
Anti-freezing flexible aqueous Zn–MnO2 batteries working at −35 °C enabled by a borax-crosslinked polyvinyl alcohol/glycerol gel electrolyte†
Minfeng
Chen
a,
Weijun
Zhou
a,
Anran
Wang
a,
Aixiang
Huang
a,
Jizhang
Chen
 *a,
Junling
Xu
b and
Ching-Ping
Wong
*bc
*a,
Junling
Xu
b and
Ching-Ping
Wong
*bc
aCollege of Materials Science and Engineering, Nanjing Forestry University, Nanjing 210037, China. E-mail: chenjizhang@njfu.edu.cn; jizhang.chen@hotmail.com
bDepartment of Electronic Engineering, The Chinese University of Hong Kong, NT, Hong Kong, China
cSchool of Materials Science and Engineering, Georgia Institute of Technology, Atlanta, USA. E-mail: cp.wong@mse.gatech.edu
First published on 11th March 2020
Abstract
Flexible aqueous zinc-ion batteries (AZIBs) are promising to satisfy the emerging wearable electronics. However, conventional hydrogel electrolytes are unable to work at subzero temperatures because they inevitably freeze. In this work, a borax-crosslinked polyvinyl alcohol (PVA)/glycerol gel electrolyte is developed, in which glycerol can strongly interact with PVA chains, thus effectively prohibiting the formation of ice crystals within the whole gel network. Thanks to this, the freezing point of this gel electrolyte is below −60 °C, which allows it to work in extremely cold environments. Even at −35 °C, it still exhibits a high ionic conductivity of 10.1 mS cm−1 and great mechanical properties. On the basis of this anti-freezing gel electrolyte, a flexible quasi-solid-state aqueous Zn–MnO2 battery is assembled and realizes an impressive energy density of 46.8 mW h cm−3 (1330 μW h cm−2) at a power density of 96 mW cm−3 (2.7 mW cm−2) at 25 °C, outperforming nearly all the reported AZIBs. More importantly, when the temperature is reduced to −35 °C, a rather high energy density (25.8 mW h cm−3, 732 μW h cm−2) can still be achieved, and 53.3% of that value can be retained when the power density is increased to about 10-fold. This battery also shows excellent cycling durability (around 90% capacity retention over 2000 cycles) and great tolerance to various extreme conditions even when the temperature is down to −35 °C. These findings provide valuable insights into designing aqueous batteries/supercapacitors that can work in cold climates and high-altitude areas.
1. Introduction
Flexible energy storage devices represent a key technology for realizing wearable electronics, which have become a hot research topic.1,2 Although Li-ion batteries (LIBs) have dominated the power source market for portable electronics and electric vehicles, they involve volatile, flammable, toxic, and expensive organic electrolytes or rigid and fragile ceramic electrolytes. Therefore, LIBs might be inappropriate for wearable electronics, especially those in close contact with human bodies.3 Thanks to their high safety, low cost, large energy/power densities, and environmental benignity, flexible aqueous zinc-ion batteries (AZIBs) are emerging as advanced and favorable energy storage devices to satisfy the ever-increasing pursuit of wearable electronics, though they are still in their infancy stage.4 As an essential part of flexible AZIBs, the gel electrolyte significantly influences device performances.5 Compared to liquid electrolytes, gel electrolytes offer the following merits. Firstly, gel electrolytes can avoid the leakage issue of liquid electrolytes. Secondly, unlike liquid electrolytes that suffer from undesired dislocation under strain, gel electrolytes can maintain physical integrity and flexibility under various mechanical deformations. Last but not least, gel electrolytes with limited free water and good adsorption affinity are able to mitigate the dissolution of active materials and inhibit the growth of zinc dendrites, therefore contributing to improved electrochemical stability.3,5–9 The general requirements for gel electrolytes are superior mechanical properties and high ionic conductivity, to achieve which the gel electrolyte components should be tuned properly. With the advantages of excellent ion transport capability, high water solubility, great swelling ability, good moisture retainability, non-toxicity, and biocompatibility, poly(vinyl alcohol) (PVA) has been reported by several groups as the polymetric framework of gel electrolytes for flexible AZIBs and aqueous zinc-ion hybrid supercapacitors (AZHSs).10–12 However, the commonly used PVA gel electrolytes prepared by simply mixing PVA solution with zinc salts would suffer from poor mechanical strength, low dimensional stability, limited elasticity, and unsatisfactory malleability.13,14Establishing cross-linked polymeric networks through physical, ionic, or covalent interactions could address the above-mentioned problem to a certain extent. For example, Niu et al. fabricated a physically cross-linked PVA/Zn(CF3SO3)2 gel electrolyte by a freezing-thawing method and assembled a high-performance integrated all-in-one AZIB.15 However, additional filter paper was employed as the separator in that study, probably due to unsatisfactory mechanical properties of PVA/Zn(CF3SO3)2 gel, which are caused by the following aspects.14,16 On the one hand, the PVA crystalline microdomains are insufficient in dissipating external energy, since they are difficult to damage. On the other hand, the steric hindrance of PVA chains impedes the formation of hydrogen bonding. Another effective strategy to enhance the mechanical properties of PVA hydrogels is employing a reversible chemically cross-linker such as borax, which has proven effective for other applications.13,17 Besides PVA, several other polymeric frameworks such as polyacrylamide (PAM),14,18 gelatin,19–21 polyacrylic acid (PAA),22 xanthan gum,23 guar gum,24 carboxymethyl cellulose sodium,25 and poly(ethylene glycol)26 have also been reported for the construction of gel electrolytes of AZIBs and AZHSs. For example, Zhi et al. incorporated a crosslinked porous gelatin/PAM gel electrolyte by grafting PAM onto gelatin chains that are supported with a polyacrylonitrile electrospun fiber membrane and realized good Zn2+ ion conductivity, great flexibility, and high mechanical strength.19 Despite great advances in recent years, the performances of AZIBs based on gel electrolytes are investigated only at room temperature in most of the reports. In fact, the average temperature in winter is below 0 °C in many regions, not to mention the lowest temperature. With massive solvent water, conventional aqueous electrolytes including liquid and gel electrolytes would inevitably freeze at subzero temperatures, thus restricting ion transport and limiting the utilization of AZIBs in cold climates. Therefore, it is necessary and urgent to improve the adaptability of gel electrolytes at low temperatures.
Recently, various freeze-tolerant functional organohydrogels were reported by adding anti-freezing agents such as glycol, glycerol, and sorbitol.27–31 For instance, Lu et al. demonstrated a mussel-inspired anti-freezing and electrically conductive hydrogel by combining the covalent crosslinked PAA-PAM network with polydopamine-decorated carbon nanotubes (CNTs) and introducing a glycerol/water binary solvent as the dispersion medium.30 The hydrogel in that study remains flexible at −20 °C. In the respect of gel electrolytes for AZIBs, Zhi et al. proposed an anti-freezing dually cross-linked gel electrolyte based on glycol-waterborne anionic polyurethane acrylate and PAM (denoted as EG-waPUA/PAM), in which the hydroxyl groups of glycol covalently bind with isocyanate groups.32 The ionic conductivity of the EG-waPUA/PAM hydrogel electrolyte is 16.8 mS cm−1 at room temperature and 86.9% of that value can be retained at −20 °C, because of which a high-performance flexible freeze-resistant Zn–MnO2 battery was realized.32 However, the synthesis procedure of the EG-waPUA/PAM hydrogel electrolyte in that study is complicated and requires N2 gas protection. In this study, we try to modify conventional PVA gel electrolytes by crosslinking PVA and glycerol in the presence of borax. The nontoxic and biocompatible glycerol can interact with water molecules strongly via hydrogen bonds. As a result, the hydrogen bonds among water molecules would be disrupted, thus hindering the formation of ice crystals and lowering the freezing point.16,29,30 It is noteworthy that the synergistic effect of borax and glycerol plays a significant role in considerably enhancing the anti-freezing and mechanical properties of the obtained gel electrolyte (denoted as PVA-B-G) over the counterparts. Benefiting from this, the quasi-solid-state aqueous Zn–MnO2 battery based on PVA-B-G delivers great performances at subzero temperatures even under mechanical deformation. Remarkably, at −35 °C, a rather high energy density of 25.8 mW h cm−3 (732 μW h cm−2) can still be realized, corresponding to 55.0% of that at 25 °C. Despite this, such a performance at −35 °C rivals most of the previously reported flexible AZIBs operated at room temperature.
2. Experimental
2.1. Preparation of the PVA-B-G gel electrolyte
4 g of PVA powder (Mw = 85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–124000, 99.0% hydrolyzed) was added into 50 mL deionized water under constant stirring at 90 °C until PVA was swelled and dissolved completely. In another beaker, 2 g glycerol was dissolved into 10 mL aqueous solution containing 2 M ZnSO4 and 0.2 M MnSO4, followed by the addition of 0.4 g borax (Na2B4O7·10 H2O) under stirring at 60 °C for 0.5 h. The above-mentioned two solutions were mixed together, heated to 90 °C, and stirred vigorously at that temperature for 2 h. After this, the mixture was poured into tailor-made moulds to generate the PVA-B-G gel electrolyte with a gelling time of 4 h. For comparison, a PVA-B gel electrolyte was synthesized by the same procedure except for that glycerol was not added and the amount of PVA was increased by 2.0 g. And the PVA/G gel electrolyte was produced without the addition of borax while other conditions were kept the same as those of PVA-B-G.
000–124000, 99.0% hydrolyzed) was added into 50 mL deionized water under constant stirring at 90 °C until PVA was swelled and dissolved completely. In another beaker, 2 g glycerol was dissolved into 10 mL aqueous solution containing 2 M ZnSO4 and 0.2 M MnSO4, followed by the addition of 0.4 g borax (Na2B4O7·10 H2O) under stirring at 60 °C for 0.5 h. The above-mentioned two solutions were mixed together, heated to 90 °C, and stirred vigorously at that temperature for 2 h. After this, the mixture was poured into tailor-made moulds to generate the PVA-B-G gel electrolyte with a gelling time of 4 h. For comparison, a PVA-B gel electrolyte was synthesized by the same procedure except for that glycerol was not added and the amount of PVA was increased by 2.0 g. And the PVA/G gel electrolyte was produced without the addition of borax while other conditions were kept the same as those of PVA-B-G.
2.2. Characterization
The samples were characterized by using an Ultima IV powder X-ray diffractometer (XRD) with a Cu Kα radiation source (λ = 0.1540598 nm, Rigaku), VERTEX 80V Fourier transform infrared spectrometer (FTIR, ATR mode, Bruker), JSM-7600F field emission scanning electron microscope (FE-SEM, JEOL), JEM-2100F transmission electron microscope (TEM, JEOL) equipped with an energy dispersive X-ray spectroscopy (EDX) detector, and UTM2502 universal testing machine (SANS). The freezing point was determined with a DSC8500 differential scan calorimeter (DSC, PerkinElmer) and Q800 dynamic thermomechanical analyzer (DMA, TA Instruments). The DSC analysis was performed from 30 to −120 °C at a cooling/heating rate of 5 °C min−1. The DMA measurements were conducted in a compression mode over the temperature range of −120 to 35 °C with a heating rate of 3 °C min−1 at γ = 1% and ω = 1 Hz. The measurements of ionic conductivity are shown in the ESI.†2.3. Electrochemical measurements
The rGO/MnO2 cathode material was synthesized by a hydrothermal method (described in the ESI†). It was mixed with Super-P carbon black and polyvinylidene fluoride (PVDF) at a weight ratio of 7:2:1 in N-methylpyrrolidone (NMP). The obtained slurry was coated onto Ti foil and put into an oven at 80 °C overnight. The mass loading of rGO/MnO2 on the Ti foil is around 4 mg cm−2. The flexible quasi-solid-state AZIBs were assembled by sandwiching gel electrolytes between the rGO/MnO2 cathode and Zn foil anode. For comparison, CR2016-type coin cells were assembled to evaluate the AZIBs with a liquid electrolyte by replacing the gel electrolyte with a separator filled with 2 M ZnSO4/0.2 M MnSO4 aqueous solution. Galvanostatic charge/discharge (GCD) measurements were carried out on a CT2001A battery testing system (Wuhan LAND). Cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) measurements were performed on a VSP-300 electrochemical workstation (Bio-Logic). All the electrochemical measurements were conducted in a BPH-060B high–low temperature chamber (Shanghai Yiheng).
3. Results and discussion
The fabrication procedure of the PVA-B-G gel electrolyte is shown in Fig. 1a. A glycerol–water binary solution containing ZnSO4 and MnSO4 is mixed with a PVA dispersion, accompanied by the employment of borax, which is readily hydrolyzed in water to dissociate into boric acid (B(OH)3) and borate ions (B(OH)4−), as shown in the following equations:| Na2B4O7 + 7H2O → 2B(OH)3 + 2B(OH)4− + 2Na+ | (1) |
| B(OH)3 + 2H2O ⇄ B(OH)4− + H3O+ | (2) |
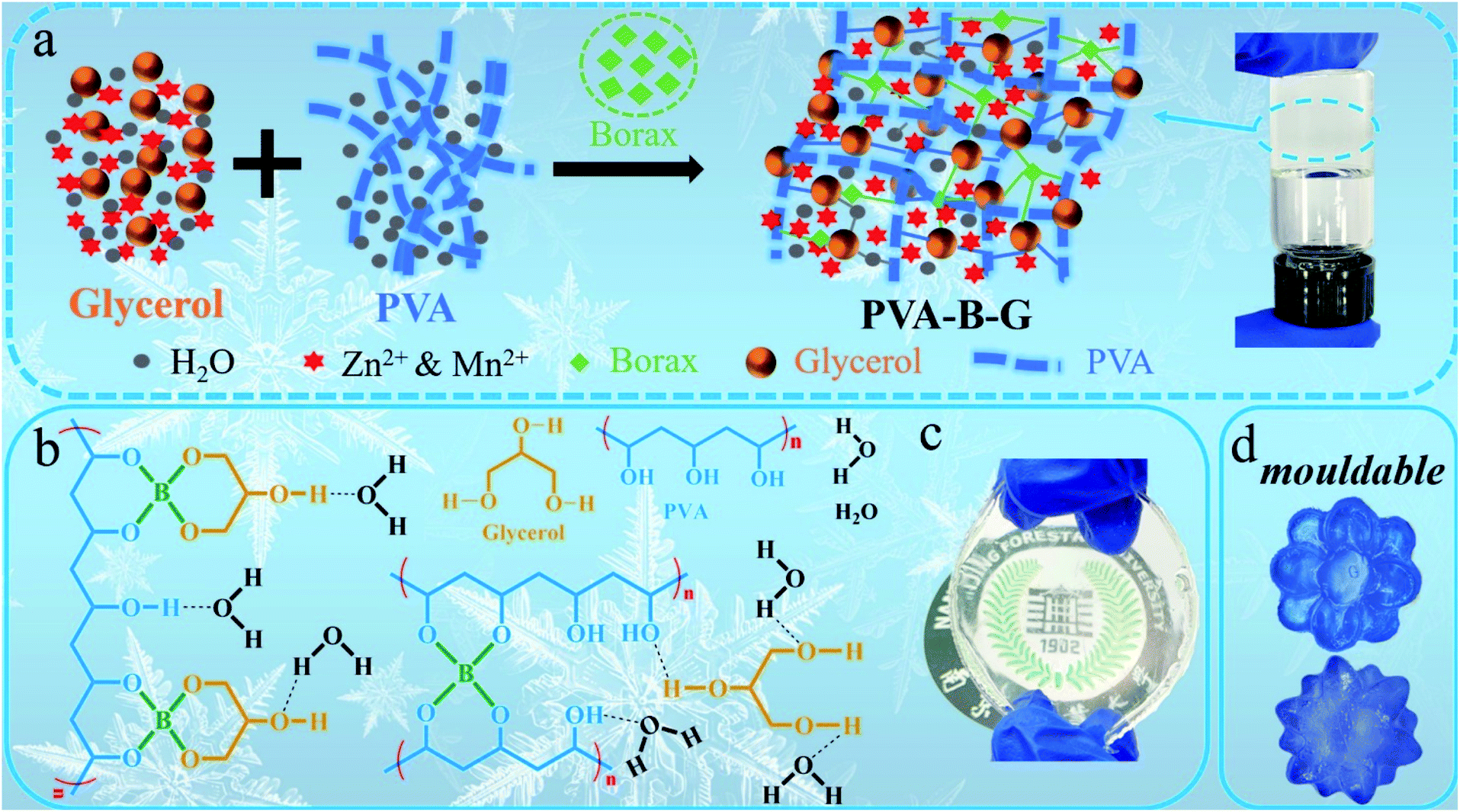 | ||
| Fig. 1 (a) The synthesis schematic of PVA-B-G. (b) Multi-complexation of the integrated 3D network within PVA-B-G. Photographs of (c) PVA-B-G film and (d) PVA-B-G moulded into desirable shapes. | ||
The B(OH)4− ions can easily crosslink PVA and glycerol via “di-diol” complexation.13,17,33 Besides, abundant hydrogen bonds are formed among PVA, glycerol, and water molecules. Thus, an integrated three-dimensional (3D) network is formed, as illustrated in Fig. 1a and b. The obtained PVA-B-G appears to be a very stable and uniform gel (see the photographs in Fig. 1a and c). It should be mentioned that the addition of MnSO4 into electrolytes of AZIBs can effectively inhibit the dissolution of MnO2 active materials during the electrochemical process, thus achieving better long-term cyclability.34 Notably, the synthesis process of PVA-B-G in this study is greatly simplified in comparison with the previously reported anti-freezing EG-waPUA/PAM gel electrolyte for AZIBs.32 We also avoided the electrolyte immersion step. In some other reports, the as-prepared hydrogels must be immersed in liquid electrolytes for a long time (usually 12–72 h) to absorb electrolyte ions.14,18,22,35 Such a time-consuming process is no longer required for PVA-B-G.
XRD and FTIR measurements were conducted to confirm the multi-complexation within PVA-B-G. The pure PVA raw material exhibits two diffraction peaks at 19.6° and 22.7° (Fig. S1†), which are assigned to the (101) semi-crystalline and (112) crystalline planes, respectively. As for PVA-B, PVA/G, and PVA-B-G, the corresponding diffraction peaks become much broader than those of PVA, indicating that either borax or glycerol can disrupt PVA–PVA interactions. Therefore, it is anticipated that the organized configuration of PVA could be considerably destroyed within PVA-B-G when both borax and glycerol are introduced. Fig. S1† also presents the XRD profile of ZnSO4/MnSO4, which was obtained by freeze drying an aqueous solution containing 2 M ZnSO4 and 0.2 M MnSO4. A series of sharp peaks are observed in ZnSO4/MnSO4, whereas these peaks are absent in the three gel electrolytes (freeze-dried before XRD measurements), indicating that Zn2+, Mn2+, and SO42− ions are homogeneously distributed in the gel electrolytes and these ions are unable to crystallize during freeze drying in the presence of the polymeric framework due to which the abundant polymer chains impede the diffusion of these ions when the ice crystals are evaporated. Similar to XRD, the FTIR response of ZnSO4/MnSO4 is also not reflected in freeze dried gel electrolytes (Fig. S2†). Furthermore, the bands arising from O–H stretching (∼3280 cm−1) and O–H bending (∼1640 cm−1) in PVA-B and PVA-B-G are dramatically weakened in comparison with those of pure PVA, indicating that borax-mediated “di-diol” complexation is rather effective.36
As can be seen from Fig. 1c, the PVA-B-G exhibits a transparent state. In contrast, the PVA-B and PVA/G are nearly lightproof (Fig. S3†), which might be ascribed to the existence of a large amount of PVA crystalline microdomains in these two samples.15,31 That is, the interaction energy within PVA molecular chains is so large that neither borax nor glycerol alone can effectively destroy hydrogen bonds within the crystalline microdomains. When both borax and glycerol are added, glycerol can interact with PVA chains strongly through borax-mediated crosslinking, therefore significantly eliminating PVA crystalline microdomains, which is beneficial for Zn2+ transport.15 The remoldability and reusability are important features for practical applications of gel electrolytes. In this respect, the PVA-B-G was pre-dyed with a blue ink for easy observation. As shown in Fig. 1d, the PVA-B-G can be remolded into complicated shapes at room temperature without heating. This benefit is derived from the dynamic and reversible borax-mediated crosslinking as well as hydrogen bonds. In particular, the “di-diol” complexation of borax-mediated crosslinking is very fast and could be readily broken and reformed when the PVA-B-G is deformed.13,17,33 The PVA-B-G was freeze dried before SEM characterization. It is seen from Fig. S4† that PVA-B-G possesses a 3D hierarchically porous architecture with open channels, which can accommodate sufficient electrolyte solvent within the gel network, therefore facilitating ion transport.
For practical applications, the gel electrolytes must be tough enough even after undergoing subzero temperatures. In our frost resistance tests, the PVA/G and PVA-B-G samples were placed at 25 or −35 °C in a high–low temperature chamber for 24 h and then tested immediately after being taken out. These two samples look similar at 25 °C, and both can be twisted easily without breakage, as shown in Fig. 2a and b. However, when the PVA/G was taken out from the −35 °C environment, it became hard and brittle, and obvious white ice crystals appeared on its surface. What's more, the ice-like PVA/G solid would be easily broken under bending or folding. On the contrary, the PVA-B-G is still soft and flexible at −35 °C and can be readily bent and folded without any cracking, demonstrating an excellent anti-freezing function under extremely cold conditions.
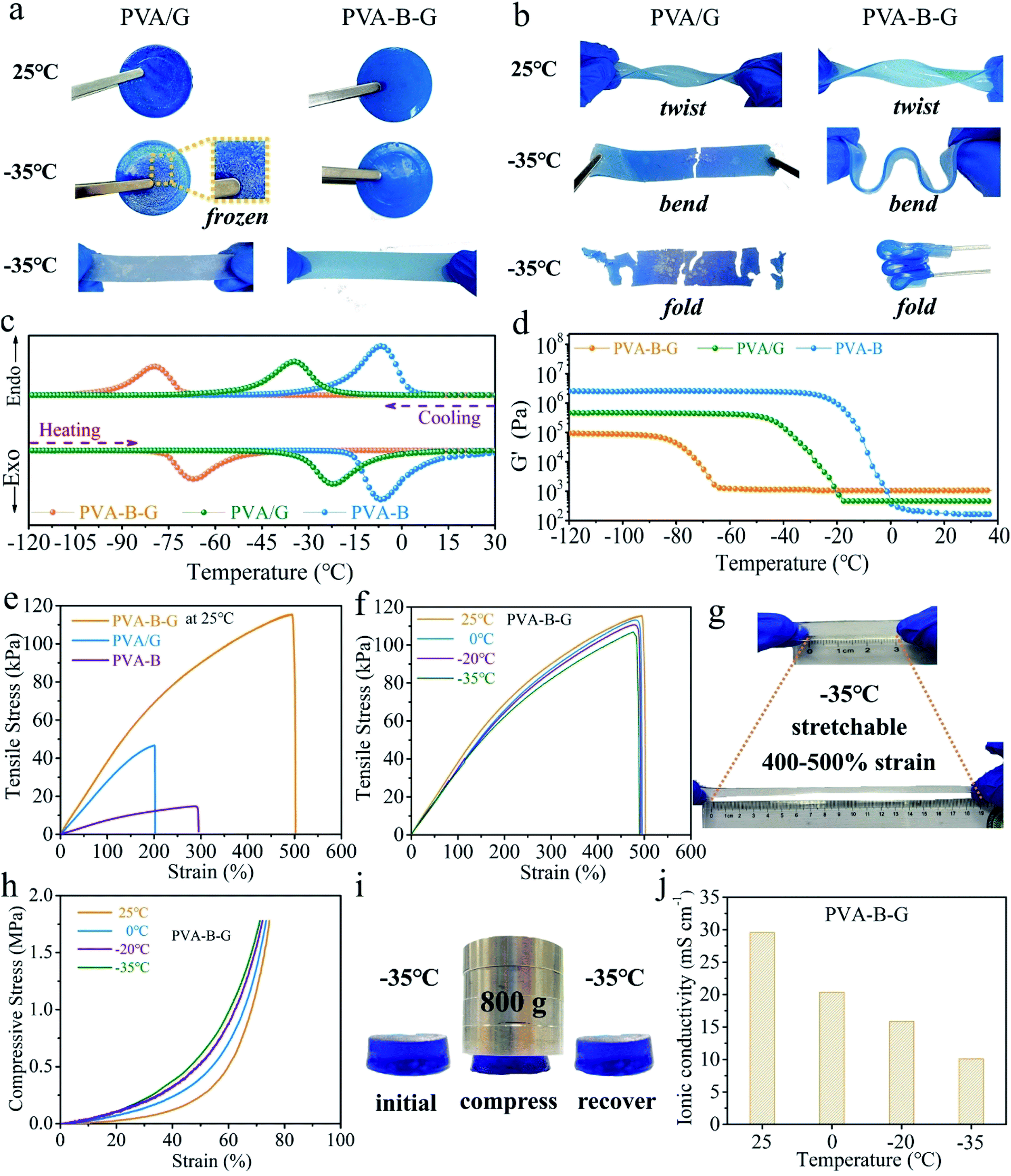 | ||
| Fig. 2 Photographs of PVA/G and PVA-B-G at 25 °C and −35 °C: (a) at the normal state and (b) under twisting, bending, and folding. (c) DSC curves, (d) G′ values with respect to the temperature, and (e) tensile stress–strain curves at 25 °C of PVA-B-G, PVA/G, and PVA-B. (f) Tensile stress–strain curves of PVA-B-G at different temperatures. (g) Stretching demonstration of PVA-B-G at −35 °C. (h) Compressive stress–strain curves of PVA-B-G at different temperatures. (i) Photographs of PVA-B-G at initial, compressed, and released states at −35 °C. (j) The ionic conductivity of PVA-B-G as a function of temperature. | ||
In order to further confirm the great anti-freezing ability of PVA-B-G, DSC and DMA measurements were conducted. The DSC curves of three samples (PVA-B-G, PVA/G, and PVA-B) are shown in Fig. 2c, in which the cooling branch is used to determine the freezing point. For PVA-B gel, a peak is observed at −7.5 °C, which is ascribed to the freezing of water in the gel. The deviation of the freezing point from conventional 0 °C originates from the following two aspects. Firstly, PVA can interact with water molecules through hydrogen bonding, although the interaction is insufficient due to the steric hindrance of PVA chains. Secondly, the presence of inorganic salts (ZnSO4 and MnSO4 in this study) can inhibit ice crystallization.29,37 On the other hand, the freezing point of PVA/G goes down to −34 °C. Such a dramatic decline is due to the decreased saturated vapor pressure of water as a result of the formation of abundant hydrogen bonds between small glycerol molecules and water molecules. Yet as discussed earlier, there exist lots of PVA crystalline microdomains in the PVA/G. The water within and surrounding these crystalline microdomains cannot form efficient hydrogen bonds with glycerol. Hence, it is possible to further decrease the freezing point if these crystalline microdomains are removed, which could be realized by the simultaneous addition of borax and glycerol. In the presence of borax, glycerol can crosslink with PVA chains to a large content, thus destroying PVA crystalline microdomains. In this way, the anti-freezing role of glycerol can be brought into full play. Consequently, the PVA-B-G exhibits an ultralow freezing point of −79 °C. It is also seen that the peak differences (ΔT) between cooling and heating branches are 12.7, 12.5, and 0.6 °C for PVA-B-G, PVA/G, and PVA-B, respectively. The larger ΔT might be ascribed to stronger hydrogen bonds between water molecules and other components. To further explore this concept, the storage modulus (G′) values of the three samples with respect to the temperature are plotted in Fig. 2d, where the G′ begins to increase abruptly indicating the beginning of water freezing. Below that temperature, the samples begin to turn into fragile ice-like solids. Interestingly, the G′ of PVA-B-G only increases by 15% when the temperature is down to −60 °C. Comparatively, the G′ of PVA-B at −20 °C is 5789 times that at 25 °C.
The mechanical properties were also evaluated. In Fig. 2e, the PVA-B presents a relatively low tensile strength of 14.7 kPa and a large fracture elongation of 295%. Such a low strength is ascribed to the large number of PVA crystalline microdomains. As for the PVA/G, the glycerol can form abundant hydrogen bonds with PVA chains. Accordingly, the tensile strength of PVA/G increases to 46.7 kPa. Nonetheless, the glycerol is composed of small molecules. As a result, the fracture elongation of PVA/G decreases to 202%. Furthermore, when both borax and glycerol are incorporated, the obtained PVA-B-G is drastically reinforced to 115.5 kPa, which is 2.47 times that of PVA/G. Besides, the fracture elongation is dramatically enhanced to 502%. Such significant improvements are attributed to the synergistic effect of borax-mediated crosslinking and H-bonding.18 As discussed earlier, the PVA crystalline microdomains are effectively relieved in the PVA-B-G, which favors strength improvement. Besides, the easy breaking and reformation of borax-crosslinking and H-bonding in the integrated 3D network provide an effective energy-dissipation mechanism. When the PVA-B-G is deformed under an external force, the network collapses rapidly while the PVA chains slip and rearrange, accompanied by quick reformation of the network, therefore acquiring great flexibility, stretchability, toughness, and mouldability.13,17,33
As shown in Fig. 2f, comparing the stress–strain curves of PVA-B-G at different temperatures, it can be found that the tensile properties evolve slightly as the temperature declines. For example, at −35 °C, the tensile strength and fracture elongation are 106.9 kPa and 490%, corresponding to 92.6% and 97.6% at 25 °C, respectively. It is clearly seen from the photograph in Fig. 2g that at −35 °C the PVA-B-G can be easily stretched to 400–500% strain with no visible cracking. Compressive measurements were also conducted (Fig. 2h). These tests were terminated when a maximum force of 500 N was reached. Obviously, only slight change of the compression behavior can be observed when the temperature was decreased from 25 to −35 °C. The above results corroborate great anti-freezing properties of PVA-B-G. To further confirm this, the PVA-B-G was loaded by 800 g weight immediately when it was taken out from the −35 °C environment (Fig. 2i). If the solvent within a gel is frozen, the gel would be hard and brittle. As a result, it would be either unchanged or crushed under compression. Encouragingly, the PVA-B-G shrank without fracture when it is compressed and recovered to its original shape once the external force was withdrawn, unveiling good elasticity at such a low temperature. Thus, on the basis of the above results, it's clearly known that PVA-B-G can still maintain an unfrozen state at −35 °C.
As mentioned above, the PVA-B-G has an ultralow freezing point. Hence, it is reasonable to anticipate high ionic conductivities at low temperatures. The ionic conductivity values of PVA-B-G as a function of the temperature were investigated by EIS measurements, and the obtained Nyquist plots are shown in Fig. S5a.† The ionic conductivity of PVA-B-G is calculated to be 29.6 mS cm−1 at 25 °C, which is much higher than that of many previously reported gel electrolytes for AZIBs/AZHSs, as shown in Table S1.† When the temperature declines to 0, −20, and −35 °C, the ionic conductivity of PVA-B-G drops to 20.4, 15.9, and 10.1 mS cm−1, respectively (Fig. 2j). Impressively, to the best of our knowledge, the ionic conductivity of PVA-B-G at −20 °C ranks the highest one among all the reported gel electrolytes for AZIBs/AZHSs at subzero temperatures. Although the conductivity retention at −35 °C is not very high (34.1% as compared to 25 °C), the conductivity value at −35 °C is even higher than that of several gel electrolytes for AZIBs/AZHSs at room temperature (see Table S1†). For comparison, the liquid electrolyte containing 2 M ZnSO4 and 0.2 M MnSO4 was also evaluated (Fig. S5b†). It is not surprising that the ionic conductivity of the liquid electrolyte at 25 °C (96.3 mS cm−1) is much higher than that of PVA-B-G. Unfortunately, the ionic conductivity of the liquid electrolyte substantially falls to merely 1.1 and 0.2 mS cm−1 at −20 and −35 °C, respectively (Fig. S6†), due to water freezing. These results firmly validate the great applicability of PVA-B-G as the anti-freezing electrolyte, while conventional liquid electrolytes would fail at low temperatures.
AZIB devices were assembled to gain more insight into the benefits of anti-freezing PVA-B-G. So far, various cathode materials have been reported for AZIBs, including Mn-based oxides,38–43 V-based materials,44–55 Prussian blue analogues,56 Mo-based compounds,57,58 polyanionic compounds,59 and organic and polymer compounds.60,61 Considering that MnO2 gives relatively high working potential and large specific capacity, we used rGO/MnO2 nanocomposites synthesized from the hydrothermal reaction as the cathode material. All the XRD peaks of rGO/MnO2 in Fig. S7† can be well indexed as α-MnO2 (JCPDS: 44-0141). The broadening of these diffraction peaks implies small sizes of α-MnO2. Owing to the low content and poor crystallinity of rGO, its existence in the nanocomposite is not evident by XRD characterization.
As illustrated in Fig. 3a, MnO2 nanobelts are grown on a curved rGO scaffold. Such a morphology and structure are verified by the SEM, TEM, and EDX mapping images in Fig. S8 and S9.† The MnO2 nanobelts are so small in size that charge storage within MnO2 is favored owing to the large contact with the electrolyte and short diffusion paths for both electrolyte ions and electrons. As shown in Fig. 3b and c, a planar thin-film quasi-solid-state battery was assembled using rGO/MnO2 as the cathode material (coated on Ti foil), PVA-B-G (or PVA, PVA/G) as the electrolyte, and Zn foil as the anode. Unlike many other reports about quasi-solid-state AZIBs that electrodeposited Zn onto an extra conductive substrate as the anode, here we directly utilized Zn foil. Given that Zn foil is much cheaper than widely used carbon cloth, it would be more meaningful if our device can provide similar or better performances in comparison with other reports. The resultant PVA-B-G battery is flexible (Fig. 3c) and merely 0.284 mm in thickness (Fig. S10†).
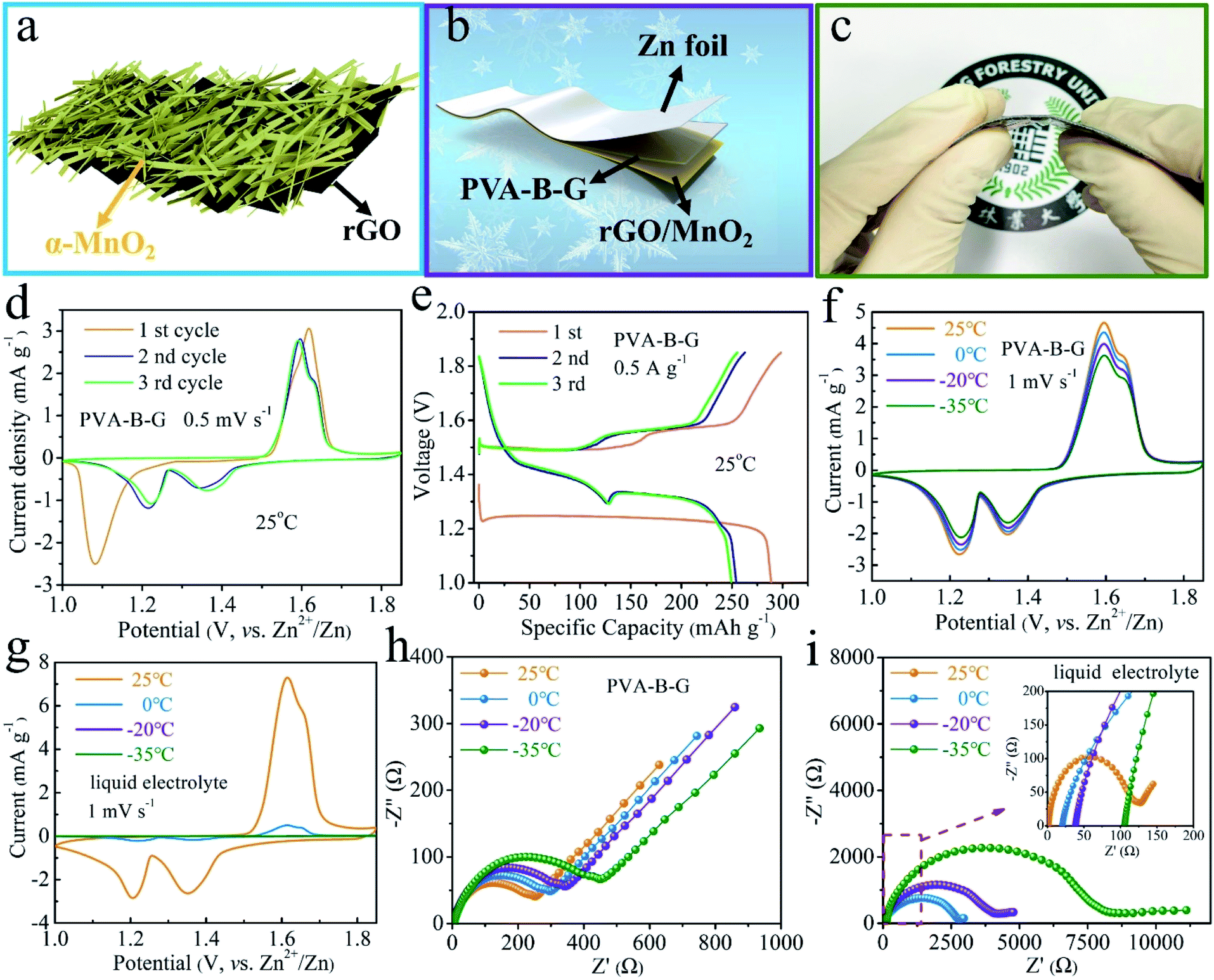 | ||
| Fig. 3 (a) Graphic illustration of the rGO/MnO2 nanocomposite. (b) Schematic diagram, (c) photograph, (d) CV curves of the initial three cycles at 0.5 mV s−1 at 25 °C, and (e) GCD curves of the initial three cycles at 0.5 A g−1 at 25 °C of the PVA-B-G battery. (f and g) CV curves at 1 mV s−1 and (h and i) Nyquist plots of (f and h) PVA-B-G battery and (g and i) liquid-electrolyte battery at different temperatures. | ||
The electrochemical behavior of the PVA-B-G battery was first studied by CV at 0.5 mV s−1 in the potential range of 1–1.85 V vs. Zn2+/Zn at 25 °C. As can be seen from Fig. 3d, a cathodic peak at 1.08 V is observed in the first cycle, while two cathodic peaks at ∼1.35 and ∼1.22 V appear in the following cycles. The change in the peak number and position is ascribed to the phase transition and morphology evolution during the first cycle.34,35,62 In the anodic branches, two overlapped peaks at 1.59 and 1.62 V are observed. These results demonstrate a two-step charge storage mechanism after the first discharging process. Besides, the CV profiles in the second and third cycles nearly coincide, indicative of great electrochemical reversibility. The GCD curves of the PVA-B-G battery in the initial three cycles at 0.5 A g−1 at 25 °C are shown in Fig. 3e and are in good consistent with the CV results in Fig. 3d. In the 2nd and 3rd cycles, the first discharge plateau at around 1.4 V results from H+ insertion, while the second discharge plateau at around 1.3 V comes from subsequent Zn2+ intercalation.34,62,63 The distinct bound between two plateaus arises from larger charge transfer resistance of Zn2+ intercalation with respective to H+. It is also seen that the PVA-B-G battery delivers an initial discharge capacity of 288.9 mA h g−1, which decreases to 254.2 and 249.5 mA h g−1 in the 2nd and 3rd cycles, respectively. Correspondingly, the coulombic efficiency stays above 96% in the initial three cycles, reflecting great electrochemical stability.
The temperature-dependent electrochemical properties of the PVA-B-G battery were first examined by CV at 1 mV s−1 (Fig. 3f). The batteries were put inside a high–low temperature chamber for tests. When the temperature was gradually decreased from 25 to −35 °C, the CV peaks remain well-resolved, the peak positions remain nearly unchanged, and the peak current only declines slightly. Impressively, around 80% of the peak current can be sustained at −35 °C compared to 25 °C. These results manifest excellent freezing tolerant ability of PVA-B-G and fast Zn2+/H+ intercalation kinetics of rGO/MnO2 at subzero temperatures. For comparison, the liquid electrolyte was also used to assemble AZIBs. In this case, CR2016-type coin cells were assembled and an extra separator was introduced. As shown in Fig. 3g, the CV peaks of the liquid-electrolyte battery are drastically weakened at 0 °C. Furthermore, these peaks vanished at −20 and −35 °C, resulting from the freezing of the liquid electrolyte. To gain more insight, EIS measurements were carried out at different temperatures, and the collected Nyquist plots are shown in Fig. 3h and i. In particular, the semicircle in the high frequency region represents charge transfer resistance (Rct) at the interface between the electrolyte and electrodes. Upon cooling from 25 to −35 °C, the Rct of the PVA-B-G battery increases from 238 to 453 Ω with 90.3% increasement. In contrast, for the liquid-electrolyte battery, the Rct increases by 6232% at −35 °C. These results lead us to conclude that the PVA-B-G can still offer high ionic conductivity and guarantee fast reaction kinetics of the electrodes when the temperature is down to −35 °C, while liquid electrolytes cannot withstand low-temperature environments.
As shown in Fig. 4a, the rate performances of AZIBs were investigated at 25 °C. It is not surprising that the liquid-electrolyte battery possesses the highest capacities at all the current densities among all four types of batteries, since the ionic conductivity of the liquid electrolyte vastly exceeds that of the other electrolytes at 25 °C. At the end of each rate, the liquid-electrolyte battery gives a high discharge capacity of 261.0 mA h g−1 at 0.5 A g−1, and 154.9 mA h g−1 is retained at 5 A g−1. However, when the temperature is 0 °C, the discharge capacity at 0.5 A g−1 of the liquid-electrolyte battery falls down to merely 33.5 mA h g−1 (Fig. 4b), which becomes negligible (0.1 mA h g−1) at −20 and −35 °C (Fig. 4c and d), due to the inevitable freezing of the liquid electrolyte at subzero temperatures. That is, conventional AZIBs with liquid electrolytes are unable to work in low-temperature environments.
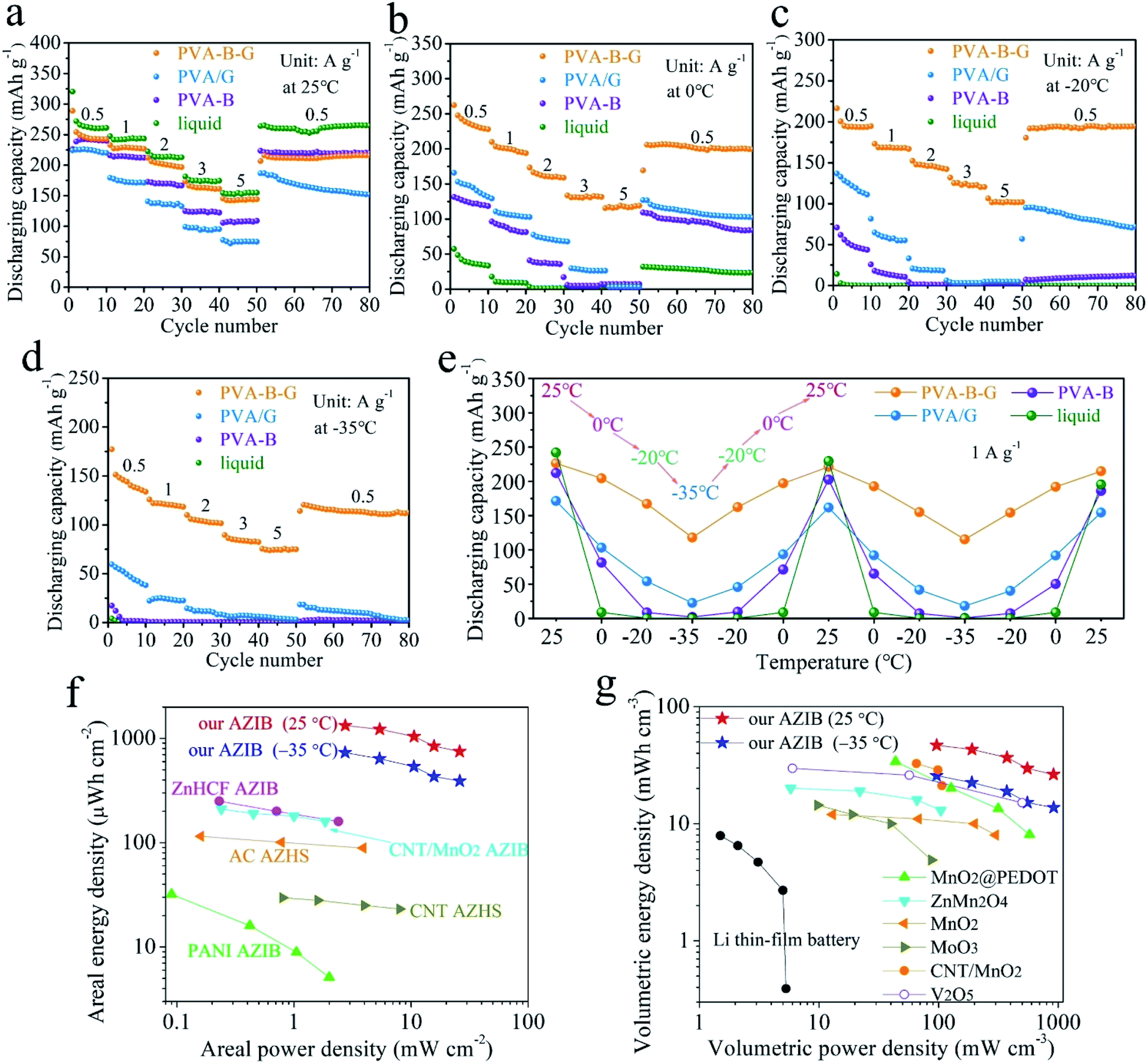 | ||
| Fig. 4 Rate performances of PVA-B-G, PVA/G, PVA-B, and liquid-electrolyte batteries: (a) at 25 °C, (b) at 0 °C, (c) at −20 oC, and (d) at −35 oC. (e) Cyclic testing results of four types of batteries at 25, 0, −20 °C, and −35 °C at 1 A g−1. (f) Areal and (g) volumetric Ragone plots of our PVA-B-G battery, in comparison with previously reported energy storage devices. | ||
As for the PVA-B-G battery, it delivers 242.5 mA h g−1 at 0.5 A g−1 at 25 °C (Fig. 4a). As the current density is progressively increased to 1, 2, 3, and 5 A g−1, the specific capacity gradually drops to 226.6, 196.6, 161.1, and 143.9 mA h g−1, respectively. That is, the capacity retention of the PVA-B-G battery approaches 60% when the current density is increased to 10-fold, disclosing rapid charge transports within both PVA-B-G and rGO/MnO2. The GCD curves of the PVA-B-G battery at different current densities are shown in Fig. S11.† The electrochemical polarization is not severe even at 5 A g−1, indicative of great kinetics. It is also seen that during the discharging process, the H+ insertion region contracts mildly as the current density increases, while the Zn2+ intercalation region severely shrinks, revealing that the H+ insertion is more kinetically favored. The electrochemical kinetics were further analyzed by CV at various scan rates (Fig. S12†), indicating that the capacitive contribution occupies more than half of the total capacity, which implies fast charge storage kinetics.
When the temperature is reduced to 0 °C, at 0.5 A g−1, the PVA-B-G battery delivers 228 mA h g−1 (Fig. 4b), which goes down to 194.6 and 133.8 mA h g−1 at −20 and −35 °C (Fig. 4c and d), corresponding to 80.2% and 55.2% at 25 °C, respectively. Such high capacity retentions at low temperatures are much superior to previously reported anti-freezing Zn-based batteries.64,65 What's more, the PVA-B-G battery can still provide great rate capability at 0, −20, and −35 °C. For example, at −35 °C, the specific capacity of the PVA-B-G battery at 1, 2, 3, and 5 A g−1 retains 88.5%, 75.9%, 61.5%, and 56.0% of that at 0.5 A g−1, respectively (Fig. 4d). Besides, after cycling back to 0.5 A g−1, the specific capacity of the PVA-B-G battery recovers to ∼120 mA h g−1, demonstrating good reversibility. It is worth mentioning that the PVA-B-G battery delivers high coulombic efficiencies of 96.82%, 96.71%, 96.56%, and 96.21% at 25, 0, −20, and −35 °C, respectively (Fig. S13†). After around 3 cycles, the coulombic efficiency begins to approach 100%, further confirming good reversibility. We can also see from Fig. 4 that the low-temperature performances of PVA-B-G significantly outperform PVA/G and PVA-B batteries, since PVA-B-G has a much lower freezing point than the two counterparts. However, the low-temperature tolerant properties of PVA/G and PVA-B batteries are obviously better than those of the liquid-electrolyte battery.
The GCD curves of the PVA-B-G battery at 1 A g−1 at different temperatures are shown in Fig. S14.† No obvious change in the plateau voltage can be observed, indicating great potential of working at subzero temperatures. To further examine the ability of enduring cold environments, the PVA-B-G battery was cycled at 1 A g−1 when the working temperature was changed among 25, 0, −20, and −35 °C in succession (10 cycles at each temperature, Fig. S15†). The specific capacities (plotted in Fig. 4e) of the PVA-B-G battery at different temperatures are similar to those in Fig. 4a–d. In addition, no evident deterioration in the capacity can be identified after the PVA-B-G battery underwent two freezing/heating cycles (totally 130 cycles), suggesting excellent anti-freezing stability and durability. Furthermore, by comparing the performances of four batteries, we can clearly see that the PVA-B-G battery manifests the best capacity restoring capability.
The areal and volumetric energy/power densities are important parameters, since the space occupied by energy storage devices is often limited. On the basis of discharge curves, the areal and volumetric Ragone plots of the PVA-B-G battery at 25 and −35 °C are calculated and presented in Fig. 4f and g, respectively. Note that the volumes of PVA-B-G, Ti foil (including rGO/MnO2 and other components), and Zn foil are all considered into the total volume of our device. It is also worth mentioning that the performances of other energy storage devices in Fig. 4f and g were measured at room temperature (probably 25 or 20 °C). It can be seen that our PVA-B-G battery exhibits an extraordinary areal energy density of 1330 μW h cm−2 when the power density is 2.7 mW cm−2, and retains 749 μW h cm−2 at 26 mW cm−2 (25 °C). When the temperature is reduced to −35 °C, our PVA-B-G battery can still deliver a marvelous areal energy density of 732 μW h cm−2 at 2.7 mW cm−2, which dramatically surpasses that of previously reported AZIB and AZHS devices, such as zinc hexacyanoferrate (ZnHCF)-based AZIBs (250 μW h cm−2 at 0.23 mW cm−2),25 CNT/MnO2-based AZIBs (210 μW h cm−2 at 0.24 mW cm−2),23 polyaniline (PANI)-based AZIBs (32 μW h cm−2 at 0.09 mW cm−2),66 activated carbon (AC)-based AZHSs (115 μW h cm−2 at 0.16 mW cm−2),67 CNT-based AZHSs (30 μW h cm−2 at 0.8 mW cm−2),12 and our recently reported AC-based AZHS (180 μW h cm−2 at 0.4 mW cm−2).36 As shown in Fig. 4g, our PVA-B-G battery also exhibits a satisfactory volumetric energy density of 46.8 mW h cm−3 when the power density is 96 mW cm−3, and retains 26.4 mW h cm−3 at 916 mW cm−3. Such a performance greatly exceeds that of Li thin-film batteries,68 our recently reported AC-based AZHS (6.2 mW h cm−3 at 13.9 mW cm−3),36 and previously reported state-of-the-art quasi-solid-state AZIB devices in the literature, including MnO2@poly(3,4-ethylenedioxythiophene) (PEDOT)-based AZIBs (34.0 mW h cm−3 at 43.7 mW cm−3),10 ZnMn2O4-based AZIBs (20.1 mW h cm−3 at 5.8 mW cm−3),69 MnO2-based AZIBs (12 mW h cm−3 at 13 mW cm−3),70 MoO3-based AZIBs (14.4 mW h cm−3 at 9.8 mW cm−3),71 CNT/MnO2-based AZIBs (32.7 mW h cm−3 at 65.4 mW cm−3),32 and V2O5-based AZIBs (29.7 mW h cm−3 at 6 mW cm−3).72 Moreover, when the temperature is decreased to −35 °C, our PVA-B-G battery is still able to provide high volumetric energy densities of 25.8 mW h cm−3 at 96 mW cm−3 and 13.8 mW h cm−3 at 918 mW cm−3, outperforming many previously reported AZIB devices operated at room temperature. The above results not only unveil the superiority of our PVA-B-G battery but also further confirm that it can work smoothly even at a rather low temperature of −35 °C.
The cycling stability of the PVA-B-G battery at 1 A g−1 at different temperatures was also evaluated, as shown in Fig. 5a. Relatively fast capacity fading is observed in the initial 100 cycles. After this, the capacity becomes rather stable in the following cycles. After 2000 cycles, the discharge capacity of the PVA-B-G battery retains 93.7%, 91.9%, 87.0%, and 89.4% that of the 1st cycle at 25, 0, −20, and −35 °C, respectively. Besides, the coulombic efficiency of the PVA-B-G battery approaches 100% throughout all the cycles at different temperatures (Fig. S16†). Compared with the PVA-B-G battery, the liquid-electrolyte battery exhibits much inferior cycling stability with a low capacity retention of 72.6% after 2000 cycles at 25 °C (Fig. S17†). Such a rapid capacity decay with the liquid electrolyte is ascribed to the following two factors. On the one hand, the heterogeneous nucleation and deposition of Zn at the interface between the anode and liquid electrolyte lead to the formation of vertical and harsh Zn dendrites.73 On the other hand, the rGO/MnO2 cathode materials would be partially dissolved in the liquid electrolyte due to the Mn3+ disproportionation reaction.34 Instead, when the PVA-B-G was used to replace the liquid electrolyte, these two effects can be considerably suppressed, because the interfacial stability between gel electrolytes and electrodes is usually superior.3–6,14 In particular, the formation of Zn dendrites can be effectively impeded by gel electrolytes to help maintain a smooth anode surface.19,23 The possible reason behind this is the high adhesive strength of optimized gel electrolytes such as PVA-B-G in this study that provides close contact between the gel electrolyte and anode. The polymer chains can be intimately adsorbed on the Zn tip and then act as an electrostatic shield, thus prohibiting further Zn deposition at that position and ensuring homogeneous Zn deposition.
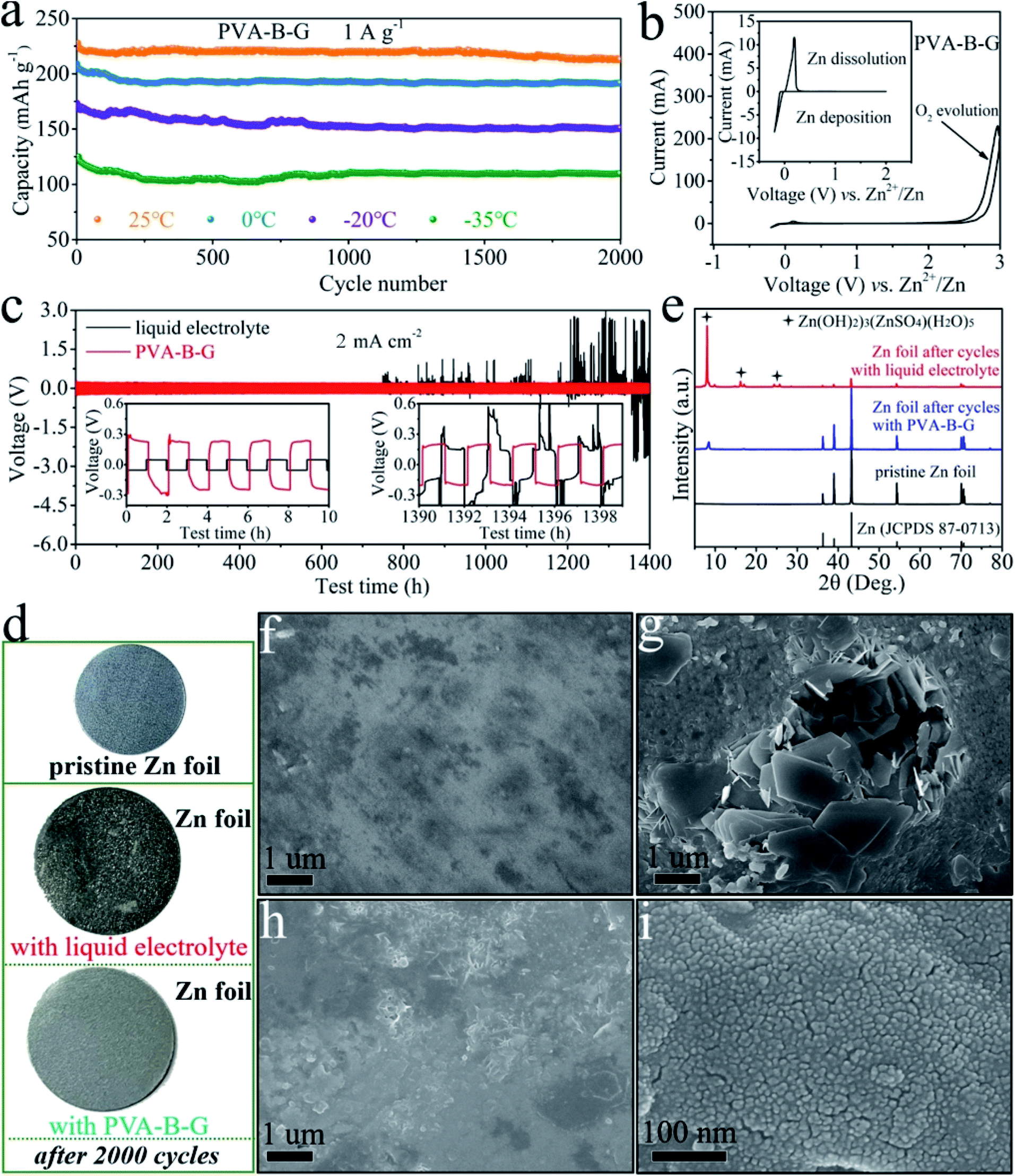 | ||
| Fig. 5 (a) Cycling performances of the PVA-B-G battery at 1 A g−1 at different temperatures. (b) CV curve of Zn deposition/dissolution in a Zn//PVA-B-G//stainless steel cell between −0.2 and 3.0 V. (c) GCD curves of Zn//Zn symmetrical cells with PVA-B-G and liquid electrolytes cycled at 2 mA cm−2 (insets show the enlarged voltage profiles of the initial and last 10 h). (d) Photographs and (e) XRD patterns of the Zn foil at the pristine state and after 2000 cycles with different electrolytes. SEM images of (f) pristine Zn foil, (g) Zn foil with the liquid electrolyte after 2000 cycles, and (h and i) Zn foil with PVA-B-G after 2000 cycles. | ||
To further elucidate the great electrochemical stability of PVA-B-G, CV measurement was carried out on a Zn//PVA-B-G//stainless steel cell between −0.2 and 3.0 V (Fig. 5b). The Zn dissolution potential is 0.19 V vs. Zn2+/Zn, indicative of small electrochemical polarization. It is also observed the response current increases abruptly after 2.2 V during the anodic scan, suggesting that the O2 evolution potential is pushed to 2.2 V, which is larger than the maximum operating potential (1.85 V) of the PVA-B-G battery. Such a large electrochemical stability window endows PVA-B-G with great reliability when used for AZIBs that work below 2.0 V. What's more, the cycling stability of Zn//Zn symmetrical cells with PVA-B-G and liquid electrolytes is compared in Fig. 5c to understand the interfacial stability. The cells were charged and discharged for 1400 h of 700 cycles at a current density of 2 mA cm−2. The insets of Fig. 5c and S18† show the expanded voltage profiles during different cycling periods for better recognition. Obviously, the PVA-B-G cell exhibits stable and flat voltage profiles over the entire 1400 h, disclosing stable Zn plating/stripping behavior. Except for the first cycle, the overpotential of the PVA-B-G cell remains at ∼0.46 V over the initial several cycles. After 700 cycles, the profile is similar to that at initial cycles, and the overpotential slightly drops to ∼0.39 V, indicating excellent electrochemical reliability. When the liquid electrolyte was used, the cell also shows great electrochemical stability and reversibility in the initial 61 cycles, during which the overpotential is much lower than that of the PVA-B-G cell due to much higher ionic conductivity of the liquid electrolyte. Unfortunately, after 61 cycles, the overpotential of the liquid-electrolyte cell suddenly increases to the same level of the PVA-B-G cell (Fig. S18†). Even worse is a significant voltage fluctuation of the liquid-electrolyte cell that is observed after 750 h testing. For example, the voltage jumps to ∼2.6 V at the 604th cycle. Such a high voltage indicates cell failure, which might result from the dynamic dendrite-induced soft short circuit. These results shed light on the exceptional compatibility of PVA-B-G with the Zn anode.
It is critical to prevent side reactions on Zn anodes.74,75 The Zn foil was taken out from the liquid-electrolyte and PVA-B-G batteries after 2000 cycles at 1 A g−1. The photographs in Fig. 5d show that the surface of Zn foil with the liquid electrolyte becomes rather rough and rugged with many black points, while the Zn foil with PVA-B-G remains smooth and looks nearly the same as pristine Zn foil. The surface composition and morphology of Zn foil were monitored by XRD and SEM analysis, as shown in Fig. 5e–i. The pristine Zn foil is made of pure metallic Zn (JCPDS: 87-0713) and nearly no impurity is visible on its surface, whereas large protuberances/dendrites are generated onto the Zn foil with the liquid electrolyte after 2000 cycles. These protuberances/dendrites are indexed as (Zn(OH)2)3(ZnSO4)(H2O)5 (zinc hydroxide sulfate hydrate, JCPDS: 78-0246), which was reported as a byproduct at the cathode of AZIBs.62 Herein, the formation of zinc hydroxide sulfate hydrate on the Zn foil is presumably related to the water splitting at the anode, producing OH− to react with Zn2+ and SO42− in the electrolyte. The insulating zinc hydroxide sulfate hydrate, in turn, would cause uneven electric fields on the surface of the Zn anode, thus leading to unstable Zn stripping/plating and exacerbating water splitting. The zinc hydroxide sulfate hydrate would also passivate the Zn surface and even penetrate through the separator, resulting in continuous capacity fading and even battery failure. In contrast, when the PVA-B-G was utilized, the Zn surface remains uniform and smooth after 2000 cycles. It is also seen from Fig. 5i that the deposited Zn is comprised of numerous tiny nanoparticles, indicative of the homogeneous Zn stripping/plating process in the PVA-B-G battery. Besides, the XRD pattern in Fig. 5e shows that a very little amount of zinc hydroxide sulfate hydrate was formed at the anode of the PVA-B-G battery after 2000 cycles. On the basis of these findings, it can be concluded that the PVA-B-G can effectively stabilize the electrolyte/anode interface during repeated cycles.
In order to demonstrate the practical application potential, the PVA-B-G batteries were used to charge a mobile phone. Given that at least 5.0 V is required, four PVA-B-G batteries were connected in series. The mobile phone can be charged successfully (see Fig. 6a and Video S1†), evidencing the high energy/power densities, shown in Fig. 4f and g. Besides, it is an essential requirement for flexible energy storage devices to retain their performances under extreme conditions. After being sealed into an ice solid in the −35 °C environment, the battery was taken out and used to power an electric timer, which can be immediately lit up (Fig. 6b). Besides, the timer can still be powered up when the battery is subjected to consecutive and violent hammering (Fig. 6c and Video S2†), indicating that the PVA-B-G battery can withstand a sudden hit that may frequently occur in a practical situation. As shown in Fig. 6d, the PVA-B-G battery can still normally power the timer when it is bent to different angles (30°, 60°, 90° and 180°). As a further demonstration, the as-assembled fresh battery was impaled with a pin (Fig. 6e and f). After 6 holes were generated, the open-circuit voltage only drops from 1.354 to 1.338 V. It is noteworthy that the pin is made of stainless steel, which would definitely cause shortcoming of the battery during the impaling process. Therefore, the decrease of open-circuit voltage is inevitable. Moreover, as shown in Fig. 6g, to further evaluate whether the PVA-B-G battery can run smoothly in extremely cold environments under deformation, the battery was cycled at 1 A g−1 at −35 °C. Impressively, when it was bent to 180° at the 21st cycle, a tiny capacity decay of 1.1% was observed in comparison with that at the 20th cycle (normal state). During the 21st–40th cycles under bending, the total capacity fading is merely 2.2 mA h g−1. Such great stability and reliability can be ascribed to the extraordinary anti-freezing and mechanical properties of PVA-B-G.
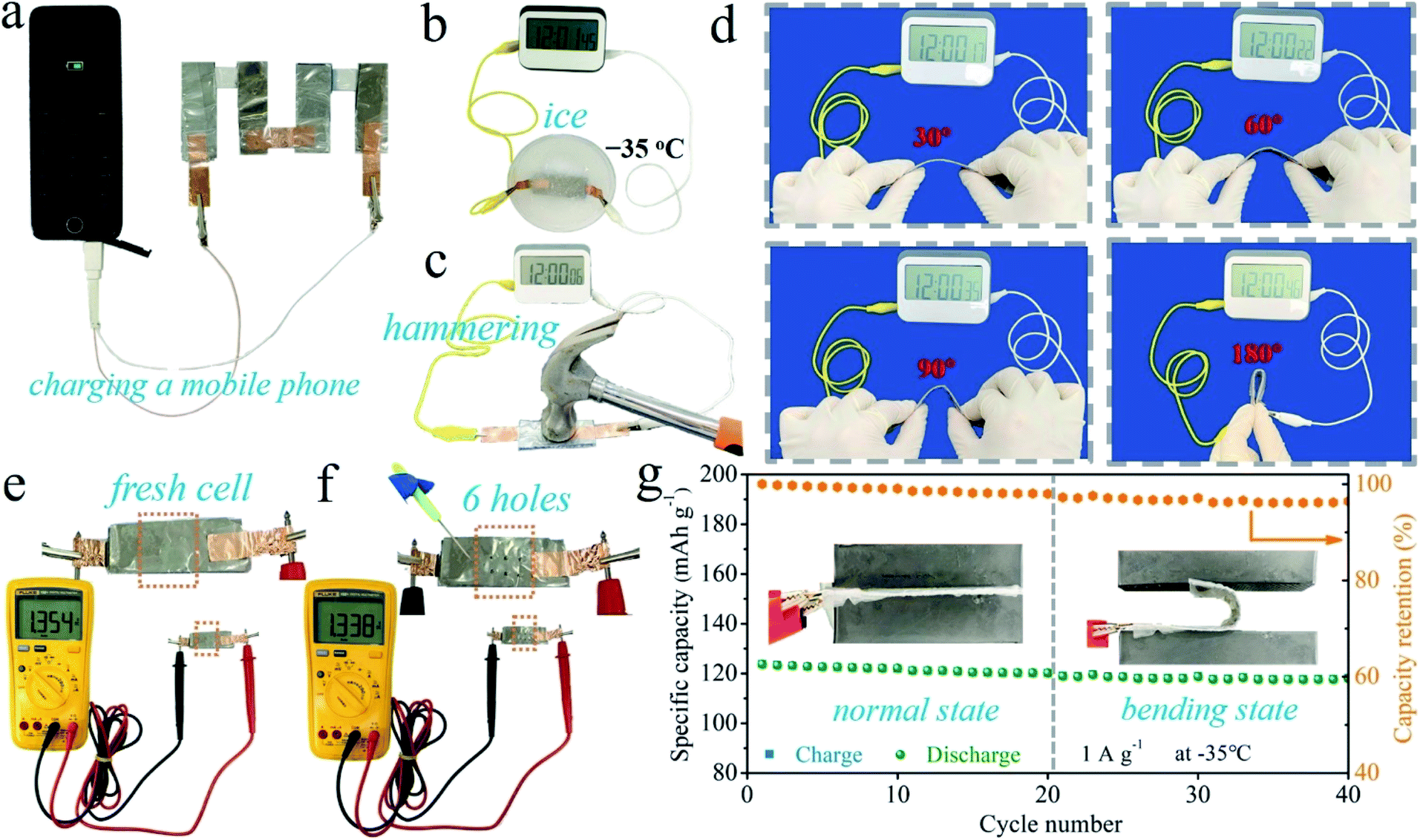 | ||
| Fig. 6 (a) Demonstration of four PVA-B-G batteries connected in series to charge a mobile phone. Demonstrations of a PVA-B-G battery powering an electric timer when (b) sealed into −35 °C ice solid, (c) hammered, and (d) subjected to different bending angles. Photographs of measuring the open-circuit voltage of a PVA-B-G battery: (e) after it is freshly assembled and (f) after impaled with a pin. (g) The specific capacity of the PVA-B-G battery vs. cycle number at −35 °C at the normal state (1st–20th cycles) and under 180° bending (21st–40th cycles). | ||
4. Conclusions
Although great advances have been made for AZIBs, conventional aqueous electrolytes would be frozen at subzero temperatures, meaning that AZIBs are unable to work in cold climates and high-altitude areas. In order to overcome this inherent shortcoming of AZIBs, we propose to crosslink PVA and glycerol with borax. In this respect, glycerol can interact with PVA chains strongly, thus significantly destroying PVA crystalline microdomains. As a result, not only can the ion transport be facilitated and mechanical properties be improved, but the ice crystal formation inside the whole gel network may also be effectively inhibited. Even at −35 °C, the PVA-B-G remains unfrozen and maintains great properties, because of which the assembled flexible quasi-solid-state aqueous PVA-B-G battery is capable of delivering excellent electrochemical performances from 25 to −35 °C. In particular, at −35 °C, a high volumetric energy density of 25.8 mW h cm−3 can be obtained at a power density of 96 mW cm−3, and 13.8 mW h cm−3 is retained at 918 mW cm−3. These values are even higher than those of many recently reported AZIBs operated at room temperature. Besides, the capacity retention is around 90% after 2000 cycles at all the temperatures, owing to the excellent compatibility of PVA-B-G with the Zn anode. The PVA-B-G battery is also found to work smoothly under various extreme conditions. This work provides new perspectives to explore low-cost anti-freezing gel electrolytes for emerging flexible power supplies.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the National Natural Science Foundation of China (No. 51902165), the Natural Science Foundation of Jiangsu Province (No. BK20170917), the Scientific Research Foundation for High-Level Talents of Nanjing Forestry University (No. GXL2016023), the Program of High-Level Talents in Six Industries of Jiangsu Province (No. XCL-040), and the Jiangsu Specially-Appointed Professor Program.References
- C. Wan, Y. Jiao and J. Li, J. Mater. Chem. A, 2017, 5, 3819–3831 RSC.
- C. Wan, Y. Jiao, D. Liang, Y. Wu and J. Li, Adv. Energy Mater., 2018, 8, 1802388 CrossRef.
- P. Yu, Y. Zeng, H. Zhang, M. Yu, Y. Tong and X. Lu, Small, 2019, 15, 1804760 CrossRef PubMed.
- Y. B. Li, J. Fu, C. Zhong, T. P. Wu, Z. W. Chen, W. B. Hu, K. Amine and J. Lu, Adv. Energy Mater., 2019, 9, 1802605 CrossRef.
- S. Huang, J. Zhu, J. Tian and Z. Niu, Chemistry, 2019, 25, 14480–14494 CrossRef CAS PubMed.
- W. J. Lu, C. X. Xie, H. M. Zhang and X. F. Li, ChemSusChem, 2018, 11, 3996–4006 CrossRef CAS PubMed.
- T. K. A. Hoang, T. N. L. Doan, C. Lu, M. Ghaznavi, H. Zhao and P. Chen, ACS Sustainable Chem. Eng., 2017, 5, 1804–1811 CrossRef CAS.
- T. K. A. Hoang, T. N. L. Doan, J. H. Cho, J. Y. J. Su, C. Lee, C. Lu and P. Chen, ChemSusChem, 2017, 10, 2816–2822 CrossRef CAS PubMed.
- C. Lu, T. K. A. Hoang, T. N. L. Doan, M. Acton, H. Zhao, W. Guan and P. Chen, J. Ind. Eng. Chem., 2016, 42, 101–106 CrossRef CAS.
- Y. X. Zeng, X. Y. Zhang, Y. Meng, M. H. Yu, J. N. Yi, Y. Q. Wu, X. H. Lu and Y. X. Tong, Adv. Mater., 2017, 29, 1700274 CrossRef PubMed.
- F. Wan, L. L. Zhang, X. Y. Wang, S. S. Bi, Z. Q. Niu and J. Chen, Adv. Funct. Mater., 2018, 28, 1804975 CrossRef.
- G. Q. Sun, H. S. Yang, G. F. Zhang, J. Gao, X. T. Jin, Y. Zhao, L. Jiang and L. T. Qu, Energy Environ. Sci., 2018, 11, 3367–3374 RSC.
- S. Spoljaric, A. Salminen, N. D. Luong and J. Seppala, Eur. Polym. J., 2014, 56, 105–117 CrossRef CAS.
- H. F. Li, Z. X. Liu, G. J. Liang, Y. Huang, Y. Huan, M. S. Zhu, Z. X. Pei, Q. Xue, Z. J. Tang, Y. K. Wang, B. H. Li and C. Y. Zhi, ACS Nano, 2018, 12, 3140–3148 CrossRef CAS PubMed.
- S. Huang, F. Wan, S. S. Bi, J. C. Zhu, Z. Q. Niu and J. Chen, Angew. Chem., Int. Ed., 2019, 58, 4313–4317 CrossRef CAS PubMed.
- S. J. Shi, X. Peng, T. Q. Liu, Y. N. Chen, C. C. He and H. L. Wang, Polymer, 2017, 111, 168–176 CrossRef CAS.
- B. L. Lu, F. C. Lin, X. Jiang, J. J. Cheng, Q. L. Lu, J. B. Song, C. Chen and B. Huang, ACS Sustainable Chem. Eng., 2017, 5, 948–956 CrossRef CAS.
- Z. F. Wang, F. N. Mo, L. T. Ma, Q. Yang, G. J. Liang, Z. X. Liu, H. F. Li, N. Li, H. Y. Zhang and C. Y. Zhi, ACS Appl. Mater. Interfaces, 2018, 10, 44527–44534 CrossRef CAS PubMed.
- H. F. Li, C. P. Han, Y. Huang, Y. Huang, M. S. Zhu, Z. X. Pei, Q. Xue, Z. F. Wang, Z. X. Liu, Z. J. Tang, Y. K. Wang, F. Y. Kang, B. H. Li and C. Y. Zhi, Energy Environ. Sci., 2018, 11, 941–951 RSC.
- Y. Lu, T. Zhu, N. Xu and K. Huang, ACS Appl. Energy Mater., 2019, 2, 6904–6910 CrossRef CAS.
- Q. Han, X. Chi, S. Zhang, Y. Liu, B. Zhou, J. Yang and Y. Liu, J. Mater. Chem. A, 2018, 6, 23046–23054 RSC.
- X. Zhang, Z. Pei, C. Wang, Z. Yuan, L. Wei, Y. Pan, A. Mahmood, Q. Shao and Y. Chen, Small, 2019, 15, 1903817 CrossRef CAS PubMed.
- S. L. Zhang, N. S. Yu, S. Zeng, S. S. Zhou, M. H. Chen, J. T. Di and Q. W. Li, J. Mater. Chem. A, 2018, 6, 12237–12243 RSC.
- Y. Huang, J. Zhang, J. Liu, Z. Li, S. Jin, Z. Li, S. Zhang and H. Zhou, Materials Today Energy, 2019, 14, 100349 CrossRef.
- Q. Zhang, C. Li, Q. Li, Z. Pan, J. Sun, Z. Zhou, B. He, P. Man, L. Xie, L. Kang, X. Wang, J. Yang, T. Zhang, P. P. Shum, Q. Li, Y. Yao and L. Wei, Nano Lett., 2019, 19, 4035–4042 CrossRef CAS PubMed.
- A. Mitha, H. Y. Mi, W. H. Dong, I. S. Cho, J. Ly, S. Yoo, S. Bang, T. K. A. Hoang and P. Chen, J. Electroanal. Chem., 2019, 836, 1–6 CrossRef CAS.
- Q. F. Rong, W. W. Lei, L. Chen, Y. A. Yin, J. J. Zhou and M. J. Liu, Angew. Chem., Int. Ed., 2017, 56, 14159–14163 CrossRef CAS PubMed.
- Q. F. Rong, W. W. Lei, J. Huang and M. J. Liu, Adv. Energy Mater., 2018, 8, 1801967 CrossRef.
- F. Chen, D. Zhou, J. Wang, T. Li, X. Zhou, T. Gan, S. Handschuh-Wang and X. Zhou, Angew. Chem., Int. Ed., 2018, 57, 6568–6571 CrossRef CAS PubMed.
- L. Han, K. Z. Liu, M. H. Wang, K. F. Wang, L. M. Fang, H. T. Chen, J. Zhou and X. Lu, Adv. Funct. Mater., 2018, 28, 1704195 CrossRef.
- C. X. Hu, Y. L. Zhang, X. D. Wang, L. Xing, L. Y. Shi and R. Ran, ACS Appl. Mater. Interfaces, 2018, 10, 44000–44010 CrossRef CAS PubMed.
- F. N. Mo, G. J. Liang, Q. Q. Meng, Z. X. Liu, H. F. Li, J. Fan and C. Y. Zhi, Energy Environ. Sci., 2019, 12, 706–715 RSC.
- X. F. Pan, Q. H. Wang, D. W. Ning, L. Dai, K. Liu, Y. H. Ni, L. H. Chen and L. L. Huang, ACS Biomater. Sci. Eng., 2018, 4, 3397–3404 CrossRef CAS.
- H. L. Pan, Y. Y. Shao, P. F. Yan, Y. W. Cheng, K. S. Han, Z. M. Nie, C. M. Wang, J. H. Yang, X. L. Li, P. Bhattacharya, K. T. Mueller and J. Liu, Nat. Energy, 2016, 1, 16039 CrossRef CAS.
- D. Wang, H. Li, Z. Liu, Z. Tang, G. Liang, F. Mo, Q. Yang, L. Ma and C. Zhi, Small, 2018, 14, 1803978 CrossRef PubMed.
- M. Chen, J. Chen, W. Zhou, J. Xu and C.-P. Wong, J. Mater. Chem. A, 2019, 7, 26524–26532 RSC.
- D. Zhou, F. Chen, S. Handschuh-Wang, T. Gan, X. Zhou and X. Zhou, Chemphyschem, 2019, 20, 2139–2154 CrossRef CAS PubMed.
- N. Zhang, F. Y. Cheng, J. X. Liu, L. B. Wang, X. H. Long, X. S. Liu, F. J. Li and J. Chen, Nat. Commun., 2017, 8, 405 CrossRef PubMed.
- D. Chao, W. Zhou, C. Ye, Q. Zhang, Y. Chen, L. Gu, K. Davey and S.-Z. Qiao, Angew. Chem., Int. Ed., 2019, 58, 7823–7828 CrossRef CAS PubMed.
- Y. Fu, Q. Wei, G. Zhang, X. Wang, J. Zhang, Y. Hu, D. Wang, L. Zuin, T. Zhou, Y. Wu and S. Sun, Adv. Energy Mater., 2018, 8, 1801445 CrossRef.
- Y. Jin, L. Zou, L. Liu, M. H. Engelhard, R. L. Patel, Z. Nie, K. S. Han, Y. Shao, C. Wang, J. Zhu, H. Pan and J. Liu, Adv. Mater., 2019, 31, 1900567 CrossRef PubMed.
- G. Fang, C. Zhu, M. Chen, J. Zhou, B. Tang, X. Cao, X. Zheng, A. Pan and S. Liang, Adv. Funct. Mater., 2019, 29, 1808375 CrossRef.
- K. W. Nam, H. Kim, J. H. Choi and J. W. Choi, Energy Environ. Sci., 2019, 12, 1999–2009 RSC.
- D. Kundu, B. D. Adams, V. Duffort, S. H. Vajargah and L. F. Nazar, Nat. Energy, 2016, 1, 16119 CrossRef CAS.
- V. Soundharrajan, B. Sambandam, S. Kim, M. H. Alfaruqi, D. Y. Putro, J. Jo, S. Kim, V. Mathew, Y. K. Sun and J. Kim, Nano Lett., 2018, 18, 2402–2410 CrossRef CAS PubMed.
- P. He, G. B. Zhang, X. B. Liao, M. Y. Yan, X. Xu, Q. Y. An, J. Liu and L. Q. Mai, Adv. Energy Mater., 2018, 8, 1702463 CrossRef.
- X. Guo, G. Z. Fang, W. Y. Zhang, J. Zhou, L. T. Shan, L. B. Wang, C. Wang, T. Q. Lin, Y. Tang and S. Q. Liang, Adv. Energy Mater., 2018, 8, 1801819 CrossRef.
- Q. Pang, C. L. Sun, Y. H. Yu, K. N. Zhao, Z. Y. Zhang, P. M. Voyles, G. Chen, Y. J. Wei and X. D. Wang, Adv. Energy Mater., 2018, 8, 1800144 CrossRef.
- D. Chao, C. Zhu, M. Song, P. Liang, X. Zhang, N. H. Tiep, H. Zhao, J. Wang, R. Wang, H. Zhang and H. J. Fan, Adv. Mater., 2018, 30, 1803181 CrossRef PubMed.
- C. Xia, J. Guo, P. Li, X. X. Zhang and H. N. Alshareef, Angew. Chem., Int. Ed., 2018, 57, 3943–3948 CrossRef CAS PubMed.
- P. Hu, T. Zhu, X. P. Wang, X. J. Wei, M. Y. Yan, J. T. Li, W. Luo, W. Yang, W. C. Zhang, L. Zhou, Z. Q. Zhou and L. Q. Mai, Nano Lett., 2018, 18, 1758–1763 CrossRef CAS PubMed.
- Y. Q. Yang, Y. Tang, G. Z. Fang, L. T. Shan, J. S. Guo, W. Y. Zhang, C. Wang, L. B. Wang, J. Zhou and S. Q. Liang, Energy Environ. Sci., 2018, 11, 3157–3162 RSC.
- C. Liu, Z. Neale, J. Zheng, X. Jia, J. Huang, M. Yan, M. Tian, M. Wang, J. Yang and G. Cao, Energy Environ. Sci., 2019, 12, 2273–2285 RSC.
- J. Ding, Z. Du, B. Li, L. Wang, S. Wang, Y. Gong and S. Yang, Adv. Mater., 2019, 31, 1904369 CrossRef CAS PubMed.
- J. Shin, D. S. Choi, H. J. Lee, Y. Jung and J. W. Choi, Adv. Energy Mater., 2019, 9, 1900083 CrossRef.
- G. Kasiri, J. Glenneberg, A. Bani Hashemi, R. Kun and F. La Mantia, Energy Storage Materials, 2019, 19, 360–369 CrossRef.
- W. Xu, C. Sun, K. Zhao, X. Cheng, S. Rawal, Y. Xu and Y. Wang, Energy Storage Materials, 2019, 16, 527–534 CrossRef.
- H. Liang, Z. Cao, F. Ming, W. Zhang, D. H. Anjum, Y. Cui, L. Cavallo and H. N. Alshareef, Nano Lett., 2019, 19, 3199–3206 CrossRef CAS PubMed.
- F. Wang, E. Y. Hu, W. Sun, T. Gao, X. Ji, X. L. Fan, F. D. Han, X. Q. Yang, K. Xu and C. S. Wang, Energy Environ. Sci., 2018, 11, 3168–3175 RSC.
- Z. W. Guo, Y. Y. Ma, X. L. Dong, J. H. Huang, Y. G. Wang and Y. Y. Xia, Angew. Chem., Int. Ed., 2018, 57, 11737–11741 CrossRef CAS PubMed.
- D. Kundu, P. Oberholzer, C. Glaros, A. Bouzid, E. Tervoort, A. Pasquarello and M. Niederberger, Chem. Mater., 2018, 30, 3874–3881 CrossRef CAS.
- J. H. Huang, Z. Wang, M. Y. Hou, X. L. Dong, Y. Liu, Y. G. Wang and Y. Y. Xia, Nat. Commun., 2018, 9, 2906 CrossRef PubMed.
- W. Sun, F. Wang, S. Y. Hou, C. Y. Yang, X. L. Fan, Z. H. Ma, T. Gao, F. D. Han, R. Z. Hu, M. Zhu and C. S. Wang, J. Am. Chem. Soc., 2017, 139, 9775–9778 CrossRef CAS PubMed.
- Q. Yang, F. Mo, Z. Liu, L. Ma, X. Li, D. Fang, S. Chen, S. Zhang and C. Zhi, Adv. Mater., 2019, 31, 1901521 CrossRef PubMed.
- H. Geng, M. Cheng, B. Wang, Y. Yang, Y. Zhang and C. C. Li, Adv. Funct. Mater., 2020, 30, 1907684 CrossRef CAS.
- S. Bi, F. Wan, S. Huang, X. Wang and Z. Niu, Chemelectrochem, 2019, 6, 3933–3939 CrossRef CAS.
- P. P. Zhang, Y. Li, G. Wang, F. X. Wang, S. Yang, F. Zhu, X. D. Zhuang, O. G. Schmidt and X. L. Feng, Adv. Mater., 2019, 31, 1806005 CrossRef PubMed.
- D. Pech, M. Brunet, H. Durou, P. H. Huang, V. Mochalin, Y. Gogotsi, P. L. Taberna and P. Simon, Nat. Nanotechnol., 2010, 5, 651–654 CrossRef CAS PubMed.
- H. Zhang, J. Wang, Q. Liu, W. He, Z. Lai, X. Zhang, M. Yu, Y. Tong and X. Lu, Energy Storage Materials, 2019, 21, 154–161 CrossRef.
- T. Zhao, G. M. Zhang, F. S. Zhou, S. Zhang and C. Deng, Small, 2018, 14, 1802320 CrossRef PubMed.
- X. J. He, H. Z. Zhang, X. Y. Zhao, P. Zhang, M. H. Chen, Z. K. Zheng, Z. J. Han, T. S. Zhu, Y. X. Tong and X. H. Lu, Adv. Sci., 2019, 6, 1900151 CrossRef PubMed.
- H. Wang, S. Zhang and C. Deng, ACS Appl. Mater. Interfaces, 2019, 11, 35796–35808 CrossRef CAS PubMed.
- W. Xu, K. Zhao, W. Huo, Y. Wang, G. Yao, X. Gu, H. Cheng, L. Mai, C. Hu and X. Wang, Nano Energy, 2019, 62, 275–281 CrossRef CAS.
- C. Lu, T. K. A. Hoang, T. N. L. Doan, H. Zhao, R. Pan, L. Yang, W. Guan and P. Chen, Appl. Energy, 2016, 170, 58–64 CrossRef CAS.
- H. B. Zhao, C. J. Hu, H. W. Cheng, J. H. Fang, Y. P. Xie, W. Y. Fang, T. N. L. Doan, T. K. A. Hoang, J. Q. Xu and P. Chen, Sci. Rep., 2016, 6, 25809 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, Fig. S1–S18, and supplementary videos. See DOI: 10.1039/d0ta01553a |
| This journal is © The Royal Society of Chemistry 2020 |