
Switching energy dissipation pathway: in situ proton-induced transformation of AIE-active self-assemblies to boost photodynamic therapy†
Jie
Li
ab,
Jianxing
Wang
ab,
Jianyu
Zhang
 c,
Xiyao
Hu
a,
Dong
Wang
*a and
Ben Zhong
Tang
c
c,
Xiyao
Hu
a,
Dong
Wang
*a and
Ben Zhong
Tang
c
aCenter for AIE Research, College of Materials Science and Engineering, Shenzhen University, Shenzhen 518060, China. E-mail: wangd@szu.edu.cn
bKey Laboratory of Optoelectronic Devices and Systems of Ministry of Education and Guangdong Province, College of Physics and Optoelectronic Engineering, Shenzhen University, Shenzhen 518060, China
cHong Kong Branch of Chinese National Engineering Research Center for Tissue Restoration and Reconstruction, Department of Chemistry, The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong, China
First published on 10th February 2021
Abstract
With the morphological transformation of fluorescent self-assembled nanostructures, their functions can be varied simultaneously. However, little attention has been paid to the function variation in this process. Herein, we present aggregation-induced emission (AIE)-active self-assembled nanospheres to investigate the transformation-induced function variation by switching the energy dissipation pathway. The self-assembled nanospheres showed strong emission under neutral conditions, indicating that radiative decay dominates the energy dissipation. Under acidic conditions, the spheres transformed to vesicles and nanotubes, in which the excited energy was largely consumed by the intersystem crossing pathway and highly efficient reactive oxygen species (ROS) generation was afforded. In particular, this morphological transformation and function variation can smoothly proceed in acidic lysosomes, thus drastically boosting photodynamic cancer therapy.
1. Introduction
Modulating transformation between different self-assembled nanostructures, e.g., spheres, vesicles, helices and nanotubes, is of great significance because it provides a fundamental understanding of biological self-assembly processes, as well as efficient strategies for constructing various supramolecular nanomaterials.1–8 Among various kinds of self-assembled systems, of particular interest are fluorescent ones, which have attracted significant scientific interest in the fields of drug delivery, biological and chemical sensors, and theranostics.9–12 Remarkably, these morphological transformations are often accompanied by quenched fluorescence emission,13–15 revealing the transformation of the energy consumption pathway from radiative to non-radiative decay. However, this energy dissipation change is usually neglected, as are the applications of the energy dissipated from non-radiative decay. As an emerging therapy for cancer via producing reactive oxygen species (ROS) upon irradiation, photodynamic therapy (PDT) exhibits distinct advantages of vascular shutdown and immune activation while being non-invasive,16–18 and the ROS generation is closely related to non-radiative pathways. Therefore, we are wondering whether the energy dissipated from non-radiative decay involved in morphological transformation could be optimally utilized?Protonation holds great potential for the variation of energy decay pathways and utilization of non-radiative decay energy.19–21 The combination of positively charged protons with chromophores is capable of improving the electron-donating–accepting (D–A) strength in structures, resulting in the decrease of the energy gap (ΔEST) between singlet and triplet excited states. Consequently, the non-radiative decay pathway of intersystem crossing (ISC) is established to consume excited-state energy, where reactive oxygen species (ROS) are significantly generated, allowing for PDT. Moreover, protonation, the strategy of regulating molecular hydrophilicity by changing pH, also presents a feasible protocol for manipulating the morphology of self-assembled nanostructures.22,23 More interestingly, as cellular organelles with acidic vesicular compartments (pH ≈ 5), lysosomes normally receive nano-assemblies via endocytosis, producing self-assembled nanostructures with promising potential for undergoing in situ morphological transformation driven by protonation in cells.24–27 In this regard, switching the energy dissipation and promoting the non-radiative decay pathway to boost PDT in proton-induced transformation assemblies would be an appealing yet significantly challenging task.
Self-assemblies constructed from traditional luminogens always suffer the inherent obstacle of aggregation-caused fluorescence quenching, which hinders their application as fluorescent materials. On the contrary, aggregation-induced emission (AIE) molecules possess fantastic emission in aggregated states because their twisted structures prevent strong π–π stacking, and thus are ideal candidates for fluorescent self-assemblies.28–31 Herein, we present a self-assembly based on AIE amphiphiles that can undergo morphological transformations under acidic conditions to comprehensively study the switching of energy dissipation and variation in the functions. The AIE amphiphile TPE-BEP was elaborately designed and synthesized with hydrophilic units of tetraethylene glycol groups and proton-binding sites of pyridine (Schemes S1–S3†). As illustrated in Fig. 1A, TPE-BEP can self-assemble into nanospheres with strong emission in water due to its typical AIE features. In dimethylformamide (DMF) solvent, TPE-BEP was well dispersed and showed very weak emission with a quantum yield (QY) of 0.9%. On increasing the fraction of H2O, the fluorescence emission intensity gradually enhanced due to the formation of aggregates, and the QY reached to 31.2% with H2O fraction of 90% (Fig. S1†). After entering the lysosomes of cells, the self-assemblies changed their morphology from nanospheres to vesicles resulting from the protonation of TPE-BEP. Concurrently, the non-radiative ISC process dominates the energy consumption of the excited state. As a result, large amounts of ROS were produced, endowing the self-assemblies with distinctive cancer cell-killing ability.
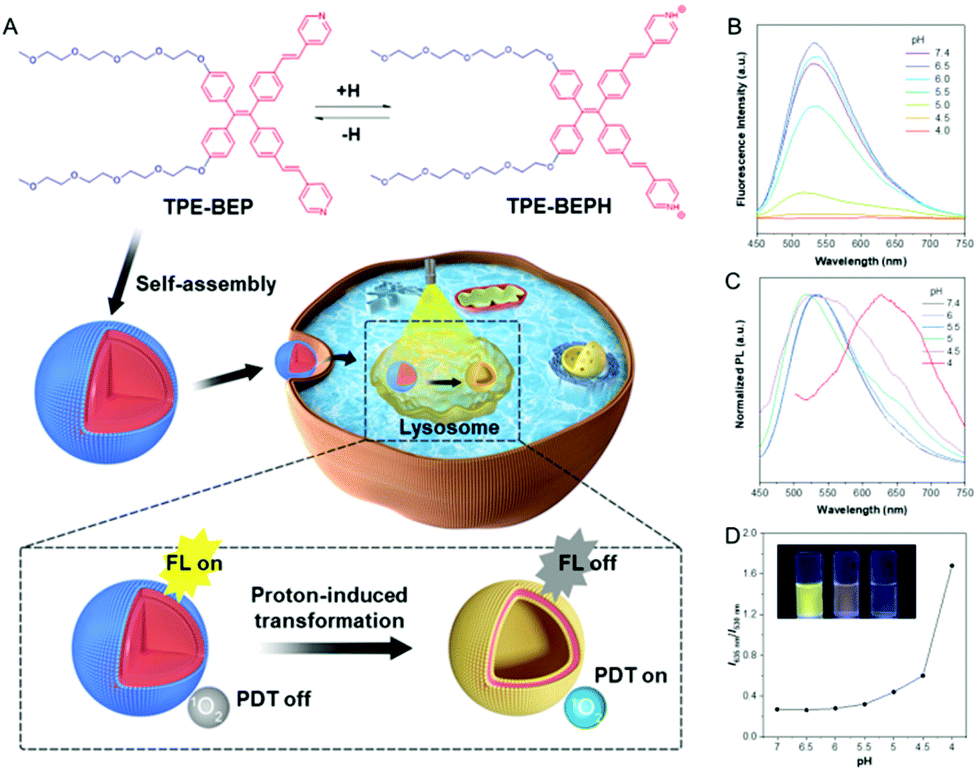 | ||
| Fig. 1 (A) Molecular structures of TPE-BEP and the protonated TPE-BEPH, and schematic illustration of in situ transformation and function variation in cells. (B) Fluorescence spectra of TPE-BEP in PBS with different pH values. (C) Normalized fluorescence spectra of TPE-BEP in PBS with different pH values. (D) Emission ratios of 635 nm (TPE-BEPH) and 530 nm (TPE-BEP) in different pH solutions. The inset in (D) is a photograph of TPE-BEP solutions with different pH (left to right: 7.4, 5.0 and 4.0). [TPE-BEP] = 10 μM. | ||
2. Experimental section
2.1. Materials
4,4′-Dibromobenzophenone, 4,4′-dimethoxybenzophenone, 4-vinylpyridine, triethylene glycol 2-bromoethyl methyl ether, and tris-o-tolylphosphine were purchased from J&K, TCI, Sigma and Adamas. 2′,7′-Dichlorodihydrofluorescein diacetate (DCFH-DA), 9,10-anthracenediyl-bis(methylene)dimalonic acid (ABDA) and Rose Bengal were purchased from Sigma-Aldrich. Phosphate-buffered saline (PBS), LysoTracker Green and MitoTracker Green were purchased from Thermo Fisher Scientific. All other chemicals were of analytical grade and were used as received without further purification. Ultrapure water was supplied by a Mill-Q Plus System (Millipore Corporation, USA).2.2. Instrumentation
1H nuclear magnetic resonance (NMR) and 13C NMR spectra were obtained using a Bruker AV 500 NMR spectrometer. High-resolution mass spectra (HRMS) were recorded on a Finnegan MAT TSQ 7000 mass spectrometer operated in matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mode. Transmission electron microscopy (TEM) images were recorded using a JEM-100 CX II transmission electron microscope (JEOL, Japan, 60 kV). Samples were prepared by dropping solutions onto copper grids and then directly observed without any staining. AFM was recorded using an atomic force microscope (Bruker Dimension Icon microscope, Germany) in tapping mode. The UV absorbance and fluorescence emission of the samples were respectively measured by UV/vis spectrophotometer (PerkinElmer Lambda 950, USA) and fluorescence spectrophotometer (Edinburgh Instruments FS5, UK). The particle size and zeta potential of the samples were determined using a Mastersizer 2000 instrument and a Zeta-sizer Nano ZS90 (Malvern, UK). CLSM images were taken on a confocal laser scanning microscope (ZEISS-LSM880, Germany).2.3. 1O2- and ROS-generation detection
DCFH-DA and ABDA were respectively utilized as indicators of 1O2 and reactive oxygen species (ROS). In the experiments, 13 μL of ABDA solution (7.5 mM) was added to 2 mL of AIE-active amphiphile water solutions (10 μM) with various pH values, then the mixed solutions were irradiated under white light (4.2 mW cm−2). The absorption of ABDA at 378 nm was recorded at different irradiation times to obtain the 1O2 generation efficiency. Similarly, to obtain the ROS-generation efficiency, activated DCFH-DA (5 μM) solution was mixed with AIE-active amphiphile solutions (2 μM) followed by irradiation with white light (WL, 4.2 mW cm−2). The fluorescence emission at 525 nm (Ex = 488 nm) was detected, and the ROS generation efficiency (I/I0) was calculated by using the fluorescence intensity (525 nm) after irradiation for the desired time and the fluorescence intensity (I0) without irradiation.2.4. Cell culture
Murine mammary carcinoma 4T1 cells, human cervical cancer HeLa cells and HepG2 cells were cultured in DMEM medium with 10% FBS and 1% penicillin/streptomycin under a humidified environment of 5% CO2 at 37 °C.2.5. Co-location cell imaging
All of the cells were incubated overnight in 35 mm Petri dishes. The amphiphile TPE-BEP was added into the medium at a dose of 10 μM and immediately incubated with the cells at 37 °C. After the desired incubation time, 200 nM of MTO or lysotracker were added and stained for 30 min. Then, the cells were washed with PBS and imaged under an inverted fluorescence optical microscope (Zeiss LSM, Eclipse Ti). Excitation filter: 405 nm for TPE-BEP, 488 nm for MTO and lysotracker; emission: 500–550 nm and 600–650 nm for TPE-BEP; 520–550 nm for lysotracker and MTO.2.6. Cell uptake
Cancer cells were seeded into confocal plates and incubated for 24 h to obtain monolayer cells. After incubation with the amphiphiles TPE-BEP (10 μM) at 37 °C for different periods (0.5, 1, 2, 3 and 4 h), the cells were washed with PBS and observed by CLSM. The exciting wavelength for TPE-BEP was 405 nm and the emission channels were 500–550 nm and 600–650 nm. The emission intensity was quantitively calculated using ImageJ.2.7. Real-time cell imaging with acetic acid
The cells were grown in 35 mm Petri dishes and incubated with amphiphile TPE-BEP (10 μM) for 2 h. After washing with PBS three times, 1 mL of PBS containing 10 μM acetic acid was added and the cells were observed immediately. Excitation filter: 405 nm. Emission: 500–550 nm and 600–650 nm.2.8. Cell imaging at various pH
The cells were grown in 35 mm Petri dishes and incubated with amphiphile TPE-BEP (10 μM) for 2 h. After being washed with PBS three times, 1 mL of PBS with various pH values (containing 10 μM nigericin and 5 μM monensin) was added and the cells were incubated for 30 min before observation. Excitation filter: 405 nm. Emission: 500–550 nm and 600–650 nm.2.9. Cell cytotoxicity
4T1, HeLa and HepG2 cells were seeded in 96-well plates at a density of 5000 cells per well and cultured overnight. Then the medium was replaced with 100 μL of fresh medium containing TPE-BEP with different concentrations. After incubation for 24 h, the medium was replaced by 100 μL of fresh medium containing 10% CCK-8 and an additional 2 h incubation was conducted for the cells. The relative cell viability was calculated according to the absorbance of the correlated cells at 450 nm by a microplate spectrophotometer with the cells only cultured with medium as a control. Each trial was performed with five wells in parallel.2.10. Cell cytotoxicity of TPE-BEP under irradiation
Cells were seeded in 96-well plates at a density of 5000 cells per well and cultured overnight. The TPE-BEP was added at different concentrations after replacing the cell medium. While being incubated for 4 h, the cells were exposed to white light (24 mW cm−2) for 30 min and 1 h, and kept in the dark as a control. Subsequently, after incubation for 20 h, the cell viability was calculated by CCK-8 according to the cell cytotoxicity method. These experiments were conducted in 4T1, HeLa and HepG2 cells with the same procedures.2.11. Generation of intracellular ROS
The generation of intracellular ROS was evaluated in living 4T1 cells using DCFH-DA as an indicator. 4T1 cells were seeded into confocal plates and incubated for 24 h. The culture mediums were replaced with 1 mL of fresh medium containing TPE-BEP (10 μM) and the cells were incubated for another 4 h at 37 °C. Then the cells were washed with PBS and incubated with 1 mL of fresh medium containing 5 × 10−6 M DCFH-DA for an additional 20 min at 37 °C. After being irradiated by white light (24 mW cm−2) for 30 min, the fluorescence signals of the cells were captured.2.12. Computational methods
All the geometric and electronic structures were determined by quantum mechanics calculation using the Gaussian 09 program. The ground (S0) and excited states (S1) were optimized using the time-dependent density functional theory (TD-DFT) method at the M06-2X/6-31G(d,p) level.32 In order to consider the bulky solvation effects of water, the polarizable continuum model (PCM) with self-consistent reaction field (SCRF) and water as the solvent was taken into consideration. The energy level of the ground state (S0) was set to zero to draw the energy diagram. The energy differences for the most relevant singlet and triplet excited states were evaluated at the same calculation level. All the frontier molecular orbitals were visualized using the IQmol molecular viewer package.3. Results and discussion
3.1. Proton-induced transformation of TPE-BEP self-assembly
NMR measurements obviously showed that all the signals of the pyridinium ring protons and ethenyl protons of TPE-BEP were shifted downfield after addition of trifluoroacetic acid (Fig. S2†). This was caused by the decrease of the electron density, suggesting protonation. After addition of triethylamine, these signals further reversed back to the initial positions, indicating deprotonation. UV-vis and fluorescence spectra also demonstrated the protonation and deprotonation processes (Fig. S3†). When TPE-BEP was exposed to an acidic environment, the UV absorbance changed from 360 to 410 nm and the fluorescence emission simultaneously moved from 530 to 635 nm. On the contrary, the absorption and fluorescence emission were recovered by adding NaOH. This was because combining H+ could prompt charge separation and lower the energy gap between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), resulting in red shifts of both absorbance and emission. Density functional theory (DFT) calculation revealed that the HOMO and LUMO of TPE-BEPH (protonated TPE-BEP) were separated to a much larger extent than in TPE-BEP: the corresponding energy gap between the HOMO and LUMO of TPE-BEPH was 4.39 eV, which was lower than the 5.14 eV for TPE-BEP (Fig. S4†). This solidly confirmed the protonation of TPE-BEP and the long emission wavelength (635 nm) of TPE-BEPH (Fig. 1C).To gain a deep insight into the protonation process, the fluorescence emission of TPE-BEP in aqueous solutions with different pH values was systematically investigated. The pKa of TPE-BEP was firstly measured to be 5.6, according to the results of UV absorbance via pH values (Fig. S5†). As depicted in Fig. 1B and D, the TPE-BEP solution showed yellow emission of 530 nm at pH = 7.4, where the TPE-BEP was seldom protonated with an extremely low I635 nm/I530 nm ratio. However, upon gradually decreasing the pH, the emission intensity at 530 nm stepwise reduced and the ratio of I635 nm/I530 nm obviously enhanced owing to the protonation of TPE-BEP. The species distributions of TPE-BEP and TPE-BEPH in Fig. S6† show the progressive decrease of TPE-BEP and growth of TPE-BEPH with decreasing pH. For example, 72% of TPE-BEP existed in the unprotonated form while 28% was protonated TPE-BEPH at pH = 6, where the I635 nm/I530 nm ratio increased to 0.28. Consequently, the solution emission turned to orange at pH = 5, and it further became red with an emission peak of 635 nm at pH 4 owing to the complete protonation.
By virtue of the amphiphilic molecular structure, TPE-BEP could spontaneously form nano-aggregates in water, and the self-assembly behavior was evaluated at various pHs. Transmission electron microscopy (TEM) and dynamic laser scattering (DLS) measurements showed that the aggregates of TPE-BEP in pH = 7.4 solution were well-defined spheres with a diameter of 220 nm (Fig. 2A and D). CLSM further demonstrated that the spherical self-assemblies emitted yellow fluorescence (Fig. S7†). Atomic force microscopy (AFM) results confirmed the spherical structure, and the height profile demonstrated that the collapsed spheres may possess a multilamellar configuration (Fig. S8†).33 Moreover, TPE-BEP was well dissolved in DMF, but after addition of H2O (Fig. S9†), pronounced red shifts of both absorbance and emission were observed, suggesting a possible J-aggregated stacking in spherical structure. Similarly, at pH = 5, the TPE-BEP assemblies were vesicle morphology with a diameter of 460 nm (Fig. 2B). AFM images also displayed the hollow vesicular structure, as well as the multilamellar structure. When the pH value was 4, red-emissive nanotubes with 100 nm length and 5 nm width were obtained (Fig. 2C).
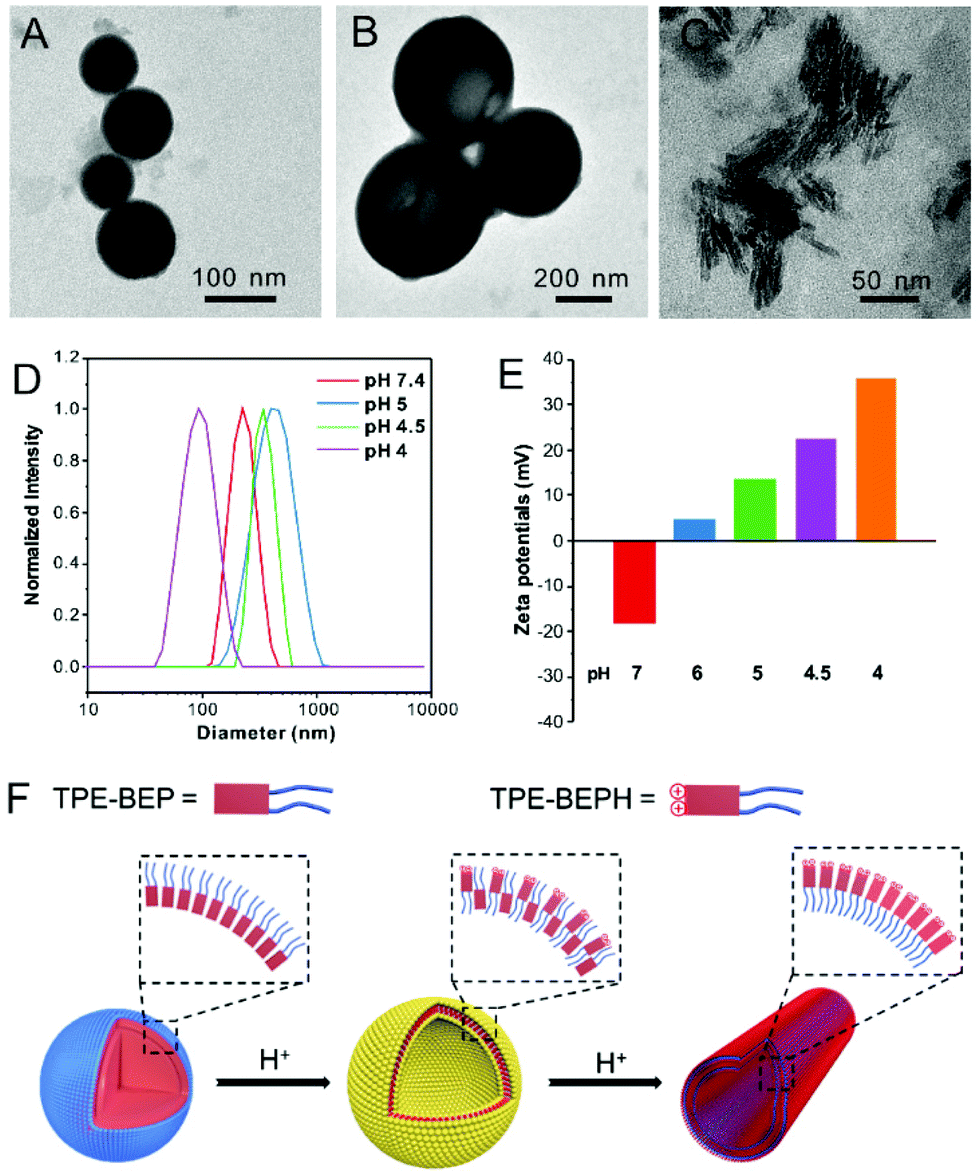 | ||
| Fig. 2 TEM images of TPE-BEP self-assemblies under various pH conditions: (A) 7.4, (B) 5.0 and (C) 4.0. (D) DLS results and (E) zeta potentials of TPE-BEP self-assemblies under various pH conditions. (F) Schematic illustration of the transformation between TPE-BEP self-assemblies driven by protons. | ||
The mechanism of the morphological transformations was further explored (Fig. 2F). The zeta potential of the TPE-BEP self-assemblies (Fig. 2E) at pH = 7.4 was determined to be −18 mV, implying that tetraethylene glycol groups are located on the outer shell of the nanospheres because hydrophilic tetraethylene glycol groups normally display negative charges in water. Nevertheless, the potential of the TPE-BEP self-assemblies changed from negative to positive on gradually decreasing the pH, and the positive potentials even increased with decreasing pH. Seeing that TPE-BEP was protonated in acidic conditions and converted to the positively charged TPE-BEPH, it was reasonable to infer that the protonated pyridinium of TPE-BEPH was positioned on the outer layer of the assemblies instead of tetraethylene glycol group, neutralizing the negative charges and further triggering the charge reversal. For example, when the pH was 5, 80% of TPE-BEP was converted to the positively charged TPE-BEPH, and positively charged pyridinium units dominated the outer shells of the vesicles, resulting in a rather positive zeta potential. The perfect overlap between yellow and red colors in the CLSM images for pH = 5 proved the coexistence of TPE-BEP and TPE-BEPH in one self-assembled nanostructure (Fig. S7†). Additionally, the zeta potential value at pH = 5 was much less than at pH = 7.4, indicating a weaker repulsive interaction at pH = 5, and the nanosphere tended to swell and gave birth to hollow vesicles. Furthermore, when all of the TPE-BEP was converted to TPE-BEPH (pH = 4), the outer layer of the self-assemblies was all pyridinium units and they thus exhibited highly positive zeta potentials, resulting in strong electrostatic repulsion and subsequent formation of nanotubes. The rapid decrease of the lifetime of TPE-BEP assemblies from pH = 7.4 (2.14 ns) to pH = 4.0 (0.07 ns) verified the loose stacking state and electrostatic repulsion of the TPE-BEP assemblies in acidic conditions (Fig. S10†).
3.2. Proton-induced conversion of excitation energy dissipation
In the morphological transformations, the fluorescence intensity and quantum yield (QY) of TPE-BEP remarkably reduced from spheres (pH 7.4, 13.2%) to vesicles (pH 5.0, 8.8%) to nanotubes (pH 4.0, 1.2%) (Fig. 3A). This suggested a critical conversion of dissipating excitation energy during the transformation.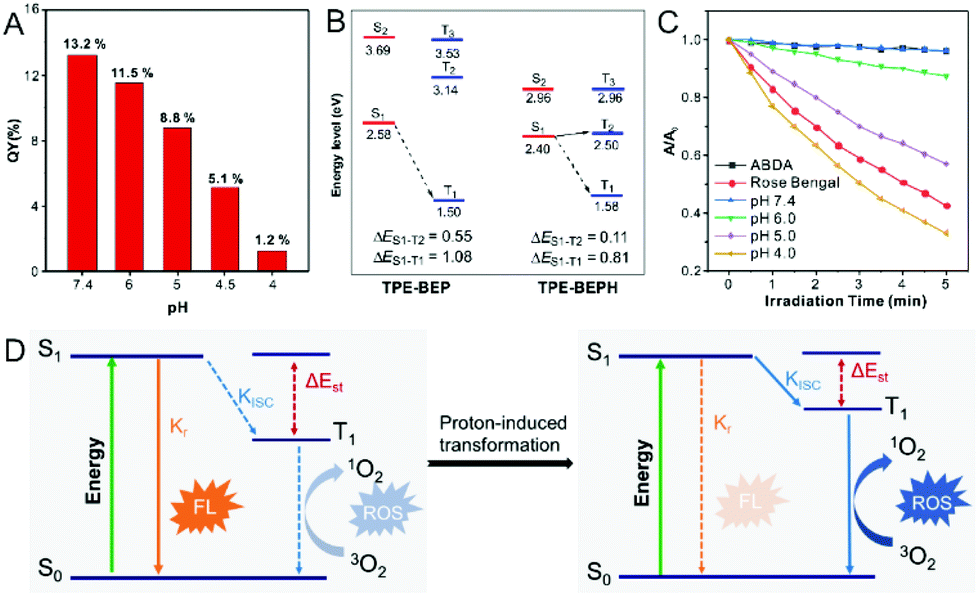 | ||
| Fig. 3 (A) Quantum yield (QY) of TPE-BEP in water solutions with different pH. (B) Calculated energy diagrams of TPE-BEP and TPE-BEPH. (C) ROS generation of TPE-BEP in different pH solutions indicated by ABDA. (D) Jablonski diagram of radiative and ISC processes before and after proton-induced transformation of TPE-BEP self-assemblies. S0: the ground state. S1: the lowest excited singlet state. T1: the lowest excited triplet state. Kr, and KISC are the rate constants of the radiative decay and the ISC process, respectively. FL: fluorescence. [TPE-BEP] = 10 μM. | ||
The energy dissipation process was further investigated. Theoretical calculation of the energy gap between singlet and triplet states was conducted.34–36 As depicted in Fig. 3B, protonated TPE-BEPH possessed a rather lower ΔEST (0.11 eV) than TPE-BEP (0.55 eV), which meant that TPE-BEPH could open up a pathway for the ISC process to dissipate the excitation energy, suggesting the potential for efficient ROS production. 9,10-Anthracenediyl-bis(methylene)dimalonic acid (ABDA) was used as the ROS indicator. As shown in Fig. 3C and S11,† the UV-vis absorption intensities of ABDA in TPE-BEP solution seldom changed at pH = 7.4 under light irradiation, which was the same as the result without TPE-BEP. This indicated that almost no singlet oxygen was produced when TPE-BEP was at pH = 7.4. However, the UV-vis absorption rapidly decreased with the irradiation time in acidic solutions, suggesting the generation of singlet oxygen. For example, the intensity of ABDA decreased by 13% at pH = 6 after 5 min irradiation, while the intensities respectively dropped by 43% and 67% when the pH of TPE-BEP was 5 and 4. This demonstrated that a lower pH induced a higher singlet oxygen yield on account that more TPE-BEP was protonated to TPE-BEPH. The performance of singlet oxygen generation at pH = 4 was even better than that of Rose Bengal. Moreover, this result was also observed when DCFH-DA acted as an ROS indicator (Fig. S12†), reflecting a promising potential for PDT application. These results solidly confirmed that protonated TPE-BEPH possesses an efficient ISC pathway to consume excited-state energy with an excellent capacity for generating ROS while TPE-BEP did not.
The energy dissipation conversion accompanying the morphological transformation is illustrated in Fig. 3D. TPE-BEP self-assembled into spheres and the luminophores tensely packed in the core, leading to the excited-state energy being dissipated through the radiative decay pathway with strong emission. When exposed to acidic conditions, owing to the protonation of TPE-BEP, the self-assemblies transferred into nanotubes with the luminophores placed in the outer shell, and the corresponding ΔEST of TPE-BEP decreased, opening the ISC pathway to consume excited energy. In brief, the radiative decay pathway converted to ISC during the morphological transformation driven by protonation.
3.3. Conversion of excitation energy dissipation and transformation of self-assemblies in cells
Cell imaging was proposed to check the proton-induced transformation of TPE-BEP in cells. As demonstrated in Fig. 4A and S13,† after treating 4T1 cancer cells with the TPE-BEP self-assemblies, bright yellow emission was observed in the cells, and the fluorescence signal was remarkably enhanced on prolonging the incubation time to 2 hours. This indicated that the TPE-BEP self-assemblies were efficiently taken up by the cells. However, no red signal, referring to TPE-BEPH, could be detected when incubated for 0.5 hours. The red emission appeared at 1 hour and its intensity further increased over time, in which the yellow emission intensity decreased simultaneously, reflecting that the spherical TPE-BEP assemblies might transform to vesicles. Co-location images further showed that both yellow and red assemblies were mainly distributed in the lysosomes (Fig. 4B and S14, S15†), suggesting that TPE-BEP self-assemblies were taken up through endocytosis and the transformation happened in the lysosomes.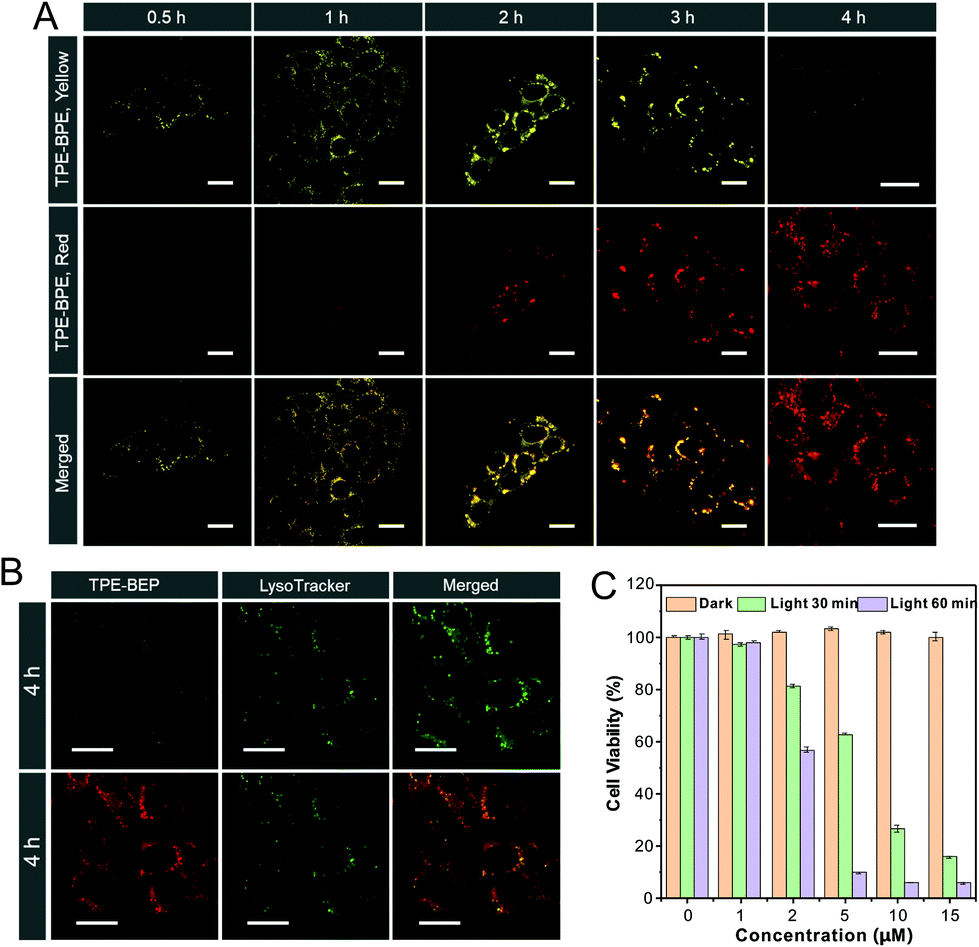 | ||
| Fig. 4 (A) Cell images of 4T1 cells after incubation with TPE-BEP self-assemblies for various periods. (B) Co-location of 4T1 cells stained with LysoTracker after incubation with TPE-BEP self-assemblies. λex: 405 nm; λem (yellow): 500–550 nm; λem (red): 600–650 nm. [TPE-BEP] = 10 μM. Scale bar is 20 μm. (C) 4T1 cell viability after incubation with different concentrations of TPE-BEP self-assemblies in the dark or under irradiation. | ||
To observe the in situ transformation in the lysosomes, intracellular pH values were regulated by various pH PBS containing nigericin (Fig. S16†). TPE-BEP assemblies were incubated for 2 hours following treatment with different pH PBS. The yellow fluorescence emission in cells decreased dramatically with the reduction of pH values, while the red emission was remarkably enhanced. This pH-dependent alteration verified the transformations induced by protonation in cells. Additionally, aiming to obtain real-time transformation in cells, acetic acid, which can permeate the cell membrane, was selected to prompt the protonation of TPE-BEP.37 As shown in Fig. S17,† the red areas greatly expanded and the red signal became much stronger over time, accompanied by the decay of the yellow fluorescence, which clearly indicated the in situ transformation in cells.
Finally, the application of ROS generated by the ISC pathway was tested in cancer cells. Compared to the negligible emission of ROS indicator dichlorofluorescein (DCFH-DA) or TPE-BEP assemblies alone, bright fluorescence was found in cells when treated with both DCFH-DA and TPE-BEP assemblies after irradiation (Fig. S18†), reflecting efficient ROS generation in cells. Moreover, when adjusting the intracellular pH to 7.4, the cells showed considerably weak emission after irradiation. Inversely, the emission at intracellular pH = 4 was evidently stronger than that of the cell without adjusting, which suggested that the ROS generation was triggered by protonation. The produced ROS could cause apoptosis of cancer cells (Fig. S19†). Dose-dependent cytotoxicity showed that cancer cell viability was gradually and rapidly decreased upon raising the TPE-BEP concentration, and the longer irradiation time caused a much lower viability (Fig. 4C and S20†). The effective PDT protocol was successfully extended to other cancer cells, such as HeLa cells and HepG2 cells (Fig. S21†). Together with their excellent photostability (Fig. S22†), TPE-BEP assemblies are excellent nano-photosensitizers for PDT applications.
4. Conclusions
We developed an AIE-active self-assembly that exhibits proton-induced morphological transformation to study the energy dissipation conversion and function variation. The self-assemblies had a spherical structure in neutral conditions and dissipated excited-state energy via the radiative pathway with strong emissions. As a result of protonation in acidic conditions, the nanospheres transformed to nanovesicles and nanotubes but with quite weak emissions, where the chromophore was located in the outer shell with incompact stacking. Concurrently, the protonation reduced the energy gap ΔEST, opening up the ISC pathway for producing ROS to consume the excited-state energy. The morphological transformation and energy dissipation pathway conversion can be achieved in acidic lysosomes, and the high-performance ROS production enables the self-assemblies to be very efficient for ablating cancer cells by means of PDT. This study thus provides an innovative insight into the relationship between the function, energy dissipation and morphology of self-assembly systems and facilitates the practical application of fluorescent self-assemblies.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the China Postdoctoral Science Foundation (2019M653005), the National Natural Science Foundation of China (21801169, 21902106), and the Developmental Fund for Science and Technology of Shenzhen government (JCYJ20190808153415062), and the Natural Science Foundation for Distinguished Young Scholars of Guangdong Province (2020B1515020011). The authors acknowledge the Instrumental Analysis Center of Shenzhen University. The 4T1, HeLa and HepG2 cell lines were purchased from the Chinese Academy of Science Cell Bank for Type Culture Collection.References
- W. Zhang and T. Aida, Science, 2012, 337, 1462–1463 CrossRef CAS.
- R. Freeman, M. Han, Z. Álvarez, J. A. Lewis, J. R. Wester, N. Stephanopoulos, M. T. McClendon, C. Lynsky, J. M. Godbe, H. Sangji, E. Luijten and S. I. Stupp, Science, 2018, 362, 808–813 CrossRef CAS.
- D. Mozhdehi, K. M. Luginbuhl, J. R. Simon, M. Dzuricky, R. Berger, H. S. Varol, F. C. Huang, K. L. Buehne, N. R. Mayne, I. Weitzhandler, M. Bonn, S. H. Parekh and A. Chilkoti, Nat. Chem., 2018, 10, 496–505 CrossRef CAS.
- C. Yuan, W. Ji, R. Xing, J. Li, E. Gazit and X. Yan, Nat. Rev. Chem., 2019, 3, 567–588 CrossRef CAS.
- L. R. MacFarlane, H. Shaikh, J. D. Garcia-Hernandez, M. Vespa, T. Fukui and I. Manners, Nat. Rev. Mater., 2021, 6, 7–26 CrossRef CAS.
- S. Chen, F. K.-C. Leung, M. C. A. Stuart, C. Wang and B. L. Feringa, J. Am. Chem. Soc., 2020, 142, 10163–10172 CrossRef CAS.
- T. Fukui, S. Kawai, S. Fujinuma, Y. Matsushita, T. Yasuda, T. Sakurai, S. Seki, M. Takeuchi and K. Sugiyasu, Nat. Chem., 2017, 9, 493–499 CrossRef CAS.
- M. Al Kobaisi, S. V. Bhosale, K. Latham, A. M. Raynor and S. V. Bhosale, Chem. Rev., 2016, 116, 11685–11796 CrossRef.
- Y. Cai, Z. Guo, J. Chen, W. Li, L. Zhong, Y. Gao, L. Jiang, L. Chi, H. Tian and W.-H. Zhu, J. Am. Chem. Soc., 2016, 138, 2219–2224 CrossRef CAS.
- J. Li, J. Wang, H. Li, N. Song, D. Wang and B. Z. Tang, Chem. Soc. Rev., 2020, 49, 1144–1172 RSC.
- W. Guan, W. Zhou, C. Lu and B. Z. Tang, Angew. Chem., Int. Ed., 2015, 54, 15160–15164 CrossRef CAS.
- Z. Wang, J. Nie, W. Qin, Q. Hu and B. Z. Tang, Nat. Commun., 2016, 7, 12033 CrossRef.
- R. Dong, B. Zhu, Y. Zhou, D. Yan and X. Zhu, Angew. Chem., Int. Ed., 2012, 51, 11633–11637 CrossRef CAS.
- J. Wang, J. Li, Y. Li, Z. Zhang, L. Wang, D. Wang, L. Su, X. Zhang and B. Z. Tang, Chem. Sci., 2020, 11, 6472–6478 RSC.
- J. Li, K. Liu, Y. Han, B. Z. Tang, J. Huang and Y. Yan, ACS Appl. Mater. Interfaces, 2016, 8, 27987–27995 CrossRef CAS.
- N. Zhang, Y. Wang, R. Wu, C. Xu, J.-J. Nie, N. Zhao, B. Yu, Z. Liu and F.-J. Xu, Small, 2019, 15, 1904017 CrossRef CAS.
- C. Xu, W. Hu, N. Zhang, Y. Qi, J.-J. Nie, N. Zhao, B. Yu and F.-J. Xu, Biomaterials, 2020, 248, 120031 CrossRef CAS.
- Y. Deng, P. Song, X. Chen, Y. Huang, L. Hong, Q. Jin and J. Ji, ACS Nano, 2020, 14, 9711–9727 CrossRef CAS.
- Q. Tang, W. Xiao, C. Huang, W. Si, J. Shao, W. Huang, P. Chen, Q. Zhang and X. Dong, Chem. Mater., 2017, 29, 5216–5224 CrossRef CAS.
- J. Tian, L. Ding, H.-J. Xu, Z. Shen, H. Ju, L. Jia, L. Bao and J.-S. Yu, J. Am. Chem. Soc., 2013, 135, 18850–18858 CrossRef CAS.
- J. F. Lovell, T. W. B. Liu, J. Chen and G. Zheng, Chem. Rev., 2010, 110, 2839–2857 CrossRef CAS.
- M. T. Fenske, W. Meyer-Zaika, H.-G. Korth, H. Vieker, A. Turchanin and C. Schmuck, J. Am. Chem. Soc., 2013, 135, 8342–8349 CrossRef CAS.
- H. Dou, M. Jiang, H. Peng, D. Chen and Y. Hong, Angew. Chem., Int. Ed., 2003, 42, 1516–1519 CrossRef CAS.
- J. Zhan, Y. Cai, S. He, L. Wang and Z. Yang, Angew. Chem., Int. Ed., 2018, 57, 1813–1816 CrossRef CAS.
- Y. Cong, L. Ji, Y.-J. Gao, F.-H. Liu, D.-B. Cheng, Z. Hu, Z.-Y. Qiao and H. Wang, Angew. Chem., Int. Ed., 2019, 58, 4632–4637 CrossRef CAS.
- M. Borkowska, M. Siek, D. V. Kolygina, Y. I. Sobolev, S. Lach, S. Kumar, Y.-K. Cho, K. Kandere-Grzybowska and B. A. Grzybowski, Nat. Nanotechnol., 2020, 15, 331–341 CrossRef CAS.
- X. Li, C. Y. Kim, S. Lee, D. Lee, H.-M. Chung, G. Kim, S.-H. Heo, C. Kim, K.-S. Hong and J. Yoon, J. Am. Chem. Soc., 2017, 139, 10880–10886 CrossRef CAS.
- H. Zhang, X. Zheng, R. T. K. Kwok, J. Wang, N. L. C. Leung, L. Shi, J. Z. Sun, Z. Tang, J. W. Y. Lam, A. Qin and B. Z. Tang, Nat. Commun., 2018, 9, 4961 CrossRef.
- H. Zhang, Z. Zhao, A. T. Turley, L. Wang, P. R. McGonigal, Y. Tu, Y. Li, Z. Wang, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Adv. Mater., 2020, 32, 2001457 CrossRef CAS.
- D. Wang and B. Z. Tang, Acc. Chem. Res., 2019, 52, 2559–2570 CrossRef CAS.
- T. Han, D. Yan, Q. Wu, N. Song, H. Zhang and D. Wang, Chin. J. Chem., 2020, 38 DOI:10.1002/cjoc.202000520.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- B.-P. Jiang, D.-S. Guo, Y.-C. Liu, K.-P. Wang and Y. Liu, ACS Nano, 2014, 8, 1609–1618 CrossRef CAS.
- G. Feng, G.-Q. Zhang and D. Ding, Chem. Soc. Rev., 2020, 49, 8179–8234 RSC.
- C. Chen, X. Ni, H.-W. Tian, Q. Liu, D.-S. Guo and D. Ding, Angew. Chem., Int. Ed., 2020, 59, 10008–10012 CrossRef CAS.
- S. Liu, H. Zhang, Y. Li, J. Liu, L. Du, M. Chen, R. T. K. Kwok, J. W. Y. Lam, D. L. Phillips and B. Z. Tang, Angew. Chem., Int. Ed., 2018, 57, 15189–15193 CrossRef CAS.
- S. Chen, Y. Hong, Y. Liu, J. Liu, C. W. T. Leung, M. Li, R. T. K. Kwok, E. Zhao, J. W. Y. Lam, Y. Yu and B. Z. Tang, J. Am. Chem. Soc., 2013, 135, 4926–4929 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic results about NMR, HR-MS, and UV-vis, fluorescence, computational data, CLSM and other supplemental figures. See DOI: 10.1039/d1bm00044f |
| This journal is © The Royal Society of Chemistry 2021 |