
Coupling efficient biomass upgrading with H2 production via bifunctional CuxS@NiCo-LDH core–shell nanoarray electrocatalysts†
Xiaohui
Deng
a,
Xiaomin
Kang
a,
Mei
Li
a,
Kun
Xiang
 a,
Cheng
Wang
a,
ZaiPing
Guo
b,
Jiujun
Zhang
c,
Xian-Zhu
Fu
*a and
Jing-Li
Luo
*ad
a,
Cheng
Wang
a,
ZaiPing
Guo
b,
Jiujun
Zhang
c,
Xian-Zhu
Fu
*a and
Jing-Li
Luo
*ad
aCollege of Materials Science and Engineering, Shenzhen University, Shenzhen, 518060, China. E-mail: xz.fu@szu.edu.cn
bInstitute for Superconducting and Electronic Materials, School of Mechanical, Materials, Mechatronic and Bio-medical Engineering, University of Wollongong, NSW 2500, Australia
cInstitute for Sustainable Energy, College of Sciences, Shanghai University, Shanghai, 200444, China
dDepartment of Chemical and Materials Engineering, University of Alberta, Edmonton, Alberta T6G 1H9, Canada. E-mail: Jingli.luo@ualberta.ca
First published on 16th September 2019
Abstract
To boost hydrogen production from water splitting, the electrochemical oxidation of biomass-derived molecules to produce valuable chemicals is regarded as a promising approach to replace the kinetically sluggish oxygen evolution reaction. Herein, copper sulfide nanowire@NiCo-layered double hydroxide (LDH) nanosheet core–shell nanoarrays are fabricated as efficient bifunctional electrocatalysts for 5-hydroxymethylfurfural (HMF) oxidation and water reduction to simultaneously produce value-added 2,5-furandicarboxylic acid (FDCA) and hydrogen fuel with less energy consumption. Benefiting from the fast charge transfer induced by the CuxS core, the Co/Ni interaction in the LDH nanosheet layer and the open nanostructure, the optimized catalysts exhibit superior electrocatalytic activity (record-high 87 mA cm−2 @ 1.3 V vs. RHE for HMF oxidation; η = 107 mV @ 10 mA cm−2 for HER) and durability; the faradaic efficiency towards FDCA and H2 is close to unity. The bifunctional two-electrode electrolyzer only requires a low voltage of 1.34 V to co-generate H2 and FDCA at 10 mA cm−2. This work highlights the significance of tuning the redox properties of transition metals and constructing nanoarray electrocatalysts towards more efficient energy utilization.
1. Introduction
In the ongoing worldwide exploration of clean energy, hydrogen (H2) is regarded as a promising candidate fuel which could be produced by electrochemical water splitting.1–4 However, the oxygen evolution reaction (OER) has been considered as the bottleneck of overall water splitting since it suffers from a high overpotential and sluggish kinetics.5 Moreover, since O2 is of limited value and the mixing of O2 with H2 gives rise to potential safety issues, it is highly desirable to replace OER with novel oxidation processes that are kinetically favorable and meanwhile, capable of producing valuable chemicals.6–8 For instance, the oxidation of hydrazine, urea, and methanol, and sulfur generation have been coupled with cathodic H2O reduction to effectively reduce energy consumption towards H2 generation.9–12Recently, the oxidation of 5-hydroxymethylfurfural (HMF) has attracted considerable attention.6,13–17 As one of the most widely studied biomass-derived platform molecules, it is in great abundance and can be upgraded to chemicals such as 5-diformylfuran (DFF), 5-hydroxymethyl-2-furancarboxylic acid (HMFCA), maleic anhydride (MA) and 2,5-furandicarboxylic acid (FDCA). In particular, FDCA is gaining intense interest as it can act as the monomer towards synthesis of renewable polymer materials.18 Compared with the traditional catalysis approach that utilizes noble metal catalysts and requires high reaction temperature, electrochemical oxidation of HMF can be performed under benign conditions without using expensive oxidants.19,20 2,2,6,6-Tetramethyl-1-piperidinyloxy (TEMPO) has been reported as a redox mediator to facilitate HMF oxidation in a photoelectrochemical cell.21 Sulfide and phosphide of Ni can act as efficient electrocatalysts for HMF oxidation and the reaction occurs at a significantly lower voltage (∼200 mV) compared with OER.6,13,14 Moreover, the reaction is highly selective and the faradaic efficiency towards FDCA production is above 97%.
The Ni catalyzed oxidation of alcohols, aldehydes and primary amines has been proposed to follow the pathway in which Ni2+ is oxidized to NiOOH in the first step, and then the formed NiOOH would chemically oxidize organic functional groups and cycle back to lower oxidation states.22,23 Recent in situ Raman studies and the appearance of surface (oxy)hydroxide species after long-term catalysis also support this reaction pathway.7,24 Due to the reason that NiOOH is a necessary prerequisite towards further oxidation reactions, the onset potential of monometallic Ni-based electrocatalysts is often limited to ∼1.36 V vs. RHE regardless of the target functional groups, and we, therefore, anticipate that tuning the redox behavior of Ni could negatively shift its oxidation potential and further enhance the H2 production efficiency.25 Furthermore, despite the fact that the construction of nanostructures is rarely targeted in this field, we consider the exposure of active sites and the mass transfer as the critical factors towards high catalytic performance since the chemical oxidation step occurs directly between the exposed NiOOH species and surface-adsorbed organic substrates.26
Herein, we report the facile preparation of copper sulfide nanowire@NiCo-layered double hydroxide nanosheet core–shell (CuxS@NiCo-LDH) nanoarrays as efficient bifunctional electrocatalysts towards simultaneous HMF oxidation and H2 evolution. By facilitating charge/mass transfer with CuxS core–NiCo-LDH shell nanostructures and tuning the electronic structure of Ni with Co, the onset potential towards HMF oxidation is negatively shifted by ∼150–200 mV compared with the typical literature value of ∼1.36 V vs. RHE, and a current density of 87 mA cm−2 is achieved at 1.3 V vs. RHE with a faradaic efficiency of nearly 100% for FDCA production. The core–shell nanoarray electrocatalysts also show outstanding hydrogen evolution activity/durability. Moreover, the electrolyzer with CuxS@NiCo-LDHs as bifunctional electrocatalysts only requires a low voltage of 1.34 V to achieve the current density of 10 mA cm−2, which illustrates their superior bifunctionality in transition metal-catalyzed biomass upgrading coupled with H2 production.
2. Experimental
2.1 Materials
2.2 Electrochemical measurements
The linear scans of electrochemical hydrogen evolution, oxygen evolution and 5-HMF oxidation reactions were conducted on a CHI760e potentiostat with a three-electrode configuration. The as-prepared composite/CF (0.5 × 0.5 cm2) was used as the working electrode and the Hg/HgO electrode was used as the reference electrode. A 1 × 1 cm2 Pt sheet was used as the counter electrode in OER and 5-HMF oxidation while a graphite rod was used in the case of HER to avoid potential contamination due to the Pt dissolution and deposition on the working electrode. 1 M KOH (pH 13.8) with or without the presence of 10 mM HMF was used as the electrolyte and the linear scans were collected with a scan rate of 5 mV s−1. For the oxidation of HMF, the working electrode was pre-conditioned by cycling between 0.6 and 1.5 V with a scan rate of 50 mV s−1 until a stable voltammogram was obtained. In order to identify current contributions from catalyst oxidation, OER and HMF oxidation, the electrodes were first tested in 1 M KOH electrolyte without the presence of HMF, afterwards, a certain amount of HMF was added to achieve a concentration of 10 mM and linear scans were collected. The electrochemical impedance spectroscopy measurements were carried out in the same configuration with a cathodic potential of −1.1 V vs. Hg/HgO. The spectra were collected in a frequency range of 0.1 to 105 Hz with an amplitude of 5 mV. The ohmic drop was determined from electrochemical impedance spectroscopy and was compensated manually.The constant potential oxidation of HMF was conducted with a Solartron SI1287 potentiostat and the IR drop was compensated automatically in the current interrupt mode (off time 27 μs, on/off ratio 255) to maintain the potential applied on the working electrode. The stability of CuxS@Ni0.75Co0.25OxHy towards HMF oxidation was evaluated by chronoamperometry at 1.32 V vs. RHE in 10 mL 1.0 M KOH with 10 mM HMF for five successive trials. For the HER stability test, the potential to reach a current density of 10 mA cm−2 was monitored for 20 h. For the two-electrode electrolyzer, CuxS@Ni0.75Co0.25OxHy/CF was employed as the cathode and anode.
2.3 Characterization and product quantification
The morphologies were characterized by field-emission scanning electron microscopy (FESEM, SU-70) and field-emission transmission electron microscopy (FETEM, JEM-3200FS). X-ray diffraction (XRD) patterns were recorded using a Bruker D8 Advance (Cu Kα, 50 kV and 360 mA). X-ray photoelectron spectroscopy (XPS) was performed on a PHI5000 VersaProbe and all the peaks were calibrated with the C 1s spectrum at a binding energy of 284.8 eV. For ICP-OES measurements, the materials were collected from the electrodes by sonication and centrifugation. Then, the powders were dissolved in aqua regia and diluted with DIW. The liquid was then analyzed with an Agilent 720. To measure the leaching losses of metal species after HER and HMF oxidation, the experiments were conducted in a two-compartment electrochemical cell separated by a Nafion 117 membrane. The electrolytes were collected, filtered and neutralized with HCl solution before being analyzed by ICP-OES. The H2 produced from the cathodic reaction was identified by gas chromatography (Agilent 7890A) analysis and the amount generated was determined with the water displacement method.The identification and quantification of 5-HMF and oxidation products were performed by high-performance liquid chromatography (HPLC) on a Shimadzu Prominence-I LC 2030 Plus at room temperature. After a certain number of charges were passed, 20 μL of the electrolyte solution was withdrawn and diluted with 980 μL DIW. The HPLC was equipped with a Shimadzu Shim-pack GIST column (5 μm C18, 4.6 × 250 mm) and a UV-VIS detector set at λ = 265 nm. A mixture of 70% 5 mM ammonium formate aqueous solution and 30% methanol was used as the eluent. An injection volume of 10 μL was applied and the identification/determination of the concentration of reactants/oxidation products were achieved from calibration curves of known concentrations of commercially purchased compounds.
The conversion (%) of 5-HMF and the yield (%) of oxidation products were calculated based on the following equations:
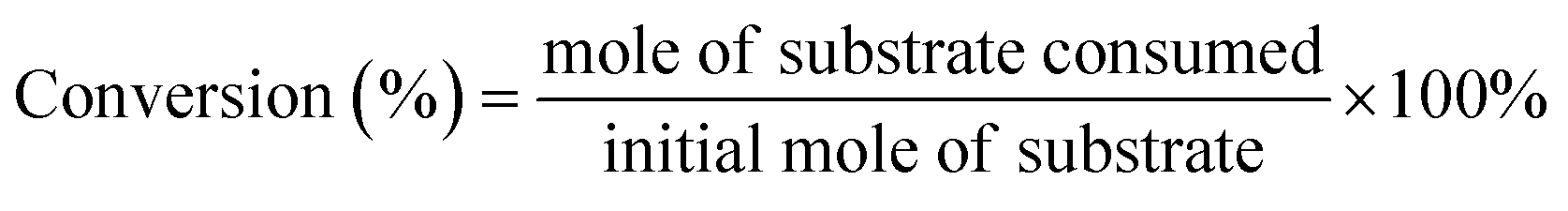
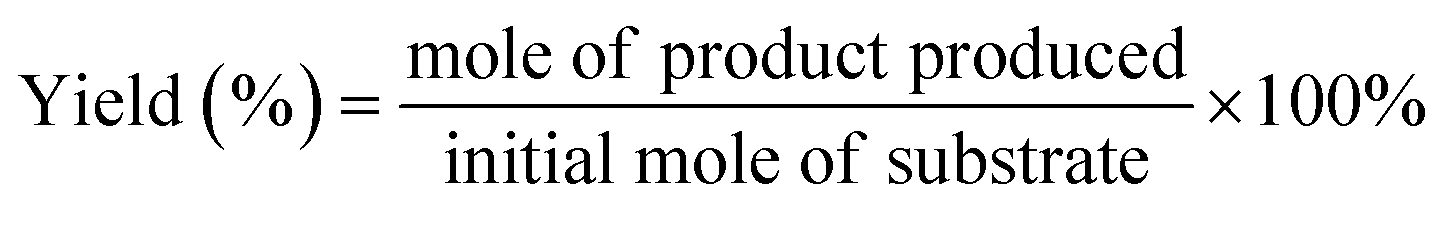
The faradaic efficiency (FE) of product formation was calculated based on the following equation:
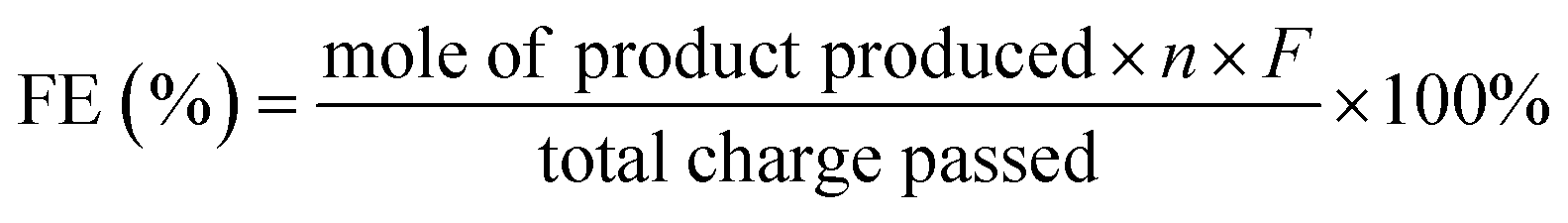
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1).
485 C mol−1).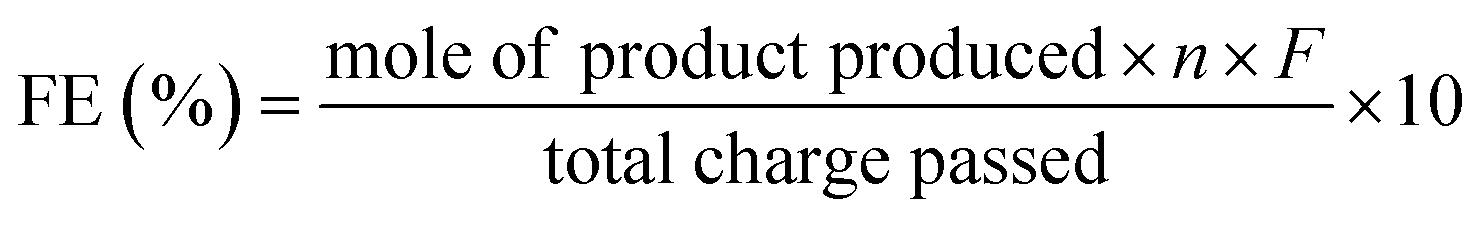
2.4 Computational method
The density of states calculations were performed by using the Vienna Ab initio Simulation Package (VASP), employing the density functional theory (DFT) and the Projected Augmented Wave (PAW) method.27–29 The Perdew–Burke–Ernzerhof (PBE) functional was used to describe the exchange and correlation effect.30 For all the geometry optimizations, the cutoff energy was set to be 450 eV. Spin polarization was included in all the cases. The GGA+U calculations were performed using the model proposed by Dudarev et al., with the Ueff (Ueff = Coulomb U − exchange J) values of 3.3 eV and 6.4 eV for Co and Ni, respectively.31 The energy and force convergence tolerances were set to be 10−5 eV and 0.01 eV Å−1, respectively.3. Results and discussion
The preparation of CuxS@NiCo-LDH core–shell nanoarrays is illustrated in Fig. 1a. Briefly, the Cu(OH)2 nanowire arrays (NAs) are first fabricated from commercially available Cu foam (Fig. S1a–c†). Afterwards, the Cu(OH)2 NAs are immersed in Na2S aqueous solution and the ion exchange process occurring at room temperature leads to the formation of CuxS NAs. The SEM images (Fig. S1d–f†) show that the nanowire morphology is well preserved after the sulfidation step while a slight increase of the diameter of nanowires (∼30 nm) can be observed, which is attributed to the increased surface roughness during the sulfide formation.32 A Cu to S ratio of close to 2 is obtained from the elemental analysis (Fig. S1g–i and Table S1†) and the XRD pattern (Fig. S2a†) suggests the dominant presence of metallic Cu (substrate), Cu31S16 and Cu2O (trace mount). Since the copper sulfide polymorphs (e.g., Cu9S5, Cu7.2S4 and Cu2S) exhibit reflections at similar positions, the copper sulfide in this work is referred to as CuxS. In the final step, the NiCo-LDHs are coated on the CuxS NAs by electrodeposition. | ||
| Fig. 1 (a) Schematic illustration of the preparation of CuxS@NiCo-LDHs core–shell composites. (LDH for layered double hydroxide, NA for nanoarrays, and RT for room temperature). (b–d) SEM images, (e) TEM image, (f) Elemental mappings of CuxS@Ni0.75Co0.25OmHn. (g) HRTEM image of ultra-thin Ni0.75Co0.25-LDH. | ||
The morphology of synthesized CuxS@NiCo-LDHs is then characterized by scanning electron microscopy (SEM). As shown in the SEM images (Fig. 1b–d and S3†), all the composite materials (with various Ni to Co ratios) show well-aligned nanoarrays coated by a 3D nanosheet-assembled porous nanostructure. Due to the nature of metal cations, the detailed structure of hydroxide species varies depending on the precursor composition.33 The porosity of deposited LDHs and vertically aligned nanoarrays could expose a large number of active sites and provide low-resistant pathways for the electrolytes and reactants, thus facilitating the mass transfer of the reactive species. The chemical composition of electrodeposited species is analyzed by ICP-OES and the Ni to Co ratio is in good agreement with the anticipated values (Table S2†). Fig. 1b–d show the SEM images of the sample with a Ni to Co ratio of 3. The core–shell nanoarray structure is homogeneously distributed on the copper foam, and the TEM image (Fig. 1e) shows that CuxS nanowires with a diameter of ∼260 nm are uniformly coated by the LDH nanosheets. Elemental mappings (Fig. 1f) confirm that Cu and S are dominantly located on the CuxS nanowires while Ni and Co are uniformly distributed on the nanosheets in the outer-shell. The HRTEM image (Fig. 1g) shows lattice fringes with the interplanar spacing of 0.23 nm, which could be assigned to the (015) planes of NiCo-LDH. The XRD patterns of as-prepared CuxS@NiCo LDHs were also collected and the reflections which can be assigned to the (110) plane of the LDH phase are marked in Fig. S2b.† Due to the ultrathin morphology and low content, the intensities of the characteristic reflections are quite weak.
The room-temperature sulfidation step is essential for the successful preparation of the core–shell structure as the metallic character of CuxS would provide sufficient electrical conductivity for the electrodeposition and more importantly, be beneficial for the transfer of charge carriers in further electrochemical applications.34–36 This key aspect is also highlighted elsewhere in the CuxS@NiFe LDH core–shell nanoarrays towards overall water splitting where the Cu core provides good electronic transport.37 However, in their report, the transformation of Cu(OH)2 NAs to Cu involves a high energy consumption step (calcination at 180 °C for 1 h) followed by an electrochemical reduction procedure. In this work, the overall preparation protocol is significantly simplified and much less energy-consuming as the sulfidation step occurs at room temperature in aqueous solution and the reduction by electricity or any other reductant is not necessary. Furthermore, the readily maintained nanoarray structure would greatly facilitate the mass transfer involved in electrochemical processes.38 Therefore, this low-cost, all solution-based synthesis protocol could potentially provide guidance for large-scale manufacture of highly active electrocatalysts. In the control experiment, the electrodeposition of LDHs on pristine Cu(OH)2 NAs is not feasible and results in large areas where LDHs are not deposited due to their poor conductivity (Fig. S4†).
The electrocatalytic performance of as-prepared CuxS@NiCo-LDHs towards HMF oxidation is evaluated under alkaline conditions. The linear scans (OER and HMF oxidation after addition of 10 mM HMF) of CuxS@NiOmHn and CuxS@CoOmHn are presented in Fig. 2a and b. It can be observed that compared with OER, in the case of Ni, the anodic current of HMF oxidation follows the Ni(OH)2/NiOOH oxidation couple in a lower potential range but with significantly enhanced current density at potentials exceeding 1.4 V. The reason is that the NiOOH generated from the oxidation of Ni(OH)2 acts as the chemical oxidant towards HMF oxidation and in this way, the Ni(OH)2 species are regenerated and therefore more charges are passed during the anodic scan.22 Recently, a new mechanism has been proposed for HMF oxidation on a nickel electrode that at higher oxidation potentials, HMF could be directly oxidized by NiOOH without involving the transformation of NiOOH to Ni(OH)2.15 However, this phenomenon is only present on thin film electrodes and not observed in our experiments, probably due to the high current densities. As is shown, CuxS@NiOmHn exhibits anodic current densities of 42.3 mA cm−2 and 187.4 mA cm−2 at 1.35 V and 1.4 V vs. RHE, respectively, and such performance is comparable to those of the recently reported benchmark electrocatalysts (NiFe LDH, Ni2P@NF, and Ni3S2@NF) towards HMF oxidation (Table S3†),6,13,17 while in the case of electrodeposited Co, the position and current of the oxidation peak (Co2+/Co3+) at 1.13 V vs. RHE are nearly identical regardless of the presence of HMF, indicating that the formed CoOOH is not able to chemically oxidize HMF efficiently.15 Interestingly, the sample exhibits an additional anodic current when the potential exceeds 1.23 V vs. RHE in comparison with that measured under OER conditions, illustrating the direct oxidation of HMF. It is worth mentioning that the CuxS@Co(OH)2 shows the lowest onset potential (1.23 V vs. RHE) compared with the recently reported Co-based electrocatalysts for HMF oxidation (Co-P/CF, 1.3 V vs. RHE; CoB/NF, ∼1.32 V vs. RHE).14,39 Furthermore, it exhibits a current density of 80 mA cm−2 at 1.4 V vs. RHE, which is significantly higher than that of the aforementioned counterparts (Table S3†). The superior catalytic activity of CuxS@Co(OH)2 is attributed to the nanowires@nanosheet core–shell array architecture and a large number of exposed active sites.
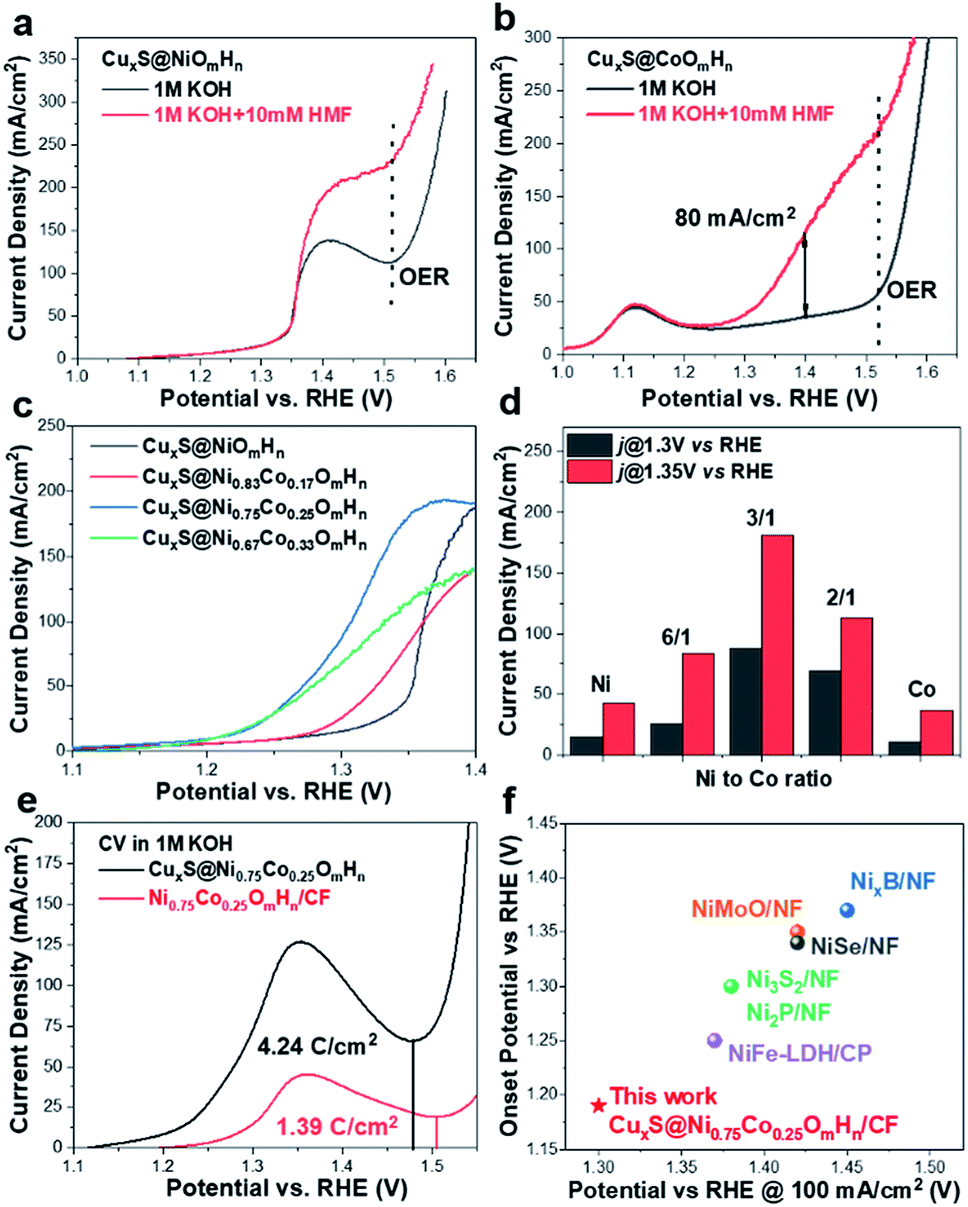 | ||
| Fig. 2 (a and b) LSV curves of CuxS@NiOmHn and CuxS@CoOmHn towards electrochemical HMF oxidation. (c) Comparison of LSV curves of CuxS@NiCo LDHs with various Ni to Co ratios. (d) Current densities of prepared electrodes at 1.3 V and 1.35 V vs. RHE. (e) CV curves of CuxS@Ni0.75Co0.25OmHn, Ni0.75Co0.25OmHn/CF in 1 M KOH and calculated charges for the oxidation of transition metal cations prior to the oxygen evolution reaction. (f) Performance comparison of CuxS@Ni0.75Co0.25OmHn with reported electrocatalysts towards electrochemical oxidation of HMF, urea and primary amine. | ||
Since the redox behavior of transition metals can be tuned by the incorporation of secondary species, it is then intriguing to investigate the activity of NiCo-LDHs.25,40 The OER polarization curves and the anodic scans in the presence of 10 mM HMF of all the mixed composites are plotted in Fig. S5.† As can be observed, all samples exhibit an enhanced anodic current before the onset of OER when HMF is present, suggesting the activity for HMF oxidation. The linear scans in 1 M KOH + 10 mM HMF are then compared and plotted in Fig. 2c. Due to the reason that under higher potentials, the mass transport of HMF under such concentration (10 mM) may hinder the overall process, the potential range of the plot is chosen between 1.1 V and 1.40 V vs. RHE. As is shown, the onset potential is clearly negatively-shifted compared with CuxS@NiOmHn and this indicates that the HMF oxidation catalyzed by CuxS@NiCo-LDHs occurs with less energy consumption. The Ni/Co ratio of 3 is shown to be the optimal composition. It can be observed from the linear scan comparison (Fig. 2c) that the Co content in the mixed LDH is critical to induce the decrease of onset potential as the electrodes with the lowest amount of Co (Ni/Co = 5) show the slightest performance enhancement. Further increasing the Co content to Ni/Co = 3 and 2 shifts the onset potential to a nearly identical value (∼1.18 V vs. RHE) and this indicates that an additional amount of Co has a negligible impact on the onset potential towards HMF oxidation after the optimal scenario. Meanwhile, the Ni content is another important parameter as we speculate that Ni acts as the dominant active species in the mixed LDHs (see the following discussion) and an excess amount of Co decreases the content of electrochemically active nickel species in the LDH and is therefore detrimental to the overall catalytic activity. To be more specific, the optimized electrode (with a Ni/Co ratio of 3) exhibits current densities of 87 mA cm−2 and 180.6 mA cm−2 at 1.3 V and 1.35 V vs. RHE, respectively, which are 5.8 and 4.3 times higher than that of CuxS@NiOmHn and also significantly greater than those of the other counterparts (Fig. 2d). The Co incorporation also shows an influence on the reaction kinetics. As plotted in Fig. S6,† the CuxS@NiOmHn displays a Tafel slope of 27 mV dec−1, indicating the fast kinetics of Ni(OH)2 being oxidized to NiOOH. On the other hand, the Tafel slopes are around ∼85 mV dec−1 for all Co-containing samples regardless of the cobalt concentration. The advantage of the core–shell nanoarray structure is clearly demonstrated when compared with the films directly deposited on copper foam. As shown in Fig. S7,† due to the relatively smaller surface area of untreated CF, the deposited electrode shows thick films with multiple cracks. To straightforwardly compare the quantity of electrochemically active species on the electrode surface, the charge consumed by the oxidation of transition metal cations is calculated from the integration of oxidation peaks as shown in the CVs (Fig. 2e). As calculated, CuxS@Ni0.75Co0.25OmHn exhibits a total anodic charge of 4.24 C cm−2 prior to the oxygen evolution reaction, which is ∼3 times higher than that of Ni0.75Co0.25OmHn deposited on copper foam (1.39 C cm−2) and this suggests greatly increased electrochemically active species in the core–shell nanoarray electrocatalyst. As a result, the catalytic activity towards HMF oxidation is substantially higher (Fig. S7†). It should be further highlighted that CuxS@Ni0.75Co0.25OmHn LDHs can be considered as the most active catalysts towards electrochemical HMF oxidation reported so far (Fig. 2f) since the lower onset potential (1.19 V vs. RHE) and the significant oxidation current at potentials more negative than 1.3 V vs. RHE represent a remarkable advance in this field. Recently, the electro-oxidation of urea and amine catalyzed by Ni-based electrocatalysts (NiMoO/NF and NiSe/NF, Fig. 3d) has also been studied and the in situ/ex situ characterization proved that such reactions follow a similar pathway to Ni-catalyzed HMF oxidation.7,41 Therefore, we believe that the strategy demonstrated in this study could potentially boost the development of efficient catalysts towards the electrochemical oxidation of small molecules in general. Pristine Cu2S NAs are also tested for HMF oxidation (Fig. S8†) and much lower anodic currents are observed. Besides, the Cu to Ni + Co ratio (full-range XPS spectra shown in Fig. S9a†) is determined to be ∼1/9 after the electrodeposition, which implies that NiCo-LDHs are responsible for the outstanding activity. The electrodeposition time is optimized to be 450 s (Fig. S10†).
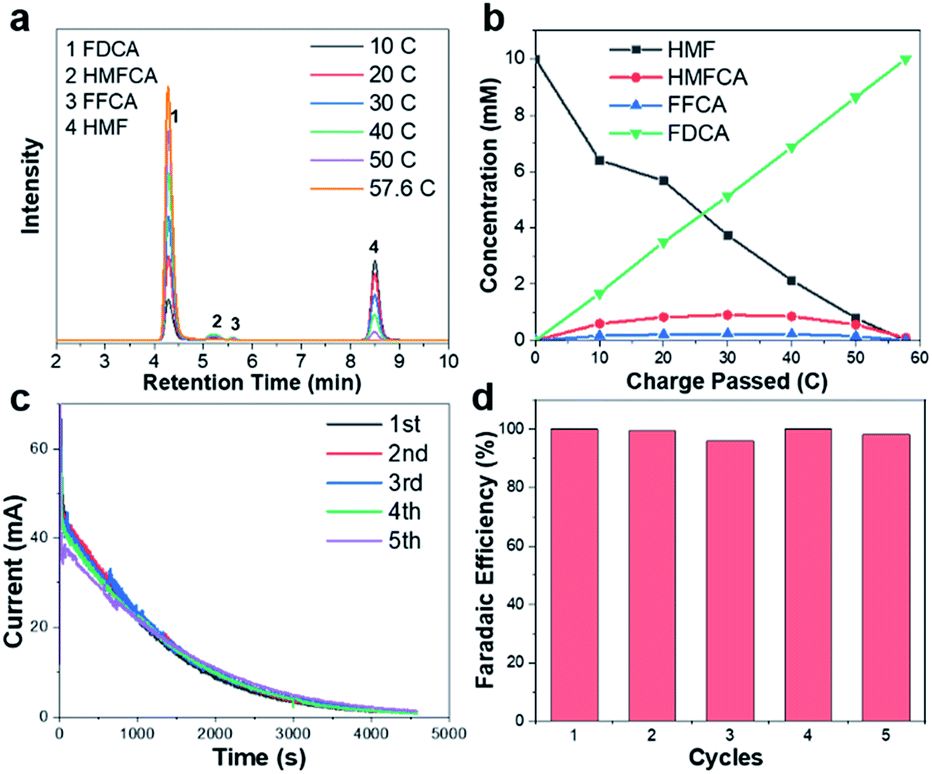 | ||
| Fig. 3 (a) HPLC chromatogram traces of electrolyte during the electrochemical oxidation of HMF catalyzed by CuxS@Ni0.75Co0.25OmHn at 1.32 V vs. RHE. (b) Concentration profiles of oxidation products with increasing charge. (c) Current–time curves and (d) faradaic efficiency towards FDCA in 5 successive runs. | ||
To identify the oxidation products and quantify the corresponding yields and faradaic efficiency, HMF oxidation catalyzed by CuxS@Ni0.75Co0.25OmHn is performed at a fixed potential of 1.32 V vs. RHE and a total charge of ∼58 C is passed.
The liquid samples are then collected after every 10 C of charge and analyzed by high-performance liquid chromatography (the spectra and corresponding calibration curves are shown in Fig. S11†). Fig. 3a shows the evolution of HPLC chromatogram traces, and the HMF/product concentration profile with increasing quantity of charge is plotted in Fig. 3b. A FDCA yield and faradaic efficiency of ∼99% are obtained at the end of electrolysis, suggesting that CuxS@Ni0.75Co0.25OmHn is highly active and selective under a potential as low as 1.32 V vs. RHE. Although the degradation of HMF under strong alkaline conditions has been reported, a highly active electrocatalyst could minimize this impact if HMF is quickly transformed to other chemicals. In the present study, ∼50 C is passed in the first 35 min at 1.32 V vs. RHE and this corresponds to an HMF conversion of 87%. Therefore, the FDCA yield loss due to HMF degradation is negligible. The formation rate of HMFCA is higher in the initial period while no DFF is detected through the electrolysis, indicating that the oxidation of HMF might follow the pathway in which HMFCA is the oxidation intermediate towards FFCA (Scheme S1†).13,24 The anodic scans of CuxS@Ni0.75Co0.25OmHn towards the oxidation of reaction intermediates (FFCA and HMFCA) are also collected and an onset potential similar to that of HMF is shown in Fig. S12;† this can be expected as the oxidation of hydroxyl and aldehyde functional groups follows the same Ni2+/NiOOH pathway. The electrodes are further tested for 5 successive runs and the current–time curves show negligible degradation (Fig. 3c). The close-to-unity faradaic efficiency in all cases (Fig. 3d) also demonstrates their superior stability towards HMF oxidation.
To further address the effect of cobalt incorporation on the catalytic activity, we theoretically investigated the electronic structure of pristine NiOOH and NiCo-LDH with a Ni to Co ratio of 3. The projected density of states (Fig. 4a) shows the electronic states of the layered double hydroxide being modulated after Co participation. The electronic interaction between Co and Ni is further evidenced by the XPS (X-ray photoelectron spectroscopy) spectra of CuxS@NiOmHn and CuxS@Ni0.75Co0.25OmHn. The O 1s spectra of both samples are plotted in Fig. S9b† and the binding energy at 531.6 eV is assigned to the oxygen in the hydroxyl group.25,42 As shown in the Ni 2p3/2 spectra (Fig. 4b), the binding energy of 855.6 eV in CuxS@NiOmHn agrees well with that of nickel hydroxide.42–44 The obvious positive shift (0.3 eV) of the Ni 2p3/2 peak in CuxS@Ni0.75Co0.25OmHn suggests a higher ratio of Ni3+/Ni2+ when Co is present in the LDH.45 As has been intensively studied in the oxygen evolution reaction, the occupancy of eg orbitals in 3d transition-metal-based electrocatalysts is considered as a general activity descriptor.46,47 The eg orbital occupancy of close to 1 for surface metal ions could increase the covalency of the metal–oxygen bonds and is regarded as the optimal electronic configuration towards the adsorption/desorption of oxygen-containing reaction intermediates. Meanwhile, recent DFT calculations show that HMF molecules tend to be adsorbed on the surface of transition metal oxides by forming bonds between the metal cations and the formyl/hydroxyl oxygen atoms.48 As illustrated in Fig. 4c, the Ni2+ is with a t62ge2g electronic configuration while Ni3+ possesses the t62ge1g configuration with a near-unity occupancy of the eg orbital. Therefore, we anticipate that the tuned electronic structure of Ni could facilitate the adsorption of HMF on the electrode surface. At the present stage, it remains unclear whether or not the Co cation in mixed LDH is active towards oxidation of HMF. Nevertheless, as suggested by the recent in situ X-ray absorption spectroscopy measurements, the oxidation state of Co cation in NiCo mixed oxides remains nearly unchanged under applied electrochemical bias.49,50 We then cautiously speculate that Ni acts as the dominant active sites in the mixed LDH whilst Co mainly promotes the oxidation of Ni(OH)2 to NiOOH and facilitates HMF adsorption by modulating the electronic configuration of Ni. Furthermore, Boettcher et al. reported that the facile oxidation of NiCo-LDH can boost its electric conductivity by two orders of magnitude at ∼1.2 V vs. RHE (compared with ∼1.35 V vs. RHE for Ni hydroxide), indicating enhanced charge transfer in the mixed hydroxides.51
 | ||
| Fig. 4 (a) Projected density of states calculations for Ni0.75Co0.25-LDH and NiOOH. (b) High resolution Ni 2p3/2 XPS spectra of CuxS@Ni0.75Co0.25OmHn and CuxS@NiOmHn. (c) Schematic illustration of the electronic configuration of the Ni cations with valence states of +2 and +3. (d) Schematic illustration of factors contributing to the high HMF oxidation activity. | ||
Based on the provided experimental evidence and discussion, we unambiguously attribute the superior HMF oxidation activity of the CuxS@Ni0.75Co0.25OmHn to the following factors (Fig. 4d): (1) the transformation of Cu(OH)2 to CuxS nanoarrays provides sufficient conductivity not only for the electrodeposition of ultra-thin NiCo-LDHs as active species but also for ensuring fast charge carrier transfer from the conductive substrate (Cu foam) to the surface of the shell catalytic layer; (2) the large surface area and open-channels of the core–shell nanostructure could expose a large number of active sites and also facilitate the mass transfer of reactants and products, assumably a more critical issue in HMF oxidation compared with O2/H2 evolution reaction where the gaseous products are neutrally charged and much easier to escape from the catalyst surface; (3) the tuned electronic structure of Ni due to Co incorporation represents an optimal construction where a higher content of Ni3+ with the eg orbital occupancy close to 1 facilitates the adsorption of reactants and intermediates on the electrode surface and moreover, the oxidation of Ni2+ to NiOOH (which is the ‘true’ active species in HMF oxidation) occurs at significantly lower potentials compared with pristine Ni(OH)2. Overall, the results presented herein demonstrate the synergy between nanostructure construction and electronic structure modulation to design more efficient electrocatalysts for biomass valorization and possibly other transition metal-catalyzed oxidation reactions.
As transition metal-based materials have been demonstrated in recent reports as efficient non-noble metal HER catalysts, it would be intriguing to integrate biomass valorization and H2 production with a bifunctional electrocatalyst.52–54 We thus evaluated the HER activity of CuxS@NiCo LDHs under alkaline conditions with a graphite rod as the counter electrode to avoid the possible contamination by Pt.37,55–57 The LSV curves are shown in Fig. 5a. Apart from CuxS@CoOmHn, all the nickel containing composite electrodes show similar, yet outstanding HER activity. The overpotential to reach a current density of 10 mA cm−2 is in the range of 108–120 mV and such performance is comparable to those of the benchmark transition metal-based HER catalysts (Table S4†). The HER performance of CuxS@Ni0.75Co0.25OmHn is then compared with those of bare CuxS, Pt mesh and Ni0.75Co0.25OmHn deposited on Cu foam. As shown in the LSV curves (Fig. 5b) and Tafel plots (Fig. 5c), the Pt mesh demonstrates HER activity with an overpotential of 79 mV (@10 mA cm−2) and a Tafel slope of 42 mV dec−1, while CuxS@Ni0.75Co0.25OmHn exhibits an overpotential of 107 mV and a remarkable Tafel slope of 35 mV dec−1. In comparison, bare CuxS and Ni0.75Co0.25OmHn/CF show sluggish HER kinetics instead. Electrochemical impedance spectroscopy (EIS) is used to further compare the reaction kinetics. As plotted in Fig. S13,† CuxS@Ni0.75Co0.25OmHn shows a much smaller charge transfer resistance of 5 Ω compared with bare CuxS and Ni0.75Co0.25OmHn/CF (∼170 Ω) at −1.1 V vs. Hg/HgO, further demonstrating the significantly enhanced charge transfer in the core–shell nanoarray electrocatalyst. It should be noted that such HER performance enhancement cannot be solely explained by the contribution from the open nanostructure and larger number of exposed active sites. As suggested by the recent reports, we speculate that Cu–S–transition metal hydroxide interfaces might play a vital role in promoting water dissociation and facilitating the formation of Had intermediates in this case.58,59 However, this requires more detailed investigation with additional theoretical insights, which will be studied further. The electrode exhibits an outstanding durability as the overpotential remains stable during 20 h of constant electrolysis (Fig. 5d) and the presence of HMF is found to have a negligible effect on the HER performance (Fig. 5e). The faradaic efficiency towards H2 evolution is measured to be 100% (inset in Fig. 5e). Encouraged by the superior bifunctionality, an electrolyzer employing CuxS@Ni0.75Co0.25OmHn as the cathode and anode is assembled and tested for simultaneous HMF oxidation and H2 production. Impressively (Fig. 5f), the electrolyzer requires a mere 1.34 V to reach the current density of 10 mA cm−2 and 1.58 V to reach 100 mA cm−2, which is ∼270 mV less than that for the overall water splitting. Moreover, such performance is superior to those of other reported bifunctional electrocatalysts in light of the unique coupling of anodic oxidation reactions and H2 production (Table S5†), further illustrating the effectiveness of the CuxS@NiCo-LDH nanoarray electrocatalysts.
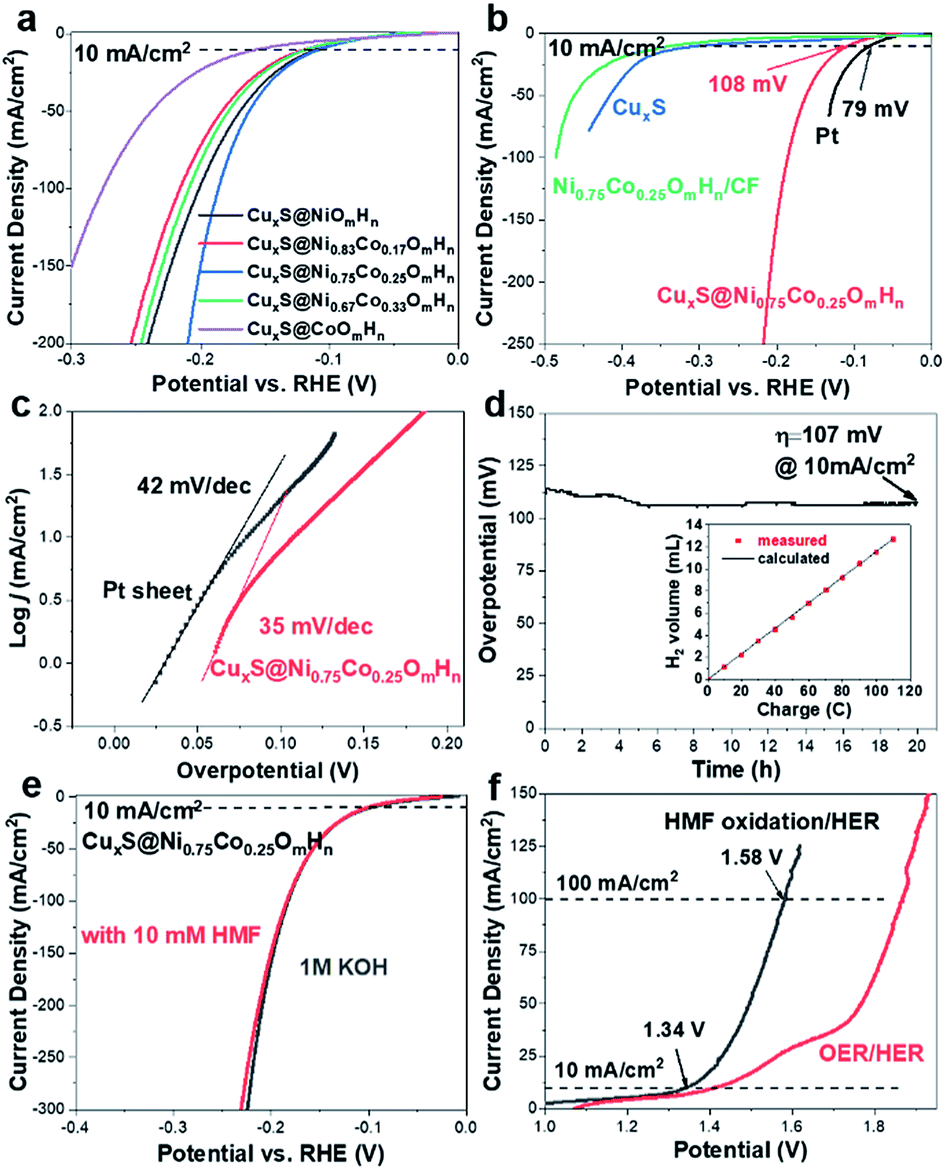 | ||
| Fig. 5 HER curve comparison of (a) core–shell composites with various Ni and Co contents and (b) CuxS, CuxS@Ni0.75Co0.25OmHn, Ni0.75Co0.25OmHn/CF and Pt sheet (1 M KOH, pH 13.8; graphite rod as the counter electrode; 5 mV s−1). (c) Tafel plots of Pt and CuxS@Ni0.75Co0.25OmHn towards HER. (d) Stability test of CuxS@Ni0.75Co0.25OmHn for 20 h (inset shows the faradaic efficiency towards H2 evolution). (e) HER curve comparison of CuxS@Ni0.75Co0.25OmHn in 1 M KOH with/without 10 mM HMF. (f) Comparison of LSV curves of overall water splitting and hydrogen evolution/HMF oxidation over the CuxS@Ni0.75Co0.25OmHn‖CuxS@Ni0.75Co0.25OmHn electrolyzer. | ||
We further conducted detailed characterization on the electrodes after catalyzing long-term HMF oxidation and hydrogen evolution. As shown in the SEM images (Fig. 6), the nanoarray structure is maintained in both cases. However, it is noticed that the electrodeposited NiCo-LDH after HMF oxidation (Fig. 6a–c) turned into a dense layered structure with significantly increased thickness and such morphology is similar to the reported hydroxides before exfoliation.60,61 This structural transformation is attributed to the phase cycling of metal hydroxides/oxyhydroxides during the oxidation of HMF, in which the transition metal hydroxides re-form at the end of each catalytic turnover. XPS results (Fig. 6g–i) show that the oxidation state of Ni remains unchanged after the stability test while an obvious negative shift (0.8 eV) of Co 2p spectra is observed along with a new peak appearing at 529.6 eV in O 1s spectra, which is then correlated with the formation of CoOOH species.43 As pointed out earlier, Co catalyzes the HMF oxidation through a dominant direct oxidation pathway and the chemical oxidation (similar to the case of Ni) occurs at a much slower rate, it could therefore be expected that the oxidation state of Co after catalysis would be close to +3. On the other hand, SEM images (Fig. 6d–f) after the long-term HER test show that the previous 3D core–shell nanostructure is maintained with little aggregation and the high-resolution Ni 2p, Co 2p and O 1s XPS spectra are nearly identical to that of the as-prepared sample. The leaching of transition metal species after catalyzing long-term HER and HMF oxidation is investigated by ICP-OES. It is shown that the content of Ni and Co (as active species) in the electrolyte in both cases is below the detection limit (0.02 mg L−1) while the Cu leaching is more severe as 0.12 mg L−1 and 11.03 mg L−1 of Cu are detected after catalyzing HER and HMF oxidation. The much higher leaching of Cu in the case of HMF oxidation corresponds to a leached Cu amount of 0.17 mg cm−2 and is attributed to the oxidation/dissolution of exposed CuxS species under anodic conditions. Nevertheless, the electrochemical stability test and post-SEM characterization show that such leaching has a negligible effect on the performance and structural stability of core–shell nanoarrays.
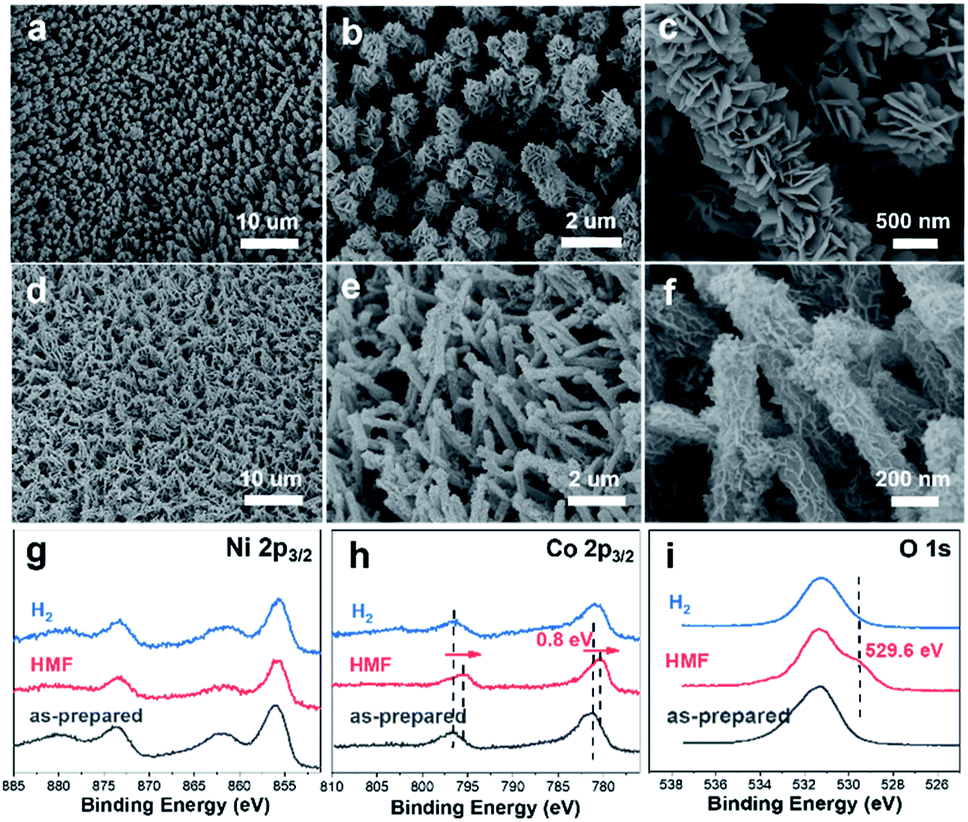 | ||
| Fig. 6 SEM images of CuxS@Ni0.75Co0.25OmHn after catalyzing long-term HMF oxidation (a–c) and HER (d–f). Ni 2p3/2 (g), Co 2p3/2 (h) and O 1s (i) XPS spectra of CuxS@Ni0.75Co0.25OmHn after catalyzing long-term HMF oxidation (labeled as ‘HMF’) and hydrogen evolution reaction (labeled as ‘H2’). | ||
4. Conclusions
To summarize, CuxS@NiCo-LDHs nanoarrays are facilely fabricated as efficient bifunctional electrocatalysts towards HMF oxidation and H2 generation. Owing to the superior conductivity of the copper sulfide core, the modulated electronic structure, the redox behavior of Ni in the catalyst layer and the nanowire@nanosheet open structure which greatly facilitates the mass transfer and exposes a large number of active sites, the optimized catalysts exhibit superior electrocatalytic activity/durability towards HMF oxidation and H2 evolution. Moreover, the faradaic efficiency towards FDCA and H2 is close to unity. With the outstanding bifunctionality, the two-electrode electrolyzer only requires a voltage of 1.34 V to achieve simultaneous biomass upgrading and H2 generation at the current density of 10 mA cm−2. Our study significantly advances research towards Ni-catalyzed biomass upgrading and more efficient H2 production from aqueous solution.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (No. 21905181 and 21975163) and China Postdoctoral Science Foundation (No. 2018M633125). The authors acknowledge Dr Wenguang Zhao from Peking University, Shenzhen Graduate School for the TEM analysis. We are also grateful to Dr Lin Lin and Materials Characterization & Preparation Center of Southern University of Science and Technology for HPLC measurements.Notes and references
- L. Han, S. J. Dong and E. K. Wang, Adv. Mater., 2016, 28, 9266–9291 CrossRef CAS PubMed.
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem., 2017, 1, 1–13 CrossRef.
- T. W. Kim and K. S. Choi, Science, 2014, 343, 990–994 CrossRef CAS PubMed.
- F. Yu, L. Yu, I. K. Mishra, Y. Yu, Z. F. Ren and H. Q. Zhou, Materials Today Physics, 2018, 7, 121–138 CrossRef.
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS PubMed.
- B. You, X. Liu, N. Jiang and Y. Sun, J. Am. Chem. Soc., 2016, 138, 13639–13646 CrossRef CAS PubMed.
- Y. Huang, X. D. Chong, C. B. Liu, Y. Liang and B. Zhang, Angew. Chem., Int. Ed., 2018, 57, 13163–13166 CrossRef CAS PubMed.
- L. Du, Y. Y. Shao, J. M. Sun, G. P. Yin, C. Y. Du and Y. Wang, Catal. Sci. Technol., 2018, 8, 3216–3232 RSC.
- J.-Y. Zhang, H. Wang, Y. Tian, Y. Yan, Q. Xue, T. He, H. Liu, C. Wang, Y. Chen and B. Y. Xia, Angew. Chem., Int. Ed., 2018, 57, 7649–7653 CrossRef CAS PubMed.
- G.-F. Chen, Y. Luo, L.-X. Ding and H. Wang, ACS Catal., 2018, 8, 526–530 CrossRef CAS.
- Q. Zhou, Z. Shen, C. Zhu, J. Li, Z. Ding, P. Wang, F. Pan, Z. Zhang, H. Ma, S. Wang and H. Zhang, Adv. Mater., 2018, 1800140 CrossRef PubMed.
- G. Wang and Z. Wen, Nanoscale, 2018, 10, 21087–21095 RSC.
- B. You, N. Jiang, X. Liu and Y. Sun, Angew. Chem., Int. Ed., 2016, 55, 9913–9917 CrossRef CAS PubMed.
- N. Jiang, B. You, R. Boonstra, I. M. Terrero Rodriguez and Y. Sun, ACS Energy Lett., 2016, 1, 386–390 CrossRef CAS.
- B. J. Taitt, D.-H. Nam and K.-S. Choi, ACS Catal., 2019, 9, 660–670 CrossRef CAS.
- S. R. Kubota and K. S. Choi, ChemSusChem, 2018, 11, 2138–2145 CrossRef CAS.
- W. J. Liu, L. N. Dang, Z. R. Xu, H. Q. Yu, S. Jin and G. W. Huber, ACS Catal., 2018, 8, 5533–5541 CrossRef CAS.
- M. E. Zakrzewska, E. Bogel-Łukasik and R. Bogel-Łukasik, Chem. Rev., 2011, 111, 397–417 CrossRef CAS PubMed.
- H. A. Rass, N. Essayem and M. Besson, ChemSusChem, 2015, 8, 1206–1217 CrossRef.
- B. Siyo, M. Schneider, J. Radnik, M. M. Pohl, P. Langer and N. Steinfeldt, Appl. Catal., A, 2014, 478, 107–116 CrossRef CAS.
- H. G. Cha and K. S. Choi, Nat. Chem., 2015, 7, 328–333 CrossRef CAS.
- M. Fleischmann, K. Korinek and D. J. Pletcher, J. Chem. Soc., Perkin Trans. 2, 1972,(10), 1396–1403 RSC.
- M. Fleischmann, K. Korinek and D. J. Pletcher, J. Electroanal. Chem. Interfacial Electrochem., 1971, 31, 39–49 CrossRef CAS.
- S. Barwe, J. Weidner, S. Cychy, D. M. Morales, S. Dieckhöfer, D. Hiltrop, J. Masa, M. Muhler and W. Schuhmann, Angew. Chem., Int. Ed., 2018, 57, 11460–11464 CrossRef CAS.
- Q. H. Zhao, J. L. Yang, M. Q. Liu, R. Wang, G. X. Zhang, H. Wang, H. T. Tang, C. K. Liu, Z. W. Mei, H. B. Chen and F. Pan, ACS Catal., 2018, 8, 5621–5629 CrossRef CAS.
- Y. Zhao, X. Zhang, X. Jia, G. I. N. Waterhouse, R. Shi, X. Zhang, F. Zhan, Y. Tao, L.-Z. Wu, C.-H. Tung, D. O'Hare and T. Zhang, Adv. Energy Mater., 2018, 8, 1703585 CrossRef.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- X. H. Xia, C. R. Zhu, J. S. Luo, Z. Y. Zeng, C. Guan, C. F. Ng, H. Zhang and H. J. Fan, Small, 2014, 10, 766–773 CrossRef CAS.
- M. S. Burke, L. J. Enman, A. S. Batchellor, S. H. Zou and S. W. Boettcher, Chem. Mater., 2015, 27, 7549–7558 CrossRef CAS.
- B. Liu, S. X. Qu, Y. Kou, Z. Liu, X. Chen, Y. T. Wu, X. P. Han, Y. D. Deng, W. B. Hu and C. Zhong, ACS Appl. Mater. Interfaces, 2018, 10, 30433–30440 CrossRef CAS PubMed.
- L. L. Feng, M. H. Fan, Y. Y. Wu, Y. P. Liu, G. D. Li, H. Chen, W. Chen, D. J. Wang and X. X. Zou, J. Mater. Chem. A, 2016, 4, 6860–6867 RSC.
- X. Long, G. X. Li, Z. L. Wang, H. Y. Zhu, T. Zhang, S. Xiao, W. Y. Guo and S. H. Yang, J. Am. Chem. Soc., 2015, 137, 11900–11903 CrossRef CAS PubMed.
- L. Yu, H. Q. Zhou, J. Y. Sun, F. Qin, F. Yu, J. M. Bao, Y. Yu, S. Chen and Z. F. Ren, Energy Environ. Sci., 2017, 10, 1820–1827 RSC.
- J. Hou, Y. Wu, B. Zhang, S. Cao, Z. Li and L. Sun, Adv. Funct. Mater., 2019, 1808367 CrossRef.
- J. Weidner, S. Barwe, K. Sliozberg, S. Piontek, J. Masa, U. P. Apfel and W. Schuhmann, Beilstein J. Org. Chem., 2018, 14, 1436–1445 CrossRef PubMed.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS PubMed.
- Z.-Y. Yu, C.-C. Lang, M.-R. Gao, Y. Chen, Q.-Q. Fu, Y. Duan and S.-H. Yu, Energy Environ. Sci., 2018, 11, 1890–1897 RSC.
- X. H. Deng, S. Ozturk, C. Weidenthaler and H. Tuysuz, ACS Appl. Mater. Interfaces, 2017, 9, 21225–21233 CrossRef CAS PubMed.
- N. S. McIntyre and M. G. Cook, Anal. Chem., 1975, 47, 2208–2213 CrossRef CAS.
- A. P. Grosvenor, M. C. Biesinger, R. S. Smart and N. S. McIntyre, Surf. Sci., 2006, 600, 1771–1779 CrossRef CAS.
- S. H. Ye, Z. X. Shi, J. X. Feng, Y. X. Tong and G. R. Li, Angew. Chem., Int. Ed., 2018, 57, 2672–2676 CrossRef CAS.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS.
- Y. Zhao, X. Jia, G. Chen, L. Shang, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung, D. O'Hare and T. Zhang, J. Am. Chem. Soc., 2016, 138, 6517–6524 CrossRef CAS.
- J. Ren, K. H. Song, Z. Li, Q. Wang, J. Li, Y. X. Wang, D. B. Li and C. K. Kim, Appl. Surf. Sci., 2018, 456, 174–183 CrossRef CAS.
- H. Y. Wang, Y. Y. Hsu, R. Chen, T. S. Chan, H. M. Chen and B. Liu, Adv. Energy Mater., 2015, 5, 1500091 CrossRef.
- M. K. Bates, Q. Jia, H. Doan, W. Liang and S. Mukerjee, ACS Catal., 2016, 6, 155–161 CrossRef CAS.
- M. B. Stevens, L. J. Enman, E. H. Korkus, J. Zaffran, C. D. M. Trang, J. Asbury, M. G. Kast, M. C. Toroker and S. W. Boettcher, Nano Res., 2019, 12, 2288–2295 CrossRef CAS.
- F. Yu, H. Zhou, Y. Huang, J. Sun, F. Qin, J. Bao, W. A. GoddardIII, S. Chen and Z. Ren, Nat. Commun., 2018, 9, 2551 CrossRef.
- J. Sun, M. Ren, L. Yu, Z. Yang, L. Xie, F. Tian, Y. Yu, Z. Ren, S. Chen and H. Zhou, Small, 2019, 15, 1804272 CrossRef.
- J. Zhang, T. Wang, P. Liu, Z. Liao, S. Liu, X. Zhuang, M. Chen, E. Zschech and X. Feng, Nat. Commun., 2017, 8, 15437 CrossRef CAS PubMed.
- X. Wang, Y. Yang, L. Diao, Y. Tang, F. He, E. Liu, C. He, C. Shi, J. Li, J. Sha, S. Ji, P. Zhang, L. Ma and N. Zhao, ACS Appl. Mater. Interfaces, 2018, 10, 35145–35153 CrossRef CAS PubMed.
- Y. Zhou, Z. Wang, Z. Pan, L. Liu, J. Xi, X. Luo and Y. Shen, Adv. Mater., 2019, 31, 18067690 Search PubMed.
- R. Chen, C. J. Yang, W. Z. Cai, H. Y. Wang, J. W. Miao, L. P. Zhang, S. L. Chen and B. Liu, ACS Energy Lett., 2017, 2, 1070–1075 CrossRef CAS.
- Y. Liu, Q. Li, R. Si, G.-D. Li, W. Li, D.-P. Liu, D. Wang, L. Sun, Y. Zhang and X. X. Zou, Adv. Mater., 2017, 29, 1606200 CrossRef PubMed.
- J.-X. Feng, J.-Q. Wu, Y.-X. Tong and G.-R. Li, J. Am. Chem. Soc., 2018, 140, 610–617 CrossRef CAS PubMed.
- M. Gao, W. Sheng, Z. Zhuang, Q. Fang, S. Gu, J. Jiang and Y. Yan, J. Am. Chem. Soc., 2014, 136, 7077–7084 CrossRef CAS PubMed.
- F. Song and X. Hu, Nat. Commun., 2014, 5, 4477 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ta06917h |
| This journal is © The Royal Society of Chemistry 2020 |