
Electron attachment to isolated and microhydrated favipiravir†
Barbora
Sedmidubská
 ab,
Thomas F. M.
Luxford
a and
Jaroslav
Kočišek
*a
ab,
Thomas F. M.
Luxford
a and
Jaroslav
Kočišek
*a
aJ. Heyrovský Institute of Physical Chemistry of the Czech Academy of Sciences, Dolejškova 3, 18223 Prague, Czech Republic. E-mail: jaroslav.kocisek@jh-inst.cas.cz
bDepartment of Nuclear Chemistry, Faculty of Nuclear Sciences and Physical Engineering, Břehová 7, 11519 Prague, Czech Republic
First published on 6th August 2021
Abstract
Electron attachment and its equivalent in complex environments, single-electron reduction, are important in many biological processes. Here, we experimentally study the electron attachment to favipiravir, a well-known antiviral agent. Electron attachment spectroscopy is used to explore the energetics of associative (AEA) and dissociative (DEA) electron attachment to isolated favipiravir. AEA dominates the interaction and the yields of the fragment anions after DEA are an order of magnitude lower than that of the parent anion. DEA primary proceeds via decomposition of the CONH2 functional group, which is supported by reaction threshold calculations using ab initio methods. Mass spectrometry of small favipiravir–water clusters demonstrates that a lot of energy is transferred to the solvent upon electron attachment. The energy gained upon electron attachment, and the high stability of the parent anion were previously suggested as important properties for the action of several electron-affinic radiosensitizers. If any of these mechanisms cause synergism in chemo-radiation therapy, favipiravir could be repurposed as a radiosensitizer.
 Barbora Sedmidubská | Barbora Sedmidubská received her undergraduate and MSc (2020) degree in the field of nuclear chemistry and now she is continuing PhD studies at the Faculty of Nuclear Sciences and Physical Engineering, Czech Technical University in Prague. Her main research interest is in the molecular understanding of radiation chemistry in living tissues. Her PhD project is focused on noncovalent interactions of radiosensitizers and Fullerenes. |
 Thomas F. M. Luxford | Thomas F. M. Luxford received his undergraduate and MSc degrees from the University of York (UK), and his PhD from Herriot-Watt University in Edinburgh (UK, 2017). He is currently a research assistant at the J Heyrovský Institute of Physical Chemistry in Prague (Czech Republic), as part of the Department of Dynamics of Molecules and Clusters. His research is primarily focused on better understanding the fundamental interactions of species in the gas phase, and he is currently studying the electron-induced reactivity of molecules. |
 Jaroslav Kočišek | Jaroslav Kočišek received PhDs in plasma physics from Comenius University in Bratislava (2010) and in photo-dissociation dynamics from Charles University in Prague (2013). After postdocs studying electron scattering at the University of Fribourg and collisions of multiply charged ions at GANIL, he started a junior research group at J. Heyrovský Institute of Physical Chemistry in 2016. Since 2019, he is holding a recognized J. Heyrovský Young Scientist position. His research is focused on environmental effects on reaction dynamics and elementary processes in radiation interaction with living tissue, studied experimentally on model systems ranging from isolated molecules and clusters to self-assembled nanostructures. |
1 Introduction
The present study is motivated by the fact that many biological processes based on reduction can be related to electron attachment (EA). This relation was explored by the Modelli and Pshenichnyuk groups for a wide range of biological processes, from metabolic pathways to the functionality of the olfactory system.1–7 The electron reduction properties can also influence the transport of drugs through biological membranes and influence their target binding properties.8,9 Recently, the RNA inhibitor efficiency of favipiravir tautomers was related to the energy difference between its highest occupied (HOMO) and lowest unoccupied (LUMO) molecular orbitals.10 Unoccupied orbitals can become singly occupied upon electron attachment and appear as so-called shape resonances in the electron scattering spectrum. A present study of the electron attachment to favipiravir may therefore provide an important key to better understand its activity.Another important motivation is based on the relation of low-energy electrons to the synergism observed in the concomitant chemo-radiation therapy of cancer.11,12 Two facts motivated a range of studies on the relation of low-energy electrons to the synergistic action of radiation with a range of chemotherapeutic and radio-sensitizing drugs.13,14 The first fact is a large amount of available secondary low-energy electrons in the irradiated tissue.15,16 The second fact is that despite the different possible modes of action,17 most of the known small molecule radiosensitizers and their important functional groups have high electron affinities.18 Processes such as the formation of reactive anions and radical species via dissociative electron attachment,19–22 enhanced linear energy transfer,23 transport properties8 due to associative electron attachment, or DNA sensitization24–30 have been proposed as possible sources of synergism observed in concomitant chemo-radiation therapy. Several of the processes were then used to suggest novel radiosensitizers.31–34 However, in many cases, the suggested molecules with “ideal” electron-attachment properties fail to exhibit the synergism35 or are biologically incompatible.36 Since drug repurposing37 is becoming still more important and antivirotics are often chosen as cancer chemo or even chemo-radio therapeutics,38–43 we explore favipiravir as a promising candidate for an electron affinic radiosensitizer.
Favipiravir (Fig. 1) has already been established as a broad-range antivirotic44–47 with good biocompatibility48 and known pharmacokinetics.49,50 Here, we explore its behavior in the reactions with low-energy electrons using electron attachment spectroscopy of the isolated and microhydrated molecule and perform basic computational modeling to support our experimental data.
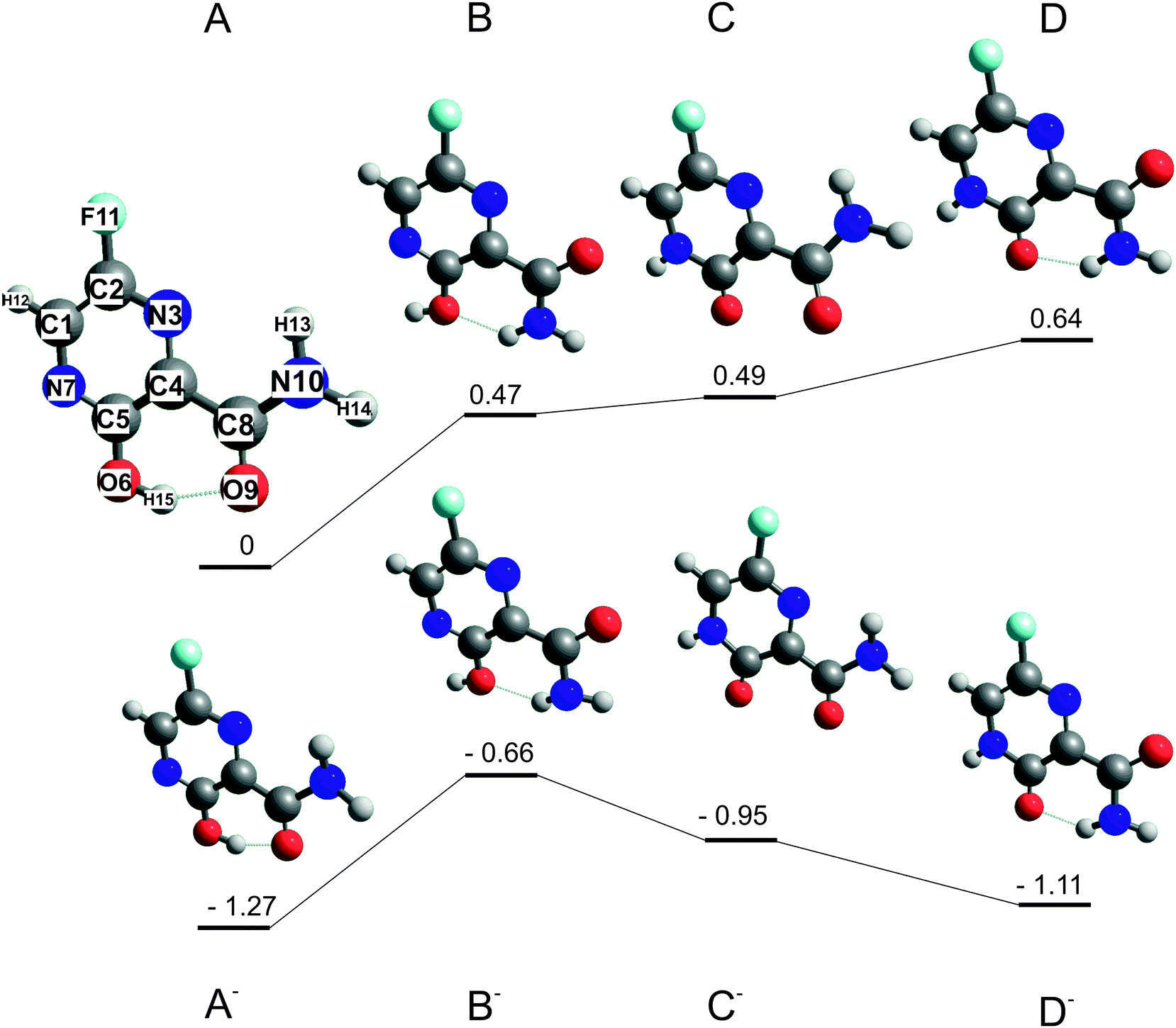 | ||
| Fig. 1 Optimized structures of stable conformers A, B and keto tautomers B, C of favipiravir and their respective anions. Energies in eV relative to the most stable neutral conformer A calculated at the B3LYP/aug-cc-pVTZ level of theory. | ||
2 Methods
2.1 Experimental
Favipiravir was purchased from Santiago Labs with stated purity of 99% and its electron-induced chemistry was explored on two experimental setups. The isolated molecule was studied using the TEM-QMS setup51 and hydration effects were studied using the CLUster Beam (CLUB) apparatus in the M. Fárník group.52The TEM-QMS apparatus is an electron attachment spectrometer combining a trochoidal electron monochromator (TEM) with a quadrupole mass spectrometer (QMS) analyser originally built in the M. Allan group.53 With the TEM,54 electrons thermo-emitted from an iridium-yttrium cathode, are selected according to their kinetic energy, narrowing the initial electron energy distribution function. In the present experiment, the electron-energy resolution was around 150 meV as estimated from the FWHM of the 0 eV peak in the anion yield for AEA to sulphur hexafluoride.55 The electrons collide with sample molecules in the reaction chamber and the formed ions are extracted towards the QMS. Two modes of operation are possible, with ion yield measurement as a function of incident electron energy at a constant m/z of QMS or ion yield measurement as a function of m/z at constant incident electron energy. Favipiravir was introduced using the direct insertion probe.56 Sample powder was loaded into a glass bulb and placed at the end of the probe, which was inserted into a resistively heated copper cylinder with the constant temperature kept at 340 K. The sublimed molecules effused into the reaction zone through a 1 cm long capillary. For calibration of the energy axis, we used SF6 and CO2 gases, which were introduced through the same inlet. The 4.3 eV resonance of O− from CO257 was used to calibrate the electron-energy axis and 0 eV resonance of SF6− from SF6 to determine the energy threshold for electron transmission through the TEM. In the present case, the threshold is around 0.1 eV and constant electron current is reached around 0.25 eV as demonstrated on SF6− signal in Fig. 2. Anion yields lower than this value presented in this work, particularly the yield of parent anion, are therefore underestimated.
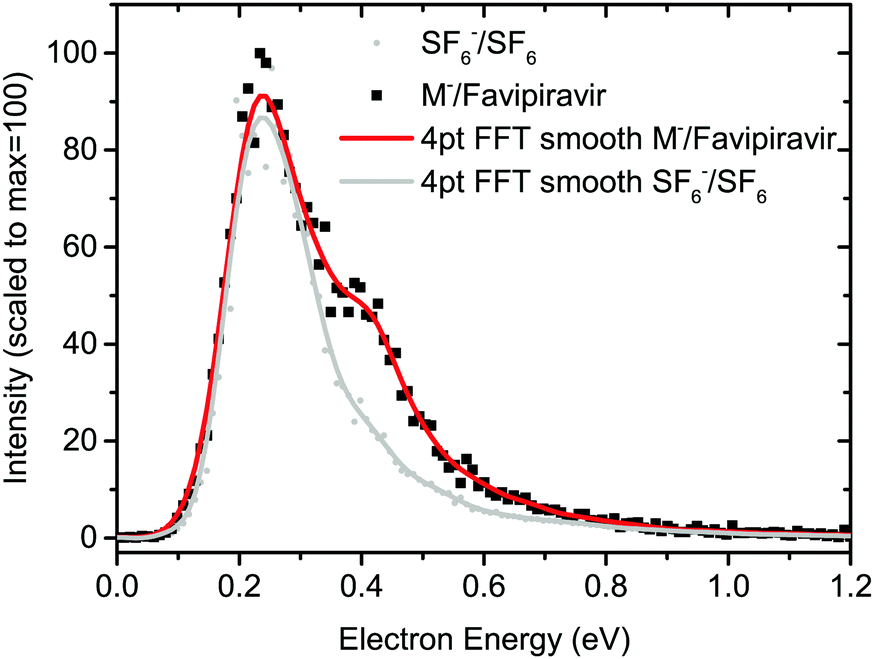 | ||
| Fig. 2 Electron energy-dependent ion yields for AEA to favipiravir and sulfur hexafluoride. The drop of the anion yield below ∼0.25 eV is caused by decrease of the incident electron current below this value. Anion yields at energies lower than ∼0.25 eV presented in this work are, therefore, underestimated. | ||
On the CLUB setup, we only used the neutral cluster source and reflectron time-of-flight mass spectrometer. Experimental details for the negative and positive mass spectra measurements can be found in ref. 58 and 59, respectively. The molecular target was prepared by co-expansion of He buffer gas and favipiravir vapors through a conical 90 μm nozzle into the vacuum. Clusters of microhydrated molecules were prepared by an approach developed in our laboratory based on the addition of a small amount of water into the buffer gas through the Nafion membrane.60 During all experiments, the sample was sublimed at a temperature of approximately 85 °C. To test the thermal stability of the molecule, we heated the sample up to 118 °C. Even at this elevated temperature, we did not observe any new product ions due to thermal decomposition in the ionization MS (see ESI†).
2.2 Ab initio calculations
Two favipiravir conformers, differing in the orientation of the CONH2 functional group, its keto form, and their respective anions, were pre-optimized at the B3LYP/6-31(d) level of theory and then optimized at the B3LYP/aug-cc-pVTZ level of theory. The structures of neutrals and respective energies of neutrals and anions optimized at the B3LYP/aug-cc-pVTZ level of theory are in Fig. 1 The order of the conformers was checked by energy calculation using G3MP261 method and method error by re-optimizing the B3LYP62 structures using M062x63 functional and the same aug-cc-PVTZ basis set. Adiabatic electron affinities were calculated as a difference of the neutral and anion energies in their optimized geometry and they are listed together with other parameters in Table 1.| Conformer | Energy B3LYP | Energy G3MP2 | Energy M062x | Dipole moment B3LYP |
|---|---|---|---|---|
| A | 0 | 0 | 0 | 3.2 |
| B | 0.47 | 0.44 | 0.79 | 5.7 |
| C | 0.49 | 0.57 | 0.34 | 6 |
| D | 0.64 | 0.72 | 1.09 | 5.3 |
| A− | −1.27 | −1.14 | −0.68 | 4.8 |
| B− | −0.66 | −0.54 | −0.3 | 7.8 |
| C− | −0.95 | −0.91 | −0.9 | 9.2 |
| D− | −1.11 | −0.73 | −0.5 | 9.1 |
Then, energetic thresholds for individual reaction channels of DEA to favipiravir were calculated at the B3LYP/aug-cc-pVTZ theory to support the experimental results. Threshold energies were obtained using the formula:
| Eth = EMa− + EMb − EM | (1) |
Finally, virtual orbital energies of favipiravir were calculated at the HF/6-31G* level of theory, using the structures optimised at the B3LYP/aug-cc-PVTZ level of theory. These energies were then scaled to obtain vertical attachment energies according to the empirical formula of Aflatooni et al.64:
| VAE = [εVO − 2.5553]/1.3749 | (2) |
All calculations were performed in Gaussian 1665 and the results were analyzed and visualized using Chemcraft.66
3 Results and discussion
3.1 Isolated molecule
Structures of the most stable isomers and keto tautomers of favipiravir are shown in Fig. 1. Conformer A is the most stable conformer of neutral favipiravir, independent of the computational method (Table 1), due to the aromatic nature of the ring and being stabilized by O(9)–H(15) hydrogen bond (atom numbers are shown on the structure of conformer A of Fig. 1). The energy of the conformer is (depending on the method) ∼0.5 eV below the other studied structures and therefore will dominate the gas phase distribution of the sublimed favipiravir. The recent work of Antonov67 also demonstrated the stability of this conformer in various solvents. We will therefore relate all energies in the following discussion to this most stable conformer A. The energy difference due to different conformers of the neutral precursor molecule may be easily estimated from Table 1.The calculated adiabatic electron affinity of the molecule is 1.3 eV (Fig. 1 and Table 1). Therefore, favipiravir can form a stable valence-bound anion, which is also close in geometry to the neutral molecule. However, in its anionic state, the energy gap between the enol A conformer and keto D conformer is much lower than in the case of the neutral molecule and therefore both forms may be present. This may influence the dissociation dynamics, as will be discussed later.
An estimated value of the vertical electron affinity at the B3LYP/aug-cc-pVTZ level of theory is 0.3 eV. The value may vary due to the limitations of the DFT approach,68 however, its positive character is clear also from our estimation of the negative value of the LUMO vertical attachment energy (Fig. 3).
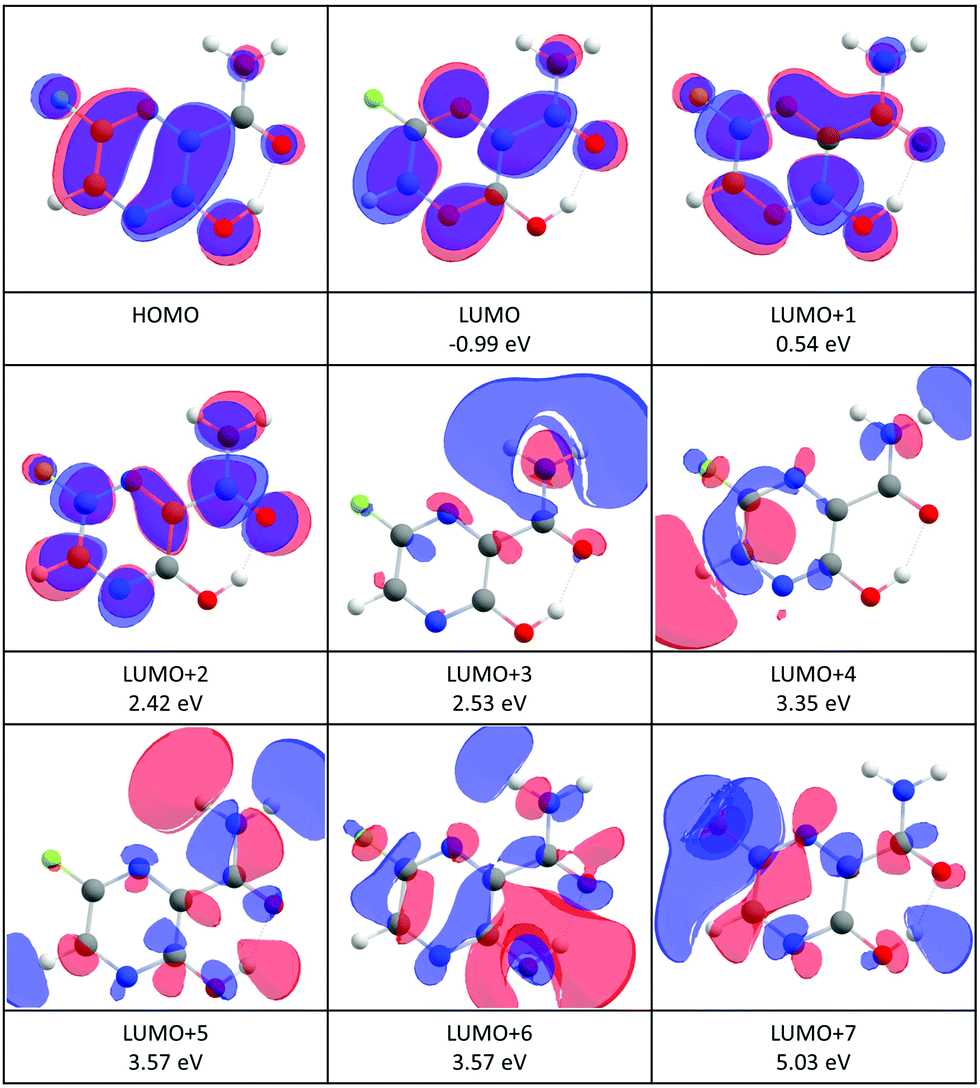 | ||
| Fig. 3 Contour plots of the highest occupied (HOMO) and low-lying virtual orbitals of favipiravir. The orbitals were calculated at the HF/6-31G(d) level of theory. The numbers in eV are vertical attachment energies according to the formula (2). | ||
Favipiravir has a large dipole moment. The most stable conformer A has the lowest dipole moment (3.3 D) of all explored conformers, above the commonly set limits for the formation of dipole supported states of the anion (ref. 69 and references cited therein).
The incident electron energy dependent ion yields for the most stable anionic products of electron attachment to isolated favipiravir are shown in Fig. 4 and relative intensities of the bands, obtained as the area of Gaussian peaks fitted to the data, are listed in Table 2. The yields peak at energies of around 0, 1, 2, 3, 4, 5, 7, and 9 eV. The 0 eV peak is observed only for the parent anion. The 2, 3, and 5 eV peaks can be assigned to shape resonances according to estimated vertical attachment energies of LUMO+2, 4, and 7 (see Fig. 3). The yields peaking at 1 or 4 eV can also be due to the attachment to virtual orbitals, however, their low energy onset may be shifted due to the reaction endothermicity, which will be discussed later. The high energy peaks in the spectrum can be formed by core excited Feshbach resonances or core excited shape resonances. Unfortunately, the photo-electron spectrum of favipiravir, which would allow us to better identify the Feshbach resonances70–72 has not yet been measured.
 | ||
| Fig. 4 Electron-energy dependent ion yields for the main products of EA to favipiravir. The contributions of ion signal from neighboring m/z due to low selectivity of QMS are marked by *. | ||
| m/z | Anion | Neutral | M + e− → products calculated threshold | Experimental peak energy (relative intensity) | ||
|---|---|---|---|---|---|---|
| 157 | C5H4FN3O2− | ∼0(100); | 2.2(0.06) | |||
| 156 | C5H3FN3O2− | 1.8(0.2); | 3.2(0.12) | |||
| [M–H]− A | H(15) | 0.64 | ||||
| [M–H]− B | H(13) | 1.03 | ||||
| [M–H]− C | H(15) | 1.03 | ||||
| [M–H]− D | H(12) | 2.05 | ||||
| [M–H]− E | H(13) | 2.22 | ||||
| [M–H]− F | H(12) | 2.50 | ||||
| 140 | C5HFN2O2− | 1.1(1.4); | 1.9(0.26) | |||
| [M–NH3]− | NH3 | 0.17 | ||||
| [M–OH]− A | OH | 2.96 | ||||
| [M–OH]− B | OH | 3.17 | ||||
| [M–OH]− C | OH | 4.92 | ||||
| [M–OH]− D | OH | 5.03 | ||||
| 113 | C4H2FN2O− | 5.0(6.0); | 6.8(1.1) | 8.5(0.67) | ||
| [M–CONH2]− A | CO + NH2 | 3.24 | ||||
| [M–CONH2]− B | CO + NH2 | 3.46 | ||||
| [M–CONH2]− C | CO + NH2 | 3.62 | ||||
| [M–CONH2]− A | CONH2 | 4.39 | ||||
| [M–CONH2]− B | CONH2 | 4.61 | ||||
| [M–CONH2]− C | CONH2 | 4.77 | ||||
| 112 | C4HFN2O− | 2.3(1.8); | 4.3(3.3) | |||
| [M–CONH3]− A | CONH3 | 0.86 | ||||
| [M–CONH3]− A | CO + NH3 | 1.31 | ||||
| [M–CONH3]− B | CONH3 | 1.96 | ||||
| [M–CONH3]− B | CO + NH3 | 2.41 | ||||
| [M–CONH3]− C | CONH3 | 2.35 | ||||
| [M–CONH3]− C | CO + NH3 | 2.8 | ||||
| [M–CONH3]− D | CONH3 | 2.4 | ||||
| [M–CONH3]− D | CO + NH3 | 2.85 | ||||
| 42 | OCN− | — | — | 5.0(1.4); | 6.6(0.4); | |
| OCN− | C4H4FN2O | 1.93 | ||||
| OCN− | H2 + C4H2FN2O A | 4.1 | ||||
| OCN− | H2 + C4H2FN2O B | 4.87 | ||||
| OCN− | H2 + C4H2FN2O C | 4.95 | ||||
| 26 | CN− | — | — | 5.0(0.17); | 8.7(0.55) | |
| CN− | H2O + C4H2FN2O A | 1.85 | ||||
| CN− | C4H4FN2O2 | 1.97 | ||||
| CN− | HF + C4H3N2O2 | 2.61 | ||||
| CN− | H2O + C4H2FN2O B | 2.62 | ||||
| CN− | H2O + C4H2FN2O C | 2.7 | ||||
| 19 | F− | — | — | 5.0(0.02); | 9.4(0.1) | |
| F− | (M–F) A | 1.72 | ||||
| F− | (M–F) B | 2.15 | ||||
| F− | (M–F) C | 2.15 | ||||
| F− | (M–F) D | 2.34 | ||||
Positions of the Feshbach resonances can be, however, estimated on the basis of known resonances for components of the molecule,56,73 such as pyrazine or small amides. From the known electron energy loss spectrum of pyrazine,74 which matches the central favipiravir ring structure, we expect that Feshbach resonances in favipiravir can occur as low as at 4 eV. Further excited states of pyrazine are available at 5 eV, 6.5 eV, and 7.5 eV. For the amide functional group, excited states may be expected at higher energies of 7 eV and 9 eV, based on electron energy loss and DEA data of small amides.75,76 However, for amides also an alternative process of core excited shape resonances was suggested,77 but has yet to be confirmed.78
The product anion yield is dominated by the parent favipiravir anion M−. Several mechanisms for the formation of the anion at low energies are possible. As we discussed in the previous paragraphs, the attachment is exothermic and therefore attachment at 0 eV can be expected. The high dipole moment and similar geometries of the neutral and anion allow for dipole-supported vibrational Feshbach resonances, which are known DEA to various molecules.79–81 The electron-energy dependent ion yield curve of the M− anion is shown in detail in Fig. 2. First, we can not see the onset at expected 0 eV but at ∼0.1 eV due to the electron transmission function of the monochromator, which can be well demonstrated on the comparison with SF6− ion yield with the known 0 eV resonance and similar onset. Second, there is a small bump at energies ∼0.4 eV, which may be an indication of vibrational structure. However, the vibrational mode can not be unambiguously assigned because the spectrum is shifted by the electron transmission function of the monochromator. In bare pyrazine (the central cyclic structure of favipiravir), the structures in electron scattering were assigned to breathing modes of the ring.82 In the present case, the energy is high, if we exclude a possibility that more structures occur in the part of the ion yield to which we are blind, the modes involved will be rather O–H or C–H stretches. However, the bump in the spectrum may also be simply an experimental artefact or indication of attachment via the low-lying π shape resonance. Vertical attachment to LUMO+1 may occur for electrons with energies ∼0.5 eV (Fig. 3) according to the scaling of Aflatooni et al.64
A clear structure around 2 eV in the spectrum of M−, in the panel a) of Fig. 4 can be also assigned to a shape resonance of LUMO+2.
We can see that for several bands in the spectrum of favipiravir, there are multiple possible explanations of the undergoing attachment mechanism. Their unambiguous identification requires further experiments. We will not speculate here and will instead focus on the exit channel of the DEA, which was directly studied in the present experiment.
The main DEA channels of favipiravir are listed in Table 2, together with their calculated threshold energies and information about the relative intensities of the main peaks in the experimentally measured ion yields (Fig. 4). As already mentioned, AEA dominates the interaction. In Table 2, where the intensity of the near 0 eV peak of M− is set to 100, the intensity of the second most intense peak in the yield of EA products to favipiravir corresponds to the loss of the CONH2 functional group at energies of incident electrons ∼5 eV, which has a relative intensity of 6. It is important to mention that the relative intensity ratios in the present experiment may be influenced by the ion optics and quadrupole mass filter transmission settings, which are mass-dependent. To minimize these effects we tuned the ion optics for maximum transmission at m/z = 79, Br− anion, which is present as a background in the TEM-QMS setup and is approximately in the center of the m/z fragment distribution. However, the relative intensities may still vary by up to ∼20%.
[M–CONH2]− can be formed by the simple breaking of the C(4)–C(8) bond. The electron energy-dependent spectrum for its formation is shown on panel d) of Fig. 4. The calculated threshold energy for this process is 4 eV (Table 2). This means that there is sufficient energy in the system for this process to occur at incident electron energies above 4 eV, while it is energetically inaccessible at incident electron energies below 4 eV. The onset of the most intense peak in the spectrum is around 3.5 eV, below the calculated reaction threshold for simple C(4)–C(8) bond cleavage, and is therefore energetically inaccessible. Only when we consider further dissociation of the CONH2 fragment into more stable CO and NH2 molecules, the calculated threshold energy shifts below the experimentally observed threshold. Additionally, there is a peak with a maximum at around 2 eV, which is an experimental artefact, caused by the low selectivity of the QMS. The low energy peak is a contribution from neighboring intense fragment ion, [M–CONH3]−, as can be read from its spectra discussed in the next paragraph.
The second most intense fragment ion is [M–CONH3]−, whose spectrum is shown in Fig. 4, panel e). In certain configurations of the anion, the H(15) hydrogen of the OH group can be easily transferred to the amino group. Then, the formation of CO and NH3 neutral fragments together with the C4HFNO− anion is possible at energies as low as 0.86 eV, explaining the first peak in the ion yield. The second peak in the spectrum can be due to anions formed after H(12) removal, which requires more than 2 eV.
The spectrum of the third most intense fragment, OCN− is shown in panel f) of Fig. 4. Due to its high electron affinity (EA), OCN− is a common ion formed by DEA to many biologically relevant molecules in the gas phase,83–90 as well as clusters58 and molecules deposited on surfaces.91,92 The high electron affinity of OCN (∼3.6 eV)93 allows for very complex rearrangement and fragmentation reactions to be induced by the attachment of low-energy electrons.83,86 In the present case, the most straightforward pathway for OCN− extraction is the cleavage of C(4)–C(8) bond followed by the elimination of H2 from the resulting CONH2− anion. As we have seen already for the [M–CONH2]− channel the C(4)–C(8) bond is strong and therefore the reaction energies for such mechanism are above 4 eV. Within experimental and computational errors, the mechanisms can still explain all the peaks observed in the OCN− spectrum. However, we also found a more energetically favorable channel, which can be opened by hydrogen transfer to C(4) carbon allowing for N(7)C(5)O(6) anion extraction at energies below 2 eV together with a C4H4FN2O neutral co-fragment.
Another intense fragmentation channel results in an ion corresponding to the neutral loss of 17 Da from the parent ion (Fig. 4, panel c)), which could correspond to either OH or NH3 loss. The calculated reaction thresholds allow us to exclude the OH loss channel, as it occurs only at energies above 2.9 eV, higher than both observed peaks in the spectrum of this anion. On the other hand, the elimination of NH3 has a threshold of only 0.17 eV, which is well below the onset of the observed signal, meaning that it is accessible.
The [M–H]− ion yield is dominated by a near 0 eV peak, which is due to the low selectivity of QMS and overlap with the neighboring signal of the M− anion, which can not be fully separated in the present experiment. Energetically, this channel is only accessible above 0.64 eV. H(15) or H(13) hydrogen loss may occur at energies of the first real peak in the spectrum with a maximum at ∼1.8 eV and cleavage of H(12) is possible at the higher energies of the second peak with a maximum at ∼3.2 eV.
Another common biomolecular fragment ion with high electron affinity, CN− can be formed from the decomposition of the CONH2 functional group or the ring of favipiravir. The energetic thresholds for both mechanisms are similar, at 1.85 eV and 1.97 eV respectively. In the first mechanism, a stable water molecule is formed as a neutral co-product. In the second mechanism, the C(1)–N(7) atoms are easiest to extract after hydrogen migration from C(1) to C(2). We also found several other possible reaction pathways that are possible for the observed CN− formation, with an onset around 4 eV (Fig. 4, panel g). In some cases, the reaction energy is lowered by closing the ring after the extraction of CN− anion forming the C3N ring. Such rearrangement, however, may proceed over a reaction barrier, which can not be identified using the simple calculations present here.
Finally, with a relative intensity of only ∼0.1% that of the parent ion signal, we observe F− (Fig. 4, panel h)). The signal levels for this ion are near the detection limits of the setup, there appear to be three peaks in the spectrum, at ∼1 eV, ∼5 eV, and ∼9.5 eV. The calculated threshold energy for simple cleavage of the C(2)–F(11) bond is 2.15 eV, which explains the two higher energy resonances, but not the low energy resonance. This unassigned low-energy signal is possibly an experimental artefact, and is most likely caused by background ions in the experiment.
3.2 Hydration
To explore environmental effects, we measured EA to favipiravir in clusters with attached water molecules, prepared in a molecular beam using the CLUB experimental setup. Negative ion spectra of the molecule under dry and hydrated conditions taken in the energy range 0.6 to 8.6 eV is shown in Fig. 5. We can see that for the dry conditions, the intensity of the DEA channels is already reduced with respect to parent anion intensity. Using the CLUB apparatus, we measured reasonable signal only for the m/z = 112, 113 fragment anions. Let's compare the relative intensities of these anions to the parent anion in the two experiments. Dividing the integral intensity of the fragment m/z = 112 + m/z = 113 ion signal by that of m/z = 157 parent ion signal from the energy dependent ion yields in Fig. 4, we obtain the ratio of fragment to parent ion signal in the QMS-TEM setup of ∼1/10. Dividing the integral intensity of the the fragment m/z = 112 + m/z = 113 peak to that of m/z = 157 parent peak in the cumulative MS in Fig. 5, we obtain the ratio of fragment to parent ion signal in CLUB experiment at dry conditions to be 1/33, which is much lower. Additionally, the ratio 1/33 is only the highest estimate, since the electron current in CLUB experiment significantly drops in the near 0 eV region of the parent ion resonance, resulting in an apparent reduction of the parent ion signal.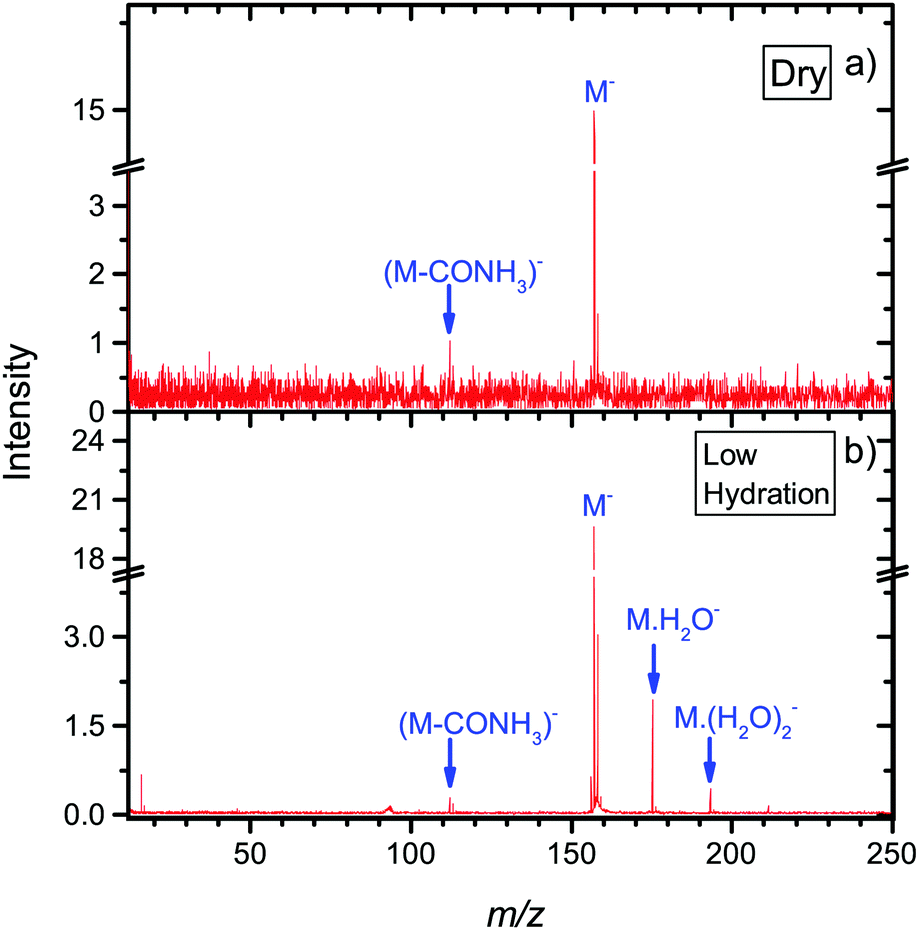 | ||
| Fig. 5 Mass spectra from the CLUB experiment of anions formed after electron attachment to isolated or microhydrated neutral favipiravir in molecular beams. Negative ion MS are prepared by summing spectra taken with 0.2 eV step in the 0.6 eV to 8.6 eV range. | ||
The main reasons for suppression of fragmentation in the CLUB experiment under dry conditions is the detection time of only tens of microseconds, in comparison to the TEM-QMS experiment, where it is hundreds of microseconds. The much shorter detection timescale of the CLUB setup means that there is less time for fragmentation or autodetachment to occur. These effects of the detection time scale were studied in detail by Asfandiarov and co-workers.94,95 Another parameter influencing the fragmentation is the temperature of the precursor molecules, which is lower in the molecular beam of the CLUB than in the effusive beam of the TEM-QMS.96
Further stabilization of parent anion with respect to both DEA and autodetachment can be induced by hydration. Under hydrated conditions the ratio of m/z =112,113 to parent anion signal further decreases to ∼1/90.
Under hydrated conditions, anions are stabilized by energy transfer to the solvent. The total energy available for transfer to surrounding water molecules and their subsequent evaporation (TET) can be written as a sum of adiabatic electron affinity (AEA) and energy of the incoming electron (Ee):
| TET = Ee + AEA | (3) |
For halouracils, energy of several eV was estimated to be transferred to the solvent after electron attachment.23 We also postulated that such energy transfer to the solvent can increase linear energy transfer (LET) value after interaction of the ionizing radiation with an environment containing electron affinic molecules and can explain their radiosensitizing action. The largest effect was previously observed for bromouracil, for which the number of evaporated water molecules after electron attachment was similar to that after electron ionization. In Fig. 6 we can see that the situation is also very similar for favipiravir. The figure shows a comparison of electron impact ionization MS of favipiravir at 70 eV and negative ion MS after electron attachment at 1.4 eV for two different hydration conditions. We can see that the observed numbers of water molecules, n, m, and p, attached to anions M(H2O)n− and cations M(H2O)m+ and M(H2O)Hp+ are similar. Despite the well-known fragmentation efficiency of electron impact ionization,97 the “soft” EA leads to a similar fragmentation, in the form of loss of neutral water molecules from the clusters. This is caused by the high electron affinity of favipiravir, which results in water evaporation from cluster after electron attachment. Therefore, if our hypothesis that energy transfer to the solvent after EA enhances LET and causes radiosensitization is correct, favipiravir should demonstrate radiosensitizing effects comparable to halogenated uracils.
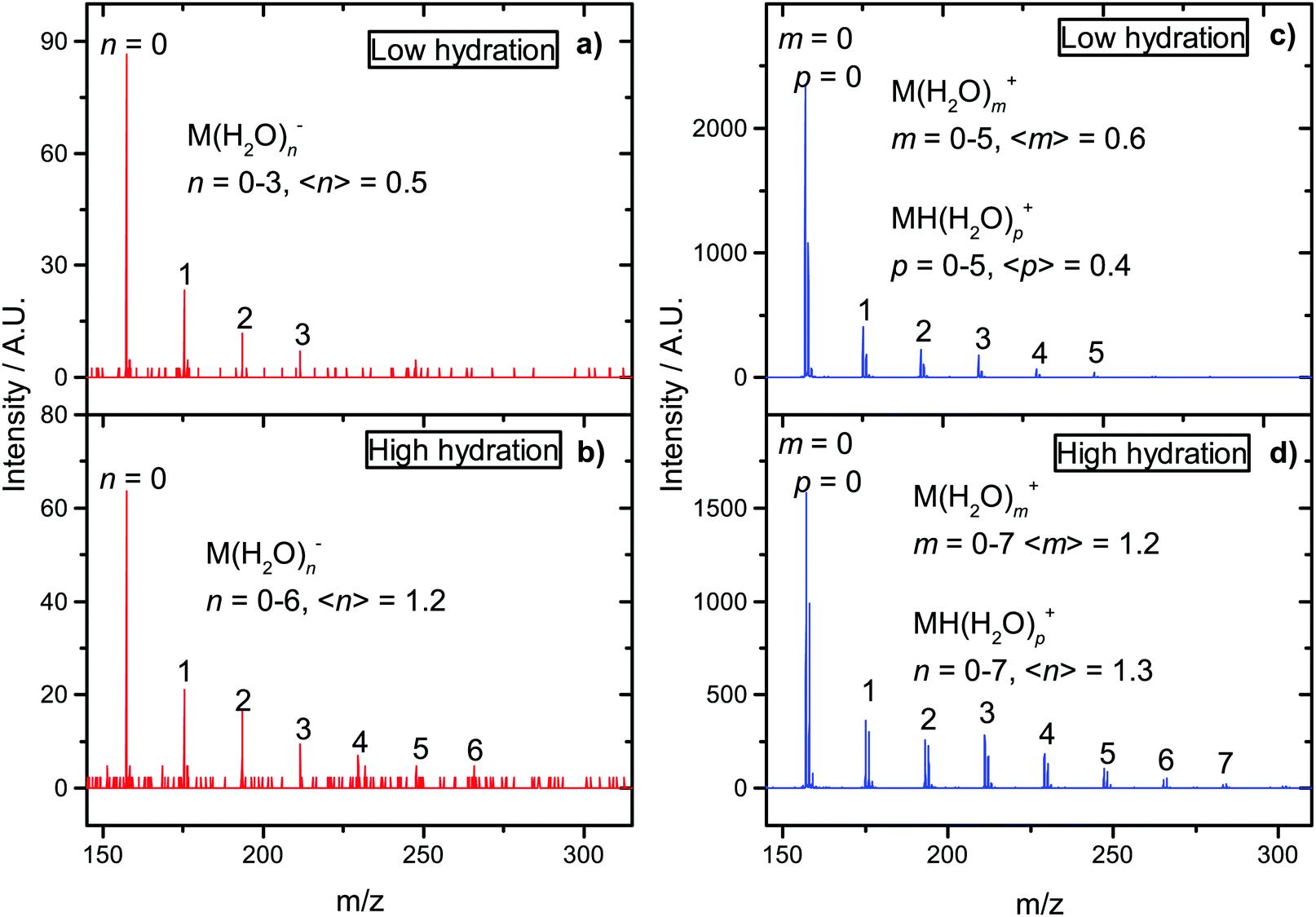 | ||
| Fig. 6 Mass spectra from the CLUB experiment for hydrated favipiravir in molecular beams. Negative ion MS (a) and (b) at the electron-energy 1.4 eV and positive ion MS (c) and (d) at the electron-energy 70 eV. The values in brackets represents peak intensity weighted average. | ||
4 Conclusions
We present an experimental study of electron attachment to favipiravir as an interesting hetero-aromatic, important biochemical, and promising radiosensitizing molecule. Electron attachment to favipiravir leads primarily to the formation of the parent anion with a long lifetime. Such stability may be important for the transport and radiosensitizing properties of the molecule8 but may also allow for multiple electron reduction.98 The threshold DFT calculations helped us to interpret the fragmentation pattern of the molecule with main fragmentation reactions occurring on the CONH2 group. The estimation of vertical attachment energies demonstrates several possible virtual states available for attachment via shape resonances that may be related to favipiravir's antiviral action.10 The mass spectrometry of small clusters of the form Fav(H2O)n, reveals that a large amount of energy is transferred to the solvent after electron attachment. This energy may contribute to favipiravir's reactivity in its reduced form but also support our hypothesis that favipiravir may be repurposed as a radiosensitizer.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors appreciate the support of the Czech Science Foundation, grant number 19-01159S. We would like to thank R. Čurík and M. Tarana for providing us with computational equipment and software available at the Department of Theoretical Chemistry of J. Heyrovský Institute of Physical Chemistry.Notes and references
- S. A. Pshenichnyuk and A. Modelli, J. Chem. Phys., 2014, 140, 034313 CrossRef PubMed.
- S. A. Pshenichnyuk and A. Modelli, in ETS and DEAS Studies of the Reduction of Xenobiotics in Mitochondrial Intermembrane Space, ed. V. Weissig and M. Edeas, Springer New York, New York, NY, 2015, pp. 285–305 Search PubMed.
- S. A. Pshenichnyuk and A. S. Komolov, J. Phys. Chem. Lett., 2015, 6, 1104–1110 CrossRef CAS PubMed.
- S. A. Pshenichnyuk, A. Modelli, E. F. Lazneva and A. S. Komolov, J. Phys. Chem. A, 2016, 120, 2667–2676 CrossRef CAS PubMed.
- S. A. Pshenichnyuk, R. G. Rakhmeyev, N. L. Asfandiarov, A. S. Komolov, A. Modelli and D. Jones, J. Phys. Chem. Lett., 2018, 9, 2320–2325 CrossRef CAS PubMed.
- S. A. Pshenichnyuk, A. Modelli and A. S. Komolov, Int. Rev. Phys. Chem., 2018, 37, 125–170 Search PubMed.
- S. A. Pshenichnyuk and A. Modelli, in Electron Attachment to Isolated Molecules as a Probe to Understand Mitochondrial Reductive Processes, ed. V. Weissig and M. Edeas, Springer US, New York, NY, 2021, pp. 101–124 Search PubMed.
- R. Meißner, J. Kočišek, L. Feketeová, J. Fedor, M. Fárník, P. Limão-Vieira, E. Illenberger and S. Denifl, Nat. Commun., 2019, 10, 2388 Search PubMed.
- C. W. Fong, Toxicology of platinum anticancer drugs: oxidative stress and antioxidant effect of stable free radical Pt-nitroxides, Eigenenergy, Adelaide, Australia research report, 2019.
- H. M. Yasir and F. H. Hanoon, IOP Conference Series: Materials Science and Engineering, 2020, vol. 928, p. 072066.
- R. Schürmann, S. Vogel, K. Ebel and I. Bald, Chem. – Eur. J., 2018, 24, 10271–10279 CrossRef PubMed.
- Y. Dong, Y. Wang, P. Zhuang, X. Fu, Y. Zheng and L. Sanche, J. Phys. Chem. B, 2020, 124, 3315–3325 CrossRef CAS PubMed.
- Advances In Atomic, Molecular, and Optical Physics, ed. E. Arimondo, C. C. Lin and S. F. Yelin, Academic Press, 2017, vol. 66, pp. 545–657 Search PubMed.
- J. D. Gorfinkiel and S. Ptasinska, J. Phys. B: At., Mol. Opt. Phys., 2017, 50, 182001 CrossRef.
- S. M. Pimblott and J. A. LaVerne, Radiat. Phys. Chem., 2007, 76, 1244–1247 CrossRef CAS.
- E. Alizadeh and L. Sanche, Chem. Rev., 2012, 112, 5578–5602 CrossRef CAS PubMed.
- C. D. Willey, E. S.-H. Yang and J. A. Bonner, in Clinical Radiation Oncology, ed. L. L. Gunderson and J. E. Tepper, Elsevier, Philadelphia, 4th edn, 2016, pp. 63–79.e4 Search PubMed.
- P. Wardman, Clin. Oncol., 2007, 19, 397–417 CrossRef CAS PubMed.
- L. Chomicz, M. Zdrowowicz, F. Kasprzykowski, J. Rak, A. Buonaugurio, Y. Wang and K. H. Bowen, J. Phys. Chem. Lett., 2013, 4, 2853–2857 CrossRef CAS.
- J. Kopyra, C. Koenig-Lehmann, I. Bald and E. Illenberger, Angew. Chem., Int. Ed., 2009, 48, 7904–7907 CrossRef CAS PubMed.
- J. Rak, L. Chomicz, J. Wiczk, K. Westphal, M. Zdrowowicz, P. Wityk, M. Żyndul, S. Makurat and Ł. Golon, J. Phys. Chem. B, 2015, 119, 8227–8238 CrossRef CAS PubMed.
- C. R. Wang, J. Nguyen and Q. B. Lu, J. Am. Chem. Soc., 2009, 131, 11320–11322 CrossRef CAS PubMed.
- J. Poštulka, P. Slavíček, J. Fedor, M. Fárník and J. Kočišek, J. Phys. Chem. B, 2017, 121, 8965–8974 CrossRef PubMed.
- M. Rezaee, D. J. Hunting and L. Sanche, Int. J. Radiat. Oncol., Biol., Phys., 2013, 87, 847–853 CrossRef CAS PubMed.
- D. Reimitz, M. Davídková, O. Mestek, J. Pinkas and J. Kočišek, Radiat. Phys. Chem., 2017, 141, 229–234 CrossRef CAS.
- Y. Dong, L. Zhou, Q. Tian, Y. Zheng and L. Sanche, J. Phys. Chem. C, 2017, 121, 17505–17513 CrossRef CAS.
- C. Wagner and H.-A. Wagenknecht, Chem. – Eur. J., 2005, 11, 1871–1876 CrossRef CAS PubMed.
- F. Xiao, X. Luo, X. Fu and Y. Zheng, J. Phys. Chem. B, 2013, 117, 4893–4900 CrossRef CAS PubMed.
- J. Rackwitz, M. L. Ranković, A. R. Milosavljević and I. Bald, Eur. Phys. J. D, 2017, 71, 32 CrossRef.
- J. Rackwitz, J. Kopyra, I. Dabkowska, K. Ebel, M. L. Ranković, A. R. Milosavljević and I. Bald, Angew. Chem., Int. Ed., 2016, 55, 10248–10252 CrossRef CAS PubMed.
- L. Chomicz, M. Zdrowowicz, F. Kasprzykowski, J. Rak, A. Buonaugurio, Y. Wang and K. H. Bowen, J. Phys. Chem. Lett., 2013, 4, 2853–2857 CrossRef CAS.
- M. Zdrowowicz, L. Chomicz, M. Żyndul, P. Wityk, J. Rak, T. J. Wiegand, C. G. Hanson, A. Adhikary and M. D. Sevilla, Phys. Chem. Chem. Phys., 2015, 17, 16907–16916 RSC.
- J. Ameixa, E. Arthur-Baidoo, R. Meißner, S. Makurat, W. Kozak, K. Butowska, F. Ferreira da Silva, J. Rak and S. Denifl, J. Chem. Phys., 2018, 149, 164307 CrossRef CAS PubMed.
- R. Meißner, S. Makurat, W. Kozak, P. Limão-Vieira, J. Rak and S. Denifl, J. Phys. Chem. B, 2019, 123, 1274–1282 CrossRef PubMed.
- P. Spisz, M. Zdrowowicz, W. Kozak, L. Chomicz-Mańka, K. Falkiewicz, S. Makurat, A. Sikorski, D. Wyrzykowski, J. Rak, E. Arthur-Baidoo, P. Ziegler, M. S. Rodrigues Costa and S. Denifl, J. Phys. Chem. B, 2020, 124, 5600–5613 CrossRef CAS PubMed.
- T. F. M. Luxford, S. A. Pshenichnyuk, N. L. Asfandiarov, T. Perečko, M. Falk and J. Kočišek, Int. J. Mol. Sci., 2020, 21, 8173 CrossRef CAS PubMed.
- S. Pushpakom, F. Iorio, P. A. Eyers, K. J. Escott, S. Hopper, A. Wells, A. Doig, T. Guilliams, J. Latimer, C. McNamee, A. Norris, P. Sanseau, D. Cavalla and M. Pirmohamed, Nat. Rev. Drug Discovery, 2019, 18, 41–58 CrossRef CAS PubMed.
- S. H. Kim, J. H. Kim, A. Kolozsvary, S. L. Brown and S. O. Freytag, J. Neuro-Oncol., 1997, 33, 189–194 CrossRef CAS PubMed.
- L. Sleire, H. E. Førde, I. A. Netland, L. Leiss, B. S. Skeie and P. Øyvind Enger, Pharmacol. Res., 2017, 124, 74–91 CrossRef CAS PubMed.
- X. Zhang, F. Wang, C. Zhang, S. Wu, X. Zheng, T. Gong, R. Ding, K. Chen and D. Bai, Inorg. Chem. Commun., 2018, 94, 92–97 CrossRef CAS.
- Z. Zhang, L. Zhou, N. Xie, E. C. Nice, T. Zhang, Y. Cui and C. Huang, Signal Transduction Targeted Ther., 2020, 5, 113 CrossRef PubMed.
- D. Bai, Y. Tian, K. Chen, X. Zhang, F. Wang, Y. Cheng, X. Zheng, K. Xiao and X. Dong, Dyes Pigm., 2020, 182, 108635 CrossRef CAS.
- S. Huq, J. Kedda, T. Zhao, R. Serra, A. Ding, M. Morales, J. Ehresman, H. Brem, G. L. Gallia, D. M. Sciubba and B. Tyler, Neurosurgery, 2020, 67, 309 Search PubMed.
- M. Van Herp, H. Declerck and T. Decroo, Lancet, 2015, 385, 2350 CrossRef.
- K. Rosenke, H. Feldmann, J. Westover, P. W. Hanley, C. Martellaro, F. Feldmann, G. Saturday, J. Lovaglio, D. Scott, Y. Furuta, T. Komeno, B. Gowen and D. Safronetz, Emerging Infectious Disease J., 2018, 24, 1696 CrossRef CAS PubMed.
- Z. F. Udwadia, P. Singh, H. Barkate, S. Patil, S. Rangwala, A. Pendse, J. Kadam, W. Wu, C. F. Caracta and M. Tandon, Int. J. Infect. Dis., 2021, 103, 62–71 CrossRef CAS PubMed.
- K. Shiraki and T. Daikoku, Pharmacol. Ther., 2020, 209, 107512 CrossRef CAS PubMed.
- Y. Furuta, K. Takahashi, Y. Fukuda, M. Kuno, T. Kamiyama, K. Kozaki, N. Nomura, H. Egawa, S. Minami, Y. Watanabe, H. Narita and K. Shiraki, Antimicrob. Agents Chemother., 2002, 46, 977–981 CrossRef CAS PubMed.
- Y.-X. Du and X.-P. Chen, Clin. Pharm. Ther., 2020, 108, 242–247 CrossRef CAS PubMed.
- M. G. Ison and M. H. Scheetz, EBioMedicine, 2021, 63, 103204 CrossRef CAS PubMed.
- J. Langer, M. Zawadzki, M. Fárník, J. Pinkas, J. Fedor and J. Kočišek, Eur. Phys. J. D, 2018, 72, 112 CrossRef.
- M. Fárník and J. Lengyel, Mass Spectrom. Rev., 2018, 37, 630–651 CrossRef PubMed.
- M. Stepanović, Y. Pariat and M. Allan, J. Chem. Phys., 1999, 110, 11376–11382 CrossRef.
- A. Stamatovic and G. J. Schulz, Rev. Sci. Instrum., 1970, 41, 423–427 CrossRef.
- L. G. Christophorou and J. K. Olthoff, J. Phys. Chem. Ref. Data, 2000, 29, 267–330 CrossRef CAS.
- M. Zawadzki, T. F. M. Luxford and J. Kočišek, J. Phys. Chem. A, 2020, 124, 9427–9435 CrossRef CAS PubMed.
- R. Dressler and M. Allan, Chem. Phys., 1985, 92, 449–455 CrossRef CAS.
- J. Kočišek, K. Grygoryeva, J. Lengyel, M. Fárník and J. Fedor, Eur. Phys J. D, 2016, 70, 98 CrossRef.
- J. Kočišek, J. Lengyel and M. Fárník, J. Chem. Phys., 2013, 138, 124306 CrossRef PubMed.
- J. Kočišek, A. Pysanenko, M. Fárník and J. Fedor, J. Phys. Chem. Lett., 2016, 7, 3401–3405 CrossRef PubMed.
- L. A. Curtiss, K. Raghavachari and J. A. Pople, J. Chem. Phys., 1993, 98, 1293–1298 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- K. Aflatooni, G. A. Gallup and P. D. Burrow, J. Phys. Chem. A, 1998, 102, 6205–6207 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian∼16 Revision C.01, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- G. Zhurko, Chemcraf – Graphical Program for Visualization of Quantum Chemistry Computations, available online: https://chemcraftprog.com.
- L. Antonov, Theor. Chem. Acc., 2020, 139, 145 Search PubMed.
- J. Simons, Annu. Rev. Phys. Chem., 2011, 62, 107–128 CrossRef CAS PubMed.
- H. Hao, J. Shee, S. Upadhyay, C. Ataca, K. D. Jordan and B. M. Rubenstein, J. Phys. Chem. Lett., 2018, 9, 6185–6190 CrossRef CAS PubMed.
- L. Sanche and G. J. Schulz, Phys. Rev. A, 1972, 5, 1672–1683 CrossRef CAS.
- D. Spence, J. Chem. Phys., 1977, 66, 669–674 CrossRef CAS.
- M. Zawadzki, M. Ranković, J. Kočišek and J. Fedor, Phys. Chem. Chem. Phys., 2018, 20, 6838–6844 RSC.
- V. S. Prabhudesai, A. H. Kelkar, D. Nandi and E. Krishnakumar, Phys. Rev. Lett., 2005, 95, 143202 CrossRef PubMed.
- C. Fridh, L. Åsbrink, B. Jonsson and E. Lindholm, Int. J. Mass Spectrom. Ion Phys., 1972, 8, 101–118 CrossRef CAS.
- M. V. Muftakhov and A. I. Fokin, Rapid Commun. Mass Spectrom., 1997, 11, 1923–1925 CrossRef CAS.
- J. Gingell, N. Mason, H. Zhao, I. Walker and M. Siggel, Chem. Phys., 1997, 220, 191–205 CrossRef CAS.
- Z. Li, M. Ryszka, M. M. Dawley, I. Carmichael, K. B. Bravaya and S. Ptasińska, Phys. Rev. Lett., 2019, 122, 073002 CrossRef PubMed.
- J. Fedor, Phys. Rev. Lett., 2020, 124, 199301 CrossRef CAS PubMed.
- R. Abouaf and H. Dunet, Eur. Phys. J. D, 2005, 35, 405–410 CrossRef CAS.
- P. D. Burrow, G. A. Gallup, A. M. Scheer, S. Denifl, S. Ptasinska, T. Märk and P. Scheier, J. Chem. Phys., 2006, 124, 124310 CrossRef CAS PubMed.
- M. Allan, M. Lacko, P. Papp, Š. Matejčík, M. Zlatar, I. I. Fabrikant, J. Kočišek and J. Fedor, Phys. Chem. Chem. Phys., 2018, 20, 11692–11701 RSC.
- I. Nenner and G. J. Schulz, J. Chem. Phys., 1975, 62, 1747–1758 CrossRef CAS.
- C. Koenig-Lehmann, J. Kopyra, I. Dabkowska, J. Kočišek and E. Illenberger, Phys. Chem. Chem. Phys., 2008, 10, 6954–6961 RSC.
- T. Hamann, A. Edtbauer, F. Ferreira da Silva, S. Denifl, P. Scheier and P. Swiderek, Phys. Chem. Chem. Phys., 2011, 13, 12305–12313 RSC.
- A. Keller, J. Kopyra, K. V. Gothelf and I. Bald, New J. Phys., 2013, 15, 083045 CrossRef CAS.
- F. Ferreira da Silva, C. Matias, D. Almeida, G. García, O. Ingólfsson, H. D. Flosadóttir, B. Ómarsson, S. Ptasinska, B. Puschnigg, P. Scheier, P. Limão-Vieira and S. Denifl, J. Am. Soc. Mass Spectrom., 2013, 24, 1787–1797 CrossRef CAS PubMed.
- M. M. Dawley and S. Ptasińska, Int. J. Mass Spectrom., 2014, 365–366, 143–151 CrossRef CAS.
- M. M. Dawley, K. Tanzer, I. Carmichael, S. Denifl and S. Ptasińska, J. Chem. Phys., 2015, 142, 215101 CrossRef PubMed.
- M. Muftakhov, P. Shchukin and R. Khatymov, Radiat. Phys. Chem., 2021, 184, 109464 CrossRef CAS.
- M. V. Muftakhov and P. V. Shchukin, Rapid Commun. Mass Spectrom., 2019, 33, 482–490 CrossRef CAS PubMed.
- M. M. Dawley, C. Pirim and T. M. Orlando, J. Phys. Chem. A, 2014, 118, 1228–1236 CrossRef CAS PubMed.
- H. Abdoul-Carime, P. Cloutier and L. Sanche, Radiat. Res., 2001, 155, 625–633 CrossRef CAS PubMed.
- S. E. Bradforth, E. H. Kim, D. W. Arnold and D. M. Neumark, J. Chem. Phys., 1993, 98, 800–810 CrossRef CAS.
- N. L. Asfandiarov, S. A. Pshenichnyuk, R. G. Rakhmeyev, R. F. Tuktarov, N. L. Zaitsev, A. S. Vorobev, J. Kočišek, J. Fedor and A. Modelli, J. Chem. Phys., 2019, 150, 114304 CrossRef CAS PubMed.
- N. L. Asfandiarov, M. V. Muftakhov, S. A. Pshenichnyuk, P. Papp, M. Danko, M. Lacko, J. Blaško, Š. Matejcik and A. Modelli, J. Chem. Phys., 2017, 147, 234302 CrossRef CAS PubMed.
- H. Pauly, Atomic and Molecular Beam Methods, vol. 1, 1998.
- J. Lengyel, A. Pysanenko, V. Poterya, J. Kočišek and M. Fárník, Chem. Phys. Lett., 2014, 612, 256–261 CrossRef CAS.
- R. Schürmann, T. F. M. Luxford, I. S. Vinklárek, J. Kočišek, M. Zawadzki and I. Bald, J. Chem. Phys., 2020, 153, 104303 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Low m/z part of the spectra depicted in Fig. 6, panels (c) and (d). Comparison of the reaction energies calculated using B3LYP and M062x functionals, Cartesian coordinates of favipiravir conformers and its DEA anion and neutral fragments at B3LYP and M062x levels of theory. See DOI: 10.1039/d1cp02686k |
| This journal is © the Owner Societies 2021 |