
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent developments in catalysis with Pickering Emulsions
Fuqiang
Chang†
 a,
Carolien M.
Vis†
b,
Wirawan
Ciptonugroho†
b and
Pieter C. A.
Bruijnincx
*a
a,
Carolien M.
Vis†
b,
Wirawan
Ciptonugroho†
b and
Pieter C. A.
Bruijnincx
*a
aOrganic Chemistry and Catalysis, Debye Institute for Nanomaterials Science, Utrecht University, Universiteitsweg 99, 3584 CG Utrecht, The Netherlands. E-mail: p.c.a.bruijnincx@uu.nl
bInorganic Chemistry and Catalysis, Debye Institute for Nanomaterials Science, Utrecht University, Universiteitsweg 99, 3584 CG Utrecht, The Netherlands
First published on 17th March 2021
Abstract
Pickering emulsions (PEs), emulsions stabilized by solid emulsifiers, are already of great importance for the food, pharmaceutical and biomedical industry. More recently, PEs are also being increasingly used as advanced catalytic systems for green chemical transformations. These efforts aim to combine the green credentials of biphasic catalysis with the intrinsic advantages offered by PEs, which include increased stability and interfacial area. Here, we provide a review of the recent advances in the field of PE catalysis, emphasizing the developments in the design of (functional) solid stabilizing particles, the range of accessible catalytic reactions and reaction conditions, as well as advances in reactor engineering, such as the application of PE catalysis in continuous flow systems.
Introduction
Emulsions are dispersions of two immiscible liquids (e.g. water and oil). These biphasic systems can be stabilized by surfactants, as is the case in classical emulsions, or by solid micro- or nanosized particles. The latter type of emulsion is a so-called Pickering emulsion (PE), named after S.U. Pickering who was second to describe them,1 a few years after Ramsden in 1903.2 Generally, two types of emulsions can be readily distinguished, water-in-oil (w/o) or oil-in-water (o/w) emulsions stabilized by hydrophilic and hydrophobic particles, respectively (Fig. 1).3 Which type of PE is formed is determined by the type of particles used, i.e. by the contact angle the particles exhibit with, e.g., the water–oil interface. Hydrophobic particles, such as carbon nanotubes or silylated silica, are more easily wetted by the oil phase than by the aqueous phase, showing contact angles of >90°, resulting in the formation of w/o PEs. Hydrophilic particles, such as bare silica and metal oxides, are more easily wetted by the aqueous phase, resulting in contact angles <90° and the formation of o/w PEs. Other types of emulsions, such as water-in-water (w/w),4 gas-in-liquid (g/w or g/o)5 or multiple emulsions, water-in-oil-in-water (w/o/w) and oil-in-water-in-oil (o/w/o),6 have been studied as well.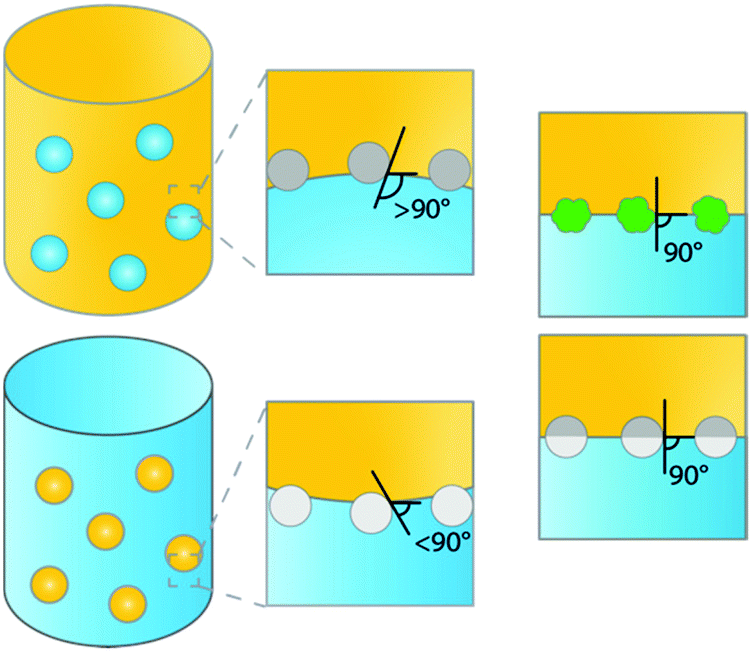 | ||
| Fig. 1 Schematic representation of o/w and w/o emulsions with corresponding contact angles measured through the aqueous phase, yellow: oil, blue: water. Top left: Water-in-oil (w/o) PE, stabilized by hydrophobic particles with contact angle >90°. Bottom left: Oil-in-water (o/w) PE, stabilized by hydrophilic particles with contact angle <90°. Right: Rough (top) and amphiphilic Janus particles (bottom) adsorb at the interface with contact angles of 90°. | ||
PEs have received much attention for application in, e.g., the food, pharmaceutical and biomedical industries, and more recently, catalysis. In the food industry, for example, edible micro- and nanoparticles, such as particles derived from starch and lipids, are used to make reconstituted milk and margarine or foams such as whipped cream.7 In the pharmaceutical and biomedical industries, PEs are mainly used for the targeted delivery of drugs through the skin.8,9 In the past few years, increased attention is being paid to the use of PEs for catalysis, i.e. as advanced biphasic reaction media. Biphasic systems (BS) are widely used to perform reactions between immiscible reagents and catalysts, to extract products from the reaction phase to a second liquid phase or to separate a homogeneous chemo- or biocatalyst from reagents and products, e.g. to improve product recovery and catalyst recyclability.10–12 Such BS thus offer clear advantages from a green chemistry perspective. Further improvements can still be made, however, as conventional BS exhibit a rather low interfacial area of the two immiscible phases, limiting reaction and extraction efficiencies. The application of stable dispersions created by emulsification of the BS is a promising strategy to further improve the efficiency of a chemical reaction.
PEs are often chosen over surfactant-based classical emulsions,13,14 because of their higher resistance against destabilization by droplet coalescence and Ostwald ripening, for example. Once the particles are adsorbed at the liquid–liquid interface, the energy required to desorb them is much higher than the thermal energy, making them highly stable. Stability in this case is often monitored as function of the height of the emulsion phase with respect to the total liquid height as function of time. Depending on the relative densities of the two liquid phases, any destabilization can then be described as creaming, i.e., the formation of a clear liquid phase under the emulsion phase, or as sedimentation, the formation of a clear liquid phase on top of the emulsion.
Indeed, the combination of a high interfacial surface area and the increased (thermal) stability that PE use brings to biphasic catalysis, offers considerable advantages. These include an enhanced extraction efficiency, a larger window of operation in terms of process parameters, the opportunity to operate in continuous flow mode and the ability to readily recover the emulsifier and/or catalyst. Moreover, taking advantage of the advances in materials science, the solid particle emulsifiers can be modified to be catalytically active and/or stimuli responsive. PE systems also don't need additional external energy input for mechanical mixing, nor rely on the hard-to-recover molecular surfactants used in classic emulsions. These particular PE characteristics thus add to the well-known green chemistry credentials of general biphasic catalysis. More generally, research on PE use for sustainable chemical manufacturing, e.g. by leveraging innovations in materials science, catalysis and chemical conversion, contributes to the efforts aimed at achieving several of the United Nations Sustainable Development Goals (SDG), in particular SDG 12 on responsible consumption and production.15 Indeed, adherence to the green chemistry principles as well as a more endemic implementation of a systems thinking approach in chemistry are essential for the transition to a more sustainable chemical industry. Provided that the products targeted are safe- and circular-by-design and that individual components used for PE catalysis are benign and sufficiently abundant, the development of PE catalysis technology can thus contribute to the sustainability of their production by improving the overall energy and resource demand of a given process.
Here, recent developments in the use of PEs as reaction media for catalytic reactions will be discussed. This nascent field was first reviewed by Nardello-Rataj et al. in 2015, who defined two types of PE catalysis: Pickering Assisted Catalysis (PAC) and Pickering Interfacial Catalysis (PIC).16 In PAC, non-catalytic particles are used to stabilize the PE, in combination with homogeneous catalysts confined in either the aqueous or the organic phase. With PIC, catalytically active particles are used as stabilizing particles. As noted above, PE systems are used in many applications and the reader is referred to several reviews covering different aspects of PE formation and use,17–19 including a very recent one on PE catalysis published while this manuscript was under review.20 Here, we review the state of the art in PE catalysis and cover the literature mostly from 2015 onwards in the context of green chemistry, with a particular focus on the developments in the design of functional solid emulsifiers, on the scope attainable in terms or types of chemical transformations and accessible process conditions and on the use of PEs in continuous flow reactors.
Advances in solid emulsifiers
Various kinds of solid particles with moderate wettability have been investigated as solid surfactants for the preparation of PEs, with silica, carbon and various polymer-based materials as notable examples. Recently, rapid developments in material science have greatly increased the variety of particle compositions and topologies available for PE formation, as well as the additional functionalities these particles can be endowed with. For example, using functional colloids, PE have been prepared with reversible properties by tuning the surface chemistry of the emulsifiers via light,21 gas,22 chemical auxiliaries,23,24 or temperature variation.25 Moreover, particles with well-designed anisotropy26–28 provide superior stability compared to traditional emulsions. These advanced materials indeed offer many opportunities for application in either a PAC or PIC strategy. Here, we discuss some of the properties of (functional) solid emulsifiers most relevant for PE use in catalysis.Wettability and surface roughness
The studies of PE stabilization of oil–water emulsions are numerous and include systems containing hydrophobized inorganic materials such as silica,29 as well as carbon particles,30 synthesized polymers such as crosslinked polystyrene,31,32 and more elaborate Janus particles (i.e. chemically anisotropic/two-sided).26,33 To understand how the physicochemical characteristics of a solid emulsifier affect PE stability, we need to know what the orientation and spatial arrangement of these materials is at the liquid–liquid interface. The configuration of the particles adsorbed at the interface depends on the chemical composition, surface morphology and geometry of the particles. Hydrophobicity is clearly crucial in dictating the emulsion type (oil-in-water or water-in-oil) and the stability against coalescence. Fig. 1 summarizes schematically some of the most common material configurations at the oil–water interface. For hydrophobic materials most of the particle will be located in the oil phase, resulting in contact angles >90°, (Fig. 1, mid top), leading to water-in-oil (w/o) type PEs, while hydrophilic particles lead to inverse o/w emulsions, i.e. showing contact angles <90° (Fig. 1, mid bottom). For example, stable oil-in-water (o/w) droplets can be easily prepared using hydrophilic goethite particles, while (w/o) droplets are obtained with hydrophobic polystyrene particles.34 Notably, while numerous examples report the use of hydrophobic silica for the generation of highly stable PEs, hydrophilic silica spheres have not been found capable of stabilizing PEs without additional surfactants.29,35 Hydrophobization of silica is often used to allow PE catalysis and the same concept has been utilized to tune the contact angle of other types of materials, including catalytically active ones for PIC. For example, hydrophilic zeolites are unable to stabilize o/w PEs, while zeolites hydrophobized by silylation proved very efficient.36The strength of adsorption of the particle at the liquid–liquid interface is governed by both the interfacial tensions and the surface morphology of the particles.37 Increasing the roughness of the surface of the solid particles results in very strong adsorption at the interface (Fig. 1, right top). Isa et al.38 illustrated this by preparing silica microparticles with tunable surface roughness and demonstrated that surface roughness caused pinning of the contact line around the adsorbed particles, consequently trapping the particle via strong adsorption and thus dramatically promoting emulsion stability. Increased surface roughness of the particle not only dramatically improves PE stability, but also offers the opportunity to produce stable o/w and w/o emulsions by dispersing the same type of particle initially into either the aqueous or oil phase.38,39 This holds the advantage of being able to choose, for example, the continuous phase in advanced flow applications (see below).
Particle geometry
The rapid developments in the synthesis of advanced materials have led to a wealth of available uniform (an)isotropic particles that can be studied as model emulsifiers to investigate the interplay between liquid–liquid confinement and particle geometry. Solid emulsifiers used for in Pickering emulsions can be categorized into three main groups when it comes to particle geometry: spheres or patchy particles featuring spherical lobes (Fig. 2a–d), one dimensional (1D) material such as rods, ellipsoid and fibers (Fig. 2e–h), and two-dimensional (2D) nanoplates or nanosheets (Fig. 2i–k). In early work, most of the particles investigated in PEs were homogeneous spheres, which have in more recent examples been endowed with more advanced attributes. For example, Yang et al. used a combination of hydrophobic (MeO)3Si(CH2)7CH3 and relatively hydrophilic (MeO)3SiCH2CH2CH2(NHCH2CH2)2NH2 grafts to turn silica microspheres into a pH-sensitive emulsion stabilizer and allow toluene–water emulsion phase inversion (Fig. 2b).40 | ||
| Fig. 2 Examples of particle geometries used as stabilizers for Pickering emulsions. (a) (Janus) spheres or patchy particles featuring spherical lobes: (b) TEM image of hydrophobic silica spheres (reprinted from ref. 40), (c) SEM image of bifunctional Janus spheres (reprinted from ref. 43), (d) SEM image of carbon–organosilica bi-component dumbbells (reprinted from ref. 27). (e) One-dimensional materials: (f) SEM image of silica rods (reprinted from ref. 49), (g) SEM image of polystyrene ellipsoid (reprinted from ref. 34), (h) SEM image of bacterial cellulose nanocrystals (reprinted from ref. 46). (i) Two-dimensional materials: (j) SEM image of nanoplates (reprinted from ref. 4), (k) SEM image of graphite oxide sheets (reprinted from ref. 53). | ||
Similar to the spheres with high surface roughness mentioned above, pinned contact lines and hence strong adsorption can also be achieved using Janus spheres,41 as the roughness at the Janus boundary is denser than at homogeneously covered particles (Fig. 1, bottom right). Binks and Fletcher reported on a theoretical comparison between spheres of uniform wettability and Janus particles with two surface regions of different wettability.33 By varying the relative surface areas or the wettability of the two surface regions on the Janus particles, they demonstrated that, unlike homogeneous particles, Janus particles retain their strong adsorption regardless of the wettability of different regions. The results suggested that Janus particles with either low or high average contact angles should be efficient emulsion stabilizers. Chemically anisotropic, amphiphilic Janus particles were synthesized by Granick et al. and indeed shown able to form emulsions that maintained their stability for an extended period of time42 and to do so more effectively than homogeneous particles. Lee et al. also showed that (highly modular) Janus spheres can indeed effectively stabilize PEs (Fig. 2c).43 Resasco et al. in turn demonstrated the first use of spherical Pd decorated Janus particles in catalysis for aldehyde hydrogenation.44 Related to the spherical Janus particles, dumbbell and snowman-like functionalized Janus particles have also been reported. Liang et al. recently synthesized snowman-like silica-polymer composites and decorated these with Au nanoparticles for catalytic nitroarene reduction at the emulsion interphase.45 Liu et al. in turn provided an example of dumbbell-shaped, carbon–organosilica based mesoporous Janus nanoparticles prepared through a wet chemical method (Fig. 2d) and loaded with Pt nanoparticles. The Pt-containing Janus catalyst not only showed excellent interfacial activity for stabilizing Pickering emulsions, but also enhanced reaction efficiency in the biphasic nitroarene hydrogenation reaction.27
With the development of new synthesis techniques, various non-spherical particles have now been produced and tested as colloidal surfactants, including particles shaped as one-dimensional fibers,46,47 ellipsoids,34 rods,48–51 two-dimensional nanosheets28,52 or plates.4 Huang et al. prepared silica microrods with aspect ratios (AR, ratio of long to short axis) varying from 1 to 16 (Fig. 2f) to stabilize hexadecane/water emulsions and found that the microrods with larger ARs exhibited superior stabilization efficiency, attributing this to their higher steric hindrance, interface adsorption energy and stronger capillary forces.49 Similarly, in a study of the effect of particle shape on emulsion stability, Vermant et al. prepared a series of polystyrene particles of different ellipticity via physical stretching (Fig. 2g).34 They found that an increase in aspect ratio, keeping all other parameters constant, led to more stable PEs. The resulting emulsions were found to be highly stable for over 10 months when AR > 4.6. One-dimensional nanofibers also exhibited outstanding stability in PE use. Capron et al. prepared monodisperse o/w droplets with 1D bacterial cellulose nanocrystal fibers, with the PE remaining stable for several months (Fig. 2h).46
Illustrative of what can be achieved with the proper particle geometry, 2D graphite oxide sheets (GO) (Fig. 2k) were found to assembly at the water–oil interface without any structural modification to create submillimeter-sized organic solvent droplets that are stable in water for months.53 Erné et al.4 and Capron et al.50 proved, respectively, that ultrathin, plate-like colloidal gibbsite particles and cellulose crystals nanofibers (Fig. 2j and h) are even able to stabilize water–water emulsions, systems that feature ultralow interfacial tension. Nanoplates also provide a versatile geometry for PE stabilization. The adsorption of such nanoplates was found to be stronger than for spheres with the same maximal cross section.4 Moreover, compared to a sphere that lies at the interface, nanoplates blocked a larger area of the liquid–liquid interface, resulting in a higher emulsion stability.
The evident advantage of anisotropic particles with high aspect ratios in stabilizing PEs is that particles tend to lie flat at the interface; hence, little weight is needed to cover a large surface area, contributing to efficient material use, this in contrast to spherical particles.54 Moreover, the adsorption of particles with a high AR is much less sensitive to the three-phase contact angle than sphere adsorption is. Lastly, since the configuration of high AR particles is relatively independent from the three-phase contact angle both stable o/w and w/o can be achieved using same type of particles,52 by tuning the ratio of the two immiscible liquids. This is an obvious advantage over spherical emulsifier particles that need to be modified to a suitable hydrophobicity.28,55,56
Stimuli-responsiveness
Stimuli-responsive materials respond sharply to small changes in physical or chemical conditions with relatively large phase or property changes, including emulsification–demulsification and phase inversion (Fig. 3a). These materials are playing an increasingly important role in a diverse range of applications, such as drug delivery, optical devices and coatings.57 Stimuli-response materials also have great potential in the application of PE systems for catalysis, meeting the strong desire to design PEs with tunable states. For example, careful control over wettability and interfacial tension is needed to facilitate mass transport processes or to obtain reversible PEs that allow easy catalyst recycling. Indeed, it is clearly advantageous for the overall economic viability and sustainability of a process, that both the formation and destabilization of a PE should be controllably accessible. Environment-responsive materials have thus recently attracted considerable interest as they provide not only the ability to stabilize emulsions, but also a tool to reconstruct the emulsion by responding to environmental changes, for example, changes in pH58 (Fig. 3b), CO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 22 or salt59 addition (Fig. 3c), temperature60 (Fig. 3d), light61 (Fig. 3e), or external fields62,63 (Fig. 3f) or even a combination of those.64 The ability to carefully control emulsion morphology and reversibility with stimuli-responsive materials can reduce surfactant use, waste generation and process remediation costs and thus increase the greenness of a process.
22 or salt59 addition (Fig. 3c), temperature60 (Fig. 3d), light61 (Fig. 3e), or external fields62,63 (Fig. 3f) or even a combination of those.64 The ability to carefully control emulsion morphology and reversibility with stimuli-responsive materials can reduce surfactant use, waste generation and process remediation costs and thus increase the greenness of a process.
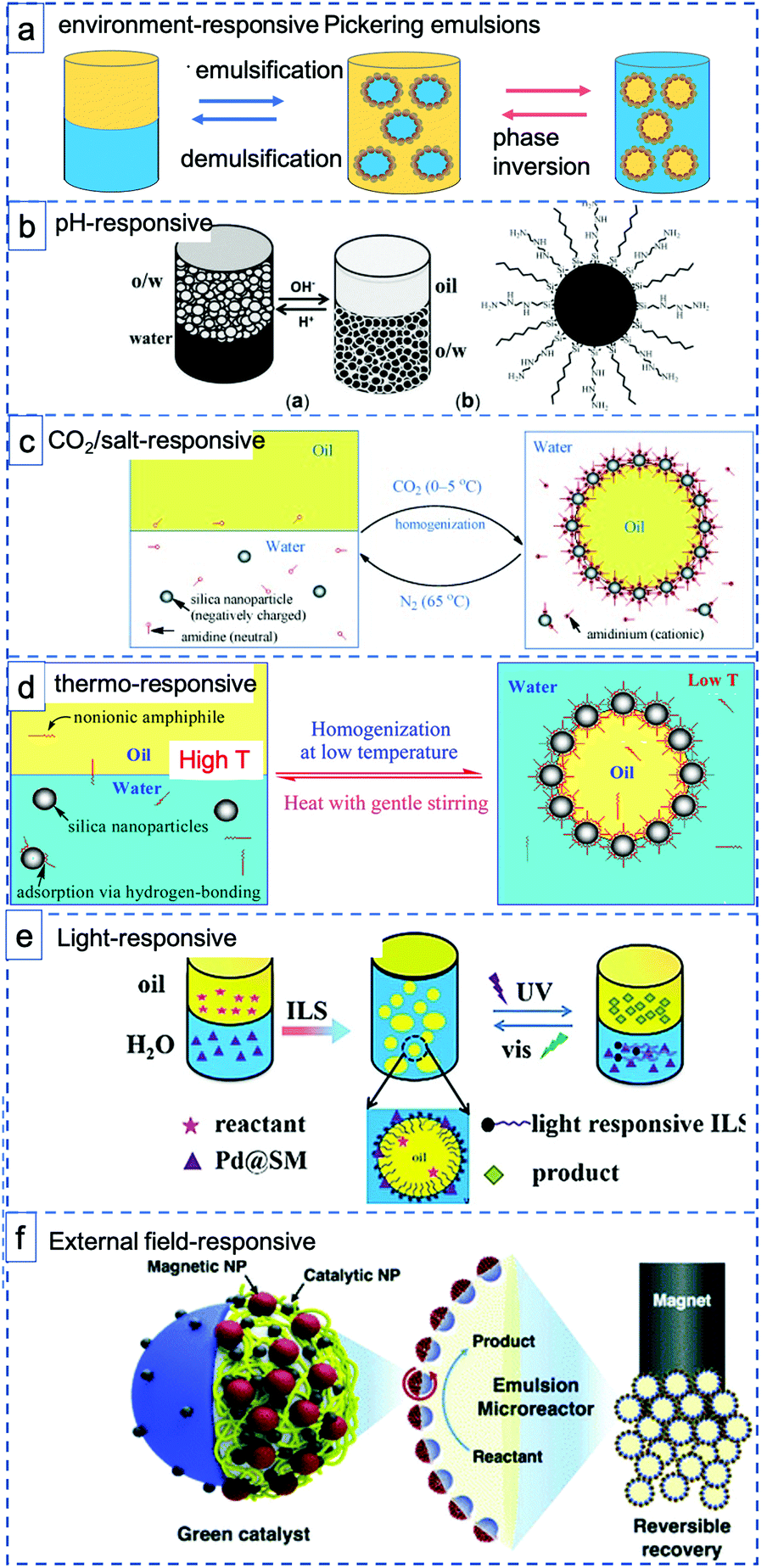 | ||
| Fig. 3 Schematic illustration of various environment-responsive Pickering emulsions: (a) two types of environment-responsive phase changes: emulsification–demulsification and phase inversion. (b) pH-Responsive toluene–water PEs prepared with hydrophobic, pH-sensitive silica sphere (reprinted from ref. 40), (c–e) CO2-responsive, thermo-responsive and light responsive PEs prepared with silica spheres and various functional surfactants (reprinted from ref. 22, 66 and 61), (f) magnetic field-responsive PEs stabilized with Janus spheres decorated with Pd NPs Fe2O3 NPs (reprinted from ref. 63). | ||
Generally, pH-responsive PEs make use of particles that have functional groups that are (de)protonated upon changing the pH. For example, Yang et al. prepared hydrophobic silica spheres decorated with amine groups as interfacially active and pH-responsive solid catalysts. The protonation and deprotonation make the hydrophilicity/hydrophobicity of the catalyst surface switchable, thus driving emulsion inversion. After depositing Pd nanoparticles onto the particles, the emulsifier catalyst exhibited excellent catalytic performance in the hydrogenation of styrene (Fig. 3b).40 Dong et al. reported a thermo-switchable PE system, consisting of an UiO-66-type NMOF (nanoscale metal–organic framework) grafted with poly(N-isopropylacrylamide) sulfide (PNIPAM) to make it thermo-responsive and loaded with Pd nanoparticles for biphasic interfacial chlorobenzene dechlorination.65 The solid emulsifier was used to trigger toluene/water emulsification at room temperature (25 °C), to perform the catalytic reaction and subsequently demulsify at a more elevated temperature of 45 °C. This process could be repeated for at least five times without any loss of its catalytic activity.
In a strategy differing from the use of grafted functional handles to install responsiveness, combinations of solid nanoparticles with additional ‘free’ functional molecules have also been used, e.g. to prepare CO2-responsive or thermoresponsive PEs by self-assembly. For example, Binks et al. used an amidine/amidinium couple to reversibly achieve emulsification/demulsification upon switching between CO2/N2 atmospheres at controlled temperatures. Upon acidification of the water phase, the amidinium surfactant and negatively charged silica components self-assemble through ionic interactions leading to emulsification (Fig. 3c).22 Building further on this strategy, Binks et al. used hydrophilic silica nanoparticles in combination with a low concentration of alkyl polyoxyethylene monododecyl ether as emulsifier to generate emulsions that are stable at room temperature but demulsify at elevated temperature. The thermal effect is achieved through reversible adsorption of the nonionic surfactant at the silica surface via hydrogen bonding at low temperature, interactions that weaken upon increasing the temperature (Fig. 3d).66 An elegant recent extension of this approach involved the use of a photoswitchable, azobenzene dye-containing ionic liquid surfactant(ILS) with which emulsification and demulsification can be triggered by alternating between UV and Vis light irradiation (Fig. 3e). The PE formed by reversible self-assembly was now also used in catalysis, with Pd/SiO2/ILS being active in styrene hydrogenation under ambient conditions (Fig. 3e).61 Phase composition could be controlled by irradiation, again facilitating product separation and catalyst reuse.
When the particles are field-responsive, magnetic for example, the position of the PE stabilized with such particles can be easily changed or the PE itself can be destabilized by applying a magnetic field to easily recycle the emulsifier particles, the reactants and products.62,67,68 Song et al. showed this by preparing Fe3O4 NP/PW12O403−-based ionic liquid composite nanosheets as magnetic field-responsive catalysts for the esterification of oleic acid and methanol.67 The amphipathic and ultrathin magnetic Janus nanosheets not only served as superior solid surfactants to stabilize PEs, but also showed good catalytic performance and reasonable recyclability (78% esterification yield after four recycles). Relatedly, Kim et al. prepared polymer-based Janus microparticles decorated with Pd or Ag nanoparticles for catalysis and Fe2O3 nanoparticles for magnetic separation as PE solid emulsifier and applied the PE systems for advancing organic reactions, such as oxidation, amination, and reduction reactions (Fig. 3f).63
Solid emulsifiers as (bifunctional) catalyst
The previous sections highlighted the opportunities available to tailor the behavior and functionality of the materials used for PE formation. Some of the examples already highlighted that the solid particles can serve not only as emulsifier, but also as catalyst. The catalytic functionality can be introduced using the various techniques available for the synthesis of supported catalysts, e.g. by grafting of organic moieties or by the (selective) deposition of nanoparticles. For example, Yang et al. synthesized Pd/SiO2 emulsifiers by depositing Pd nanoparticles on hydrophobized mesoporous silica.69 These materials acted as interfacial nanoparticle catalysts in a w/o PE and showed significantly enhanced catalytic activity in olefin hydrogenation compared to a conventional BS using Pd on hydrophilic mesoporous silica or a commercial Pd/C catalyst. An example of surface modification by grafting was provided Nardello-Rataj et al.70 who used amphiphilic, acidic silica nanoparticles by introducing inert alkyl chains (C3, C8 and C18) and active propylsulfonic acids on the silica NPs. After optimizing the ratio between the inert and the active surface functional groups (Cn:SO3H molar ratio = 54:46), stable methanol/triglyceride oil PEs could be obtained. The acid groups were introduced via silane thiol grafting, followed by oxidation to yield the sulfonic acids, a strategy that has been often used for monophasic solid acid catalysis. The solid catalysts did not only provide stable MeOH/oil PEs but proved to be highly efficient in the transesterification between methanol and the vegetable oil.
Endowing the particles with two different catalysts offers potential for more versatile PE catalysis. The Pd/NMOF reported by Dong et al. and discussed above in relation to its pH sensitivity, for example, contains both Lewis acidic Zr and Pd hydrogenation sites, allowing tandem PIC, in this case a one-pot biphasic Knoevenagel condensation–hydrogenation reaction.71 In such tandem reactions, the intermediate product produced in the first step can be directly used as the substrate for the next reaction, eliminating the need for separation, purification, and transfer, thereby saving energy and chemicals. It also helps to reduce byproducts and waste generated in separate single-step reactions. For catalysts that are compatible with each other, a combination of the traditional synthesis strategies used for mono-catalyst particles is sufficient as is the case for the noble metal nanoparticles supported on the MOF.72 For catalysts that are incompatible, such as acid and base catalysts, spatial separation of the catalysts and alternative synthesis methods are necessary to avoid active site quenching.
A classical strategy to spatially separate (antagonistic) catalysts is to use stacked-shell structures such as core–shell, yolk–shell or multi-shelled hollow structures (Fig. 4).73,74 For example, Tang et al. spatially separated complementary catalysts in a core–shell Pd@IRMOF-3 nanostructure, synthesizing a single Pd nanoparticle core surrounded by an amino-functionalized IRMOF-3 shell that could efficiently catalyze a Knoevenagel condensation–hydrogenation tandem catalysis using 4-nitrocinnamaldehyde and malononitrile as substrates (Fig. 4a).75 Gu et al. in turn combined otherwise incompatible catalytic functionalities in hyper-cross-linked polystyrene hollow nanospheres, by positioning acidic (sulfonic acid) and basic (amine) groups in the microporous organic structure.76 Because of the nanosphere's geometry, most of the sulfonic acid groups were located on the external shell of the sphere, while the amine precursors were located preferentially on the inside (Fig. 4b). The resulting acid–base catalyst was utilized in the one-pot tandem catalytic hydrolysis-Henry and hydrolysis–Knoevenagel condensation reactions using 2-(2-bromophenyl)-1,3-dioxolane as substrate. Similarly, Weck et al. developed a strategy for the compartmentalization of incompatible catalysts, using core–shell cross-linked micelles. Combining two block copolymers, Co-porphyrin and HNTf2 were located in the hydrophobic core and Rh-TsDPEN and HCOONa in the hydrophilic shell in site-isolated microenvironments.77 The micelles could catalyze an enantioselective tandem reaction consisting of Co-catalyzed hydration and Rh-catalyzed asymmetric transfer hydrogenation, and its efficiency was attributed to fast intraparticle diffusion of the intermediates.
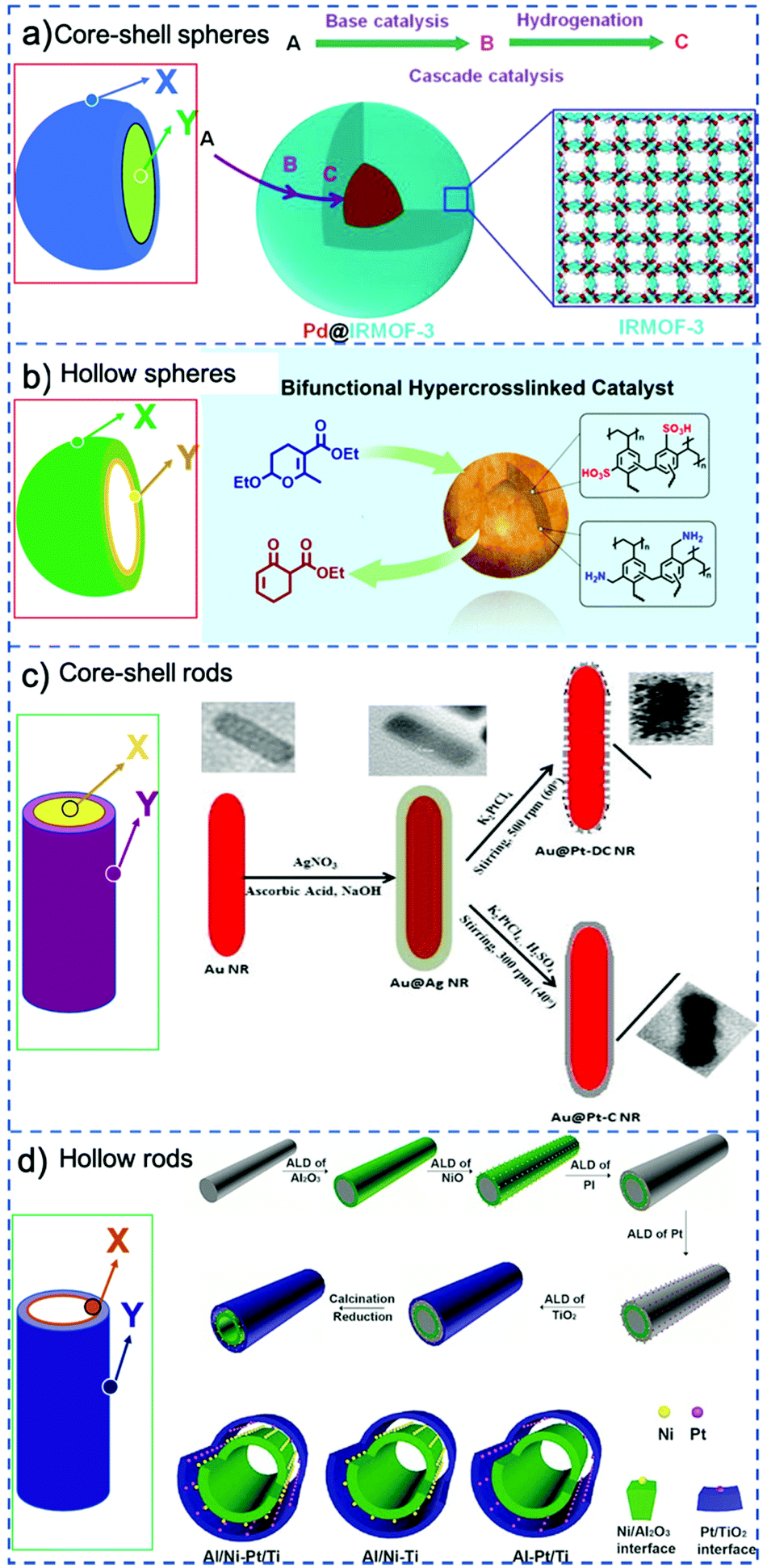 | ||
| Fig. 4 Schematic representation of various possible configurations of stacked-shell structures: for convenience, a general spherical and rod-like morphology is adopted in all cases. X and Y represent different catalysts that are incorporated at different locations. (a) Core–shell spheres. Reprinted from ref. 75; (b) double-shelled hollow spheres. Reprinted from ref. 76; (c) core–shell rods. Reprinted from ref. 78; (d) double-layered tubes. Reprinted ref. 79. | ||
Stacked-shell structures were also exploited in other geometries to control the catalysts’ location. For example, Siril et al. prepared bi-metallic Au–Pt nanorods and these core–shell nanorods exhibit better efficiency in reduction of methylene blue by NaBH4 compared to Au nanorods (Fig. 4c).78 Qin et al. integrated two distinct metal–oxide interfaces based on a tube-in-tube nanostructure using template-assisted atomic layer deposition (Fig. 4d),79 in which Ni nanoparticles are supported on the outer surface of the inner Al2O3 nanotube and Pt nanoparticles are attached to the inner surface of the outer TiO2 nanotube. The confined space favors the instant transfer of intermediates between the two metal–oxide interfaces. This tandem catalyst showed remarkably high catalytic efficiency in nitrobenzene hydrogenation with hydrogen formed by in situ decomposition of N2H4·H2O.
An alternative strategy to make particles with multiple functionalities that are spatially separated is to use patchy particles such as bifunctional Janus sheets,28 spheres80 or rods.81 If the distinct faces of the Janus particles can (both) separately be decorated, this would allow compartmentalization of the catalysts in either one of the PE solvents. Similarly, a catalyst and a functional trigger could be combined on the same particle.
Zhang et al. reported that Janus nanosheets, grafted on one side with nonpolar phenyl groups and on the other side with a polar ionic liquid, were utilized to achieve ionic liquid (IL) in oil PEs with excellent emulsion stability.82 Using triethoxy[(3-(2-imidazolin-1-yl)propyl)]silane and triethoxyphenylsilane as comonomers, paraffin/silica core–shell Janus microspheres were made with imidazoline grafts on the side oriented towards to the aqueous phase and phenyl grafts on the inside. Janus sheets were then obtained by crushing the hollow spheres in an ultrasonic cell crusher. The imidazoline grafts were subsequently alkylated, followed by anion-exchange to finally yield the (monofunctional) catalytic Janus materials. The resulting materials showed excellent performance in the desulfurization of dibenzothiophene and proved to be reusable.
The latter example is typical of almost all recently reported catalytic Janus spheres and dumbbells in the sense that at least one patch is catalytically “inert” and, hence, cannot be used yet for tandem catalysis.27,45,83 It is indeed a considerable challenge to synthesize bifunctional catalytic Janus particles. Recently, bifunctional particles with non-catalytic, but chemically orthogonal patches were successfully developed, for applications other than Pickering emulsion catalysis. For example, Yang et al. developed a versatile method to prepare inorganic silica Janus nanosheets by crushing the corresponding parent Janus hollow spheres. The synthesized bifunctional nanosheets exhibit superior PEs stability and could be easily further functionalized to be paramagnetic with Fe3O4 nanoparticles to give Janus composite nanosheets.28 Weck et al. developed bifunctional anisotropic particles featuring chlorine-functionalized patches and carboxylated domains.84 Kegel et al. prepared patchy particles with tunable geometry that carry chemical handles facilitating independent and orthogonal surface modification using Atom Transfer Radical Polymerization (ATRP) and thiol–yne click chemistry or silane chemistry.80,85
These various approaches offer versatile synthesis strategies to expand the combinations of catalysts that such bifunctional Janus particles can be endowed with. In a more general sense, the opportunities that Janus particles offer for PE stabilization, by pinning the solids at the interface, and for catalysis, by orthogonally addressable patches, holds great promise for their future application. Very recently, Bruijnincx et al.86 synthesized monodisperse, amphiphilic bifunctional Janus silica spheres decorated with sulfonic acid and basic amine groups (Janus-A–B) through a solidified wax Pickering emulsion method. As a proof of concept, they demonstrated that the Janus-A–B could be used as Pickering stabilizers and heterogeneous catalysts for the tandem catalytic dehydration–Knoevenagel condensation reaction of fructose. By tuning the acid/base catalyst ratio with a physical mixture of Janus-A–B and SiO2–NH2–OSi(CH3)3 as solid catalysts, the yield of 5-HMF-DEM could be optimized. Notably, a PE stabilized with monofunctional SiO2–NH2–OSi(CH3)3 and HCl as acid catalyst dissolved in the water phase showed no catalytic activity, as a result of neutralization of the active species, indicative of the advantages bifunctional Janus particles can offer for antagonistic PE catalysis.
Advances in catalysis
Having highlighted the various advances made in the design of functional materials for use in PEs and their most important properties, here we highlight the recent developments and strategies adopted for performing catalysis in PEs. Fig. 5 schematically shows the different possible strategies for PAC and PIC, exemplified for w/o PEs.16 PAC strategies include the use of homogeneous chemo- or biocatalysts, confined in either the dispersed or the continuous phase of the PE (Fig. 5a and b), depending on the nature of the liquid phases and the solubility of the catalyst. Another possibility is to use a combination of catalysts confined in the aqueous as well as organic phase (Fig. 5c) for tandem or cascade reactions. PIC strategies in turn comprise all catalytic reactions in PEs that use catalytically active stabilizing particles. This can be done using catalysts on an inert support, using one type of emulsifier (Fig. 5d) or a physical mixture of particles with different catalytic functionalities (Fig. 5e). Another option is to use non-supported bulk catalyst particles as PE stabilizer (Fig. 5f).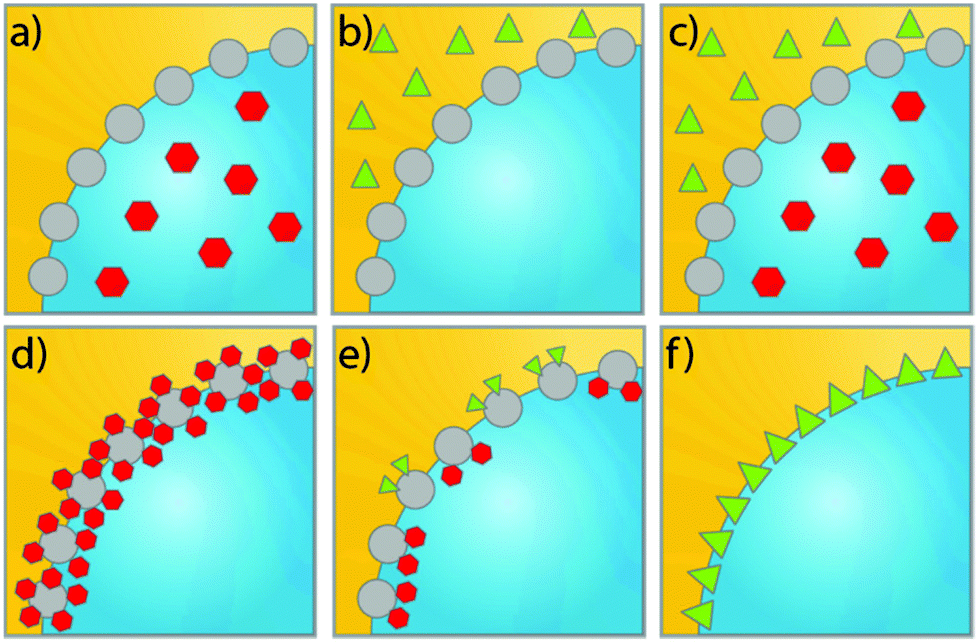 | ||
| Fig. 5 Schematic representation of PAC and PIC in w/o PEs. Yellow: organic phase, blue: aqueous phase. (a–c) PAC approach with (a) catalyst confined in aqueous phase, (b) catalyst confined in organic phase and (c) catalysts confined in both the aqueous and organic phase. (d–f) PIC approach with (d) one catalyst supported on (or in) inert material used as Pickering stabilizer and (e) physical mixture of particles with two distinct catalysts on (or in) an inert material used as Pickering stabilizer and (f) catalyst particles used as Pickering stabilizing particles. | ||
Advances in PAC
One of the reasons for PE use is the high interfacial area that is beneficial for the rate of reactions taking place at the liquid–liquid interface,16 but also for reactions that require extraction of products or intermediates from one phase to the other. One such an example was given by Zhang et al., who employed PEs as a reaction medium for the acid-catalyzed dehydration of fructose to 5-(hydroxymethyl)furfural (5-HMF).87 This reaction is known to be very susceptible to side-product formation as the 5-HMF easily reacts in the presence of acid to small organic acids or polymers. Therefore, quite some studies have been performed on the extraction of 5-HMF during reaction to prevent this side-product formation, with methyl isobutyl ketone (MIBK) having been identified as a suitable extracting solvent.88However, HMF extraction had only been performed in normal BS and not in a PE. Zhang et al. prepared MIBK formulated PEs stabilized with hydrophobic silica particles to perform fructose dehydration reactions.87 The use of these PEs, made with acidic water saturated with NaCl as aqueous phase and MIBK as organic phase, resulted in suppression of side product formation and, consequently, an increase in yield and selectivity (Table 1, entry 1). Notably, the reactions were performed under conditions that are rather demanding for a PE, i.e. at 100 °C for 2 h. Due to the low concentration of solid particles (0.33 wt% with respect to MIBK) emulsification was not complete and stirring was required to keep the high interfacial area. However, the addition of more emulsifier did enhance the yield and selectivity from 57% to 73% and from 60% to 90%, respectively.

|
Whereas most PAC examples use liquid–liquid–solid PE systems to increase the interfacial area to enhance mass transport between the two immiscible phases, this approach can be extended to reactions that require gasses, such as hydroformylation or hydrogenation reactions. Addition of gasses make the systems more complex as the mass transfer of the gas should also be taken into account. For o/w emulsions, the mass transfer coefficient of the gas is similar to that in the pure aqueous phase.89 Therefore, the PE system should principally not affect the reaction efficiency other than the effect of the increased interfacial area when compared to the usual biphasic reaction. For w/o emulsions, the volume fraction of oil is proportionately related to the mass transfer coefficient. Therefore, the mass transfer coefficient can be calculated as the mean of the coefficients in the pure components weighted by the respective volume fractions. The group of von Klitzing et al. studied hydroformylation of 1-dodecene in o/w PEs.90 Hydroformylation reactions are commonly catalyzed by a homogeneous rhodium complex, in the presence of syngas. Third generation hydroformylation processes, such as the one run by Rhone-Poulenc, make use of a biphasic setup and have the catalyst confined to the aqueous phase to allow easy reuse. One of the major issues for biphasic hydroformylation reactions is that longer hydrocarbons are not sufficiently soluble in the aqueous phase and are thus not converted in standard BS systems. Studying PE as alternative reaction medium, the BS system was compared to an o/w PE formed using Halloysite nanotubes (HNTs) or silica as stabilizers, with reactions performed at 100 °C, and 15 bar of syngas (Table 1, entries 2 and 3). Interestingly, the activity of the most stable PE, the one stabilized by HNTs, was low (TOF of 18 h−1), but a high aldehyde selectivity of 86% was obtained. The emulsions formulated without any particles were, not surprisingly, the least stable, but gave a higher TOF of 26 h−1 and an aldehyde selectivity of 84%. The PEs stabilized by silica particles, had the highest activity, a TOF of 55 h−1 was measured, but with the lowest aldehyde selectivity (77%). The use of PEs was thus not intrinsically beneficial for the performance of the catalytic reaction, as the highest TOF was obtained by BS with similar aldehyde selectivity as the HNT-stabilized PE. This could be due to the fact that the reaction mixtures were stirred at 1200 rpm, enhancing the interfacial area of the non-stabilized system as well.
The group of Yang et al. used a different type of PE for the hydroformylation of 1-dodecene, namely an IL/o PE stabilized by C18-functionalized mesoporous silica (Table 1, entry 4).91 The solubility of olefins is higher in ILs than in water and ILs also have a higher boiling point than water. Hydroformylation reactions were run at 110 °C and with 20 bar of syngas, stirring the system at 1000 rpm. The IL system proved superior, combining an excellent TOF of 413 h−1 with a much higher selectivity than a w/o PE (94% vs. 69% at similar conversions). This was ascribed to the higher solubility of substrate and of CO in the IL. The particles could be recycled for five runs without significant loss of reactivity.
Bruijnincx et al. showed that the solid particles in PEs cannot only serve as stabilizing components but also aid in compartmentalizing antagonistic catalysts.92 They formulated w/o PEs containing both homogeneous acid and base catalysts. The acid catalyst was confined within the aqueous phase while the organic acid was dissolved within the oil phase (Table 1, entry 5). These catalysts were used to perform a tandem deacetalization–Knoevenagel condensation of benzaldehyde dimethyl acetal. In the single, acid-catalyzed, deacetalization of benzaldehyde dimethyl acetal, the PEs already outperformed the BS in terms of reaction rate. In the PE, the tandem catalytic reaction could be performed with a yield of final product of 76%, while in the BS no tandem reaction was observed, which was attributed to quenching of the acid and base catalysts.
Generally, for PAC where the homogeneous catalysts are dissolved in the aqueous phase and the solid particles located at the liquid–liquid interface serve only as emulsifiers, the major challenge for long term catalytic stability and catalysts recycle is the loss, by leaching or quenching in case of antagonistic catalysts, of the catalysts. The extent of surface coverage will influence this and can be controlled by the choice of solid emulsifier used; e.g., materials with high aspect ratio such as nanofibers or nanosheets can be applied instead of spheres to physically increase the surface coverage. Mass transport across the boundary will in general also be affected naturally, something that can impact the efficiency of the process, depending on the strategy employed. Adverse electrostatic interactions between solid and soluble catalyst could lead to increased stability of the system. Alternatively, porous materials with well-defined pore size allowing the transport of the substrates but not the (bio)catalysts could also minimize the loss of catalyst. In addition, minimization of catalyst leaching should also be a consideration in the choice of solvent combination used; for example, the use of could improve retention of a (ionically-tagged) catalyst.
Advances in PIC
In the past five years, considerable advances have been made in the field of PIC.93 One example being the heterogenization of biocatalysts, in which enzymes were immobilized on or in the stabilizing materials.94–96 These are often lipases, such as the one from Candida rugosa (CRL), which in combination with the metal–organic framework (MOF) ZIF-8 was shown to form an o/w PE with an aqueous buffer phase and heptane. Two different examples of incorporating the enzyme in the MOF were reported, one being the immobilization of CRL on the outer surface of ZIF-8 crystals after synthesis (Table 1, entry 6)94 and the other being encapsulation of CRL inside the ZIF-8 crystals by addition during MOF synthesis (Table 1, entry 7).95 These catalysts were both tested for the hydrolysis of oil-soluble p-nitrophenyl palmitate (p-NPP) to form and extract the water-soluble reporter product p-nitrophenol (p-NP) at 25 °C. The catalyst with the CRL immobilized on the outside of the ZIF-8 crystals was the most active with a specific activity of 0.25 μmol min−1 mg−1 against 0.16 μmol min−1 mg−1 for the catalyst with the enzymes incorporated inside the ZIF-8 crystals. It was also the most stable, as 83% of the initial activity was retained after 13 recycling runs while the encapsulated enzyme lost all activity after four runs. Furthermore, the specific activity of the immobilized enzyme catalyst was similar to that of the free enzyme, but the storage stability of the free enzyme was much lower (85% for the heterogeneous catalyst vs. 60% for the free enzyme).The same enzyme was successfully adsorbed on carbonaceous microspheres (CMs) in the work of Yang et al. and used for the hydrolysis of olive oil in a o/w PE (Table 1, entry 8).96 This reaction was studied as function of interfacial area and interfacial enzyme concentration. An increase of the interfacial area usually leads to an increase in reaction rate due to shortening the distance between substrate and enzyme. However, at a constant enzyme concentration an increase in interfacial area also results in a decrease of interfacial enzyme concentration, leading to a reduced reaction rate. As a result of these two competing effects, no significant influence of the change in interfacial area on the reaction rate was observed.
Heterogeneous, microporous titanosilicates (TS-1) were used for liquid-phase oxidation and epoxidation reactions. Wu et al. published the preparation of a TS-1/mesoporous silica composite material as catalyst for the epoxidation of 1-hexene (Table 1, entry 9).97 The formation of the composite material was necessary to stabilize PEs as the TS-1 itself is not amphiphilic enough. In this case the increase in interfacial area did have a positive effect, with the PE system showing a considerably higher TON than a non-PE benchmark (7.74 vs. 0.58 mol (mol Ti)−1). However, the activity decreased drastically upon recycling with only 50% of the activity remaining after four runs.
An improved way of using TS-1 as catalyst for 1-hexene epoxidation was shown by Binks et al., making use of hollow, amphiphilic TS-1 (HTS-1) particles synthesized by post-synthesis desilication and silylation (Table 1, entry 10).98 Using the same conditions as Wu et al. a TOF of 20.6 h−1 was obtained, with the almost threefold improvement in activity being attributed to improved mass transfer. The catalyst particles also proved more resilient towards recycling, resulting in 95% of the starting activity after five runs. These examples thus show the power of using the broad range of catalyst synthesis option available to tailor the material's properties to meet the specific requirements for PE catalysis.
Another direction in the application of PIC was taken by Clacens et al.99 in the preparation of bio-based surfactants by acid-catalyzed etherification of glycerol and dodecanol, two immiscible reactants. Glycerol was mixed with dodecanol to form a decanol–in-glycerol–in-decanol PE (d/g/d) stabilized by polystyrene-grafted silica particles functionalized with sulfuric acid groups utilized as PIC (Table 1, entry 11). After reaction for 24 h at 150 °C, a dodecanol conversion of 43% and a dodecyl glyceryl ether (DGE) yield of 34% was obtained. The PE showed slight destabilization, even though a synergistic stabilization between particles and surfactants might have been expected.100 The solid catalyst outperformed benchmark acid catalysts such as p-toluenesulfonic acid (PTSA) and dodecylbenzenesulfonic acid (DBSA) which were not able to form a stable emulsion at all, showing the benefit of using PEs as reaction media for reactions using immiscible substrates.
As noted above for PAC, considerable advances have been made in heterogeneously catalyzed gas–liquid phase PIC reactions. Several different approaches for hydrogenation in PEs have been published by the group of Yang et al.101–103 They created two pH switchable systems stabilized by Pd/SiO2 particles, one being a w/o PE,101 the other one being a g/w PE.58 By increasing the pH to ∼7 a stable emulsion was created, while lowering the pH by addition of HCl led to destabilization and therefore easy recovery of the catalysts. The g/w PE was shown to be stable at room temperature, however, no stability data during reaction was given. More generally, one can say that (operando) studies of PE stability under the actual working conditions requires more attention. The w/o and g/w PE were utilized for the hydrogenation of oil-soluble and water-soluble substrates, respectively, at relatively low temperatures (≤40 °C) and pressures (max. 3.5 bar, Table 1, entries 12 and 13). The PE system outperformed the conventional BS in terms of conversion, however no yield or selectivity was reported.58 Catalyst stability in the liquid phase was high as after 15 runs a yield of 90% was still obtained. In the g/w system the stability was lower, however; already in the second run longer reaction times were needed to obtain full conversion, which was attributed to aggregation of the Pd particles.
Ru-Catalyzed selective hydrogenation of benzene was performed in a o/w and w/o PE stabilized by Ru/TiO2 nanoparticles, varying the type of PEs by altering the hydrophilicity of the TiO2 particles (Table 1, entries 14 and 15).103 The o/w PE gave somewhat better results than both the conventional BS and the w/o PE, because of the low solubility of H2 in water. The selectivity towards cyclohexene was 51% at 84% conversion while, at the same conversion level, only 44 and 46% was reached in the biphasic and the w/o PE, respectively. Recycling of the particles resulted in a gradually drop of the activity, claimed to be a result of loss of material due to multiple transfer and separation steps. These results show that the type of PE (w/o or o/w) should be carefully chosen dependent on the targeted reaction and the reactants involved. In this work, the effect of the droplet size on the performance of the o/w PE as reaction medium was investigated. The droplet size was decreased by adjusting the shear rate during PE preparation. Decreasing the droplet size from 270 to 60 μm resulted in an increase in both conversion, from 68 to 84%, and yield, from 29 to 44%.
The examples highlighted in this section show PEs to be versatile reaction media for catalysis, offering the option to compartmentalize catalysts, to improve reactivity between immiscible substrates as well as opportunities for efficient substrate and product recovery as well as catalyst reuse. The main advantage of the use of PAC is the ability to have molecularly defined catalysts fully dissolved in either phase, thus not having to compromise on catalytic performance by immobilization. The use of heterogeneous catalysts of PIC, for which the synthesis may sometimes be more involved, holds the advantage of having the catalyst located exactly at the liquid–liquid interface. The activity of the latter catalytic systems is generally higher than the PAC systems. Notably, the window of operation of PEs as reaction media has been expanded in the past five years by performing reactions requiring elevated temperatures, using enzymes immobilized on and in support materials and by the use of pH switchable PEs. It should also be noted, though, that many of the reported examples of PE catalysis lack PE stability data, leaving the question open if catalysis is really occurring in a PE system. This in turn makes firm attributions of how PE systems affect catalysis difficult. PE stability is not always easy to investigate under reaction conditions, especially when working with closed vessels such as autoclaves. Many instruments/reaction cells and spectroscopic methods have over the past years been developed, however, for operando spectroscopy studies of catalytic reactions and it would be of interest to see which of these can be applied to the opaque, strongly scattering PE systems.104 In any case, the influence of (high concentrations of) substrate, product and additives and elevated temperatures or high pressures on emulsion stability requires attention. Another important factor that deserves more attention is the effect of droplet size on the catalytic performance. As the PEs are produced by applying high shear to the mixtures it is hard to study this systematically. However, other methods for studying this could be considered, such as systematically changing the concentration of solid stabilizing particles or altering the volume fractions of the liquids.
Advances in PE reactor engineering
All previous examples make use of PE as reaction media in batch reactions. However, the ability of PE to encapsulate the water or oil phase in a stable droplet in a continuous phase offers the opportunity to use PE as method for catalyst immobilization in a flow reactor. Such a combination of continuous flow and catalyst immobilization provides an opportunity for facile product separation together with catalyst recovery, resembling heterogeneous catalyst use. Furthermore, performing reaction in flow can also favorably increase the selectivity due to the shortened contact between product and catalyst preventing the formation of side products. Several catalytic reactions have been performed in Flow Pickering Emulsion (FPE) demonstrating high activity as well as (enantio)selectivity, as summarized in Table 2. A first step towards a continuous flow PE system was provided by Yang et al., with the semi-continuous Pickering emulsion/organic biphasic system (PEOBS) concept that combines facile product separation with facile recycling of the nanoparticle catalysts.106 PEOBS consist of a Pickering emulsion phase (w/o; the lower layer) with an excessive organic phase (the upper layer). During the catalytic reaction, the organic substrates in the upper layer (as a reservoir) can rapidly exchange with the organic compounds in the continuous phase of the lower Pickering emulsion. At the end of reaction, the final product that has accumulated in the upper organic phase could be straightforwardly separated simply by liquid transfer, and the solid catalysts recycled in situ without the need for demulsification or another separation procedure.

|
The first implementation of a packed bed-like flow system was reported by Yang et al.107 In their seminal contribution, they demonstrated that a cascade reaction involving two incompatible catalysts could be run by compartmentalizing those substances in PEs.
The reaction was carried out by continuously feeding benzaldehyde dimethyl acetal dissolved in the continuous non-polar solvent phase to a glass column reactor hosting two layers of w/o emulsion beds (Fig. 6a). On a first layer of PE containing the acid catalyst, compartmentalized inside the water droplets, the second layer was stacked carrying NaBH4 inside its water droplets. Having the reagents immobilized in the aqueous droplets, neutralization of the catalysts could thus be prevented by the PE. The non-polar benzaldehyde dimethyl acetal, dissolved in the continuous organic phase, could pass the interfacial barrier and be converted to benzaldehyde by the acid catalyst. Further transport of benzaldehyde via the continuous phase to the sodium borohydride containing aqueous droplets in the second layer resulted in reduction to benzyl alcohol. This system shows the ability of PEs to enable the diffusion of non-polar molecules in the continuous phase passing static catalyst droplets packed along a reaction volume, thus resembling a classical continuous plug flow reactor (PFR). This result consequently motivated a number of investigations to understand and implement flow PEs (FPEs) in a continuous reaction system.
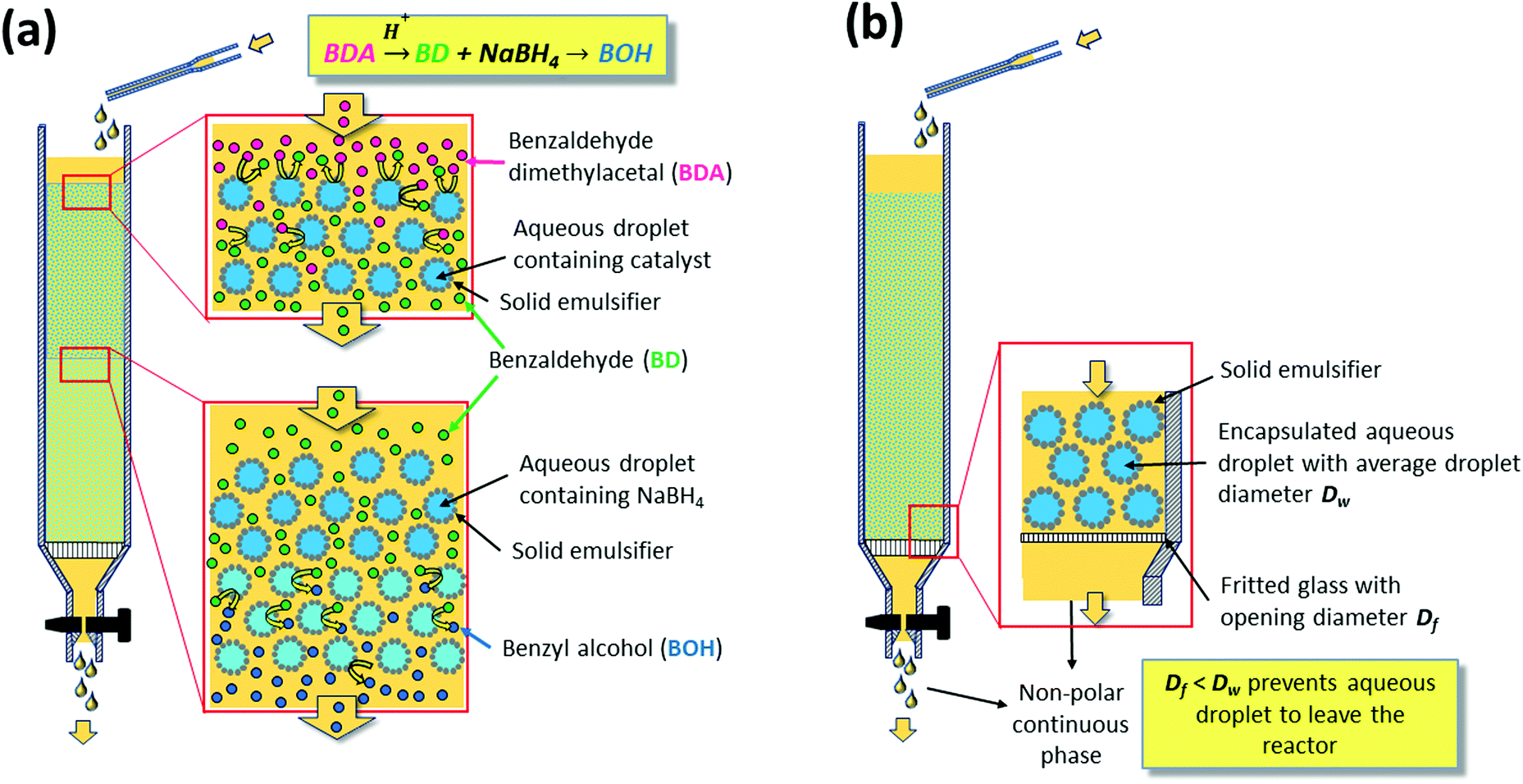 | ||
| Fig. 6 (a) Illustration of the lamination technique used to compartmentalize a substance inside a droplet in a Pickering emulsion (b) realization of flow Pickering emulsions. | ||
Molecular transport in FPE was first studied in detail by Yang et al. in 2016.108 To realize a continuous operation, n-octane was constantly fed to a column filled with w/o PE with different solid emulsifier concentrations (Fig. 6b).107 A steady flow rate was achieved for 360 h, highlighting the stability of the PE against gravity and flow shear force. PEs possessing different water droplet diameters (DW) were successfully prepared by altering the amphiphilic silica concentrations. Higher solid emulsifier concentrations produced smaller DW, in turn resulting in a low flow rate and higher residence time because of the low permeability in the bed. Additionally, a smaller Dw enlarged the ratio between interfacial area and reaction volume (a/V), which is potentially beneficial for catalytic efficiency (CE). Contrarily, at lower silica concentrations, high flow rates were observed due to the formation of larger Dw resulting in higher permeability. This behavior suggests that water droplets in the PE resemble the solid particles in a catalyst bed in a packed column used for classical liquid or gas phase catalytic flow reactions. A theoretical understanding of the effect of DW on the flow rate in PE was sought by applying Darcy's law for laminar fluid permeation through densely packed porous media, as expressed in eqn (1).
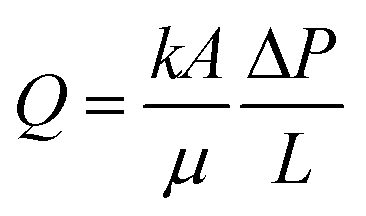 | (1) |
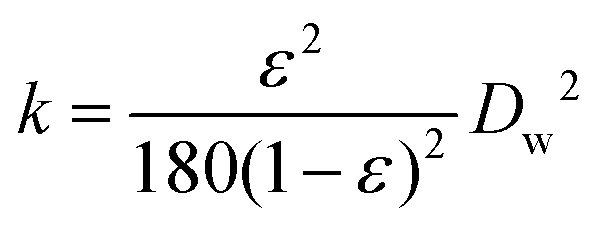 | (2) |
For a given cross sectional area (A), fluid viscosity (μ), total pressure drop (ΔP) and thickness of the bed (L), the discharge volumetric rate (Q) is proportional to the permeability (k) which is a function of porosity (ε) and Dw in PE (eqn (2)). Darcy's law fitted fairly well at smaller DW (30–70) μm. However, a large difference is observed between the calculated and measured flow rate as the droplets grow larger than 70 μm, probably caused by the broad distribution of DW. Moreover, a residence time distribution (RTD) plot, assessing the resemblance of FPE to the plug flow model showed a narrow sharp distribution curve implying that the permeation of n-octane is close to ideal plug-flow with negligible dead zone and channeling. Furthermore, a theoretical model was developed to describe the stability of PEs under flow conditions. This was realized by calculating the adsorption force (Fa) holding particles at w/o interface and the drag force of the running fluid (Fd) attempting to detach the particles from the interface. According to the calculation, Fa is considerably larger than Fd, 5.2 × 10−9 N and 7.47 × 10−17 N respectively, suggesting that the stress caused by the flowing fluid has a negligible effect on breaking PEs.
FPE were furthermore exploited to mediate catalytic additions of alcohol to 3,4-dihydro-2H-pyran. The reactions were performed by feeding non-polar substrate in toluene through a w/o emulsion encapsulating the homogeneous H2SO4 catalyst. For comparison, a biphasic system used in batch showed a catalytic activity of 0.011 mol mol cat.−1 h−1, which could be increased to 0.024 mol mol cat.−1 h−1 after adding hydrophobic silica to form a w/o PE. Switching the system to continuous FPE mode, led to an order of magnitude performance enhancement at 0.235 mol mol cat.−1 h−1, providing a powerful example of the possible advantages offered by FPE catalysis. Variation of the emulsifier concentration again influenced Dw, with higher concentrations generating smaller droplets, ultimately leading to a further increase in CE. This was attributed to the better contact between catalyst and substrates and the longer residence time as a result of the larger pressure drop over the bed that accompanies a smaller DW. However, it is important to note that excessive emulsifier concentrations negatively affected CE due to the formation of multiparticle layers. These layers create a diffusion barrier for the substrate to reach the active w/o interface buried underneath, resulting in a slower reaction. A first example has also been reported of the use of FPE to increase stereoselectivity, in this case in the cyclization of citronellal over a H3PW12O40 catalyst to form isopulegol.109 Citronellal is slightly water miscible and can therefore be converted both on the interface and in the aqueous droplets. Interestingly, the authors observed an increase in diastereoselectivity upon changing from a simple batch BS to a PE system, a selectivity that was further improved with the application of flow FPE catalysis. This was attributed to the microenvironment at the water interface being dramatically different to the bulk phase due to the presence of unique hydrogen bonding interactions. An o/w FPE has also been successfully implemented for the reduction of polar compounds (nitrophenol, methylene blue and methyl orange) with NaBH4 and Au or Pd particles supported on aldehyde-decorated cellulose nanofibers (ANCF) as catalyst. The FPE system showed stable operation for over 50 h time on stream at (close to) complete conversion. With the reductant and substrate both being dissolved in the continuous phase, the reaction in this case takes place at the liquid–liquid interface, where the immobilized metal nanoparticles are located. The hexadecane droplets in this case just serve to offer a large liquid–liquid interphase area.110
The two examples above show the benefits of FPE in promoting interfacial catalysis. However, under flow conditions away from equilibrium, only those catalytically active species close to or at the w/o interface contribute to the reaction, while most of the molecular catalysts used so far in FPE are present throughout the droplet. A microscale study provided further insight into the extent of catalyst participation in the reaction. In this study, Yang et al. demonstrated that enantiomeric transesterification of alcohols with chiral esters can be successfully performed in an ([BMIM]PF6) IL/octane system catalyzed by CLAB lipase.111 IL were chosen as dispersed phase rather than water, as they are also immiscible with the organic continuous phase, but do dissolve a wider range of organic substrates than water. Using Fick's 2nd law to solve the mass balance of limiting reactant and product, concentration profiles of reactant and product were obtained inside the droplets (Fig. 7a). The profiles suggest that the transport of reactant and product molecules is driven by the diffusion of these compounds and that the reaction takes place in the droplet only a very short distance away from the shell. As a result, there is an optimum in IL droplet diameter (DIL) providing the highest CE for the reaction (Fig. 7b). Principally, when DIL is too large, the contribution of catalyst located far inside the droplet will be small leading to a lower CE. On the other hand, if the DIL is too small, most reactant molecules will reside too shortly in the droplet, also causing CE to drop. Droplet size optimization was also applied in an example of continuous cyanosilylation of ketones over the [BMIM]Cl as catalyst encapsulated in [BMIM]BF4 IL droplets with n-octane as continuous phase. Excitingly, this FPE showed no signs of deactivation over a 250 h run.112 The sensitivity of catalyst performance to droplet size was also noted in an example using [BMIM]BF4 IL as dispersed phase stabilized by H3PW12O40 functionalized Janus nanoparticles in toluene used for the acylation of toluene with acetic anhydride. In this case, the activity and selectivity of the acylation could be improved by introducing a smaller amount of IL to decrease the droplet size and increase the higher interfacial area.113
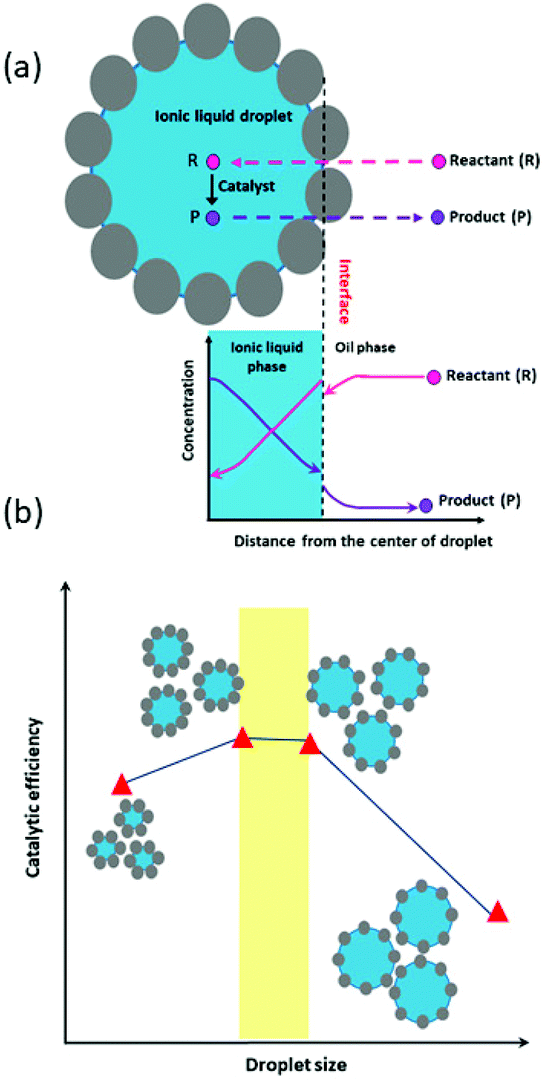 | ||
| Fig. 7 (a) Concentration gradient of reactant and product in the IL droplet and continuous phase affected by diffusional transport and chemical reaction. (b) The influence of droplet size on catalytic efficiency, demonstrating the effect of droplet size on efficiency. Adapted from ref. 113. | ||
Another challenge posed by the soft characteristics of the PE droplets is that tight packing of tiny droplets is required in macrofluidic continuous flow operations to achieve good emulsion stability, a requirement that comes at the expense of high viscosity. Also, practically, charging a reactor column having a large height to diameter ratio with a thick emulsion to produce the PE analogue of a fixed bed is not trivial and can potentially lead to local heterogeneities. If packing is indeed heterogeneous, this may contribute to irreproducibility in performance, e.g. as a result of channeling and/or dead zones along the bed. The filling technique as well as longer term macroscopic stability of the bed therefore requires considerable attention, as it does in traditional packed bed reactors filled with solid catalysts. For the latter systems, mechanical stability and mass transfer considerations dictate that in such classical packed bed reactors, shaping of powdered heterogeneous catalysts is crucial to reduce attrition and the pressure drop over catalyst bed. This then needs to be done without significantly reducing the effectiveness factor of the catalyst. Such traditional fixed bed reactors furthermore offer the flexibility of easy control over the residence time inside the reactor by changing the gas or weight hourly space velocity, i.e. the flow rate. The ability to produce stable and homogeneous reactor beds and to easily and systematically vary process conditions such as residence time offered by traditional fixed bed operations are not that readily replicated with the soft droplets used in FPEs.
In general, for FPE beds homogeneity and stability depend on how the emulsification method and amount and type of solid emulsifier used affect the droplet size and polydispersity. For example, a pressure drop in the emulsion bed can be reduced by using a lower emulsifier concentration resulting in bigger droplets, but larger droplets tend to be more unstable. Enlarging the droplets also shortens the residence time and decreases the surface to volume ratio resulting in lower conversion. This sensitivity and interdependence are also encountered when considering residence time. Flow can, for example, be enhanced by applying additional pressure to the system, but this could at the same time promote coalescence of the droplets, affecting FPE performance. Moreover, attention must be given to the fact that particles can tend to detach from the interface. These unbound particles could deposit on the filter grid creating a cake that can potentially impose additional pressure build up in the system. An example of how this can be prevented by judicious choice of PE solid emulsifier is given below. The development and application of amphiphilic materials with a hollow structure, larger size and rigid shell-crust, for example, are interesting approaches to address this problem. Notably, the stability of a IL/o FPE system could be improved by fortifying IL droplets in n-octane stabilized by hydrophobic silica nanoparticles with a silica crust grown by sol–gel condensation of trimethoxysilane (TMS).114 The silica crust thickness can be adjusted by altering TMS concentration during the synthesis. N2-Physisorption confirmed the structure's mesoporosity, an advantageous attribute to avoid or limit mass transfer limitations. These IL/o PEs with the silica crust maintained their stability under continuous flow at 120 °C and 1.0 MPa for 70 h. In the absence of the silica crust the droplets collapsed already after 0.5 h under 0.3 MPa. Moreover, this stable material combined a high catalytic efficiency with high ee for asymmetric transesterifications, ring opening of epoxides and Tsuji–Trost reactions even after being exposed to an n-octane flow for at least 100 h.
So far, the FPE discussed above were all performed using a glass column hosting a PE bed with continuous addition of the organic continuous phase. A different approach to FPE was studied by the group of Drews. In their study, Stöber silica was silylated with trimethoxy-octadecyl silane to create amphiphilic spherical particles to compartmentalize water droplets under a flow of organic phase in a continuous stirred tank reactor (Fig. 8). Owing to the increased viscosity of the emulsion, N2 pressure and elevated temperature were applied simultaneously to readily squeeze the organic phase out of a membrane mounted at the bottom of the vessel.115 Interestingly, the applied pressure positively affected the flux of organic phase leaving the vessel. Optical micrographs confirmed that the increased flux along with the pressure was caused by the droplet breakage. This FPE system was tested in the transesterification of vinyl-butyrate with phenyl ethanol catalyzed by lipase Ca1A.116 Flow behavior in the presence and absence of lipase Ca1A were firstly studied in an unreactive system before advancing to the continuous reaction. Notably, the addition of lipase Ca1A, which is a surface-active enzyme, to the FPE system appreciably altered the amount of solid emulsifier on the interface because of competitive adsorption of solid emulsifier and lipase. This competitive adsorption led to particle detachment from the w/o interface with the unbound particles getting dispersed in the organic phase, ultimately leading to a build-up of a cake layer covering the membrane surface responsible to the reduced permeability. The rate of the organic phase leaving the vessel declined with time, as the pressure drop gradually increased as a result of more dense droplet packing on the membrane. This decrease in flow did, however, lead to longer residence times, enhancing enhanced product yield in this case. The issue of membrane blockage was addressed by addition of amphiphilic colloidal silica; this type of silica could prevent accumulation of residual unbound particles on the membrane by creating a particle network dispersed in the continuous phase leading to improved flux stability and reproducibility.117 Moreover, the colloidal silica provided droplets with a smaller mean diameter, leading to higher activity compared to the spherical one.
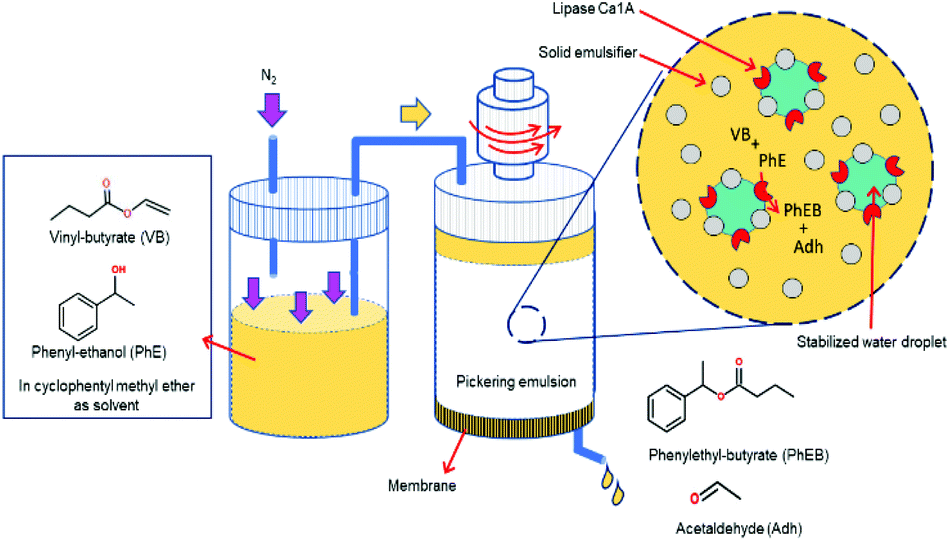 | ||
| Fig. 8 Enzymatic transesterification in a flow Pickering emulsion performed in a continuous stirred tank reactor. | ||
Finally, in addition to the macrofluidic flow systems discussed above, microfluidics also offers interesting advantages for FPE application. Bruijnincx et al. first reported on microfluidic PE catalysis with a tandem deacetalization–Knoevenagel condensation of benzaldehyde-dimethyl acetal to benzylidene–malononitrile using hydrophilic FEP tube to facilitate FPE (Fig. 9).118 To create the w/o emulsion, the organic phase containing silica emulsifier, the substrate benzaldehyde dimethyl acetal, and 2-(1-ethylpropyl)piperidine as the base catalyst was led to a T-junction, allowing it to mix with an aqueous phase containing HCl. Consequently, a single aqueous droplet stabilized by silica emulsifier was observed leaving the junction separated uniformly from the next droplet by the dispersed organic phase. The transparent tubing allowed operando monitoring of the reaction via Raman spectroscopy providing real-time kinetic information at different locations along the tube. This feature was utilized to follow the kinetics of the first deacetalization step, demonstrating that the addition of silica resulted in a noticeably increased reaction rate along with a higher yield of benzaldehyde in comparison to a biphasic flow system without silica. This improvement was attributed to the enlargement of interfacial area accompanied with improved droplets stability. The addition of silica in the FPE was also beneficial for the acid–base catalyzed tandem reaction, as this decreased to amount of acid–base quenching and therefore increased product yield.
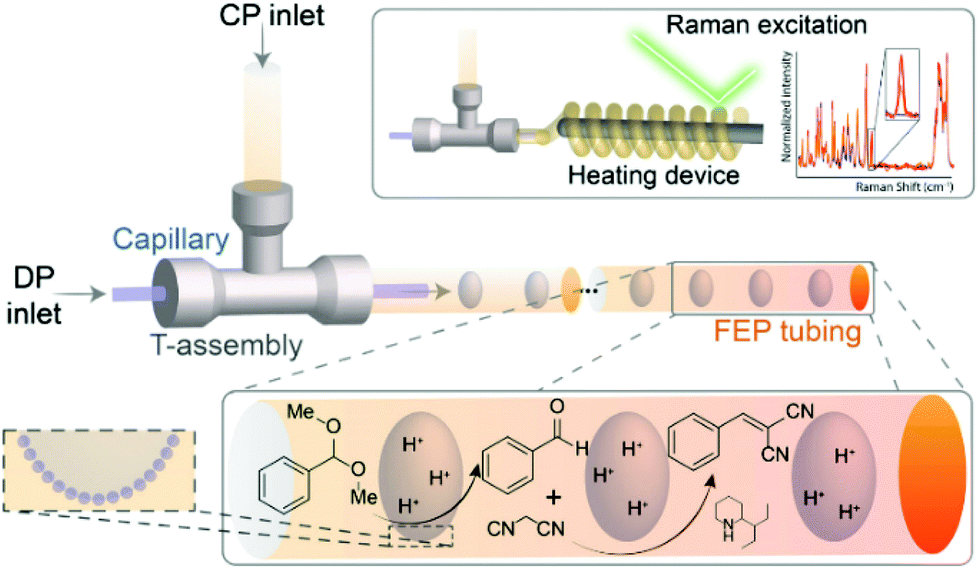 | ||
| Fig. 9 PE for antagonistic tandem FPE catalysis in a microchannel. Reprinted with permission from ref. 118. | ||
General sustainability considerations
As noted throughout the different sections above, Pickering emulsion catalysis can offer considerable advantages from a green chemistry perspective, i.e. with respect to resources used, energy consumption and process intensification.93,119–121 Provided that the reactions that are catalyzed are in and of themselves sound (i.e. the ‘right thing’ is to target a chemical conversion based on renewables and with benign reagents), PE catalysis can ensure that “the right thing is done right”, i.e. with the proper approach in terms of process operation.122Regardless of actual composition, PEs offer several advantages over conventional BS, as noted above. These include improved interfacial area, hence improved mass-transfer properties,103 and easy recovery of the emulsifier.70,123 Most importantly, PE systems exhibit long term emulsion stability, a property crucial for their application in flow.
These advantages do not matter, however, if the components of the PE itself are not sustainable. Clearly, key considerations here are the solvents chosen to make the biphasic (of multiphasic in case of reactions that require gases) and the solids used as emulsifiers/catalysts.
Solvents in PE
To truly make PEs a green alternative for more conventional reaction media, the choice of organic solvent requires careful consideration. In Tables 1 and 2 the organic solvents used to formulate the PE have been highlighted based on their classification according to the CHEM21 selection guide.105 A considerable number of the commonly used solvents had to be classified as problematic. For those examples where the organic phase was purely used as solvent and not, as in some examples that are run neat, also as reactant, a more sustainable and greener alternative is suggested below (Table 3). For example, toluene is one of the most commonly used organic solvents in PEs, as advantage can be taken from it being highly insoluble in water and its ability to solubilize many substrates and products. Toluene use has been ranked as problematic, however, given that it is fossil-based and has carcinogenic and toxic properties. A biobased alternative for toluene was proposed by Hunt et al. with 2,2,5,5-tetramethyltetrahydrofuran (TMTHF),124 which can be synthesized from renewable resources and has similar solvent properties as toluene. Although it is an ether, it cannot form hazardous peroxides, thus providing a greener and safer alternative for toluene. n-Heptane, a solvent often used in enzyme-catalyzed reactions, is also considered problematic, being very toxic for aquatic life with long term effects. In this case, an alternative class of solvents was suggested by Bergbreiter et al., in the form of hydrocarbon oligomers, the so-called poly(α-olefin)s or PAOs.125 These PAOs can be used in combination with polar solvents without mixing. They are non-volatile, non-toxic and have similar solubility properties as heptane. Finally, the classification as problematic of cyclo-pentyl methyl ether (CPME) is not based on health or environmental issues, but on the fact that it has a relatively low auto-ignition point (180 °C). While this temperature is far from those reached in the FPE examples shown, which are typically executed at room temperature, this may become an issue to consider when the window of operation of PE in catalysis is widened.| Solvent used | CHEM21 classification | Alternative solvent |
|---|---|---|
| Toluene | Problematic | 2,2,5,5-Tetramethyloxolane |
| n-Heptane | Problematic | Poly(α-olefin)s (PAOs) |
| CPME | Problematic | — |
Ionic liquids have also proven very versatile for application in PE catalysis and have the potential of minimizing the environmental impact of some organic solvents.126 Naturally, PE catalysis can benefit from the classical merits of IL use, i.e. negligible vapor pressure, excellent thermal stability as well as outstanding solvation properties compared to conventional organic solvents. Moreover, ILs can serve both as polar and apolar phase in PE and can thus be applied to prepare different types of emulsions including aqueous-IL, oil-IL or even IL-oil–water systems.127
Solid emulsifiers in PE
Any green chemistry considerations related to the solid emulsifiers have mainly to do with composition, synthesis and stability under use. Naturally, materials made from abundant resources are preferred, as are scalable and efficient synthesis routes.120,128 Currently, those routes may often still be cumbersome and wasteful, in particular when the more advanced solid emulsifiers are concerned. For example, the use of Janus particles as advanced emulsifiers offers many opportunities for PE catalysis, having both hydrophilic and hydrophobic faces, similar to molecular surfactants.17,43,113 Indeed, the formulation of new ‘smart’ Janus particles allowing control over emulsion morphology on demand with external controllable stimuli using thermo-, pH-, light-, or electro/magnetic-responsive catalytic nanoparticles provides an exciting research direction.24,68,129 But synthesizing such asymmetric particles and functionalizing the individual patches selectively is often an elaborate (and wasteful) synthetic exercise130 and it is therefore also essential to explore how such engineered, colloidal particles can be made efficiently in a scalable manner, from green, sustainable materials.120,128–130Furthermore, the functionalities of a given emulsifier need to be stable, so that the corresponding properties (wetting, stimulus-response, catalysis, etc.) are maintained. This requires the materials to show long-term stability under reaction conditions for both flow and batch systems, as well as under recovery and re-emulsification conditions for those used in batch. For PAC systems, this requires attention, e.g. for the fate of the functional groups introduced to tailor wettability or stimulus response of the solid emulsifiers and for PIC systems for the catalytically active phase, as well. In general, more attention needs to be paid to the characterization of a used PE solid emulsifier. Leaching of any grafted or deposited functionality, changes in surface composition or structure as well as poisoning or fouling of (components of) the material by reaction-derived carbonaceous deposits can all possibly lead to loss of performance. This becomes increasingly relevant when the operational window of PE catalysis, i.e. the severity of conditions to which the solids are exposed, becomes wider. If chemical changes do occur on the (catalytic) solid emulsifier, regeneration or reactivation protocols need to be adopted to allow repeated reuse. The sensitive nature of components of many of the emulsifiers, e.g. the organic moieties grafted on the solids for wettability, stimulus response or catalysis, preclude the application of some of the more standard means of catalyst regeneration used in traditional heterogeneous catalysis (e.g. a simple coke burn off). In this light, if properties such as strong adhesion to the interface are intrinsic to the (inorganic) solid, e.g. as a result of particle geometry or morphology, rather than the result of an added (sensitive) component, then this could greatly benefit not only the ease of preparation, but also the extent of possible long-term use of the material.
As in all branches of (materials) science, the application of the tools of machine learning, or artificial intelligence in general, can guide the way to discovery of new advanced materials for application in PE catalysis.131 The search for new materials should then explicitly incorporate sustainability considerations for materials fabrication in the algorithm to arrive at solid emulsifiers that cannot only do the right things right, but also reduce the environmental burden by being designed and synthesized right.132
Engineering in PE
In addition to a judicious choice of solvent as well as of composition and preparation of the solid emulsifier, chemical (reactor) engineering considerations also greatly impact the overall green chemistry credentials of a given process, as they affect energy and resource consumption, productivity, process intensification as well as downstream processing requirements in terms of product separation and substrate and solid reuse.The developments in FPE application are particularly interesting here for chemical manufacturing, with both packed bed and CSTR like configurations now having been demonstrated. The possibility to carry out multiphase organic synthesis in such a continuous manner clearly holds potential to produce fine, specialty and perhaps even commodity chemicals in a time-, energy- and cost-efficient manner. Further study of the long-term stability and homogeneity of the emulsion bed, also at more severe conditions, and of mass and heat transport phenomena, residence time control and, more generally, the hydrodynamics of FPE will undoubtedly lead to further improvements in the energy- and resource efficiency and scope of these continuous operations. Process intensification by tandem catalysis in FPE also warrants further exploration. The use of laminated PEs in flow for incompatible reagent/catalyst use provides an elegant example of the potential of this direction.107 Inspiration can also be taken from the many developments in the application of conventional flow chemistry for the multiple step synthesis of organic compounds, which has moved from a laboratory technique to industrial application.133 The highly modular approach of flow chemistry is increasingly adopted in the manufacturing of fine chemicals and pharmaceuticals for reasons of chemical control, affordability and environmental footprint reduction.134 Indeed, FPE can become a new tool in the flow chemistry toolbox for sustainable chemical synthesis. Opportunities for further development here lie, for example, in the radiation sources in FPE catalysis to target specific compounds. Examples of this have been shown in the review by Noël et al. discussing photochemical reactions in flow.135 Naturally, these opportunities also extend to studying the chemical conversions in FPE with operando spectroscopy.
Conclusions
Considerable progress has been made in PE catalysis in terms of solid emulsifier design, scope of reactions that can be catalyzed, as well as in the engineering aspects of PE catalysis. In terms of material design, the shift to more advanced emulsifiers, be it those endowed with stimuli responsiveness or with catalytic bifunctionality, is striking and given the wealth of synthesis methods available for structured (nano)materials many more new contributions can be expected here in the near future. Efforts should then focus on combining PE stability, as exemplified by the pinning of particles that are either rough or have Janus properties, with dynamic and/or catalytic functionality. The scope of reactions that can be performed is also rapidly expanding. Specially, attention should now be paid to expanding the operating window in PE catalysis to shift to more practical and commercially relevant conversions, accompanied by operando experiments to really explore the dynamics of PE systems. Next to the choice of solid emulsifier and type of catalyst, the solvent combinations that form stable PEs can also be expanded, keeping the solvent selection guides based on the green chemistry principles in mind. The development of PE catalysis in flow (FPE) is highly exciting, both from a catalytic efficiency and process engineering point of view. The opportunities offered by this approach for combining (homogeneous) catalyst immobilization with continuous processing are numerous, although the interplay between droplet size, flow rate, residence time, diffusion and stability needs to be further systematically explored. Ultimately, for catalytic PEs to become industrially viable, PEs with uniform and controllable droplet size need to be reproducibly accessible at large scale and these PEs should be amenable to use in different reactor configurations (batch, packed bed, CSTR) depending on the specific process requirements. In any case, the rapid developments in PE catalysis show that these advanced biphasic systems hold great promise for the development more sustainable, green chemical conversion processes.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work is part of an NWO VIDI project with project number 723.013.001, which is financed by the Dutch Research Council (NWO).References
- S. U. Pickering, J. Chem. Soc. Trans., 1907, 91, 2001–2021 RSC.
- W. Ramsden, Proc. R. Soc. London, 1903, 72, 156–164 CAS.
- Y. Chevalier and M.-A. A. Bolzinger, Colloids Surf., A, 2013, 439, 23–34 CrossRef CAS.
- M. Vis, J. Opdam, I. S. J. van ‘t Oor, G. Soligno, R. van Roij, R. H. Tromp and B. H. Erné, ACS Macro Lett., 2015, 4, 965–968 CrossRef CAS.
- D. G. Shchukin, K. Köhler, H. Möhwald and G. B. Sukhorukov, Angew. Chem., Int. Ed., 2005, 44, 3310–3314 CrossRef CAS PubMed.
- R. Pal, Curr. Opin. Colloid Interface Sci., 2011, 16, 41–60 CrossRef CAS.
- E. Dickinson, Curr. Opin. Colloid Interface Sci., 2010, 15, 40–49 CrossRef CAS.
- J. Frelichowska, M.-A. Bolzinger, J.-P. Valour, H. Mouaziz, J. Pelletier and Y. Chevalier, Int. J. Pharm., 2009, 368, 7–15 CrossRef CAS PubMed.
- J. Marto, A. Ascenso, S. Simoes, A. J. Almeida and H. M. Ribeiro, Expert Opin. Drug Delivery, 2016, 13, 1093–1107 CrossRef CAS PubMed.
- H. Hugl and M. Nobis, Top Organomet Chem, Springer, Berlin, 2008, 23, 1–17 Search PubMed.
- C. Li and Y. Liu, Bridging Heterogeneous and Homogeneous Catalysis: Concepts, Strategies, and Applications, Wiley-VCH, Weinheim, Germany, 2014 Search PubMed.
- L. Gonsalvi, Catalysts, 2018, 8, 1–3 CrossRef.
- S. M. Kelly and B. H. Lipshutz, Org. Lett., 2014, 16, 98–101 CrossRef CAS PubMed.
- M. Cortes-Clerget, N. Akporji, J. Zhou, F. Gao, P. Guo, M. Parmentier, F. Gallou, J. Y. Berthon and B. H. Lipshutz, Nat. Commun., 2019, 10, 2169 CrossRef PubMed.
- T. Welton, Curr. Opin. Green Sustain. Chem., 2018, 13, A7–A9 CrossRef.
- M. Pera-Titus, L. Leclercq, J.-M. Clacens, F. De Campo and V. Nardello-Rataj, Angew. Chem., Int. Ed., 2015, 54, 2006–2021 CrossRef CAS PubMed.
- Y. Yang, Z. Fang, X. Chen, W. Zhang, Y. Xie, Y. Chen, Z. Liu and W. Yuan, Front. Pharmacol., 2017, 8, 1–20 Search PubMed.
- H. Jiang, Y. Sheng and T. Ngai, Curr. Opin. Colloid Interface Sci., 2020, 49, 1–15 CrossRef CAS PubMed.
- C. L. G. Harman, M. A. Patel, S. Guldin and G.-L. Davies, Curr. Opin. Colloid Interface Sci., 2019, 39, 173–189 CrossRef CAS.
- A. M. B. Rodriguez and B. P. Binks, Soft Matter, 2020, 16, 10221–10243 RSC.
- Z. Chen, L. Zhou, W. Bing, Z. Zhang, Z. Li, J. Ren and X. Qu, J. Am. Chem. Soc., 2014, 136, 7498–7504 CrossRef CAS PubMed.
- J. Jiang, Y. Zhu, Z. Cui and B. P. Binks, Angew. Chem., Int. Ed., 2013, 52, 12373–12376 CrossRef CAS PubMed.
- Y. Zhu, J. Jiang, K. Liu, Z. Cui and B. P. Binks, Langmuir, 2015, 31, 3301–3307 CrossRef CAS PubMed.
- F. Tu and D. Lee, J. Am. Chem. Soc., 2014, 136, 9999–10006 CrossRef CAS PubMed.
- F. Chen, C. Dong, C. Chen, W. D. Yin, W. Zhai, X. Y. Ma and B. Wei, Ultrason. Sonochem., 2019, 58, 104705 CrossRef CAS PubMed.
- T. Tanaka, M. Okayama, H. Minami and M. Okubo, Langmuir, 2010, 26, 11732–11736 CrossRef CAS PubMed.
- T. Yang, L. Wei, L. Jing, J. Liang, X. Zhang, M. Tang, M. J. Monteiro, Y. I. Chen, Y. Wang, S. Gu, D. Zhao, H. Yang, J. Liu and G. Q. M. Lu, Angew. Chem., Int. Ed., 2017, 56, 8459–8463 CrossRef CAS PubMed.
- F. Liang, K. Shen, X. Qu, C. Zhang, Q. Wang, J. Li, J. Liu and Z. Yang, Angew. Chem., Int. Ed., 2011, 50, 2379–2382 CrossRef CAS PubMed.
- B. P. Binks and S. O. Lumsdon, Langmuir, 2000, 16, 2539–2547 CrossRef CAS.
- P. Venkataraman, B. Sunkara, J. E. St. Dennis, J. He, V. T. John and A. Bose, Langmuir, 2012, 28, 1058–1063 CrossRef CAS PubMed.
- A. D. Dinsmore, M. F. Hsu, M. G. Nikolaides, M. Marquez, A. R. Bausch and D. A. Weitz, Science, 2002, 298, 1006–1009 CrossRef CAS PubMed.
- O. D. Velev, K. Furusawa and K. Nagayama, Langmuir, 1996, 12, 2374–2384 CrossRef CAS.
- B. P. Binks and P. D. I. Fletcher, Langmuir, 2001, 17, 4708–4710 CrossRef CAS.
- B. Madivala, S. Vandebril, J. Fransaer and J. Vermant, Soft Matter, 2009, 5, 1717 RSC.
- B. P. Binks and C. P. Whitby, Colloids Surf., A, 2005, 253, 105–115 CrossRef CAS.
- P. A. Zapata, J. Faria, M. P. Ruiz, R. E. Jentoft and D. E. Resasco, J. Am. Chem. Soc., 2012, 134, 8570–8578 CrossRef CAS PubMed.
- Y. Nonomura, S. Komura and K. Tsujii, J. Phys. Chem. B, 2006, 110, 13124–13129 CrossRef CAS PubMed.
- M. Zanini, C. Marschelke, S. E. Anachkov, E. Marini, A. Synytska and L. Isa, Nat. Commun., 2017, 8, 1–9 CrossRef PubMed.
- A. San-Miguel and S. H. Behrens, Langmuir, 2012, 28, 12038–12043 CrossRef CAS PubMed.
- H. Yang, T. Zhou and W. Zhang, Angew. Chem., Int. Ed., 2013, 52, 7455–7459 CrossRef CAS PubMed.
- X. Wang, M. In, C. Blanc, P. Malgaretti, M. Nobili and A. Stocco, Faraday Discuss., 2016, 191, 305–324 RSC.
- S. Jiang, Q. Chen, M. Tripathy, E. Luijten, K. S. Schweizer and S. Granick, Adv. Mater., 2010, 22, 1060–1071 CrossRef CAS PubMed.
- L. C. Bradley, K. J. Stebe and D. Lee, J. Am. Chem. Soc., 2016, 138, 11437–11440 CrossRef CAS PubMed.
- J. Faria, M. P. Ruiz and D. E. Resasco, Adv. Synth. Catal., 2010, 352, 2359–2364 CrossRef CAS.
- Y. Liu, J. Hu, X. Yu, X. Xu, Y. Gao, H. Li and F. Liang, J. Colloid Interface Sci., 2017, 490, 357–364 CrossRef CAS PubMed.
- I. Kalashnikova, H. Bizot, B. Cathala and I. Capron, Langmuir, 2011, 27, 7471–7479 CrossRef CAS PubMed.
- Z. Hu, S. Ballinger, R. Pelton and E. D. Cranston, J. Colloid Interface Sci., 2015, 439, 139–148 CrossRef CAS PubMed.
- I. Capron and B. Cathala, Biomacromolecules, 2013, 14, 291–296 CrossRef CAS PubMed.
- F. Lou, L. Ye, M. Kong, Q. Yang, G. Li and Y. Huang, RSC Adv., 2016, 6, 24195–24202 RSC.
- K. R. Peddireddy, T. Nicolai, L. Benyahia and I. Capron, ACS Macro Lett., 2016, 5, 283–286 CrossRef CAS.
- S. V. Daware and M. G. Basavaraj, Langmuir, 2015, 31, 6649–6654 CrossRef CAS PubMed.
- Q. B. Meng, P. Yang, T. Feng, X. Ji, Q. Zhang, D. Liu, S. Wu, F. Liang, Z. Zheng and X.-M. Song, J. Colloid Interface Sci., 2017, 507, 74–82 CrossRef CAS PubMed.
- J. Kim, L. J. Cote, F. Kim, W. Yuan, K. R. Shull and J. Huang, J. Am. Chem. Soc., 2010, 132, 8180–8186 CrossRef CAS PubMed.
- J. S. Guevara, A. F. Mejia, M. Shuai, Y.-W. Chang, M. S. Mannan and Z. Cheng, Soft Matter, 2013, 9, 1327–1336 RSC.
- C. Yu, L. Fan, J. Yang, Y. Shan and J. Qiu, Chem. – Eur. J., 2013, 19, 16192–16195 CrossRef CAS PubMed.
- Y. Nonomura and N. Kobayashi, J. Colloid Interface Sci., 2009, 330, 463–466 CrossRef CAS PubMed.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- J. Huang, F. Cheng, B. P. Binks and H. Yang, J. Am. Chem. Soc., 2015, 137, 15015–15025 CrossRef CAS.
- C. Zhao, J. Tan, W. Li, K. Tong, J. Xu and D. Sun, Langmuir, 2013, 29, 14421–14428 CrossRef CAS PubMed.
- B. P. Binks, R. Murakami, S. P. Armes and S. Fujii, Angew. Chem., Int. Ed., 2005, 44, 4795–4798 CrossRef CAS PubMed.
- Z. Li, Y. Shi, A. Zhu, Y. Zhao, H. Wang, B. P. Binks and J. Wang, Angew. Chem., Int. Ed., 2021, 60, 3928–3933 CrossRef CAS PubMed.
- S. Lam, E. Blanco, S. K. Smoukov, K. P. Velikov and O. D. Velev, J. Am. Chem. Soc., 2011, 133, 13856–13859 CrossRef CAS PubMed.
- J. Cho, J. Cho, H. Kim, M. Lim, H. Jo, H. Kim, S.-J. Min, H. Rhee and J. W. Kim, Green Chem., 2018, 20, 2840–2844 RSC.
- Z. Zhao, F. Liang, G. Zhang, X. Ji, Q. Wang, X. Qu, X. Song and Z. Yang, Macromolecules, 2015, 48, 3598–3603 CrossRef CAS.
- B.-J. Yao, Q.-J. Fu, A.-X. Li, X.-M. Zhang, Y.-A. Li and Y.-B. Dong, Green Chem., 2019, 21, 1625–1634 RSC.
- Y. Zhu, T. Fu, K. Liu, Q. Lin, X. Pei, J. Jiang, Z. Cui and B. P. Binks, Langmuir, 2017, 33, 5724–5733 CrossRef CAS PubMed.
- D. Xue, Q. B. Meng and X.-M. Song, ACS Appl. Mater. Interfaces, 2019, 11, 10967–10974 CrossRef CAS PubMed.
- H. Kim, J. Cho, J. Cho, B. J. Park and J. W. Kim, ACS Appl. Mater. Interfaces, 2018, 10, 1408–1414 CrossRef CAS PubMed.
- L. Fu, S. Li, Z. Han, H. Liu and H. Yang, Chem. Commun., 2014, 50, 10045–10048 RSC.
- B. Yang, L. Leclercq, J.-M. Clacens and V. Nardello-Rataj, Green Chem., 2017, 19, 4552–4562 RSC.
- W.-L. Jiang, Q.-J. Fu, B.-J. Yao, L.-G. Ding, C.-X. Liu and Y.-B. Dong, ACS Appl. Mater. Interfaces, 2017, 9, 36438–36446 CrossRef CAS PubMed.
- S. Luo, Z. Zeng, G. Zeng, Z. Liu, R. Xiao, M. Chen, L. Tang, W. Tang, C. Lai, M. Cheng, B. Shao, Q. Liang, H. Wang and D. Jiang, ACS Appl. Mater. Interfaces, 2019, 11, 32579–32598 CrossRef CAS PubMed.
- G. Yang, C. Xing, W. Hirohama, Y. Jin, C. Zeng, Y. Suehiro, T. Wang, Y. Yoneyama and N. Tsubaki, Catal. Today, 2013, 215, 29–35 CrossRef CAS.
- J. Zhang, Z. Yu, Z. Gao, H. Ge, S. Zhao, C. Chen, S. Chen, X. Tong, M. Wang, Z. Zheng and Y. Qin, Angew. Chem., Int. Ed., 2017, 56, 816–820 CrossRef CAS PubMed.
- M. Zhao, K. Deng, L. He, Y. Liu, G. Li, H. Zhao and Z. Tang, J. Am. Chem. Soc., 2014, 136, 1738–1741 CrossRef CAS PubMed.
- Z. Jia, K. Wang, B. Tan and Y. Gu, ACS Catal., 2017, 7, 3693–3702 CrossRef CAS.
- J. Lu, J. Dimroth and M. Weck, J. Am. Chem. Soc., 2015, 137, 12984–12989 CrossRef CAS PubMed.
- V. Sharma, N. Sinha, S. Dutt, M. Chawla and P. F. Siril, J. Colloid Interface Sci., 2016, 463, 180–187 CrossRef CAS PubMed.
- H. Ge, B. Zhang, X. Gu, H. Liang, H. Yang, Z. Gao, J. Wang and Y. Qin, Angew. Chem., Int. Ed., 2016, 55, 7081–7085 CrossRef CAS PubMed.
- F. Chang, B. G. P. van Ravensteijn, K. S. Lacina and W. K. Kegel, ACS Macro Lett., 2019, 8, 714–718 CrossRef.
- J. Yan, K. Chaudhary, S. C. Bae, J. a. Lewis and S. Granick, Nat. Commun., 2013, 4, 1516 CrossRef.
- L. Xia, H. Zhang, Z. Wei, Y. Jiang, L. Zhang, J. Zhao, J. Zhang, L. Dong, E. Li, L. Ruhlmann and Q. Zhang, Chem. – Eur. J., 2017, 23, 1920–1929 CrossRef CAS PubMed.
- W. Cao, R. Huang, W. Qi, R. Su and Z. He, ACS Appl. Mater. Interfaces, 2015, 7, 465–473 CrossRef CAS PubMed.
- X. Zheng, Y. Wang, Y. Wang, D. J. Pine and M. Weck, Chem. Mater., 2016, 28, 3984–3989 CrossRef CAS.
- F. Chang, S. Ouhajji, A. Townsend, K. S. Lacina, B. G. P. van Ravensteijn and W. K. Kegel, J. Colloid Interface Sci., 2021, 582, 333–341 CrossRef CAS PubMed.
- C. M. Vis, Pickering Emulsions as Compartmentalized Reaction Media for Catalysis, PhD thesis, Utrecht University, 2020 Search PubMed.
- S. P. Teong, G. Yi, H. Zeng and Y. Zhang, Green Chem., 2015, 17, 3751–3755 RSC.
- J. N. Chheda, Y. Roman-Leshkov and J. A. Dumesic, Green Chem., 2007, 9, 342–350 RSC.
- V. Linek and P. Beneš, Chem. Eng. Sci., 1976, 31, 1037–1046 CrossRef CAS.
- D. Stehl, N. Milojević, S. Stock, R. Schomäcker and R. Von Klitzing, Ind. Eng. Chem. Res., 2019, 58, 2524–2536 CrossRef CAS.
- L. Tao, M. Zhong, J. Chen, S. Jayakumar, L. Liu, H. Li and Q. Yang, Green Chem., 2018, 20, 188–196 RSC.
- C. M. Vis, L. C. J. Smulders and P. C. A. Bruijnincx, ChemSusChem, 2019, 12, 2176–2180 CrossRef CAS PubMed.
- W. Zhang, L. Fu and H. Yang, ChemSusChem, 2014, 7, 391–396 CrossRef CAS PubMed.
- J. Shi, X. Wang, S. Zhang, L. Tang and Z. Jiang, J. Mater. Chem. B, 2016, 4, 2654–2661 RSC.
- L. Qi, Z. Luo and X. Lu, ACS Sustainable Chem. Eng., 2019, 7, 7127–7139 CrossRef CAS.
- L. Liu, L. Jiang, X. Xie and S. Yang, ChemPlusChem, 2016, 81, 629–636 CrossRef CAS PubMed.
- Y. Yang, W. J. Zhou, A. Liebens, J. M. Clacens, M. Pera-Titus and P. Wu, J. Phys. Chem. C, 2015, 119, 25377–25384 CrossRef CAS.
- G. Lv, F. Wang, X. Zhang and B. P. Binks, Langmuir, 2018, 34, 302–310 CrossRef CAS PubMed.
- H. Shi, Z. Fan, V. Ponsinet, R. Sellier, H. Liu, M. Pera-Titus and J. M. Clacens, ChemCatChem, 2015, 7, 3229–3233 CrossRef CAS.
- B. P. Binks and J. A. Rodrigues, Langmuir, 2007, 23, 3626–3636 CrossRef CAS PubMed.
- J. Huang and H. Yang, Chem. Commun., 2015, 51, 7333–7336 RSC.
- J. Huang, F. Cheng, B. P. Binks and H. Yang, J. Am. Chem. Soc., 2015, 137, 15015–15025 CrossRef CAS PubMed.
- Y. Zhang, M. Zhang and H. Yang, ChemCatChem, 2018, 10, 5224–5230 CrossRef CAS.
- In-Situ Spectroscopy of Catalysts, ed. B. M. Weckhuysen, American Scientific Publishers, 2004 Search PubMed.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC.
- H. Liu, Z. Zhang, H. Yang, F. Cheng and Z. Du, ChemSusChem, 2014, 7, 1888–1900 CrossRef CAS PubMed.
- H. Yang, L. Fu, L. Wei, J. Liang and B. P. Binks, J. Am. Chem. Soc., 2015, 137, 1362–1371 CrossRef CAS PubMed.
- M. Zhang, L. Wei, H. Chen, Z. Du, B. P. Binks and H. Yang, J. Am. Chem. Soc., 2016, 138, 10173–10183 CrossRef CAS PubMed.
- H. Chen, H. Zou, Y. Hao and H. Yang, ChemSusChem, 2017, 10, 1989–1995 CrossRef CAS PubMed.
- F. Peng, J. Xu, X. Zeng, G. Feng and H. Bao, Part. Part. Syst. Charact., 2020, 37, 1900382 CrossRef CAS.
- M. Zhang, R. Ettelaie, T. Yan, S. Zhang, F. Cheng, B. P. Binks and H. Yang, J. Am. Chem. Soc., 2017, 139, 17387–17396 CrossRef CAS PubMed.
- Z. Meng, M. Zhang and H. Yang, Green Chem., 2019, 21, 627–633 RSC.
- X. Tang, Y. Hou, Q. B. Meng, G. Zhang, F. Liang and X.-M. Song, Colloids Surf., A, 2019, 570, 191–198 CrossRef CAS.
- X. Zhang, Y. Hou, R. Ettelaie, R. Guan, M. Zhang, Y. Zhang and H. Yang, J. Am. Chem. Soc., 2019, 141, 5220–5230 CrossRef CAS PubMed.
- T. Skale, D. Stehl, L. Hohl, M. Kraume, R. von Klitzing and A. Drews, Chem. Ing. Tech, John Wiley & Sons, Ltd., 2016, vol. 88, pp. 1827–1832 Search PubMed.
- A. Heyse, C. Plikat, M. Grün, S. Delaval, M. Ansorge-Schumacher and A. Drews, Process Biochem., 2018, 72, 86–95 CrossRef CAS.
- A. Heyse, C. Plikat, M. Ansorge-Schumacher and A. Drews, Catal. Today, 2019, 331, 60–67 CrossRef CAS.
- C. M. Vis, A.-E. Nieuwelink, B. M. Weckhuysen and P. C. A. Bruijnincx, Chem. – Eur. J., 2020, 26, 15099–15102 CrossRef CAS PubMed.
- S. Yu, D. Zhang, J. Jiang, Z. Cui, W. Xia, B. P. Binks and H. Yang, Green Chem., 2019, 21, 4062–4068 RSC.
- K. Kanomata, N. Fukuda, T. Miyata, P. Y. Lam, T. Takano, Y. Tobimatsu and T. Kitaoka, ACS Sustainable Chem. Eng., 2020, 8, 1185–1194 CrossRef CAS.
- L. Ni, C. Yu, Q. Wei, J. Chang and J. Qiu, Green Chem., 2020, 22, 5711–5721 RSC.
- P. T. Anastas and J. B. Zimmerman, Curr. Opin. Green Sustain. Chem., 2018, 13, 150–153 CrossRef.
- S. Yu, H. Zhang, J. Jiang, Z. Cui, W. Xia and B. P. Binks, Green Chem., 2020, 22, 5470–5475 RSC.
- F. Byrne, B. Forier, G. Bossaert, C. Hoebers, T. J. Farmer, J. H. Clark and A. J. Hunt, Green Chem., 2017, 19, 3671–3678 RSC.
- M. L. Harrell, T. Malinski, C. Torres-López, K. Gonzalez, J. Suriboot and D. E. Bergbreiter, J. Am. Chem. Soc., 2016, 138, 14650–14657 CrossRef CAS PubMed.
- Z. Lei, B. Chen, Y.-M. Koo and D. R. MacFarlane, Chem. Rev., 2017, 117, 6633–6635 CrossRef PubMed.
- M. Hejazifar, O. Lanaridi and K. Bica-Schröder, J. Mol. Liq., 2020, 303, 112264 CrossRef CAS.
- L. Qi, Z. Luo and X. Lu, Green Chem., 2019, 21, 2412–2427 RSC.
- S. Berger, A. Synytska, L. Ionov, K. J. Eichhorn and M. Stamm, Macromolecules, 2008, 41, 9669–9676 CrossRef CAS.
- A. Kirillova, C. Marschelke and A. Synytska, ACS Appl. Mater. Interfaces, 2019, 11, 9643–9671 CrossRef CAS.
- A. Tkatchenko, Nat. Commun., 2020, 11, 4125 CrossRef CAS.
- R. Hardian, Z. Liang, X. Zhang and G. Szekely, Green Chem., 2020, 22, 7521–7528 RSC.
- R. L. Hartman, Curr. Opin. Chem. Eng., 2020, 29, 42–50 CrossRef.
- S. V. Ley, Y. Chen, D. E. Fitzpatrick and O. S. May, Curr. Opin. Green Sustain. Chem., 2020, 25, 100353 CrossRef.
- C. Sambiagio and T. Noël, Trends Chem., 2020, 2, 92–106 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |