative potential by band-tuning. For this reason, Ishitani et al. developed the oxyfluoride Pb2Ti2O5.4F1.2 as a semiconductor (Table 7). Normally, photocatalysis using oxyfluorides is unsuitable given the highest electronegativity of fluorine, which is located at the lower part of the valence band. This oxyfluoride photocatalyst was mainly developed for the evolution of H2 but with modification via the linkage with RuRu′, it is also capable to reduce CO2 to formate with high selectivity (99%) and a TONFA of 26.102 In terms of oxynitrides, it was already proven that these semiconductors were able to reduce CO2 to FA when RuRu′ was present.98 However, Ishitani et al. proposed that 2D layered oxynitrides may improve the catalytic reactivity compared to 3D layered oxynitrides (e.g. CaTaO2N). Therefore, they synthesized a 2D layered Li2LaTa2O6N perovskite and investigated its photocatalytic activity by forming a linkage with the RuRu′ supramolecular complex (Table 7). FA was produced with a selectivity of 97% with a TONFA > 50. This experiment was repeated using Ag-deposited Li2LaTa2O6N and the reactivity was increased by a factor of two (TONFA of 110) while maintaining high selectivity (99%).103 The use of oxysulfides for CO2 reduction started with the use of rare-earth-based semiconductors as photocatalysts for water splitting.104 It was also reported that rare-earth species were effective modifiers for the reduction of CO2 over Ag/Ga2O3.105 This triggered the interest of Ishitani et al. to use rare-earth metal-based semiconductors (LnTaOxNy, where Ln = rare-earth metal) in a Z-scheme fashion for CO2 reduction. Several LnTaOxNy based semiconductors were linked to RuRu′ for investigation and the catalytic activity was found to be strongly dependent on the Ln element (Table 7). When Ln represented Nd or Sm, no FA was found, whereas FA was observed when Ln was represented by Gd, Tb, Dy or Ho. Among these, Tb provided the highest reactivity with a selectivity of >99%. The low reactivity of Nd and Sm was explained by their low crystallinity, which enhanced electron–hole recombination processes.106
In general, most of the semiconductors absorb wavelengths shorter than 500 nm. Synthesizing semiconductors which absorb at longer wavelengths is important to expand the use of a broader range of the visible spectrum. In 2018, Ishitani et al. reported the use of a Ta3N5 semiconductor for the photoreduction of CO2 when it was linked to RuRu′ (Table 7). FA was produced in high selectivity (93%) at a wavelength of >480 nm, although with lower activity. The activity was increased further using Ag-doped Ta3N5 linked to RuRu′, while at the same time the selectivity was increased to 98%.107
In conclusion, these heterogenized catalysts possess excellent selectivity for the photoreduction of CO2 to formic acid. However, the use of heterogeneous catalysts decreases the contact surface of CO2 and the active catalytic centre, which translated to a decrease in the efficiency of this catalytic cycle. This method has more potential, since only a few homogeneous catalysts have been anchored onto solid semi-conductor supports. Specifically, when the semi-conductor is doped with Ag-particles, reports have proven that the efficiency can be enhanced tremendously.
C3N4 as a metal-free semiconductor
The use of metal-free, active, stable and inexpensive semiconductors that are capable of being used in a Z-scheme system for the conversion of CO2 was first introduced by Ishitani et al. in 2013. In a previous research study, it was reported that carbon nitride (C3N4) was successfully used as a photocatalyst for water reduction and oxidation.108 As a result, Ishitani et al. developed a hybrid photocatalyst consisting of a Ru-based metal complex (Table 8; 87) absorbed on graphitic C3N4 (g-C3N4). This hybrid system was able to reduce CO2 to FA photochemically with high selectivity (>80%) and a TONFA of >200. However, a significant amount of hydrogen gas was also produced.109|
|
|||||||
|---|---|---|---|---|---|---|---|
| Cat. | SC | Solv. | Irr. t (h) | Sel. (%) | TONFA | λ (nm) | Ref. |
| Cat. represents catalyst; SC represents semiconductor; Solv. represents solvent; Irr. t represents irradiation time; Sel. represents selectivity; TONFA represents turnover number for formic acid generation; λ represents wavelength of the incoming radiation; Ref. represents reference; N.A. represents not applicable. Selectivities and TONs were determined after the corresponding irradiation times unless stated otherwise. — = not mentioned. | |||||||
| 87 | g-C3N4 | MeCN | 20 | >80 | 200 | >400 | 109 |
| 88 | g-C3N4 | MeCN | 20 | >80 | 100 | 400 | 111 |
| 88 | g-C3N4 | DMA | 20 | >80 | 1070 | 400 | 111 |
| 86 | g-C3N4 | DMA | 5 | 52 | 43 | >400 | 112 |
| 88 | Ag-g-C3N4 | DMA | 5 | 98 | 1428 | >400 | 112 |
| 86 | Ag-g-C3N4 | DMA | 5 | >99 | 3110 | >400 | 112 |
| 86 | Ag-g-C3N4 | (COOK)2 | 15 | 83 | 586 | >400 | 112 |
| 86 | Ag-NS-C3N4 | K2CO3 | 15 | 90 | 2090 | >400 | 113 |
| 88 | Ag-g-C3N4 | DMA | 5 | >99 | 650 | >400 | 116 |
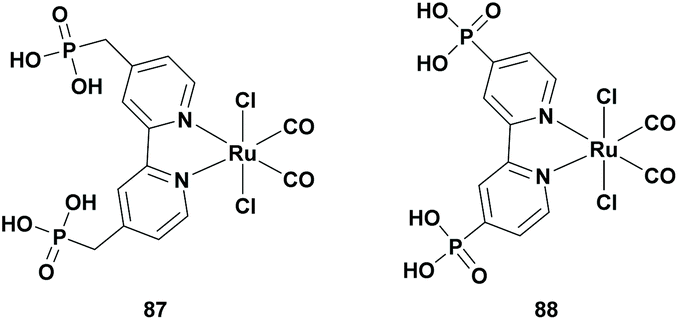
Despite the promising use of this hybrid system as a photocatalyst for the reduction of CO2, no optimization studies were carried out. Later, Ishitani et al. improved their catalytic system by optimizing the Ru-based complex. Based on their previous report,110 they investigated different trans(Cl)-[Ru(bpyX2)(CO)2Cl2] complexes, where X = H, Me, PO3H2 or CH2PO3H2. The RuP (Table 8; 88) and RuCP (Table 8; 87) complexes were absorbed quantitatively, whereas RuH and RuMe were not absorbed at all as they do not possess an anchoring group. The catalytic reactivity of these hybrid catalysts was investigated. In both of the cases, FA was selectively formed with the highest TONFA of approximately 200 for the g-C3N4/RuP hybrid photocatalyst. Secondly, the effect of the solvent on the catalytic activity was investigated and the best result was obtained when DMA was used as the solvent (TONFA of 1070).111
In the meantime, it was shown that supramolecular complexes exhibit higher reactivity.89–93 For this reason, Ishitani et al. continued their research based on the deposition of a supramolecular complex on the g-C3N4 semiconductor. When the binuclear RuRu′ (Table 7; 86) complex was immobilized on g-C3N4 and the resulting hybrid structure was used as a photocatalyst for the reduction of CO2, FA was produced in low selectivity (52%) and with a lower TONFA (43) compared to the RuP/g-C3N4 hybrid catalyst (Table 8). Furthermore, it was proven that the deposition of silver particles on a TaON semiconductor improved the catalytic activity.98 This concept was then employed for the RuP/C3N4 hybrid catalyst (Table 8; 88) and the RuCP/C3N4 hybrid catalyst (Table 8; 87). It was found that Ag modification improved the reactivity of the RuP/Ag-g-C3N4 catalyst immensely and it also improved the selectivity to 98%. An even higher increase in reactivity was observed using the RuCP/Ag-g-C3N4 catalyst.112
Ideally, heterogenized photocatalysts exhibit higher reactivity and selectivity in aqueous solutions, where water acts as a solvent and as an electron donor. Despite the excellent reactivity and selectivity of the binuclear complex RuRu′ in DMA/TEOA, the reduction in aqueous solutions was moderate with a selectivity of around 80% and a TONFA of 586.112 In 2017, Ishitani et al. performed research on carbon nitride nanosheets (NS-C3N4) instead of graphitic carbon nitride. On this semiconductor, Ag was loaded and RuRu′ (Table 7; 86) was absorbed. This hybrid catalyst, in the presence of EDTA·2Na solution (pH 4.3), exhibited reactivity for the reduction of CO2 to FA with a selectivity of 76%. Notably, when NaH2PO4 and Na2HPO4 were added, the pH increased to 6.1 and parallelly the reactivity and selectivity were also increased. The selectivity and activity were increased further (TONFA of 2090) when the mixture had a pH between 6.5 and 7 (addition of K2CO3) (Table 8). However, the evolution of hydrogen decreased in neutral pH solutions. This pH dependency was explained by the fact that the semiconductor desorbed RuRu′ in more acidic environments, since the interaction between NS-C3N4 and RuRu′ was based on hydrogen bonding.113
It was clear that silver species, deposited on semiconductors, promoted the photocatalytic reduction of CO2 with higher reactivity. It was explained that silver trapped electrons to mediate the charge transfer from the semiconductor to the photosensitizer unit of the RuRu′ complex. However, the local structure of the loaded silver species in hybrid systems and its photocatalytic properties for CO2 reduction were not investigated in detail. Therefore, no guideline to improve the photocatalytic reactivity was available regarding the structure of the silver species. As a result, Ishitani et al. investigated the structure of silver-modified NS-C3N4 to determine the impact on the photocatalytic reactivity of the loaded Ag-species. At first, they investigated silver-modified NS-C3N4 which was prepared by an impregnation–H2 reduction method of the RuP/Ag-NS-C3N4 hybrid catalyst. Here, the reactivity for the formation of formate was improved with the increase of Ag loading up to 2 wt% at an impregnation temperature of 473 K. Moreover, the reactivity was strongly temperature dependent with the highest reactivity at an impregnation temperature of 473 K. Spectroscopic studies revealed that the silver particles were close in resemblance to Ag2O when a low amount of Ag was loaded on the semiconductor, but metallic silver particles were more pronounced when a larger amount was loaded on the semiconductor. Moreover, the impregnation temperature determines the form as well, where higher temperatures yield more metallic silver particles. Thus, in the case of the 2 wt% impregnation–H2 reduction method at 473 K, Ag2O was dominant, whereas in the 2 wt% impregnation–H2 reduction method at 623 K, Ag0 was formed to a greater extent. It was also shown that a higher dispersion enhanced the reactivity, while aggregates or excess of Ag0-particles provided lower reactivity. However, whether Ag0 had a positive effect on the reactivity could not have been drawn here. Therefore, further experiments were performed, where silver-modified NS-C3N4 was prepared from impregnation-air heating (minimalize Ag0 formation) or where it was prepared from in situ photodeposition (Ag0 has to be dominant). Both of the samples were used for the reduction of CO2 when RuP was absorbed and both provided lower reactivity. However, the sample prepared via photodeposition (widely distributed Ag0) was much more reactive than the sample prepared via the H2 reduction method at 623 K (dispersedly divided Ag0 + Ag2O). This result indicated that Ag0 had a better promotional effect than Ag2O-like species. As a result, finely dispersed Ag0 species would be key to promote the activity of hybrid systems.114
Synthetic procedures for the preparation of C3N4 usually require hard-template methods with multistep procedures. For this reason, the desire of synthesizing carbon nitride in a more effective and less expensive way is of great interest. It was shown by Chen et al. that g-C3N4 could be synthesized from the pyrolysis of urea.115 In fact it had been proven that urea-derived C3N4 showed improved reactivity for the reduction of CO2.113 However, thermal decomposition of urea to form C3N4 at different temperatures should affect its physicochemical properties, which has not been studied before. Therefore, research was performed on this topic by Ishitani et al. by synthesizing urea at different temperatures for the photocatalytic reduction of CO2 when RuP was added as the catalyst. Spectroscopic techniques exhibited an increase of crystallized C3N4 formation at higher temperatures. However, at elevated temperatures, the formed C3N4 decomposed. It was also found that at 773 K a lot of voids were formed in the graphitic structure, whereas a nanosheet-like structure was formed at 823 K or higher temperatures. Afterwards, photoreductions were carried out using the synthesized C3N4 structures at different temperatures in a TEOA/DMA solvent mixture. Here, Ag was deposited and the RuP (Table 8; 88) catalyst was absorbed on the semiconductor. With an increase in the deposition temperature, more formate was formed with a maximum TONFA of 650 at 873–923 K. The catalytic reactivity was explained by the structure of the synthesized C3N4: the C3N4 at elevated temperatures absorbed visible light better and had a higher specific surface area for the catalyst and Ag dispersion.116
As stated before, heterogeneous catalysts have the main advantage that they can be recycled and used in subsequent reactions. When metal-free semiconductors are used, the economic advantage increases even more, since metal-free semiconductors are generally cheaper compared to their metal counterparts. From the reported procedures, it can be concluded that g-C3N4 is an extremely viable semi-conductor for the photocatalytic reduction of CO2 to FA. Specifically, when it is doped with Ag-particles, the TONFA is comparable with mononuclear Ru-complexes. Since the concept of using C3N4 is relatively new, many reports are expected in the future.
Conclusion and outlook
CO2 photoreduction to FA using transition metal complexes has been summarized in this review. From the discussion, it is clear that visible light promoted CO2 reductions to FA has become a topic of great interest in green chemistry. The use of ruthenium-based metal complexes as homogeneous catalysts has been well-explored. Although great TONs for the production of HCOOH could be observed, scientists searched for Earth abundant metal complexes such as Co-, Mn-, Fe- and Mb-based catalysts, as well as metal-free catalysts. Furthermore, it was shown that photoreductions only proceeded in the presence of a photosensitizer. This led to the linkage of a photosensitiser and a catalyst in a supramolecular complex, which resulted in higher activities since electron exchange from the photosensitizer to the catalyst was facilitated.In addition, this review shows that heterogeneous photocatalysts are desirable for the photoreduction of CO2 to FA as recyclable catalysts in green chemistry.117,118 Here, silver-doped semiconductors linked to supramolecular complexes in a Z-scheme heterojunction are the most noteworthy catalysts. Although the visible-light promoted reduction of CO2 to FA by (heterogenized) homogeneous catalysis has made excellent progress over the last few decades, only a few cases yielded TONFA values greater than 1000 and still, a lot of metal complexes remain unexplored for this application. We hope that this review will encourage scientists to design new homogeneous metal complexes and explore these complexes in the visible-light promoted CO2 reduction to FA. Additionally, we hope that the heterogenized concept discussed in this review will inspire chemists to develop and explore new functionalised semiconductors. Moreover, the efforts will also be not limited to metal-based complexes, rather they will also focus on some metal-free catalysts owing to their cheaper price and easy availability.119–135
Although extensive efforts are being made to achieve high reactivity, metal-based catalysts still have some drawbacks. Firstly, the reported results vary immensely, even when the same metal centre has been used. As a result, it is impossible to establish a general structure–activity relationship on which researchers can rely in the future. Secondly, many of the reported catalysts lose reactivity under the photocatalytic conditions, since they are not stable enough. Thirdly, the reported catalytic systems are not sustainable and economically feasible to perform on a larger scale. Fourthly, the use of heterogenized catalysts requires the diffusion of CO2 through the catalysts. Up to now, no study has been performed on the adsorption behaviour of CO2 in these catalysts. Finally, in general, the systems are not efficient enough in terms of activity and selectivity.
To overcome the abovementioned problems, extensive research studies on the photocatalytic reduction of CO2 to FA have to be performed in the coming years. One way of doing so is by varying the PC in the catalytic systems. In such a way, efficient cycles can be found. Additionally, this can provide a better understanding of the structure–activity relationship of the used catalysts for the photoreduction of CO2 to FA. Another way is the development of new PSs, especially, Earth abundant metal based PSs, based on organic molecules and inorganic semiconductors. In this way, the absorption of visible light can be enhanced allowing the optimum use of the incident energy. Secondly, the stability of the catalytic system can be examined by performing recycling experiments. Additionally, by using 13CO2, the carbon source of FA can be verified, since the catalyst or other organic materials in the catalytic system can decompose themselves. Thirdly, the photocatalytic systems should be optimised to avoid sacrificial electron donors such as TEOA or BIH. This can be done by coupling the system to the formation of value added organic compounds via an oxidation process or by the coupling of the system to water oxidation. In fact, the coupling of CO2 reduction with H2O oxidation has already been performed by Li et al. when using heterogeneous Cu2O-Pt/SiC/IrOx composites with excellent efficiency.136 In summary, extensive research studies on the photocatalytic reduction of CO2 to FA have great potential to achieve high activities and selectivities in a sustainable and economically favourable way.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Fond der Chemie Industrie (Liebig fellowship to SD), Francqui Foundation (Francqui lecturer award to SD) and Dehausse fellowship (to RC). Special thanks to Dr Yu Zhang and Mr Tong Zhang for their kind help during the preparation of this manuscript.Notes and references
- T. A. Carleton and S. M. Hsiang, Science, 2016, 353, aad9837 CrossRef PubMed.
- https://www.iea.org/reports/global-energy-co2-status-report-2019 .
- M. Bui, C. S. Adjiman, A. Bardow, E. J. Anthony, A. Boston, S. Brown, P. S. Fennell, S. Fuss, A. Galindo, L. A. Hackett, J. P. Hallett, H. J. Herzog, G. Jackson, J. Kemper, S. Krevor, G. C. Maitland, M. Matuszewski, I. S. Metcalfe, C. Petit, G. Puxty, J. Reimer, D. M. Reiner, E. S. Rubin, S. A. Scott, N. Shah, B. Smit, J. P. M. Trusler, P. Webley, J. Wilcox and N. Mac Dowell, Energy Environ. Sci., 2018, 11, 1062 RSC.
- J. Tollefson, Nature, 2018, 558, 173 CrossRef CAS PubMed.
- S. Sorcar, J. Thompson, Y. Hwang, Y. H. Park, T. Majima, C. A. Grimes, J. R. Durrant and S.-I. In, Energy Environ. Sci., 2018, 11, 3183 RSC.
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed.
- https://setis.ec.europa.eu/related-jrc-activities/jrc-setis-reports/carbon-capture-and-storage-technology-information-sheet .
- H. Seo, M. H. Katcher and T. F. Jamison, Nat. Chem., 2017, 9, 453 CrossRef CAS PubMed.
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709 CrossRef CAS PubMed.
- S. Ye, C. Ding, R. Chen, F. Fan, P. Fu, H. Yin, X. Wang, Z. Wang, P. Du and C. Li, J. Am. Chem. Soc., 2018, 140, 3250 CrossRef CAS PubMed.
- P. F. Wareing, M. M. Khalifa and K. J. Treharne, Nature, 1968, 220(5166), 453 CrossRef CAS PubMed.
- Y. Wang, J. Liu, Y. Wang, Y. Wang and G. Zheng, Nat. Commun., 2018, 9, 5003 CrossRef PubMed.
- D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef PubMed.
- R. J. Detz, J. N. H. Reek and B. C. C. van der Zwaan, Energy Environ. Sci., 2018, 11, 1653 RSC.
- S. Lin, C. S. Diercks, Y.-B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang, O. M. Yaghi and C. J. Chang, Science, 2015, 349, 1208 CrossRef CAS PubMed.
- J. J. Leung, J. Warnan, K. H. Ly, N. Heidary, D. H. Nam, M. F. Kuehnel and E. Reisner, Nat. Catal., 2019, 2, 354 CrossRef CAS.
- A. K. Singh, J. H. Montoya, J. M. Gregoire and K. A. Persson, Nat. Commun., 2019, 10, 443 CrossRef CAS PubMed.
- X. Li, Y. Sun, J. Xu, Y. Shao, J. Wu, X. Xu, Y. Pan, H. Ju, J. Zhu and Y. Xie, Nat. Energy, 2019, 4, 690 CrossRef CAS.
- J. L. White, M. F. Baruch, J. E. Pander, Y. Hu, I. C. Fortmeyer, J. E. Park, T. Zhang, K. Liao, J. Gu, Y. Yan, T. W. Shaw, E. Abelev and A. B. Bocarsly, Chem. Rev., 2015, 115, 12888 CrossRef CAS PubMed.
- S. Sato, T. Arai, T. Morikawa, K. Uemura, T. M. Suzuki, H. Tanaka and T. Kajino, J. Am. Chem. Soc., 2011, 133, 15240 CrossRef CAS PubMed.
- K. Sekizawa, K. Maeda, K. Domen, K. Koike and O. Ishitani, J. Am. Chem. Soc., 2013, 135, 4596 CrossRef CAS PubMed.
- S. Matsuoka, K. Yamamoto, T. Ogata, M. Kusaba, N. Nakashima, E. Fujita and S. Yanagida, J. Am. Chem. Soc., 1993, 115, 601 CrossRef CAS.
- S. Moret, P. J. Dyson and G. Laurenczy, Nat. Commun., 2014, 5, 4017 CrossRef CAS PubMed.
- A. Boddien, D. Mellmann, F. Gärtner, R. Jackstell, H. Junge, P. J. Dyson, G. Laurenczy, R. Ludwig and M. Beller, Science, 2011, 333, 1733 CrossRef CAS PubMed.
- D. Mellmann, P. Sponholz, H. Junge and M. Beller, Chem. Soc. Rev., 2016, 45, 3954 RSC.
- C. Fellay, P. J. Dyson and G. Laurenczy, Angew. Chem., Int. Ed., 2008, 47, 3966 CrossRef CAS PubMed.
- https://www.mordorintelligence.com/industry-reports/formic-acid-market .
- U. Fegade and G. Jethave, in Conversion of Carbon Dioxide into Hydrocarbons Vol. 2 Technology, ed. Inamuddin, A. M. Asiri and E. Lichtfouse, Springer International Publishing, Cham, 2020, pp. 91–110, DOI:10.1007/978-3-030-28638-5_4.
- E. Fujita, Coord. Chem. Rev., 1999, 185–186, 373 CrossRef CAS.
- T. Inoue, A. Fujishima, S. Konishi and K. Honda, Nature, 1979, 277, 637 CrossRef CAS.
- N. Li, J. Liu, J.-J. Liu, L.-Z. Dong, Z.-F. Xin, Y.-L. Teng and Y.-Q. Lan, Angew. Chem., Int. Ed., 2019, 58, 5280 CrossRef.
- D. Wang, R. Huang, W. Liu, D. Sun and Z. Li, ACS Catal., 2014, 4, 4254 CrossRef CAS.
- Z.-H. Yan, M.-H. Du, J. Liu, S. Jin, C. Wang, G.-L. Zhuang, X.-J. Kong, L.-S. Long and L.-S. Zheng, Nat. Commun., 2018, 9, 3353 CrossRef PubMed.
- J. Hawecker, J.-M. Lehn and R. Ziessel, ChemComm, 1983, 536, 10.1039/C39830000536.
- J. Hawecker, J.-M. Lehn and R. Ziessel, ChemComm, 1985, 56, 10.1039/C39850000056.
- J.-M. Lehn and R. Ziessel, J. Organomet. Chem., 1990, 382, 157 CrossRef CAS.
- H. Ishida, T. Terada, K. Tanaka and T. Tanaka, Inorg. Chem., 1990, 29, 905 CrossRef CAS.
- J. R. Pugh, M. R. M. Bruce, B. P. Sullivan and T. J. Meyer, Inorg. Chem., 1991, 30, 86 CrossRef CAS.
- A. Paul, D. Connolly, M. Schulz, M. T. Pryce and J. G. Vos, Inorg. Chem., 2012, 51, 1977 CrossRef CAS PubMed.
- Y. Kuramochi, M. Kamiya and H. Ishida, Inorg. Chem., 2014, 53, 3326 CrossRef CAS PubMed.
- Y. Kuramochi, J. Itabashi, K. Fukaya, A. Enomoto, M. Yoshida and H. Ishida, Chem. Sci., 2015, 6, 3063 RSC.
- A. Rosas-Hernández, H. Junge and M. Beller, ChemCatChem, 2015, 7, 3316 CrossRef.
- T. Morimoto, C. Nishiura, M. Tanaka, J. Rohacova, Y. Nakagawa, Y. Funada, K. Koike, Y. Yamamoto, S. Shishido, T. Kojima, T. Saeki, T. Ozeki and O. Ishitani, J. Am. Chem. Soc., 2013, 135, 13266 CrossRef CAS PubMed.
- T. Asatani, Y. Nakagawa, Y. Funada, S. Sawa, H. Takeda, T. Morimoto, K. Koike and O. Ishitani, Inorg. Chem., 2014, 53, 7170 CrossRef CAS PubMed.
- J. Rohacova, A. Sekine, T. Kawano, S. Tamari and O. Ishitani, Inorg. Chem., 2015, 54, 8769 CrossRef CAS PubMed.
- J. Rohacova and O. Ishitani, Chem. Sci., 2016, 7, 6728 RSC.
- S. K. Lee, M. Kondo, G. Nakamura, M. Okamura and S. Masaoka, ChemComm, 2018, 54, 6915 RSC.
- S. K. Lee, M. Kondo, M. Okamura, T. Enomoto, G. Nakamura and S. Masaoka, J. Am. Chem. Soc., 2018, 140, 16899 CrossRef CAS PubMed.
- Y. Hameed, G. K. Rao, J. S. Ovens, B. Gabidullin and D. Richeson, ChemSusChem, 2019, 12, 3453 CrossRef CAS PubMed.
- Y. Arikawa, T. Nakamura, S. Ogushi, K. Eguchi and K. Umakoshi, Dalton Trans., 2015, 44, 5303 RSC.
- Y. Arikawa, I. Tabata, Y. Miura, H. Tajiri, Y. Seto, S. Horiuchi, E. Sakuda and K. Umakoshi, Chem. – Eur. J., 2020, 26, 5603 CrossRef CAS PubMed.
- D. Behar, T. Dhanasekaran, P. Neta, C. M. Hosten, D. Ejeh, P. Hambright and E. Fujita, J. Phys. Chem. A, 1998, 102, 2870 CrossRef CAS.
- D. Lexa, J. M. Savéant and J. P. Soufflet, J. Electroanal. Chem. Interfacial Electrochem., 1979, 100, 159 CrossRef CAS.
- J. Grodkowski and P. Neta, J. Phys. Chem. A, 2000, 104, 1848 CrossRef CAS.
- Z. Guo, G. Chen, C. Cometto, B. Ma, H. Zhao, T. Groizard, L. Chen, H. Fan, W.-L. Man, S.-M. Yiu, K.-C. Lau, T.-C. Lau and M. Robert, Nat. Catal., 2019, 2, 801 CrossRef CAS.
- M. Bourrez, F. Molton, S. Chardon-Noblat and A. Deronzier, Angew. Chem., Int. Ed., 2011, 50, 9903 CrossRef CAS PubMed.
- H. Takeda, H. Koizumi, K. Okamoto and O. Ishitani, ChemComm, 2014, 50, 1491 RSC.
- P. L. Cheung, C. W. Machan, A. Y. S. Malkhasian, J. Agarwal and C. P. Kubiak, Inorg. Chem., 2016, 55, 3192 CrossRef CAS PubMed.
- M. Stanbury, J.-D. Compain, M. Trejo, P. Smith, E. Gouré and S. Chardon-Noblat, Electrochim. Acta, 2017, 240, 288 CrossRef CAS.
- J.-X. Zhang, C.-Y. Hu, W. Wang, H. Wang and Z.-Y. Bian, Appl. Catal., A, 2016, 522, 145 CrossRef CAS.
- X. Zhang, M. Cibian, A. Call, K. Yamauchi and K. Sakai, ACS Catal., 2019, 9, 11263–11273 CrossRef CAS.
- H. Takeda, H. Kamiyama, K. Okamoto, M. Irimajiri, T. Mizutani, K. Koike, A. Sekine and O. Ishitani, J. Am. Chem. Soc., 2018, 140, 17241 CrossRef CAS PubMed.
- L. Chen, Z. Guo, X.-G. Wei, C. Gallenkamp, J. Bonin, E. Anxolabéhère-Mallart, K.-C. Lau, T.-C. Lau and M. Robert, J. Am. Chem. Soc., 2015, 137, 10918 CrossRef CAS PubMed.
- T. Fogeron, T. K. Todorova, J.-P. Porcher, M. Gomez-Mingot, L.-M. Chamoreau, C. Mellot-Draznieks, Y. Li and M. Fontecave, ACS Catal., 2018, 8, 2030 CrossRef CAS.
- T. Fogeron, P. Retailleau, L.-M. Chamoreau, Y. Li and M. Fontecave, Angew. Chem., Int. Ed., 2018, 57, 17033 CrossRef CAS PubMed.
- S. Sato, T. Morikawa, T. Kajino and O. Ishitani, Angew. Chem., Int. Ed., 2013, 52, 988 CrossRef CAS PubMed.
- S. Sato and T. Morikawa, ChemPhotoChem, 2018, 2, 207 CrossRef CAS.
- Y.-H. Luo, L.-Z. Dong, J. Liu, S.-L. Li and Y.-Q. Lan, Coord. Chem. Rev., 2019, 390, 86–126 CrossRef CAS.
- K. Kamada, J. Jung, T. Wakabayashi, K. Sekizawa, S. Sato, T. Morikawa, S. Fukuzumi and S. Saito, J. Am. Chem. Soc., 2020, 142, 10261 CrossRef CAS PubMed.
- J. Hawecker, J.-M. Lehn and R. Ziessel, J. Chem. Soc., Chem. Commun., 1983, 536–538, 10.1039/C39830000536.
- Y. Hameed, P. Berro, B. Gabidullin and D. Richeson, ChemComm, 2019, 55, 11041 RSC.
- S. Matsuoka, H. Fujii, C. Pac and S. Yanagida, Chem. Lett., 1990, 19, 1501 CrossRef.
- S. Tazuke and H. Ozawa, ChemComm, 1975, 237, 10.1039/C39750000237.
- S. Matsuoka, T. Kohzuki, C. Pac and S. Yanagida, Chem. Lett., 1990, 19, 2047 CrossRef.
- B. J. Fisher and R. Eisenberg, J. Am. Chem. Soc., 1980, 102, 7361 CrossRef CAS.
- S. Matsuoka, K. Yamamoto, C. Pac and S. Yanagida, Chem. Lett., 1991, 20, 2099 CrossRef.
- J.-M. Lehn and R. Ziessel, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 701–704 CrossRef CAS PubMed.
- T. Hideyuki, O. Masatoshi, T. Yoshiki, W. Yoshinori, K. Masa and C. Koji, Bull. Chem. Soc. Jpn., 1990, 63, 3233 CrossRef.
- I. Willner and D. Mandler, J. Am. Chem. Soc., 1989, 111, 1330 CrossRef CAS.
- T. Ogata, Y. Yamamoto, Y. Wada, K. Murakoshi, M. Kusaba, N. Nakashima, A. Ishida, S. Takamuku and S. Yanagida, J. Phys. Chem., 1995, 99, 11916 CrossRef CAS.
- D. J. Boston, C. Xu, D. W. Armstrong and F. M. MacDonnell, J. Am. Chem. Soc., 2013, 135, 16252 CrossRef CAS PubMed.
- A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. DuBois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille, P. J. A. Kenis, C. A. Kerfeld, R. H. Morris, C. H. F. Peden, A. R. Portis, S. W. Ragsdale, T. B. Rauchfuss, J. N. H. Reek, L. C. Seefeldt, R. K. Thauer and G. L. Waldrop, Chem. Rev., 2013, 113, 6621 CrossRef CAS PubMed.
- Z. Han, W. R. McNamara, M.-S. Eum, P. L. Holland and R. Eisenberg, Angew. Chem., Int. Ed., 2012, 51, 1667 CrossRef CAS PubMed.
- Z. Han, L. Shen, W. W. Brennessel, P. L. Holland and R. Eisenberg, J. Am. Chem. Soc., 2013, 135, 14659 CrossRef CAS PubMed.
- S. E. Lee, A. Nasirian, Y. E. Kim, P. T. Fard, Y. Kim, B. Jeong, S.-J. Kim, J.-O. Baeg and J. Kim, J. Am. Chem. Soc., 2020, 142, 19142 CrossRef CAS PubMed.
- H. Takeda and O. Ishitani, Coord. Chem. Rev., 2010, 254, 346 CrossRef CAS.
- S. Sato, K. Koike, H. Inoue and O. Ishitani, Photochem. Photobiol. Sci., 2007, 6, 454 CrossRef CAS PubMed.
- Y. Tamaki, K. Watanabe, K. Koike, H. Inoue, T. Morimoto and O. Ishitani, Faraday Discuss., 2012, 155, 115 RSC.
- A. Nakada, K. Koike, T. Nakashima, T. Morimoto and O. Ishitani, Inorg. Chem., 2015, 54, 1800 CrossRef CAS PubMed.
- Y. Tamaki, T. Morimoto, K. Koike and O. Ishitani, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15673 CrossRef CAS PubMed.
- Y. Tamaki, K. Koike and O. Ishitani, Chem. Sci., 2015, 6, 7213 RSC.
- Y. Tamaki and O. Ishitani, Faraday Discuss., 2017, 198, 319 RSC.
- D. C. Fabry, H. Koizumi, D. Ghosh, Y. Yamazaki, H. Takeda, Y. Tamaki and O. Ishitani, Organometallics, 2020, 39, 1511 CrossRef CAS.
- S. Sato, T. Morikawa, S. Saeki, T. Kajino and T. Motohiro, Angew. Chem., Int. Ed., 2010, 49, 5101 CrossRef CAS PubMed.
- T. M. Suzuki, H. Tanaka, T. Morikawa, M. Iwaki, S. Sato, S. Saeki, M. Inoue, T. Kajino and T. Motohiro, ChemComm, 2011, 47, 8673 RSC.
- M. S. Wrighton, A. B. Ellis, P. T. Wolczanski, D. L. Morse, H. B. Abrahamson and D. S. Ginley, J. Am. Chem. Soc., 1976, 98, 2774 CrossRef CAS.
- T. Arai, S. Sato, T. Kajino and T. Morikawa, Energy Environ. Sci., 2013, 6, 1274 RSC.
- F. Yoshitomi, K. Sekizawa, K. Maeda and O. Ishitani, ACS Appl. Mater. Interfaces, 2015, 7, 13092 CrossRef CAS PubMed.
- M. Liu, W. You, Z. Lei, G. Zhou, J. Yang, G. Wu, G. Ma, G. Luan, T. Takata, M. Hara, K. Domen and C. Li, ChemComm, 2004, 2192, 10.1039/B407892F.
- K. Muraoka, H. Kumagai, M. Eguchi, O. Ishitani and K. Maeda, ChemComm, 2016, 52, 7886 RSC.
- A. Nakada, R. Kuriki, K. Sekizawa, S. Nishioka, J. J. M. Vequizo, T. Uchiyama, N. Kawakami, D. Lu, A. Yamakata, Y. Uchimoto, O. Ishitani and K. Maeda, ACS Catal., 2018, 8, 9744 CrossRef CAS.
- R. Kuriki, T. Ichibha, K. Hongo, D. Lu, R. Maezono, H. Kageyama, O. Ishitani, K. Oka and K. Maeda, J. Am. Chem. Soc., 2018, 140, 6648 CrossRef CAS PubMed.
- T. Oshima, T. Ichibha, K. S. Qin, K. Muraoka, J. J. M. Vequizo, K. Hibino, R. Kuriki, S. Yamashita, K. Hongo, T. Uchiyama, K. Fujii, D. Lu, R. Maezono, A. Yamakata, H. Kato, K. Kimoto, M. Yashima, Y. Uchimoto, M. Kakihana, O. Ishitani, H. Kageyama and K. Maeda, Angew. Chem., Int. Ed., 2018, 57, 8154 CrossRef CAS PubMed.
- A. Ishikawa, T. Takata, T. Matsumura, J. N. Kondo, M. Hara, H. Kobayashi and K. Domen, J. Phys. Chem. B, 2004, 108, 2637 CrossRef CAS.
- H. Tatsumi, K. Teramura, Z. Huang, Z. Wang, H. Asakura, S. Hosokawa and T. Tanaka, Langmuir, 2017, 33, 13929 CrossRef CAS PubMed.
- K. Muraoka, M. Eguchi, O. Ishitani, F. Cheviré and K. Maeda, J. Energy Chem., 2021, 55, 176 CrossRef.
- K. Muraoka, T. Uchiyama, D. Lu, Y. Uchimoto, O. Ishitani and K. Maeda, Bull. Chem. Soc. Jpn., 2019, 92, 124 CrossRef CAS.
- K. Maeda, X. Wang, Y. Nishihara, D. Lu, M. Antonietti and K. Domen, J. Phys. Chem. C, 2009, 113, 4940 CrossRef CAS.
- K. Maeda, K. Sekizawa and O. Ishitani, ChemComm, 2013, 49, 10127 RSC.
- H. Takeda, K. Koike, H. Inoue and O. Ishitani, J. Am. Chem. Soc., 2008, 130, 2023 CrossRef CAS PubMed.
- R. Kuriki, K. Sekizawa, O. Ishitani and K. Maeda, Angew. Chem., Int. Ed., 2015, 54, 2406 CrossRef CAS PubMed.
- R. Kuriki, H. Matsunaga, T. Nakashima, K. Wada, A. Yamakata, O. Ishitani and K. Maeda, J. Am. Chem. Soc., 2016, 138, 5159 CrossRef CAS PubMed.
- R. Kuriki, M. Yamamoto, K. Higuchi, Y. Yamamoto, M. Akatsuka, D. Lu, S. Yagi, T. Yoshida, O. Ishitani and K. Maeda, Angew. Chem., Int. Ed., 2017, 56, 4867 CrossRef CAS PubMed.
- K. Maeda, D. An, C. S. Kumara Ranasinghe, T. Uchiyama, R. Kuriki, T. Kanazawa, D. Lu, S. Nozawa, A. Yamakata, Y. Uchimoto and O. Ishitani, J. Mater. Chem. A, 2018, 6, 9708 RSC.
- J. Liu, T. Zhang, Z. Wang, G. Dawson and W. Chen, J. Mater. Chem., 2011, 21, 14398 RSC.
- K. Maeda, D. An, R. Kuriki, D. Lu and O. Ishitani, Beilstein J. Org. Chem., 2018, 14, 1806 CrossRef CAS PubMed.
- A. Karakulina, A. Gopakumar, İ. Akçok, B. L. Roulier, T. LaGrange, S. A. Katsyuba, S. Das and P. J. Dyson, Angew. Chem., Int. Ed., 2016, 55, 292 CrossRef CAS PubMed.
- Y. Zhang, N. Hatami, N. S. Lange, E. Ronge, W. Schilling, C. Jooss and S. Das, Green Chem., 2020, 22, 4516 RSC.
- P. Hirapara, D. Riemer, N. Hazra, J. Gajera, M. Finger and S. Das, Green Chem., 2017, 19, 5356 RSC.
- D. Riemer, P. Hirapara and S. Das, ChemSusChem, 2016, 9, 1916 CrossRef CAS PubMed.
- D. Riemer, B. Mandaviya, W. Schilling, A. C. Götz, T. Kühl, M. Finger and S. Das, ACS Catal., 2018, 8, 3030 CrossRef CAS.
- W. Schilling and S. Das, ChemSusChem, 2020, 13, 6246 CAS.
- F. D. Bobbink, S. Das and P. J. Dyson, Nat. Protoc., 2017, 12, 417 CrossRef CAS.
- S. Das, Y. Li, L.-Q. Lu, K. Junge and M. Beller, Chem. – Eur. J., 2016, 22, 7050 CrossRef CAS PubMed.
- W. Schilling and S. Das, Tetrahedron Lett., 2018, 59, 3821 CrossRef CAS.
- Y. Zhang, T. Zhang and S. Das, Green Chem., 2020, 22, 1800 RSC.
- M. Hulla, S. M. A. Chamam, G. Laurenczy, S. Das and P. J. Dyson, Angew. Chem., Int. Ed., 2017, 56, 10559 CrossRef CAS PubMed.
- D. Riemer, W. Schilling, A. Goetz, Y. Zhang, S. Gehrke, I. Tkach, O. Hollóczki and S. Das, ACS Catal., 2018, 8, 11679 CrossRef CAS.
- Y. Zhang, D. Riemer, W. Schilling, J. Kollmann and S. Das, ACS Catal., 2018, 8, 6659 CrossRef CAS.
- W. Schilling, D. Riemer, Y. Zhang, N. Hatami and S. Das, ACS Catal., 2018, 8, 5425 CrossRef CAS.
- J. Kollmann, Y. Zhang, W. Schilling, T. Zhang, D. Riemer and S. Das, Green Chem., 2019, 21, 1916 RSC.
- T. Zhang, Y. Zhang and S. Das, ChemCatChem, 2020, 12, 6173 CrossRef CAS.
- Y. Zhang, W. Schilling, D. Riemer and S. Das, Nat. Protoc., 2020, 15, 822 CrossRef CAS.
- W. Schilling, Y. Zhang, D. Riemer and S. Das, Chem. – Eur. J., 2020, 26, 390 CrossRef CAS.
- Y. Zhang, W. Schilling and S. Das, ChemSusChem, 2019, 12, 2898 CrossRef CAS.
- Y. Wang, X. Shang, J. Shen, Z. Zhang, D. Wang, J. Lin, J. C. S. Wu, X. Fu, X. Wang and C. Li, Nat. Commun., 2020, 11, 3043 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |