
A review of phosphorus and phosphides as anode materials for advanced sodium-ion batteries
Guoliang
Chang
a,
Yufeng
Zhao
*a,
Li
Dong
a,
David P.
Wilkinson
b,
Lei
Zhang
ac,
Qinsi
Shao
a,
Wei
Yan
a,
Xueliang (Andy)
Sun
 *ad and
Jiujun
Zhang
*ab
*ad and
Jiujun
Zhang
*ab
aInstitute for Sustainable Energy/College of Sciences, Shanghai University, Shanghai, 200444, China. E-mail: yufengzhao@shu.edu.cn; xsun9@uwo.ca; jiujun.zhang@i.shu.edu.cn
bDepartment of Chemical and Biochemical Engineering, University of British Columbia, Vancouver, BC V6T 1W5, Canada
cEnergy, Mining, and Environment, National Research Council of Canada, Vancouver, BC V6T 1W5, Canada
dDepartment of Mechanical and Materials Engineering, University of Western Ontario, London, Ontario N6A 3K7, Canada
First published on 5th February 2020
Abstract
Sodium-ion batteries (SIBs) are promising low-cost alternatives to lithium-ion batteries (LIBs) in energy storage applications because of the natural abundance of sodium as compared with lithium. However, the radius of Na+ ions is ∼50% larger than that of Li+ ions, leading to challenging sodiation/desodiation at the anodes of SIBs and reduced performances. Therefore, the exploration of novel SIB anode materials to facilitate sodiation/desodiation has become a leading area of research. Here, cost-effective phosphorus and metal/nonmetal phosphides, possessing high theoretical gravimetric capacities and volumetric specific capacities, have emerged as new and promising anode materials for SIBs. Based on this, this review will provide a comprehensive summary of the recent progress in the development of phosphorus and metal/nonmetal phosphide materials and corresponding composite SIB anodes in terms of material synthesis, characterization, sodium storage mechanism and performance validation in SIB apparatuses. In addition, challenges associated with the development of such advanced materials are summarized and analyzed. Finally, to facilitate the research and development of these emerging SIB anode materials, future research directions are proposed to overcome challenges toward the practical commercialization of sodium-ion batteries.
 Guoliang Chang | Guoliang Chang is currently a PhD candidate under the supervision of Prof. Jiujun Zhang at the College of Sciences/Institute for Sustainable Energy, Shanghai University. His research interests include electrochemical energy storage and conversion, e.g., sodium-ion batteries and photoelectrochemical water splitting. |
 Yufeng Zhao | Dr Yufeng Zhao is currently working as a professor at Shanghai University. She received her B.S. and M.S. degrees from Tianjin University, China, in 2000 and 2003 respectively. She obtained her PhD from Nanyang Technological University, Singapore in 2006. Afterwards, she worked at Deakin University, Australia (2006–2008) and Phillips University Marburg, Germany (2008–2009) as a research scientist. She was also a visiting professor at Northwestern University (2014–2015). Her research mainly focuses on energy storage materials and devices, such as nanocarbon materials, supercapacitors, and electrocatalysts. |
 Xueliang (Andy) Sun | Prof. Xueliang (Andy) Sun holds a Canada Research Chair in the Development of Nanomaterials for Clean Energy, Fellow of the Royal Society of Canada and Canadian Academy of Engineering and Full Professor at the University of Western Ontario, Canada. Dr Sun received his PhD in materials chemistry in 1999 from the University of Manchester, UK, which he followed up by working as a postdoctoral fellow at the University of British Columbia, Canada and as a Research Associate at L'Institut National de la Recherche Scientifique (INRS), Canada. His current research interests are focused on advanced materials for electrochemical energy storage and conversion, including solid-state batteries and electrocatalysis in fuel cells. |
 Jiujun Zhang | Dr Jiujun Zhang is a Professor at the College of Sciences/Institute for Sustainable Energy at Shanghai University, a former Principal Research Officer (Emeritus) at the National Research Council of Canada (NRC). Dr Zhang received his B.S. and M.Sc. in Electrochemistry from Peking University in 1982 and 1985, respectively, and his PhD in Electrochemistry from Wuhan University in 1988. Dr Zhang serves as an Editor-in-Chief of Electrochemical Energy Reviews (Springer Nature) and Associate Editor of Green Energy & Environment (KeAi), and Editorial Board Member for several international journals as well as Editor for a book series of Electrochemical Energy Storage and Conversion (CRC). Dr Zhang's expertise areas are electrochemistry, electrocatalysis, fuel cells, batteries, supercapacitors, and water/CO2 electrolysis. |
1 Introduction
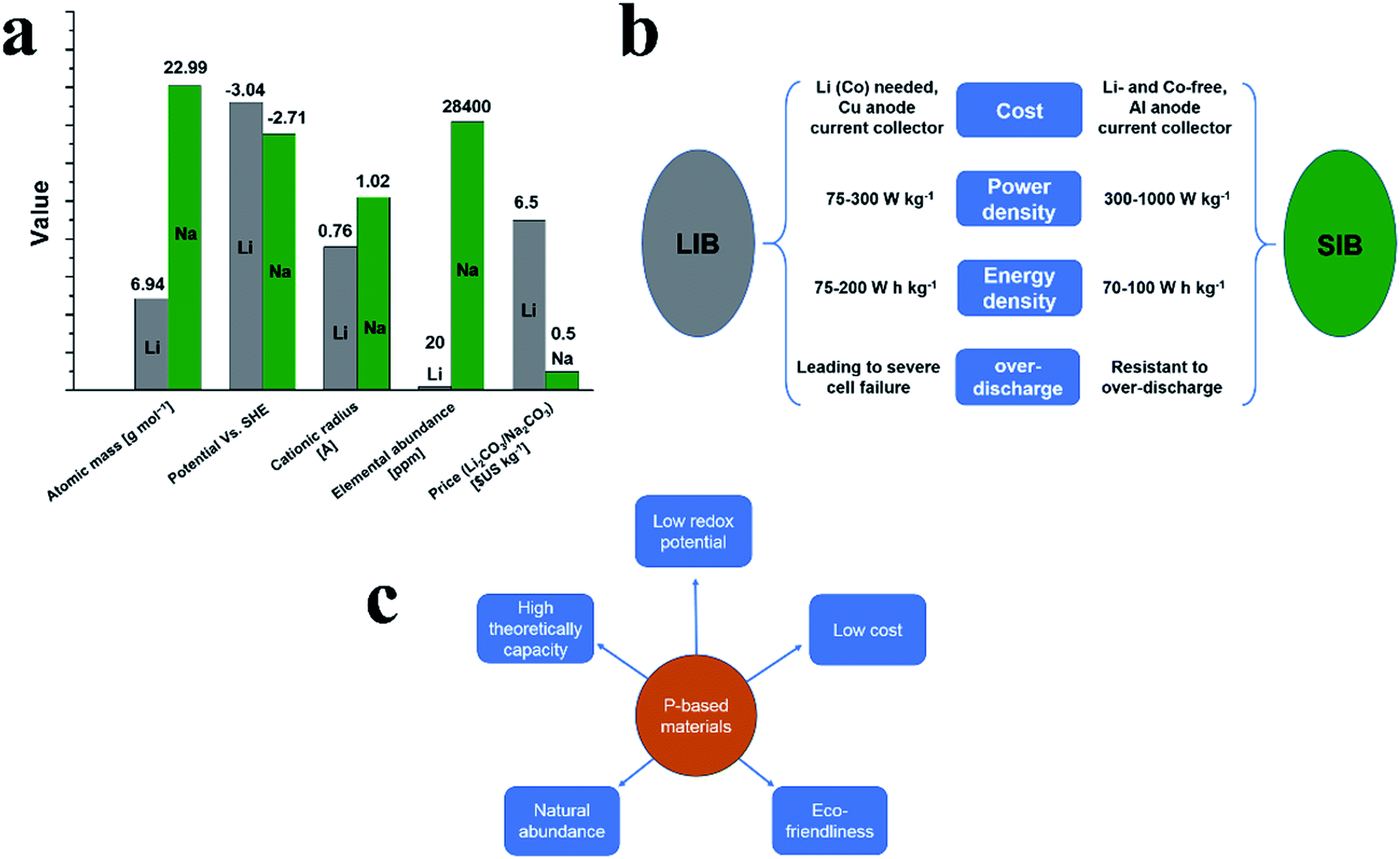
Global demand for advanced energy storage and conversion technologies, especially electrochemical energy technologies such as batteries, is rapidly growing and among various types of batteries, Li-ion batteries (LIB) have shown success in many applications.1,2 However, although LIBs can meet the tough demands of energy storage and conversion applications, the limited abundance of lithium (only 20 ppm) needs to be considered in terms of sustainability.3 Therefore, the exploration of alternatives to replace lithium ion batteries is necessary. Fortunately, sodium has been identified as a suitable alternative to lithium as demonstrated by the practical feasibility of sodium-ion batteries (SIBs).4Fig. 1a shows the comparison between lithium and sodium in terms of their atomic masses, electrode potentials, cationic radiuses, elemental abundances, and prices. It can be seen that sodium is the second-lightest and smallest alkali metal next to lithium and is one of the most abundant elements in Earth's crust with an average abundance of 28![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 ppm.5 And in battery applications, sodium can provide a suitable electrochemical redox potential of −2.71 V vs. the standard hydrogen electrode (SHE), which is only 0.3 V less negative than that of lithium. Furthermore, sodium is much cheaper than lithium. Due to these promising properties, SIBs are promising alternatives to LIBs, in particular for large-scale applications such as energy storage systems for power grids.5
400 ppm.5 And in battery applications, sodium can provide a suitable electrochemical redox potential of −2.71 V vs. the standard hydrogen electrode (SHE), which is only 0.3 V less negative than that of lithium. Furthermore, sodium is much cheaper than lithium. Due to these promising properties, SIBs are promising alternatives to LIBs, in particular for large-scale applications such as energy storage systems for power grids.5
 | ||
| Fig. 1 (a) Major property differences between lithium and sodium; (b) main differences between lithium-ion batteries (LIBs) and sodium-ion batteries (SIBs); (c) main advantages of phosphorus/phosphides as anode materials for SIBs. | ||
However, the ionic radius of Na is larger than that of Li (1.02 Å for Na+vs. 0.76 Å for Li+), which results in sluggish reaction kinetics and poor electrochemical performances in corresponding batteries. To address this, great efforts have been undertaken and in recent years, significant progress has been made for the cathodes of SIBs with a variety of materials being demonstrated as suitable cathodes for sodium ion storage.6–11Fig. 2b summarizes the main difference between lithium-ion batteries (LIBs) and sodium-ion batteries (SIBs) in terms of their cost, power density, energy density and overcharge resistibility. In general, the exploration of low-cost anodes for both LIBs and SIBs to match their corresponding cathodes as well as the corresponding synthesis strategies remain challenging. Similar to LIBs, carbonaceous materials such as hard carbon,12–19 graphitic carbon,20–22 and graphene23–28 along with novel materials such as metals,29–35 metal oxides,36–41 metal sulfides,42–48 metal selenides,49–54 metal alloys55–64 and organic compounds65–69 have all been explored as anode materials for SIBs. Most recently, phosphorus (P) and phosphides70–78 have also been extensively explored for Na storage. Among these P-based materials, red phosphorus (RP) was the first to attract attention from researchers due to favorable properties as anode materials in SIBs, including a highest theoretical specific capacity of ∼2600 mA h g−1 and a suitable insertion voltage of ∼0.4 V vs. Na/Na+.79 The major advantages of SIBs are summarized in Fig. 1b. Particularly, RP is abundant, easily available and nontoxic, being identified as the most promising anode candidate material for practical SIBs (Fig. 1c). However, RP also possesses drawbacks such as low electrical conductivity (∼10−14 S cm−1)80 and large volume expansion after full sodiation (300–500%),81–83 leading to shortened cycle lifespans and poor rate performances in sodium storage applications; in addition, the sodiated product Na3P also shows a low electrical conductivity and the surface of Na3P is highly reactive which may cause a severe electrolyte decomposition.84 To overcome these issues, great efforts have been made and considerable progress has been achieved. Many composites such as amorphous RP/carbon,79,82 RP/carbon nanotubes,85 RP/Ni–P86 and hollow RP nanospheres87 have been prepared to improve the Na storage performance of RP anodes, which could buffer the volume expansion of RP during cycles and/or enhance the conductivity of electrodes. In addition to RP, black phosphorus (BP) is also gaining prominence due to a similarly high theoretical specific capacity to RP (∼2600 mA h g−1) and a higher electronic conductivity (∼300 S m−1).88 However, large volume expansion, sluggish reaction kinetics and rigorous synthesis conditions also hinder the application of BP in Na storage devices. Phosphorene is one or few layers of BP and has also attracted much attention as a promising anode material due to its unique 2D structure (similar to a graphene).89 Furthermore, metal phosphides, which usually possess higher electrical conductivities than phosphorus,77 can easily be synthesized into various nanostructures and are also promising anode materials for SIBs. Some unique metal phosphide nanostructures such as CoP4 nanoarrays,90 yolk–shell Sn4P3@C nanospheres91 and monodisperse Ni2P nanocrystals92 have been prepared and showed excellent Na-ion storage performance as anode materials. Finally, non-metal phosphides and ternary phosphides as supplemental materials to metal phosphides have also experienced great progress for practical application in SIBs.
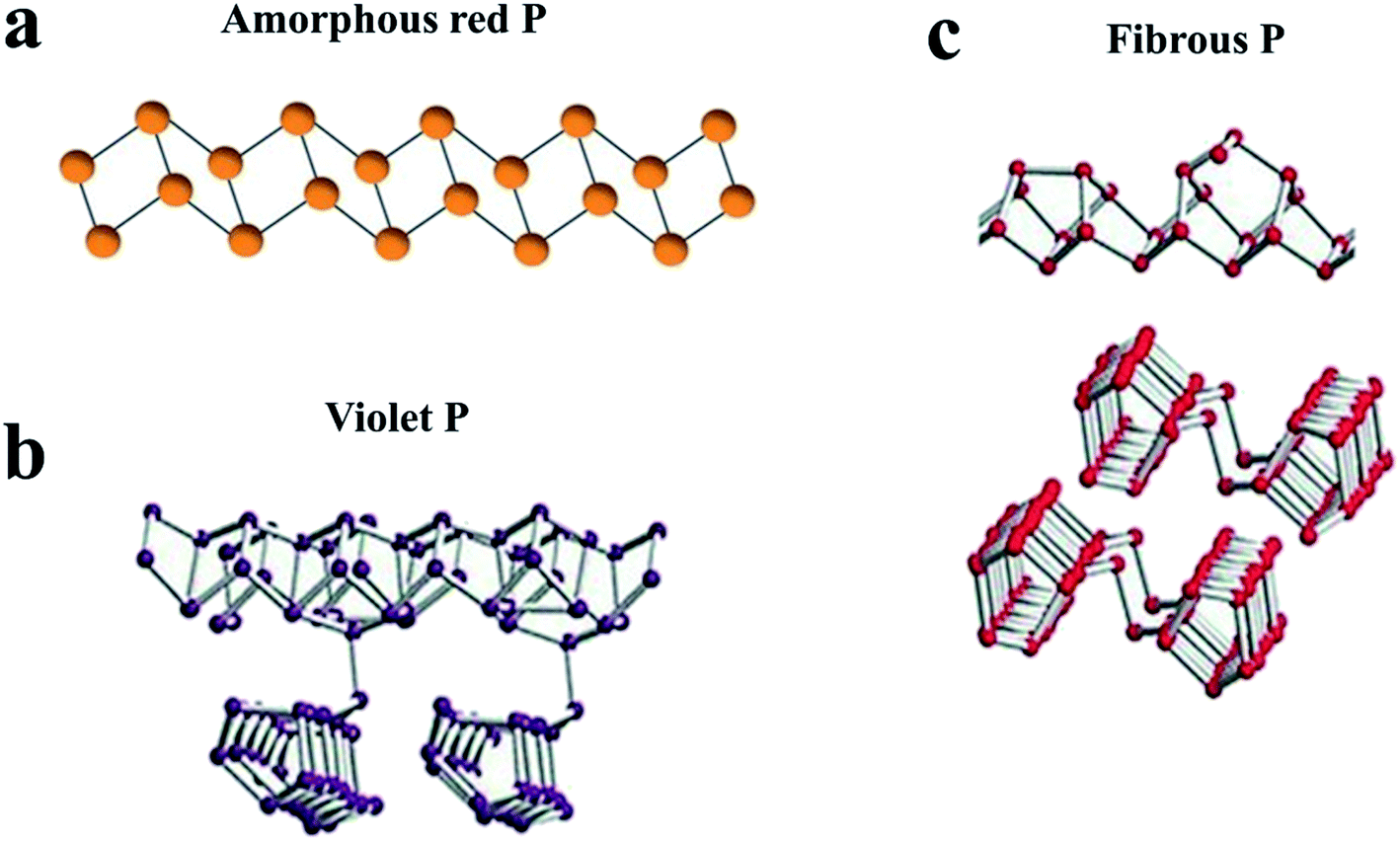 | ||
| Fig. 2 Characteristic structural fragments of (a) amorphous RP; (b) violet P (Hittorf's P); (c) fibrous P. (a) Reproduced with permission.100 Copyright 2019, WILEY-VCH. (b, c) Reproduced with permission.113 Copyright 2017, Wiley-VCH. | ||
Because of all these advancements, this review will comprehensively summarize the recent progress in the research and development of phosphorus and phosphide composites for SIB anodes. It covers the structural properties and sodium storage mechanisms of these composites as well as the design strategies. In addition, the advantages and limitations of current synthesis methods for the preparation of phosphorus/phosphide anodes are also discussed. And to facilitate future research efforts, associated challenges are also presented and analyzed as well as possible research directions to address these challenges.
2 Phosphorus anodes for SIBs
In general, elemental phosphorus exists in three allotropes, namely, red, black and white phosphorus. White phosphorus is flammable and toxic, and not suitable for SIBs, whereas RP and BP can react with sodium to form Na3P and present high theoretical capacities of ∼2600 mA h g−1 as electrochemical sodium storage materials.93 Compared with BP, which is difficult to synthesize, RP is more commercially available, and thus is regarded as the most promising candidate for SIB anode materials. In addition, phosphorene (one or several layers of BP) has also attracted great interest for Na-ion storage because of its unique 2D structure, high electronic conductivity, and other advantages. Recently, some new allotropic modifications of P have been experimentally produced, such as blue phosphorus94 and ring-shaped phosphorus within carbon nanotube nanoreactors;95 nevertheless, these allotropes have not been experimentally applied in batteries yet.96 So in this section, we will present recent advances in RP, BP and phosphorene anodes for SIBs. And Table 1 summarizes these recent developments.| Material | Synthetic method | Cycling performance | Rate capability | P content | Initial coulombic efficiency | Ref. |
|---|---|---|---|---|---|---|
| RP/CNT | Hand grinding | 1283 mA h g−1, at 143 mA g−1, 10th | — | 70% | 69% | 121 |
| RP/graphene | Ball milling | 1700 mA h g−1, at 520 mA g−1, 60th | 520 mA h g−1, 5200 mA g−1 | 70% | 83% | 73 |
| RP/CNT/PDA | Ball milling | 470 mA h g−1, at 5200 mA g−1, 5000th | 1060 mA h g−1, 2600 mA g−1 | 66.04% | ∼60% | 122 |
| RP/C | Ball milling/spray drying | 1408 mA h gp−1, at 0.1 A g−1, 300th | 986 mA h gp−1, 2000 mA g−1 | 42.5% | 86.2% | 125 |
| RP-graphene | Ball milling | 2172 mA h g−1, at 250 mA g−1, 150th | 342 mA h g−1, 2600 mA g−1 | — | — | 126 |
| P–TiO2–C | Ball milling | 632 mA h g−1, at 500 mA g−1, 100th | 958.9 mA h g−1, 1000 mA g−1 | 60% | 77% | 130 |
| RP/boron nitride/Gr | Ball milling | 947 mA h g−1, at 50 mA g−1, 100th | 776 mA h g−1, 300 mA g−1 | 42% | 64% | 179 |
| Fe3O4/C/RP | Vaporization/condensation | 1390 mA h g−1, at 200 mA g−1, 200th | 692 mA h g−1, 2000 mA g−1 | 25% | 80.4% | 129 |
| RP/C | Vaporization/condensation | 700 mA h g−1, at 2 A g−1, 920th | 500 mA h g−1, 10 A g−1 | ∼60% | 69.9% | 134 |
| P@RGO | Vaporization/condensation | 914 mA h g−1, at 1594 mA g−1, 300th | 519 mA h g−1, 31.88A g−1 | 61.4% | 75.2% | 132 |
| RP/C | Vaporization/condensation | 580 mA h g−1, at 2500 mA g−1, 800th | 430 mA h g−1, 8000 mA g−1 | ∼40% | 69.8% | 117 |
| RP@RGO | Vaporization/condensation | 1625 mA h g−1, at 1000 mA g−1, 200th | 679 mA h g−1, 6000 mA g−1 | 57.9% | 73.2% | 133 |
| RP@YP | Vaporization/condensation | 1064 mA h gp−1, at 420 mA g−1, 100th | 601 mA h g−1, 6780 mA gp−1 | 48% | 68% | 136 |
| 533 mA h gp−1, at 420 mA g−1, 1000th | ||||||
| Hollow P sphere | Solvothermal | 1500 mA h g−1, at 1300 mA g−1, 80th | 278 mA h g−1, 10.4 A g−1 | 99.9% | 77.3% | 87 |
| 970 mA h g−1, at 2600 mA g−1, 600th | ||||||
| Porous RP/GO | Boiling method | 1250 mA h g−1, at 173 mA g−1, 150th | 657 mA h g−1, 3465 mA g−1 | 67.6% | 78.5% | 144 |
| I-Doped hollow porous RP | Redox reaction | 1658 mA h g−1, at 260 mA g−1, 100th | 760 mA h g−1, 5200 mA g−1 | ∼100% | 75.6% | 145 |
| Boiling process | ||||||
| RPQDs/rGO | Hydrothermal | 900 mA h g−1, at 250 mA g−1, 250th | 193 mA h g−1, 4000 mA g−1 | 55.7% | 55.5% | 146 |
| RP@C | Shear emulsifying electrospinning | 821 mA h g−1, at 1000 mA g−1, 300th | 676 mA h g−1, 5000 mA g−1 | 51% | 75% | 127 |
| NTO/C-BP | Ball milling | 225 mA h g−1, at 20 mA g−1, 55th | — | 20% | 42% | 177 |
| BP-CNT | Ball milling | 1092 mA h g−1, at 520 mA g−1, 200th | 441 mA h g−1, 11.7 A g−1 | 74.5% | 87.7% | 174 |
| BP/G | Sonication | 1297 mA h g−1, at 100 mA g−1, 100th | 623 mA h g−1, 500 mA g−1 | 50% | 75.6% | 175 |
| Electrophoretic deposition | ||||||
| BP/C-MWNT | Ball milling | 1700 mA h g−1, at 1300 mA g−1, 100th (for BP) | 928 mA h g−1, 3000 mA g−1 | 70% | 91.1% | 173 |
| Phosphorene-C | Liquid-phase exfoliation | 2080 mA h g−1, at 50 mA g−1, 100th | 645 mA h g−1, 26 A g−1 | 48.3% | 80% | 72 |
| Phosphorene | Electrochemical exfoliation | ∼1190 mA h g−1, at 100 mA g−1, 50th | 591 mA h g−1, 1500 mA g−1 | 100% | ∼50% | 188 |
2.1 Red phosphorus
RP is the most abundant and easily available allotrope of elemental phosphorus,97,98 and is chemically stable and nontoxic to the environment.99 Commercial RP exhibits an amorphous form or an intermediate state between amorphous and crystalline.79,100,101 RP has been widely used in our daily life and industry, such as safety matches,102 flame retardants103 and chemical analysis.104 Recently, the application of RP has extended to the field of semiconductors,105 rechargeable batteries82 and photocatalysts106 with the progress of nanotechnology. And it is necessary to understand the structural information of RP for further research. Roth et al.107 proposed five distinct allotropes of RP, four crystalline (Types II to V) and one amorphous (Type I). The exact structures of these four crystalline types can be determined by X-ray diffraction (XRD) or other methods. For example, Type IV (fibrous RP) and Type V (violet P or Hittorf's P) were confirmed to consist of tubes with a pentagonal cross section, which in turn are a regular sequence of three different building units (Fig. 2b and c).100,108–110,113 As for amorphous RP, the exact structure remained unclear until Zhang et al.100 investigated some of its basic properties by several experimental methods, including scanning tunneling microscopy (STM), gel permeation chromatography (GPC) and single-molecule atomic-force microscopy (AFM). Based on the results, they proposed that amorphous RP should be a linear inorganic polymer with a broad molecular weight distribution (mainly at 31000 ± 20000 Da) and with a zig-zag ladder chain structure (85 ± 55 nm), as shown in Fig. 2a.100 Compared with crystalline RP, amorphous RP seems more advantageous in accommodating the volumetric expansion during Li+/Na+ insertion–extraction cycles. In fact, most RP anodes for Li/Na-ion batteries in the literature are in the amorphous form, especially for Na-ion batteries.82,111 However, because RP anodes usually exhibit an amorphous form and most of the sodiated RP phases are also amorphous,82 they are difficult to investigate in terms of sodiation and desodiation mechanisms.82 Nevertheless, it is acknowledged that the sodiation of RP mainly occurs between 0.5 V and 0 V to form Na3P, while the desodiation process involves a stepwise sodium extraction from Na3P with possible intermediates such as Na2P, NaP and NaP7.76,79,87
Although RP possesses great potential for practical application in SIBs due to high theoretical capacity, low redox potential (<0.4 V),82 natural abundance and eco-friendliness, major drawbacks such as low electronic conductivity (∼10−14 S cm−1),80 large volume expansion during sodiation (300–500%)81,82,112 and low initial coulombic efficiency hinder its application. There are several approaches to overcome these drawbacks: one is to combine RP nanostructures with electronic conductive materials and supporting matrices including carbon-based materials, conductive polymers, metals, metal oxides or metal phosphides; another one is to synthesize novel or self-supported RP nanostructures, and particle surface modification may also be a potential method to address these issues.
As for the synthesis of RP anode composites, two conventional methods are mainly used, including ball milling (or other mechanical milling methods) and vaporization/condensation methods. Ball milling (high energy) is a mechanochemical process that is facile and cost-effective to produce a wide range of nano-powders from bulk raw materials. During a high energy and long-term ball milling process, chemical reactions and structural changes can occur and allow for the formation of new phases or complex composites.114 The vaporization–condensation method is also an effective approach to prepare RP/carbon composites.115 In a typical process, RP is first sublimated by heating and the gaseous P subsequently diffused into the pores or on the surface of the carbon matrix through capillary forces and pressure differences, and then the P is adsorbed and deposited onto the internal surface to form solid RP as the temperature drops down.81,116 Compared with mechanical strategies, the vaporization–condensation method can ensure the uniform dispersal or adsorption of RP into carbon matrices and facilitate intimate contact between RP and host materials.116 However, the RP content in the composite synthesized by this method was usually not high. Some other methods such as solvothermal and hydrothermal have also been applied to prepare RP nanostructure/nanocomposite, which will be fully discussed in latter sections. Among these various synthesis methods, the high energy ball milling method is the most widely used approach to prepare RP-based anodes for both LIBs and SIBs.
RP was used as the anode material in LIBs earlier than in SIBs,115,118,119 and researchers found that the lithium ion storage capacity of RP depended dramatically on its crystalline structure and morphology, and a better lithium ion storage performance could be obtained when the crystalline and large-size RP was transformed into amorphous RP nanoparticles.120 Soon after these reports, Qian et al. and Kim et al.79,82 first introduced RP into SIBs, and respectively prepared an amorphous RP/carbon nanocomposite as an anode by simply ball milling commercially available amorphous RP and Super P (7:3). They found that the micro-sized commercially available RP could not react with Na reversibly in spite of a large initial specific capacity obtained, while the ball-milled RP nanoparticles show a much better Na-ion storage performance. During this long-term and high-energy ball milling process, bulk commercial phosphorus particles were crushed into amorphous nanoparticles under a strong mechanical force, dispersing into the conductive carbon matrix. Due to the short electron/ion transfer route and the amorphous form of RP, these P/C nanocomposites exhibited high reversible capacity and high rate capability, for example, a rate capability of 1540 mA h g−1 at a current density of 2.86 A g−1 could be obtained.82 Interestingly, Li et al.121 demonstrated that commercial micro-sized RP could also deliver a high reversible capacity after being mixed with carbon nanotubes (CNTs) by simple hand milling. And the authors proposed that a good electronic conductivity is essential for RP to reversibly react with Na. This indicates that increasing the electronic conductivity of RP is an effective way to improve the Na storage performance of RP anodes in terms of either the rate capability and the specific capacity. As the combination of RP with CNTs shows noticeable success, predictably, ball milling of RP and carbon materials with high mechanical strength such as CNTs or graphene may produce amazing results. Song et al.73 found that after a ball milling process of RP and graphene stacks, the graphene stacks were mechanically exfoliated to nanosheets, and more importantly, these nanosheets were chemically bonded with the surfaces of RP particles. Benefiting from this robust and intimate contact, the graphene at the particle surfaces could help to maintain electrical contact and stabilize the solid electrolyte interphase upon the large volume change of RP during cycling, leading to an improved cycling stability.
However, these simply ball milled RP anodes often suffered from insufficient rate capability and long-term stability, mainly caused by the large and rough particle size for this method, as the larger particle size means a longer diffusion length for electrons/ions, leading to unsatisfactory rate performance. And it also causes severe pulverization of RP after repeated large volume changes between P and Na3P, and as a result, lots of pulverized RP particles are electrically isolated in the anode,83 and a continuous solid electrolyte interphase (SEI) growth occurs, leading to fast capacity decay. To address these issues, combination with conductive polymers is an effective strategy. For example, Liu et al.122 recently designed a strategy to in situ coat ball milled P/C hybrids (phosphorus-carbon nanotube, P-CNT) with a polydopamine layer (Fig. 3a–d). They proposed that the coating layer could provide an elastic buffer to accommodate active material volume change and prevent their direct contact with electrolytes. As a result, the P-CNT@PD (PD, polydopamine) composite anode presented high rate capacities of 1060 mA h g−1 in the second discharge and 730 mA h g−1 after 2000 cycles at 2.6 A g−1 and high cycling stabilities with 470 mA h g−1 capacity retained after 5000 cycles at 5.2 A g−1 (Fig. 3e and f), and this is the longest cycle lifespan that has ever been reported for RP-based materials.
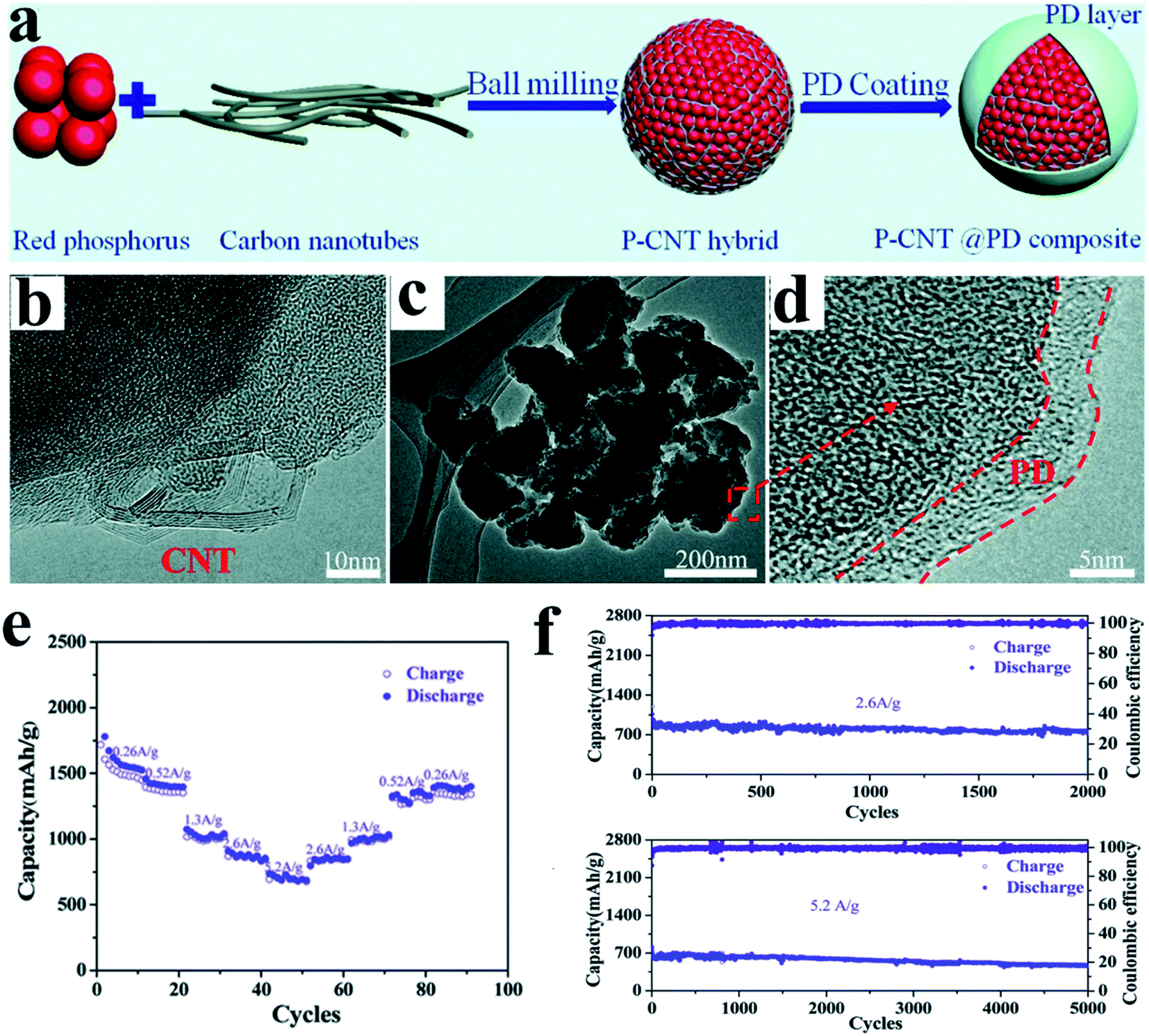 | ||
| Fig. 3 (a) Schematic of the synthesis of the P-CNT@PD composite. (b) HRTEM image of the P-CNT hybrid. (c) TEM and (d) HRTEM images of the P-CNT@PD composite. (e) Rate performances at various current densities from 0.26 A g−1 to 5.2 A g−1 of the P-CNT@PD composite between 0.001 and 2.0 V (vs. Na+/Na). (f) Long-term cycling performances of the P-CNT@PD composite at 2.6 A g−1 and 5.2 A g−1 between 0.001 and 2.0 V (vs. Na+/Na). Reproduced with permission.122 Copyright 2018, The Royal Society of Chemistry. | ||
In another exmaple,123 a conductive polymer of sulfurized polyacrylonitrile (SPAN) was ball milled with RP, forming a chemically bonded RP/SPAN hybrid. The functional conductive matrix with robust P–S bonds linked to RP could accommodate the large volume change during cycles and enhance the conductivity of the anode. Consequently, this hybrid anode delivered a capacity of ∼1300 mA h g−1 at 520 mA g−1 and good cycling performance (91% capacity retention after 100 cycles).
Song et al.124 developed a novel cross-linked polymer binder and found that it could effectively maintain the structural stability of RP anodes during cycling. They first prepared a chemically bonded RP-carbon nanotube hybrid (P-CNT) as an active material through ball milling, and then, this cross-linked sodium carboxymethyl cellulose–citric acid was used as a binder to fabricate a robust RP electrode, in which citric acid functioned as a cross-linker to form a 3D network when drying the electrode at 150 °C for 2 h. As a result, a high reversible capacity of 2134 mA h g−1 and good cycling stability (∼91% capacity retention after 100 cycles) were achieved, with a high initial coulombic efficiency of 84.7%. Similarly, Xu et al.125 used sodium alginate as a binder to synthesize an affordable phosphorus/carbon (APC) anode and obtained an improved electrochemical performance. APC nano/microspheres were prepared by ball milling of RP, flake graphite, CNTs, Super P® carbon and sodium alginate (SA, a biomass binder), followed by a closed spray drying process (Fig. 4a). The researchers proposed that this binder-filled phosphorus/carbon structure could maintain the structural integrity after repetitive volume variation and enhance the overall electrical conductivity of the RP anode due to its porous structure and strong surface interactions (Fig. 4b). As a result, this anode delivered a high reversible capacity of 1408 mA h gP−1 at 100 mA g−1 with a capacity retention of 82.6% for over 300 cycles and showed superior rate capabilities, demonstrating that this industrially adaptable process and the application of sodium alginate binders are promising methods for RP-based practical SIBs (Fig. 4c). However, the low RP content in this anode (∼42.5%) and the massive usage of carbon-based materials may cause issues with respect to gravimetric/volumetric capacity for practical application.
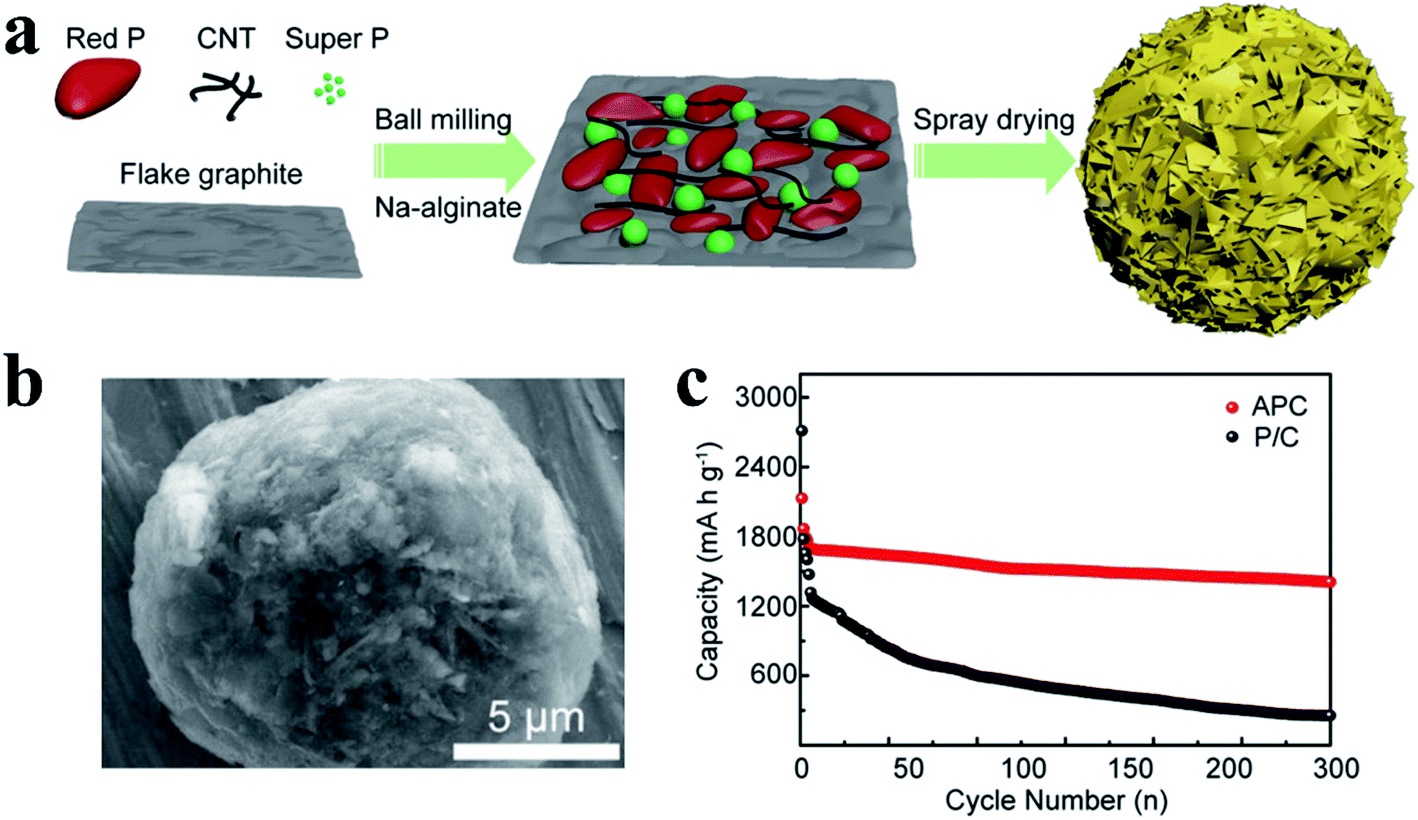 | ||
| Fig. 4 (a) Schematic of the synthetic process of phosphorus/carbon (APC) nano/microspheres. (b) SEM image of the typical morphology of the APC nano/microspheres. (c) Cycling performance of the anode at a constant current density of 100 mA g−1. P/C refers to the control material without SA binders. Reproduced with permission.125 Copyright 2018, American Chemical Society. | ||
To investigate whether the carbon defects of the carbon scaffold in a anode have an effect on the electrochemical performance, Li et al.126 designed a carbon scaffold containing controlled single-walled carbon nano-horn (SWCNH) and single-walled carbon nanotube (SWCNT) compositions, and combined this carbon scaffold with RP by hand milling. The researcher chose these two kinds of carbon for the reason that SWCNHs exhibit a significant presence of sp3 hybridized carbon defects embedded into the sp2 hybridized carbon matrix while SWCNTs are fully based on defect-free sp2 hybridized carbon species, and thus carefully controlled defect density composites can be obtained. Here, they reported that the stable alloy region in which NaP and Na5P4 were formed between 0.40 and 0.15 V versus Na/Na+ was mostly independent of carbon composite matrix chemistry and that an unstable alloying region (0–0.15 V) where defects could facilitate the continuous formation of irreversible Na3P products over the carbon surface. This work elucidates the mechanism why sp2 hybridized carbon is better suited as a reversible host material for RP anodes, and suggests that future effort can leverage surface engineering routes to further improve performance in this unstable alloying regime.
Interestingly, besides this ball milling method, Liu et al.127 broke RP particles into smaller clusters by using a high-shear emulsifying machine, and embedded these RP nanoparticles (∼97 nm) homogeneously in porous nitrogen-doped carbon nanofibers (RP@C) by a electrospinning technique (Fig. 5a). They first ball milled commercial RP into RP particles, and then these RP particles were broken into smaller clusters under a high energy shear force during the emulsifying process. After standing, the RP with small sizes (∼97 nm) in the upper liquid were collected and were homogeneously embedded in porous nitrogen-doped carbon nanofibers via a electrospinning process and a heat treatment (Fig. 5b and c). Although the yield of these small RP particles may be low as they only exist in the upper liquid, this method is still interesting because of its uniform particle size, and the larger RP particles in the sediment can be recycled for further preparation. In addition, these RP embedded nanofibers could be fabricated as a binder- and current collector-free anode, which could not only increase the P content in the anode, but also reduce the fabrication cost for practical application. The anode showed a high reversible Na storage capacity of 1308 mA h g−1 at 200 mA g−1, exceptionally high rate capabilities of 637 mA h g−1 at 5000 mA g−1 and 343 mA h g−1 at 10000 mA g−1 and long cycling stabilities of ∼81% capacity retention over 1000 cycles (Fig. 5d and e). Moreover, researchers also assembled a soft package Na-ion full battery using RP@C as the anode and Na3V2(PO4)2F3/C as the cathode and obtained an operational voltage of ∼3.65 V and a SIB energy density of 161.8 W h kg−1.
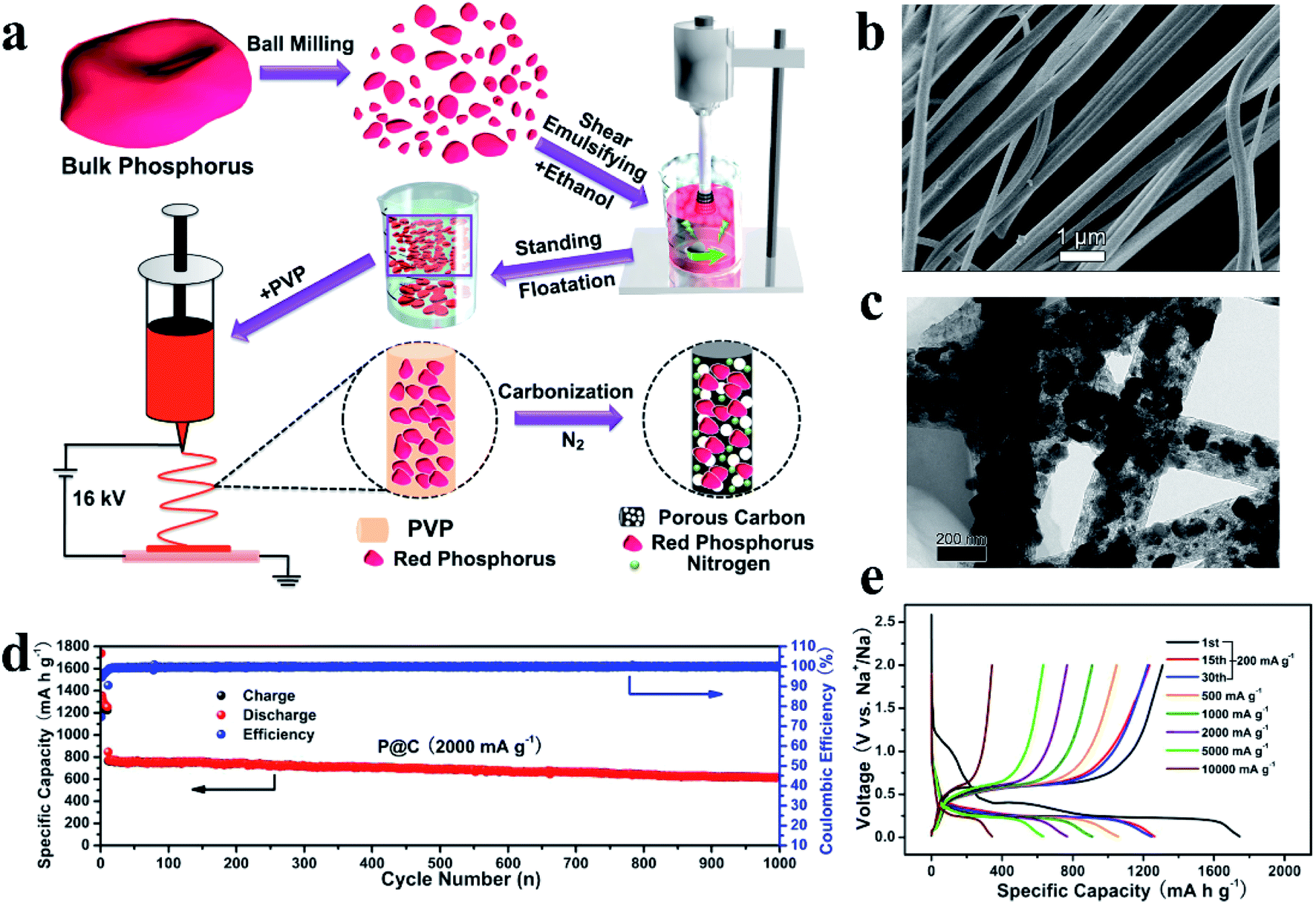 | ||
| Fig. 5 (a) Schematic illustration of the preparation process for RP@C nanofibers. (b, c) SEM and TEM images of red P@C nanofibers. (d) Long-term cycling stability of the P@C electrode at a current density of 2000 mA g−1. (e) Charge/discharge profiles obtained at various rates of the P@C electrode in the voltage range of 0.01–2 V vs. Na+/Na. Reproduced with permission.127 Copyright 2017, Elsevier B.V. | ||
Aside from ball milling or other mechanical force methods, the vaporization–condensation method is also an effective approach to prepare RP/carbon composites.115 In a typical process of this method, RP is first sublimated by heating and subsequently diffused into the pores or surface of a carbon matrix through capillary forces and pressure differences, and then the gaseous P is adsorbed and deposited onto the internal surface.81,116 As compared with the mechanical strategy, the vaporization–condensation method can ensure the uniform dispersal or adsorption of RP into carbon matrices and can obtain nanosized RP.76,116 As a result, the P/C composite fabricated by this method usually shows better long-term stability than that by the ball milling method. For the first time, Marino et al.115 prepared a carbon–phosphorus composite using this method for application in LIBs. Subsequently, this method was widely applied in SIBs, for example, Zhu et al.85 prepared a RP/single-walled carbon nanotube composite (P/SWCNT) as an anode, in which RP was uniformly distributed between tangled SWCNT bundles with an average particle size of ∼5 μm and a P content of ∼40%. Benefiting from this mild and nondestructive preparation process, uniform mixing and intimate contact between RP and SWCNTs could be achieved, and the mechanically strong SWCNT network was preserved, which enhanced the conductivity of the composite, helped to maintain the integrality of the anode, and stabilized the solid electrolyte interphase, leading to a stable long-term cycling performance with 80% capacity retention after 2000 cycles. However, because the particle size was quite large (∼5 μm), the rate capability of the anode was not sufficient (∼300 mA h g−1 at 2000 mA g−1), and the high cost of SWCNTs is also a concern for practical application. In another study, ordered mesoporous carbon (commercial CMK-3) with a pore size of ∼4 nm was also used as a carbon host material to load RP. In this RP/CMK-3 composite, the nanoscale of RP and the interconnected pores of the CMK-3 matrix provide many advantages for Na storage, such as accommodating the volume change of RP during cycling, reducing the diffusion length of Na+/e− and leading to an enhanced Na storage performance.128 However, the P content of this RP/CMK-3 composite was not high (∼31%), and the other RP/C composites prepared by this method in the literature also suffered from this same issue.117,129 The large amount of carbon in the composite is also a concern, which may lead to low volumetric capacity, low coulombic efficiency and other negative effects.130 Gao et al.131 developed a novel strategy and designed a unique structure: coating a 3D porous graphene-hydrogel/RP precursor with a poly-pyrrole layer, and thus the P content in the composite could be effectively maintained during the following vaporization–condensation–redistribution process. This coated composite showed a P content of ∼47.2%, which was more than twice that of the sample without a carbon layer (22.8%). In addition, the researchers proposed that the unique nanostructure of this 3D porous carbon matrix with RP particles (10–20 nm) uniformly distributed and firmly sealed in it was an ideal design for an anode material in SIBs.131
Recently, Liu et al.132 developed a method to densely and uniformly deposit RP nanodots onto reduced graphene oxide sheets (P@RGO) with RP nanodot sizes ranging from tens to several hundred nanometers (Fig. 6a–c). Here, the researchers suggested that this unique size and structure could minimize sodium ion diffusion lengths and sodiation/desodiation stress whereas the RGO network could serve as an efficient pathway for electron transfer and accommodate the volume variations of phosphorus particles. As a result, the P@RGO flexible anode reportedly achieved specific charge capacities of 1165.4, 510.6 and 135.3 mA h g−1 at 159.4, 31878.9, and 47818.3 mA g−1 respectively along with a 914 mA h g−1 capacity after 300 deep cycles at a current density of 1593.9 mA g−1, which was much better than that of conventional RP anodes for SIBs (Fig. 6d–f).
 | ||
| Fig. 6 (a) Schematic of the P@RGO synthesis. (b, c) SEM and TEM images of the P@RGO composite. (d) Cyclic voltammetry of the P@RGO anode at a scan rate of 0.1 mV s−1 between 0 and 3.0 V vs. Na/Na+. (e) Rate performance of the P@RGO anode. (f) Cycling performance of the P@RGO anode at a charge/discharge current density of 1593.9 mA g−1. Reproduced with permission.132 Copyright 2017, American Chemical Society. | ||
Furthermore, Zhou et al.133 developed a facile single-step flash-heat treatment to obtain reduced graphene oxide and deposit RP onto the rGO sheets simultaneously. This single-step method is energy-saving and is beneficial to preparation on a large scale. The resulting RP/rGO flexible film anode for SIBs produced an average capacity of 1625 mA h g−1 after 200 cycles at a charge/discharge current density of 1 A g−1 and also showed an excellent rate capability with average charge capacities of 1786, 1597, 1324 and 679 mA h g−1 at 1, 2, 4 and 6 A g−1 current densities respectively.
In addition to the application of these traditional carbon materials, the design and fabrication of novel carbon hosts with high electronic conductivity, high mechanical strength and porous morphology to load RP is an important topic for this vaporization–condensation method to further improve the Na storage performance. Based on this, Sun et al.134 used this approach to prepare composites of RP encapsulated into porous multichannel carbon nanofibers as a flexible anode material for SIBs and reported a rate capability of 500 mA h g−1 at 10 A g−1 and long cycle lifespans with 700 mA h g−1 capacity retention at 2 A g−1 after 920 cycles. The researchers suggested that the novel structure of the synthesized composite could reduce ion and electron diffusion barriers, accommodate volume variations during sodiation/desodiation, improve electrode mechanical strength, and thus lead to excellent rate capabilities and cycling performances. Liu et al.117 designed a carbon nanotube-backboned mesoporous carbon (TBMC) material as a matrix for RP (Fig. 7a). In their preparation process, resorcinol-formaldehyde resin/SiO2 was first grown onto carbon nanotubes, with tetraethyl orthosilicate (TEOS) used as the SiO2 precursor and resorcinol-formaldehyde (RF) resin as the carbon source, and then the resin was carbonized and the SiO2 was removed to obtain TBMC. The researchers reported that the competitive growth of SiO2 and RF resin during the synthesis process could lead to various SiO2 distributions and thus different final structures. Consequently, TBMC, hollow TBMC and deflated TBMC could be obtained based on high, medium and low dosages of RF respectively (Fig. 7a). These various TBMCs were subsequently infiltered with RP using the vaporization/condensation method to form P@TBMC composites. The researchers found that the TBMC with a high dosage of RF showed the best Na storage performance, providing a high reversible specific capacity of ∼1000 mA h g−1 at 0.05 A g−1, a superior rate performance of ∼430 mA h g−1 at 8 A g−1 and a long cycle lifespan with no capacity decay for 800 cycles at 2.5 A g−1 (Fig. 7b–d). They proposed that the multi-walled carbon nanotubes could facilitate electron transfer due to the high content of sp2 carbon whereas the mesoporous carbon layers provide voids to load P and ensure adequate spacing to accommodate large volume variations of P during cycles, leading to an excellent Na storage performance.
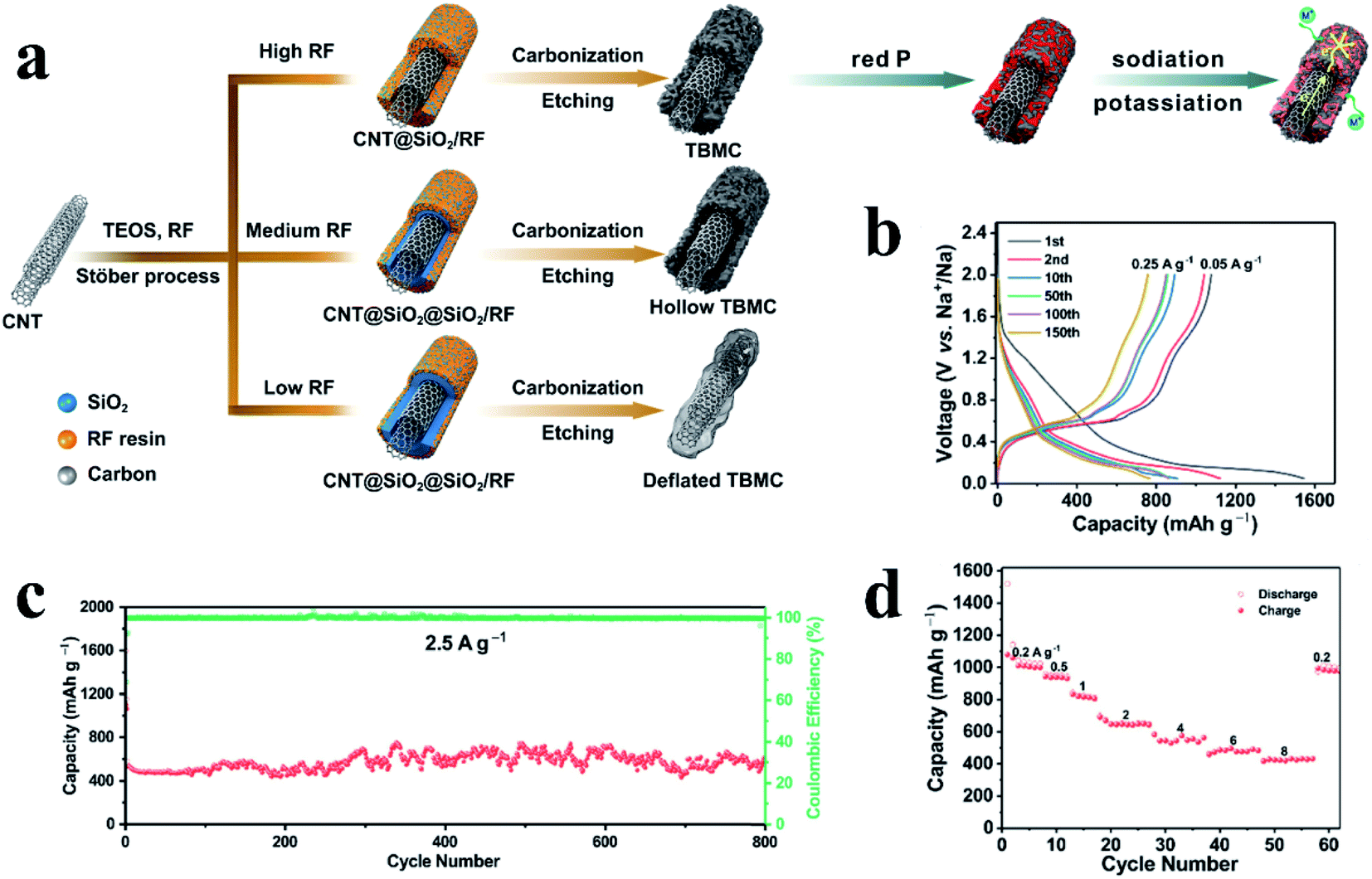 | ||
| Fig. 7 (a) Schematic of the synthesis of nanotube-backboned mesoporous carbon (TBMC), hollow TBMC and deflated TBMC and the sodiation/potassiation of the P@TBMC composite. (b) Representative galvanostatic discharge/charge voltage profiles of the P@TBMC electrode at 0.25 A g−1 after the initial two-cycle activation at 0.05 A g−1. (c) Long-term cycling performance of P@TBMC at 2.5 A g−1 and (d) rate capability of P@TBMC. Reproduced with permission.117 Copyright 2018, Elsevier Ltd. | ||
Yao et al.135 also designed novel hollow porous carbon nanospheres (HPCNSs) as host structures for the vaporization/condensation synthesis of P/C composites. Through the methods of molecular dynamic simulations and density functional theory calculations, they found that the morphology of polymeric P4 and the P loading in the P/C composite were mainly dependent on the pore size and surface conditions of the carbon support. The micropores of 1–2 nm in diameter and oxygenated functional groups attached on the carbon surface were essential for achieving high P loadings and high structural stability. This HPCNS/amorphous RP composite with enhanced structural/functional features presented a low volume expansion of ∼67.3% during anode cycles, much smaller than that of the theoretical value of commercial RP (≈400%), and showed remarkable long-life cycling stabilities with a capacity retention of over 76% after 1000 cycles. This study signified the importance of the rational design of electrode materials to obtain cost-effective P/C composite anodes for SIBs. Furthermore, Li et al.76 reported that a composite of confined amorphous RP with particle sizes of less than 1 nm in MOF-derived N-doped microporous carbon also produced promising performances.
These studies indicate that the design of favorable carbon hosts for RP is an effective approach for improving the Na storage performance. In addition, facile preparation methods for these materials are also necessary for practical applications.
Compared with the ball milling method, the vaporization/condensation method is a mild and nondestructive process; however, the phase-transformation process of P will inevitably generate white phosphorus, which is toxic and highly active.116 To address this, Yu et al.136 developed a modified vaporization–deposition–conversion method in which a multi-step heating/cooling process was used to avoid the generation of white phosphorus. By using this refined strategy, RP nanoparticles were adsorbed and confined within the nanopores of commercial microporous carbon YP-80F (P@YP) (Fig. 8a), ensuring high reversible capacity, excellent rate capability and long-term cycling stability with a superior capacity retention of 92% after 100 cycles and 46% after 1000 cycles (Fig. 8b–d). And as compared with P@YP, a P@CNT composite without confined P was deposited onto the surface of carbon nanotubes and presented rapid capacity decay with a capacity retention of only ∼40.6% after 100 cycles. Here, the researchers suggested that the nanostructure of the P–C composite with P confined in the porous carbon is key to enhanced electrochemical performances and long-term cycling stabilities.136
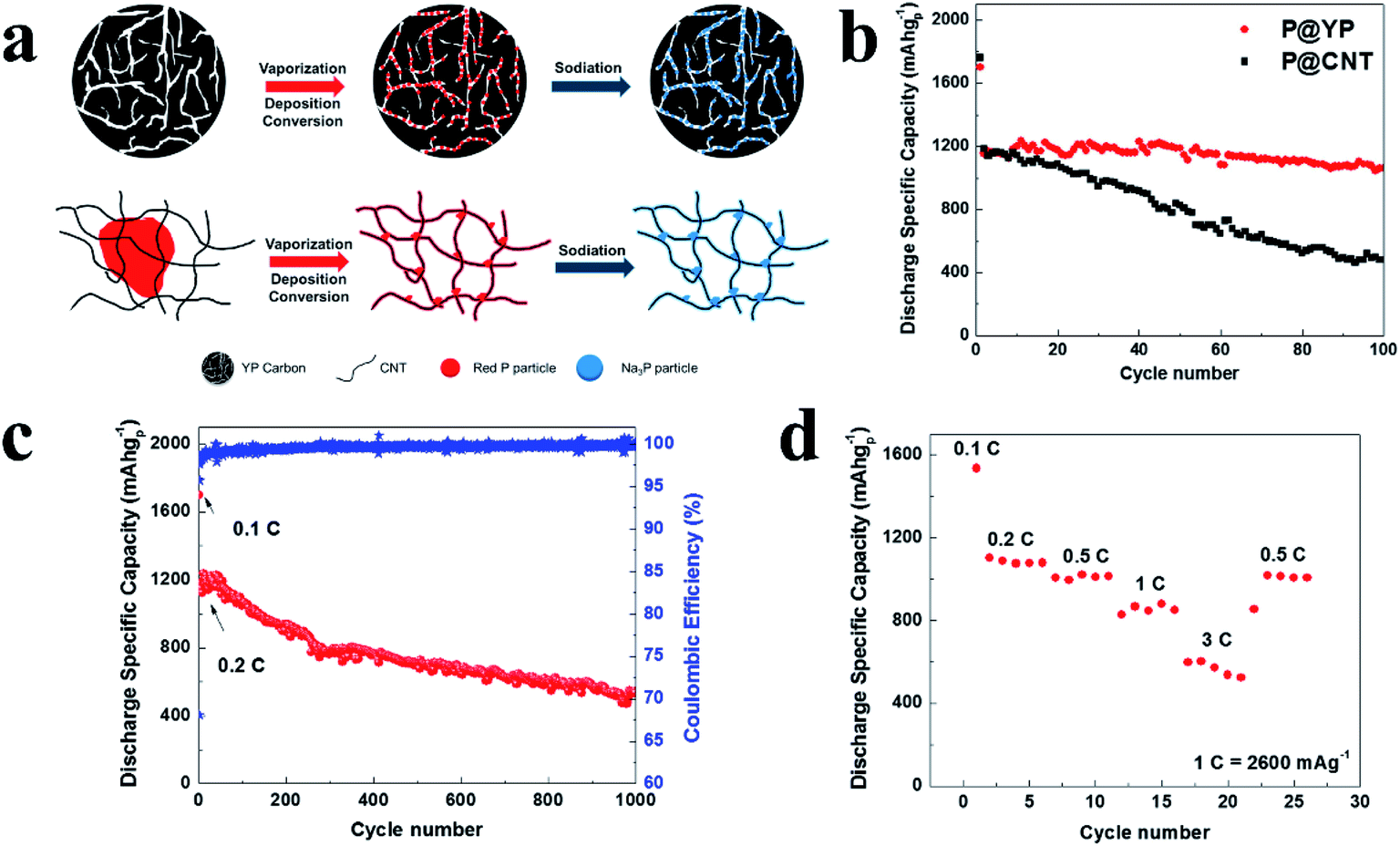 | ||
| Fig. 8 (a) Schematics of the preparation and sodiation of P@YP and P@CNT composites. (b) Cycling performances of P@YP and P@CNT composites within 100 cycles. (c) Long term cycling performance of the P@YP composite. (d) Rate performance of the P@YP composite at C-rates between 0.1C and 3C (1C = 2600 mA g−1). Reproduced with permission.136 Copyright 2017, Elsevier Ltd. | ||
In fact, the combination of RP with other non-carbon materials, such as metals, metal oxides or metal phosphides also demonstrates improved Na storage performance. For example, Chin et al.142 mixed commercial RP with appropriate amounts of iron using high-energy ball milling to obtain a RP/Fe composite. As a result, they found that the RP/Fe anode showed a higher sodium diffusion coefficient than pure RP anodes, and improved electrochemical performance of 1582, 1419, 884 and 676 mA h g−1 specific desodiation capacities at 0.2, 0.6, 6 and 10 A g−1 current densities respectively were obtained for the 9.1% Fe-containing RP composite. Although the cycle lifespan of this anode may not be long because of the pulverization of RP during charge/discharge processes, the addition of low cost iron is still a novel and effective approach to improve the electrochemical performance of RP. Based on the idea that the functional synergy of higher capacity of P with high-rate capability and high cyclability of Sb may obtain a compelling electrochemical performance, Walter et al.143 reported a P/Sb/Cu composite anode synthesized by simple mechanic mixing of monodisperse Sb nanocrystals with RP particles and Cu nanowires. Benefiting from the good electronic conductivity of Sb and Cu and the mechanical strength of Cu nanowires, this P/Sb/Cu anode showed much greater cycling stability than pure RP anodes, providing a capacity of above 1100 mA h g−1 after 50 cycles at a current density of 125 mA g−1, and an excellent rate capability of more than 900 mA h g−1 at a high current density of 2000 mA g−1. Compared with the addition of Fe within RP, the use of Sb and Cu in this study may increase the cost for practical application, as the P content in this P/Sb/Cu anode is low (40%). Nevertheless, the mechanical strength of Cu nanowires along with the matrix of Sb can ensure a longer cycle lifespan for this P/Sb/Cu anode.
Lan et al.130 prepared a phosphorus–titania–carbon (P–TiO2–C) composite as an anode for SIBs by ball milling of RP, titanium oxide and carbon powder. The researchers suggested that the P–TiO2–C composites could alter local chemistries in both titanium oxide and phosphorus such as the doping of TiO2 by P and/or C, as well as stabilization of P by the Ti–O–P and/or P–C chemical environment, and thus lead to an improved electronic conductivity and enhanced stability during repeated charge/discharge cycles. The resulting composite anode delivered a high reversible capacity of 1147 mA h g−1 at a current density of 100 mA g−1 with good cyclability and a high rate performance with 81.5% of its original capacity being maintained as discharge rates increased from 100 to 1000 mA g−1. In addition, a chain-like Fe3O4/C/RP (C means carbon) composite was also prepared by introducing a magnetic stimulus source during the vaporization/condensation process and also demonstrated high cycling performances and superior rate capabilities.129 Interestingly, this assisted magnetic field could cause a high pressure driven force to uniformly deposit RP onto the Fe3O4/C surface, and consequently increase the P loading and enhance the contact between Fe3O4/C and RP. Although the content of RP in this anode is quite low (25%) and the RP is only loaded on the surface of the conductive carbon chain, the addition of RP can obviously increase the specific capacity and shows little negative effects on the cycling stability. This indicates that the combination of low-cost metal oxides and RP may be an effective approach to apply in practical SIBs; however, the low electronic conductive of metal oxides is still a concern and more effort should be dedicated to addressing this issue.
Liu et al.86 designed RP@Ni–P core–shell nanostructures as anodes for SIBs through a two-step approach: electroless deposition of Ni on RP particles and then chemical dealloying of this RP@Ni compound, in which the Ni–P shell thicknesses could be controlled by regulating the dealloying time (Fig. 9a). Here, Liu et al.86 suggested that in situ generated Ni2P on RP particle surfaces could facilitate intimate contact between RP and the amorphous Ni–P outer shell which was mechanically strong and highly electron conductive, thus ensuring strong electrode structural integrity, stable solid electrolyte interphases and fast electronic transport. As a result, the RP@Ni–P composite with a dealloying time of 8 h displayed a super high capacity of 1256.2 mA h g−1 after 200 cycles at 260 mA g−1, a superior rate capability of 491 mA h g−1 at 5200 mA g−1 and an long cycle lifespan with a capacity of 409.1 mA h g−1 retained after 2000 cycles at 5000 mA g−1 (Fig. 9b and c). When the RP particle size is below 200 nm, the outer protective shell is a key factor in obtaining long cycle stability, which can effectively prevent the pulverization of RP during cycles. In addition, the facile process of in situ growth and the low-cost of Ni make it a promising strategy for practical SIBs. In another study, Kim et al.139 synthesized a P–TiP2–C nanocomposite as an anode material for SIBs using a facile and simple one-step high energy mechanical milling process in which the resulting composite anode (Fig. 9d–f) displayed an initial desodiation capacity of 755 mA h g−1 with a coulombic efficiency of 80% and a good capacity retention of 80% after 100 cycles. Here, the researchers suggested that the greatly improved electrochemical performances could be ascribed to the unique nanostructure in which RP and crystalline TiP2 were homogeneously embedded into a conductive carbon network. They also suggested that in this phosphorus-based composite, the conductive TiP2 could provide structural stability as well as high conductivity during cycling, leading to better SIB cycling stabilities and rate capabilities as compared with nanocomposites without TiP2.
 | ||
| Fig. 9 (a) Schematics of the electroless deposition of Ni on RP nanoparticles (i–iii) and the evolution of the RP@Ni–P core@shell nanostructure through chemical dealloying (iv–viii). (b) Long-term cycling performance of the 8 h RP@Ni–P electrode at 5 A g−1 between 0.05 and 2.0 V. (c) Galvanostatic charge–discharge voltage profiles of the 8 h RP@Ni–P anode between 0.05 and 2.0 V at a current density of 260 mA g−1. Reproduced with permission.86 Copyright 2017, The Royal Society of Chemistry. (d) Low magnification TEM image of the P–TiP2–C composite, (e) cycle performances and (f) rate capabilities of the P–C and P–TiP2–C composite electrodes in sodium half cells. The specific capacity was calculated based on the total mass of the composite materials. Reproduced with permission.139 Copyright 2016, American Chemical Society. | ||
 | (1) |
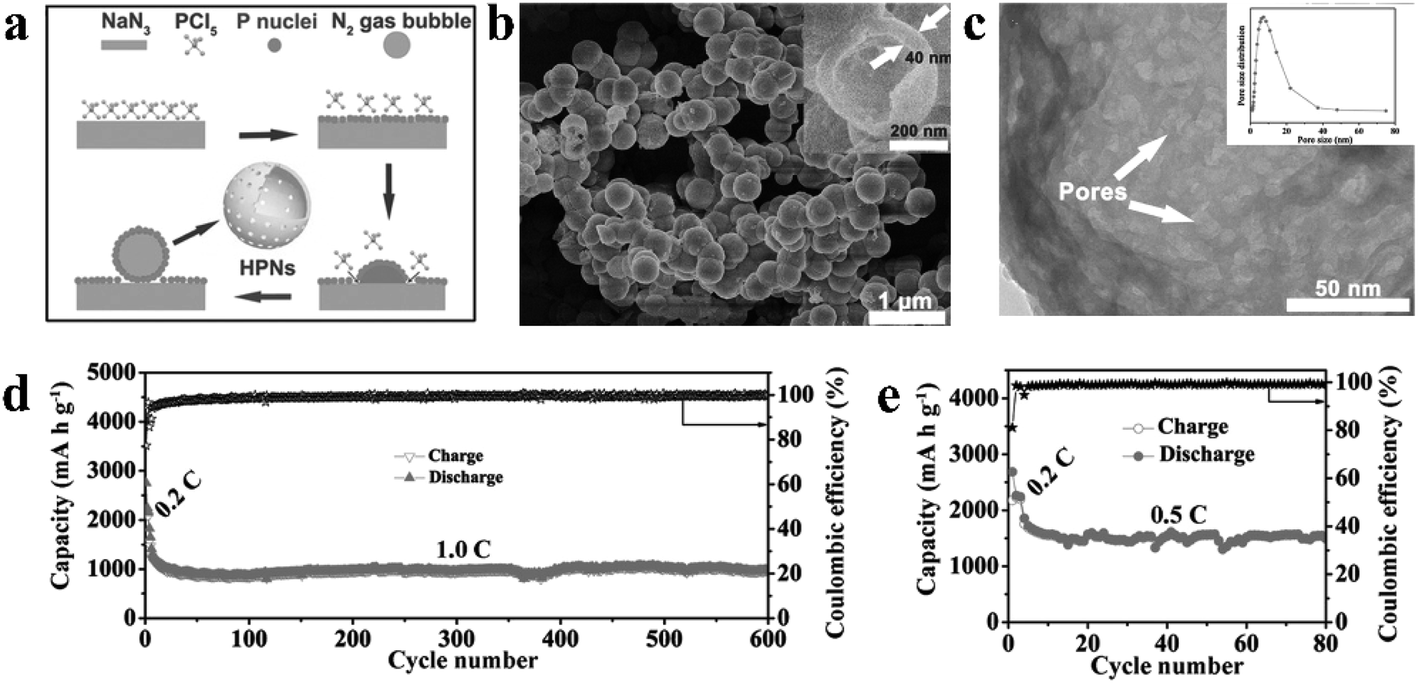 | ||
| Fig. 10 (a) Schematic of the proposed formation mechanism of hollow phosphorus nanospheres (HPNs). (b) SEM image of the prepared HPNs; inset shows the SEM image of a broken nanosphere. (c) TEM image of the HPNs under higher magnification; inset shows the pore-size distribution of the HPNs. Cycling stability and coulombic efficiency of the HPNs at (d) 1.0C for 600 cycles and (e) at 0.5C for 80 cycles. 1C = 2.6 A g−1. Reproduced with permission.87 Copyright 2017, WILEY-VCH. | ||
However, this pure phase RP nanostructure may suffer from insufficient electronic conductivity. Furthermore, the use of toxic and explosive NaN3 in the preparation process is also a concern. In another study, iodine-doped RP nanoparticles with size ranging from 100 to 200 nm have also been synthesized by a simple and large-scale solution phase method.140 During the reaction process, the PI3 precursor was reduced with ethylene glycol and simultaneously RP nanoparticles (RPNPs) were formed in the presence of CTAB under an ambient environment. The conductivity of these I-doped RPNPs was measured to be between 2.62 × 10−3 and 1.81 × 10−2 S m−1, which is close to that of semiconductor germanium (1.02 × 10−2 S m−1). Although the authors only investigated the Li-ion storage performance of these RPNPs, the unique morphology/nanostructure and the novel synthesis method are all progressive for the application of RP.
Based on this novel solution phase method, Liu et al.144 synthesized nanoporous RP on reduced graphene oxide as a SIB anode (NPRP@RGO) by a boiling method. In brief, the RP@RGO precursor was first synthesized involving several steps, including the reduction of phosphorus triiodide (PI3) to produce RP, the uniform mixing of the as-prepared RP with GO in a solution, and a further reduction of RP@GO to RP@RGO. Then this reduced RP@RGO was subsequently subjected to centrifugation and the concentrated RP@RGO solution was heated in a vacuum drying chamber at 200 °C to allow the RP to assemble around gas bubbles. And with the growth of these bubbles, increasing amounts of disordered RP particles became dispersed onto the surface of the gas bubbles and the final NPRP@RGO was obtained after the gas bubbles were detached (Fig. 11a). Here, the researchers reported that the unique nanostructure of the nanoporous RP with sizes mainly in the range of 20–100 nm (Fig. 11b) was able to accommodate volume change and minimize ion diffusion lengths and that the highly conductive RGO network sheets could facilitate fast electron and ion transportation. As a result, this anode exhibited a high capacity of 1249.7 mA h g−1 after 150 cycles at 173.26 mA g−1, a superior rate capability of 656.9 mA h−1 at 3465.28 mA g−1 based on the anode composite and a long cycle lifespan of 1500 cycles with a capacity of 775.3 mA h g−1 at 5.12 A g−1 based on the RP mass (Fig. 11c and d).144 In addition, in their another study,145 iodine-doped hollow nanoporous RP (HNPRP) was prepared by a similar preparation method. When tested as an anode for SIBs,140 this HNPRP exhibited a high capacity of 1658.2 mA h g−1 after 100 cycles at 0.26 A g−1, superior rate capability (759.6 mA h g−1 at 5.2 A g−1) and ultralong cycle-life (857.3 mA h g−1 after 1000 cycles at 2.6 A g−1).
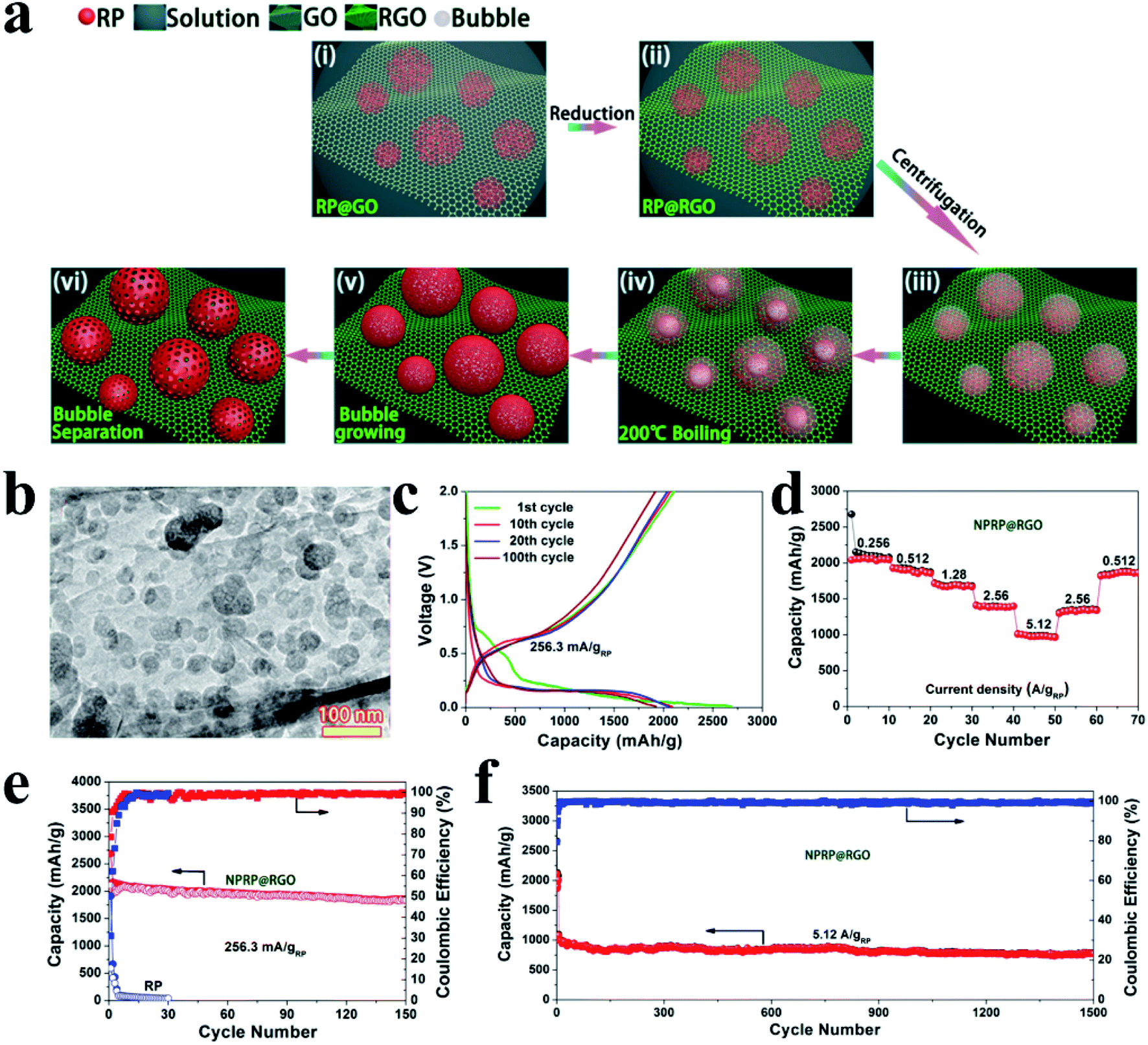 | ||
| Fig. 11 (a) Schematic of the preparation of NPRP@RGO. (b) TEM image of NPRP@RGO. (c) Galvanostatic charge/discharge curves of the NPRP@RGO electrode at 256.3 mA gRP−1 (173.26 mA gcomposite−1) within a voltage range of 0.01–2 V. (d) Rate capability of the NPRP@RGO electrode from 0.256 to 5.12 A gRP−1. (e) Cycling performances of NPRP@RGO and commercial RP electrodes at 256.3 mA gRP−1 within the voltage range of 0.01–2 V. (f) Long-term cycling performance of the NPRP@RGO electrode at 5.12 A gRP−1 (3465.28 mA gcomposite−1) between 0.01 and 2.0 V. Current density and specific capacity were calculated based on the mass of NPRP. Reproduced with permission.144 Copyright 2018, American Chemical Society. | ||
Sun et al.101 further fabricated ultrafine RP particles with an average size of ∼10 nm embedded in a 3D carbon framework through the carbothermic reduction of P2O5 and reported that these small sized phosphorus particles could accommodate large stress without cracking, decrease electron/ion diffusion lengths and connect strongly with the carbon framework. And as a result, their SIB anode provided a reversible specific capacity of 1027 mA h g−1 and a high capacity retention of 88% over 160 cycles. RP quantum dots (RPQDs) were also prepared using a reliable hydrothermal method with commercial RP as the only phosphorus source.146 Although the yield of RPQDs in this synthesis process was low, this method was quite simple, and the size of these quantum dots was at the nano-level with an average size of 4.0 nm. And a hybrid of these dots with rGO (RPQDs/rGO) delivered an initial specific capacity of 1161 mA h g−1 and a low capacity deterioration rate of less than 0.12% per cycle after 250 cycles at 200 mA g−1.
2.2 Black phosphorus and phosphorene
BP is another important allotrope of phosphorus and is most thermodynamically stable among these allotropes.88,147,148 Due to its layered structure and other properties, BP attracts much attention and shows great potential for many applications such as batteries,72,149 field-effect transistors,150 and photodetectors.149 BP possesses three crystalline modifications induced by different temperature and pressure,151 including orthorhombic, rhombohedral, and cubic, and also can present an amorphous form.152,153 Under normal temperature and pressure, BP usually shows an orthorhombic crystallinity with a layered structure, as shown in Fig. 12a and d.89 Here, phosphorus atoms are covalently bonded with three adjacent phosphorus atoms to form a puckered honeycomb structure in a single layer of BP,89,150 and these individual atomic layers are stacked together by van der Waals interactions, similar to graphite (Fig. 12a),148,150,154 giving a calculated density of ∼2.69 g cm−3. The layer-to-layer spacing is about 5.3 Å, and there are no contacts shorter than 3.59 Å for atoms between successive layers.154 BP shows good electronic conductivities (∼66–300 S m−1vs. ∼10−12 S m−1 for RP)155,156 and has a suitable operating potential (≈0.45 V vs. Na/Na+) for Na storage.157 In addition, the interlayer channel size of BP is ∼3.08 Å, which is suitable for the storage of both lithium (1.52 Å) and sodium (2.04 Å) ions in the channels (Fig. 12a).148,158 However, sluggish reaction kinetics and large volume changes (300–500%) during desodiation/sodiation hinder its application as a SIB anode.158,159 Furthermore, the lack of effective, low-cost synthesis methods to produce BP on a large scale is a major concern for its practical application. Currently, the main strategies for preparing BP include high-energy mechanical milling,147 mineralizer-assisted gas-phase transformation160 and high-pressure (or with high-temperature) conversion from RP.161,162 All of these methods are critically dependent on the preparative conditions. With respect to this, some progress for novel methods to synthesize BP has been made recently.163–166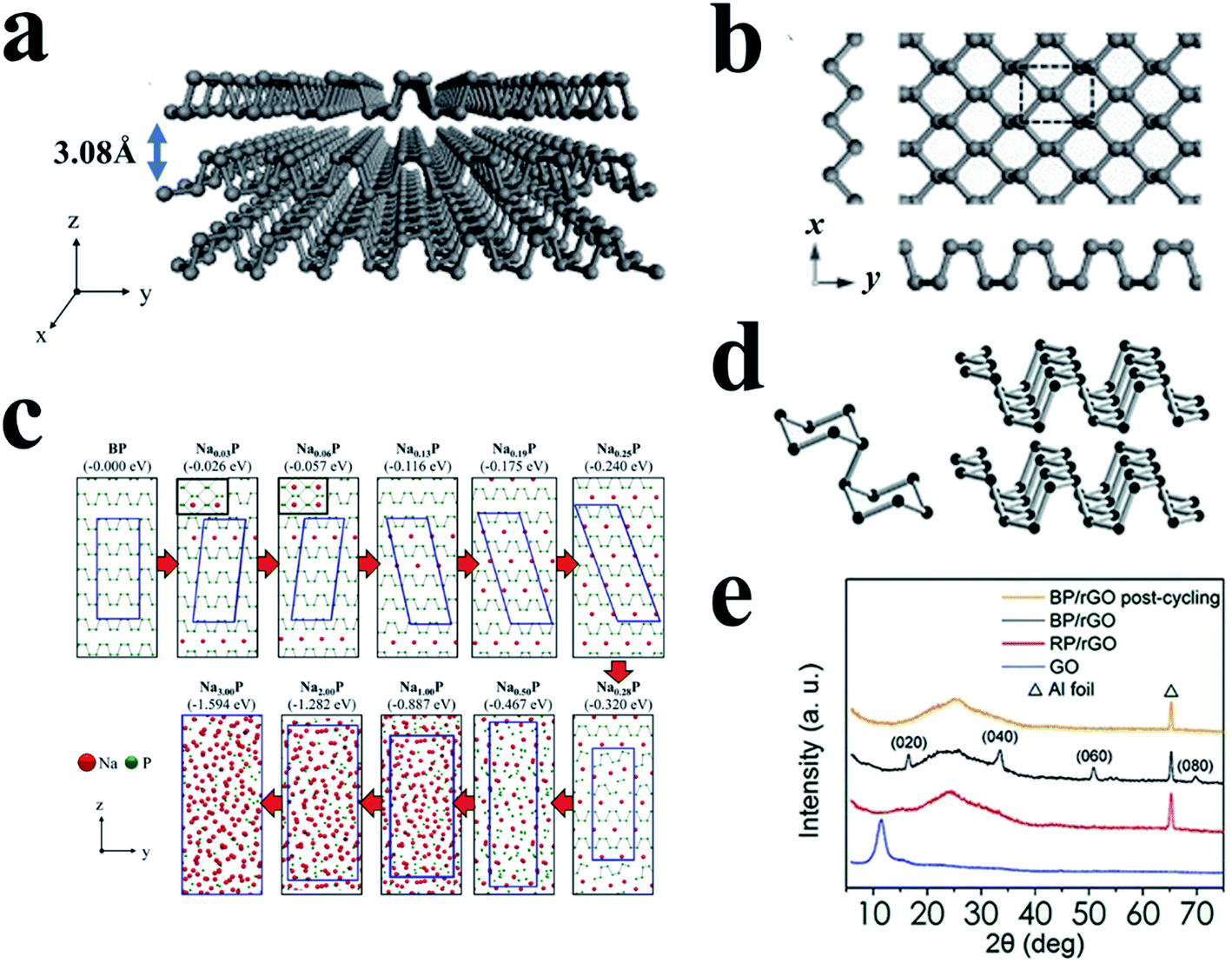 | ||
| Fig. 12 (a) The puckered structure of a few-layer BP sheet. (b) Top view of monolayer BP. The x and y axes are along zigzag and armchair directions, respectively. Reproduced with permission.89 Copyright 2015, Wiley-VCH. (c) Sodiation mechanism of black phosphorus. The numbers in parentheses indicate calculated formation energies of NaxP structures. Reproduced with permission.167 Copyright 2015, American Chemical Society. (d) Characteristic structural fragments of BP. Reproduced with permission.113 Copyright 2017, Wiley-VCH. (e) XRD patterns of the GO, RP/rGO precursor, as-prepared and post-cycling BP/rGO samples. Reproduced with permission.162 Copyright 2018, American Chemical Society. | ||
Compared with RP, the electrochemical mechanism of BP to store Na is easier to unravel due to its crystalline structure. And a thorough understanding of the chemistry that occurs in sodiation and desodiation can elucidate the structure–performance relationships responsible for high capacities and help to devise strategies to mitigate the performance degradation.168 Many theoretical calculations have been carried out to understand the process of sodiation for BP.167,169–171 Based on these investigations, sodiation of BP includes two steps: (1) an intercalation process occurring in the interlayer channels of BP (along the x-axial) until forming the composition of Na0.25P (diffusion barrier of 0.18 eV); and (2) an alloying process to form a layered NaxP structure and eventually an Na3P phase. After the intercalation process (step 1) was completed, one Na atom tended to bind with four P atoms, resulting in sliding of the phosphorene layers (Fig. 12c). At Na levels beyond Na0.25P, alloying with P could lead to the breaking of P–P bonds, in which the staggered P–P bonds were preferentially broken rather than the planar P–P bonds, generating P2 dumbbells. As sodiation proceeded further, most of the P2 dumbbells became isolated P atoms. Thus, after full sodiation to form the amorphous Na3P phase, only low-coordinate P components such as isolated atoms (primarily) and dumbbells could be found.
However, the mechanism of the desodiation process was little reported in the literature. In addition, it seems that the crystalline BP was converted into an amorphous form during cycling, and this has been experimentally evidenced. In a study, Liu et al.162 found that the XRD characteristic peaks of BP disappeared after repeated charge and discharge (Fig. 12e), suggesting that the BP did not recover to its crystalline phase after desodiation, which was consistent with the results of Raman patterns, TEM and fast Fourier transform (FFT) images. If the BP was amorphized during cycling, it was difficult to explain why the cycling performance of anodes prepared from BP could continue to exceed that of anodes prepared from RP. To answer this question, researchers speculated that phosphorus (like C, H2O, and silicate glasses) might be polyamorphous and that amorphous P formed from BP could retain both structural and electronic advantages over RP or amorphous products formed from RP, and BP-graphite/graphene composites might play a role in maintaining the layered structures of BP.72,162 In addition, it was proposed that during the charge–discharge process, the electrolyte molecules could intercalate into the layer spaces and then lead to exfoliation of the layered structures and thus BP-graphite/graphene composites or pillared BP might be good options to improve the stability of the layered structure of BP during sodiation.167,169–171
Recently, Marbella et al.168 investigated phosphorus intermediates in BP anodes during the sodiation and desodiation process on a molecular level by a combined experimental and theoretical approach, involving techniques such as solid-state NMR (ssNMR), powder X-ray diffraction (XRD), and DFT and NMR calculations. Specifically, individual P binding sites within the amorphous NaxP phases that were formed during cycling can be probed with 2D 31P phase-adjusted spinning sideband (PASS) ssNMR experiments, thus providing information on the geometry of chemical bonding environments and structures of molecular fragments. In their study, ex situ31P magic-angle spinning (MAS) NMR spectra were acquired at different stages of (de)sodiation to probe the structural features of various NaxP phases in BP anodes (Fig. 13a). During the first sodiation, 31P NMR shifts consistent with P helices/zig-zags were present as early as 0.60 V (Na0.52P), suggesting that P–P cleavage began at this composition. The ex situ XRD results also indicated that an amorphous or highly disordered phase was present in this stage in addition to residual BP. P helices were the predominant structural motifs that were subsequently formed after this stage. In addition, P helices and zig-zags (which are likely truncated helices) might allow some of the original alternating planar/staggered P–P bonds found in pristine BP to persist while also forming an energetically favorable NaxP phase. As BP was further sodiated to 0.38 V, the amorphous NaxP phase (780 mA h g−1, approximate composition of NaP) showed 31P sites consistent with P helices and terminal P units from short P chains (four-membered P zig-zags), as shown in Fig. 13b–e. The corresponding ex situ XRD results also indicated that this phase was amorphous (a-NaxP). In addition, the researchers proposed that dumbbell-containing structures previously predicted by others did not form in a measurable quantity in these SIBs during cycling. Once the potential dropped below 0.22 V, the formation of a lower energy crystalline Na3P–P63cm structure occurred. And a plateau at 0.21 V (ranging from 1358–2340 mA h g−1, corresponding x to 1.57–2.70) was observed and could be attributed to the formation of c-Na3P–P63cm, which is the most stable crystal structure of crystalline Na3P upon sodiation based on DFT calculations. During desodiation, the c-Na3P–P63cm31P resonance at −207 ppm persisted until >0.60 V (>1490 mA h g−1, approximately Na1.28P), as shown in Fig. 13a. At 0.8 V (1730 mA h g−1, approximately NaP), P motifs of helices could again be observed in a-NaP, but with longer average Na–P interatomic distances (Fig. 13g and h). Increased structural disorder was also observed and might be due to a larger range of clusters that were formed from the isolated P ions in Na3P, as opposed to the original P–P bonded BP phase. At the end of desodiation, the researchers found that BP was not re-formed, which might contribute to the poor capacity retention observed in the Na–P system. And the results of NMR also indicated that the Na ions were not completely removed during desodiation and/or were consumed in deleterious side reactions.
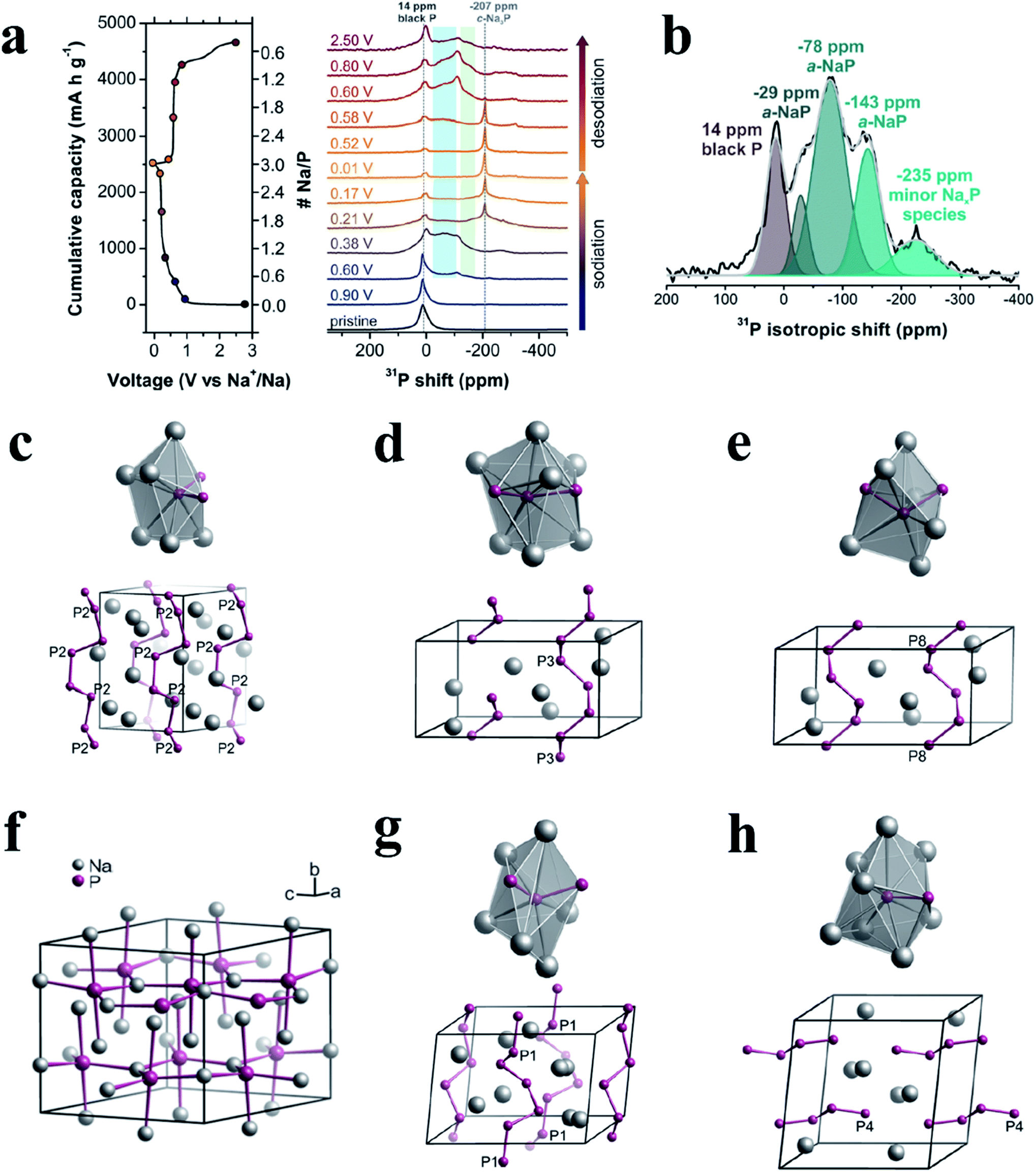 | ||
| Fig. 13 (a) Ex situ31P MAS NMR spectra of BP recorded at various stages of sodiation/desodiation (right) and the corresponding electrochemistry (left). 31P chemical shift regions corresponding to P helical motifs and P near the end of chains are shaded in blue and green respectively. (b) Deconvolution of the 31P isotropic projection from 2D 31P phase-adjusted spinning sideband (PASS) experiments (black line) performed on a sample of approximate composition NaP, extracted from a SIB at 0.38 V (780 mA h g−1) during sodiation. Five regions with centers-of-mass at 14 (BP), −29, −78, −143, and −235 ppm are observed. (c–e) The corresponding P sites (top) and extended P structures (bottom) of sideband patterns (calculated) for different isotropic chemical shifts, δiso = −29 (c), −78 (d), and −143 ppm (e), extracted from a SIB at 0.38 V (780 mA h g−1) during sodiation. (f) Crystal structures of Na3P–P63cm (calculated). (g, h) The corresponding P sites (top) and extended P structures (bottom) of sideband patterns (calculated) for δiso = −29 (g) and −78 (h), extracted from a SIB at 0.80 V (1730 mA h g−1) during desodiation. The symbol of Px, x = 2, 3, 8, 1, 4 (c–e, g and h), denotes individual 31P sites in certain structural models. Reproduced with permission.168 Copyright 2018, American Chemical Society. | ||
Compared with pure theoretical calculations, the use of experimental methods can make it more authentic and concrete for understanding the mechanism of Na storage, and more leveraging of advanced and suitable detecting instruments to study the behavior of active materials in SIBs is necessary.
To solve the issue of sluggish reaction kinetics, Dou et al.159 utilized nanoscale surface engineering to homogeneously deposit highly electron conductive poly(3,4-ethylenedioxythiophene) (PEDOT) nanofibers onto special surface-modified BP nanosheets (Fig. 14a) and reported that this design could enhance charge transfer kinetics and super surface wettability with electrolytes, leading to enhancement in the reaction kinetics of sodium storage (Fig. 14b and c). In another study, Wang et al.172 used 4-nitrobenzenediazonium (4-NBD) to modify BP and further made it chemically bond with reduced graphene oxide to prepare a hybrid as an anode material. The researchers reported that due to enhanced interactions between the chemically functionalized BP and graphene, the cycling performance of the resulting SIB anode was significantly improved, exhibiting a specific capacity of 1472 mA h g−1 at 0.1 A g−1 in the 50th cycle and 650 mA h g−1 at 1 A g−1 after 200 cycles. The results further indicated that these interactions could improve BP stability during long term cycling, reduce surface energies and increase BP thicknesses, all of which enabled the enlargement of channels between BP nanosheets and graphene, allowing more sodium ions to be stored for improved cycling performances. According to these studies, it can be seen that surface engineering is an effective way to improve the reaction kinetics of BP and thus can enhance the rate capability.
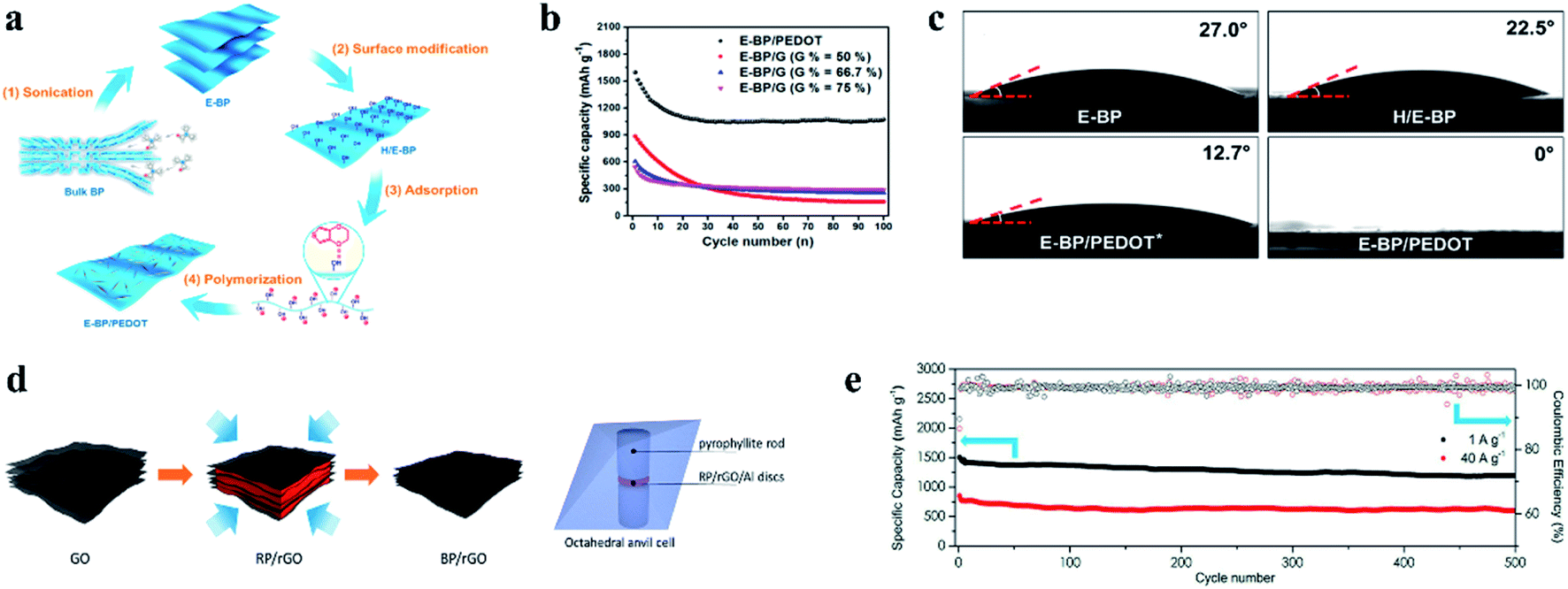 | ||
| Fig. 14 (a) Schematic of the preparation of the E-BP/PEDOT architecture. (b) Cycling life of E-BP/PEDOT and E-BP/graphene hybrid electrodes with different ratios at 100 mA g−1. (c) Characterization of wetting properties and optical images of contact angles between electrolytes and E-BP, H/E-BP, E-BP/PEDOT * (less PEDOT grown on BP nanosheets) and E-BP/PEDOT electrodes. (a–c) Reproduced with permission.159 Copyright 2017, Elsevier Ltd. (d) Schematic description of the BP/rGO synthesis. (e) Cycling performance of the BP/rGO anodes at charge and discharge current densities of 1 and 40 A g−1 in black and red, respectively. (d, e) Reproduced with permission.162 Copyright 2018, American Chemical Society. | ||
Similar to RP, direct combination with carbon-based materials is an effective approach to overcome the drawbacks of BP for Na storage. For example, Xu et al.173 reported that a composite of ball milled BP/Ketjen-black/multiwalled carbon nanotubes (BPC) could exhibit high performances: a first-cycle reversible capacity of 2011.1 mA h g−1, with an initial coulombic efficiency of 91.1% and a capacity of 1700 mA h g−1 over 100 cycles at a current of 1.3 A g−1. Similarly, Haghighat-Shishavan et al.174 also prepared a composite of BP and multiwalled carbon nanotubes (BP-CNTs) through ball milling process; in addition, they found that appropriate air exposure could successfully alter BP powder from a naturally hydrophobic form to a desired hydrophilic form, which is essential to the formation of stable bonds between BP and functionalized CNTs during ball milling. In this study, a binary polymeric binder of sodium carboxyl methyl cellulose–poly acrylic acid (NaCMC–PAA) was used to fabricate an anode; as a result, the resulting anode delivered a first discharge capacity of 2073 mA h g−1 at 0.2C for SIBs, with a high capacity retention of 75.3% after 200 cycles, and a rate capacity of 609 mA h g−1 at 2C was obtained. Here, the researchers suggested the strong bonds between BP and the hydroxyl groups of NaCMC and carboxylic acid groups of PAA played an important role for the improved Na storage performance of BP.174 In another study, Li et al.175 reported a BP/graphene (BP/G) 2D macroscopic heterostructure prepared by a co-exfoliation/electrophoretic deposition process. When used as a binder- and additive-free anode for SIBs, this composite demonstrated superior specific capacities of 2365 mA h gP−1, 1894 mA h gP−1 and 1456 mA h gP−1 at 100, 200 and 500 mA gP−1 rates respectively and showed a stable cycling performance. This novel preparation method ensures the intimate contact of BP and graphene, and thus a binder- and additive-free anode can be obtained, not only increasing the specific capacity but also reducing the cost for practical application. Recently, a BP-graphene layered structure was synthesized directly from a RP-graphene precursor by a low-cost and scalable pressurization (8 GP) process at room temperature (Fig. 14d).162 These hybrids could also be directly used as anode materials without a conductive additive and binder for SIBs and showed excellent rate capabilities and cycling stabilities (∼1250 mA h g−1 at 1 A g−1 and ∼640 mA h g−1 at 40 A g−1 after 600 cycles) (Fig. 14e). This study combines the synthesis of BP and the preparation of a BP-graphene hybrid anode in one step, offering excellent possibility for scalable production of composites for practical SIBs.
Besides carbon materials to be combined with BP, researchers have also explored composites with other types of materials. For example, Meng et al.176 prepared a composite of BP quantum dots (BPQDs) anchored onto Ti3C2 nanosheets (TNSs) as an anode material for SIBs by a assembling process with the assistance of sonication and long-term stirring. They reported that due to the addition of TNSs, the BPQDs were endowed with improved conductivity and reduced stress during cycling, allowing for a high initial discharge capacity of ∼723 mA h g−1 and outstanding long-term cycling stabilities up to 1000 cycles with a capacity retention of nearly 100%. In another study, Song et al.177 employed a simple high-energy mechanical milling approach to fabricate a novel and stable sodium titanate/carbon-BP (Na2Ti3O7/C-BP) hybrid as a SIB anode material and found that the BP nanoparticles could interconnect with bare NTO through P–O–Ti bonds and/or P–C bonds. As a result, the NTO/C-BP hybrid delivered specific capacities of ∼225 mA h g−1 at 20 mA g−1 after 55 cycles and ∼183 mA h g−1 after 100 cycles at 100 mA g−1. The researchers also suggested that the formation of stable P–C bonds and P–O–Ti bonds was a promising strategy to fabricate other Ti-based and P-based electrode materials for practical application in SIBs. However, the BP content in this composite is quite low (20%) which results in a reduced special capacity, and the large specific surface area of these ball milled hybrids causes a low initial coulombic efficiency (42%), which is not suitable for practical applications. Xia's group178 synthesized a composite of sulfur-doped BP-TiO2 (TiO2-BP-S) as an anode material for SIBs using a feasible and large-scale high energy ball milling method and obtained a discharge specific capacity of up to 490 mA h g−1 after 100 cycles at 50 mA g−1 and good cycling stabilities with 290 mA h g−1 capacity retention after 600 cycles at 500 mA g−1. In another study, Ding et al.179 also designed a strategy to produce BP composites in which they first ball milled amorphous RP with cubic boron nitride to obtain a BP/c-BN composite, and then wrapped this composite by nanoporous graphene networks via a sonication method. In this composite, the super hard c-BN could not only reduce the size of RP during ball milling, but also act as a mechanical skeleton to suppress and buffer the large volume expansion of BP during the sodiation process. As a result, this BP/c-BN/Gr composite anode demonstrated high cyclability with a stable specific capacity of 947 mA h g−1 and a ∼90% capacity retention after 100 cycles at a current density of 50 mA g−1. This is the first time that a super hard material was introduced into P-based anodes for SIBs; however, the high electron resistance of such c-BN may cause insufficient rate capability, and finding materials that possess both high mechanical strength and high electronic conductivity to host BP is a promising strategy for practical SIBs.
Phosphorene is a new 2D material that contains one or few layers of BP (Fig. 12a and b)89 and can be obtained from bulk BP through various methods180–183 or even from white phosphorus.164 These 2D phosphorene materials possess unique properties such as enlarged specific surface areas, increased interlayer distances,72 and fast electron transfer and Na diffusivity (energy barrier of only 0.04 eV for monolayer BP),184,185 and have attracted significant attention and witnessed development in SIB applications. For example, Sun et al.72 synthesized a hybrid with several layers of phosphorene sandwiched between graphene layers and obtained a specific capacity of 2440 mA h g−1 (based on phosphorus) at a current density of 50 mA g−1 and a 83% capacity retention after 100 cycles. The results of in situ TEM and ex situ XRD indicated a dual Na storage mechanism involving the intercalation of sodium ions along the x-axis of phosphorene and the formation of Na3P, which is consistent with the theoretical calculations carried out by other researchers.167 Furthermore, the graphene layers in this hybrid were found to serve as a mechanical backbone and an electrical highway to accommodate the anisotropic expansion of phosphorene layers along the y- and z-axial directions for stable cycling operations, leading to excellent stability.72
The lack of large-scale and low-cost methods to prepare phosphorene hinders its practical applications; however, developments have been made after continuous efforts, such as mechanical exfoliation and liquid-phase peeling.180,182,183,186,187 Recently, Huang et al.188 developed a electrochemical cationic intercalation method to prepare phosphorene from BP crystals and applied it as an anode material for SIBs (Fig. 15a). Here, tetraalkylammonium cations were used to intercalate BP, and FL-P-5 meant that a potential of −5 V was applied to exfoliate bulk BP; similarly, FL-P-10 and FL-P-15 correspond to potentials of −10 and −15, respectively. The researchers found that the number of BP layers (from 2 to 11 layers) could be manipulated by changing the applied potential and that the BP layers were free of surface functional groups (Fig. 15b–d). As a result, this phosphorene anode showed excellent performances for SIBs, delivering a capacity of 1968 mA h g−1 after 15 cycles and ∼1190 mA h g−1 after 50 cycles at a current density of 100 mA g−1 (Fig. 15f and g).
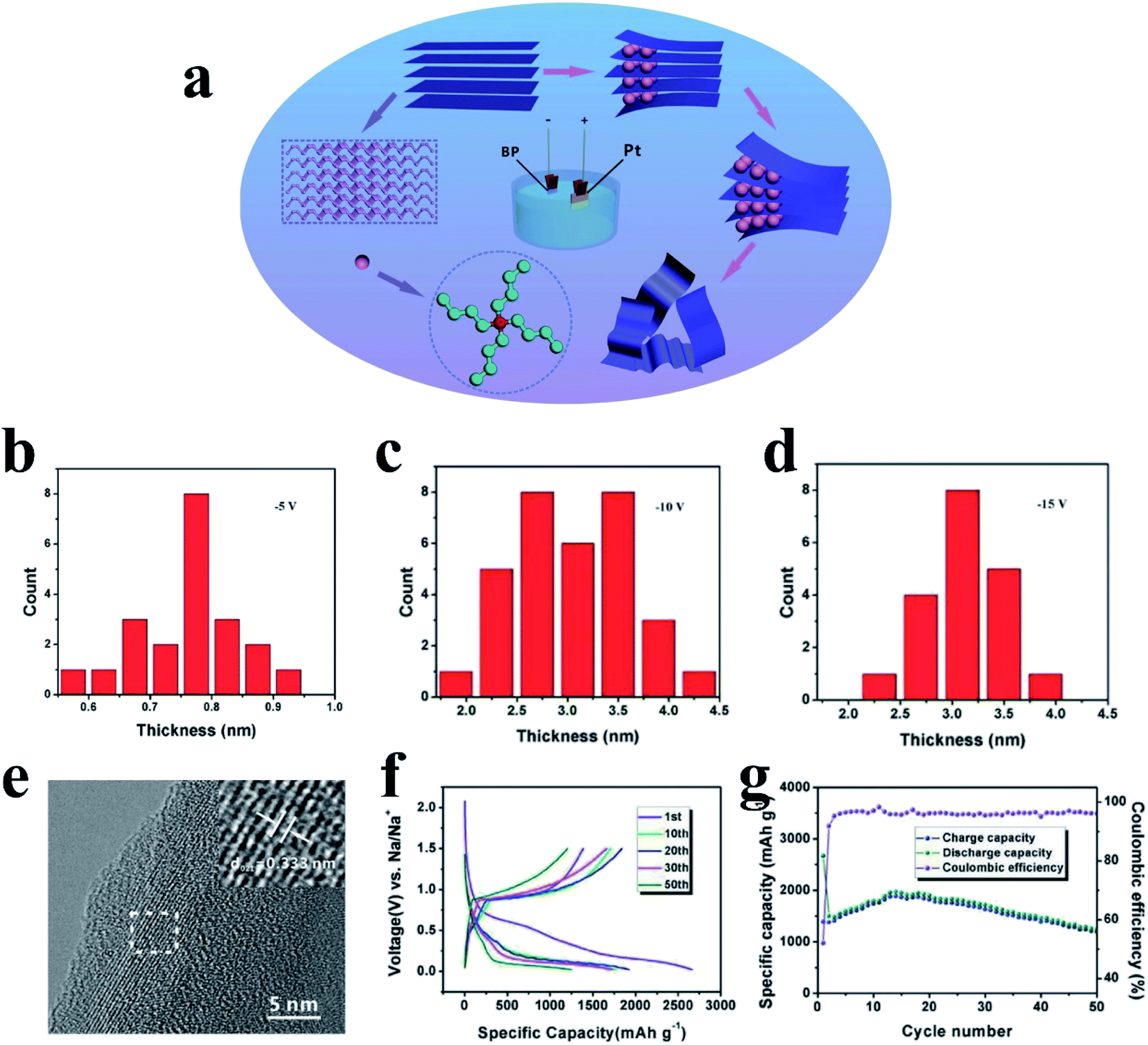 | ||
| Fig. 15 (a) Schematic of the electrochemical exfoliation process of phosphorene. (b–d) The corresponding thickness distribution charts of FL-P-5. FL-P-10, and FL-P-15, respectively. (e) HRTEM image of FL-P-5. (f) Charge and discharge profiles of different cycles at 100 mA g−1. (g) Cycling performances and coulombic efficiencies at 100 mA g−1. Reproduced with permission.188 Copyright 2017, WILEY-VCH. | ||
3 Metal phosphides
Metal phosphides (MPs) are also promising anode materials for SIBs due to their noticeably high gravimetric and volumetric specific capacities, appropriate redox potentials and higher electrical conductivities as compared with pure phosphorus.77,189 The higher performance of MPs as SIB anodes can mainly be attributed to the presence of metal atoms acting as electronic pathways that drastically enhance the electronic conductivity.190 In addition, conductive metal particles can be formed in MP anodes during battery cycling which further increases the conductivity of electrodes.91,189 Furthermore, in some MPs, metal nanocrystals are embedded in phosphorus matrices during cycling and thus inhibit the agglomeration of metals and improve the cycling performance of MP anodes. However, there are also drawbacks of MP anodes, including large volume variations and poor diffusion kinetics during cycling which is similar to phosphorus. To overcome these disadvantages and allow MPs to become more practical for SIBs, extensive investigations have been conducted and various MP nanostructures have been prepared, such as yolk–shell Sn4P3@C nanospheres,91 CoP nanowires,191 monodisperse Ni2P particles92 and hollow FeP@carbon nanocomposites,192 all of which possess unique nanostructures or conductive matrices that could accommodate volume expansion and offer more active sites for Na insertion/extraction and thus high electrochemical performances.In general, the reaction mechanisms of MPs are more complex than those of pure phosphorus. Based on whether the metal elements in MPs can electrochemically react with sodium, MPs can be divided into metal-active phosphides and metal-inactive phosphides. For metal-active phosphides, both the metal element and P phase can react with Na, and in a typical reaction process, crystalline metal and P phases are formed after the first cycle through a conversion process and react with sodium during the subsequent cycles through a reversible alloying process respectively, as expressed as follows:74,77,91,193,194
| MPx + (3x + y)Na+ + (3x + y)e− → NayM + xNa3P | (2) |
| NayM ↔ M + yNa+ + ye− | (3) |
| Na3P ↔ 3Na+ + P + 3e− | (4) |
Examples of metal-active phosphides include Sn4P3 (ref. 91 and 195) and GeP5.193
For another kind of metal-active phosphide, crystalline metal and P phases can reform the original phase of MPs after desodiation, such as GeP3 (ref. 196) and GeP,197 and the process can be expressed as follows:
| MPx + (3x + y)Na+ + (3x + y)e− ↔ NayM + xNa3P | (5) |
Similarly, metal-inactive phosphides also proceed through two main types of reactions: one allows for the reconversion to the original MP phase after desodiation whereas the other one cannot. In the former reaction, metallic and NaxP phases are formed after sodiation and the metal and P atoms can re-bond to form MPs during desodiation. Examples of this type of material include CuP2,189 Cu3P,198 NiP3 (ref. 199) and CoP3 (ref. 200) and the reaction process can be presented as eqn (5):
| MPx + 3xNa+ + 3xe− ↔ xNa3P + M | (6) |
As for the latter reaction, nanosized metallic nanocrystals and NaxP phases are formed after metal–P bonds break during the first sodiation process. In the following desodiation process, a pure P phase is formed after Na-extraction and undergoes a reversible conversion reaction with Na in the subsequent cycles. Examples of this type of material include FeP201,202 and CoP203,204 and the reaction process can be expressed as follows:
| MPx + 3xNa+ + 3xe− → xNa3P + M | (7) |
| Na3P ↔ 3Na+ + P + 3e− | (8) |
Overall, details of Na-storage mechanisms for MPs have yet to be fully understood and more efforts and new advanced characterization methods are needed.
3.1 Tin phosphides
Tin phosphides such as Sn4P3 and SnP have attracted considerable attention because of their high theoretical volumetric specific capacity (6650 mA h cm−3 for Sn4P3vs. 5710 mA h cm−3 for P)77 and high electronic conductivity (30.7 S cm−1 for Sn4P3vs. ∼10−14 S cm−1 for RP).77,80 Sn4P3 exhibits a rhombohedral lattice (JCPDS No. 71-2221) with a layered structure consisting of Sn layers and P layers packed alternately (Fig. 16a).205,206 Kim et al.77 for the first time studied the electrochemical performance of Sn4P3 as a SIB anode obtained through ball milling and reported a high reversible capacity of 718 mA h g−1 and a high stability with negligible capacity fading over 100 cycles. In addition, the Sn4P3 sample in this study also showed an appropriately low redox potential of ∼0.3 V vs. Na/Na+, allowing the anode to achieve high energy densities in full cells. According to the reported literature,74,91,195 researchers suggested that the better cycle performance of Sn4P3 compared with Sn could be attributed to the confinement effect of Sn nanocrystallites in the amorphous phosphorus matrix during cycling. And the Na storage mechanism of Sn4P3 could be summarized based on their results,74,91,195 as expressed as follows:| Sn4P3 + 24Na+ + 24e− → Na15Sn4 + 3Na3P | (9) |
| Na15Sn4 ↔ 4Sn + 15Na + 15e− | (10) |
| Na3P ↔ 3Na+ + P + 3e− | (11) |
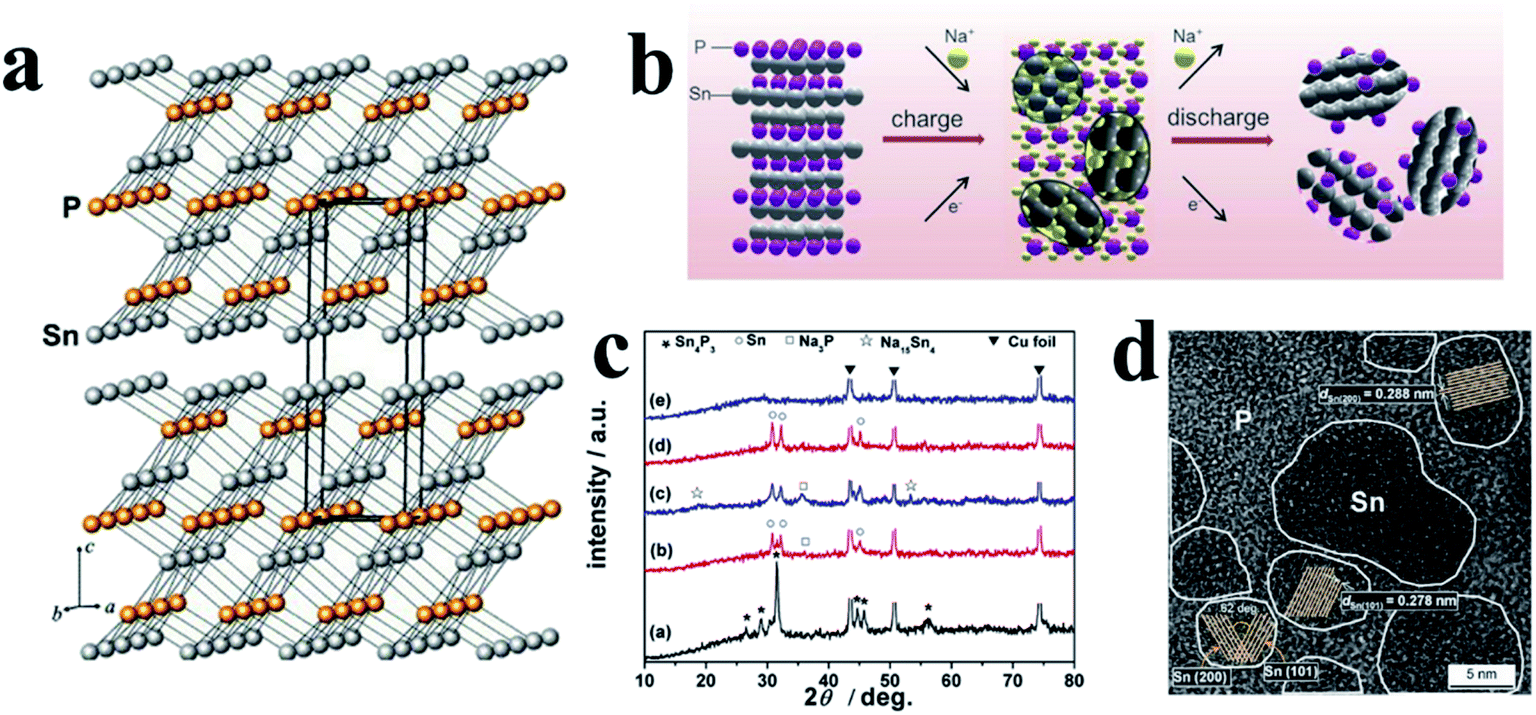 | ||
| Fig. 16 (a) Optimized structure of hexagonal crystalline Sn4P3; orange and gray balls represent the Sn and P atoms, respectively. Reproduced with permission.206 Copyright 2006, Elsevier Inc. (b) Schematic illustration of the Na storage mechanism in the Sn4P3 electrode. Phase transformation of Sn4P3 by sodiation and desodiation (discharge) reactions. (c) Ex situ XRD patterns of the Sn4P3/C electrode in different charge and discharge states: (a) fresh electrode; (b) after the 1st charge to 0.25 V; (c) after completion of the 1st charge to 0.01 V; (d) after the 1st discharge to 0.5 V; and (e) full discharge to 2.0 V. Here the charge process represents sodiation. (c, d) Reproduced with permission.74 Copyright 2014, American Chemical Society. (d) High magnification TEM image of Sn4P3 after the first desodiation (in an ionic liquid electrolyte). Reproduced with permission.208 Copyright 2017, American Chemical Society. | ||
According to the literature reported by Jung et al.,207 the sodiation/desodiation processes of Sn4P3 can be depicted as follows: in the initial stage of sodiation, the Na ions break the Sn–P bonds of Sn4P3 and react with P to form NaxP due to the strong preference of Na for P over Sn; meanwhile, the Sn atoms are separated from P atoms and then join together to form local Sn particles. The NaxP phase can surround the Sn particles because of its volume expansion with Na alloying, or Sn nanoparticles are dispersed in the amorphous-like P matrix. As sodiation continues, the NaxP phase evolves into Na3P, and the Sn particles evolve into NaySn by reacting with Na ions and eventually into Na15Sn4.74 After full sodiation, Sn4P3 is changed to a morphology where Na15Sn4 particles are confined in a Na3P matrix. After Na-extraction of these products, low crystallinity Sn nanoparticles are formed and highly dispersed in the amorphous P host matrix. In the subsequent sodiation/desodiation cycles, P and Sn undergo reversible alloying and de-alloying reactions, respectively. This is well consistent with the experimental results (Fig. 16c and d). In addition, this Na storage mechanism can well explain the excellent cycle performance of Sn4P3. The core (Na15Sn4)/matrix (Na3P) morphology formed by initial sodiation has high structural integrity because the bulk modulus of Na3P is 71% higher than that of Na15Sn4. Thus, Na3P is much better able to withstand deformation than Na15Sn4, and effectively prevent the aggregation of the Sn particles in the next charge–discharge cycle, and this is largely responsible for the excellent cycle performance and high coulombic efficiency of Sn4P3.74,77,208 Furthermore, the Sn phase can change its morphology during sodiation–desodiation in response to P's volume changes; consequently, the void spaces could be filled by their morphological changes, preventing the electrical isolation of the active material layer.209 The structure of crystalline Sn4P3, the schematic illustration of the Na storage mechanism, and the experimental results are shown in Fig. 16a and b.
