
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProtease-responsive mass barcoded nanotranslators for simultaneously quantifying the intracellular activity of cascaded caspases in apoptosis pathways†
Hongmei
Xu
,
Xiaodan
Huang
,
Zhenzhen
Zhang
,
Xuemeng
Zhang
,
Qianhao
Min
 * and
Jun-Jie
Zhu
* and
Jun-Jie
Zhu
State Key Laboratory of Analytical Chemistry for Life Science, Chemistry and Biomedicine Innovation Center, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210023, P. R. China. E-mail: minqianhao@nju.edu.cn
First published on 4th May 2020
Abstract
Quantitatively delineating the activation network of multiple proteases that participate in cellular processes is highly essential for understanding the physiological and pathological states of cells. In this study, protease-responsive mass barcoded nanotranslators (PRMNTs) were engineered for revealing the activity of cascaded caspases in apoptosis in a multiplex and quantitative manner. In the PRMNTs, a series of mass tag-decorated gold nanoparticles (AuNPs) were tethered onto magnetic Fe3O4 nanospheres via a linker containing the substrate peptide of the target protease to form a “one-to-many” core–satellite structure. This nanostructure was internalized into the cells, underwent an enzymatic reaction within the cells, and allowed post-reaction mass spectrometry (MS) interrogation after magnetic separation from the cells. In the presence of intracellular caspases, enzymatic cleavage of the linker could be translated to the decreased ion signals of the mass tags on the remaining AuNPs in the PRMNTs by MS decoding. Benefiting from the multiplexing capability of MS, the intracellular activity of caspase-3, -8 and -9 that orchestrate the apoptotic process was simultaneously quantified at any given time. Kinetic analysis of caspase activity under stimulation of diverse anticancer drugs revealed that programmed cell death followed individual apoptosis pathways, differing in the activation degree and sequence of the caspase cascade. This work represents a modality that interfaces nanotechnology with MS for quantitatively probing the intracellular activity of multiple proteases, which opens up new avenues for revealing the apoptosis mechanism and developing innovative drugs.
Introduction
Apoptosis is a genetically directed suicide program occurring in all animal cells, and is triggered, executed and regulated by the activation of a family of cysteine proteases designated as caspases.1,2 It is generally accepted that there are two distinct apoptosis pathways involving caspase activation.3–5 One is the death receptor pathway, in which ligation with death receptors can trigger the autoproteolysis of caspase-8 and initiate the subsequent interaction with adapter molecules.4 The other is the mitochondrial pathway that releases the cytochrome C to trigger the formation of an apoptosome complex and promote the activation of caspase-9.6 The activated initiator caspases, such as caspase-8 and caspase-9, can in turn activate caspase-3 or other effector caspases to induce cell death. Given the participation of multiple caspases in the apoptosis signaling network, many anti-cancer drugs have been developed to target them to potentiate therapeutic effects.7,8 More importantly, their activation reveals typical pro-apoptotic mechanisms which also constitute a fundamental part of drug research and development.9 Therefore, simultaneously assaying the activity of cascaded caspases is extremely necessary for both understanding the apoptosis pathways and developing target therapeutics.To date, various strategies including colorimetric assays,10 electrochemical methods,11 fluorescence techniques12,13 and surface-enhanced Raman scattering (SERS)14 have emerged as potent tools for the detection of active caspases. Specifically, by virtue of real-time visualization and high sensitivity, a series of fluorescence probes equipped with substrate peptides have been applied in imaging the activated caspases in cell apoptosis.15–18 However, the band overlap of fluorescent chromophores and the susceptibility of fluorescence to intracellular environmental changes limited their applications in quantitatively profiling the cascaded caspases with signal multiplexing and high accuracy.19 Characterized by high mass resolution and outstanding qualitative and quantitative precision, the mass spectrometry (MS)-based strategy may be able to overcome the above limitation in multiplex protease assays.20–23 Since MS is an invasive analytical method for characterizing living cells, thus far most MS-based approaches towards enzyme assay have been restricted to cell extracts, rather than the intracellular physiological environment.20,23–25 Due to the difference in proteolytic reaction conditions, the activity of proteases measured in cell lysates cannot reveal their actual function and behavior in cells. A proteomics experiment could profile cellular proteolytic processing events at the level of neo-N-terminal peptides, but tedious pretreatment procedures and complicated data analysis are always unavoidable.26–28 Thus, it is urgently necessary to develop a simple and multiplex quantitative MS-based approach that can truly reflect the intracellular activity of proteases.
Mass barcodes, a series of small tag molecules, featuring similar mass fingerprints and high ionization efficiency in MS, have been widely employed to label nanoparticles for the detection of multiple disease-associated biomolecules.29–34 In general, mass barcoded nanoprobes specifically bound with target biomolecules (e.g. oligonucleotides, thrombin, membrane proteins, and glycans on cell surfaces) that were pre-anchored on a chip or a plate convert biological recognition events to the amplified ion signals of mass tags upon ionization.29–31,35,36 Inspired by chip or plate-based mass barcoding strategies, we consider the possibility of transporting mass barcoded nanoscale assay platforms into cells, which may offer the opportunity to gain an insight into cellular processes from the perspective of intracellular bioactive molecules. In this sense, these mass barcoded nanoplatforms would be the feasible solution to study the intracellular functions of biomolecules by follow-up MS analysis.
Herein, protease-responsive mass barcoded nanotranslators (PRMNTs) with a core–satellite structure were developed to convert protease activity to MS signals for assaying intracellular cascaded caspases in cell apoptosis by matrix assisted laser desorption/ionization mass spectrometry (MALDI MS) (Scheme 1). The core–satellite PRMNTs were fabricated by coupling streptavidin-capped magnetic Fe3O4 nanospheres (Fe3O4@strep) with satellite gold nanoparticles co-decorated with mass tags (MTs) and biotinylated polyethylene glycol (PEG)–substrate peptide conjugates (AuNPs@Con-MT) (Scheme 1a). After being internalized into cells, the PRMNTs underwent enzymatic reactions catalysed by multiple caspases, resulting in cleavage of the substrate peptide linkers and consequent release of the corresponding AuNPs@Con-MT. Since the MTs can detach from the satellite AuNPs to generate ion signals upon laser ablation, the degree of intracellular proteolytic reactions could be reflected by the varied MALDI MS signal intensity of ionized MTs from the PRMNTs. To obtain the protease activity in a quantitative format, AuNPs modified with HS-PEG-biotin and internal standard mass tags (AuNPs@PEG-IS) were additionally incorporated into the PRMNTs to generate the internal standard ion signal. The activities of multiple caspases involved in apoptosis pathways were accurately translated to the decreased ion intensity ratios of MTs to IS on the PRMNTs (Scheme 1b). In this manner, the quantitative information about the intracellular activity of apoptosis-associated caspases could be precisely imprinted on the mass barcoded nanotranslators in cells and quickly retrieved by MS decoding after rapid magnetic separation. Moreover, the proposed nanotranslators were further applied to explore the mechanism of apoptotic cell death induced by diverse anti-cancer drugs based on the kinetic analysis of activated caspases, thus paving the way for deciphering apoptosis signaling pathways and screening therapeutic strategies.
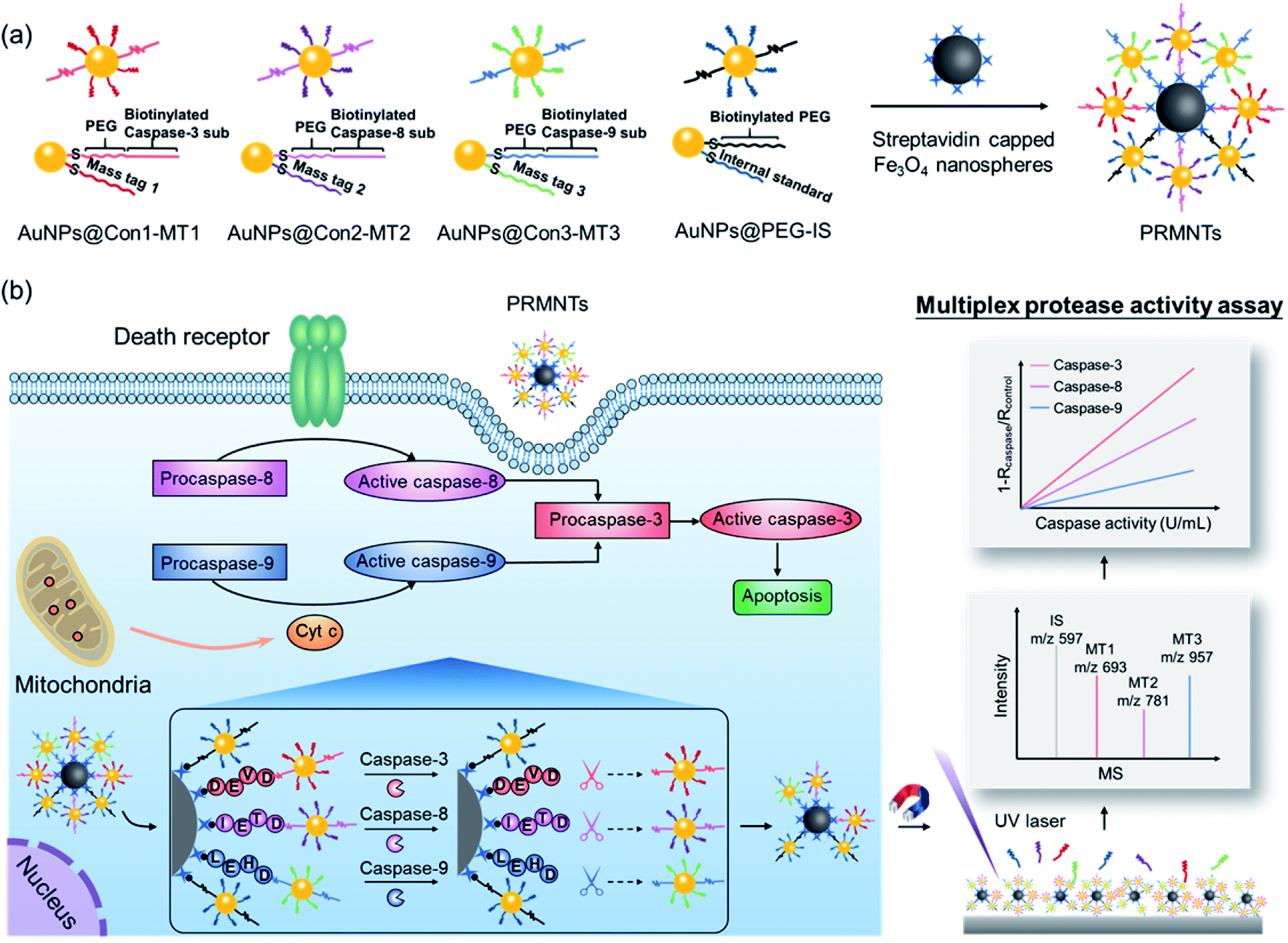 | ||
| Scheme 1 Schematic design of the core–satellite PRMNTs for activity assay of intracellular caspases in apoptosis. (a) Preparation of the PRMNTs by coupling streptavidin capped magnetic Fe3O4 nanospheres with a series of satellite AuNPs co-decorated with mass tags and biotinylated linkers. (b) The principle of quantifying the intracellular activity of cascaded caspases in apoptosis pathways by using the core–satellite PRMNTs. | ||
Results and discussion
Preparation and characterization of the core–satellite PRMNTs
In principle, the core–satellite PRMNTs were constructed by coupling streptavidin-modified Fe3O4 nanospheres with mass tag-grafted AuNPs using the substrate peptides of target proteases as the linker. Amine-functionalized magnetic cores with a particle size of around 195 nm were obtained using a solvothermal reaction (Fig. S1a and b†), followed by surface activation with glutaraldehyde for streptavidin conjugation (Fig. S1c and d†). In our experiments, a series of molecular homologues HS-(CH2)11(OCH2CH2)nOH (MT1: HS-(CH2)11(OCH2CH2)3OH, MT2: HS-(CH2)11(OCH2CH2)4OH, MT3: HS-(CH2)11(OCH2CH2)6OH) were employed as the mass tags due to their ideal ionization efficiency. Meanwhile, three biotinylated HS-PEG-substrate conjugates (denoted as biotin-sub-PEG-SH, including Con1: biotin-GGDEVDGKK-PEG-SH, Con2: biotin-GGIETDGKK-PEG-SH, and Con3: biotin-GGLEHDGKK-PEG-SH) respectively responsive to caspase-3, -8 and -9 were prepared and verified by MALDI MS (Fig. S2†). Thereafter, the satellite AuNPs (named AuNPs@Con1-MT1, AuNPs@Con2-MT2, and AuNPs@Con3-MT3, shown in Scheme 1a) were constructed by co-assembling thiol-terminated mass tags and biotin-sub-PEG-SH onto the surface of 13 nm AuNPs (Fig. S3a†). The surface assembly with these thiols was also proved by the increased hydrodynamic size of AuNPs (Fig. S3b†). In addition, internal standard satellite AuNPs (AuNPs@PEG-IS) were built by adopting HS-PEG-biotin as the enzymatically inert linker and mercaptohexadecanoic acid (HS-(CH2)15-COOH) as the internal standard mass tag. Both the photographs and UV-vis absorption spectra demonstrated that the four types of satellite AuNPs showed good stability in both water and PBS, in comparison with the aggregation of undecorated AuNPs in PBS (Fig. S3c and d†). Furthermore, the signal output of the satellite AuNPs was checked by MALDI MS. In comparison with 1,5-diaminonaphthalene (1,5-DAN), 2,5-dihydroxybenzoic acid (DHB) was selected as matrix substance for better sensitivity and less interference in the low mass range (Fig. S4†). As shown in Fig. S5,† the distinct MS peaks at m/z 597, 693, 781 and 957 corresponding to [2IS − H2 + Na]+, [2MT1 − H2 + Na]+, [2MT2 − H2 + Na]+ and [2MT3 − H2 + Na]+ were respectively assigned as the signature of AuNPs@PEG-IS, AuNPs@Con1-MT1, AuNPs@Con2-MT2, and AuNPs@Con3-MT3, further corroborating the existence of mass tags on the satellite AuNPs. The core–satellite PRMNTs were obtained by coupling Fe3O4@strep with the satellite AuNPs in a “one-to-four” format via biotin–streptavidin interaction. As illustrated in Fig. 1a and b, the PRMNTs were characterized as core–satellite nanostructures with AuNPs evenly distributed on the surface of Fe3O4 nanospheres. The stepwise decreasing zeta potential and gradually increasing average hydrodynamic diameter also evidenced the coating of streptavidin and subsequent satellite AuNPs on the magnetic cores (Fig. 1c). In the PRMNTs, streptavidin and the flexible PEG–peptide linker between Fe3O4 nanospheres and AuNPs could create a dynamic gap for the access of caspases, thus allowing the subsequent caspase-mediated peptide cleavage. The magnetic hysteresis loops were measured and the saturation magnetization of the PRMNTs was calculated to be 74.4 emu g−1 (Fig. 1d), which would be contributing to the quick magnetic separation from the cell lysates for MS analysis. According to the MALDI mass spectra of PRMNTs, the ion signals from MT1, MT2, MT3 and IS appeared at the mass locations identical with those obtained from the mixture of individual satellite AuNPs (Fig. 1c and d), thus confirming the inclusion of the four types of satellite AuNPs in the PRMNTs. The good reproducibility of the PRMNTs was also proved by the low relative standard deviation (<10%) of the peak intensity ratio of MTs to IS from batch to batch (Fig. S6 and Table S1†). To verify the connection between magnetic cores and satellite AuNPs arising from specific biotin–streptavidin interaction, we prepared the satellite AuNPs without biotin-sub-PEG-SH (AuNPs@MT1, AuNPs@MT2, and AuNPs@MT3) to investigate their adsorption behavior on Fe3O4@strep. It was found that the absence of biotin-sub-PEG-SH led to the disappearance of signature MS peaks from the three corresponding mass tags (Fig. S7†), implying that biotin-sub-PEG-SH is the determining factor in the linkage to form the core–satellite PRMNTs. To further investigate the stability of PRMNTs in PBS, we incubated PRMNTs-Casp-3 in PBS for one week and monitored the MS peak intensity of MT1 and IS every day. As shown in Fig. S8,† the ion signals of each MT demonstrated no obvious variation, manifesting ignorable degradation during storage. Besides, the non-specific adsorption of ligands on PRMNTs and satellite AuNPs was minimized by repeated washing procedures and checked by MALDI MS (Fig. S9, Tables S2 and S3†).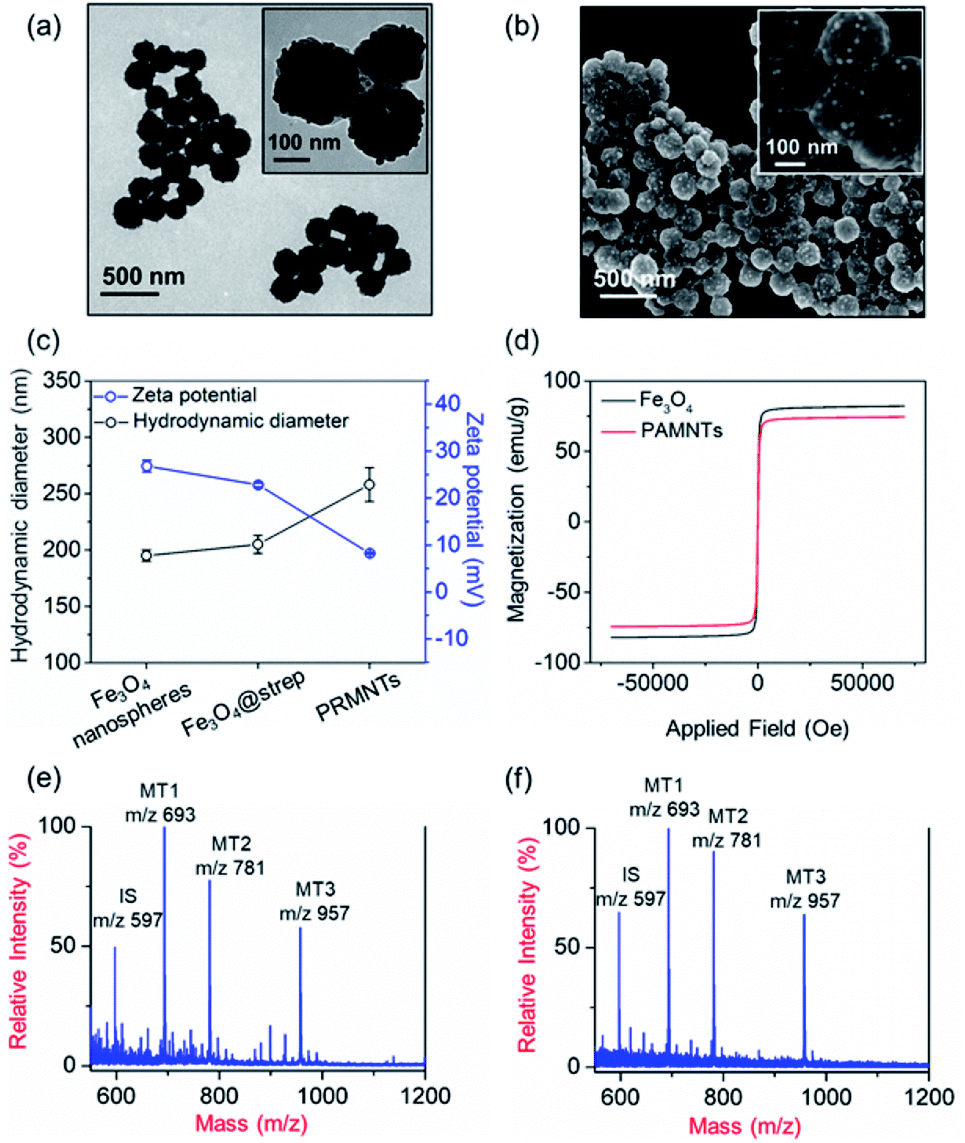 | ||
| Fig. 1 Characterization of the core–satellite PRMNTs. (a) TEM and (b) SEM images of the core–satellite PRMNTs. Insets show the high resolution TEM image and magnified SEM image. (c) The variations in hydrodynamic size distribution and zeta potential during the assembly of PRMNTs. (d) The magnetic hysteresis loops of Fe3O4 nanospheres and PRMNTs. MALDI mass spectra of mixed satellite AuNPs (e) and intact core–satellite PRMNTs (f). m/z 597: [2IS − H2 + Na]+, m/z 693: [2MT1 − H2 + Na]+, m/z 781: [2MT2 − H2 + Na]+, and m/z 957: [2MT3 − H2 + Na]+. | ||
Optimization of the core–satellite PRMNTs for caspase activity assay
To ensure high responsiveness to the target proteases, several parameters in the construction of the core–satellite PRMNTs have been systematically optimized. The PRMNTs specific for caspase-3 (PRMNTs-Casp-3) were fabricated by assembling AuNPs@Con1-MT1 and AuNPs@PEG-IS onto Fe3O4@strep and employed as the model system to perform the optimization. To maximize the loading of satellite AuNPs, the concentration of streptavidin for Fe3O4 nanosphere decoration was adjusted to 140 μg mL−1, in which case the adsorption of streptavidin reached saturation (Fig. S10†). The coverage density of MT1 and Con1 on satellite AuNPs was speculated to exert great influence on the responsiveness and sensitivity of caspase-3 activity assay. Excessive MT1 could negatively affect the binding efficiency of satellite AuNPs to the magnetic cores, while insufficient MT1 modification would weaken the effect of signal conversion and amplification. As shown in Fig. S11 and S12,† the response to caspase-3 was defined as the variation of the MS peak intensity ratio of MT1 to IS. The response value peaked when the concentration ratio of MT1 to Con1 was adjusted to 0.3 (Fig. S11†), and their optimal dosages were determined to be 24 μM and 80 μM (Fig. S12c†). The influence of the AuNP size on the responsiveness was also investigated. As shown in Fig. S12d,† the optimal response to caspase-3 was achieved upon using 13 nm AuNPs, which can be ascribed to the sufficient coverage density of MT1 and Con1 and the resultant adequate satellite AuNPs linked to Fe3O4 nanospheres. Under the optimized decoration conditions, it was seen that there were about 119 Con1 and 442 MT1 molecules immobilized on each single 13 nm AuNP (described in Fig. S13†). Furthermore, the concentration ratio of AuNPs@Con1-MT1 to AuNPs@PEG-IS for the assembly of core–satellite nanostructures was also finely tuned. As shown in Fig. S14,† the response to caspase-3 was progressively elevated by increasing this ratio until the response value reached its maximum at 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3. Consequently, the optimized parameters for ratios of MT1 to Con1 on satellite AuNPs and AuNPs@Con1-MT1 to AuNPs@PEG-IS on the PRMNTs were fixed and applied in the following experiments.
:3. Consequently, the optimized parameters for ratios of MT1 to Con1 on satellite AuNPs and AuNPs@Con1-MT1 to AuNPs@PEG-IS on the PRMNTs were fixed and applied in the following experiments.
Quantitative evaluation of the activity of caspase-3
To demonstrate the applicability of the PRMNTs to activity quantification of caspases, the PRMNTs-Casp-3 were first utilized to assess the caspase-3 activity, in which the MS peak intensity ratio of MT1 to IS was appointed as the quantitative index. As shown in Fig. 2a, after incubation with 25 U mL−1 caspase-3 for 1.5 h, a dramatic decrease was observed in the MS peak intensity of MT1 to IS, thus reflecting the successful detachment of AuNPs@Con1-MT1 from the PRMNTs-Casp-3 owing to the cleavage of the substrate mediated by caspase-3. The caspase-triggered dissociation of the core–satellite structure was also verified by the TEM and SEM images (Fig. S15†). Compared to the initial PRMNTs-Casp-3 without treatment, a significant reduction in the loading of satellite AuNPs was observed upon caspase-3 treatment, along with the appearance of purple gold colloids in the supernatant (Fig. S15†). The proteolysis of the substrate peptide linker was further confirmed by the MALDI mass spectra (Fig. S16†), which ensured that the prepared PRMNTs-Casp-3 showed specific responsiveness to caspase-3. To accurately quantify the activity of cellular activated caspase-3, the calibration curve was plotted by recording the relative variation in the intensity ratio (1 − Rcaspase-3/Rcontrol) with different concentrations of caspase-3. As illustrated in Fig. 2b, the relative variation in the intensity ratio was proportional to the activity of caspase-3 ranging from 2.5 to 30 U mL−1, and the regression equation was represented as 1 − Rcaspase-3/Rcontrol = 1.54Acaspase-3 − 1.98 with an R-squared value of 0.996, where Acaspase-3 represents the activity of caspase-3.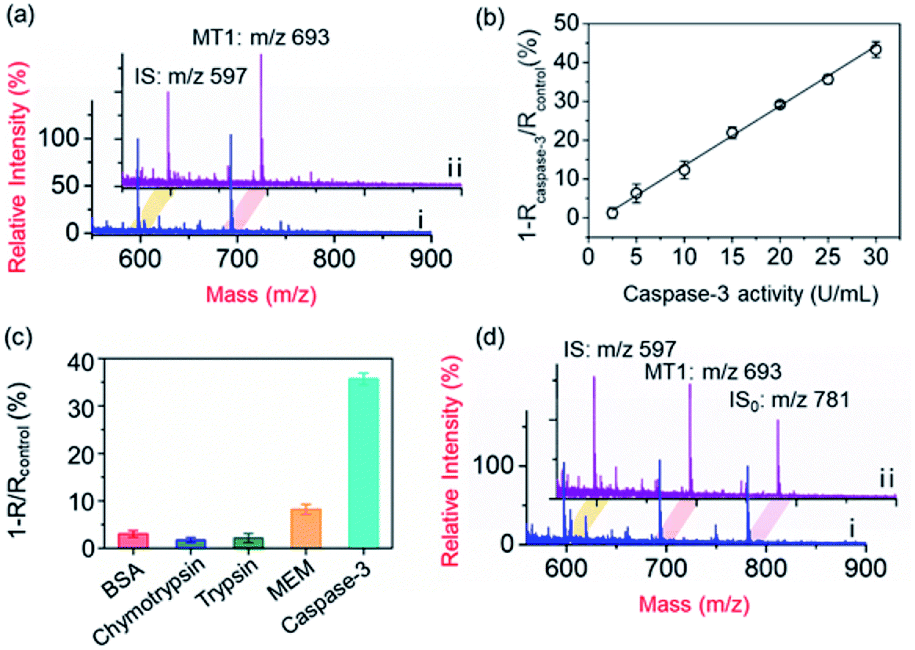 | ||
| Fig. 2 Quantification of caspase-3 activity. (a) MALDI mass spectra of the PRMNTs-Casp-3 incubated with 25 U mL−1 caspase-3 (i) or the blank solution (ii) at 37 °C for 1.5 h. (b) The linear correlation between the relative variation of the intensity ratio and caspase-3 activity. The peak intensity ratio of MT1 to IS of the PRMNTs-Casp-3 incubated with caspase-3 was denoted as Rcaspase-3, and that incubated with the blank solution was denoted as Rcontrol. (c) The relative variation of the intensity ratio (1 − R/Rcontrol) in PRMNTs-Casp-3 after incubation with 25 U mL−1 caspase-3 for 1.5 h or 4 mM GSH, 50 μg mL−1 BSA, 10 μg mL−1 chymotrypsin, 10 μg mL−1 trypsin or MEM buffer for 12 h. (d) MALDI mass spectra of the PRMNTs-Casp-3 with (i) or without (ii) incubation of 4 mM GSH at 37 °C for 12 h. Just before the sample loading for MALDI MS analysis, AuNPs@MT2 (ion signal denoted as IS0) were added as the reference to assess the relative ion signals of MT1 and IS. | ||
Considering the feasibility of the nanotranslators under intracellular conditions, the specificity of PRMNTs-Casp-3 was investigated by incubating the nanotranslators in 25 U mL−1 caspase-3 or other matrices that simulated cytoplasmic and lysosomal environments. The results shown in Fig. 2c reveal that only caspase-3 could induce a distinct increase in the relative variation of the intensity ratio, affirming the high specificity of the nanotranslators to the target caspase. In these nanotranslators, another merit of incorporating AuNPs@PEG-IS is to minimize the interference from high-level cellular biothiols. These nonprotein thiols may substitute the thiol-terminated species including mass tags and PEG–substrate conjugates on AuNPs, thus lowering the ion signal output. It was observed in Fig. 2d and Table S4† that the respective MS peak intensity of MT1 and IS saw a decrease by one third after the treatment with 4 mM GSH, similar to the intracellular reductive environments, while the intensity ratio of MT1 to IS remained almost constant during this process. Thus, the signal loss in quantification resulting from the detachment of thiol-terminated species could be circumvented by additionally loading AuNPs@PEG-IS on the PRMNTs-Casp-3.
Quantitative caspase-3 activity assay in HeLa cells
Our aim is to truly profile the protease activity in intracellular milieu by locating the PRMNTs in live cells. So first of all, the introduction of PRMNTs should ideally have the least effect possible on cellular functions and processes. Accordingly, the cytotoxicity of the PRMNTs-Casp-3 and satellite AuNPs was evaluated by the MTT assay. As shown in Fig. S17a and b,† both PRMNTs and satellite AuNPs showed low cytotoxicity within the concentration range used in this study. For profiling the intracellular activity of caspase-3, the dosage of PRMNTs was finely tuned so that the cellular uptake of the nanotranslators measured by ICP-MS could be consistent with that used to establish the calibration curve for in vitro protease activity assay. Therefore, the concentration of PRMNTs was fixed at 40 μg mL−1 in cell incubation for the determination of intracellular protease activity (Fig. S18†).In our design, the core–satellite nanostructures were anticipated to be disassembled within the cells containing activated caspases. To verify this process, bio-TEM was used to monitor the morphological variation of the PRMNTs in the cytoplasm after internalization into HeLa cells. As illustrated in Fig. 3a, the core–satellite structure of PRMNTs-Casp-3 was maintained in the cells without any stimulation, and few AuNPs were detached from the Fe3O4 nanospheres. In comparison, after introducing 2 μM staurosporine (STS) to induce apoptosis via caspase-3 activation, the core–satellite topology of PRMNTs-Casp-3 was observed to be broken up, with a mass of satellite AuNPs being liberated from the magnetic cores (Fig. 3b). To confirm that the structure disintegration originated from the specific cleavage of the substrate, a caspase-3 inhibitor, N-acetyl-Asp-Glu-Val-Asp-CHO (Ac-DEVD-CHO), was pre-incubated with HeLa cells before stimulation by STS. As expected, the bio-TEM image showed only a small number of dissociated satellite AuNPs (Fig. 3c), proving that the detachment of AuNPs@Con1-MT1 from the PRMNTs-Casp-3 was mediated by STS-stimulated caspase-3 activation. Dark field microscopy (DFM) was used to inspect the distribution of PRMNTs in live cells. The magnetic PRMNTs were internalized and randomly distributed within the cells as single particles or few-particle aggregates, indicated with the bright scattering light in the DFM images (Fig. 3d). The bright field (BF) image in Fig. 3e reveals the co-localization of the single-particle or aggregated PRMNTs with the scattering spots in DFM images, implying that a considerable amount of PRMNTs could be internalized into live cells for the intracellular proteolytic reactions governed by caspases.
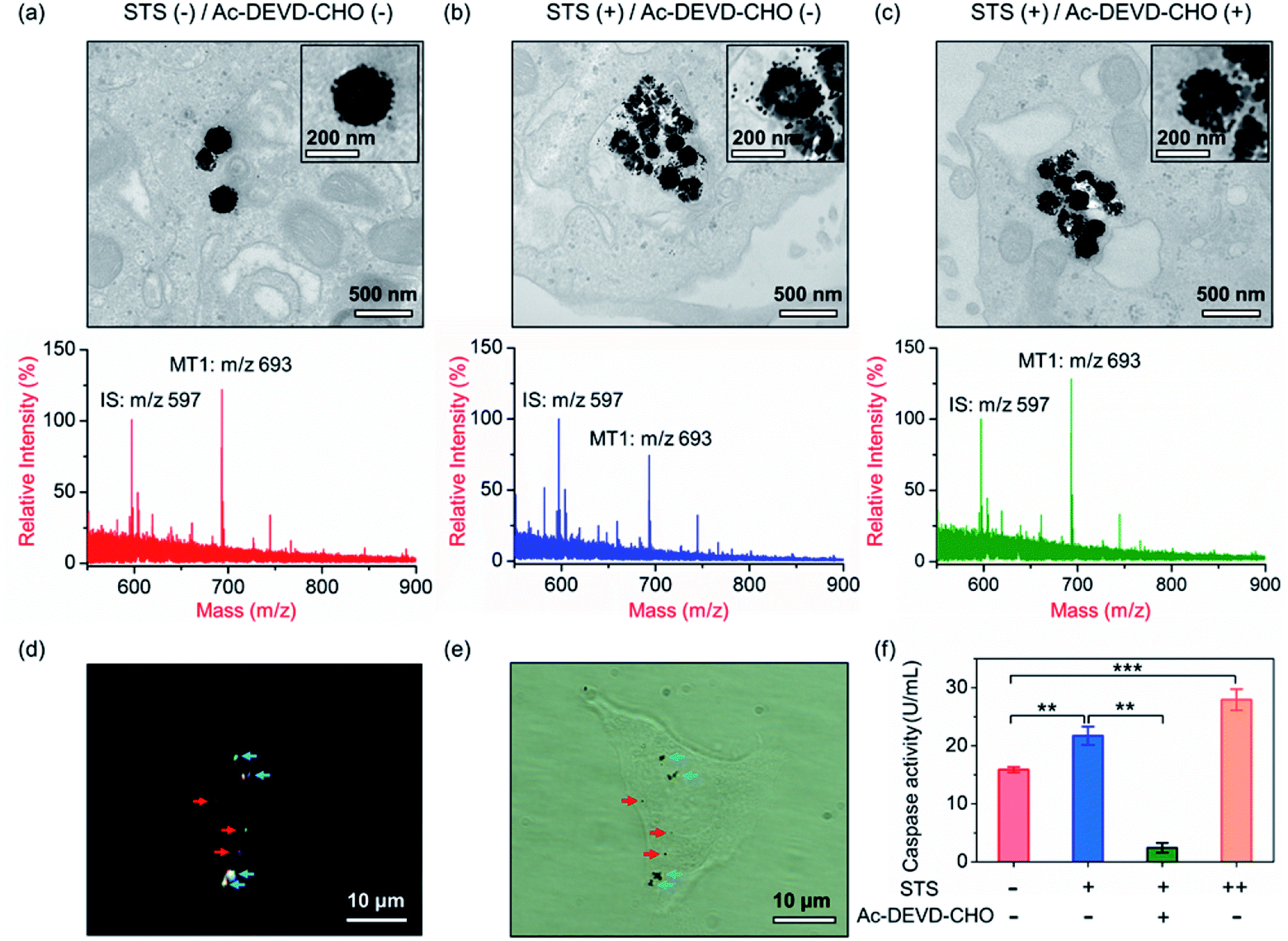 | ||
| Fig. 3 (a–c) Bio-TEM images and MALDI mass spectra of PRMNTs-Casp-3 internalized into HeLa cells with different stimulations. (a) Without stimulation; (b) 2 μM STS; (c) 2 μM STS and 100 μM Ac-DEVD-CHO. Insets are the magnified single-particle TEM images of PRMNTs-Casp-3. (d) DFM and (e) BF images of HeLa cells incubated with the PRMNTs for 12 h. Red arrows: single particles; blue arrows: few-particle aggregates. (f) Quantification of the activity of caspase-3 in HeLa cells with different stimulations. Statistical analysis: t-test (**p < 0.01, ***p < 0.001). | ||
Having verified that the dissociation of the core–satellite structures was induced by caspase-3 in live cells, we next attempted to decode the protease activity information carried on the PRMNTs-Casp-3 by MALDI MS. For caspase-3 activity assay, the engulfed PRMNTs-Casp-3 were recovered from the cells by magnetic separation immediately after the cells were lysed at a given time. The intracellular activity of caspase-3 was acquired by recording the MS peak intensity ratio of MT1 to IS of PRMNTs-Casp-3 with reference to the calibration curve. As displayed in the MALDI mass spectra of Fig. 3a and b, STS treatment resulted in an apparent decline in the intensity ratio of MT1 to IS as compared to those internalized into cells without stimulation. Caspase-3 activity calculated from the variation of ion signal ratios was also plotted in Fig. 3f, showing that caspase-3 got more activated upon incubation with 4 μM STS. Further addition of Ac-DEVD-CHO gave rise to an elevation in the MS intensity ratio of MT1 to IS due to the suppression of caspase-3 activity (Fig. 3c), corresponding to the observation that Ac-DEVD-CHO decelerated the dissociation process of the PRMNTs-Casp-3. The applicability of this approach in quantifying the intracellular activity of caspase-3 in other cell lines was further investigated. As shown in Fig. S19,† the activity of caspase-3 in MCF-7 cells under different treatments was accurately profiled, which showed a similar tendency to that detected in HeLa cells. All these data showed that the PRMNTs were able to effectively reflect the intracellular activity of proteases in cells via ion signal intensity ratios. Furthermore, the reliability of the PRMNT-based quantitative activity assay of caspase-3 was validated by comparison with a commercial activity assay kit. The activity of caspase-3 in the cell lysates was separately quantified by using the caspase-3 activity assay kit and the PRMNT-based approach. As listed in Table S5,† the relative deviation of the two quantitative methods was less than 15%, confirming the quantification accuracy of our proposed strategy.
Multiplex quantitative assessment of the activity of cascaded caspases
The activation of the caspase cascade has been proved to be involved in the development of diseases by regulating cellular signaling networks.9,37 Due to the mutual activation of caspases and crosstalk of signalling pathways, understanding the function of each caspase and exploring the activation of cascades show great significance in unraveling the process of programed cell death.38,39 In the extrinsic apoptosis pathway, the death-inducing signaling complex on the cell membrane recruits procaspase-8, resulting in the activation of caspase-3 and caspase-7.40 The intrinsic apoptosis pathway starts with the release of Cyt c from mitochondria, involves the recruitment and activation of procaspase-9, and finally converges with the extrinsic pathway at caspase-3 (Scheme 1b).41–43 Considering their crucial roles in apoptosis, caspase-3, -8 and -9 were employed to demonstrate the applicability of the PRMNTs in multiplex quantitative protease activity assay. Due to the overlapping consensus substrate sequences of caspases,16,44,45 the substantial cross-reactivity among caspases should be first minimized. As shown in Fig. S20,† when the loading concentration of the four satellite AuNPs (AuNPs@Con1-MT1, AuNPs@Con2-MT2, and AuNPs@Con3-MT3, and AuNPs@PEG-IS) was optimized to be 2:1:1:3, the MS signal outputs demonstrated a specific response in the presence of individual caspases. Based on this, the activities of caspase-3, -8 and -9 could be respectively represented by the varied intensity ratios of MT1, MT2, and MT3 to IS in a multi-channel mode. After treating the PRMNTs with mixed caspases, the MS peak intensity ratios respectively indicative of caspase activities decreased with different degrees (Fig. 4a), reflecting the cleavage of the multiple substrates on the PRMNTs by target caspases. The calibration curves were also plotted to quantify the activity of intrinsic caspases in HeLa cells. As illustrated in Fig. 4b, the relative variation of MS peak intensity ratios increased linearly with the increasing caspase activity, and the regression equations for caspase-3, -8 and -9 were represented as 1 − Rcaspase-3/Rcontrol = 2.25Acaspase-3 − 4.15, 1 − Rcaspase-8/Rcontrol = 1.42Acaspase-8 − 2.62, and 1 − Rcaspase-9/Rcontrol = 1.64Acaspase-9 − 0.46 with R-squared values over 0.99, respectively. Besides, the interference from other components including biothiols in the cellular matrices was also investigated. As shown in Fig. S21,† all the interfering agents only generated relative variations less than 10%, exerting negligible influence on the detection of the target caspases. To demonstrate the multiplex quantification accuracy and anti-interference ability of the proposed method, the activity of caspases in the cell lysates was determined by using the PRMNT-based mass spectrometric approach and commercial activity assay kit, respectively. As summarized in Table S6,† the activities obtained by the two methods only demonstrated relative deviations less than 15% for individual caspases, implying the feasibility of our presented method even in the real case of biological complexity.
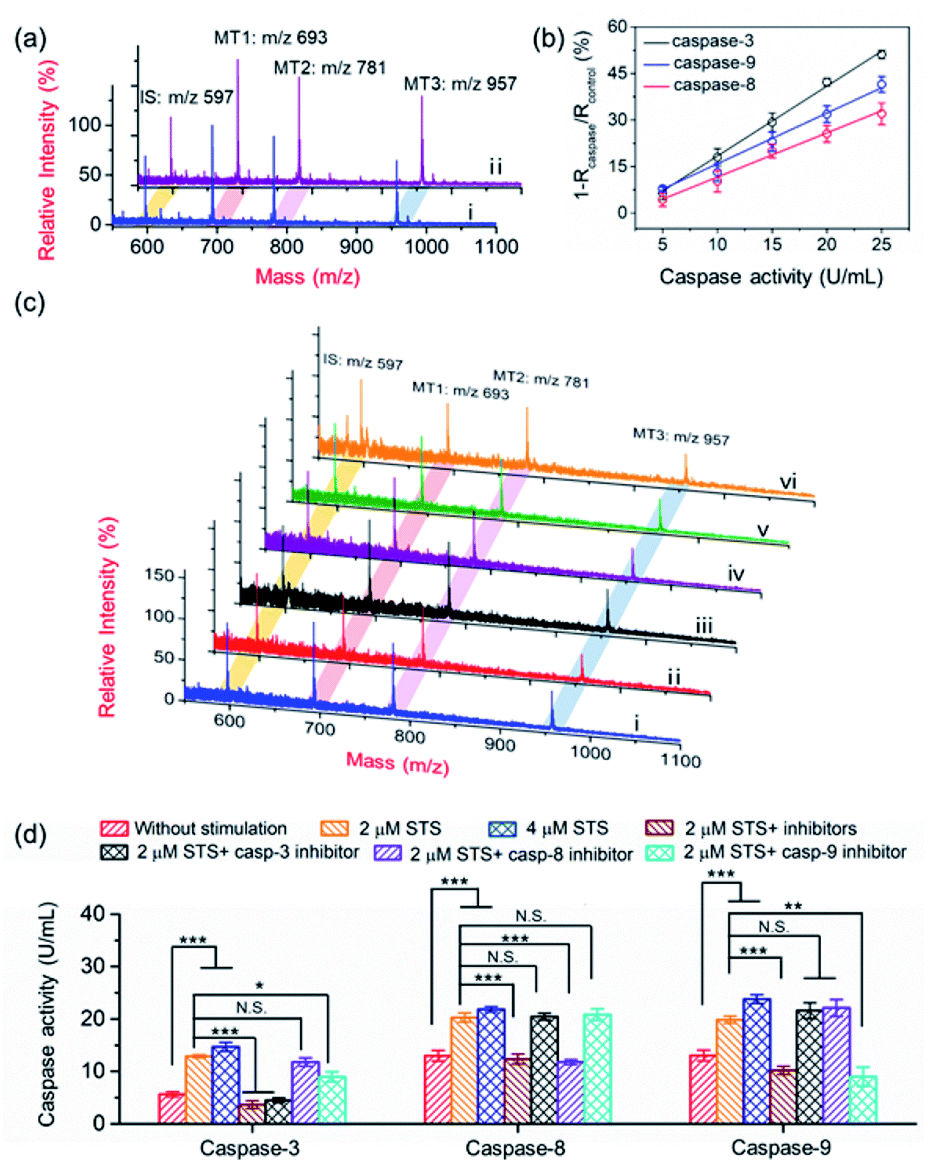 | ||
| Fig. 4 (a) MALDI mass spectra of PRMNTs incubated with 15 U mL−1 caspase-3, 20 U mL−1 caspase-8 and caspase-9 (i) and the blank solution (ii) at 37 °C for 1.5 h. (b) The linear correlation of relative variation of the MS peak intensity ratio with the activity of caspases. The intensity ratio of MT1, MT2 and MT3 to IS of PRMNTs incubated with mixed caspases was collectively denoted as Rcaspase, and the intensity ratio of them in the PRMNTs treated with the blank solution was denoted as Rcontrol. (c) MALDI mass spectra of the PRMNTs recovered from HeLa cells with different stimulations ((i) without stimulation; (ii) 2 μM STS; (iii) 2 μM STS and 50 μM Ac-DEVD-CHO, 50 μM Z-IETD-FMK, and 50 μM Z-LEHD-FMK; (iv) 2 μM STS and 50 μM Ac-DEVD-CHO, (v) 2 μM STS and 50 μM Z-LEHD-FMK; and (vi) 2 μM STS and 50 μM Z-IETD-FMK). (d) The quantification of the activities of caspase-3, -8 and -9 in HeLa cells with different stimulations. Statistical analysis: t-test (*p < 0.05; **p < 0.01; ***p < 0.001; N.S., not significant). | ||
The core–satellite PRMNTs were internalized into HeLa cells, and the triplex activity assay of caspase-3, -8 and -9 was implemented by measuring the corresponding intensity ratios referred to the fitted standard curve. As shown in Fig. 4c and d, all three caspases were activated after incubation with 2 μM STS, which was reflected by the decreased MS peak intensity ratio of MTs to IS in the PRMNTs isolated from the cells (Fig. 4c). Furthermore, increasing the dosage of STS spurred the activation of all three caspases at higher levels (Fig. 4d). To elucidate the mechanism of STS-induced apoptosis, HeLa cells were pretreated with different caspase inhibitors before exposure to STS. As shown in Fig. 4d, the activities of all three caspases were simultaneously down-regulated once applying the mixture of their inhibitors. To further differentiate the functions of the cascade caspases in the cellular signal relay, the activities of caspase-3, -8 and -9 were monitored after separately introducing their inhibitors, Ac-DEVD-CHO for caspase-3, benzyloxycarbonyl Ile-Glu(O-ME)-Thr-Asp(O-Me) fluoromethyl ketone (Z-IETD-FMK) for caspase-8, and benzyloxycarbonyl Leu-Glu(O-Me)-His-Asp(O-Me) fluoromethyl ketone (Z-LEHD-FMK) for caspase-9. Since caspase-3 resides at the downstream of the cascade in apoptosis, its inhibitor appeared to exclusively deactivate caspase-3, having little effect on caspase-8 and -9 activity. In comparison, the caspase-9 inhibitor led to degradative activity of both caspase-9 and caspase-3, which emphasized the contribution of the caspase-9 involved intrinsic pathway to STS-induced apoptosis. However, the caspase-8 inhibitor only exhibited suppression specific to caspase-8, while no significant activity variation was detected for the downstream executioner caspase-3. These outcomes indicated that the mitochondrial caspase cascade may play the major role in the execution of STS-induced programmed death, consistent with the conclusions of previous research.46–48 Collectively, relying on the ability of signal acquisition in multiple mass channels, the PRMNTs have been proved to be a powerful probe to characterize the activation of a hierarchy of caspases and expound their functions in apoptosis.
Kinetic analysis of activated caspases in chemical-induced cell apoptosis
To date, numerous anticancer drugs have been developed by treating caspases as the therapeutic targets to trigger the two or either one of the apoptosis pathways to accelerate cell death. Revealing how caspases are implicated in the regulation of cell apoptosis initiated by different chemicals is highly desirable to rational screening of therapeutic drugs. In this regard, we first profiled the activity of caspase-3, -8 and -9 in cells exposed to diverse chemicals by using the PRMNTs. As shown in Fig. 5a, the activation state of the caspase family varied with different chemicals, including commercial carcinostatic agents such as doxorubicin (DOX), (−)-epigallocatechin-3-O-gallate (EGCG), paclitaxel (PTX), and diosgenin, and the oxidative agent H2O2. Exposure to EGCG caused activation of all three caspases, coinciding with previous reports which showed that EGCG triggered the activation of initiator caspase-8 and -9 followed by activation of effector caspase-3.49 As DOX has been proved to cause oxidative DNA damage through H2O2 generation, the activity of both caspase-8 and -9 displayed dramatic enhancements in the presence of DOX or H2O2, subsequently leading to caspase-3 activation for cell death. In contrast, some other drugs showed preference for inducing apoptosis through a specific pathway. Typically, PTX brought about no significant variation of caspase-9 activity, showing that PTX mainly chose the caspase-8 pathway to induce the activation of caspase-3. In contrast, diosgenin-induced cell death was responsible for the caspase-9 activation in the mitochondrial pathway.50 These results indicated that the programmed cell self-destruction differed in the apoptosis pathway according to diverse external stimuli.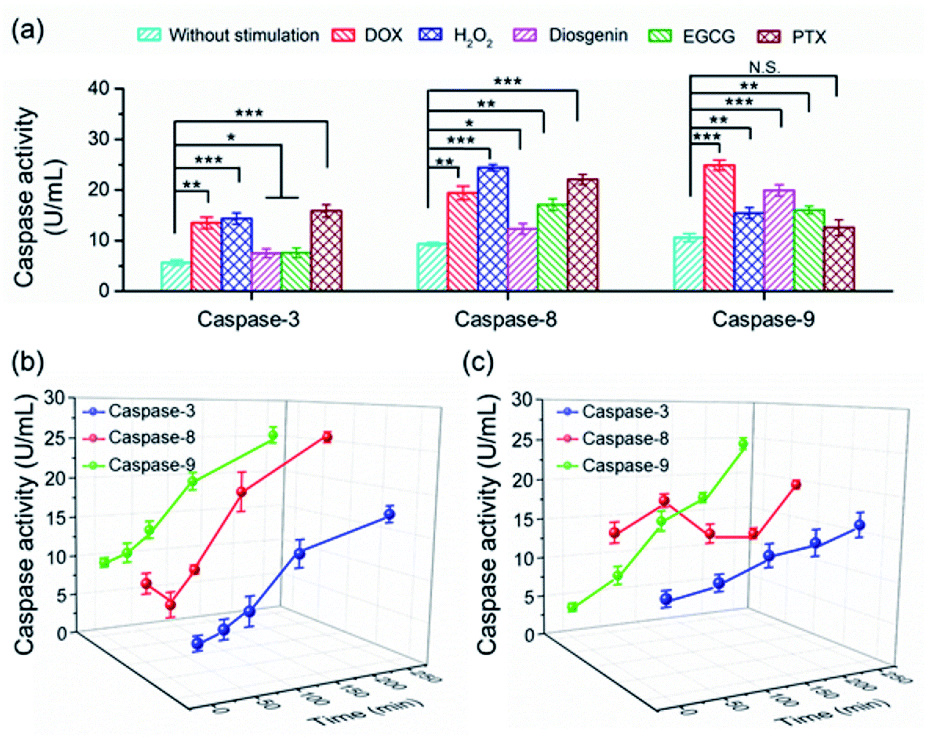 | ||
| Fig. 5 (a) The quantification of the activities of caspase-3, -8 and -9 in HeLa cells with different stimulations including 5 μM DOX, 1.3 mM H2O2, 10 μM diosgenin, 300 μg mL−1 EGCG or 30 μg mL−1 PTX. Statistical analysis: t-test (*p < 0.05; **p < 0.01; ***p < 0.001; N.S., not significant). Kinetic analysis of activated caspases in HeLa cells treated with (b) 5 μM DOX and (c) 10 μM diosgenin. | ||
To understand the activation sequence of multiple caspases correlated in the apoptotic events, we further looked into the time-dependent cellular caspase activity in drug-induced apoptosis. As can be seen in Fig. 5b, caspase-9 was first activated after being stimulated with 5 μM DOX for 30 min, and the activity rose steadily with the prolonged time. In comparison, the activity of caspase-8 appeared to fluctuate during the first 60 min, after which DOX started to cause a continuous increase in caspase-8 activation. This indicated that DOX induced apoptotic cell death via both intrinsic and extrinsic apoptotic pathways, and the onset activation of caspase-8 was marginally delayed compared with that of caspase-9. In another case, diosgenin gave rise to a different time course of cellular caspase activity. As shown in Fig. 5c, the intracellular activity of caspase-8 relatively stabilized around 15 U mL−1 after the treatment with diosgenin, which means exposure to this drug cannot upgrade the activation level of caspase-8. Unlike diosgenin-insensitive caspase-8, caspase-9 showed an upward trend in activity, which contributed to the continuous activation of caspase-3 to induce cell death. Thanks to the multiple signalling channels indicating the activity of a series of proteases in parallel, the PRMNTs allow the quantitative activity assay of cascaded caspases involved in apoptotic progress in a dynamic and multiplex manner. This advantage dramatically expands the possibility of deeply understanding cellular machinery, thus offering guidance for pharmacological research and personalized medication.
Conclusions
In summary, a mass spectrometric approach for simultaneous quantification of the intracellular activity of cascaded caspases implicated in cell apoptosis was established by using core–satellite mass barcoded nanotranslators. In the core–satellite PRMNTs, substrate peptides corresponding to different target caspases serve as the linker to couple magnetic Fe3O4 nanospheres with MT encoded satellite AuNPs to form a “one-to-many” nanostructure. Conversion of enzymatic cleavage events to varied ion signals of MTs immobilized on satellite AuNPs allowed MS interpretation of protease activity, and the introduction of IS-capped satellite AuNPs diminished the quantification inaccuracy caused by biothiols in the cellular environment. By MS-decoding the caspase activity information carried on the PRMNTs that underwent an in-cell proteolytic reaction, the intracellular activity of caspase-3, -8 and -9 actuated in diverse apoptosis pathways of HeLa cells could be quantitatively profiled in one single mass spectrum from multiple ion signal channels. In addition, the differential responses of the caspase family to various cancer therapeutic agents were globally characterized in a dynamic and multiplexed manner, thus helping deepen the understanding of how chemical drugs act upon a cascade of caspases in drug-induced apoptotic processes. Taken together, the proposed PRMNT-facilitated mass spectrometric enzyme assay offers a promising methodology for exploring the orchestration of proteases in cellular signaling networks and revealing pharmacological mechanisms in cancer medication.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was financially supported by the National Natural Science Foundation of China (No. 21622505, 21974062, 21575061, and 21834004), the Fundamental Research Funds for the Central Universities (020514380141), and the program B for Outstanding Ph.D. candidate of Nanjing University (201901B025).Notes and references
- A. G. Porter and R. U. Jänicke, Cell Death Differ., 1999, 6, 99–104 CrossRef CAS PubMed.
- S. McComb, P. K. Chan, A. Guinot, H. Hartmannsdottir, S. Jenni, M. P. Dobay, J.-P. Bourquin and B. C. Bornhauser, Sci. Adv., 2019, 5, eaau9433 CrossRef PubMed.
- Y. Wu, D. Wang, X. Wang, Y. Wang, F. Ren, D. Chang, Z. Chang and B. Jia, Cell. Physiol. Biochem., 2011, 27, 539–546 CrossRef CAS PubMed.
- H. Li, H. Zhu, C.-j. Xu and J. Yuan, Cell, 1998, 94, 491–501 CrossRef CAS.
- R. V. Talanian, K. D. Brady and V. L. Cryns, J. Med. Chem., 2000, 43, 3351–3371 CrossRef CAS.
- Y. Li, M. Zhou, Q. Hu, X.-c. Bai, W. Huang, S. H. W. Scheres and Y. Shi, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1542 CrossRef CAS PubMed.
- C. Holohan, S. Van Schaeybroeck, D. B. Longley and P. G. Johnston, Nat. Rev. Cancer, 2013, 13, 714–726 CrossRef CAS PubMed.
- S. H. Kaufmann and W. C. Earnshaw, Exp. Cell Res., 2000, 256, 42–49 CrossRef CAS.
- L. Galluzzi, L. Senovilla, I. Vitale, J. Michels, I. Martins, O. Kepp, M. Castedo and G. Kroemer, Oncogene, 2011, 31, 1869 CrossRef PubMed.
- Y. Pan, M. Guo, Z. Nie, Y. Huang, Y. Peng, A. Liu, M. Qing and S. Yao, Chem. Commun., 2012, 48, 997–999 RSC.
- S. Takano, S. Shiomoto, K. Y. Inoue, K. Ino, H. Shiku and T. Matsue, Anal. Chem., 2014, 86, 4723–4728 CrossRef CAS PubMed.
- H. Wang, Q. Zhang, X. Chu, T. Chen, J. Ge and R. Yu, Angew. Chem., Int. Ed., 2011, 50, 7065–7069 CrossRef CAS.
- Y. Xie, R. Zhao, Y. Tan, X. Zhang, F. Liu, Y. Jiang and C. Tan, ACS Appl. Mater. Interfaces, 2012, 4, 405–410 CrossRef CAS PubMed.
- S. Wen, Y. Su, R. Wu, S. Zhou, Q. Min, G.-C. Fan, L.-P. Jiang, R.-B. Song and J.-J. Zhu, Biosens. Bioelectron., 2018, 117, 260–266 CrossRef CAS.
- X. Zhang, N. Liao, G. Chen, A. Zheng, Y. Zeng, X. Liu and J. Liu, Nanoscale, 2017, 9, 10861–10868 RSC.
- X. Huang, M. Swierczewska, K. Y. Choi, L. Zhu, A. Bhirde, J. Park, K. Kim, J. Xie, G. Niu, K. C. Lee, S. Lee and X. Chen, Angew. Chem., Int. Ed., 2012, 51, 1625–1630 CrossRef CAS PubMed.
- M. Luan, J. Chang, W. Pan, Y. Chen, N. Li and B. Tang, Anal. Chem., 2018, 90, 10951–10957 CrossRef CAS PubMed.
- X. Liu, X. Song, D. Luan, B. Hu, K. Xu and B. Tang, Anal. Chem., 2019, 91, 5994–6002 CrossRef CAS.
- L. Zhang, J. Lei, J. Liu, F. Ma and H. Ju, Chem. Sci., 2015, 6, 3365–3372 RSC.
- J. Hu, F. Liu and H. Ju, Angew. Chem., Int. Ed., 2016, 55, 6667–6670 CrossRef CAS PubMed.
- H. Xu, M. Liu, X. Huang, Q. Min and J.-J. Zhu, Anal. Chem., 2018, 90, 9859–9867 CrossRef CAS PubMed.
- A. Sanchez-Ruiz, S. Serna, N. Ruiz, M. Martin-Lomas and N. C. Reichardt, Angew. Chem., Int. Ed., 2011, 50, 1801–1804 CrossRef CAS PubMed.
- M. D. Cabezas, C. A. Mirkin and M. Mrksich, Nano Lett., 2017, 17, 1373–1377 CrossRef CAS PubMed.
- J. Su, T. W. Rajapaksha, M. E. Peter and M. Mrksich, Anal. Chem., 2006, 78, 4945–4951 CrossRef CAS PubMed.
- D.-H. Min, J. Su and M. Mrksich, Angew. Chem., Int. Ed., 2004, 43, 5973–5977 CrossRef CAS PubMed.
- M. Coradin, K. R. Karch and B. A. Garcia, Expert Rev. Proteomics, 2017, 14, 409–418 CrossRef CAS PubMed.
- A. P. Wiita, J. E. Seaman and J. A. Wells, Methods Enzymol., 2014, 544, 327–358 CAS.
- S. Mahrus, J. C. Trinidad, D. T. Barkan, A. Sali, A. L. Burlingame and J. A. Wells, Cell, 2008, 134, 866–876 CrossRef CAS.
- S. Xu, W. Ma, Y. Bai and H. Liu, J. Am. Chem. Soc., 2019, 141, 72–75 CrossRef CAS.
- Y. Wang, R. Du, L. Qiao and B. Liu, Chem. Commun., 2018, 54, 9659–9662 RSC.
- R. Ahmad, H. Jang, B. S. Batule and H. G. Park, Anal. Chem., 2017, 89, 8966–8973 CrossRef CAS.
- S. Hou, K. N. Sikora, R. Tang, Y. Liu, Y.-W. Lee, S. T. Kim, Z. Jiang, R. W. Vachet and V. M. Rotello, ACS Nano, 2016, 10, 6731–6736 CrossRef CAS PubMed.
- A. L. M. Marsico, G. S. Elci, D. F. Moyano, G. Yesilbag Tonga, B. Duncan, R. F. Landis, V. M. Rotello and R. W. Vachet, Anal. Chem., 2015, 87, 12145–12150 CrossRef CAS PubMed.
- Z.-J. Zhu, P. S. Ghosh, O. R. Miranda, R. W. Vachet and V. M. Rotello, J. Am. Chem. Soc., 2008, 130, 14139–14143 CrossRef CAS PubMed.
- W. Ma, S. Xu, H. Nie, B. Hu, Y. Bai and H. Liu, Chem. Sci., 2019, 10, 2320–2325 RSC.
- Z. Wu, L. Lin, M. Khan, W. Zhang, S. Mao, Y. Zheng, Z. Li and J.-M. Lin, Chem. Commun., 2018, 54, 11546–11549 RSC.
- E. A. Slee, M. T. Harte, R. M. Kluck, B. B. Wolf, C. A. Casiano, D. D. Newmeyer, H. G. Wang, J. C. Reed, D. W. Nicholson, E. S. Alnemri, D. R. Green and S. J. Martin, J. Cell Biol., 1999, 144, 281–292 CrossRef CAS.
- B. A. Smith and B. D. Smith, Bioconjugate Chem., 2012, 23, 1989–2006 CrossRef CAS PubMed.
- L. Galluzzi, A. López-Soto, S. Kumar and G. Kroemer, Immunity, 2016, 44, 221–231 CrossRef CAS PubMed.
- J. Yuan, A. Najafov and B. F. Py, Cell, 2016, 167, 1693–1704 CrossRef CAS PubMed.
- C. Pop, J. Timmer, S. Sperandio and G. S. Salvesen, Mol. Cell., 2006, 22, 269–275 CrossRef CAS PubMed.
- C. Du, M. Fang, Y. Li, L. Li and X. Wang, Cell, 2000, 102, 33–42 CrossRef CAS.
- S. M. Man and T.-D. Kanneganti, Nat. Rev. Immunol., 2016, 16, 7–21 CrossRef CAS PubMed.
- M. Poreba, A. Szalek, P. Kasperkiewicz, W. Rut, G. S. Salvesen and M. Drag, Chem. Rev., 2015, 115, 12546–12629 CrossRef CAS PubMed.
- M. Poreba, P. Kasperkiewicz, S. J. Snipas, D. Fasci, G. S. Salvesen and M. Drag, Cell Death Differ., 2014, 21, 1482–1492 CrossRef CAS.
- C. A. Belmokhtar, J. Hillion and E. Ségal-Bendirdjian, Oncogene, 2001, 20, 3354–3362 CrossRef CAS.
- A. C. Johansson, H. Steen, K. Öllinger and K. Roberg, Cell Death Differ., 2003, 10, 1253–1259 CrossRef CAS.
- X. D. Zhang, S. K. Gillespie and P. Hersey, Mol. Cancer Ther., 2004, 3, 187–197 CAS.
- K. Hastak, S. Gupta, N. Ahmad, M. K. Agarwal, M. L. Agarwal and H. Mukhtar, Oncogene, 2003, 22, 4851–4859 CrossRef CAS.
- Y. Yuan, C.-J. Zhang, R. T. K. Kwok, D. Mao, B. Z. Tang and B. Liu, Chem. Sci., 2017, 8, 2723–2728 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc01534b |
| This journal is © The Royal Society of Chemistry 2020 |