However, in the study carried out by Kim et al.,77 the results of ex situ TEM and XRD analyses indicated that the conversion reaction of Sn4P3 to NaxP and Na15Sn4 was reversible (Fig. 17). It is possible that the conversion reaction may be partially reversible, similar to the electrochemical behavior of Sn4P3 in LIBs.210
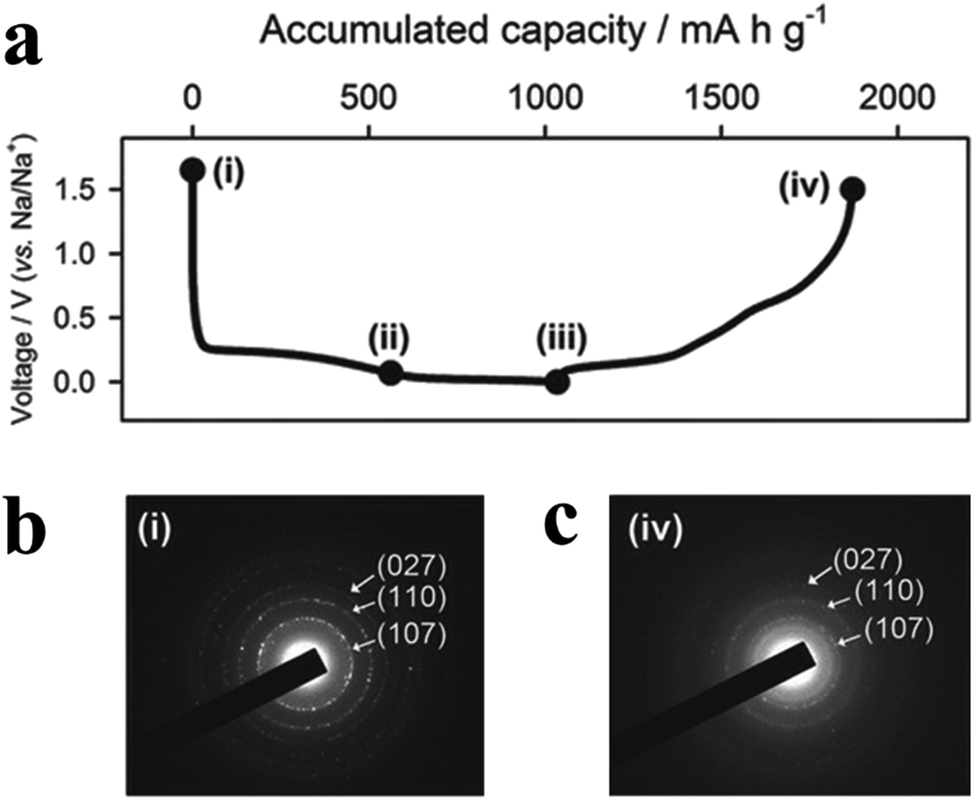 | ||
| Fig. 17 (a) First galvanostatic charge and discharge profiles of the Sn4P3 electrode obtained without an FEC additive. The different points indicate (i) pristine, (ii) 0.07 V, (iii) 0 V and (iv) 1.5 V. (b, c) Electron diffraction (ED) patterns at the points (i) and (iv) in (a), respectively. Reproduced with permission.77 Copyright 2014, WILEY-VCH. | ||
In addition to the better cycle performance, Sn4P3 could also show excellent rate performance, due to not only its intrinsic high electronic conductivity, but also the Sn phase formed during the sodiation/desodiation processes, which could compensate for the poor electronic conductivity of the Na3P phase.74,207,211 Moreover, Sn4P3 allows fast Na ion transport. The Na diffusivities in the intermediate product of amorphous NaxSn4P3 (x = 1.33–24) were found to be between 3.0 × 10−8 and 1.0 × 10−7 cm2 s−1, and for amorphous Na15Sn4 and Na3P, they are 1.1 × 10−7 and 1.6 × 10−8 cm2 s−1, respectively.207 It was indicated that the Sn-rich environment around Na could effectively enhance the Na ion conductivity in Sn4P3 because Na ions could more easily break the weak Na–Sn bonds than the strong Na–P bonds during their transport.207 As a result, the fast electron/Na-ion transport could enable an excellent rate performance of Sn4P3.
After Kim's pioneering study, a two phase nanocomposite of Sn4+xP3@Sn–P comprised of Sn4+xP3 crystals (size, ∼30 nm) with an amorphous Sn–P coating layer (thickness, ∼3 nm) has also been prepared by this ball milling method and showed an excellent Na storage performance.195 In another study, Wang et al.212 found that Sn agglomeration in pristine Sn4P3 particles during anode sodiation/desodiation was the major cause of performance fading. And they found that Sn agglomeration could be effectively suppressed through the introduction of TiC into Sn4P3 by a ball milling method. As a result, a significantly improved cycling stability has been obtained, delivering a capacity of 300 mA h g−1 over 100 cycles at a current density of 100 mA g−1 (with 30 wt% TiC). It is noticeable that the performance of Sn4P3 in this study was different from that reported by Kim's group,77 which was probably caused by the different preparation conditions such as the molar ratio of precursors (Sn:P, 4:3 vs. 6:4) and the ball milling time (1 hour vs. 30 hours) and other reasons. Xu et al.213 also prepared a Sn4P3–P@graphene nanocomposite (Sn:P = 1:3) using ball milling and reported high rate capability retentions of >550 and 371 mA h g−1 at current densities of 1 and 2 A g−1 respectively over 1000 cycles as well as high rate capacities of >815, ∼585 and ∼315 mA h g−1 at 0.1, 2 and 10 A g−1 respectively. In this study, the ball milling process involved two steps: the mixing and milling of tin and RP powders under Ar atmosphere followed by the mixing and milling of the as-synthesized SnP3 with graphene stacks under the same conditions to obtain the final Sn4P3–P@graphene nanocomposite.
However, similar to the preparation of RP, it is hard to obtain a desired small-size morphology or refined nanostructure for Sn4P3via this mechanical approach. Nevertheless, many new nanostructured Sn4P3 samples have been reported by various preparation methods recently. For example, in situ phosphidation is often used to obtain a Sn4P3 nanostructure with Sn or SnO2 as precursors. In most of the phosphidation processes for synthesizing metal phosphides (MPs), PH3 or gaseous RP is often used which can be generated from NaH2PO2 (NaH2PO2·H2O) or solid RP under an inert atmosphere and high temperature conditions.90,202,214 Interestingly, researchers found that Sn could be phosphorized at low temperature by a solvothermal method. For instance, RP powder could be soluble in ethylenediamine during a solvothermal treatment, and then, the dissolved P species reacted with solid Sn and diffused towards the interior of the Sn particles to form Sn–P alloys.206,215 This mild preparation method avoids the toxic PH3 product or the white P by-product, during which lower temperature is needed. However, this solvothermal method could only be used to phosphorize the Sn precursor, and a traditional phosphorization approach with NaH2PO2 (NaH2PO2·H2O) as the P source was often used for a SnO2 precursor. For example, Liu et al.91 synthesized uniform yolk–shell Sn4P3@C nanospheres as anode materials for SIBs by a stepwise preparation approach, involving a hydrothermal method to obtain a SnO2 template, a reduction process to obtain Sn, and a phosphorization process to transform Sn into Sn4P3 (Fig. 18a). During the reduction and phosphorization process, the hollow SnO2 template transformed into one big nanoparticle and several small nanoparticles of Sn which were encapsulated in each carbon nanocage, and evenly formed a yolk–shell Sn4P3@C nanostructure. Due to the unique nanostructure, this Sn4P3@C anode exhibited a high reversible capacity of 790 mA h g−1 at 100 mA g−1, superior rate capacities of 720, 651, 581, 505 and 421 mA h g−1 at 0.2C, 0.4C, 0.8C, 1.5C and 3C respectively (1C = 1000 mA g−1) and stable cycling performances with a high capacity of 360 mA h g−1 at 1.5C after 400 cycles. Similarly, Ma et al.216 designed an integrated anode material of Sn4P3@C with a yolk–shell nanocube structure for SIBs by phosphorization of a Sn precursor and found that the voids and spaces between Sn4P3 and carbon nanocubes could effectively buffer volume expansion during charge/discharge and that the highly conductive carbon material could promote fast electron transfer. As a result, this SIB anode exhibited a high discharge capacity of 701 mA h g−1 at 0.1 A g−1 after 50 cycles, remarkable rate capability of 508 mA h g−1 even at 2.0 A g−1 and highly stable cycling performances with 516 and 368 mA h g−1 capacity retention after 500 cycles at 1.0 and 2.0 A g−1 respectively. Furthermore, Choi et al.217 fabricated size-adjustable core–shell Sn4P3–C nanospheres using a carbonization/reduction and phosphorization process and reported that among the different sizes of Sn4P3–C nanospheres, the 140 nm Sn4P3–C nanosphere electrode demonstrated the highest reversible capacity and rate capability, with an ultra-long cycle stability with 420 mA h g−1 capacity retention after 2000 cycles at a current density of 2000 mA g−1. In addition, researchers have also reported that the cycling life of Sn4P3/C could be largely extended through the reduction of particle size to the nanoscale which could reduce the pulverization of anodes.217
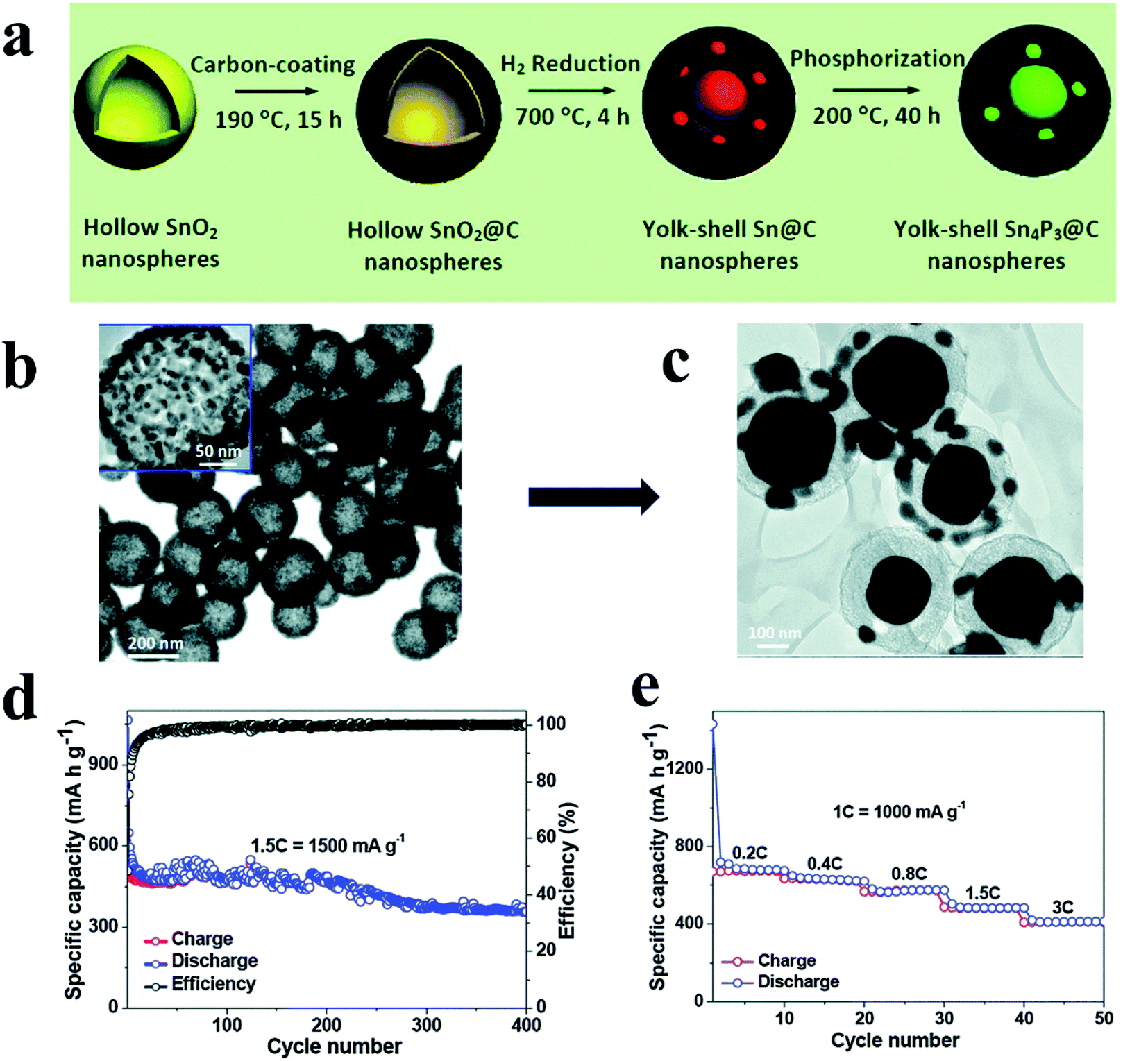 | ||
| Fig. 18 (a) Schematic of the fabrication of uniform yolk–shell Sn4P3@C nanosphere anodes. (b) TEM image of uniform SnO2 hollow nanosphere templates; the inset shows a typical SnO2 hollow nanosphere with a shell thickness of about ∼25 nm. (c) TEM images of a uniform yolk–shell Sn@C nanosphere; (d) Long cycling performance of yolk–shell Sn4P3@C nanospheres at 1.5C. (e) Rate capability at different rates (increased from 0.2C to 3C). 1C = 1000 mA g−1. Reproduced with permission.91 Copyright 2015, The Royal Society of Chemistry. | ||
These studies indicate that the combination of Sn4P3 with carbon materials is also an effective strategy to improve the performance of Na storage, and the refined nanostructures can give significant advantages for practical SIBs.
Shin et al.213 also prepared Sn–P compounds (Sn4P3, Sn4−xP3 or Sn4P3+x) as anode materials for SIBs by this simple solvothermal method and found that the temperature and time period used for solvothermal treatment played a critical role in determining the phase and composition of the Sn–P compound and thus its electrochemical behavior. When solvothermal synthesis was conducted at lower or higher temperatures than a certain critical value (e.g., 200 °C), the resulting compound contained unreacted (residual) Sn or excess P species. The Sn4P3 compounds (Sn4P3 mixed with ∼1.7% Sn) prepared with the optimized solvothermal parameters (T = 200 °C and t = 40 h) showed the best electrochemical Na storage performance. Yin et al.218 designed a Sn4P3/reduced graphene oxide nanohybrid (Sn4P3/RGO) similarly by this low-temperature solution-based phosphorization reaction route. In the preparation process, NaBH4 was used as the reducing agent to reduce Sn2+ and GO, with a Sn/RGO nanohybrid formed, and then the Sn/RGO nanostructures were used as the precursor and RP as the P source to synthesize Sn4P3/RGO by a solvothermal approach at 200 °C. Due to the hybrid structure and the high electronic conductivity of RGO, this nanohybrid SIB anode exhibited a high reversible capacity, an enhanced rate capability, and a superior long cycling life (362 mA h g−1 after 1500 cycles at a current density of 1.0 A g−1). In another study, a novel morphology of pure phase and size-controllable Sn4P3 nanotops was developed through a novel facile solution chemical method using Sn and trioctylphosphine (TOP) as raw materials.219 This Sn4P3 nanotop anode showed a good electrochemical performance for SIBs, providing an initial capacity of 719.8 mA h g−1 at a current density of 50 mA g−1, an initial coulombic efficiency of ∼72.7%, and good cyclability and stability (543 mA h g−1 at 200 mA g−1 for 80 cycles).
Alternatively, a high temperature atmosphere phosphidation approach with SnO2 as the precursor is another effective way to prepare Sn4P3 nanostructures. Compared with the solvothermal method, which often involves low temperature but a long process time (T = 200 °C and t = 40 h), the atmosphere phosphidation approach usually needs higher temperature but a shorter process time (∼300 °C, less than a few hours) with NaH2PO2 (NaH2PO2·H2O) as the P source.
As the initial coulombic efficiency (CE) of Sn4P3/C nanocomposites was found to be inefficient (40–50%)91 and it is challenging to achieve both long cycle life and high CE for high capacity tin phosphide anodes, Fan et al.205 reported an anode composed of pomegranate-structured Sn4P3@C spheres synthesized using a facile aerosol spray-pyrolysis-phosphidation method. In brief for this process, the transparent precursor containing SnSO4 and sucrose was atomized into microsized droplets (0.1–2 μm) and then these droplets were pyrolyzed in a 600 °C furnace and transformed into SnO2/C spheres (0.1–2 μm); finally, pomegranate-structured Sn4P3@C spheres were obtained after a phosphidation process with NaH2PO2 as the P source (Fig. 19a and c). It can be seen from the TEM images (Fig. 19b) that Sn4P3 nanoparticles with a size of 20–40 nm were encapsulated by thin carbon layers. The Sn4P3@C anode was able to provide a reversible capacity of ∼800 mA h g−1 with a record-high initial CE (>90%) and high cycle CE (∼99.9%) for over 120 cycles at a current of 100 mA g−1 in an ether-based electrolyte (Fig. 19d). Here, the researchers suggested that to achieve both long cycling life and high CE, ideal anode materials should possess microsized primary particles consisting of nanosized secondary high capacity active materials and nanopores in a carbon matrix, allowing for the formation of a SEI on only the primary particle surface and the maintenance of the electronically conductive nanoporous composite integrity during charge/discharge cycles. Based on this, the pomegranate-structured Sn4P3@C spheres synthesized in this study were in accordance with the ideal anode they referred to.
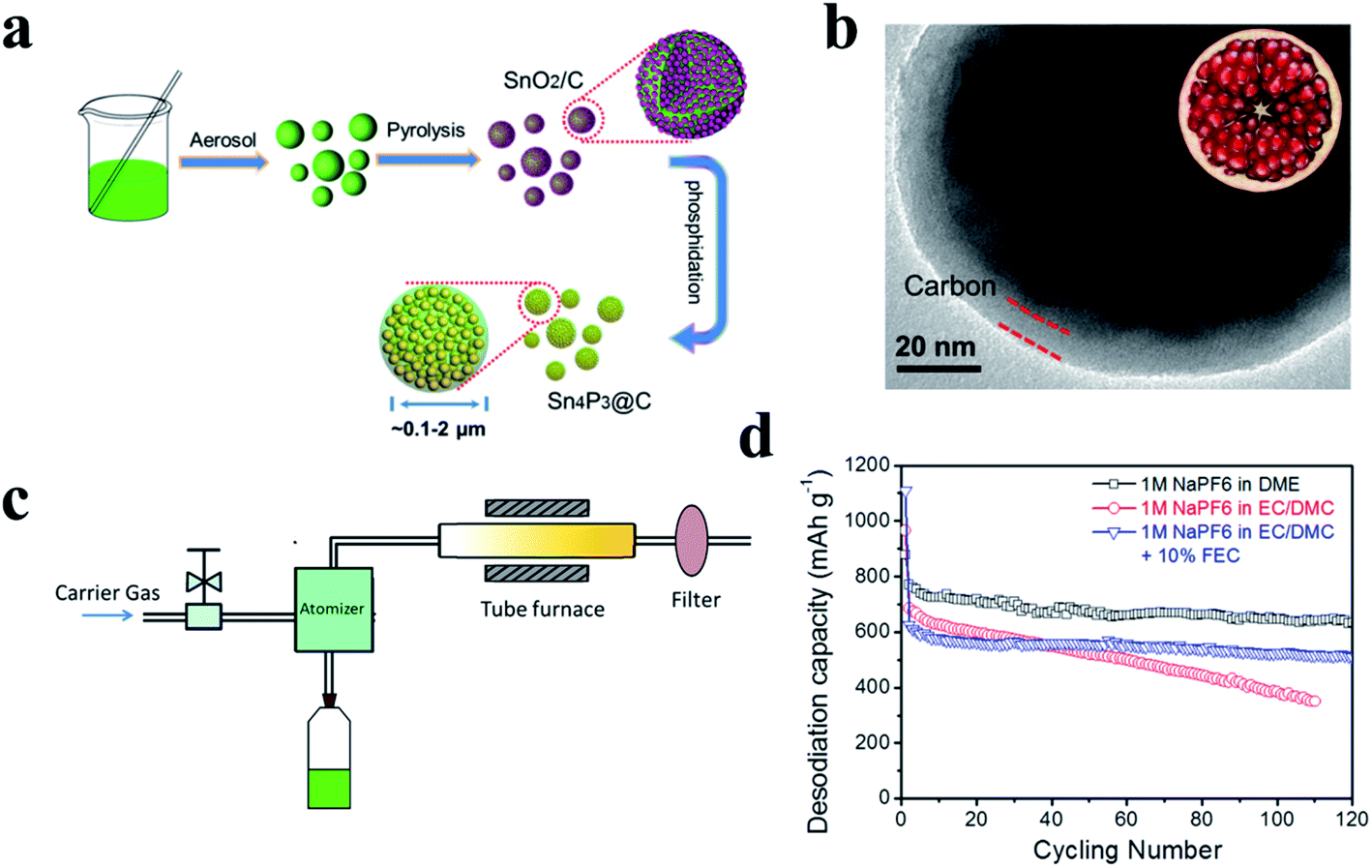 | ||
| Fig. 19 (a) Schematic illustration of the fabrication process for Sn4P3@C pomegranates. (b) TEM image of the as-synthesized pomegranate-structured Sn4P3@C sphere; inset of (b) shows the ideal pomegranate structure. (c) Schematic illustration of the fabrication of the SnO2/C spherical composite using the aerosol-spray-pyrolysis method. (d) Cycling performances of the pomegranate-structured Sn4P3@C nanosphere at 100 mA using different electrolytes. Reproduced with permission.205 Copyright 2017, Elsevier Ltd. | ||
Recently, Sn4P3 microspheres encapsulated in hollow carbon spheres (Sn4P3@C) were prepared by this phosphorization route using SnO2@C as the precursor and NaH2PO2 as the P source. The void volume of this nanostructure and the elasticity of the protective carbon spherical shell could efficiently mitigate the volume change of active Sn4P3 microspheres (200 nm) during charge/discharge processes and form a stable solid electrolyte interface film on the surface of the carbon shell. As a result, the Sn4P3@C anode for SIBs showed a stable specific capacity of 420 mA h g−1 after 300 cycles at a current density of 0.2 A g−1, as well as excellent rate capabilities of 253, 175, 111, 78, and 50 mA h g−1 at densities of 1, 2, 5, 10, and 20 A g−1, respectively. More importantly, this anode also showed an ultralong cycling performance, delivering stable capacities of 205 and 103 mA h g−1 at large current densities of 2 and 5 A g−1 after 4000 cycles, respectively.
In addition to Sn4P3, tin monophosphide (SnP) also shows promise as anode materials for SIBs, possessing a high theoretical capacity of 1209 mA h g−1 due to its high P ratio. SnP exhibits a trigonal crystallographic phase and shows a layered structure consisting of Sn–P–P–Sn sandwiches that are stacked on top of each other.220 However, SnP has not yet been extensively explored in energy storage devices due to its challenging synthesis conditions. To address this, Liu et al.190 synthesized spherical SnP nanocrystals (NCs) with a size of 34 ± 5 nm through a liquid phase synthesis method. And a SIB based on the SnP NC anode with sodium(I) bis(fluorosulfonyl)imide (NaFSI) as the electrolyte exhibited a high reversible capacity of 600 mA h g−1 at 100 mA g−1 and enhanced cycling stabilities for over 200 cycles. Here, the researchers suggested that the high cycling performance was associated with both the small size of the crystal domain and the particular composition and phase of SnP, which could prevent mechanical disintegration and major phase separation during charge/discharge.
Another tin phosphide material SnP3, which has the highest P mass ratio among tin phosphides such as Sn4P3 and SnP, exhibits the highest theoretical capacity of 1616 mA h g−1.194 Fan et al.194 prepared a composite of SnP3/C by a simple ball milling method and demonstrated that SnP3/C could reversibly react with Na through conversion and alloy reaction processes as SIB anodes. The corresponding anode could provide a high capacity of ∼810 mA h g−1 at a current density of 150 mA g−1 without capacity fading after 150 cycles and was also able to retain 400 mA h g−1 even at 2560 mA g−1. Furthermore, detailed analysis using SEM, TEM and XPS revealed that the strong bonding interaction between Sn and P could successfully mitigate the pulverization and aggregation of Sn and P, allowing for the partial self-healing of the SnP3/C composite structure and enhanced cycling stabilities.
3.2 Cobalt phosphides
Cobalt phosphides can also electrochemically react with Na.90,203 Although metallic Co is inactive with Na, researchers reported that it can act as a conductive matrix to buffer volume expansion and enhance electron transfer. CoP is the most commonly used cobalt phosphide for SIBs and usually exhibits an orthorhombic phase, as shown in Fig. 20a.221,222 Liu et al.203 synthesized nanosized CoP particles as an anode for SIBs through ball milling metal Co and commercial RP. The anode delivered a high initial specific capacity of 770 mA h g−1 and high rate capabilities, demonstrating that CoP is a promising anode candidate for sodium ion storage. In addition, ex situ XPS and STEM were carried out to investigate the sodium storage mechanism of CoP in this study and revealed that the sodium storage of CoP was based on a reaction between P and Na and can be summarized as follows:| CoP + 3Na+ + 3e− → Co + Na3P (first discharge) | (12) |
| Na3P ↔ 3Na+ + P + 3e− (subsequent cycles) | (13) |
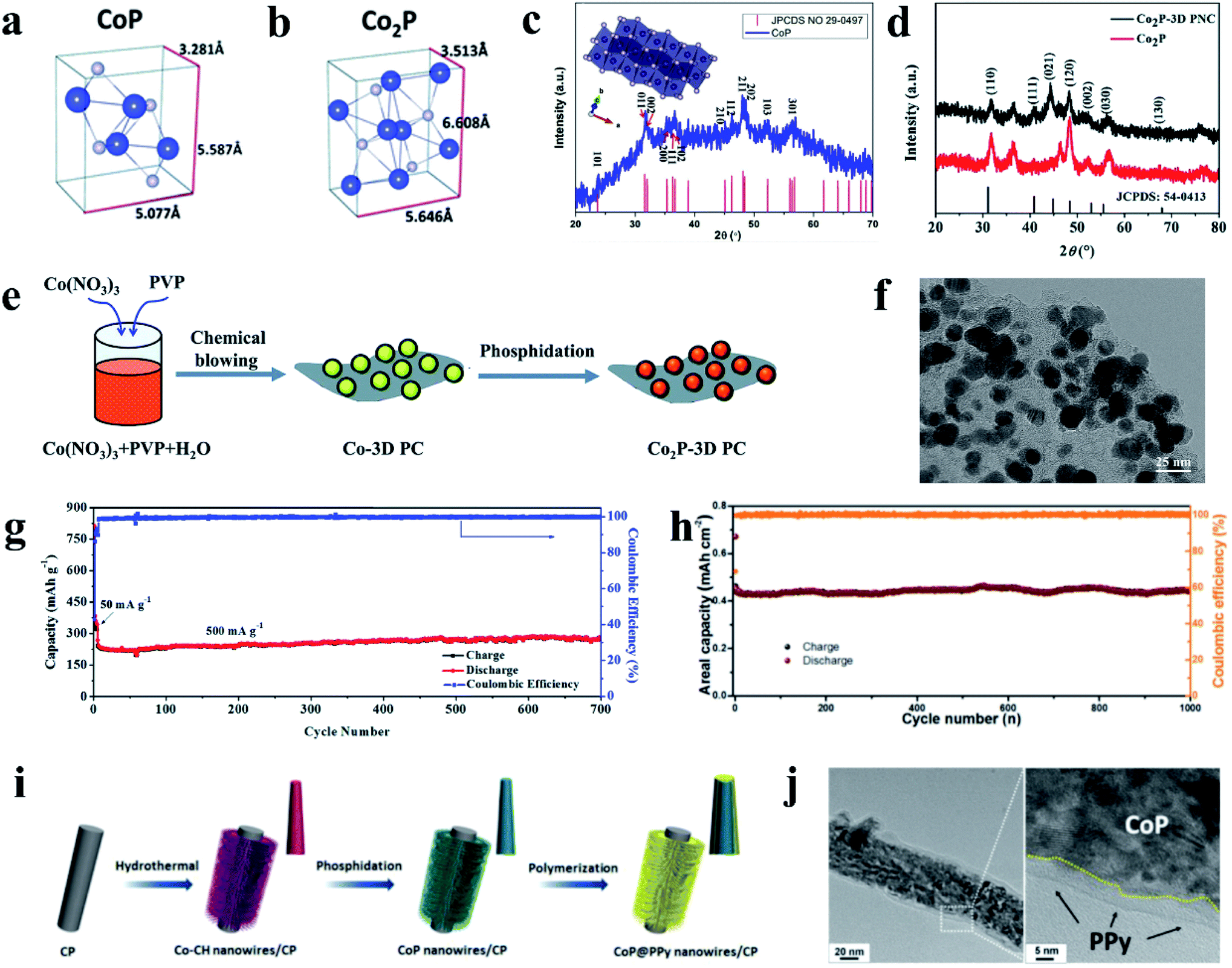 | ||
| Fig. 20 (a, b) Crystal structures of orthorhombic CoP and Co2P. (c, d) XRD pattern of crystalline CoP and Co2P. (e) Schematic representation of the synthesis process of the Co2P-3D PNC composite. (f) TEM image of the Co2P-3D PNC composite. (g) Long-term cycling performance of the Co2P-3D PNC composite at 500 mA g−1. (h) Long-term cycling stability of the CoP@PPy NWs/CP electrode at 1.5 mA cm−2. (i) Schematic diagram of the fabrication process of the CoP@PPy nanowires/CP electrode. (j) TEM images of CoP@PPy NWs. (a, b) Reproduced with permission.221 Copyright 2011, The Royal Society of Chemistry. (c) Reproduced with permission.203 Copyright 2015, Elsevier B.V. (d–g) Reproduced with permission.224 Copyright 2018, The Royal Society of Chemistry. (h–j) Reproduced with permission.191 Copyright 2018, WILEY-VCH. | ||
It means that CoP decomposed into Co and P after the initial charge–discharge process and in the next cycles, element P reversibly reacts with Na to form Na3P with intermediate phases (Na2P, NaP and NaP7), being consistent with other literature.204Table 2 summarizes the recent progress of phosphides used as an anode for SIBs.
| Materials | Synthetic method | Cycling performance | Rate capability | Initial coulombic efficiency | Ref. |
|---|---|---|---|---|---|
| Sn4P3 powder | Ball milling | ∼718 mA h g−1, at 100 mA g−1, 100th | — | ∼78% | 77 |
| Sn4P3@C | Hydrothermal/phosphorization | 360 mA h g−1, at 1000 mA g−1, 400th | 360 mA h g−1, 3000 mA g−1 | — | 91 |
| Sn4P3/TiC | Ball milling | 300 mA h g−1, at 100 mA g−1, 100th | — | 85.5% | 212 |
| Sn4P3–P@graphene | Ball milling | 607 mA h g−1, at 1000 mA g−1, 800th | 371 mA h g−1, 1000 mA g−1 | 73% | 213 |
| Sn4P3 nanotop | Solution chemistry method | 543 mA h g−1, at 200 mA g−1, 80th | 399 mA h g−1, 1000 mA g−1 | 72.7% | 219 |
| Sn4P3–C sphere | Hydrothermal/phosphorization | 420 mA h g−1, at 2000 mA g−1, 2000th | 260 mA h g−1, 4000 mA g−1 | 60% | 217 |
| Sn4P3@C sphere | Aerosol spray-pyrolysis | 700 mA h g−1, at 100 mA g−1, 120th | ∼500 mA h g−1, 800 mA g−1 | 90.7% | 205 |
| Sn4P3@C yolk–shell | Liquid phase/sintering/solvothermal | 701 mA h g−1, at 100 mA g−1, 50th | 508 mA h g−1, 2000 mA g−1 | 63.7% | 216 |
| SnP3@C | Ball milling | 810 mA h g−1, at 150 mA g−1, 150th | ∼400 mA h g−1, 2560 mA g−1 | 71.2% | 194 |
| SnP nanocrystal | Liquid phase synthesis | 600 mA h g−1, at 100 mA g−1, 200th | 396 mA h g−1, 2500 mA g−1 | — | 190 |
| CoP | Ball milling | 315 mA h g−1, at 100 mA g−1, 25th | 80 mA h g−1, 2000 mA g−1 | 65.2% | 203 |
| CoP3@C | Ball milling | 144 mA h g−1, at 100 mA g−1, 320th | 136.4 mA h g−1, 250 mA g−1 | 87% | 200 |
| CoP@C polyhedron | Calcination/phosphidation | 473 mA h g−1, at 100 mA g−1, 100th | 253.6 mA h g−1, 800 mA g−1 | 47.3% | 204 |
| CoP/CNS | Calcination | 386 mA h g−1, at 1000 mA g−1, 900th | 174 mA h g−1, 20 A g−1 | 64.6% | 223 |
| Co2P-3D PNC | Blowing method/phosphidation | 271 mA h g−1, at 500 mA g−1, 700th | 179 mA h g−1, 3000 mA g−1 | 41% | 224 |
| CoP4/CF | Hydrothermal/phosphidation | 851 mA h g−1, at 300 mA g−1, 300th | 535 mA h g−1, 4000 mA g−1 | 53% | 90 |
| FeP nanoparticle | Ball milling | 321 mA h g−1, at 50 mA g−1, 60th | 60 mA h g−1, 500 mA g−1 | 60.2% | 201 |
| FeP nanoarray | Hydrothermal/phosphidation | 548 mA h g−1, at 200 mA g−1, 100th | 184 mA h g−1, 2000 mA g−1 | 55.7% | 202 |
| CNT@FeP@C | Solution phase | 415 mA h g−1, at 100 mA g−1, 100th | 268 mA h g−1, 1500 mA g−1 | 59% | 214 |
| FeP4 | Ball milling | 1000 mA h g−1, at 89 mA g−1, 30th | 900 mA h g−1, 3578 mA g−1 | 84% | 70 |
| H–FeP@C@GR | Solvothermal/phosphidation | 446 mA h g−1, at 100 mA g−1, 100th | 237 mA h g−1, 1600 mA g−1 | 74% | 192 |
| NiP3 | Ball milling | 1079 mA h g−1, at 176 mA g−1, 15th | 720 mA h g−1, 1760 mA g−1 | — | 199 |
| Yolk–shell Ni2P | Hydrothermal/phosphidation | 181 mA h g−1, at 200 mA g−1, 100th | 101 mA h g−1, 2000 mA g−1 | 52% | 225 |
| NiP3-CNT | Ball milling | 668 mA h g−1, at 200 mA g−1, 120th | 494 mA h g−1, 3200 mA g−1 | 71.3% | 226 |
| Ni2P/C | Solution phase/heat treatment | 361 mA h g−1, at 100 mA g−1, 300th | 100 mA h g−1, 5000 mA g−1 | 76.7% | 92 |
| Ni2P/graphene/Ni2P | Hydrothermal/phosphidation | 188 mA h g−1, at 500 mA g−1, 300th | 152 mA h g−1, 2000 mA g−1 | 69% | 227 |
| CuP2/C | Ball milling | ∼450 mA h g−1, at 50 mA g−1, 30th | 178 mA h g−1, 2000 mA g−1 | ∼62% | 189 |
| Carbon sheet-CuP2 | Solution phase/phosphidation | 410 mA h g−1, at 80 mA g−1, 200th | 152 mA h g−1, 2560 mA g−1 | 87% | 228 |
| Cu3P/C | Solution phase/annealing | 221 mA h g−1, at 100 mA g−1, 100th | 142 mA h g−1, 2000 mA g−1 | 73% | 198 |
| GeP5/C | Ball milling | 1220 mA h g−1, at 100 mA g−1, 60th | 900 mA h g−1, 1500 mA g−1 | 92% | 193 |
| GeP5/AB/p-rGO | Ball milling | 400 mA h g−1, at 500 mA g−1, 50th | 175 mA h g−1, 5000 mA g−1 | 60% | 229 |
| GePx | High temperature solvothermal | 704 mA h g−1, at 240 mA g−1, 100th | 130 mA h g−1, 18 A g−1 | 82.6% | 230 |
| MoP nanorod/C | Solid phase reaction | 398 mA h g−1, at 240 mA g−1, 100th | 115.6 mA h g−1, 1600 mA g−1 | ∼40% | 231 |
| Zn3P2 nanowires | Chemical vapor deposition | ∼600 mA h g−1, at 100 mA g−1, 85th | 280 mA h g−1, 5000 mA g−1 | 55% | 232 |
| InP | Ball milling | 500 mA h g−1, at 50 mA g−1, 100th | — | — | 209 |
| SiP2 | Ball milling | 572 mA h g−1, at 100 mA g−1, 15th | — | 72% | 233 |
| Se4P4 | Ball milling | 804 mA h g−1, at 50 mA g−1, 60th | 332 mA h g−1, 3000 mA g−1 | 70.6% | 234 |
| P2S5/C | Ball milling | 394 mA h g−1, at 100 mA g−1, 100th | 114 mA h g−1, 2000 mA g−1 | — | 235 |
| NiPS3 nanosheet | Solution phase/phosphidation | 902 mA h g−1, at 50 mA g−1, 4th | 316 mA h g−1, 5000 mA g−1 | 70% | 236 |
| Cu4SnP10 nanowires | High temperature solution phase | 512 mA h g−1, at 100 mA g−1, 100th | 412 mA h g−1, 1000 mA g−1 | 63% | 237 |
| FeSi4P4 | Ball milling | 180 mA h g−1, at 100 mA g−1, 100th | 54 mA h g−1, 4000 mA g−1 | 72% | 238 |
| SnPO/rGO | Hydrothermal | 420 mA h g−1, at 50 mA g−1, 100th | 200 mA h g−1, 2000 mA g−1 | 53.2% | 239 |
| RP@Ni–P core@shell | Solution phase | 1256 mA h g−1, at 260 mA g−1, 200th | 491 mA h g−1, 5200 mA g−1 | 88.2% | 86 |
Similar to RP, CoPs obtained from ball milling methods often suffered from fast capacity fading during cycling and inferior rate performance due to the large and uneven particle size; however, complex and ultrafine nanostructures could be synthesized by methods such as hydrothermal reactions and solution phase processes for CoP, giving an effective approach to solve these issues. For example, Ge et al.204 developed a strategy to synthesize MOF derived core/shell structured CoP@C polyhedrons anchored onto 3D reduced graphene oxide (RGO) on nickel foam (NF) as a binder-free SIB anode. Here, the unique core/shell CoP@C polyhedral structure was obtained through an in situ low-temperature phosphidation process derived from ZIF-67 and was further combined with RGO/NF which could efficiently act as a binder and electrical conductor. As a result, the corresponding anode exhibited remarkable electrochemical performances and delivered a specific capacity of 473.1 mA h g−1 at a current density of 100 mA g−1 after 100 cycles. In another example, Kang et al.223 designed a nanostructure of small size CoP nanoparticles uniformly embedded in N-doped C nanosheets (CNSs) by a simple one-step calcination process with a Co-based MOF and RP as precursors. Due to the small CoP particles (∼11.3 nm), the strong P–C bonds and the high conductivity of the CNSs, a resulting anode using this composite delivered a Na-storage capacity of 598 mA h g−1 at 100 mA g−1 (pure CoP, 831 mA h g−1), a high rate capability of 174 mA h g−1 at 20 A g−1 and a long-term cyclability of 98.5% capacity retention after 900 cycles at 1 A g−1. Furthermore, Zhang et al.191 introduced a conducting polymer (poly-pyrrole, PPy) coating layer to protect 1D nanostructured CoP nanowires and fabricated a freestanding binder free SIB anode. Here, the synthesis process involved three steps, including the initial growth of 1D Co-based precursors on carbon paper using a hydrothermal method, a phosphidation process to obtain CoP nanowires and then a coating of the polymer PPy onto the surface of the CoP nanowires through polymerization and oxidation of pyrrole monomers (Fig. 20i and j). The researchers proposed that the obtained 1D core–shell CoP@PPy NWs/CP not only enhanced charge transfer and ion diffusion but also buffered volume expansion during cycling. As a result, these anodes exhibited superb electrochemical performance, delivering a high areal capacity of 0.521 mA h cm−2 at 0.15 mA cm−2 after 100 cycles and 0.443 mA h cm−2 at 1.5 mA cm−2 even after 1000 cycles (Fig. 20h), and a significant areal discharge capacity of 0.285 mA h cm−2 even at a high current density of 3 mA cm−2.
Other cobalt phosphides such as Co-rich phase Co2P (Fig. 20b) and phosphorus rich phases CoP3 and CoP4 have also been reported as viable anode materials for SIBs. For example, Zhou et al.224 prepared a composite of Co2P nanoparticle encapsulated 3D porous N-doped carbon nanosheet networks (3D-PNC) using a blowing method and an in situ phosphidation process (Fig. 20e). This unique Co2P/3D-PNC nanostructure exhibited enhanced cycling stability and rate capability with a high initial discharge capacity of 827 mA h g−1 at 50 mA g−1 and 271 mA h g−1 after 700 cycles at 500 mA g−1 (Fig. 20g). Jin et al.240 utilized a cobalt-based metal organic framework (ZIF-67) as a self-template to obtain a nanostructure of Co2P nanoparticles hybridized with N-doped carbon matrices (Co2P@N–C). For the preparation process, they first prepared a CoP@N–C nanostructure, and then combined this CoP@N–C with reduced graphene oxide by a simple hydrothermal method to form a Co2P@N–C@rGO composite. The anode of this composite exhibited an excellent sodium storage performance with a high reversible capacity of 225 mA h g−1 at 50 mA g−1 after 100 cycles. In addition, they proposed that Co2P underwent a Na storage mechanism similar to that of CoP,and decomposed into Co and P after the first discharge process, and then the P reversibly reacted with Na during the subsequent charge/discharge processes. In another study, Zhang et al.200 synthesized carbon-coated CoP3 (CoP3@C) nanocomposites as anodes for SIBs using a high energy mechanical milling method. Compared with plain CoP3, this carbon-coated CoP3 exhibited larger discharge capacities, enhanced cycling lifespans and better rate capabilities with a capacity of 212 mA h g−1 after 80 cycles at a current of 40 mA h g−1. Sun et al.90 fabricated a binder-free anode for SIBs by growing mesoporous CoP4 nanoarrays onto carbon felt by a hydrothermal and a phosphidation process and reported that this unique mesoporous nanoarray contained a secondary structure of ∼10 nm ultra-small primary particles. With the 3D carbon felt as a conductive network, this nanostructure demonstrated a capacity of 851 mA h g−1 after 300 cycles with an average coulombic efficiency of above 98%, a long cycling lifespan of up to 1000 cycles (>90% retention rate) and a high rate capability of 535 mA h g−1 at a current density of 4 A g−1. This CoP4/CF anode was also used in a full SIB with a Na3V2(PO4)2F3 cathode and an average operating voltage of ≈3.0 V, a reversible capacity of 553 mA h g−1 and a high energy density of ≈280 W h kg−1 were achieved.
In summary, these studies have shown the possible application of cobalt phosphides as anodes in SIBs, and refined Co–P nanostructures such as polyhedrons, nanowires, and nanoarrays often exhibit better Na storage performance than ball milled Co–P particles. However, the high cost of Co may hinder their practical application, especially for large-scale energy storage systems.
3.3 Iron phosphides
Iron phosphides such as FeP, FeP2 and FeP4 are also considered as promising anode materials for SIBs because of their high abundance, low-cost and environmentally friendly characteristics. Although Fe in FePx is electrochemically nonreactive towards Na, it can be used as a conductive matrix to relieve the volumetric expansion of P.214 Because of this, great efforts have been devoted to the exploration of FePx as SIB anode materials. Among these various iron phosphides, FeP is most commonly used in SIBs and exhibits an orthorhombic phase (Fig. 21a)241,242 and possesses a theoretical capacity of 924 mA h g−1. Li et al.201 for the first time synthesized a low-cost FeP anode for SIBs with a high initial capacity of 764.7 mA h g−1 through simple ball milling. The result of ex situ XRD and TEM indicated that the sodium storage mechanism of FeP was based on a conversion reaction, which can be expressed as follows:| FeP + 3Na+ + 3e− → Fe + Na3P | (14) |
| Na3P ↔ 3Na+ + P + 3e− | (15) |
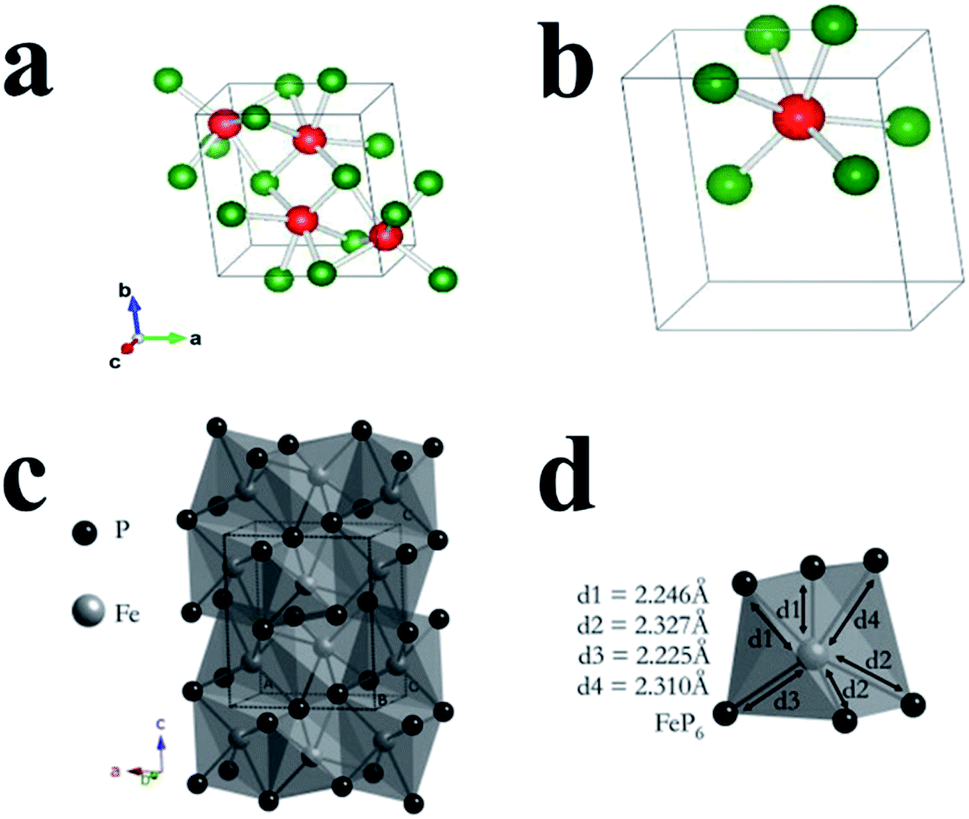 | ||
| Fig. 21 (a, b) FeP crystal structure has an orthorhombic structure, and the iron atom in FeP has only one type of coordination: six. Red and orange atoms represent Fe and green atoms indicate P. Reproduced with permission.241 Copyright 2018, Elsevier Ltd. (c) Crystal structure of orthorhombic FeP in another form. (d) FeP6 polyhedron in orthorhombic FeP. The grey and black spheres represent Fe and P atoms, respectively. Reproduced with permission.242 Copyright 2015, Elsevier B.V. | ||
The Na storage mechanism of FeP is similar to that of CoP: after the initial discharge process, FeP is converted into Co and P, and then element P reversibly reacts with Na to form Na3P in the subsequent charge/discharge processes.
In another study, Wang et al.202 reported a SIB anode composed of FeP nanorod arrays on carbon cloth (FeP NAs/CC) (Fig. 22a), which delivered a high capacity of 829 mA h g−1 at 0.1 A g−1 and a high stability of 548 mA h g−1 at 0.2 A g−1 after 100 cycles with a capacity retention of 99.8%, and an excellent rate and cycle performance (Fig. 22b and c). The synthesis process was simple, involving growth of Fe2O3 nanorod arrays onto carbon cloth through a hydrothermal method followed by a phosphidation process to transform Fe2O3 into FeP (Fig. 22a). In another study, Han et al.214 developed a synthetic strategy to construct carbon-coated FeP with an amorphous and mesoporous framework anchored onto carbon nanotubes as a SIB anode. The synthesis process involved a silica coating as a sacrificial internal spacer, which appeared to be key in the formation of a mesoporous framework. As a result, this FeP-based anode demonstrated a reversible capacity of 415 mA h g−1 after 100 cycles at 100 mA g−1 and a long-life cycling capability of 295 mA h g−1 after 500 cycles at 500 mA g−1. Furthermore, Wang et al.192 fabricated graphene encapsulated with a hollow FeP@carbon nanocomposite (H–FeP@C@GR) through a combination of solvothermal and hydrothermal methods with carbothermic reactions and phosphidation treatments (Fig. 22d) and reported that the synthesized H–FeP nanospheres were highly uniform with an average diameter of 300–400 nm and that the thickness of the carbon layer was ∼4 nm (Fig. 22e). Here, the researchers also reported that such a 3D hierarchical architecture could exhibit high electrochemical performances where a reversible capacity of 446 mA h g−1 after 100 cycles and high rate capacities and stabilities were achieved (Fig. 22f and g), suggesting that the 3D conductive network of the GR skeleton and the carbon coating of FeP were key factors in the enhancement of Na storage performances, which could not only provide open pathways for electron/ion transport, but also prevent the continual reforming of a SEI on the surface of hollow FeP.
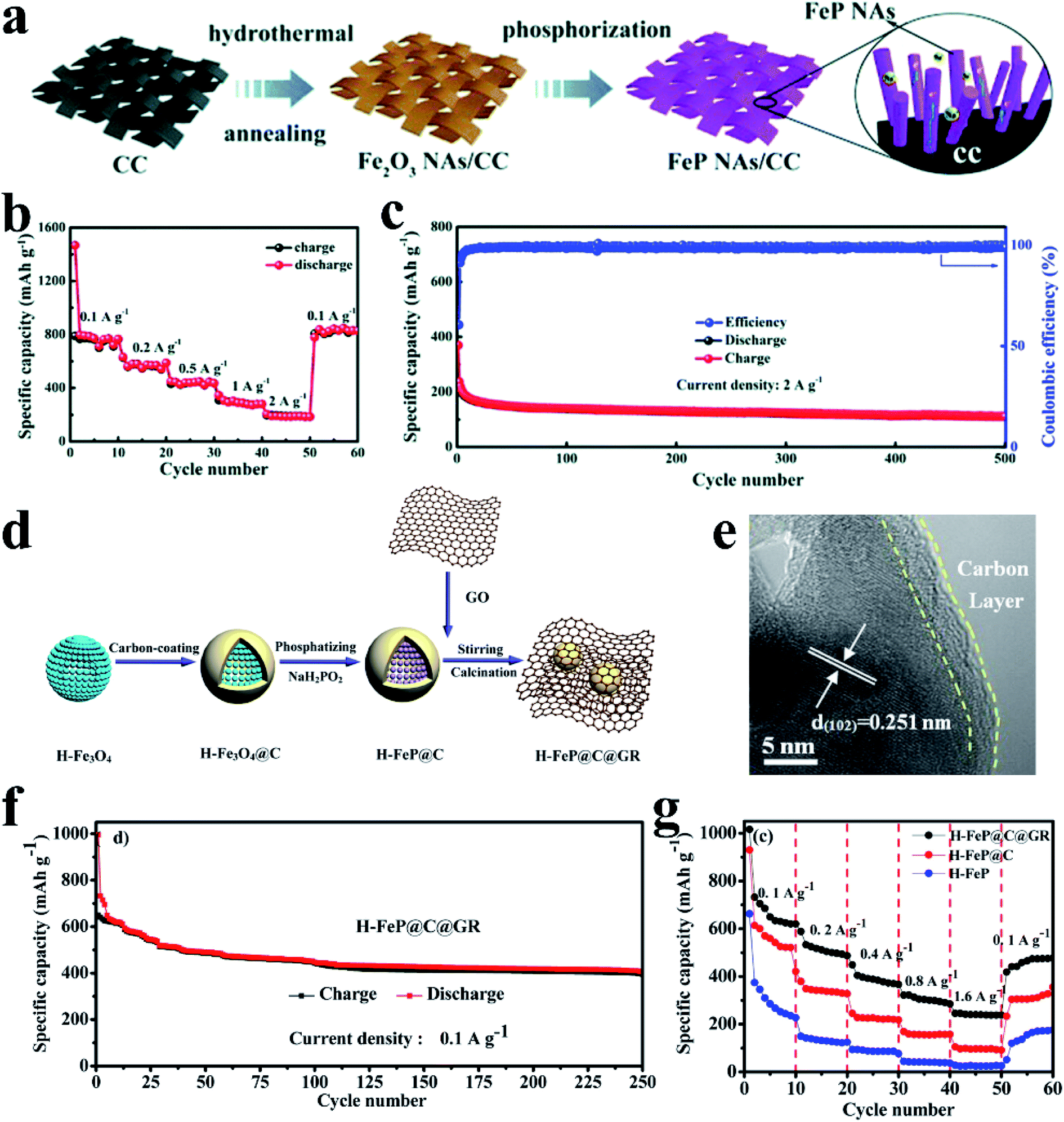 | ||
| Fig. 22 (a) Schematic of the preparation of FeP NAs/CC. (b) Rate performance of the FeP NAs/CC electrode for SIBs. (c) Long-term cycling at 2 A g−1. Reproduced with permission.202 Copyright 2016, The Royal Society of Chemistry. (d) Schematic of the synthesis of H–FeP@C@GR nanocomposites. (e) HRTEM image of the H–FeP nanospheres. (f) Long-term cycling and (g) rate performance of a H–FeP@C@GR anode at a current density of 0.1 A g−1. Reproduced with permission.192 Copyright 2017, American Chemical Society. | ||
FeP4 exhibits a monoclinic structure and also can be used as an anode for SIBs. Zhang et al.70 synthesized iron phosphides of FeP2 and FeP4 as SIB anodes by a simple ball milling method and found that the FeP4 anode demonstrated a highly reversible sodiation capacity of 1137 mA h g−1 with a coulombic efficiency of 84.0% during the first cycle at 89 mA g−1 and high stability with a capacity of ∼1000 mA h g−1 after 30 cycles along with good rate capabilities. However, the researchers also reported that FeP2 showed no significant electrochemical reactivities as a SIB anode possibly because of its low operating potential (≤0 V vs. Na/Na+).243
Overall, iron phosphides can show great potential as anodes for practical SIBs due to their low-cost and the facile synthesis of these ultrafine Fe–P nanostructures. However, FeP composites often exhibit low initial Coulomb efficiency (55–75%) and insufficient cycle life-span, which is a concern for practical application. Although ball milled FeP4 possesses a higher P content and has shown the highest gravimetric specific capacity as well as the highest initial coulombic efficiency (84%) among those Fe–P composites, the same issue of insufficient cycle stability hiders its application. In addition, its low electronic conductivity (∼3 × 10−3 S m−1 for FeP4vs. ∼1.3 × 106 S m−1 for FeP) is also a concern. It's presumed that FeP4 may undergo a similar capacity fading mechanism to amorphous RP because of its high P content. Furthermore, as ball milled FeP4 shows a large particle size (1–10 μm), it is more likely to be pulverized, leading to insufficient cycle stability. With respect to this, more detailed and concrete Na storage mechanisms should be investigated, and the synthesis of novel ultrafine Fe–P nanostructures/composites especially for FeP4 is also necessary for their practical application.
3.4 Nickel phosphides
Nickel phosphides also have many advantages in Na storage, such as high electronic conductivity (∼106 S m−1 for Ni2P and Ni3P)244,245 and low cost. In addition, various morphologies or refined nanostructures can be obtained for nickel phosphides. For example, Ni2P nanoarrays,227 nanosheets,246 nanorods,247 nanocrystals,92 and Ni3P films248 and particles226 were reported as anodes for Na-ion or Li-ion batteries. The crystal structures of cubic Ni3P and hexagonal Ni2P are shown in Fig. 23a. A nickel phosphide as a SIB anode material was first reported by Fullenwarth et al.199 in 2014 when they demonstrated the great potential of NiP3 in the Na storage field. In their study, two kinds of NiP3 compounds were synthesized via high temperature treatment or ball milling methods, respectively, both with metal Ni and commercial RP as raw materials. And the Na-storage mechanism of NiP3 was also investigated, and proposed as follows:| NiP3 + 9Na+ + 9e− → Ni + Na3P (first discharge) | (16) |
| Na3P ↔ 3Na+ + P + 3e− (subsequent cycles) | (17) |
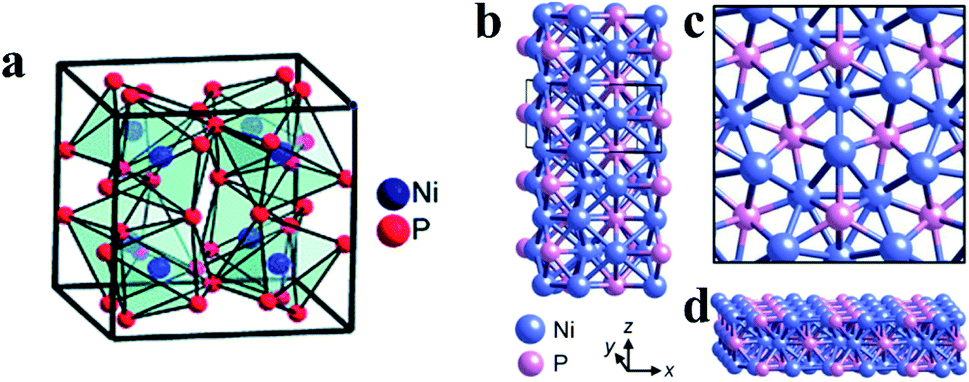 | ||
| Fig. 23 (a) Crystal structure of cubic Ni3P. Reproduced with permission.199 Copyright 2014, The Royal Society of Chemistry. (b) Crystal structure of hexagonal Ni2P: four unit cells stacked on top of one another, with a single unit cell outlined. (c) Top-down view of the Ni2P (001) surface. (d) A two-dimensional slice of Ni2P, showing the (001) surface on top. Reproduced with permission.249 Copyright 2013, American Chemical Society. | ||
Later, Kim et al.226 proposed an approach to synthesize NiP3 particles to allow for chemical bonding with functionalized CNTs through a ball milling process (Fig. 24a). The corresponding anode delivered a high initial reversible capacity of 853 mA h g−1 with more than 80% capacity retention after 120 cycles at 200 mA g−1 and a high rate capacity of 363.8 mA h g−1 after 200 cycles at 1600 mA g−1 (Fig. 24b). The results of physical characterization indicated that the P–C and P–O–C bonds between NiP3 and the functional groups on CNTs were generated during the ball milling process and played an important role in the enhancement of NiP3 anode performances in SIBs. Wu et al.225 also developed an assembly and self-template strategy for the synthesis of a 3D yolk–shell-like nanostructure in which Ni2P nanoparticles with a size of ∼24 nm were embedded into porous graphene networks. The overall fabrication process involved two main steps, an assembly step of NiNH4PO4·H2O nanorods with graphene oxide (GO), and a following annealing step under a reducing atmosphere (H2). Here, the researchers reported that the Ni2P anode for SIBs delivered a reversible initial capacity of 181 mA h g−1 at 0.2 A g−1 and maintained a capacity retention of 89% over 100 cycles. In addition, Dong et al.227 designed a sandwich-like nanoarchitecture hybrid of Ni2P nanoarrays and nitrogen-doped graphene (Ni2P/NG/Ni2P) as a SIB anode and reported that the as-prepared anode delivered a high capacity of 188 mA h g−1 (57% of initial capacity) at 0.5 A g−1 over 300 cycles. Shi et al.92 also designed a covalent heterostructure of monodisperse Ni2P nanocrystals with sizes of 10–30 nm immobilized on N, P co-doped carbon nanosheets as a SIB anode (Fig. 24c–e) and reported that the anode showed enhanced cycling stabilities and high rate performances, maintaining 361 mA h g−1 at 100 mA g−1 after 300 cycles and 181 mA h g−1 at 500 mA g−1 after 1200 cycles (Fig. 24f and g). It is acknowledged that serious agglomeration during sodiation/desodiation of transition metal phosphides (TMPs) leads to challenging kinetic issues and rapid capacity fading. Here, the researchers reported that an agglomeration free Na storage process was achieved and could be visualized using in situ TEM. In addition, the strong bonding between Ni2P and NPC was evidenced through DFT calculations, which could stabilize Ni2P nanocrystals and simultaneously enable synergistically enhanced charge storage by providing exponentially increased electronic states around the Fermi level.
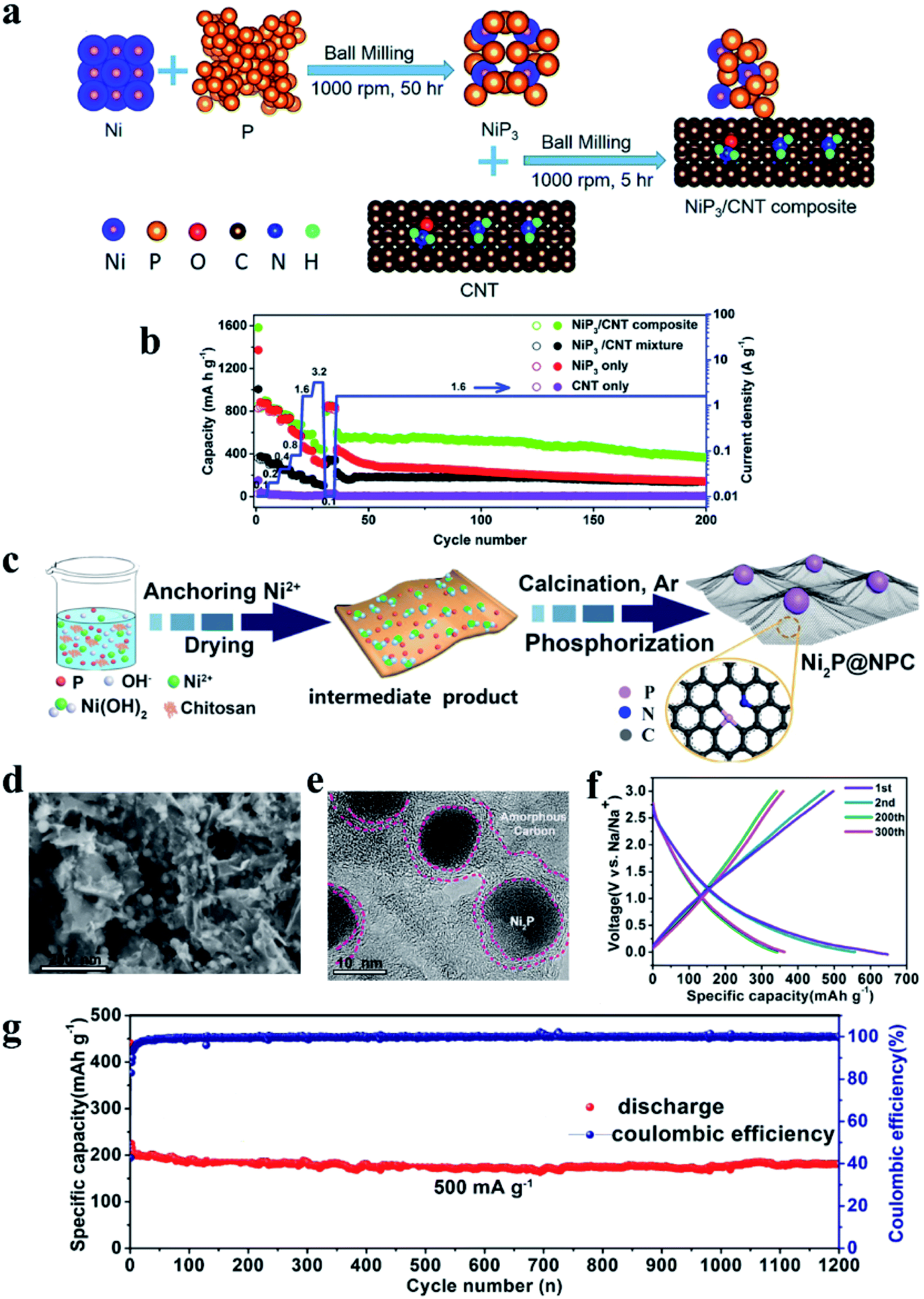 | ||
| Fig. 24 (a) Schematic of the synthesis of the NiP3/CNT composite. (b) Comparison of the cycling performances of CNT, NiP3, NiP3/CNT mixture and NiP3/CNT composite electrodes. Reproduced with permission.226 Copyright 2018, The Royal Society of Chemistry. (c) Schematic of the synthesis of Ni2P@NPC. (d) SEM and TEM images of Ni2P@NPCS. (e) HR-TEM image of Ni2P@NPC. (f) Galvanostatic charge/discharge profiles of the Ni2P@NPC SIB anode for the 1st, 2nd, 200th and 300th cycles respectively. (g) Long-term cycling stability and coulombic efficiency of the Ni2P@NPC composite at 500 mA g−1. Reproduced with permission.92 Copyright 2018, Elsevier Ltd. | ||
Recently, a microporous structure of Ni2P@C–N polyhedrons embedding in highly rough carbon fiber was designed as a self-supporting anode material for SIBs.250 Due to the ultra-small size of Ni2P particles (∼5 nm) and the unique host nanostructure, the corresponding anode delivered a long-term cycling stability (196.8 mA h g−1 at 1000 mA g−1 over 1000 cycles with a capacity fading of 0.04% per cycle) and superior rate capability (197.1 mA h g−1 at 2000 mA g−1, and returned to 752.5 mA h g−1 at 100 mA g−1). Moreover, a flexible half-cell and a full cell based on this electrode could also give an excellent Na storage performance.
Guo et al.251 synthesized tailored carbon-coated hollow Ni12P5 nanocrystals with an average size of 35 nm that were in situ grown on reduced graphene oxide nanosheets (Ni12P5@C/GNS) using a template-free refluxing method. The corresponding composite anode showed good Na storage performances due to the finely designed dual carbon shell nanostructure and was found to be able to accommodate volume expansion, enhance ion/electron transfer, prevent Ni12P5 nanocrystal aggregation and thus deliver a reversible capacity of 235 mA h g−1 at 100 mA g−1.
To sum up, nickel phosphides such as NiP3, Ni2P, and Ni12P5 can show great potential as anodes for practical SIBs, among which NiP3 possesses the highest P content as well as the highest theoretical gravimetric specific capacity. Although the pure NiP3 anode can give a high specific capacity, the insufficient cycle stability hinders its application. In contrast, the NiP3-CNT anode enables longer cycle lifespan, but a concern that the use of CNTs may increase the fabrication cost may be noticed. The high content of Ni in Ni2P or Ni12P5 leads to smaller theoretical gravimetric specific capacity than that of NiP3. Nevertheless, the high content of Ni may play an important role in achieving long cycle stability. For all these Ni–P anodes, the initial coulombic efficiency is not high (50–80%), and the specific capacities are also very low, far from the theoretical values (1591 mA h g−1 for NiP3 and 1333 mA h g−1 for Ni2P). With respect to this, more effort should be made, including design and synthesis of novel nanostructures/composites and exploration of facile and large-scale preparation methods (especially for NiP3, which has only been prepared by ball milling or high temperature methods).
3.5 Copper phosphides
Copper phosphides also possess various advantages in electrochemical Na storage applications. Among these copper phosphides, CuP2 and Cu3P are often used as anodes for Na-ion batteries. CuP2 can exhibit a monoclinic structure while Cu3P usually has a hexagonal structure. In the early stage of the study of these copper phosphides, ball milling is the most commonly used method to prepare anode materials. For example, Zhao et al.189 synthesized a composite consisting of crystalline CuP2 cores coated with carbon black nanoparticles (CuP2/C) through two steps of ball milling processes, and reported that this structure could enable fast and reversible sodiation/desodiation, allowing a capacity of more than 500 mA h g−1 at 50 mA g−1, a high rate capacity of 178 mA h g−1 at 2000 mA g−1 and decent short-term cycling stability. Then a chemically bonded CuP2/C hybrid was also prepared by a one-step ball milling method,252 and the strong P–O–C bond and the stable nanoscale conducting framework in this hybrid offered an enhanced Na storage performance. According to these studies and other literature,189,228,252 the electrochemical reaction mechanism of CuP2 can be expressed as follows:| CuP2 + 6Na+ + 6e− ↔ Cu + 2Na3P | (18) |
It means that crystalline CuP2 can be recovered after one discharge/charge cycle and shows a high theoretical capacity of 1282 mA h g−1 based on this conversion and alloy reaction.
Recently, other methods to prepare CuP2 anodes such as the solution phase method coupled with a heat treatment have been developed.198,228 For example, Yu et al.228 designed a nanocomposite of cross-linked hollow carbon sheet encapsulated CuP2 nanoparticles (CHCS–CuP2) prepared in several steps including surfactant-assisted wet-chemical precipitation, carbon coating and in situ phosphorization (Fig. 25a). Here, the researchers reported that the hollow structure and carbon shells could mitigate large volume change, prevent CuP2 nanoparticle aggregation and significantly enhance electron/ion transport (Fig. 25b–d). In addition, the connectivity of the cross-linked nanocomposite could enhance structural stability and resist inner stress, allowing for longer lifespans during cycling. As a result, the corresponding SIB anode delivered a high reversible capacity of 451 mA h g−1 at a current density of 80 mA g−1 with a retention of 91% after 200 cycles, and an excellent rate performance was also obtained (Fig. 25e and f). Furthermore, a full cell made based on this anode also provided an excellent Na storage performance, showing great potential for practical SIBs. Similarly, Duan et al.253 reported a CuCl2/chitosan monolith derived CuP2/C composite where CuP2 nanoparticles were uniformly embedded in the carbon matrix. The strong chemical bonding between electron rich groups in chitosan and the heavy metal ion (Cu2+) played a key role in the synthesis of this homogeneous monolithic composite. In addition, the chitosan derived carbon could prevent Cu and CuP2 particles from aggregation upon the subsequent thermal reduction and phosphorization. Benefiting from the synergistic effect of the small particle size and conductive carbon matrix, the CuP2/C composite anode for a SIB could deliver a high reversible capacity of 630 mA h g−1 at a current density of 100 mA g−1 and a capacity retention ratio of 91% after 200 cycles, while bare CuP2 showed a rapid capacity decay within 50 cycles.
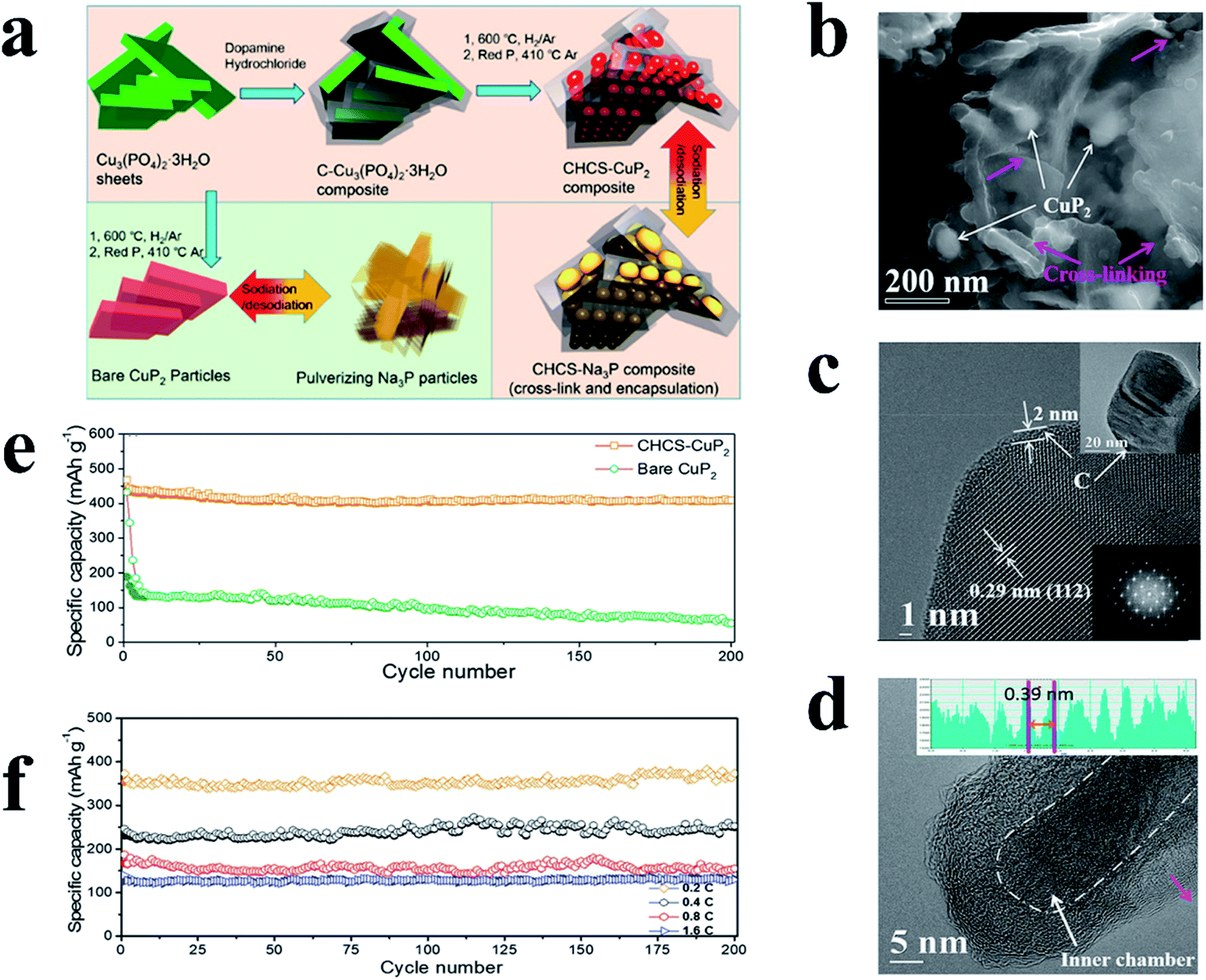 | ||
| Fig. 25 (a) Schematic image of the preparation of the CHCS–CuP2 composite and bare CuP2 particles, as well as the reversible reactions with Na+ ions of these anodes. (b) SEM images of CHCS–CuP2 composites. (c) HRTEM image of the CHCS–CuP2 composite and its corresponding Fourier transformation in the inset, showing a thin carbon layer (∼2 nm) and the (110) planes of CuP2. (d) HRTEM image of a hollow carbon sheet with a wide interlayer distance (∼0.39 nm), showing an apparent inner chamber allowing for volume changes of CuP2 during cycling. (e) Cycling performance of bare CuP2 and CHCS–CuP2 composites at 80 mA g−1. (f) Cycling performance of CHCS–CuP2 composites at 0.2, 0.4, 0.8, and 1.6C (1C = 800 mA g−1). Reproduced with permission.228 Copyright 2018, American Chemical Society. | ||
Cu3P is also often used as an anode material for SIBs. Although Cu is inactive with Na+ and thus shows a relatively low theoretical capacity of 363 mA h g−1 (similar to that of graphite), other advantages make it suitable as an anode for SIBs, such as air stability, high electronic conductivity, safe intercalation potential, and a high theoretical volumetric capacity of 4732 mA h cm−3 (830 mA h cm−3 for graphite).254 Fan et al.78 reported an additive-free Cu3P nanowire (CPNW) anode directly grown on a copper current collector via an in situ growth and a subsequent phosphidation process. Due to its structure features, the CPNW anode demonstrated highly stable cycling ability with an ∼70% retention in capacity after 260 cycles. According to the literature, a reversible sodiation/desodiation of Cu3P proceeds via a conversion mechanism, which is similar to that of CuP2, and can be expressed as follows:
| Cu3P + 3Na+ + 3e− ↔ 3Cu + Na3P | (19) |
After this study, many Cu3P composites have been successfully prepared by various methods. Inspired by metal–organic framework (MOF) derived strategies to synthesize metal phosphides/carbon (MP@C) nanostructures, Kong et al.198 designed a nanostructure of Cu3P nanoparticles (∼50 nm) coated with N, P-codoped carbon matrices using a metal–organophosphine framework (MOPF) derived strategy (Fig. 26a–d), where PTA represents 1,3,5-triaza-7-phospha-adamantane. This approach is relatively eco-friendly with no need for an additional P source, thus avoiding the release of harmful PH3 which is inevitably produced in the phosphorization process for most MOF-derived MP@C nanostructures.204,255,256 Due to this unique nanostructure, the corresponding SIB anode of this composite exhibited a high initial reversible capacity of 332 mA h g−1 at 50 mA g−1, a rate capacity of 142.5 mA h g−1 at 2000 mA g−1 and good cycling performances with a retention of 85% after 100 cycles (Fig. 26e–g). Zhu et al.257 reported a composite of 3D Cu3P@C nanosheets synthesized by heating a mixture of copper foam and phosphorus-containing resin. During the heat treatment, an epitaxial phosphidation growth coupled with a carbon deposition process occurred on the surface of copper foam, and the 3D Cu3P@C nanosheets were formed. In each individual nanosheet, the thin carbon shell could serve as an electron conductor and accommodate the volume change of the Cu3P single-crystalline nanosheet. Due to its unique 3D nanoarchitecture, the Cu3P@C anode delivered a high capacity retention of 286 mA h g−1 after 300 cycles at 0.1 A g−1 and 156 mA h g−1 after 1000 cycles at 1 A g−1.
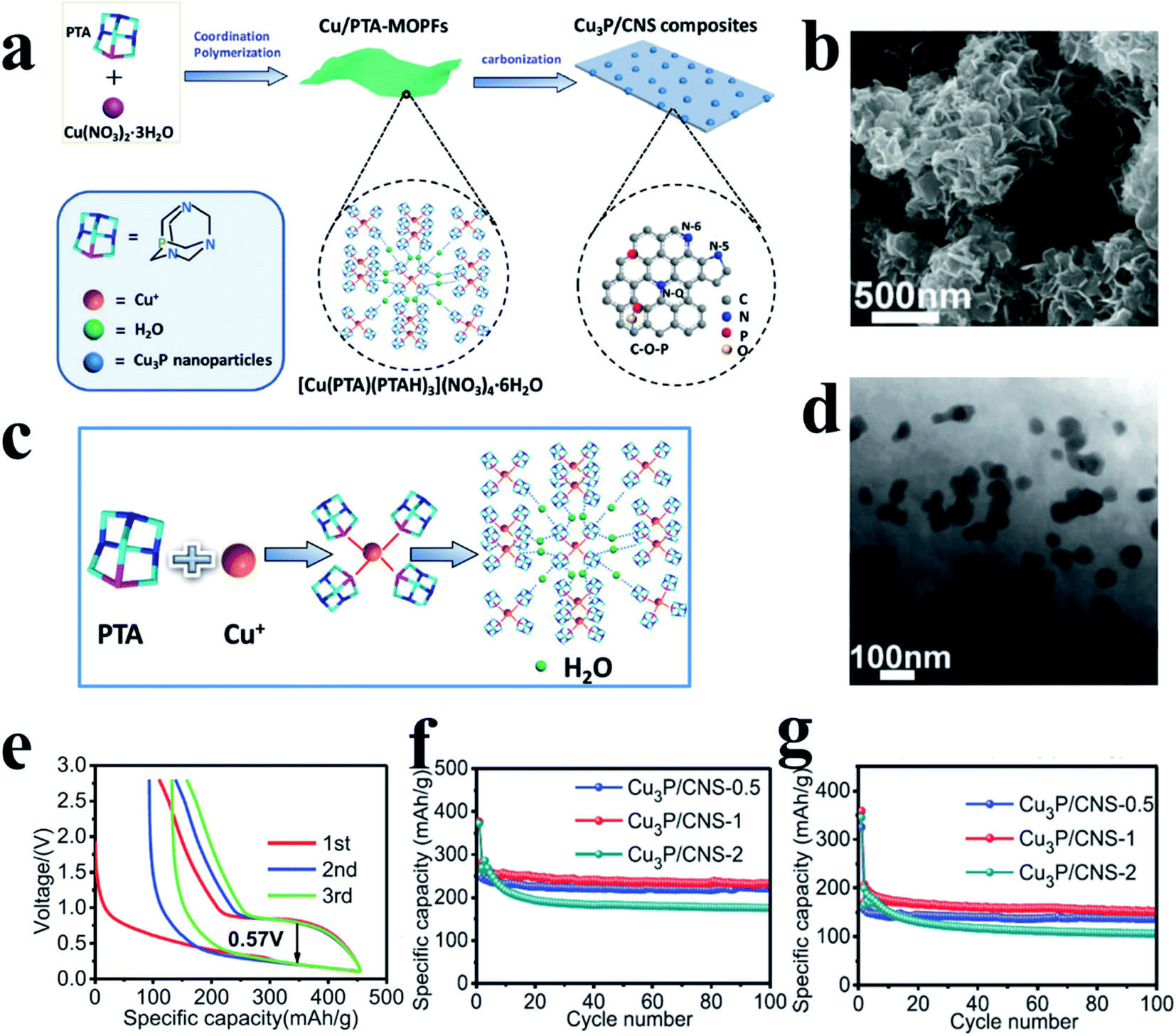 | ||
| Fig. 26 (a) Schematic illustration of the evolution process from Cu/PTA-MOPFs to Cu3P/CNS composites. (b) SEM images of Cu/PTA-MOPF-1 (molar ratio of PTA and Cu(NO3)2 is 1:1). (c) Schematic illustration of the coordination between PTA molecules and Cu ions in the precursors. (d) TEM images of the Cu3P/CNS-1 composite. (e) The initial three charge/discharge curves of the Cu3P/CNS-1 composite at 50 mA g−1. (f, g) Cycling performances of three Cu3P/CNS composites at 100 and 1000 mA g−1, respectively (CNS-0.5, CNS-1, and CNS-2 represent the molar ratio of PTA and Cu(NO3)2 of 0.5, 1, and 2, respectively). Reproduced with permission.198 Copyright 2018, WILEY-VCH. | ||
Recently, Cu2P7 as an anode material for SIBs has also been reported and showed excellent electrochemical performance. For example, Li et al.258 successfully prepared a Cu2P7/CuP2/graphite composite by a facile ball milling method and used it as an anode material for SIBs. Benefiting from a synergistic effect of the high P concentration, the layered structure and heterointerfaces between the different phases, the corresponding anode could provide capacities of 1254 mA h g−1 after 100 cycles at 200 mA g−1 and 200 mA h g−1 at 5 A g−1 in a rate capability test. Similarly, a composite of Cu2P7-BP-MWCNT (MWCNT, multiwalled carbon nanotube) was also prepared by a mechanical milling method.259 When used as an anode for Na-ion batteries, the composite delivered high comprehensive performances: initial coulombic efficiency up to 84%, 1170 mA h g−1 after 200 cycles, and 580 mA h g−1 at 5 A g−1.
In summary, copper phosphides, including CuP2, Cu3P and Cu2P7, have also attracted much attention because of their low-cost, high electronic conductivity and other advantages. Although the ball milling method to prepare CuP2 composites is facile, the insufficient cycle life-span of these anodes is a concern for practical application. In contrast, CuP2/C nanoparticles prepared by the solution phase method (assisted by other processes) show better cycle stability. However, these CuP2 anodes all suffer from insufficient gravimetric specific capacity and long-cycle lifespan, which need to be addressed in future research. Various Cu3P nanostructures/composites such as nanowires, nanoparticles, and nanosheets have been prepared, and some high sodium storage performances are achieved in terms of rate capability and long-cycle stability. However, the low theoretical specific capacity may hinder their application, especially for electric vehicles or even large-scale energy storage systems. The Cu2P7 composite has been recently reported as an anode for SIBs and exhibited high specific capacities due to the high P content of Cu2P7. In addition, Cu2P7 promises high rate capability due to its layered structure and high electronic conductivity, which favor Na+/e− transportation. All of this makes Cu2P7 a promising candidate as an anode material for practical SIBs.
3.6 Germanium phosphides
Germanium phosphides (GePx) such as GeP5 and GeP3 have recently been extensively investigated as anode materials for SIBs. GeP5 possesses a large theoretical specific gravimetric capacity of 1888 mA h g−1 due to the highest P atomic percentage among all the binary phosphides, and a high theoretical volumetric capacity of 6891 mA h cm−3 which is the best record in anodes for SIBs. In addition, rhombohedral GeP5 exhibits a layered structure similar to those of black P and graphite (Fig. 27a), with a high electrical conductivity of ∼106 S m−1, 4 orders of magnitude higher than that of black P and similar to that of graphite (XRD pattern is shown in Fig. 27b). All of this indicates that GeP5 is another promising candidate as an anode material for SIBs. Li et al.193 synthesized a GeP5/C composite by ball milling GeP5 and commercial graphite and reported a low potential of ∼0.4 V vs. Na+/Na with a smooth charge/discharge profile. This GeP5/C anode showed an excellent Na storage performance, delivering a large reversible capacity of 1250 mA h g−1 with an initial coulombic efficiency of 93% and considerable cycling stabilities with 98% capacity retention after 60 cycles (Fig. 27d). Here, the synergetic effects between the sodiation of Ge and P reportedly leveled multistep plateaus smoothly and effectively reduced polarization between charge and discharge, leading to high coulombic efficiencies. Moreover, a full SIB cell using this anode coupled with a Na3V2(PO4)3/C cathode provided a large capacity of 800 mA h g−1 with a high average output voltage of 2.65 V. The researchers also investigated the electrochemical Na storage mechanism of GeP5, which can be described by equations as shown in the following:| GeP5 + 16Na+ + 16e− → 5Na3P + NaGe (first discharge) | (20) |
| Na3P ↔ 3Na+ + P + 3e− (subsequent cycles) | (21) |
| NaGe ↔ Ge + Na+ + 3e− | (22) |
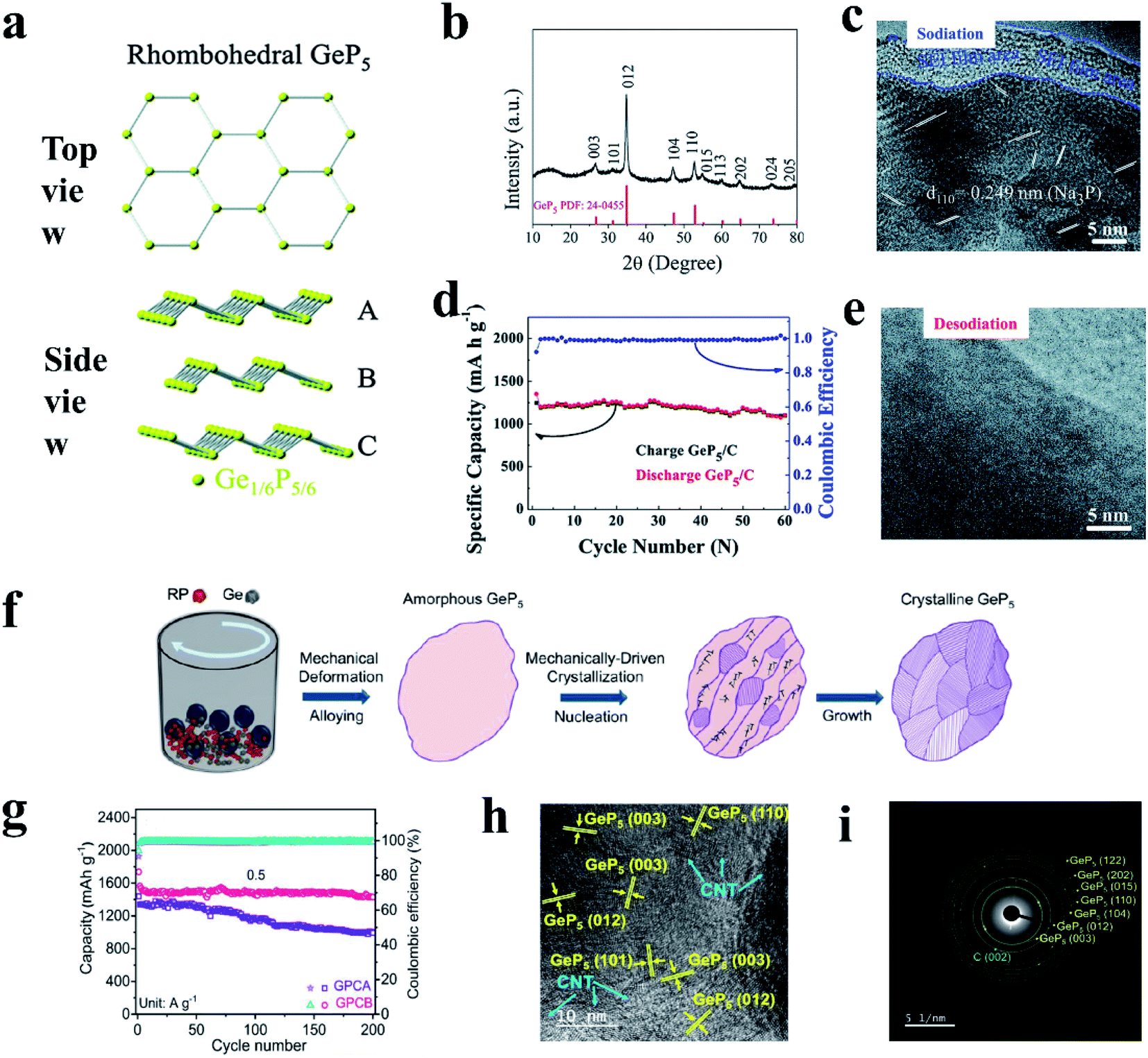 | ||
| Fig. 27 (a) Crystal structure of rhombohedral GeP5. Reproduced with permission.261 Copyright 2015, The Royal Society of Chemistry. (b) XRD patterns of the as-synthesized GeP5/C composite. (c, e) HRTEM of the GeP5/C electrode in the full sodiation state (discharged to 0.005 V), and in the full desodiation state (charged to 3.0 V), respectively. (d) The cycle performance and corresponding coulombic efficiency of the as-synthesized GeP5/C composite. (b–e) Reproduced with permission.193 Copyright 2017, The Royal Society of Chemistry. (f) Schematic explanation of mechanically induced crystallization that occurs during the milling of Ge and RP elemental powders. (g) Comparison of the cycling stability of GPCA and GPCB electrodes in the voltage range of 0.01–2.00 V at a current density of 0.50 A g−1 for 200 cycles (GPCA and GPCB represent the amorphous and crystalline GeP5-based composites, respectively). (h, i) HRTEM micrographs and SAED patterns of GPCB after the 50th cycle in the charged state. (f–i) Reproduced with permission.262 Copyright 2019, American Chemical Society. | ||
It was confirmed that GeP5 experienced conversion-alloying type sodium storage during an electrochemical reduction process. GeP5 was converted into Na3P and NaGe after the initial discharge process, and Na3P and NaGe respectively underwent desodiation/sodiation processes during the next cycles. This mechanism could be evidenced by the TEM images of the sodiated and desodiated states of GeP5 (Fig. 27c and d). Actually, in the theoretical capacity of GeP5 (1888 mA h g−1), Ge element can contribute 118 mA h g−1 and 5 P atoms donate the remaining 1770 mA h g−1.
GeP5 could also be obtained through the ball milling of high-purity Ge powder and commercial RP, and the composite of this GeP5 with carbon (GeP5/C) also demonstrated good Na storage performances.260 Ning et al.229 also prepared this GeP5 by ball milling Ge powder and RP, and further designed a dual-carbon conductive network enhanced GeP5 composite by a ball milling method (GeP5/AB/p-rGO; AB represented acetylene black, and p-rGO was partially reduced graphene oxide). They suggested that the dual carbon network not only provided more electron transport pathways but also relaxed the huge volume change during charge/discharge. As a result, this composite anode delivered a high reversible capacity of 597.5 and 175 mA h g−1 at current densities of 0.1 and 5.0 A g−1 respectively and high stability with 400 mA h g−1 capacity retention at 0.5 A g−1 after 50 cycles. Furthermore, Tseng et al.230 prepared mesoporous germanium phosphide (MGePx) microspheres as a SIB anode material through a high-temperature solvothermal method. These MGePx microspheres with diameters ranging from 0.5 to 1.5 μm were composed of 10 nm nanoparticles with a narrow pore size distribution of ∼4 nm and possessed an amorphous structure with a weight ratio of P to Ge of 38.3 to 61.7. This unique porous nanostructure demonstrated high performance in sodium storage including high energy density, ultrahigh rate capability, and superior cyclability, showing a high reversible capacity of 704 mA h g−1 after 100 cycles at 240 mA g−1 and stable cycling performances of 278 mA g−1 at 1200 mA g−1 after 200 cycles.
Very recently, Cho et al.260 synthesized two kinds of GeP5, crystalline GeP5 (the GeP5 anodes reported by others were all of this type) and amorphous GeP5 (Fig. 27f); furthermore, the authors also prepared hybrids of GeP5 and multiwalled carbon nanotubes (GeP5/CNT). The researchers found that the crystalline GeP5 exhibited better Na storage performance than the amorphous one and proposed that the crystalline GeP5 exhibited a higher electronic conductivity, more facility for the insertion of Na+, and smaller volume expansion after sodiation. As a result, this crystalline GeP5/CNT anode showed a high initial CE of 94.13% and a high capacity of 1430 mA h g−1 after 200 cycles at 0.5 A g−1, corresponding to a capacity retention of 91.43% from the second cycle (Fig. 27g). Interestingly, the authors suggested that GeP5 could recover to its crystalline phase after desodiation, as evidenced by the TEM images (Fig. 27h and i), which was contradictory to other reported literature.193,260 Therefore, further investigation about this issue is needed.
Kim et al.263 introduced GeP3 as a promising anode material for SIBs and carried out a comparative investigation of GeP3 and GeP5. In their study, GeP3 was obtained by ball milling of Ge and RP, and then the GeP3/C compound was obtained by further ball milling of GeP3 and carbon (super P) for electrochemical measurements. The GeP5/C composite was synthesized by a similarly procedure. It was confirmed that GeP3 could give a rhombohedral crystal phase similar to GeP5 (Fig. 28a), but with a larger cell volume and average bonding distance compared with GeP5. Based on theoretical calculations, they also found that the bonding strength of P–P was higher than that of P–Ge, and the elastic modulus of GeP3 showed significantly softer features than that of GeP5, indicating that the softer feature of Ge–P bonding could enable more effective accommodation for mechanical stress and deformation during charge/discharge cycles than P–P bonding. This insight could be experimentally confirmed: the synthesized GeP3 compound maintained a high discharge capacity of 1274 mA h g−1 after 150 cycles, while the GeP5 compound with more P–P bonding displayed an inferior cycling stability (Fig. 28b). Furthermore, the morphology and volume change of the GeP5 electrode seemed more serious compared to those of the GeP3 electrode during charge/discharge (Fig. 28c).
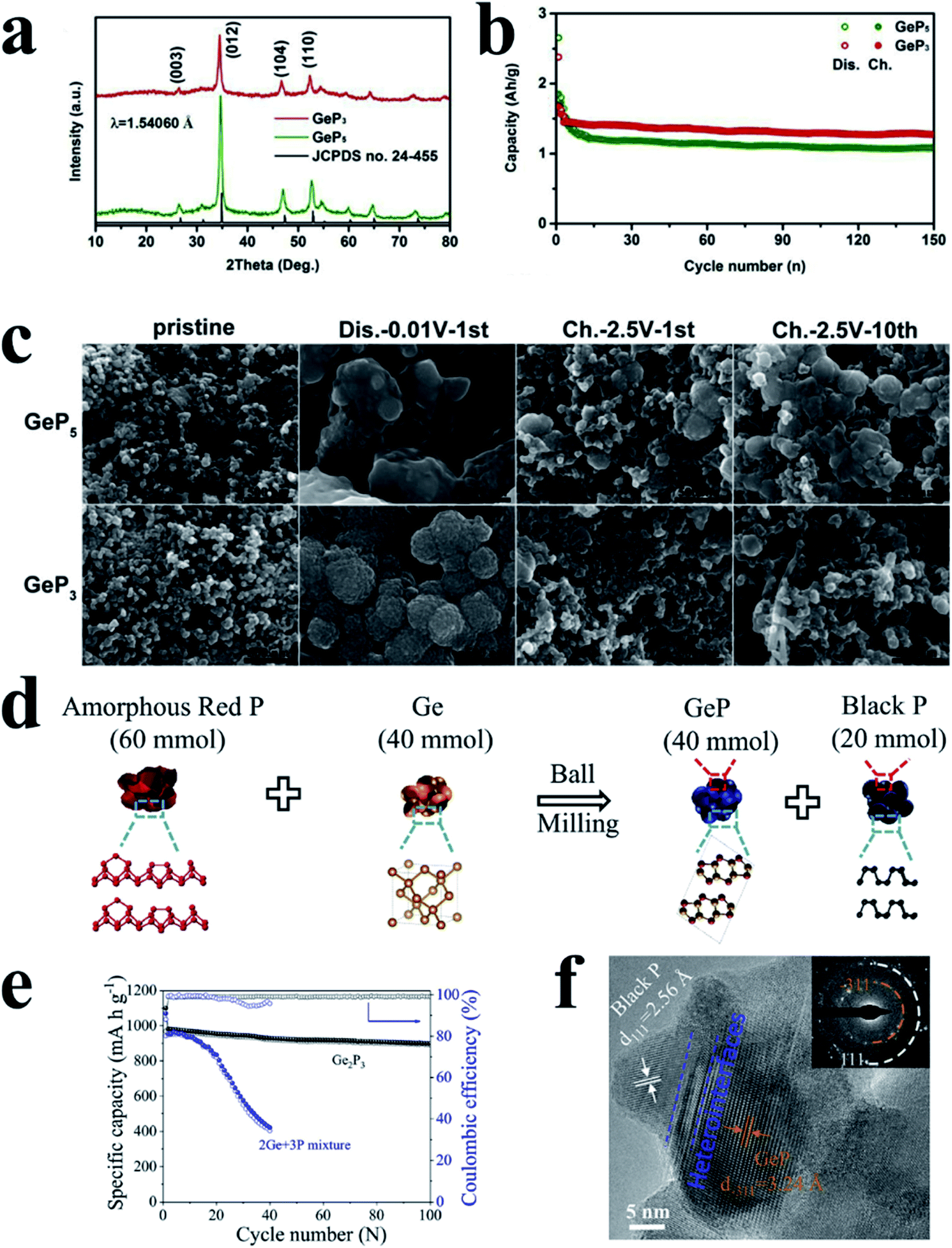 | ||
| Fig. 28 (a) XRD patterns of the as-prepared GeP3 and GeP5 samples. (b) Cycling performance of GeP3 and GeP5. (c) A comparison of morphological changes for GeP5 (up) and GeP3 (down) compounds during the first 10 cycles. Reproduced with permission.263 Copyright 2018, Elsevier Ltd. (d) Illustrated preparation process of the layered GeP-black P composite (Ge2P3). (e) Cycle stability of the Ge2P3 composite. (f) High-resolution transmission electron microscopy (HR-TEM) image and the inset shows the selected area electron diffraction of the Ge2P3 composite. Reproduced with permission.264 Copyright 2019, Elsevier Ltd. | ||
In another study, Nam et al.196 used commercial Ge and synthesized black P as raw materials to synthesize crystalline GeP3 through a high-power ball-milling (HPBM) method, and further prepared a GeP3/carbon (GeP3/C) nanocomposite by ball milling this GeP3 with Super P. The GeP3/C anode showed a highly reversible initial capacity of 984 mA h g−1 with a high initial CE of 81.3%, a very stable capacity retention of 94.9% after 30 cycles, and a high rate capacity of 520 mA h g−1 at 2C (Fig. 29e and f). Furthermore, the researchers investigated the Na storage mechanism of GeP3 by using various techniques, such as extended X-ray absorption fine structure (EXAFS) (Fig. 29b and c), X-ray diffraction (XRD), high-resolution transmission electron microscopy (HR-TEM) (Fig. 29d), and differential capacity (dQ/dV) and cyclic voltammetry (CV). It was indicated that the reaction mechanism involved one-step conversion during Na-insertion and two-step recombination/topotactic-transition during Na-extraction, as shown by the following equations:
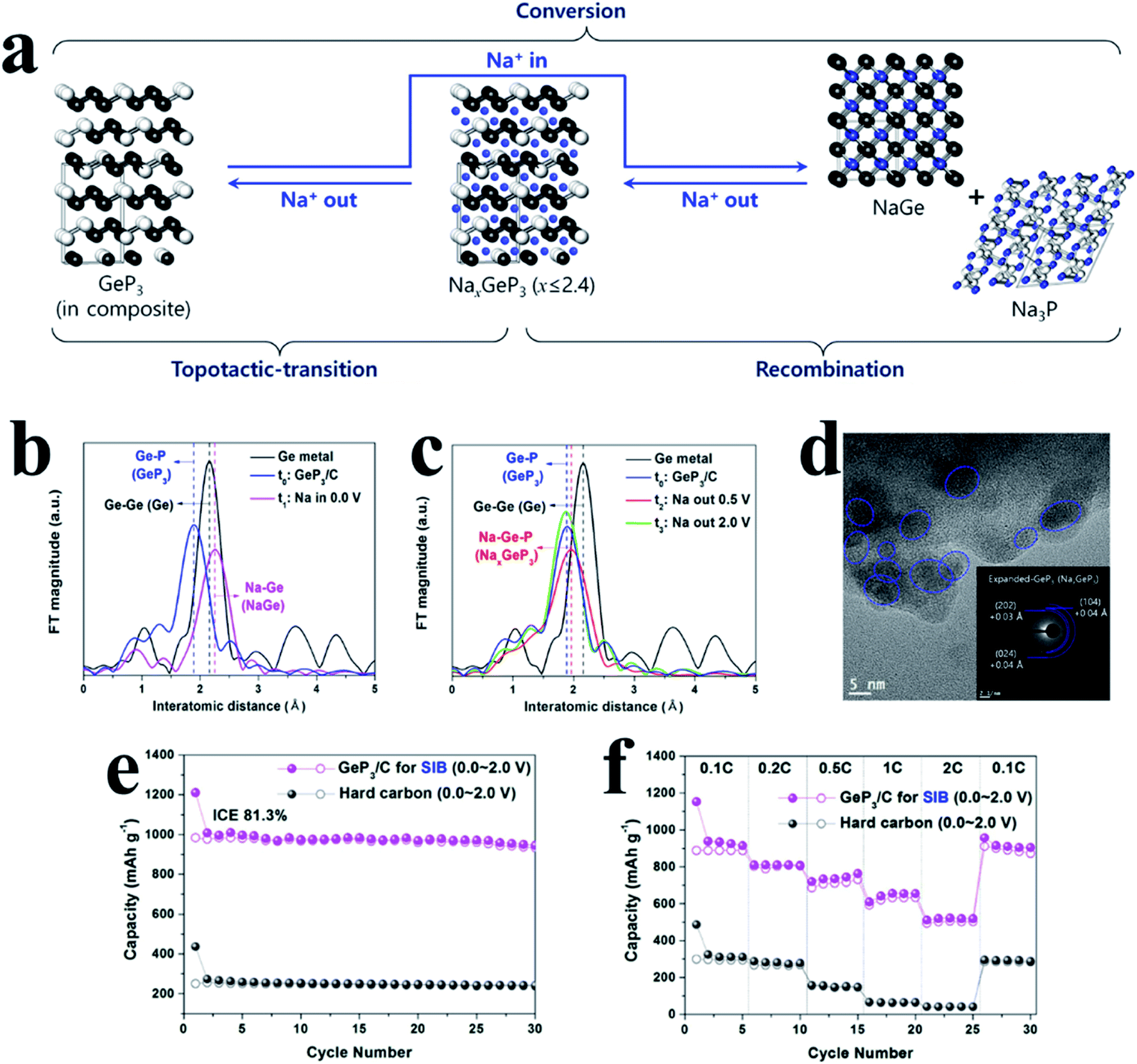 | ||
| Fig. 29 (a) Schematic diagram of the electrochemically driven crystallographic phase-change mechanism of GeP3 during Na-insertion/extraction. (b, c) Ex situ EXAFS spectra of the GeP3-based nanocomposite electrode during first Na insertion and first Na-extraction, respectively. (d) Ex situ HR-TEM image with the corresponding diffraction patterns in the Na-extracted state of 0.5 V for the GeP3-based nanocomposite electrode. (e) Cycling performance of the GeP3-based nanocomposite and hard carbon (cycling rate: 50 mA g−1). (f) Rate capabilities of the GeP3-based nanocomposite (1C = 950 mA h g−1) and hard carbon (1C = 250 mA h g−1) for the SIB. Reproduced with permission.196 Copyright 2018, Elsevier B.V. | ||
Na-insertion:
| GeP3 → NaGe + Na3P | (23) |
Na-extraction:
| NaGe + Na3P → NaxGeP3 + (x ≤ 2.4) → GeP3 | (24) |
Total:
| GeP3 + 4Na+ + 4e− ↔ NaGe + Na3P | (25) |
During Na-insertion, the layer structured GeP3 can be converted to NaGe and Na3P. During Na-extraction, however, Na3P and NaGe are transformed into a NaxGeP3 (x ≤ 2.4) phase, which is then recovered to pristine GeP3 with a topotactic-transition (Fig. 29a).
Recently, a layered GeP-black P composite (Ge2P3) as a binary-phase anode for SIBs was prepared by a simple ball milling method264 and also showed an excellent Na storage performance. During the milling process, element Ge and commercial RP were converted into GeP and BP (black P); in the meantime, these two products were hybridized to form a layered GeP-BP composite (Fig. 29d and f). The authors proposed that the numerous heterointerfaces between layered GeP and BP could increase the number of reaction sites and enhance the Na-ion and electron transport capability. Coupled with the intrinsic excellent Na storage capability of GeP and BP, this binary-phase anode showed a reversible capacity of 970 mA h g−1 at 50 mA g−1 with an initial coulombic efficiency of 88%, and retained 890 mA h g−1 after 100 cycles (Fig. 29e). Moreover, it also exhibited a rate capacity of 275 mA h g−1 at 5 A g−1, which could recover back nearly to its initial value when the current density was changed back to the initial value of 50 mA g−1. This study shows a simple and cost-efficient strategy to prepare GeP and BP in one step, offering excellent possibility for application in practical SIBs. In another study, a GeP anode with a flexible layered structure was successfully synthesized by a simple mechanochemical method.197 When tested as the anode for a SIB, the GeP anode delivered an initial discharge/charge capacity of 900/840 mA h g−1 with an initial coulombic efficiency of up to 93% and a rate capacity of 360 mA h g−1 at a current density of 5 A g−1. Similar to GeP3, GeP underwent a reversible Na-storage process that involved intercalation, conversion, and alloying. The peculiar self-healing phenomenon was derived from its ultra-low formation energy (0.19 eV) for the reconstruction of the original layered structure, as confirmed by first-principles calculations. The low formation energy also enabled the formation of chemical bonding between layered GeP and graphite, thus achieving an excellent performance as an anode for SIBs.
Overall, germanium phosphides also attract much attention and show great potential as anode materials for SIBs. Despite the high cost of Ge, these Ge–P anodes may still be affordable for large scale application, especially GeP5 which has a low molar ratio of Ge. Although high performances of these GeP5 composite anodes have been achieved in terms of initial coulombic efficiency and specific capacity (especially for GeP5/CNT), up to now, all these GeP5 composites are prepared by the ball milling method and show a large particle size. The large particle size might be a major reason for their insufficient long-cycle stability. In addition, the poor rate capability is also a concern for practical application. The MGePx (x ≈ 1.6) composite reported by Tseng et al.230 is the only case where a germanium phosphide anode was prepared by a solvothermal method. Due to the ultrafine nanostructure of this MGePx, a better rate capability could be obtained. Although the P content in GeP3 is lower than that in GeP5, it was reported that a superior sodium storage performance could be achieved for GeP3 compared to GeP5 in terms of specific capacity and cycling stability. GeP and its composites also exhibit high performance for Na storage; however, the high molar ratio of Ge may increase the fabrication cost of these GeP anodes, and thus may hinder their practical application.
3.7 Other metal phosphides
Other binary metal phosphides have also been studied for SIB anodes. For example, Li et al.232 synthesized a well-aligned Zn3P2 nanowire-assembly bundle/Ti foil integrated anode through a facile chemical vapor deposition method and reported that this anode showed high sodium storage performances, delivering a reversible capacity of ∼600 mA h g−1 at 100 mA g−1 and a high rate capability of 280 mA h g−1 at 5 A g−1. Huang et al.231 also successfully prepared molybdenum phosphide (MoP) nanorods wrapped within a thin carbon layer as a SIB anode material and reported that this MoP electrode exhibited high cycling performances with a discharge capacity of 398.4 mA h g−1 at a current density of 100 mA g−1 after 800 cycles and a remarkable rate capability of 104.5 mA g−1 at 1600 mA h g−1 after 10000 cycles. These low-cost metals such as Zn and Mo give a competitive option for metal phosphides in fabricating large-scale SIB anodes. In another study, Usui et al.209 studied sodiation/desodiation reactions in indium phosphide (InP) anodes which were obtained by ball milling in ionic liquid electrolytes and found that InP could provide phase separation in the first sodiation to form elemental In and P, which could subsequently react with Na individually. And in the following desodiation process, Na-extraction of NaIn and Na3P occurred and a nanostructure of crystalline In nanoparticles dispersed in an amorphous-like P matrix was formed. Based on this Na storage mechanism, InP can provide a theoretical capacity of 730 mA h g−1 (Fig. 30a–c). This mechanism was also observed in Sn4P3 anodes.208 These researchers also reported that the In phase in this InP anode could relax strain induced by P volume change and prevent electrical isolation of active material layers, and in addition, the high conductivity could compensate for the poor electronic conductivity of Na3P phases created by P sodiation, allowing for a high capacity of 500 mA h g−1 after 100 cycles at a current of 50 mA g−1 (Fig. 30d).
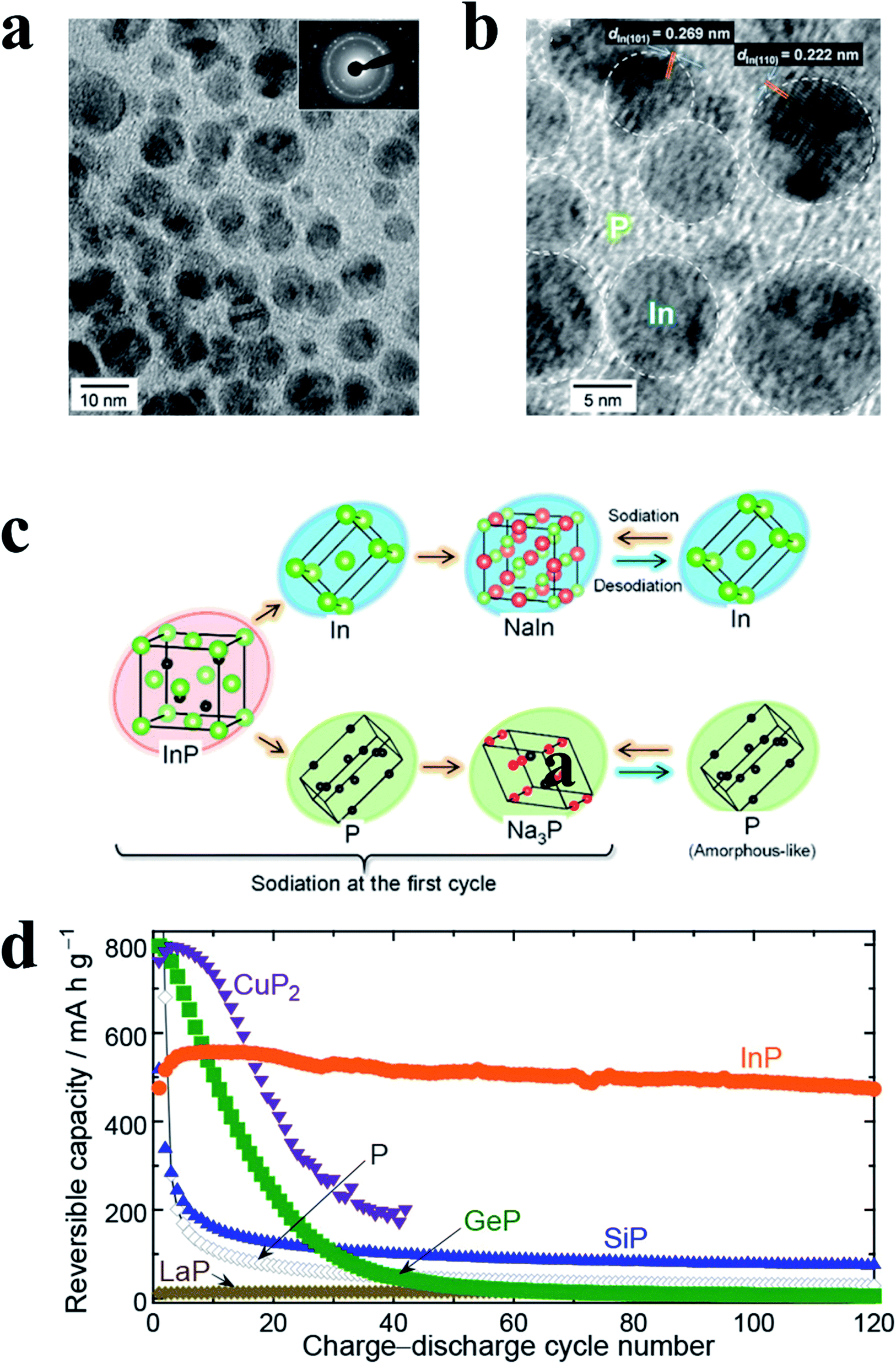 | ||
| Fig. 30 (a) TEM image of InP after the first desodiation. The particle size was 9.3 ± 1.7 nm. (b) High-magnification TEM image showing crystalline In nanoparticles dispersed in an amorphous-like P matrix. (c) Phase changes of InP in charge/discharge reactions. (d) Anode performances of various phosphide electrodes cycled at a current density of 50 mA g−1 in an ionic liquid electrolyte (NaFSA/Py13-FSA). For comparison, results are also shown for the electrode of P alone. Reproduced with permission.209 Copyright 2018, American Chemical Society. | ||
4 Non-metal phosphides
Many non-metal phosphides also possess the ability to store Na ions, such as SiP2 and Se4P4.233,234 Duveau et al.233 prepared a SiP2 material through simple ball milling of silicon and phosphorus powders at room temperature and demonstrated potential as a novel anode for SIBs in which the corresponding anode provided a sustained capacity of 572 mA h g−1 after 15 cycles. Although the reversible capacity is much lower than the theoretical capacity of 1780 mA h g−1 (considering that P can react with 3 Na to form Na3P and that Si has no electrochemical activity toward sodium),265 this pioneering study demonstrates the great potential of SiP2 as a SIB anode material. Recently, Saddique et al.266 synthesized a SiP2/C composite by a facile ball milling method (Fig. 31a). The well-constructed composite comprised of uniformly distributed SiP2 nanocrystallites within a conductive carbon framework could greatly enhance the electrode conductivity and structural compatibility for repeated sodiation/desodiation conversions, demonstrating a high capacity of 410 mA h g−1 at 50 mA g−1 after 100 cycles (Fig. 31f), and a largely improved cyclability with 80% capacity retention for 500 cycles. Furthermore, the authors elucidated the reaction mechanism of SiP2 in SIBs by various investigation methods and found that Si could also electrochemically react with Na, as expressed in the following equations:| SiP2 → Na6Si + Na3P (discharge) | (26) |
| Na6Si + Na3P → (SiP2)crystalline/amorphous + P (charge) | (27) |
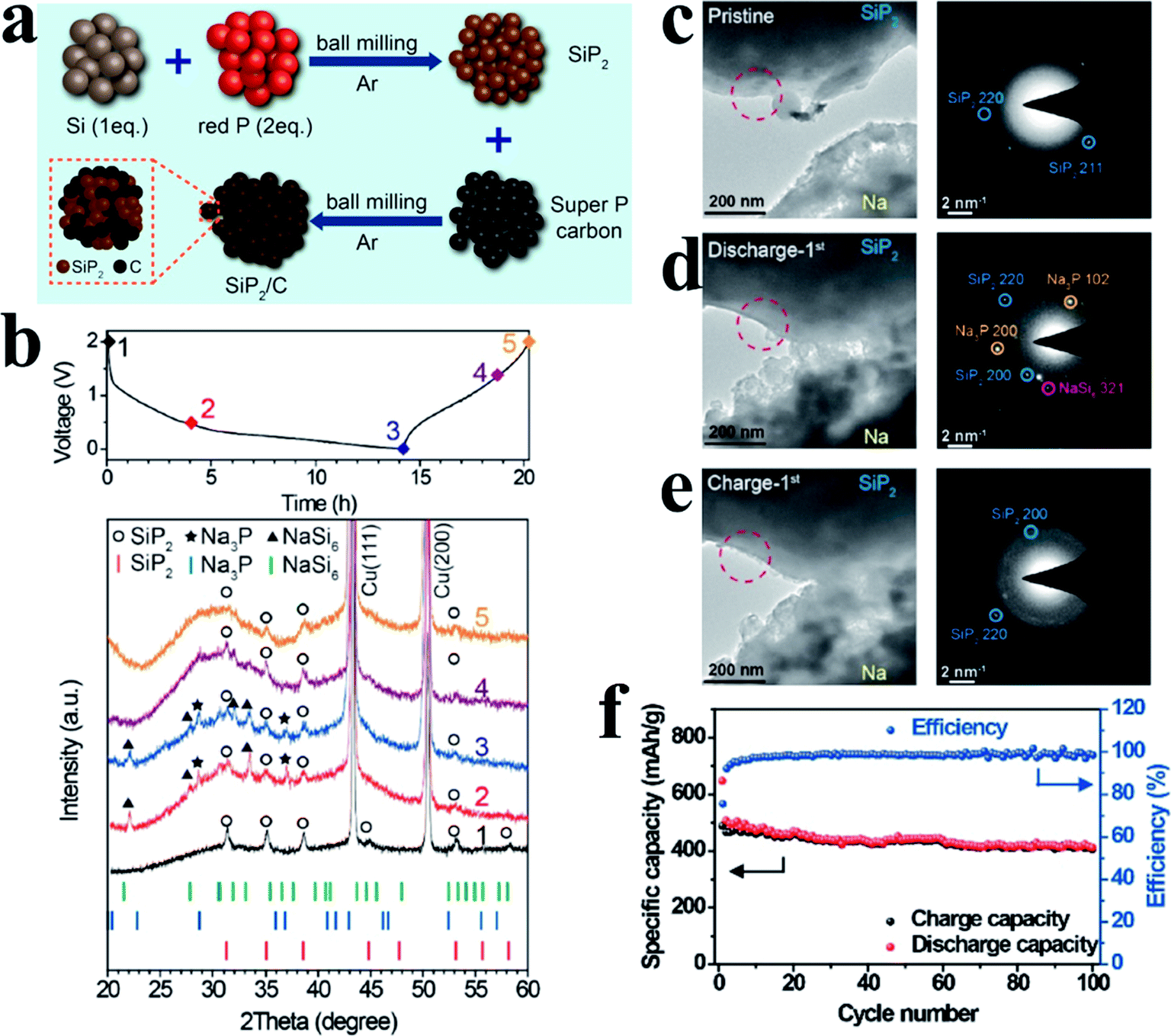 | ||
| Fig. 31 (a) Schematic illustration of the synthesis process of the SiP2/C composite. (b) Ex situ XRD of the SiP2/C electrode in different discharge/charge states in the first cycle. (c–e) In situ TEM investigation of a SiP2–Na nanobattery. TEM images of electrodes and the corresponding SAED patterns of the SiP2 electrode in selected regions acquired before battery working (c), when discharged at −3 V (d), and when further charged at +3 V (e). (f) Cycle performance of the SiP2/C electrode acquired at a current density of 50 mA g−1. Reproduced with permission.266 Copyright 2019, American Chemical Society. | ||
This reaction mechanism was evidenced by the ex situ XRD and TEM images (Fig. 31b–e). During the first discharge process, crystalline SiP2 was gradually transformed into Na6Si and Na3P because of Na insertion. In the charge process, only parts of Na6Si and Na3P could reversibly form SiP2, resulting in amorphization of part of SiP2 after full charge. This could be evidenced by the XRD result where the SiP2 peaks could not recover their initial intensities after full charge (Fig. 31b). Although Na storage performance is insufficient, this study demonstrates that SiP2 can also reversibly react with Na. And in view of the low cost and environmental friendliness of both Si and P, these studies offer the possibility for scalable application of SiP2 or other silicides in practical SIBs.
Se4P4 is another attractive anode material for SIBs because selenium is active towards Na and possesses good electrical conductivity and Se4P4 provides a theoretical capacity of 1217 mA h g−1 in reactions with 20 Na ions during sodiation.234,267 For example, Lu et al.234 synthesized an amorphous Se4P4 composite through a facile mechanical milling technique and reported that this Se4P4 anode exhibited a high reversible capacity of 1048 mA h g−1 at 50 mA g−1, good cycling stabilities of 804 mA h g−1 after 60 cycles and outstanding rate capabilities of 724 mA h g−1 at 500 mA g−1 and 332 mA h g−1 at 3000 mA g−1 respectively, which were superior to those of both P and Se. Furthermore, ex situ XRD, TEM and in situ Raman analysis revealed that the reaction mechanism during the charge/discharge of Se4P4 was as follows:
| Se4P4 + 20Na+ + 20e− ↔ 4Na3P + Na2Se | (28) |
Se4P4 was converted into Na2Se and Na3P after full discharge and subsequently reformed to amorphous Se4P4 in the subsequent charging process. Furthermore, a full cell with a Se4P4 anode and a Na3(VO0.5)2(PO4)2F2/C cathode delivered a discharge capacity of 985 mA h g−1 (based on the mass of Se4P4) and an average output voltage of ∼2.5 V. Cao et al.268 also successfully synthesized P4Se3 microparticles (higher P content as compared with Se4P4) through simple heating of selenium and amorphous RP at low temperatures (Fig. 32a and b) and reported that these orthorhombic-phase microparticles as a SIB anode could exhibit a high reversible capacity of 654 mA h g−1 at 200 mA g−1 after 70 cycles and high rate capabilities with a reversible capacity of 486 mA h g−1 at 3 A g−1 (Fig. 32c and d). Here, the researchers attributed this high electrochemical performance to the formation of more conductive Na2Se and Se (∼10−6 and ∼10−5 S cm−1) during cycling and the dominance of capacitive behavior, all of which could accelerate kinetic behaviors.
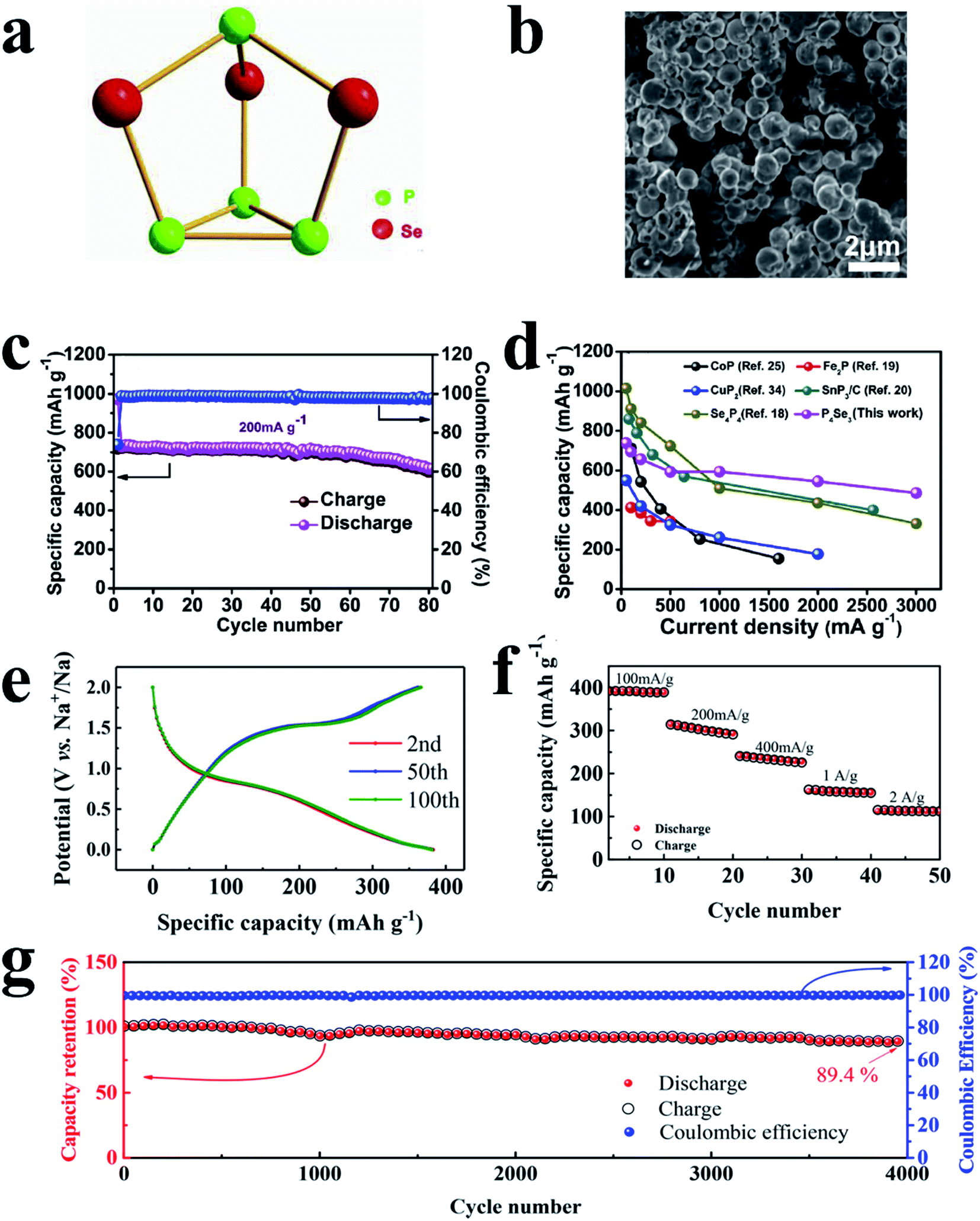 | ||
| Fig. 32 (a) Illustration of the cage structure of P4Se3. (b) SEM image of P4Se3. (c) Cycling stability tested at 200 mA g−1 and coulombic efficiency of P4Se3 anodes. (d) Rate capability comparisons of reported studies and this study. Reproduced with permission.268 Copyright 2018, Elsevier B.V. (e and f) Electrochemical performances of the P2S5/C composite after 70 hours of ball milling. (e) Typical potential profiles of the second, fifth and 100th cycles. (f) Rate capability of the P2S5/C composite. (g) Performance and coulombic efficiency at 2000 mA g−1. Reproduced with permission.235 Copyright 2017, American Chemical Society. | ||
In another example, Li et al.235 prepared a P2S5/C composite using a simple and low-cost high energy ball milling method and investigated its reaction mechanisms with Na. Here, the researchers found that during the ball milling process, crystalline P2S5 transformed into amorphous P2S5 to form an amorphous P2S5/C composite that could exhibit a safe average potential of 0.82 V, and the reaction mechanism of the first discharge process may be based on conversion and alloying processes as follows:
Conversion:
| 10Na+ + 10e− + P2S5 → 5Na2S + 2P | (29) |
Alloying:
| 6Na+ + 6e− + 2P → 5Na3P | (30) |
As a result, an anode of this P2S5/C material delivered a high reversible capacity of 396 mA h g−1 at 100 mA g−1, a high capacity retention of 99.5% after 100 cycles at 100 mA g−1 and a moderate capacity of 114 mA h g−1 at 2 A g−1 at a high rate (Fig. 32e and f). In addition, a high capacity retention of 89.4% was achieved even after the amorphous P2S5/C was cycled over 4000 times at 2 A g−1 (Fig. 32g). However, the initial coulombic efficiency (ICE) of this anode is not high (<40%), which is insufficient for practical SIBs. Even so, the low cost and facile P2S5 still shows significant potential for sodium storage if the issue of low ICE can be well addressed.
Theoretical calculations and predictions are also important in the search for new materials for SIB anode applications. For example, Zhao et al.269 identified a hitherto unknown P3C monolayer with a puckered honeycomb structure using first-principles swarm-intelligence structural calculations in which they predicted that the theoretical capacity of P3C as a SIB anode could reach 1022 mA h g−1 and that the acceptable barrier energy and lower open-circuit voltage could ensure good rate capability and safety in practical applications. Here, the researchers attributed the high performances to the unique arrangement of two types of hexagon rings that could produce highly cohesive energy and thermodynamic stability, making P3C monolayers a promising 2D material for SIB applications.
5 Ternary phosphides
Ternary phosphides are a class of novel phosphide materials in which the synergistic effects of different elements may enhance electron/ion conductivity and mechanical stability, and provide richer redox reactions, or bring about other beneficial effects on SIB anode performances.270 For example, Liang et al.236 prepared 2D MPS3 (M = Fe, Co, Ni) nanosheets using an efficient solid-state method in which the MPS3 single-crystal nanosheets were exposed at the (00l) facet and possessed an average lateral size of ≈200 nm with an average thickness of ≈18 nm. And due to the layered structure, good crystallinity, large surface area, excellent electrical conductivity and high pseudocapacitance from surface reactions, the MPS3 nanosheets provided superior performances as anode materials in SIBs; for example, NiPS3 nanosheets could deliver a high initial coulombic efficiency of 70% and a reversible capacity of 910 and 316 mA h g−1 at current densities of 0.05 and 5 A g−1, respectively, and a capacity retention of 88% over 200 cycles at 2.0 A g−1. In addition, the interlayer spacing of these MPX3 nanosheets could be expanded by intercalating guest propylamine molecules, leading to an improved Na storage performance (1090 and 536 mA h g−1 at 0.05 and 5.0 A g−1 respectively for intercalated NiPS3 nanosheets). These superior performances as well as the facile preparation method and the low cost of Fe, Ni, P and S indicate that these 2D MPS3 (M = Fe, Co, Ni) nanosheet composites are promising candidates for practical SIBs. In another example, Marino et al.271 synthesized a ternary Ni2SnP composite as an anode material for SIBs using a simple ball milling method and reported that Ni2SnP only exhibited a reversible specific charge capacity of ∼200 mA h g−1 in SIBs, which was much lower than that of the corresponding Li-ion battery. The researchers found that in SIBs, only phosphorus was active in the electrochemical reaction, unlike in the case of binary Sn-based phosphides such as Sn3P4 (ref. 91) in which Sn was also active towards Na. Here, the researchers suggested that this reaction mechanism could explain the poor cyclability of anode materials for SIBs and that in particular, the low conductivity of P could result in the formation of only small proportions of Na3P. The insufficient sodium storage performance of the Ni2SnP composite may hinder its application in SIBs; however, this study gives a suggestion that the total or partial recombination of the pristine phase is crucial to the electrochemical performance of ternary electrode materials. Lan et al.237 prepared pure phase ternary phosphide-copper tin phosphide (Cu4SnP10) nanowires by a solution-liquid-solid growth method and investigated the performance of these composites as SIB anodes. The researchers found that Cu incorporation could effectively alleviate Sn aggregation in anodes during charge/discharge cycles, as Sn aggregation was considered to be a major reason for the reduced cycling performances in Sn–P compounds. And a composite of Cu4SnP10 nanowires combined with multiwalled carbon nanotubes delivered a stable capacity of 512 mA h g−1 after 100 cycles at a current density of 100 mA g−1 and a rate capacity of 412 mA h g−1 at 1 A g−1. Furthermore, Liu et al.238 synthesized a ball milled FeSi4P4 composite as a SIB anode material and reported a reversible capacity of 180 mA h g−1 at 100 mA g−1 with a capacity retention of 99% after 100 cycles. Although the specific capacity is low, the high short-term cycle stability is quite surprising.In another study, Wang et al.270 prepared a composite of holey-structured 2D Ni1.5Co0.5Px nanosheets as a SIB anode material through a simple solution method and a subsequent phosphorization process. Then these as-prepared 2D holey-structured Ni1.5Co0.5Px nanosheets were subsequently hybridized with surface-functionalized carbon nanotubes (CNTs) to form a lightweight, free-standing and binder-free integrated flexible electrode (Fig. 33a). The researchers reported that the synergistic effects of Co and Ni atoms could enhance charge transfer and promote the release of more capacity as compared with single metal phosphides. In addition, they also suggested that the connected CNT network could affect the formation of conversion reaction products by forming a film structure rather than big-sized microspheres, which could effectively alleviate volumetric changes. As a result, this flexible anode achieved a reversible capacity of 496.4 mA h g−1 at 0.5C and a good rate capacity of 276.1 mA h g−1 at 8C. However the high cost of CNTs may increase the fabrication cost of this composite and thus hinder its practical application especially for large-scale energy storage systems. Nevertheless, the flexibility of this anode makes it very suitable for wearable products. In addition, this free-standing and binder-free integrated electrode is also beneficial to increasing the volumetric specific capacity and reducing the fabrication cost. Cheng et al.239 designed a strategy to in situ coat a tin–phosphorus–oxygen (SnPO) composite with reduced graphene oxide (rGO) and reported that the coating protection could not only increase electrical conductivity, but also buffer volume expansion and suppress the pulverization of electrode materials during cycling. As a result, the SnPO/rGO composite delivered an initial discharge/charge capacity of 843 mA h g−1/450 mA h g−1 at a current density of 50 mA g−1 and retained a reversible discharge capacity of ∼420 mA h g−1 after 100 cycles with a rate performance with a capacity of 200 mA h g−1 at a current density of 2000 mA g−1.
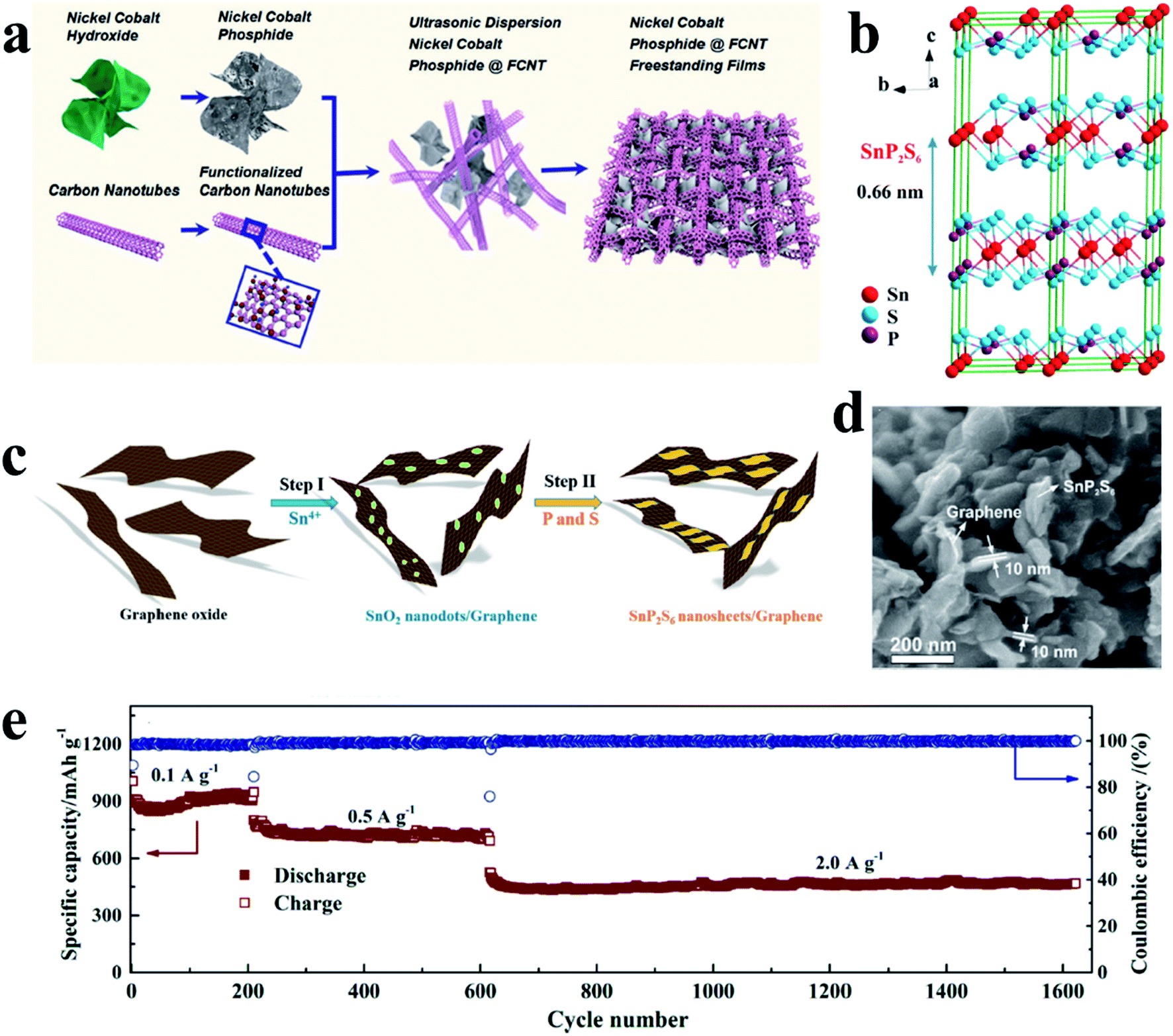 | ||
| Fig. 33 (a) Schematic diagram of the synthesis of NCP@FCNT-FS free-standing films. Reproduced with permission.270 Copyright 2018, WILEY-VCH. (b) Crystal model showing the asymmetric-layered structure of SnP2S6. (c) Schematic illustration of the in situ growth of ultrathin SnP2S6 nanosheets on graphene. (d) SEM image of the SPS/G hybrid. (e) Long-term cycling performance at current densities of 0.1, 0.5, and 2.0 A g−1. Reproduced with permission. Copyright 2018, American Chemical Society. | ||
Recently, a composite of 2D ternary tin thiophosphate (SnP2S6) nanosheets (∼10 nm thickness) grown on graphene (denoted as the SPS/G hybrid) was also demonstrated as an intriguing anode for SIBs (Fig. 33b–d).272 Benefiting from the 2D hetero-structural nanojunction and compositional advantages, the SPS/G hybrid showed good electrical conductivity and mechanical resilience to realize rapid ion and electron transfer as well as accommodation of volume change during the electrochemical process. As a result, the SPS/G hybrid anode exhibited a high reversible capacity (1230 mA h g−1 at 50 mA g−1), superior rate capability (200 mA h g−1 at 15 A g−1), and an exceptional capacity retention of 76% after 1000 cycles at 2.0 A g−1 (Fig. 33e). A full cell based on this anode also exhibited an excellent performance, affording a capacity of 470 mA h g−1 at 30 mA g−1 (on the basis of anode weight) and a good cycling capacity of 360 mA h g−1 at 150 mA g−1 after 50 cycles corresponding to a retention of 90.6%. Due to the superior sodium storage performance, the low-cost of Sn, P and S, and the facile and scalable solid-state preparation route, this SPS/G hybrid can have great potential for practical SIBs.
The combination of different kinds of metal phosphides is an interesting and effective strategy to improve performances as SIB anodes. For example, Li et al.75 synthesized a unique core–shell porous FeP@CoP phosphide microcube interconnected with reduced graphene oxide (RGO) nanosheets (RGO@CoP@FeP) through a solution method combined with a low-temperature phosphorization process. The researchers reported that the FeP@CoP core–shell structure could provide enough cushion space for volume change and shorten Na+ diffusion paths. In addition, the interconnected RGO nanosheets and carbon layers wrapped on the FeP core cubes could build a conductive highway to enhance charge transfer kinetics, leading to a reversible capacity of 551.4 mA h g−1 in the first cycle and a capacity of 456.2 mA h g−1 maintained in the 200th cycle. In this study, the combination strategy enables facile preparation of this sophisticated core–shell structure, and thus provides an improved sodium storage performance. Zhang et al.273 ball milled a mixture of elementary Sn, Sb, P, and C, and obtained a Sn5SbP3/C powder composite which consisted of micrometric agglomerates of active Sn4P3, SnSb and Sn nanoparticles. Here, the researchers reported that the carbon in the composites not only acted as a conducting matrix, but also enhanced the ball milling efficiencies and extended electrode cycle lifespans. In addition, each of the active Sn4P3, SnSb and Sn phases in the composite could function mutually as a buffer for one another. And as a result, this ternary composite SIB anode delivered a capacity of 352 mA h g−1 at a current density of 2 A g−1, which was notably higher than that of binary Sn4P3/C and SnSb/C composites produced under the same conditions. In another study,274 a self-supporting three-dimensional (3D) composite of Cu3P and Co2P interconnected by N-doped C fibers (Cu3P–Co2P/N–C) was prepared by a simple electrospinning method. The synergistic effects between Cu3P, Co2P, and N-doped carbon could increase the electrical conductivity and active sites, ensuring more ion storage. The Cu3P–Co2P/N–C anode for SIBs delivered high initial discharge/charge capacities of 429.5/321.2 mA h g−1 accompanied by a high ICE of 74.8%, as well as a stable capacity of around 166.4 mA h g−1 after 50 cycles. As the Na storage performance of this anode was inferior to its Li storage performance, it seemed to be more suitable for application in LIBs rather than in SIBs. Nevertheless, this novel nanostructure as well as the corresponding preparation method can still give inspiration for future design of SIB anode materials. Table 3 summarizes the advantages and disadvantages of different preparation methods for RP anodes. Table 4 summarizes some basic properties of RP, BP and metal phosphides, as well as the advantages and disadvantages of these P-based materials used as anodes for SIBs.
| Preparation methods | Advantages | Disadvantages | Reference |
|---|---|---|---|
| Ball milling | Simple/facile | Uneven/big particle size (∼μm) | 82 and 121 |
| Large-scale | |||
| Vaporization–condensation | Uniform/small size | Low P loading ratio | 76, 85 and 131 |
| Intimate contact with host materials | Uncontrollable distribution | ||
| Well-controlled nanostructure | White P remnants | ||
| Novel methods (solution method and solvothermal method | Controllable size | Simplex morphology (particle or sphere) | 87, 127, 144 and 145 |
| Carbothermic reduction) | Refined nanostructure | Toxic reagent involved |
| Materials | Crystal structure | Electronic conductivity | Reaction potential (vs. Na/Na+) | theoretical gravimetric capacity (mA h g−1) | Highest initial CE ever reported | Advantages | Disadvantages | Reference |
|---|---|---|---|---|---|---|---|---|
| a The advantages/disadvantages of all these P-based anode materials such as a suitable operating potential, large volume change, and insufficient cycle stability during cycles are not listed in this table. | ||||||||
| RP (amorphous) | Polymer | ∼10−12 S m−1 | ∼0.4 V | 2596 | 86.2% | Natural abundance, eco-friendliness | Low electronic conductivity | 82 and 125 |
| High theoretical gravimetric capacity | Low initial CE | |||||||
| BP | Orthorhombic | ∼3 × 102 S m−1 | ∼0.45 V | 2596 | 87.7% | High electronic conductivity | Difficult to prepare | 157 and 174 |
| Good maintenance of the structure during cycles | ||||||||
| Sn4P3 | Hexagonal | ∼3 × 103 S m−1 | ∼0.3 V | 1132 | 90.7% | High electronic conductivity | Low initial CE | 77 and 217 |
| Good maintenance of the structure | ||||||||
| High theoretical volumetric capacity | ||||||||
| Facile synthesis | ||||||||
| Fast Na diffusion | ||||||||
| Low cost of Sn and P | ||||||||
| SnP | Trigonal | — | — | 1209 | — | 190 | ||
| SnP3 | Rhombohedral | — | ∼0.3 V | 1616 | 72% | High theoretical volumetric capacity | Fast capacity decay | 194 |
| High P content, reversibility of the conversion reaction | ||||||||
| CoP | Orthorhombic | — | ∼0.2 V | 894 | 77.1% | — | High cost of Co | 204 |
| Co2P | Orthorhombic | — | 540 | 41% | — | Low initial CE | ||
| Low P content | ||||||||
| High cost of Co | ||||||||
| CoP3 | Cubic | ∼1.8 × 103 S m−1 | <0.4 V | 1588 | 87% | High P content | 200, 228 and 275 | |
| FeP | Orthorhombic | ∼1.3 × 106 S m−1 | ∼0.4 V | 924 | 74% | Low cost | 201 and 276 | |
| FeP4 | Monoclinic | ∼3 × 10−3 S m−1 | ∼0.25 V | 1790 | 84% | High P content, low cost | Low electronic conductivity | 70, 192 and 277 |
| NiP3 | Cubic | ∼2 × 106 S m−1 | ∼0.2 V | 1591 | 71.3% | High P content, low cost | — | 226 and 245 |
| High electronic conductivity | ||||||||
| NiP2 | Hexagonal | ∼3 × 106 S m−1 | ∼0.1 V | 1333 | 76.7% | High electronic conductivity | — | 92, 245 and 250 |
| High P content | ||||||||
| Low cost | ||||||||
| CuP2 | Monoclinic | ∼103 S m−1 | ∼0.5 V | 1282 | 87% | Low cost | — | 278 |
| Cu3P | Hexagonal | ∼2 × 106 S m−1 | ∼0.4 V | 363 | 80.6% | Low cost | Low theoretical gravimetric capacity | 78, 257 and 279 |
| Low P content | ||||||||
| GeP5 | Rhombohedral | ∼106 S m−1 | ∼0.4 V | 1888 | 94.1% | High initial CE | High cost of Ge | 193 and 280 |
| High P content | ||||||||
| High electronic conductivity | ||||||||
| GeP3 | Rhombohedral | — | — | 1619 | 81.3% | High cycle stability | Low initial CE | 196 and 263 |
| GeP | Monoclinic | 5 × 103 S m−1 | ∼0.25 | 1033 | 93% | High initial CE | High Ge ratio (high cost) | 197 and 281 |
| Low P content | ||||||||
6 Summary, challenges and future research directions
6.1 Summary
Sodium-ion batteries are promising alternatives to lithium-ion batteries and have attracted considerable attention due to the abundant availability of sodium and similar electrochemical properties to lithium as well as their great potential to meet the increasing demand for large-scale energy storage systems. Recently, phosphorus-based materials such as red phosphorus and metal/nonmetal phosphides have been considered as the most promising candidates as anodes for SIBs and attracted much research effort. Therefore, to facilitate the further research and development of these materials, this review has comprehensively discussed the status of phosphorus-based materials as anodes for sodium-ion batteries, including red/black phosphorus, phosphorene, metal/nonmetal-phosphides and ternary phosphides, in which the material design, synthesis strategy, structural/morphology characterization, sodium storage mechanism and electrochemical performance were analyzed. In general, the combination of red phosphorus with carbon or other materials is an effective approach to enhance the conductivity of the anode and to accommodate large volume change during charge/discharge cycles, thus leading to improved specific capacities, rate performances and durability. Various red phosphorus nanostructures/composites can be obtained through conventional ball milling or vaporization/condensation techniques and new strategies such as solution-based methods with well-controlled sizes/morphologies are emerging. Layer-structured black phosphorus and phosphorene also show great potential as anode materials as 2D materials, and many nanostructures/composites of BP/phosphorene materials have been explored for sodium storage and a comprehensive Na storage mechanism of BP was presented. Metal phosphides show higher electronic conductivities than red phosphorus and are more available than BP/phosphorene. Many nanostructures/composites of metal phosphides, including Fe–P, Co–P, Ni–P, Sn–P, Cu–P and Ge–P, have been successfully synthesized and demonstrate excellent rate capabilities and cycle stabilities. Non-metal phosphides and ternary phosphides including metal–metal phosphides and metal–nonmetal phosphides have also been explored as anodes of sodium-ion batteries with some promising results obtained.6.2 Challenges and future research directions
With the rapid advancement of different nanotechnologies, great progress has been achieved in the development of phosphorus-based composites as anode materials for sodium-ion batteries. However, several challenges must be overcome before realizing their practical application, such as (1) fast degradation of sodium-ion batteries caused by large volume change of anode materials during charge and discharge; (2) low rate capability, low initial coulombic efficiency (60–85%), and reversible capacity caused by the low electronic conductivity and/or poor diffusion kinetics of sodium ions in phosphorus-based materials.To overcome these challenges, several possible research directions are proposed:
(1) Further exploration of new phosphorus-composited materials as well as corresponding synthetic methods to obtain novel nanostructures/composites to accommodate large volume change during charge/discharge cycles. Here, the optimization of the structure/morphology/size and the introduction of counter materials with high conductivity and mechanical strengthen are promising approaches.
(2) Further improvements to the rate performance of phosphorus-based anode materials by solving the issues of poor diffusion kinetics, slow reaction kinetics and low electronic conductivity through surface engineering, compositing strategies, doping and other methods/strategies. And further improving the initial coulombic efficiency through rational composition/structure/morphology design and particle surface modification.
(3) Increasing the P content in P-composite anodes to further improve their gravimetric/volumetric specific capacities. Enhancing the phosphorus content in the composite (>70%) or constructing a self-supported anode (binder-/conductive additive-free anode) are all effective strategies to achieve this goal. In addition, fabricating P-based anodes for flexible SIBs is also interesting and meaningful; searching for large-scale, high yield and low-cost synthesis methods to prepare P-based anode materials is also important for practical applications.
(4) Combining experimental characterization with theoretical studies to obtain fundamental understandings of the relationship between the sodium storage performance and composition/structure as well as sodium storage mechanisms. This will allow for the prediction of new anode materials and guide the optimization of the design, synthesis and performance of phosphorus-composited materials for sodium-ion batteries.
(5) Further exploration of the degradation mechanisms of sodium-ion battery anode materials during charge and discharge through both theoretical and experimental approaches along with the development of mitigation strategies to enhance stability and durability.
(6) Further optimization of the design and fabrication of sodium-ion batteries using matching phosphorus-composited anodes, cathodes and electrolytes with respect to battery performance towards large-scale application in energy storage systems.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge the support from the Shanghai Education Ministry and Shanghai University PhD Program, National Natural Science Foundation of China (51774251), the Hebei Science Foundation for Distinguished Young Scholars (B2017203313), the Hundreds of Innovative Talents in Hebei Province (SLRC2017057), and the opening project of the state key laboratory of Advanced Chemical Power Sources (SKL-ACPS-C-11).References
- J. Lu, Z. Chen, F. Pan, Y. Cui and K. Amine, Electrochem. Energy Rev., 2018, 1, 35–53 CrossRef CAS.
- H. Zhang, H. Zhao, M. A. Khan, W. Zou, J. Xu, L. Zhang and J. Zhang, J. Mater. Chem. A, 2018, 6, 20564–20620 RSC.
- M. S. Dresselhaus and I. L. Thomas, Nature, 2001, 414, 332–337 CrossRef CAS.
- Y. Fang, L. Xiao, Z. Chen, X. Ai, Y. Cao and H. Yang, Electrochem. Energy Rev., 2018, 1, 294–296 CrossRef CAS.
- M. D. Slater, D. Kim, E. Lee and C. S. Johnson, Adv. Funct. Mater., 2013, 23, 947–958 CrossRef CAS.
- D. Tie, G. Gao, F. Xia, R. Yue, Q. Wang, R. Qi, B. Wang and Y. Zhao, ACS Appl. Mater. Interfaces, 2019, 11, 6978–6985 CrossRef CAS PubMed.
- J. Ding, Y.-C. Lin, J. Liu, J. Rana, H. Zhang, H. Zhou, I.-H. Chu, K. M. Wiaderek, F. Omenya, N. A. Chernova, K. W. Chapman, L. F. J. Piper, S. P. Ong and M. S. Whittingham, Adv. Energy Mater., 2018, 8, 1800221 CrossRef.
- M. H. Han, E. Gonzalo, G. Singh and T. Rojo, Energy Environ. Sci., 2015, 8, 81–102 RSC.
- Y. You and A. Manthiram, Adv. Energy Mater., 2018, 8, 1701785 CrossRef.
- G. Gao, D. Tie, H. Ma, H. Yu, S. Shi, B. Wang, S. Xu, L. Wang and Y. Zhao, J. Mater. Chem. A, 2018, 6, 6675–6684 RSC.
- J.-M. Tarascon, Nat. Chem., 2010, 2, 510 CrossRef CAS.
- R. Alcantara, J. M. J. Mateos and J. L. Tirado, J. Electrochem. Soc., 2002, 149, A201–A205 CrossRef CAS.
- W. B. Xing and J. R. Dahn, J. Electrochem. Soc., 1997, 144, 1195–1201 CrossRef CAS.
- D. A. Stevens and J. R. Dahn, J. Electrochem. Soc., 2000, 147, 1271–1273 CrossRef CAS.
- L. Xiao, H. Lu, Y. Fang, M. L. Sushko, Y. Cao, X. Ai, H. Yang and J. Liu, Adv. Energy Mater., 2018, 8, 1703238 CrossRef.
- F. A. Soto, P. Yan, M. H. Engelhard, A. Marzouk, C. Wang, G. Xu, Z. Chen, K. Amine, J. Liu, V. L. Sprenkle, F. El-Mellouhi, P. B. Balbuena and X. Li, Adv. Mater., 2017, 29, 1606860 CrossRef PubMed.
- Z. Zhu, F. Liang, Z. Zhou, X. Zeng, D. Wang, P. Dong, J. Zhao, S. Sun, Y. Zhang and X. Li, J. Mater. Chem. A, 2018, 6, 1513–1522 RSC.
- S. Huang, Z. Li, B. Wang, J. Zhang, Z. Peng, R. Qi, J. Wang and Y. Zhao, Adv. Funct. Mater., 2018, 28, 1706294 CrossRef.
- Y. Cao, L. Xiao, M. L. Sushko, W. Wang, B. Schwenzer, J. Xiao, Z. Nie, L. V. Saraf, Z. Yang and J. Liu, Nano Lett., 2012, 12, 3783–3787 CrossRef CAS PubMed.
- B. Jache and P. Adelhelm, Angew. Chem., Int. Ed., 2014, 53, 10169–10173 CrossRef CAS PubMed.
- Y. Wen, K. He, Y. Zhu, F. Han, Y. Xu, I. Matsuda, Y. Ishii, J. Cumings and C. Wang, Nat. Commun., 2014, 5, 4033–4042 CrossRef CAS.
- H. Kim, J. Hong, Y.-U. Park, J. Kim, I. Hwang and K. Kang, Adv. Funct. Mater., 2015, 25, 534–541 CrossRef CAS.
- Y. Wang, S. Chou, H. Liu and S. Dou, Carbon, 2013, 57, 202–208 CrossRef CAS.
- N. A. Kumar, R. R. Gaddam, S. R. Varanasi, D. Yang, S. K. Bhatia and X. S. Zhao, Electrochim. Acta, 2016, 214, 319–325 CrossRef CAS.
- Y. Yan, Y.-X. Yin, Y.-G. Guo and L.-J. Wan, Adv. Energy Mater., 2014, 4, 1301584 CrossRef.
- Y.-X. Wang, S.-L. Chou, H.-K. Liu and S.-X. Dou, Carbon, 2013, 57, 202–208 CrossRef CAS.
- X. Wang, G. Li, F. M. Hassan, J. Li, X. Fan, R. Batmaz, X. Xiao and Z. Chen, Nano Energy, 2015, 15, 746–754 CrossRef CAS.
- J. Xu, M. Wang, N. P. Wickramaratne, M. Jaroniec, S. Dou and L. Dai, Adv. Mater., 2015, 27, 2042–2048 CrossRef CAS PubMed.
- C. Wang, L. Wang, F. Li, F. Cheng and J. Chen, Adv. Mater., 2017, 29, 1702212 CrossRef PubMed.
- L. Wu, X. Hu, J. Qian, F. Pei, F. Wu, R. Mao, X. Ai, H. Yang and Y. Cao, Energy Environ. Sci., 2014, 7, 323–328 RSC.
- S. Liu, J. Feng, X. Bian, J. Liu and H. Xu, Energy Environ. Sci., 2016, 9, 1229–1236 RSC.
- Z. Li, J. Ding and D. Mitlin, Acc. Chem. Res., 2015, 48, 1657–1665 CrossRef CAS PubMed.
- W. Tian, S. Zhang, C. Huo, D. Zhu, Q. Li, L. Wang, X. Ren, L. Xie, S. Guo, P. K. Chu, H. Zeng and K. Huo, ACS Nano, 2018, 12, 1887–1893 CrossRef CAS PubMed.
- H. Ying, S. Zhang, Z. Meng, Z. Sun and W.-Q. Han, J. Mater. Chem. A, 2017, 5, 8334–8342 RSC.
- N. Wang, Z. Bai, Y. Qian and J. Yang, Adv. Mater., 2016, 28, 4126–4133 CrossRef CAS PubMed.
- J. Pan, N. Wang, Y. Zhou, X. Yang, W. Zhou, Y. Qian and J. Yang, Nano Res., 2017, 10, 1794–1803 CrossRef CAS.
- Y. Liu, X. Fang, M. Ge, J. Rong, C. Shen, A. Zhang, H. A. Enaya and C. Zhou, Nano Energy, 2015, 16, 399–407 CrossRef CAS.
- K. Lee, S. Shin, T. Degen, W. Lee and Y. S. Yoon, Nano Energy, 2017, 32, 397–407 CrossRef CAS.
- W. Chen, K. Song, L. Mi, X. Feng, J. Zhang, S. Cui and C. Liu, J. Mater. Chem. A, 2017, 5, 10027–10038 RSC.
- D. Ma, Y. Li, H. Mi, S. Luo, P. Zhang, Z. Lin, J. Li and H. Zhang, Angew. Chem., Int. Ed., 2018, 57, 8901–8905 CrossRef CAS PubMed.
- T. Li, A. Qin, L. Yang, J. Chen, Q. Wang, D. Zhang and H. Yang, ACS Appl. Mater. Interfaces, 2017, 9, 19900–19907 CrossRef CAS.
- D. Y. W. Yu, P. V. Prikhodchenko, C. W. Mason, S. K. Batabyal, J. Gun, S. Sladkevich, A. G. Medvedev and O. Lev, Nat. Commun., 2013, 4, 2922–2931 CrossRef PubMed.
- X. Xiong, G. Wang, Y. Lin, Y. Wang, X. Ou, F. Zheng, C. Yang, J.-H. Wang and M. Liu, ACS Nano, 2016, 10, 10953–10959 CrossRef CAS PubMed.
- Y. Xiao, S. H. Lee and Y.-K. Sun, Adv. Energy Mater., 2017, 7, 1601329 CrossRef.
- S. Wang, Y. Fang, X. Wang and X. W. D. Lou, Angew. Chem., Int. Ed., 2019, 58, 760–763 CrossRef CAS.
- F. Xie, L. Zhang, Q. Gu, D. Chao, M. Jaroniec and S.-Z. Qiao, Nano Energy, 2019, 60, 591–599 CrossRef CAS.
- D. Chao, B. Ouyang, P. Liang, T. T. T. Huong, G. Jia, H. Huang, X. Xia, R. S. Rawat and H. J. Fan, Adv. Mater., 2018, 30, 1804833 CrossRef PubMed.
- H. Park, J. Kwon, H. Choi, D. Shin, T. Song and X. W. D. Lou, ACS Nano, 2018, 12, 2827–2837 CrossRef CAS PubMed.
- S. Yuan, Y. H. Zhu, W. Li, S. Wang, D. Xu, L. Li, Y. Zhang and X. B. Zhang, Adv. Mater., 2017, 29, 1602469 CrossRef PubMed.
- F. Zhang, C. Xia, J. Zhu, B. Ahmed, H. Liang, D. B. Velusamy, U. Schwingenschlögl and H. N. Alshareef, Adv. Energy Mater., 2016, 6, 1601188 CrossRef.
- K. Zhang, Z. Hu, X. Liu, Z. Tao and J. Chen, Adv. Mater., 2015, 27, 3305–3309 CrossRef CAS.
- Z. Ali, T. Tang, X. Huang, Y. Wang, M. Asif and Y. Hou, Energy Storage Mater., 2018, 13, 19–28 CrossRef.
- H. Fan, H. Yu, Y. Zhang, J. Guo, Z. Wang, H. Wang, N. Zhao, Y. Zheng, C. Du, Z. Dai, Q. Yan and J. Xu, Energy Storage Mater., 2018, 10, 48–55 CrossRef.
- X. Ou, C. Yang, X. Xiong, F. Zheng, Q. Pan, C. Jin, M. Liu and K. Huang, Adv. Funct. Mater., 2017, 27, 1606242 CrossRef.
- L. Ji, M. Gu, Y. Shao, X. Li, M. H. Engelhard, B. W. Arey, W. Wang, Z. Nie, J. Xiao, C. Wang, J.-G. Zhang and J. Liu, Adv. Mater., 2014, 26, 2901–2908 CrossRef CAS PubMed.
- M. Walter, S. Doswald and M. V. Kovalenko, J. Mater. Chem. A, 2016, 4, 7053–7059 RSC.
- L. Xiao, Y. Cao, J. Xiao, W. Wang, L. Kovarik, Z. Nie and J. Liu, Chem. Commun., 2012, 48, 3321–3323 RSC.
- Y.-M. Lin, P. R. Abel, A. Gupta, J. B. Goodenough, A. Heller and C. B. Mullins, ACS Appl. Mater. Interfaces, 2013, 5, 8273–8277 CrossRef CAS PubMed.
- J.-H. Choi, C.-W. Ha, H.-Y. Choi, J.-W. Seong, C.-M. Park and S.-M. Lee, J. Power Sources, 2018, 386, 34–39 CrossRef CAS.
- J. Qin, T. Wang, D. Liu, E. Liu, N. Zhao, C. Shi, F. He, L. Ma and C. He, Adv. Mater., 2018, 30, 1704670 CrossRef PubMed.
- W. Ma, K. Yin, H. Gao, J. Niu, Z. Peng and Z. Zhang, Nano Energy, 2018, 54, 349–359 CrossRef CAS.
- H. Gao, J. Niu, C. Zhang, Z. Peng and Z. Zhang, ACS Nano, 2018, 12, 3568–3577 CrossRef CAS.
- B. Farbod, K. Cui, W. P. Kalisvaart, M. Kupsta, B. Zahiri, A. Kohandehghan, E. M. Lotfabad, Z. Li, E. J. Luber and D. Mitlin, ACS Nano, 2014, 8, 4415–4429 CrossRef CAS PubMed.
- L. O. Vogt and C. Villevieille, J. Mater. Chem. A, 2017, 5, 3865–3874 RSC.
- S. Wang, L. Wang, Z. Zhu, Z. Hu, Q. Zhao and J. Chen, Angew. Chem., Int. Ed., 2014, 53, 5892–5896 CrossRef CAS.
- C. Luo, G. L. Xu, X. Ji, S. Hou, L. Chen, F. Wang, J. Jiang, Z. Chen, Y. Ren, K. Amine and C. Wang, Angew. Chem., Int. Ed., 2018, 57, 2879–2883 CrossRef CAS PubMed.
- H. Zhu, J. Yin, X. Zhao, C. Wang and X. Yang, Chem. Commun., 2015, 51, 14708–14711 RSC.
- C. Luo, J. Wang, X. Fan, Y. Zhu, F. Han, L. Suo and C. Wang, Nano Energy, 2015, 13, 537–545 CrossRef CAS.
- H. J. Lee, J. Shin and J. W. Choi, Adv. Mater., 2018, 30, 1705851 CrossRef PubMed.
- W. Zhang, M. Dahbi, S. Amagasa, Y. Yamada and S. Komaba, Electrochem. Commun., 2016, 69, 11–14 CrossRef CAS.
- C. Zhang, X. Wang, Q. Liang, X. Liu, Q. Weng, J. Liu, Y. Yang, Z. Dai, K. Ding, Y. Bando, J. Tang and D. Golberg, Nano Lett., 2016, 16, 2054–2060 CrossRef CAS PubMed.
- J. Sun, H.-W. Lee, M. Pasta, H. Yuan, G. Zheng, Y. Sun, Y. Li and Y. Cui, Nat. Nanotechnol., 2015, 10, 980–986 CrossRef CAS PubMed.
- J. Song, Z. Yu, M. L. Gordin, S. Hu, R. Yi, D. Tang, T. Walter, M. Regula, D. Choi, X. Li, A. Maniyannan and D. Wang, Nano Lett., 2014, 14, 6329–6335 CrossRef CAS PubMed.
- J. Qian, Y. Xiong, Y. Cao, X. Ai and H. Yang, Nano Lett., 2014, 14, 1865–1869 CrossRef CAS PubMed.
- Z. Li, L. Zhang, X. Ge, C. Li, S. Dong, C. Wang and L. Yin, Nano Energy, 2017, 32, 494–502 CrossRef CAS.
- W. Li, S. Hu, X. Luo, Z. Li, X. Sun, M. Li, F. Liu and Y. Yu, Adv. Mater., 2017, 29, 1605820–1605827 CrossRef PubMed.
- Y. Kim, Y. Kim, A. Choi, S. Woo, D. Mok, N.-S. Choi, Y. S. Jung, J. H. Ryu, S. M. Oh and K. T. Lee, Adv. Mater., 2014, 26, 4139–4144 CrossRef CAS PubMed.
- M. Fan, Y. Chen, Y. Xie, T. Yang, X. Shen, N. Xu, H. Yu and C. Yan, Adv. Funct. Mater., 2016, 26, 5019–5027 CrossRef CAS.
- J. Qian, X. Wu, Y. Cao, X. Ai and H. Yang, Angew. Chem., Int. Ed., 2013, 52, 4633–4636 CrossRef CAS PubMed.
- P. Extance and S. R. Elliott, Philos. Mag. B, 1981, 43, 469–483 CAS.
- Y. Fu, Q. Wei, G. Zhang and S. Sun, Adv. Energy Mater., 2018, 8, 1703058 CrossRef.
- Y. Kim, Y. Park, A. Choi, N. S. Choi, J. Kim, J. Lee, J. H. Ryu, S. M. Oh and K. T. Lee, Adv. Mater., 2013, 25, 3045–3049 CrossRef CAS.
- N. Wu, H.-R. Yao, Y.-X. Yin and Y.-G. Guo, J. Mater. Chem. A, 2015, 3, 24221–24225 RSC.
- M. Dahbi, N. Yabuuchi, M. Fukunishi, K. Kubota, K. Chihara, K. Tokiwa, X.-f. Yu, H. Ushiyama, K. Yamashita, J.-Y. Son, Y.-T. Cui, H. Oji and S. Komaba, Chem. Mater., 2016, 28, 1625–1635 CrossRef CAS.
- Y. Zhu, Y. Wen, X. Fan, T. Gao, F. Han, C. Luo, S.-C. Liou and C. Wang, ACS Nano, 2015, 9, 3254–3264 CrossRef CAS PubMed.
- S. Liu, J. Feng, X. Bian, J. Liu, H. Xu and Y. An, Energy Environ. Sci., 2017, 10, 1222–1233 RSC.
- J. Zhou, X. Liu, W. Cai, Y. Zhu, J. Liang, K. Zhang, Y. Lan, Z. Jiang, G. Wang and Y. Qian, Adv. Mater., 2017, 29, 1700214 CrossRef PubMed.
- A. Morita, Appl. Phys. A, 1986, 39, 227–242 CrossRef.
- J. Wu, N. Mao, L. Xie, H. Xu and J. Zhang, Angew. Chem., Int. Ed., 2015, 54, 2366–2369 CrossRef CAS PubMed.
- D. Sun, X. Zhu, B. Luo, Y. Zhang, Y. Tang, H. Wang and L. Wang, Adv. Energy Mater., 2018, 8, 1801197 CrossRef.
- J. Liu, P. Kopold, C. Wu, P. A. van Aken, J. Maier and Y. Yu, Energy Environ. Sci., 2015, 8, 3531–3538 RSC.
- S. Shi, Z. Li, Y. Sun, B. Wang, Q. Liu, Y. Hou, S. Huang, J. Huang and Y. Zhao, Nano Energy, 2018, 48, 510–517 CrossRef CAS.
- J. M. Sangster, J. Phase Equilib. Diffus., 2009, 31, 62–67 CrossRef.
- J. L. Zhang, S. Zhao, C. Han, Z. Wang, S. Zhong, S. Sun, R. Guo, X. Zhou, C. D. Gu, K. D. Yuan, Z. Li and W. Chen, Nano Lett., 2016, 16, 4903–4908 CrossRef CAS PubMed.
- J. Zhang, D. Zhao, D. Xiao, C. Ma, H. Du, X. Li, L. Zhang, J. Huang, H. Huang, C. L. Jia, D. Tomanek and C. Niu, Angew. Chem., Int. Ed., 2017, 56, 1850–1854 CrossRef CAS.
- Q. Peng, Z. Wang, B. Sa, B. Wu and Z. Sun, ACS Appl. Mater. Interfaces, 2016, 8, 13449–13457 CrossRef CAS.
- W. Roth, T. DeWitt and A. J. Smith, J. Am. Chem. Soc., 1947, 69, 2881–2885 CrossRef CAS.
- M. Ceppatelli, R. Bini, M. Caporali and M. Peruzzini, Angew. Chem., Int. Ed., 2013, 52, 2313–2317 CrossRef CAS PubMed.
- J. A. Young, J. Chem. Educ., 2004, 81, 945 CrossRef CAS.
- S. Zhang, H. J. Qian, Z. Liu, H. Ju, Z. Y. Lu, H. Zhang, L. Chi and S. Cui, Angew. Chem., Int. Ed., 2019, 58, 1659–1663 CrossRef CAS PubMed.
- J. Sun, H.-W. Lee, M. Pasta, Y. Sun, W. Liu, Y. Li, H. R. Lee, N. Liu and Y. Cui, Energy Storage Mater., 2016, 4, 130–136 CrossRef.
- M. Scheer, G. Balazs and A. Seitz, Chem. Rev., 2010, 110, 4236–4256 CrossRef CAS PubMed.
- U. Braun and B. Schartel, Macromol. Chem. Phys., 2004, 205, 2185–2196 CrossRef CAS.
- K. Sladkova, J. Houska and J. Havel, Rapid Commun. Mass Spectrom., 2009, 23, 3114–3118 CrossRef CAS PubMed.
- B. Weber, S. Mahapatra, H. Ryu, S. Lee, A. Fuhrer, T. Reusch, D. Thompson, W. Lee, G. Klimeck and L. C. Hollenberg, Science, 2012, 335, 64–67 CrossRef CAS PubMed.
- F. Wang, W. K. H. Ng, C. Y. Jimmy, H. Zhu, C. Li, L. Zhang, Z. Liu and Q. Li, Appl. Catal., B, 2012, 111, 409–414 CrossRef.
- W. L. Roth, T. W. DeWitt and A. J. Smith, J. Am. Chem. Soc., 1947, 69, 2881–2885 CrossRef CAS.
- H. Thurn and H. Krebs, Angew. Chem., Int. Ed., 1966, 78, 1101–1102 Search PubMed.
- H. Thurn and H. Krebs, Acta Crystallogr., Sect. B: Struct. Sci., 1969, 25, 125–135 CrossRef CAS.
- M. Ruck, D. Hoppe, B. Wahl, P. Simon, Y. Wang and G. Seifert, Angew. Chem., Int. Ed., 2005, 117, 7788–7792 CrossRef.
- W. Li, Z. Yang, Y. Jiang, Z. Yu, L. Gu and Y. Yu, Carbon, 2014, 78, 455–462 CrossRef CAS.
- N. Yabuuchi, Y. Matsuura, T. Ishikawa, S. Kuze, J.-Y. Son, Y.-T. Cui, H. Oji and S. Komaba, ChemElectroChem, 2014, 1, 580–589 CrossRef.
- R. Gusmao, Z. Sofer and M. Pumera, Angew. Chem., Int. Ed., 2017, 56, 8052–8072 CrossRef CAS PubMed.
- M. Sopicka-Lizer, High-Energy Ball Milling: Mechanochemical Processing of Nanopowders, Elsevier Science, Woodheam Publishing Limited, Great Abington, Cambridge, UK, 2010 Search PubMed.
- C. Marino, A. Debenedetti, B. Fraisse, F. Favier and L. Monconduit, Electrochem. Commun., 2011, 13, 346–349 CrossRef CAS.
- F. Yang, H. Gao, J. Chen and Z. Guo, Small Methods, 2017, 1, 1700216 CrossRef.
- D. Liu, X. Huang, D. Qu, D. Zheng, G. Wang, J. Harris, J. Si, T. Ding, J. Chen and D. Qu, Nano Energy, 2018, 52, 1–10 CrossRef CAS.
- L. Wang, X. He, J. Li, W. Sun, J. Gao, J. Guo and C. Jiang, Angew. Chem., Int. Ed., 2012, 51, 9034–9037 CrossRef CAS PubMed.
- C. M. Park and H. J. Sohn, Adv. Mater., 2007, 19, 2465–2468 CrossRef CAS.
- J. Qian, D. Qiao, X. Ai, Y. Cao and H. Yang, Chem. Commun., 2012, 48, 8931–8933 RSC.
- W. J. Li, S. L. Chou, J. Z. Wang, H. K. Liu and S. X. Dou, Nano Lett., 2013, 13, 5480–5484 CrossRef CAS PubMed.
- W. Liu, X. Yuan and X. Yu, Nanoscale, 2018, 10, 16675–16682 RSC.
- Y. Hu, B. Li, X. Jiao, C. Zhang, X. Dai and J. Song, Adv. Funct. Mater., 2018, 28, 1801010 CrossRef.
- J. Song, Z. Yu, M. L. Gordin, X. Li, H. Peng and D. Wang, ACS Nano, 2015, 9, 11933–11941 CrossRef CAS.
- Q. Xu, J. K. Sun, F. S. Yue, J. Y. Li, G. Li, S. Xin, Y. X. Yin and Y. G. Guo, ACS Appl. Mater. Interfaces, 2018, 10, 30479–30486 CrossRef CAS.
- M. Li, R. Carter, L. Oakes, A. Douglas, N. Muralidharan and C. L. Pint, J. Mater. Chem. A, 2017, 5, 5266–5272 RSC.
- Y. Liu, N. Zhang, X. Liu, C. Chen, L.-Z. Fan and L. Jiao, Energy Storage Mater., 2017, 9, 170–178 CrossRef.
- W. Li, Z. Yang, M. Li, Y. Jiang, X. Wei, X. Zhong, L. Gu and Y. Yu, Nano Lett., 2016, 16, 1546–1553 CrossRef CAS PubMed.
- G. Qin, J. Duan, Y. Yang and F. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 6441–6452 CrossRef CAS PubMed.
- D. Lan and Q. Li, ACS Appl. Energy Mater., 2018, 2, 661–667 CrossRef.
- H. Gao, T. Zhou, Y. Zheng, Y. Liu, J. Chen, H. Liu and Z. Guo, Adv. Energy Mater., 2016, 6, 1601037 CrossRef.
- Y. Liu, A. Zhang, C. Shen, Q. Liu, X. Cao, Y. Ma, L. Chen, C. Lau, T. C. Chen, F. Wei and C. Zhou, ACS Nano, 2017, 11, 5530–5537 CrossRef CAS PubMed.
- Y. Liu, A. Zhang, C. Shen, Q. Liu, J. Cai, X. Cao and C. Zhou, Nano Res., 2018, 11, 3780–3790 CrossRef CAS.
- X. Sun, W. Li, X. Zhong and Y. Yu, Energy Storage Mater., 2017, 9, 112–118 CrossRef.
- S. Yao, J. Cui, J. Huang, J.-Q. Huang, W. G. Chong, L. Qin, Y.-W. Mai and J.-K. Kim, Adv. Energy Mater., 2018, 8, 1702267 CrossRef.
- Z. Yu, J. Song, D. Wang and D. Wang, Nano Energy, 2017, 40, 550–558 CrossRef CAS.
- J. Hanawalt, H. Rinn and L. Frevel, Ind. Eng. Chem., Anal. Ed., 1938, 10, 457–512 CrossRef CAS.
- D. Yan, C. Yu, X. Zhang, J. Li, J. Li, T. Lu and L. Pan, Electrochim. Acta, 2017, 254, 130–139 CrossRef CAS.
- S.-O. Kim and A. Manthiram, Chem. Mater., 2016, 28, 5935–5942 CrossRef CAS.
- W. C. Chang, K. W. Tseng and H. Y. Tuan, Nano Lett., 2017, 17, 1240–1247 CrossRef CAS PubMed.
- I. T. Kim, E. Allcorn and A. Manthiram, J. Power Sources, 2015, 281, 11–17 CrossRef CAS.
- L.-C. Chin, Y.-H. Yi, W.-C. Chang and H.-Y. Tuan, Electrochim. Acta, 2018, 266, 178–184 CrossRef CAS.
- M. Walter, R. Erni and M. V. Kovalenko, Sci. Rep., 2015, 5, 8418–8424 CrossRef CAS PubMed.
- S. Liu, H. Xu, X. Bian, J. Feng, J. Liu, Y. Yang, C. Yuan, Y. An, R. Fan and L. Ci, ACS Nano, 2018, 12, 7380–7387 CrossRef CAS.
- S. Liu, H. Xu, X. Bian, J. Feng, J. Liu, Y. Yang, C. Yuan, Y. An, R. Fan and L. Ci, J. Mater. Chem. A, 2018, 6, 12992–12998 RSC.
- G. Zeng, X. Hu, B. Zhou, J. Chen, C. Cao and Z. Wen, Nanoscale, 2017, 9, 14722–14729 RSC.
- C.-M. Park and H.-J. Sohn, Adv. Mater., 2007, 19, 2465–2468 CrossRef CAS.
- N. S. G. Ralph Hultgren and B. E. Warren, J. Chem. Phys., 1935, 3, 351–355 CrossRef.
- M. Huang, M. Wang, C. Chen, Z. Ma, X. Li, J. Han and Y. Wu, Adv. Mater., 2016, 28, 3481–3485 CrossRef CAS PubMed.
- L. Li, Y. Yu, G. J. Ye, Q. Ge, X. Ou, H. Wu, D. Feng, X. H. Chen and Y. Zhang, Nat. Nanotechnol., 2014, 9, 372–377 CrossRef CAS PubMed.
- H. Liu, Y. Du, Y. Deng and P. D. Ye, Chem. Soc. Rev., 2015, 44, 2732–2743 RSC.
- A. J. Mannix, B. Kiraly, M. C. Hersam and N. P. Guisinger, Nat. Rev. Chem., 2017, 1, 0014 CrossRef CAS.
- X. Ling, H. Wang, S. Huang, F. Xia and M. S. Dresselhaus, Proc. Natl. Acad. Sci., 2015, 112, 4523–4530 CrossRef CAS.
- S. R. Allan Brown, Acta Cryst., 1965, 19, 684–685 CrossRef.
- J. C. Slater, G. F. Koster and J. H. Wood, Phys. Rev., 1962, 126, 1307–1317 CrossRef CAS.
- R. W. Keyes, Phys. Rev., 1953, 92, 580–584 CrossRef CAS.
- Y. Zhang, Y. Zheng, K. Rui, H. H. Hng, K. Hippalgaonkar, J. Xu, W. Sun, J. Zhu, Q. Yan and W. Huang, Small, 2017, 13, 1700661 CrossRef PubMed.
- L. Li, Y. Zheng, S. Zhang, J. Yang, Z. Shao and Z. Guo, Energy Environ. Sci., 2018, 11, 2310–2340 RSC.
- Y. Zhang, W. Sun, Z.-Z. Luo, Y. Zheng, Z. Yu, D. Zhang, J. Yang, H. T. Tan, J. Zhu, X. Wang, Q. Yan and S. X. Dou, Nano Energy, 2017, 40, 576–586 CrossRef CAS.
- M. Köpf, N. Eckstein, D. Pfister, C. Grotz, I. Krüger, M. Greiwe, T. Hansen, H. Kohlmann and T. Nilges, J. Cryst. Growth, 2014, 405, 6–10 CrossRef.
- L.-Q. Sun, M.-J. Li, K. Sun, S.-H. Yu, R.-S. Wang and H.-M. Xie, J. Phys. Chem. C, 2012, 116, 14772–14779 CrossRef CAS.
- Y. Liu, Q. Liu, A. Zhang, J. Cai, X. Cao, Z. Li, P. D. Asimow and C. Zhou, ACS Nano, 2018, 12, 8323–8329 CrossRef CAS.
- Q. Jiang, L. Xu, N. Chen, H. Zhang, L. Dai and S. Wang, Angew. Chem., Int. Ed., 2016, 55, 13849–13853 CrossRef CAS PubMed.
- B. Tian, B. Tian, B. Smith, M. C. Scott, Q. Lei, R. Hua, Y. Tian and Y. Liu, Proc. Natl. Acad. Sci., 2018, 115, 4345–4350 CrossRef CAS PubMed.
- V. Eswaraiah, Q. Zeng, Y. Long and Z. Liu, Small, 2016, 12, 3480–3502 CrossRef CAS.
- X. Li, B. Deng, X. Wang, S. Chen, M. Vaisman, S.-i. Karato, G. Pan, M. Larry Lee, J. Cha, H. Wang and F. Xia, 2D Mater., 2015, 2, 031002 CrossRef.
- K. P. S. S. Hembram, H. Jung, B. C. Yeo, S. J. Pai, S. Kim, K.-R. Lee and S. S. Han, J. Phys. Chem. C, 2015, 119, 15041–15046 CrossRef CAS.
- L. E. Marbella, M. L. Evans, M. F. Groh, J. Nelson, K. J. Griffith, A. J. Morris and C. P. Grey, J. Am. Chem. Soc., 2018, 140, 7994–8004 CrossRef CAS.
- S. Mukherjee, L. Kavalsky and C. V. Singh, ACS Appl. Mater. Interfaces, 2018, 10, 8630–8639 CrossRef CAS PubMed.
- X. Liu, Y. Wen, Z. Chen, B. Shan and R. Chen, Phys. Chem. Chem. Phys., 2015, 17, 16398–16404 RSC.
- L. Wang, Z. Jiang, W. Li, X. Gu and L. Huang, J. Phys. D: Appl. Phys., 2017, 50, 165501 CrossRef.
- H. Liu, L. Tao, Y. Zhang, C. Xie, P. Zhou, H. Liu, R. Chen and S. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 36849–36856 CrossRef CAS.
- G. L. Xu, Z. Chen, G. M. Zhong, Y. Liu, Y. Yang, T. Ma, Y. Ren, X. Zuo, X. H. Wu, X. Zhang and K. Amine, Nano Lett., 2016, 16, 3955–3965 CrossRef CAS.
- S. Haghighat-Shishavan, M. Nazarian-Samani, M. Nazarian-Samani, H.-K. Roh, K.-Y. Chung, B.-W. Cho, S. F. Kashani-Bozorg and K.-B. Kim, J. Mater. Chem. A, 2018, 6, 10121–10134 RSC.
- M. Li, N. Muralidharan, K. Moyer and C. L. Pint, Nanoscale, 2018, 10, 10443–10449 RSC.
- R. Meng, J. Huang, Y. Feng, L. Zu, C. Peng, L. Zheng, L. Zheng, Z. Chen, G. Liu, B. Chen, Y. Mi and J. Yang, Adv. Energy Mater., 2018, 8, 1801514 CrossRef.
- T. Song, H. Chen, Q. Xu, H. Liu, Y. G. Wang and Y. Xia, ACS Appl. Mater. Interfaces, 2018, 10, 37163–37171 CrossRef CAS PubMed.
- T. Song, H. Chen, Z. Li, Q. Xu, H. Liu, Y. Wang and Y. Xia, Adv. Funct. Mater., 2019, 1900535 CrossRef.
- X. Ding, Y. Huang, G. Li, Y. Tang, X. Li and Y. Huang, Electrochim. Acta, 2017, 235, 150–157 CrossRef CAS.
- J. R. Brent, N. Savjani, E. A. Lewis, S. J. Haigh, D. J. Lewis and P. O'Brien, Chem. Commun., 2014, 50, 13338–13341 RSC.
- A. Favron, E. Gaufres, F. Fossard, A. L. Phaneuf-L'Heureux, N. Y. Tang, P. L. Levesque, A. Loiseau, R. Leonelli, S. Francoeur and R. Martel, Nat. Mater., 2015, 14, 826–832 CrossRef CAS PubMed.
- P. Yasaei, B. Kumar, T. Foroozan, C. Wang, M. Asadi, D. Tuschel, J. E. Indacochea, R. F. Klie and A. Salehi-Khojin, Adv. Mater., 2015, 27, 1887–1892 CrossRef CAS PubMed.
- Q. Zhou, Q. Chen, Y. Tong and J. Wang, Angew. Chem., Int. Ed., 2016, 55, 11437–11441 CrossRef CAS PubMed.
- R. Fei, A. Faghaninia, R. Soklaski, J. A. Yan, C. Lo and L. Yang, Nano Lett., 2014, 14, 6393–6399 CrossRef CAS PubMed.
- V. V. Kulish, O. I. Malyi, C. Persson and P. Wu, Phys. Chem. Chem. Phys., 2015, 17, 13921–13928 RSC.
- Y. Zhao, H. Wang, H. Huang, Q. Xiao, Y. Xu, Z. Guo, H. Xie, J. Shao, Z. Sun, W. Han, X. F. Yu, P. Li and P. K. Chu, Angew. Chem., Int. Ed., 2016, 55, 5003–5007 CrossRef CAS PubMed.
- C. R. Ryder, J. D. Wood, S. A. Wells, Y. Yang, D. Jariwala, T. J. Marks, G. C. Schatz and M. C. Hersam, Nat. Chem., 2016, 8, 597–602 CrossRef CAS PubMed.
- Z. Huang, H. Hou, Y. Zhang, C. Wang, X. Qiu and X. Ji, Adv. Mater., 2017, 29, 1702372 CrossRef PubMed.
- F. Zhao, N. Han, W. Huang, J. Li, H. Ye, F. Chen and Y. Li, J. Mater. Chem. A, 2015, 3, 21754–21759 RSC.
- J. Liu, S. Wang, K. Kravchyk, M. Ibáñez, F. Krumeich, R. Widmer, D. Nasiou, M. Meyns, J. Llorca, J. Arbiol, M. V. Kovalenko and A. Cabot, J. Mater. Chem. A, 2018, 6, 10958–10966 RSC.
- J. Zhang, K. Zhang, J. Yang, G.-H. Lee, J. Shin, V. Wing-hei Lau and Y.-M. Kang, Adv. Energy Mater., 2018, 8, 1800283 CrossRef.
- X. Wang, K. Chen, G. Wang, X. Liu and H. Wang, ACS Nano, 2017, 11, 11602–11616 CrossRef CAS.
- W. Li, L. Ke, Y. Wei, S. Guo, L. Gan, H. Li, T. Zhai and H. Zhou, J. Mater. Chem. A, 2017, 5, 4413–4420 RSC.
- X. Fan, J. Mao, Y. Zhu, C. Luo, L. Suo, T. Gao, F. Han, S.-C. Liou and C. Wang, Adv. Energy Mater., 2015, 5, 1500174 CrossRef.
- W. Li, S. L. Chou, J. Z. Wang, J. H. Kim, H. K. Liu and S. X. Dou, Adv. Mater., 2014, 26, 4037–4042 CrossRef CAS.
- K.-H. Nam, K.-J. Jeon and C.-M. Park, Energy Storage Mater., 2019, 17, 78–87 CrossRef.
- W. Li, X. Li, J. Liao, B. Zhao, L. Zhang, Y. Liu, L. Huang, Y. Li and M. Liu, Energy Storage Mater., 2019, 20, 380–387 CrossRef.
- M. Kong, H. Song and J. Zhou, Adv. Energy Mater., 2018, 8, 1801489 CrossRef.
- J. Fullenwarth, A. Darwiche, A. Soares, B. Donnadieu and L. Monconduit, J. Mater. Chem. A, 2014, 2, 2050–2059 RSC.
- W. Zhao, X. Ma, G. Wang, X. Long, Y. Li, W. Zhang and P. Zhang, Appl. Surf. Sci., 2018, 445, 167–174 CrossRef CAS.
- W. J. Li, S. L. Chou, J. Z. Wang, H. K. Liu and S. X. Dou, Chem. Commun., 2015, 51, 3682–3685 RSC.
- Y. Wang, C. Wu, Z. Wu, G. Cui, F. Xie, X. Guo and X. Sun, Chem. Commun., 2018, 54, 9341–9344 RSC.
- W.-J. Li, Q.-R. Yang, S.-L. Chou, J.-Z. Wang and H.-K. Liu, J. Power Sources, 2015, 294, 627–632 CrossRef CAS.
- X. Ge, Z. Li and L. Yin, Nano Energy, 2017, 32, 117–124 CrossRef CAS.
- X. Fan, T. Gao, C. Luo, F. Wang, J. Hu and C. Wang, Nano Energy, 2017, 38, 350–357 CrossRef CAS.
- K. A. Kovnir, Y. V. Kolen'ko, S. Ray, J. Li, T. Watanabe, M. Itoh, M. Yoshimura and A. V. Shevelkov, J. Solid State Chem., 2006, 179, 3756–3762 CrossRef CAS.
- S. C. Jung, J.-H. Choi and Y.-K. Han, J. Mater. Chem. A, 2018, 6, 1772–1779 RSC.
- H. Usui, Y. Domi, K. Fujiwara, M. Shimizu, T. Yamamoto, T. Nohira, R. Hagiwara and H. Sakaguchi, ACS Energy Lett., 2017, 2, 1139–1143 CrossRef CAS.
- H. Usui, Y. Domi, R. Yamagami, K. Fujiwara, H. Nishida and H. Sakaguchi, ACS Appl. Energy Mater., 2018, 1, 306–311 CrossRef CAS.
- Y. Ding, Z.-F. Li, E. V. Timofeeva and C. U. Segre, Adv. Energy Mater., 2018, 8, 1702134 CrossRef.
- H. Usui, T. Sakata, M. Shimizu and H. Sakaguchi, Electrochem, 2015, 83, 810–812 CrossRef CAS.
- W. Wang, J. Zhang, D. Y. W. Yu and Q. Li, J. Power Sources, 2017, 364, 420–425 CrossRef CAS.
- Y. Xu, B. Peng and F. M. Mulder, Adv. Energy Mater., 2018, 8, 1701847 CrossRef.
- F. Han, C. Y. J. Tan and Z. Gao, ChemElectroChem, 2016, 3, 1054–1062 CrossRef CAS.
- H. S. Shin, K. N. Jung, Y. N. Jo, M. S. Park, H. Kim and J. W. Lee, Sci. Rep., 2016, 6, 26195 CrossRef CAS PubMed.
- L. Ma, P. Yan, S. Wu, G. Zhu and Y. Shen, J. Mater. Chem. A, 2017, 5, 16994–17000 RSC.
- J. Choi, W.-S. Kim, K.-H. Kim and S.-H. Hong, J. Mater. Chem. A, 2018, 6, 17437–17443 RSC.
- Q. Li, Z. Li, Z. Zhang, C. Li, J. Ma, C. Wang, X. Ge, S. Dong and L. Yin, Adv. Energy Mater., 2016, 6, 1600376 CrossRef.
- D. Lan, W. Wang, L. Shi, Y. Huang, L. Hu and Q. Li, J. Mater. Chem. A, 2017, 5, 5791–5796 RSC.
- J. Gullman, J. Solid State Chem., 1990, 87, 202–207 CrossRef CAS.
- D.-H. Ha, L. M. Moreau, C. R. Bealing, H. Zhang, R. G. Hennig and R. D. Robinson, J. Mater. Chem., 2011, 21, 11498 RSC.
- Q. Zhou, Z. Shen, C. Zhu, J. Li, Z. Ding, P. Wang, F. Pan, Z. Zhang, H. Ma, S. Wang and H. Zhang, Adv. Mater., 2018, 30, 1800140 CrossRef PubMed.
- K. Zhang, M. Park, J. Zhang, G.-H. Lee, J. Shin and Y.-M. Kang, Nano Res., 2017, 10, 4337–4350 CrossRef CAS.
- D. Zhou and L.-Z. Fan, J. Mater. Chem. A, 2018, 6, 2139–2147 RSC.
- C. Wu, P. Kopold, P. A. van Aken, J. Maier and Y. Yu, Adv. Mater., 2017, 29, 1604015 CrossRef PubMed.
- M. Ihsan-Ul-Haq, H. Huang, J. Cui, S. Yao, J. Wu, W. G. Chong, B. Huang and J.-K. Kim, J. Mater. Chem. A, 2018, 6, 20184–20194 RSC.
- C. Dong, L. Guo, Y. He, C. Chen, Y. Qian, Y. Chen and L. Xu, Energy Storage Mater., 2018, 15, 234–241 CrossRef.
- S. Chen, F. Wu, L. Shen, Y. Huang, S. K. Sinha, V. Srot, P. A. van Aken, J. Maier and Y. Yu, ACS Nano, 2018, 12, 7018–7027 CrossRef CAS PubMed.
- Q. L. Ning, B. H. Hou, Y. Y. Wang, D. S. Liu, Z. Z. Luo, W. H. Li, Y. Yang, J. Z. Guo and X. L. Wu, ACS Appl. Mater. Interfaces, 2018, 10, 36902–36909 CrossRef CAS.
- K.-W. Tseng, S.-B. Huang, W.-C. Chang and H.-Y. Tuan, Chem. Mater., 2018, 30, 4440–4447 CrossRef CAS.
- Z. Huang, H. Hou, C. Wang, S. Li, Y. Zhang and X. Ji, Chem. Mater., 2017, 29, 7313–7322 CrossRef CAS.
- X. Li, W. Li, J. Yu, H. Zhang, Z. Shi and Z. Guo, J. Alloys Compd., 2017, 724, 932–939 CrossRef CAS.
- D. Duveau, S. S. Israel, J. Fullenwarth, F. Cunin and L. Monconduit, J. Mater. Chem. A, 2016, 4, 3228–3232 RSC.
- Y. Lu, P. Zhou, K. Lei, Q. Zhao, Z. Tao and J. Chen, Adv. Energy Mater., 2017, 7, 1601973 CrossRef.
- X. Li, S. Guo, K. Jiang, Y. Qiao, M. Ishida and H. Zhou, ACS Appl. Mater. Interfaces, 2018, 10, 16–20 CrossRef CAS.
- Q. Liang, Y. Zheng, C. Du, Y. Luo, J. Zhang, B. Li, Y. Zong and Q. Yan, Small Methods, 2017, 1, 1700304 CrossRef.
- D. Lan, W. Wang and Q. Li, Nano Energy, 2017, 39, 506–512 CrossRef CAS.
- Q. Liu, J. Wang, Y. Luo, L. Miao, Y. Yan, L. Xue and W. Zhang, Electrochim. Acta, 2017, 247, 820–825 CrossRef CAS.
- N. Cheng, L. Fan, Z. Liu, S. Chen, E. Zhang, J. Zhao, H. Yang, X. Yu and B. Lu, Mater. Today Energy, 2019, 11, 174–181 CrossRef.
- R. Jin, X. Li, Y. Sun, H. Shan, L. Fan, D. Li and X. Sun, ACS Appl. Mater. Interfaces, 2018, 10, 14641–14648 CrossRef CAS PubMed.
- G. Cho, H. Kim, Y. S. Park, Y.-K. Hong and D.-H. Ha, Int. J. Hydrogen Energy, 2018, 43, 11326–11334 CrossRef CAS.
- H. Yan, Comp. Mater. Sci., 2015, 107, 204–209 CrossRef CAS.
- S. Boyanov, D. Zitoun, M. Ménétrier, J. C. Jumas, M. Womes and L. Monconduit, J. Phys. Chem. C, 2009, 113, 21441–21452 CrossRef CAS.
- I. Shirotani, E. Takahashi, N. Mukai, K. Nozawa, M. Kinoshita, T. Yagi, K. Suzuki, T. Enoki and S. Hino, Jpn. J. Appl. Phys., 1993, 32, 294 CrossRef CAS.
- G. Samsonov, Y. B. Paderno, V. Lazorenko and P. Vityaz', Powder Metall. Met. Ceram., 1971, 10, 565–567 Search PubMed.
- Y. Lu and J.-p. Tu, CrystEngComm, 2012, 14, 8633–8641 RSC.
- Q. Li, J. Ma, H. Wang, X. Yang, R. Yuan and Y. Chai, Electrochim. Acta, 2016, 213, 201–206 CrossRef CAS.
- J. Xiang, X. Wang, X. Xia, J. Zhong and J. Tu, J. Alloys Compd., 2011, 509, 157–160 CrossRef CAS.
- E. J. Popczun, J. R. McKone, C. G. Read, A. J. Biacchi, A. M. Wiltrout, N. S. Lewis and R. E. Schaak, J. Am. Chem. Soc., 2013, 135, 9267–9270 CrossRef CAS.
- H. Li, S. Hao, Z. Tian, Z. Zhao and X. Wang, Electrochim. Acta, 2019, 321, 134624 CrossRef CAS.
- H. Guo, C. Chen, K. Chen, H. Cai, X. Chang, S. Liu, W. Li, Y. Wang and C. Wang, J. Mater. Chem. A, 2017, 5, 22316–22324 RSC.
- S. O. Kim and A. Manthiram, Chem. Commun., 2016, 52, 4337–4340 RSC.
- J. Duan, S. Deng, W. Wu, X. Li, H. Fu, Y. Huang and W. Luo, ACS Appl. Mater. Interfaces, 2019, 11, 12415–12420 CrossRef CAS.
- A. Zhou, B. Yang, W. Wang, X. Dai, M. Zhao, J. Xue, M. Han, C. Fan and J. Li, RSC Adv., 2016, 6, 26800–26808 RSC.
- R. Wang, X. Y. Dong, J. Du, J. Y. Zhao and S. Q. Zang, Adv. Mater., 2018, 30, 1703711 CrossRef PubMed.
- X. Y. Peilei He and X. Wen Lou, Angew. Chem., Int. Ed., 2017, 56, 3897–3900 CrossRef PubMed.
- J. Zhu, Q. He, Y. Liu, J. Key, S. Nie, M. Wu and P. K. Shen, J. Mater. Chem. A, 2019, 7, 16999–17007 RSC.
- X. Li, J. Liao, P. Shen, X. Lia, Y. Li, N. Li, Z. Li, Z. Shi, H. Zhang and W. Li, J. Alloys Compd., 2019, 803, 804–811 CrossRef CAS.
- W. Li, P. Shen, J. Liao, J. Yu, X. Li, Y. Liu, Y. Li, Z. Shi and H. Zhang, Mater. Lett., 2019, 253, 263–267 CrossRef CAS.
- Y. Liu, X. Xiao, X. Fan, M. Li, Y. Zhang, W. Zhang and L. Chen, J. Alloys Compd., 2018, 744, 15–22 CrossRef CAS.
- W. Li, H. Li, Z. Lu, L. Gan, L. Ke, T. Zhai and H. Zhou, Energy Environ. Sci., 2015, 8, 3629–3636 RSC.
- S. Haghighat-Shishavan, M. Nazarian-Samani, M. Nazarian-Samani, H. K. Roh, K. Y. Chung, S. H. Oh, B. W. Cho, S. F. Kashani-Bozorg and K. B. Kim, ACS Appl. Mater. Interfaces, 2019, 11, 32815–32825 CrossRef CAS PubMed.
- D. Kim, K. Zhang, J.-M. Lim, G.-H. Lee, K. Cho, M. Cho and Y.-M. Kang, Mater. Today Energy, 2018, 9, 126–136 CrossRef.
- X. Li, W. Li, P. Shen, L. Yang, Y. Li, Z. Shi and H. Zhang, Ceram. Int., 2019, 45, 15711–15714 CrossRef CAS.
- C.-Y. Chou, M. Lee and G. S. Hwang, J. Phys. Chem. C, 2015, 119, 14843–14850 CrossRef CAS.
- J. Saddique, X. Zhang, T. Wu, X. Wang, X. Cheng, H. Su, S. Liu, L. Zhang, G. Li, Y. Zhang and H. Yu, ACS Appl. Energy Mater., 2019, 2, 2223–2229 CrossRef CAS.
- A. Abouimrane, D. Dambournet, K. W. Chapman, P. J. Chupas, W. Weng and K. Amine, J. Am. Chem. Soc., 2012, 134, 4505–4508 CrossRef CAS PubMed.
- Y. Cao, M. K. Majeed, Y. Li, G. Ma, Z. Feng, X. Ma and W. Ma, J. Alloys Compd., 2019, 775, 1286–1292 CrossRef CAS.
- Z. Zhao, T. Yu, S. Zhang, H. Xu, G. Yang and Y. Liu, J. Mater. Chem. A, 2019, 7, 405–411 RSC.
- X.-W. Wang, H.-P. Guo, J. Liang, J.-F. Zhang, B. Zhang, J.-Z. Wang, W.-B. Luo, H.-K. Liu and S.-X. Dou, Adv. Funct. Mater., 2018, 28, 1801016 CrossRef.
- C. Marino, N. Dupré and C. Villevieille, J. Power Sources, 2017, 365, 339–347 CrossRef CAS.
- Q. Liang, Y. Zheng, C. Du, Y. Luo, J. Zhao, H. Ren, J. Xu and Q. Yan, ACS Nano, 2018, 12, 12902–12911 CrossRef CAS PubMed.
- W. Zhang, J. Mao, W. K. Pang, Z. Guo and Z. Chen, Electrochim. Acta, 2017, 235, 107–113 CrossRef CAS.
- J. Li, X. Li, P. Liu, X. Zhu, R. N. Ali, H. Naz, Y. Yu and B. Xiang, ACS Appl. Mater. Interfaces, 2019, 11, 11442–11450 CrossRef CAS PubMed.
- A. W. J. Ackermann, J. Phys. Chem. Solids, 1977, 38, 1013–1016 CrossRef.
- M. V. David Bellavance, B. Morris and A. Wold, J. Solid State Chem., 1969, 1, 82–87 CrossRef.
- N. K. M. Sugitan and M. Koizumi, J. Solid State Chem., 1978, 26, 195–201 CrossRef.
- S. S. J. P. Odile, C. A. Castro and A. Wold, Inorg. Chem., 1978, 17, 283–286 CrossRef.
- G. S. D. S. Robertson and H. Webber, J. Mater. Sci., 1980, 15, 256–258 CrossRef.
- H. S. Y. P. C. Donohue, J. Solid State Chem., 1970, 1, 143–149 CrossRef.
- H. Shen, Z. Ma, B. Yang, B. Guo, Y. Lyu, P. Wang, H. Yang, Q. Li, H. Wang, Z. Liu and A. Nie, J. Power Sources, 2019, 433, 126682 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |