
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent developments in nickel-catalyzed intermolecular dicarbofunctionalization of alkenes
Joseph
Derosa
,
Omar
Apolinar
,
Taeho
Kang
,
Van T.
Tran
and
Keary M.
Engle
 *
*
Department of Chemistry, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037, USA. E-mail: keary@scripps.edu
First published on 6th March 2020
Abstract
Nickel-catalyzed three-component alkene difunctionalization has rapidly emerged as a powerful tool for forging two C–C bonds in a single reaction. Building upon the powerful modes of bond construction in traditional two-component cross-coupling, various research groups have demonstrated the versatility of nickel in enabling catalytic 1,2-dicarbofunctionalization using a wide range of carbon-based electrophiles and nucleophiles and in a fully intermolecular fashion. Though this area has emerged only recently, the last few years have witnessed a proliferation of publications on this topic, underscoring the potential of this strategy to develop into a general platform that offers high regio- and stereoselectivity. This minireview highlights the recent progress in the area of intermolecular 1,2-dicarbofunctionalization of alkenes via nickel catalysis and discusses lingering challenges within this reactivity paradigm.
Introduction
Transition-metal-catalyzed cross-coupling has served as a powerful engine for the rapid build-up of molecular complexity through C–C bond formation using organohalides and organometallic reagents.1,2 Despite the efficiency of different transition metal catalysts, including palladium, nickel, and copper, in mediating C(sp2)–C(sp2) bond formation, C(sp3)–X and C(sp3)–M reagents have only recently become viable coupling partners with advances in mechanistic understanding and catalyst design.3 A central challenge when using C(sp3) coupling partners is the propensity of the resulting alkylmetal intermediate to undergo β-hydride elimination, generating a highly reactive metal–hydride that can take part in various off-cycle pathways that preclude the desired C–C bond-forming event. To suppress β-hydride elimination in two-component cross-couplings, different approaches have been pursued, including developing effective ligand scaffolds and employing transition metals that are resistant to β-hydride elimination, such as nickel.4 Indeed, several recent developments in nickel-catalyzed C(sp3)–C(sp3) cross-coupling have taken advantage of the unique characteristics of nickel compared to palladium, such as faster rates for oxidative addition and migratory insertion, sluggish rates for β-hydride elimination and propensity to engage in single-electron transfer (SET), to enable otherwise difficult transformations.5Decades of development in the cross-coupling arena have yielded a highly modular toolkit for bond construction in organic synthesis. Nevertheless, the introduction of an alkene as a third component has only recently emerged as a viable catalytic strategy.6 In these transformations, two carbogenic fragments are delivered across the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond, resulting in 1,2-dicarbofunctionalization. These reactions are often referred to as “conjunctive cross-coupling” in the literature due to the integration of a third “conjunctive” component that can interlink with the two familiar fragments used in classical cross-couplings. Such methods offer strategic advantages in synthesis yet present several interrelated challenges in selectivity control. As such, they are at the forefront of modern catalysis research.
C bond, resulting in 1,2-dicarbofunctionalization. These reactions are often referred to as “conjunctive cross-coupling” in the literature due to the integration of a third “conjunctive” component that can interlink with the two familiar fragments used in classical cross-couplings. Such methods offer strategic advantages in synthesis yet present several interrelated challenges in selectivity control. As such, they are at the forefront of modern catalysis research.
As with two-component cross-couplings, nickel offers advantages as a catalyst in intermolecular 1,2-dicarbofunctionalization of alkenes, namely: increased resistance to β-hydride elimination, increased propensity for oxidative addition, and ability to react with radical intermediates. These properties are ideal for preventing undesired chain-walking processes in alkene functionalization and expanding the scope of compatible coupling partners. This minireview focuses on the recent flurry of developments in nickel-catalyzed intermolecular 1,2-dicarbofunctionalization of alkenes (Scheme 1). Mechanistically, this family of reactions involves a shared sequence of elementary steps. First the alkene substrate is intercepted with an initial coupling partner (C–Y) in a net carbometallation which is followed by incorporation of the second coupling partner (C–Z) via association to the metal and reductive elimination. In most cases, a transmetalating agent (i.e., C–B(OR)2 or C–ZnX) is used to incorporate one of the carbon fragments. However, reductive couplings involving two electrophiles will also be discussed. An important aspect of this family of reactions is the difficulty of achieving high levels of regio-, stereo-, and chemoselectivity. In most cases, this is dependent on the mechanism by which the nickel catalyst interacts with the coupling partners (single- or two-electron processes) and the alkene carbometallation pathway (syn- or anti-insertion). While the intricacies of the reaction mechanisms will be described for individual reactions highlighted below, these transformations typically proceed via one of two main pathways for carbometallation: (A) syn-1,2-migratory insertion of the organonickel species directly into the C–C π-bond, or (B) single-electron transfer-initiated radical addition to the C–C π-bond followed by radical recombination of the resulting alkyl radical to produce an organonickel species.
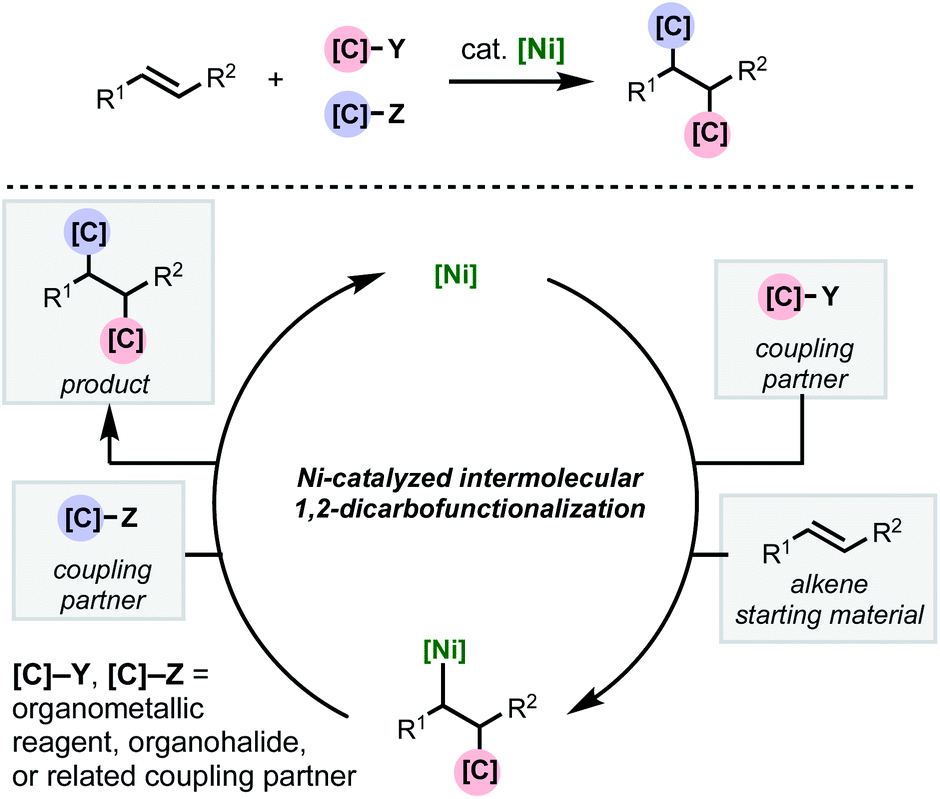 | ||
| Scheme 1 General scheme depicting nickel-catalyzed intermolecular 1,2-dicarbofunctionalization of alkenes. | ||
This review is organized based on the type of alkene employed: (1) acrylate-type substrates, (2) heteroatom-substituted alkenes, (3) styrenes, (4) strained alkenes, and (5) non-conjugated (and unstrained) alkenes (Scheme 2). Across the examples that are covered, we emphasize different strategies that prevent β-hydride elimination and control regioselectivity, noting that in many cases multiple effects operate in concert to enable the transformation. Furthermore, we highlight the different types of carbon-based coupling partners used, and include insights on the specific redox manifold of the nickel catalyst in the reaction. 1,2-Dicarbofunctionalization using other transition metals7 and nickel-catalyzed intramolecular 1,2-dicarbofunctionalization8 are beyond the scope of this article and will not be discussed. Overall, this minireview serves to showcase exciting developments in this area and to discuss current limitations, challenges, and opportunities moving forward.
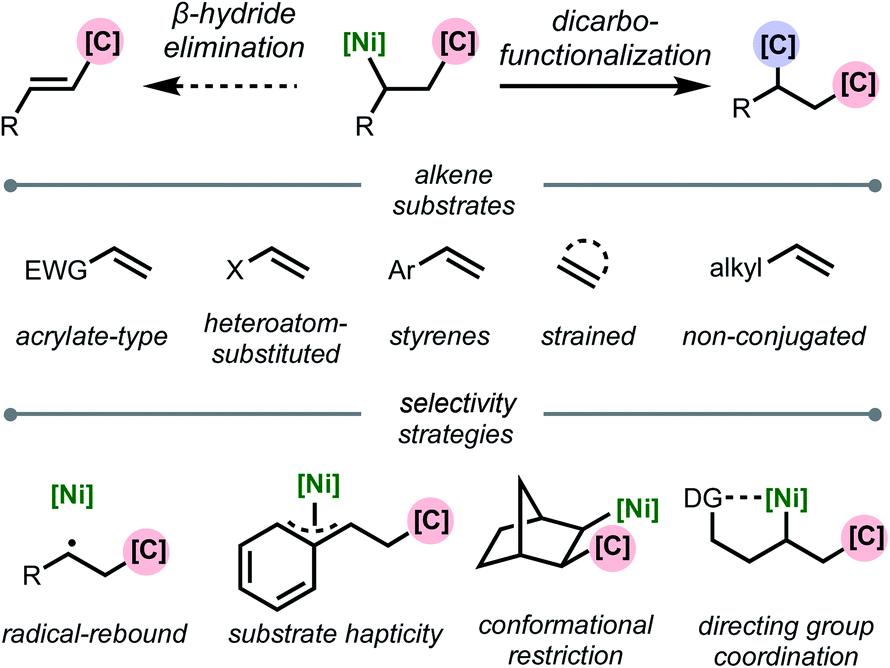 | ||
| Scheme 2 Organization of topics in this review based on alkene substrate and stabilization strategy. | ||
Acrylate-type substrates
Conjugated alkenes such as acrylates can readily undergo Giese addition with radical coupling partners usually leading to hydrofunctionalized products. However, the generated secondary alkyl radical intermediates can alternatively be trapped with organonickel species en route to 1,2-dicarbofunctionalization. In 2016, Baran and coworkers demonstrated one of the first examples of nickel-catalyzed intermolecular 1,2-dicarbofunctionalization using benzyl acrylate, redox-active esters (RAEs), and phenyl zinc halide salts as transmetalating agents (Fig. 1).9 Using 20 mol% NiCl2·glyme with 40 mol% bipyridyl ligand L1, a wide range of tertiary alkyl RAEs including heterocycles, strained carbocycles, and heteroatom-containing alkyl chains are tolerated. The authors propose an initial SET that results in decarboxylation to generate a tertiary alkyl radical. This radical can engage with benzyl acrylate in a Giese-type fashion, followed by radical recombination with the aryl nickel species generated in situ. The resulting Ni(Ar)(alkyl) species undergoes reductive elimination to yield the desired 1,2-dicarbofunctionalized product.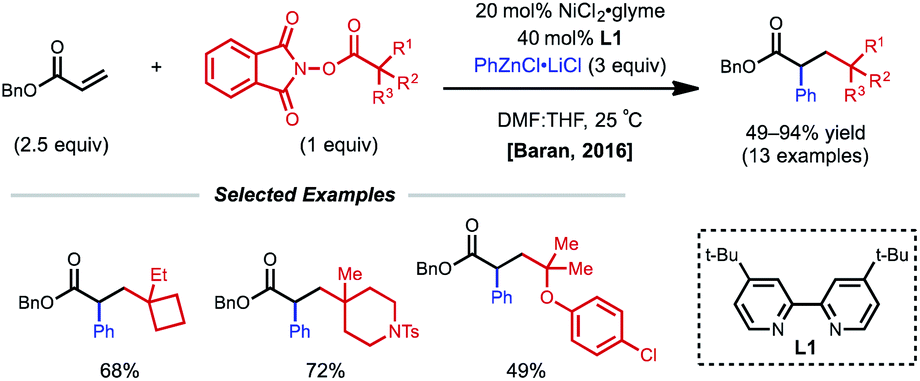 | ||
| Fig. 1 1,2-Dicarbofunctionalization of benzyl acrylate using phenyl zinc chloride and RAEs. | ||
In 2017, the Nevado lab reported a reductive 1,2-arylalkylation method. Compatible alkenes included various acrylate-type substrates (Fig. 2A), as well as three classes of allylic compounds (not shown).10 The authors employed 10 mol% NiCl2(py)4 and 10 mol% bipyridyl ligand L1 with aryl iodides and tertiary alkyl iodides using tetrakis(dimethylamino)ethylene (TDAE) as the reductant. Various control experiments such as radical-traps shed light on the radical-based nature of the reaction mechanism and the capability of TDAE in reducing Ni(II) to low-valent nickel species under the reaction conditions. The authors propose a catalytic cycle involving the reduction of Ni(II) to Ni(0) using TDAE which can then undergo disproportionation to yield Ni(I) (Fig. 2B). This Ni(I) species engages in SET with the corresponding alkyl halide, generating an alkyl radical that performs radical addition to the alkene substrate generating a new secondary alkyl radical. The newly formed Ni(II) species undergoes a second reduction event by TDAE to yield Ni(0) that can perform oxidative addition to the aryl halide coupling partner. The Ni(II)–aryl species is then intercepted by the secondary alkyl radical generated in the earlier step, resulting in a secondary alkyl Ni(III)–aryl that undergoes a facile reductive elimination to regenerate Ni(I) and close the catalytic cycle. In 2019, Nevado and coworkers expanded this reductive 1,2-alkylarylation reaction to a variety of terminal non-conjugated alkenes.11
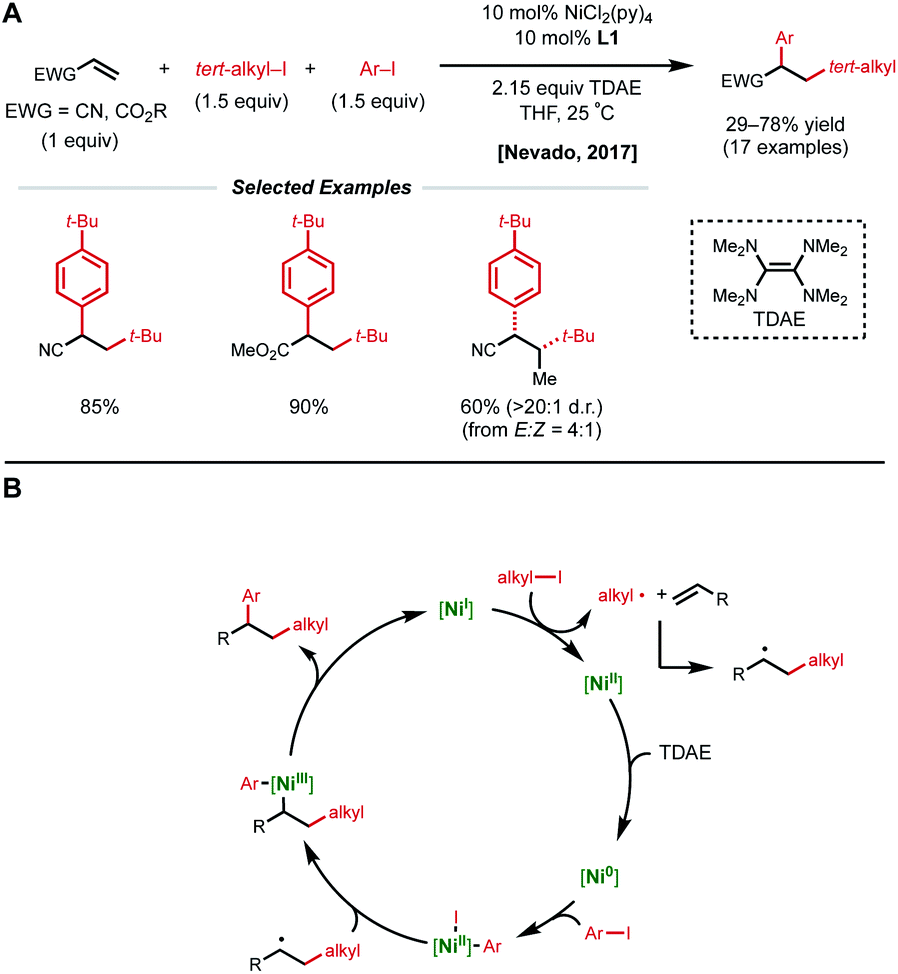 | ||
| Fig. 2 (A) Reductive 1,2-dicarbofunctionalization of acrylate-type compounds. (B) Proposed catalytic cycle. | ||
Heteroatom-substituted alkene substrates
The presence of a heteroatom on an alkene provides electronic stabilization of a Ni(II) or α-radical intermediate, allowing for facile 1,2-dicarbofunctionalization. In 2016, Zhang and coworkers reported a 1,2-difluoroalkylarylation reaction of enamides using aryl boronic acids and difluoroalkyl bromides (Fig. 3).12 This reaction employs 5 mol% NiCl2·glyme with 5 mol% bipyridyl ligand L2 and showcases a broad scope for both coupling partners. The alkene scope is limited to terminal N-vinyl-pyrrolidinones; and closely related substrates; for example, the corresponding oxazolidinone, phthalimide, and acyclic acetamide13 also react to deliver the desired products. The authors performed radical clock experiments that provide evidence for the presence of radical species in related systems. The authors propose that there is a directing effect from the carbonyl group (via a four-membered nickelacycle intermediate). Modest enantioselectivity is achieved using a chiral diamine ligand in a single example (18% ee). In 2019, the Zhang group developed a complementary 1,2-difluoroalkylalkylation reaction using the same substrate class with dialkyl zinc reagents.14 In this report, the authors conduct the reaction with an N-vinyl pyrrolidine and do not observe reactivity, supporting a nickel-chelation hypothesis. Later in 2019, the Morken group reported an asymmetric 1,2-dialkylation and 1,2-arylalkylation of alkenyl boronic acid pinacol esters (BPin) (Fig. 4A).15 Taking advantage of the stability of the α-boryl radical,16 alkyl zinc halides and tertiary alkyl halides are effective coupling partners using 10 mol% NiBr2·glyme and 13 mol% chiral diamine ligand L3. Interestingly, the use of pinacol as boron protecting group was necessary to achieve high ee. Based on earlier precedents invoking radical species in nickel-catalyzed cross-coupling and simple TEMPO inhibition experiments, the authors conclude that this reaction likely proceeds through a radical-addition-based mechanism (Fig. 4B). Since this report, alkenyl boron reagents have proven to be highly versatile conjunctive reagents, particularly when employed under dual nickel/photoredox catalysis.17 Also, it is appropriate to note that similar 1,2-dicarbofunctionalized products can also be obtained via nickel-catalyzed metalate rearrangement pathways.18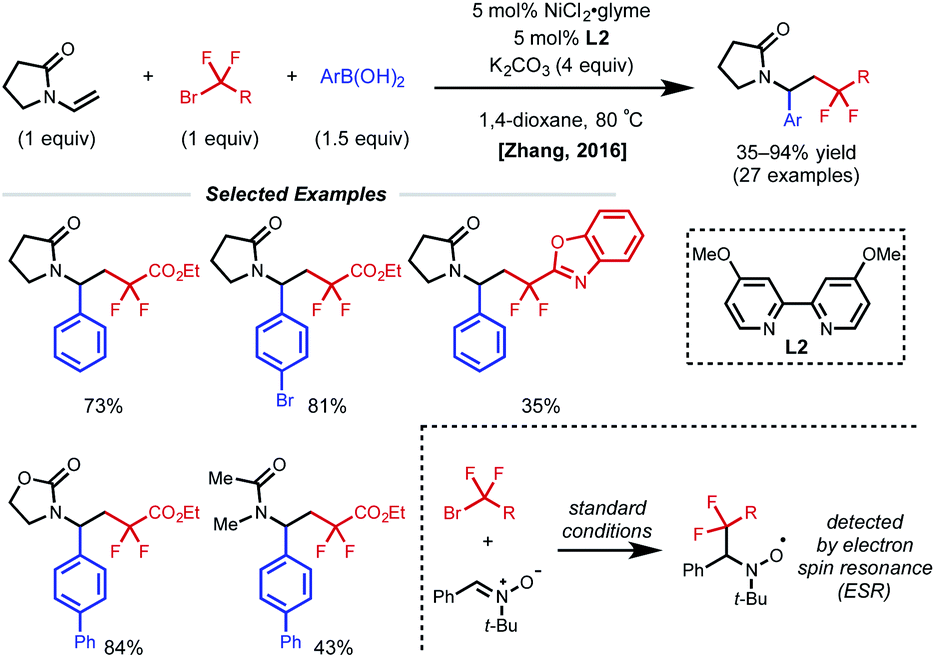 | ||
| Fig. 3 1,2-Difluoroalkylarylation of enamides. | ||
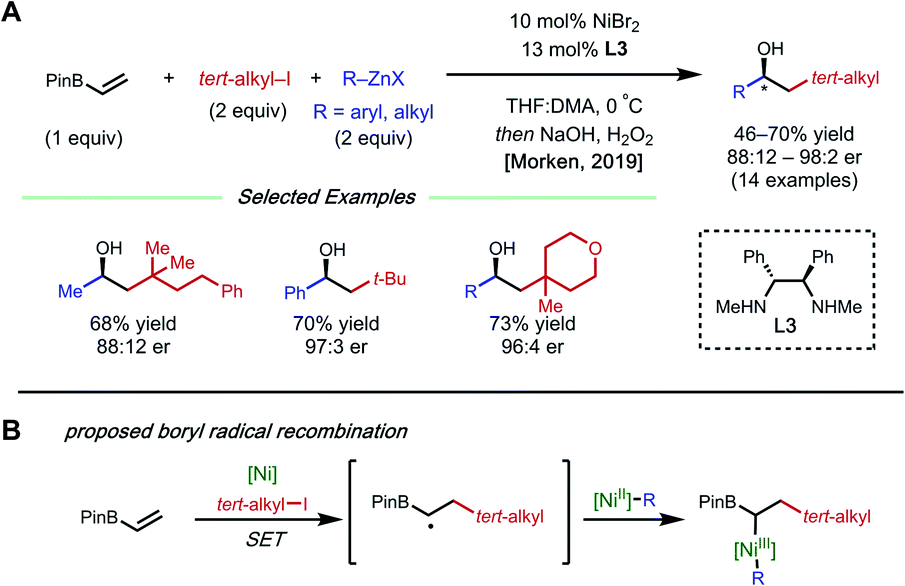 | ||
| Fig. 4 (A) Asymmetric 1,2-dicarbofunctionalization of vinyl-Bpin. (B) Proposed mechanism. | ||
Styrene substrates
Due to their resonance stabilized benzylic positions, styrenes are able to stabilize Ni(I/II/III) intermediates. This property of styrenyl substrates enables excellent regioselectivity in 1,2-dicarbofunctionalization. In 2017, the Giri laboratory developed a 1,2-diarylation of terminal styrenes using pendant imines as directing groups for stabilization of the alkylnickel intermediate (Fig. 5).19 Under optimized conditions, 2–10 mol% Ni(cod)2 with aryl zinc halides and aryl halides delivered the desired imine-containing products that were subsequently unmasked to the corresponding benzaldehyde with an acidic work-up. In contrast to previous examples, the authors propose a reaction mechanism involving nickel-mediated oxidative addition to the aryl halide followed by carbometallation, assisted by coordination to the imine directing group. At this point, the authors propose a transmetalation with aryl zinc halide and reductive elimination to forge the final C–C bond. A control experiment revealed that 2-vinylbenzaldehyde was unreactive under optimized conditions, indicating that the imine group is necessary for productive catalysis. The reaction also proceeded if the imine substrate was derived from aniline instead of benzaldehyde. In 2018, Giri reported an analogous approach toward the functionalization of dimethylpyridylvinylsilane.20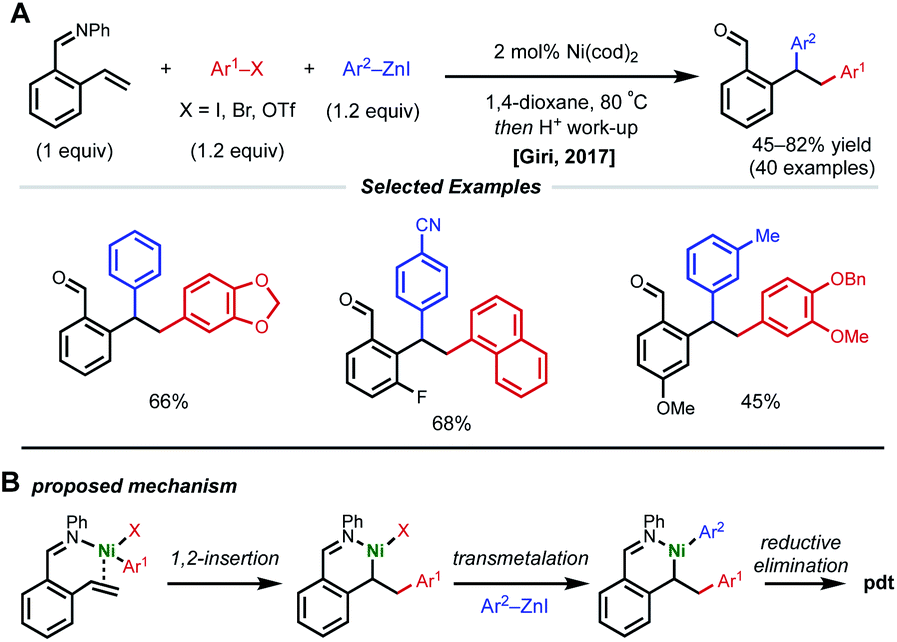 | ||
| Fig. 5 1,2-Diarylation of styrenyl aldimines. | ||
Only one year later, both the Giri and Brown labs reported 1,2-dicarbofunctionalization reactions of vinyl arenes without the use of a directing group. The Giri group employed 5 mol% NiBr2 with primary/secondary alkyl halides (or 5 mol% NiCl2(PPh3)2 with tertiary alkyl halides) and aryl zinc reagents for 1,2-alkylarylation of terminal vinyl arenes (Fig. 6A).21 Mechanistic experiments established that Ni(II) is reduced to Ni(0) in situ by the excess aryl zinc reagent and that radicals are present throughout the course of the reaction, supporting a radical addition pathway. Reaction progress kinetic analysis revealed positive order kinetics in the alkyl halide coupling partner, suggesting its involvement in the rate-determining step. Giri and coworkers also reported the utility of this method toward the synthesis of FLAP inhibitors.22
 | ||
| Fig. 6 1,2-Dicarbofunctionalization of alkenyl arenes reported by (A) Giri and coworkers and (B) Brown and coworkers. | ||
The Brown lab simultaneously reported a 1,2-diarylation of terminal and internal vinyl arenes using 7.5 mol% NiCl2 with aryl bromides and aryl boronic acid neopentyl glycol esters (ArB(nep)) (Fig. 6B).23 It is important to note that internal conjugated substrates were tolerated and gave desired products with high regio- and stereoselectivity. The authors propose a syn-insertion mechanism, which is supported by the formation of a single diastereomer with a cyclic internal substrate. Interestingly, 0.1 equiv. of B2Pin2 is utilized for the reduction of Ni(II) to Ni(0). In addition, the reaction is highly scalable and was applied in the synthesis of rac-lasofoxifene.
Despite the rapid growth in this area, the identification of a suitable asymmetric variant of this 1,2-dicarbofunctionalization cascade remained elusive. In 2019, Diao and coworkers developed the first nickel-catalyzed asymmetric reductive 1,2-homodiarylation of styrenes.24 In this reaction manifold, 10 mol% NiBr2·glyme, 20 mol% chiral bi-oxazoline L4, 8 mol% ABNO additive, excess aryl bromide, and zinc dust generated the desired homo-diarylation product in high ee (Fig. 7A). Interestingly, an N-oxyl radical additive was found to be crucial for obtaining high ee. Though the authors do not explicitly discuss the role of the additive, they propose that it may act as a ligand on the Ni-catalyst at some point given the importance of the near 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of ABNO:Ni. The choice of ligand L4 was also critical to achieving the desired reactivity and stereocontrol, with more common ligands in asymmetric nickel catalysis resulting in low or negligible ee.
:1 ratio of ABNO:Ni. The choice of ligand L4 was also critical to achieving the desired reactivity and stereocontrol, with more common ligands in asymmetric nickel catalysis resulting in low or negligible ee.
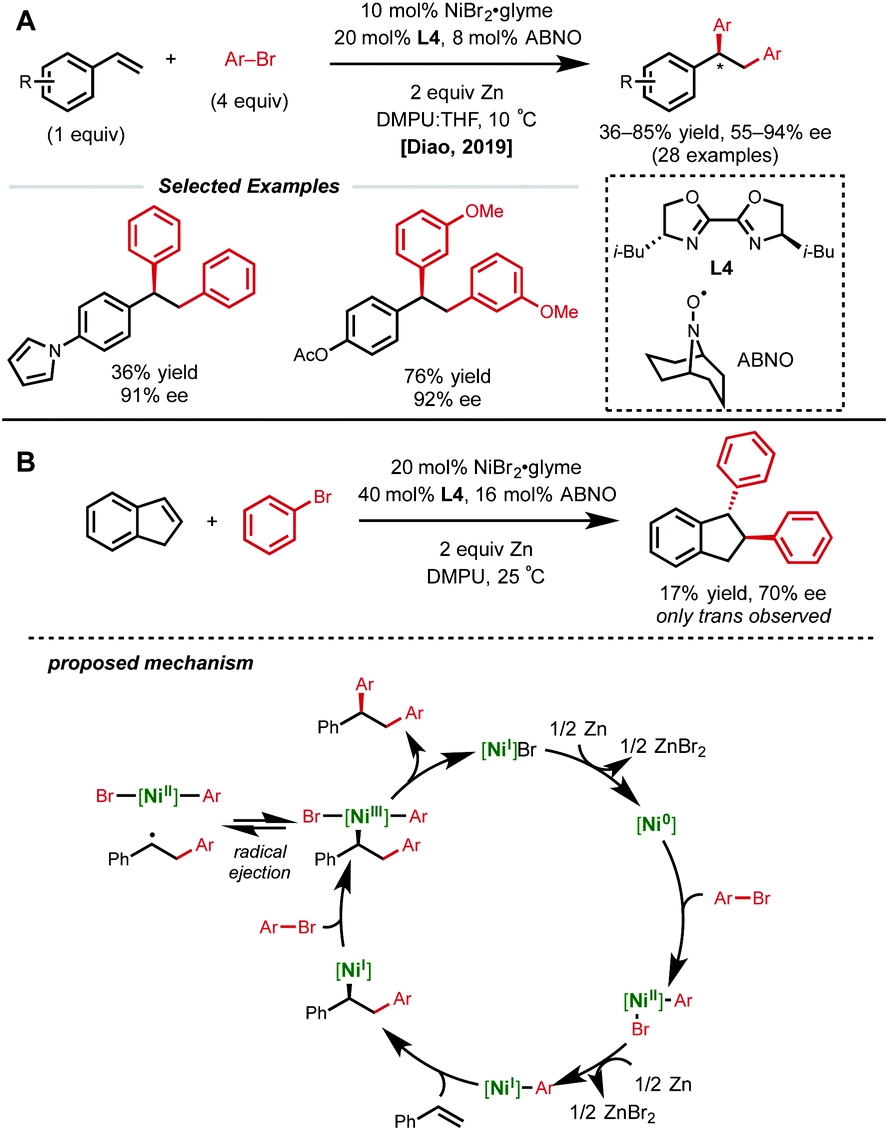 | ||
| Fig. 7 (A) Asymmetric reductive 1,2-homodiarylation of vinyl arenes. (B) Key mechanistic experiment and proposed catalytic cycle. | ||
Several experiments were conducted to help elucidate the mechanistic underpinnings of this nickel-catalyzed asymmetric 1,2-diarylation (Fig. 7B). The authors first examined the Ni/ligand ratio and observed a linear correlation, indicating that there is likely one equivalent of ligand on nickel in the enantiodetermining step. Additionally, indene was subjected to the reaction conditions and only the trans product was obtained in 70% ee. The authors conclude that this may be evidence of an initial syn-insertion followed by reversible radical ejection that selectively scrambles the stereochemistry, positioning nickel on the less hindered face. Other experiments pointing to radical species included the inhibition of the reaction by excess ABNO, and the detection of benzylic homodimers, albeit in low yield (2%).
Strained alkenes
Strained alkenes, such as norbornene, undergo strain release upon migratory insertion or radical addition, furnishing conformationally constrained alkylnickel intermediates that cannot readily undergo requisite C–C bond rotation to allow β-H elimination. As a consequence, these substrates are also competent in 1,2-dicarbofunctionalization under nickel catalysis. In 2019, Stanley and coworkers reported a syn-selective intermolecular 1,2-carboacylation procedure with norbornenes, N,N-substituted benzamide electrophiles, and tetraarylboron nucleophiles (Fig. 8).25 The transformation was found to take place under the action of 10 mol% Ni(cod)2 and 2 equiv. of H3BO3, tolerating a wide array of norbornene-type alkenes and coupling partners. The proposed catalytic cycle begins with oxidative addition of the Ni(0) catalyst to N,N-substituted benzamide, which affords a Ni(II)(acyl)(amido) species. Migratory insertion of norbornene to the Ni(II)(acyl)(amido) species forms a Ni(II)(alkyl)(amido) species. Subsequent transmetalation with in situ formed triarylborane generates a Ni(II)(alkyl)(aryl) species. Finally, reductive elimination turns over the catalytic cycle and forms the 1,2-carboacylated product.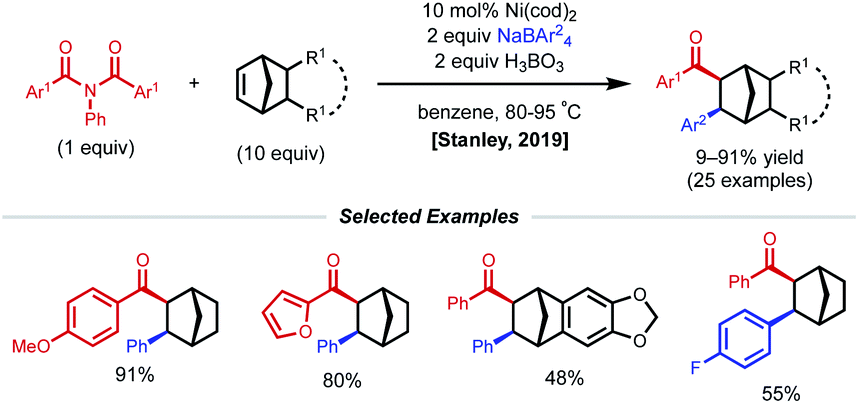 | ||
| Fig. 8 1,2-Carboacylation of norbornenes. | ||
Non-conjugated alkene substrates
In acyclic, unactivated alkenes, selective intermolecular 1,2-dicarbofunctionalization becomes a significant challenge. Despite the value of employing non-conjugated alkenes as starting materials, achieving desired reactivity is mired by two main factors: (1) migratory insertion is more difficult and (2) undesired β-hydride elimination is more prevalent. While overcoming these obstacles may seem daunting, various groups have made significant progress in providing a general platform for nickel-catalyzed 1,2-dicarbofunctionalization of non-conjugated alkenes.In 2017, Engle and coworkers implemented a directing group strategy to achieve regio- and diastereoselective 1,2-arylalkylation of β,γ-unsaturated alkenyl carbonyl compounds (Fig. 9A).26 Using a removable 8-aminoquinoline (AQ) bidentate auxiliary, a wide range of internal and α-substituted substrates could be effectively difunctionalized with organozinc reagents and aryl iodides using 10 mol% Ni(cod)2. Internal alkenes give a single diastereomer, suggesting that a syn-insertion mechanism is operative. Initial oxidative addition to the aryl iodide generates an arylnickel(II) intermediate that can coordinate AQ and undergo syn-insertion with the pendant alkene, placing the aryl fragment at the γ-position. The authors propose that the bidentate auxiliary is key to enabling this reactivity, resulting in a five-membered nickelacycle after insertion that is less prone to β-hydride elimination (Fig. 9B). At this stage, transmetalation with organozinc and reductive elimination yields the desired product. After the desired 1,2-difunctionalization, these products could undergo hydrolysis to remove the AQ auxiliary and unmask the free carboxylic acid.
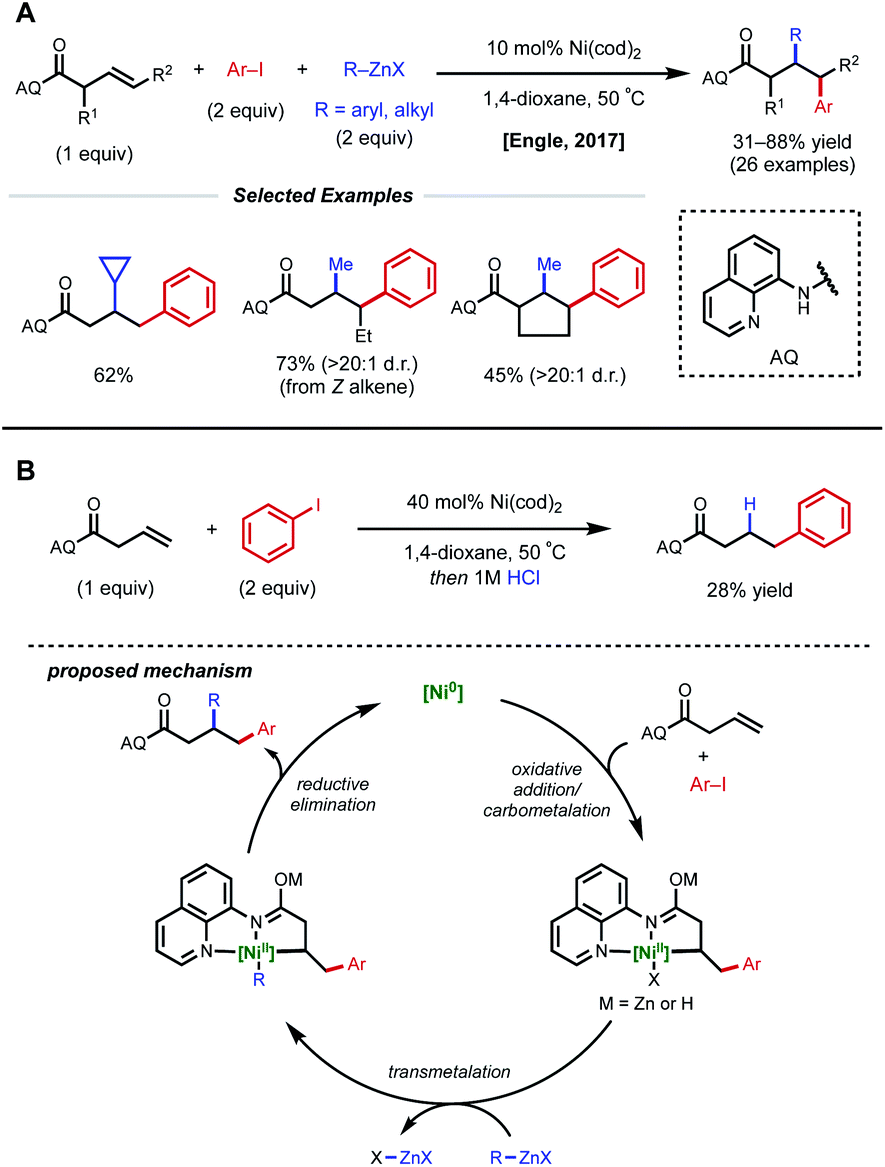 | ||
| Fig. 9 (A) Directed 1,2-arylalkylation of β,γ-unsaturated alkenyl carbonyl compounds. (B) Key mechanistic experiment and proposed catalytic cycle. | ||
Using a similar directing group strategy, the Engle laboratory reported a 1,2-dialkylation in 2018 using alkyl zinc reagents and alkyl halides, marking the first example of differentiable 1,2-dialkylation of non-conjugated alkenes (Fig. 10).27 While the standard AQ-substrate remained the same, this reaction gave the opposite regioselectivity (alkyl nucleophile at the γ-position) compared to the previous report in which aryl iodides were used as the electrophilic coupling partner. Interestingly, this reaction tolerates an even broader range of alkene coupling partners, including trisubstituted and extended chain γ,δ-unsaturated alkenes. Similarly, only a single diastereomer was detected in the case of internal alkenes, suggesting a syn-insertion mechanism. Radical-trap experiments shed light on the potential for radical species to be involved in catalysis. The authors suggest that this reaction operates through a SET pathway with different redox at nickel compared to the earlier example (Ni(I)/Ni(III) vs. Ni(0)/Ni(II)) based on the abundance of Ni(I)/Ni(III) precedent in the context of C(sp3)–X cross-coupling literature. A plausible mechanism involving the intermediacy of Ni(I) is proposed. First, transmetalation/carbometallation yields a putative chelated nickelacycle. A subsequent SET event with the alkyl halide followed by radical recombination and reductive elimination gives the desired product. This SET mode of nickel catalysis using AQ-bound substrates was later extended to a complementary 1,2-carboamination system.28 Very recently, the Lin and Yao groups applied this AQ-directed system toward a reductive 1,2-dialkynylation of non-conjugated alkenes.29
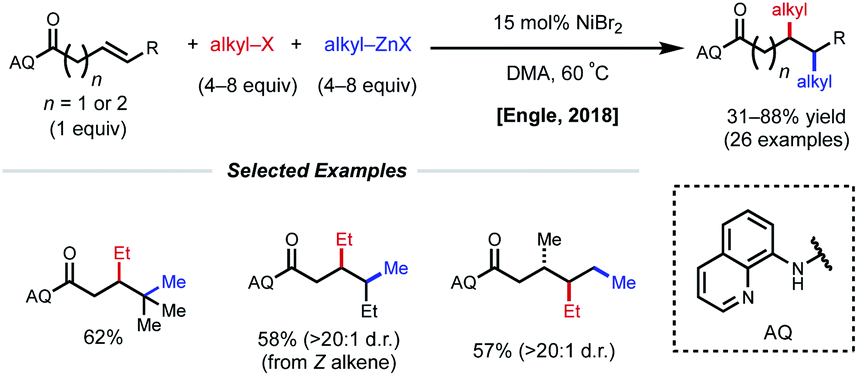 | ||
| Fig. 10 Directed 1,2-dialkylation of β,γ- and γ,δ-unsaturated alkenyl carbonyl compounds. | ||
In 2018, the Zhao group reported a series of 1,2- and 1,3-dicarbofunctionalization reactions using a 2-aminopyrimidine directing group derived from allyl amine with C(sp)/C(sp2)–X electrophiles and C(sp2)–boronic acids as nucleophiles (Fig. 11).30 In this case, the regioselectivity of the reaction was based on the combination of electrophile/nucleophile. For example, vinyl bromide electrophiles led to incorporation of the electrophile at the terminal position, and alkynyl halides lead to incorporation of the electrophile at the internal position. Interestingly, the use of an aryl electrophile led exclusively to 1,3-dicarbofunctionalized products, pointing to the challenge associated with β-hydride elimination. N-Allyl benzamides were shown to be compatible substrates in 1,2-alkynylarylation.
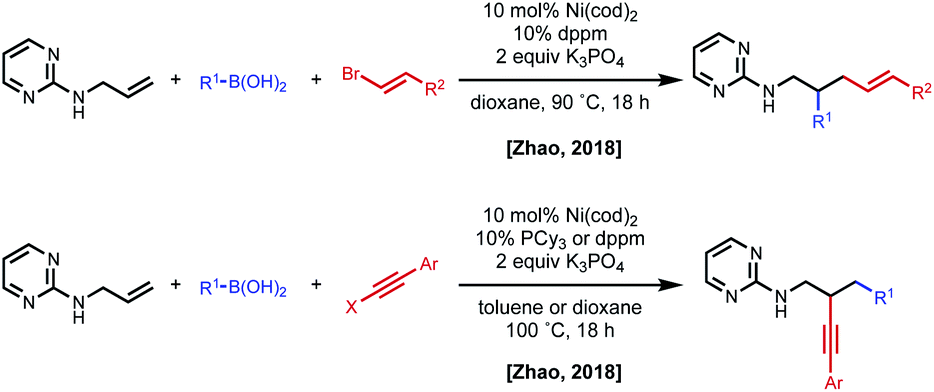 | ||
| Fig. 11 Divergent regioselectivity in 1,2-dicarbofunctionalization of allyl amines using pyrimidine directing groups. | ||
In 2018, the Giri lab extended their imine-directed diarylation reaction using aryl zinc reagents and aryl halides to linear, non-conjugated terminal alkenes (Fig. 12). In their initial attempt to obtain the desired γ,δ-diarylated products, only undesired Heck product was observed. Upon modifying reaction conditions, they found that 1,3-dicarbofunctionlization is possible with 5 mol% NiBr2 catalyst and 5 mol% (PhO)3P as ligand (Fig. 12A).31 The authors propose that reduction of the Ni(II) species to Ni(0) by the aryl zinc reagent occurs first. After oxidative addition and coordination to the alkene and imine directing group by the nickel center, the phosphite ligand facilitates the contraction of the transient 6-membered nickelacycle to a 5-membered nickelacycle via a β-hydride elimination/Ni–H reinsertion event. Lastly, transmetalation and reductive elimination afford the reported β,δ-diarylketones. Following this report, the Giri lab developed a complementary method to achieve γ,δ-diarylketone products (Fig. 12B).32 In this case, 5 mol% Ni(cod)2 catalyst without ancillary ligand was employed; however, AgBF4 or CuI were crucial halide scavenger additives. The authors claim that the abstraction of halide ligand from Ni(II) after the oxidative addition step produces a cationic aryl nickel species that can readily undergo migratory insertion, transmetalation, and reductive elimination without undergoing β-hydride elimination. In 2019, Giri reported a similar 1,3-dicarbofunctionalization using more weakly coordinating alkenyl cyanoesters as substrates with vinyl triflates and diaryl zinc reagents as coupling partners (Fig. 12C).33 The optimal conditions for this β,δ-vinylarylation use 5 mol% NiCl2 and 1.5 equiv. of both KPF6 and 4-phenylpyridine (4-PhPy). The authors postulate a Ni(I)/Ni(III) catalytic cycle where NiCl2 is first reduced to a cationic Ni(I) species that is bound to the substrate and 4-PhPy. Transmetalation with Ar2Zn then fosters a neutral Ni(I) species, allowing alkene insertion to subsequently occur. Next, the anionic 6-membered Ni(I)-enolate metallacyle contracts to form an anionic 5-membered Ni(I)-enolate metallacyle via a β-hydride elimination/Ni–H reinsertion sequence. Oxidative addition to the vinyl triflate electrophile and subsequent reductive elimination yields the β,δ-vinylarylated products.
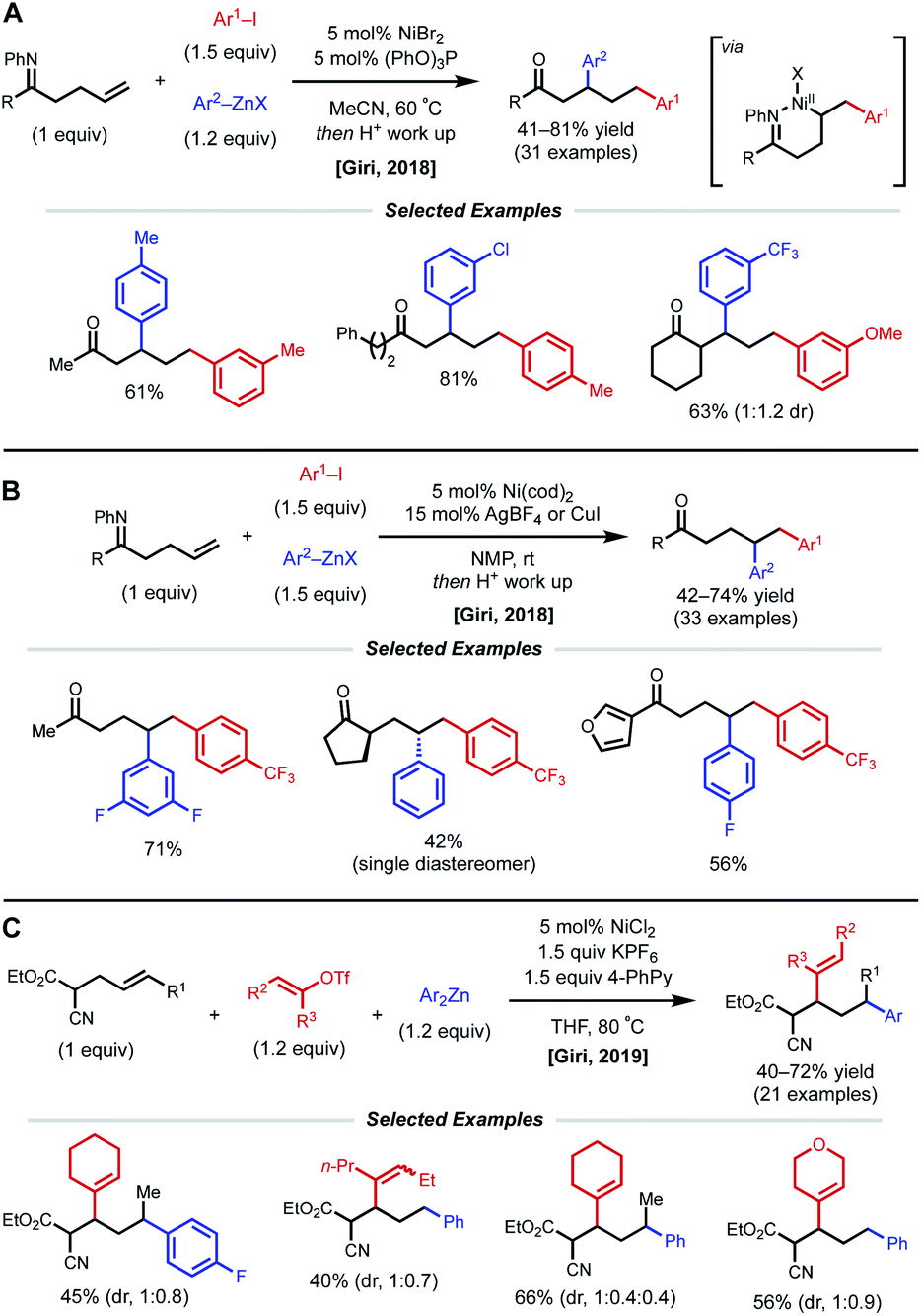 | ||
| Fig. 12 (A and B) Imine-directed 1,3- and 1,2-diarylation of γ,δ-unsaturated ketimines. (C) 1,3-Vinylarylation of γ,δ-unsaturated α-cyanoesters. | ||
A more general radical addition strategy toward reacting allylic carbonates with acyl chlorides and perfluoroalkyl halides in a reductive manifold was employed by Chu and coworkers in 2018 (Fig. 13A).34 Key to the success of this reaction was the careful positioning of a coordinating group in the substrate to allow for the generation of a stabilized 6-membered nickelacycle upon radical recombination. A series of mechanistic studies including substrate modification and TEMPO experiments support the directing group effect and radical addition pathway, respectively. The authors propose a catalytic cycle similar to that of the Nevado group.10 First, SET from low-valent nickel generates a perfluoroalkyl radical that adds to the allylic substrate (Fig. 13B). This newly generated secondary alkyl radical recombines with a Ni(II)–acyl intermediate to form a secondary Ni(III)(alkyl)(acyl) species that is stabilized by substrate coordination to allow for productive reductive elimination.
 | ||
| Fig. 13 (A) Reductive 1,2-perfluoroalkylacylation of allylic substrates. (B) Proposed catalytic cycle. | ||
Concurrent with these developments in 2018, the Engle laboratory reported a conjunctive cross-coupling method of β,γ- and γ,δ-unsaturated alkenyl amides with native monodentate amide functional groups instead of strong bidentate auxiliaries (Fig. 14).35 Using 15 mol% Ni(cod)2 with dimethyl fumarate (dmfu), an electron-deficient olefin (EDO) ligand, a wide range of aryl boronic acid neopentyl glycol esters and aryl iodides could be incorporated. Additionally, a broad scope of amides was tolerated including allyl amines with carbonyl-containing protecting groups. The authors propose a Ni(0)/Ni(II) mechanism analogous to that of the examples above. Experimental and computational evidence suggests that the substrate binds the nickel catalyst at the carbonyl oxygen rather than the amide nitrogen and that the EDO ligand greatly accelerates the key reductive elimination step.
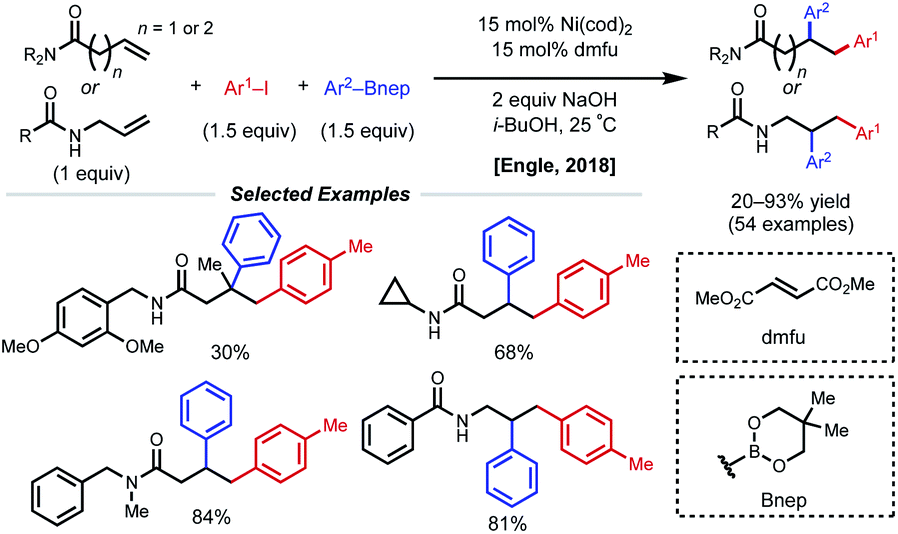 | ||
| Fig. 14 Simple amide-directed 1,2-diarylation. | ||
The Engle lab also developed a nickel-catalyzed 1,2-allylmethylation of N-allyl heterocycles (Fig. 15).36 The use of presumably labile monodentate directing groups enables a unique three-component nickel-catalyzed conjunctive cross-coupling that incorporates an allyl electrophile while preserving the resulting alkene product. The authors demonstrate compatibility with a variety of heterocycle directing groups such as saccharin, phthalimide, pyridone, pyrazole, triazole, tetrazole, and benzoxazole. Whether the reaction occurs through a Ni(0)/Ni(II) or Ni(I)/Ni(III) cycle was not confidently determined. Computational efforts underscored the importance of nickel binding to the pendant alkene of the allyl fragment after the key insertion step, providing valuable insight for future endeavors using weakly coordinating, native directing groups.
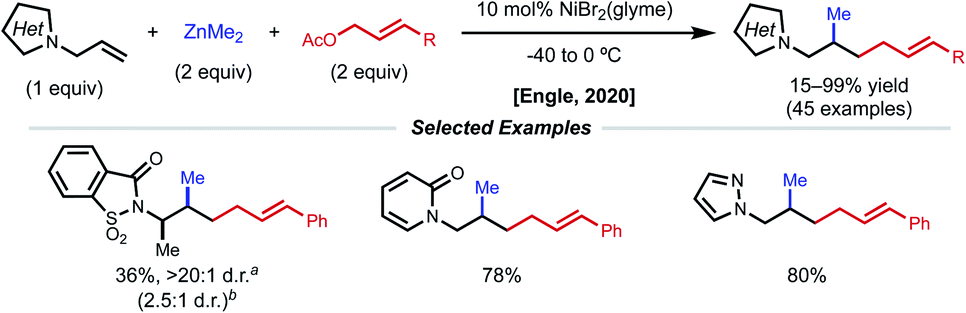 | ||
| Fig. 15 1,2-Allylmethylation of N-allyl heterocycles. | ||
In a further effort to expand conjunctive cross-coupling using native directivity, Engle and coworkers recently developed free carboxylic acid directed catalytic 1,2-diarylation and 1,2-arylalkenylation reactions of unactivated alkenes (Fig. 16).37 Aryl iodides and aryl/alkenyl boronic acid neopentyl glycol esters are used as coupling partners with 15 mol% Ni(cod)2 under “ligand-free” conditions. Notably, the use of ancillary ligands such as dmfu, bipyridine, triphenylphosphine significantly decreases yield of desired product. Internal alkenes give single regio- and diastereomers, albeit in low yields. By taking advantage of versatile carboxylic acids, the authors demonstrated rapid diversification of 1,2-diarylated products to afford 1,2,3-trifunctionalized building blocks via one-electron and two-electron synthetic logic.
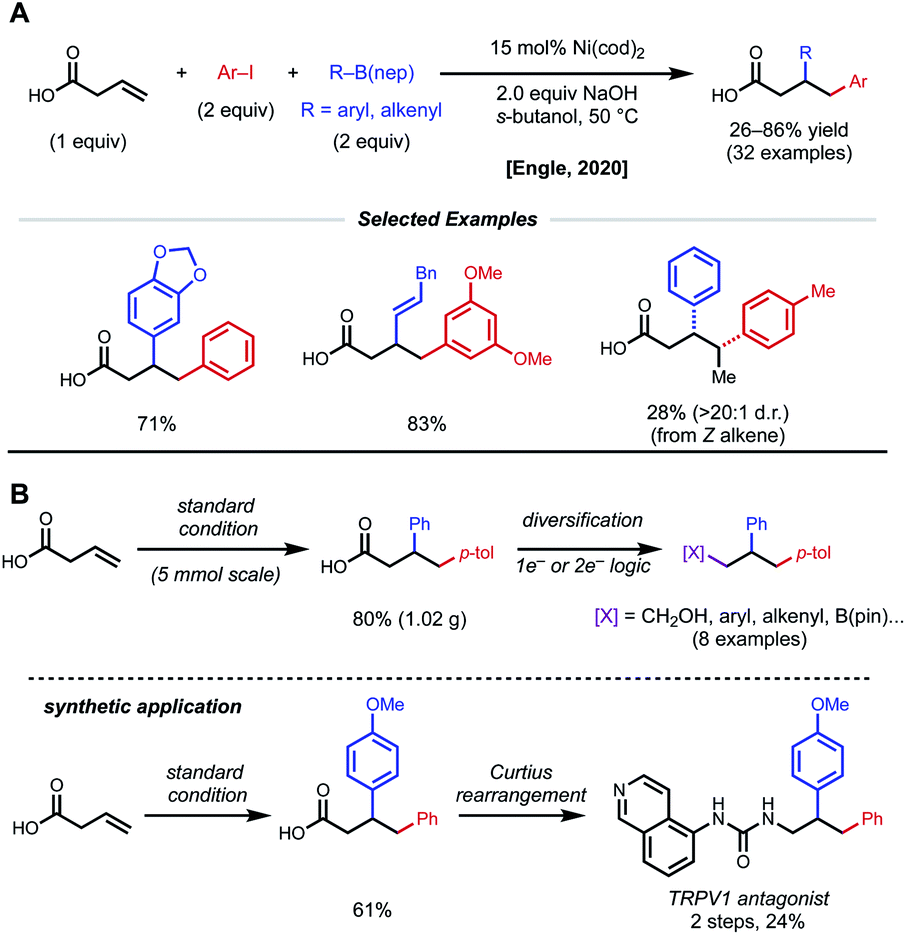 | ||
| Fig. 16 1,2-Diarylation of β,γ-unsaturated alkenyl carboxylic acids and synthetic applications. | ||
Another strategy toward nickel-catalyzed alkene difunctionalization involves the utilization of a co-catalytic photosensitizer. In 2019, Chu and coworkers reported a regioselective 1,2-alkylarylation of non-conjugated alkenes with tertiary alkyl oxalate reagents via synergistic photoredox/nickel catalysis. This transformation employs 3 mol% Ir-1 catalyst, 20 mol% NiCl2·DME, 20 mol% dtbbpy ligand, and 90 W blue LED light (Fig. 17A).38 The method tolerates alkenes bearing esters, carbamates, and carbonates, along with simple aliphatic chains. Notably, reactivity with 2-phenyl-1-butene demonstrates that proximal heteroatoms are not required. They found that the developed protocol works with both electron-deficient and electron-rich alkenes, but 1,1-disubstituted and 1,2-disubstituted alkenes are unreactive. Aryl bromides with electron-withdrawing groups and various pyridinyl bromides give moderate to good yields, while electron-rich and electron-neutral aryl iodides afford the desired products in slightly diminished yields. With respect to the alkyl oxalate scope, 6-membered oxalates perform better in comparison with 4- to 7-membered oxalates. The authors mention that the use of tertiary alkyl oxalates is critical to outcompete the two-component coupling between oxalates and aryl halides. The Chu group also conducted experiments to gain insight into the reaction mechanism. The use of stoichiometric amounts of TEMPO completely inhibited the reaction, and the use of cyclopropyl-substituted styrene gave a ring-opened product, indicating a radical relay process is present in the mechanism. A stoichiometric experiment with preformed aryl–Ni(II)–Br complex did not afford the desired difunctionalized product when subjected to the reaction conditions with either stoichiometric or catalytic amounts of Ir-1 catalyst. From these results, Chu and coworkers propose that the mechanism begins with formation of a tertiary alkyl radical via the oxidation of the oxalate by the photoexcited Ir-1 catalyst. The tertiary alkyl radical then adds onto the alkene forming a secondary alkyl radical, which is captured by the Ni(0) species to form an alkyl–Ni(I) intermediate. Oxidative addition of the alkyl–Ni(I) intermediate with an aryl halide generates an alkyl–Ni(III)–aryl intermediate. The high valent Ni(III) species undergoes rapid reductive elimination to afford the 1,2-alkylarylated product and Ni(I)–Br. Reduction of the Ni(I)–Br species by Ir(II) regenerates the ground-state Ir-1 catalyst, closing the dual catalytic cycles.
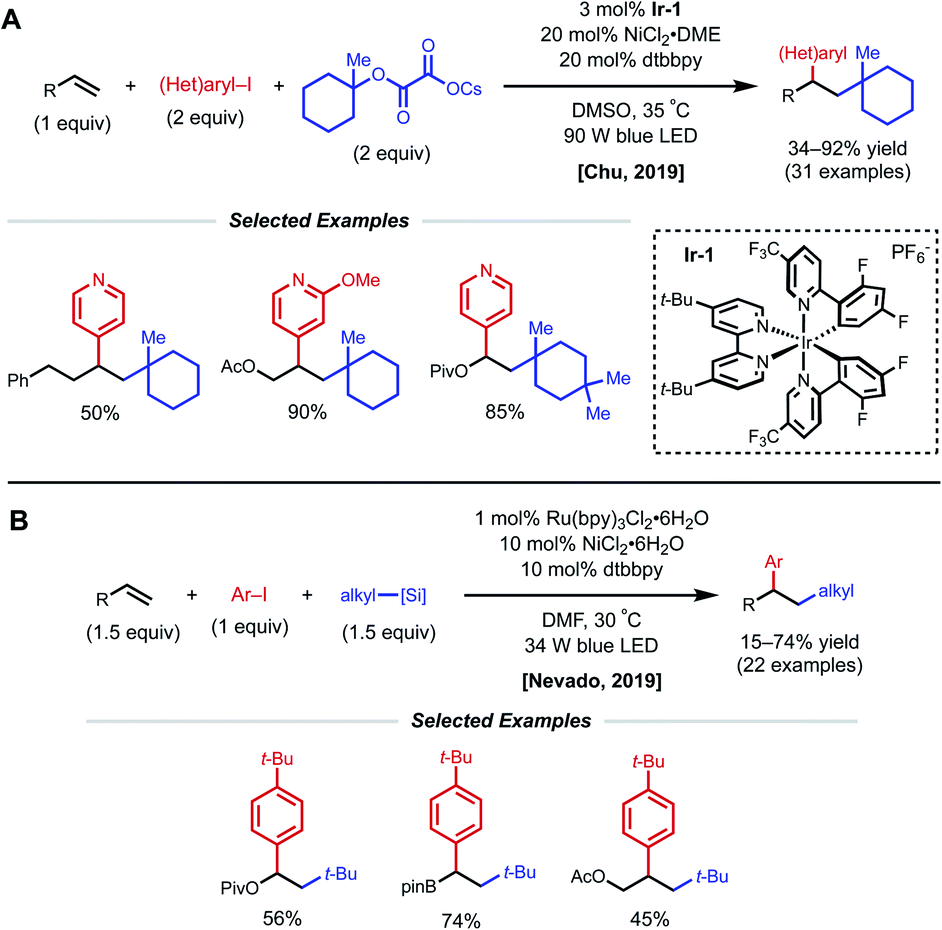 | ||
| Fig. 17 Photocatalytic 1,2-arylalkylation of alkenes reported by (A) Chu and coworkers and (B) Nevado and coworkers. | ||
Concurrent work under the same photocatalytic paradigm was done by the Nevado group to accomplish 1,2-alkylarylation of alkenes (Fig. 17B).39 Although most substrates are conjugated alkenes, a handful of non-conjugated alkenes are also viable. This transformation is achieved using aryl iodides, alkyl silicates, 1 mol% Ru(bpy)3Cl2·6H2O, 10 mol% NiCl2·6H2O, and 10 mol% dtbpy (L1) with irradiation by 34 W blue LED light. Under the optimized reaction conditions, non-conjugated alkenes such as vinyl pivalate, vinyl boronic acid pinacol ester, and allyl acetate using tertiary alkyl silicates afforded the 1,2-alkylarylated products. Within the same published work, the Nevado group additionally optimized a carbosulfonylation reaction with similar conditions using conjugated alkene substrates. The proposed mechanism for these reactions is comparable to that proposed by the Chu group.
Conclusions
In recent years, the surge of developments in nickel-catalyzed intermolecular 1,2-dicarbofunctionalization of alkenes has allowed for the progression of a wide range of elegant strategies toward achieving a general platform for universal alkene difunctionalization. While these notable efforts have laid a strong foundation for the field, several challenges have yet to be overcome. One such challenge is the expansion of the alkene scope. To date, terminal alkene substrates rarely show comparable reactivity in a regio- and stereoselective manner to their more substituted alkene counter parts (i.e. 1,1-, 1,2-disubstituted, tetrasubstituted). While strong directing groups enable this type of reactivity, we anticipate that more synthetically relevant functional groups in combination with novel ligand scaffolds will bridge the gap in this chemical space. A second unmet challenge is achieving asymmetric 1,2-dicarbofunctionalization in an intermolecular fashion. In combination with a broader alkene scope, this will undoubtedly serve as a powerful strategy for building complex molecular scaffolds. We anticipate that the combined progress on these and other fronts will ultimately result in complete regio- and stereocontrol in all different types of alkene dicarbofunctionalization.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was financially supported by the National Science Foundation (CHE-1800280), the Alfred P. Sloan Fellowship Program, and the Camille Dreyfus Teacher-Scholar Program. We further acknowledge the NSF for Graduate Research Fellowships (DGE-1346837, J. D. and DGE-1842471, O. A.) and the Kwanjeong Educational Foundation for a Graduate Fellowship (T. K.). We thank Raul Martin-Montero for carefully proofreading this manuscript.Notes and references
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef CAS PubMed.
- For reviews on C(sp3)–C(sp3) cross-coupling, see: (a) M. R. Netherton and G. C. Fu, Adv. Synth. Catal., 2004, 346, 1525–1532 CrossRef CAS; (b) A. C. Frisch and M. Beller, Angew. Chem., Int. Ed., 2005, 44, 674–688 CrossRef CAS PubMed; (c) A. Rudolph and M. Lautens, Angew. Chem., Int. Ed., 2009, 48, 2656–2670 CrossRef CAS PubMed; (d) R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1417–1492 CrossRef CAS PubMed; (e) X. Hu, Chem. Sci., 2011, 2, 1867–1886 RSC.
- S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299–309 CrossRef CAS PubMed.
- (a) J. B. Diccianni and T. Diao, Trends Chem., 2019, 1, 830–844 CrossRef; (b) B.-L. Lin, L. Liu, Y. Fu, S.-W. Luo, Q. Chen and Q.-X. Guo, Organometallics, 2004, 23, 2114–2123 CrossRef CAS; (c) V. P. Ananikov, ACS Catal., 2015, 5, 1964–1971 CrossRef CAS.
- (a) J. Derosa, V. T. Tran, V. A. van der Puyl and K. M. Engle, Aldrichimica Acta, 2018, 51, 21–32 CAS; (b) R. Giri and S. KC, J. Org. Chem., 2018, 83, 3013–3022 CrossRef CAS PubMed; (c) J.-S. Zhang, L. Liu, T. Chen and L.-B. Han, Chem.–Asian J., 2018, 13, 2277–2291 CrossRef CAS PubMed.
- For representative examples of 1,2-dicarbofunctionalization with other metals, see: (a) T.-H. Huang, H.-M. Chang, M.-Y. Wu and C.-H. Cheng, J. Org. Chem., 2002, 67, 99–105 CrossRef CAS PubMed; (b) L. Liao, R. Jana, K. B. Urkalan and M. S. Sigman, J. Am. Chem. Soc., 2011, 133, 5784–5787 CrossRef CAS PubMed; (c) K. M. Logan, K. B. Smith and M. K. Brown, Angew. Chem., Int. Ed., 2015, 54, 5228–5231 CrossRef CAS PubMed; (d) J. Terao, Y. Kato and N. Kambe, Chem.–Asian J., 2008, 3, 1472–1478 CrossRef CAS PubMed.
- For representative examples of nickel-catalyzed intramolecular 1,2-dicarbofunctionalization, see: (a) V. B. Phapale, E. Buñuel, M. García-Iglesias and D. J. Cárdenas, Angew. Chem., Int. Ed., 2007, 46, 8790–8795 CrossRef CAS PubMed; (b) S. KC, P. Basnet, S. Thapa, B. Shrestha and R. Giri, J. Org. Chem., 2018, 83, 2920–2936 CrossRef CAS PubMed; (c) K. Wang, Z. Ding, Z. Zhou and W. Kong, J. Am. Chem. Soc., 2018, 140, 12364–12668 CrossRef CAS PubMed; (d) Y.-L. Zheng and S. G. Newman, Angew. Chem., Int. Ed., 2019, 58, 18159–18164 CrossRef CAS PubMed; (e) D. Huang, D. Olivieri, Y. Sun, P. Zhang and T. R. Newhouse, J. Am. Chem. Soc., 2019, 141, 16249–16254 CrossRef CAS PubMed.
- T. Qin, J. Cornella, C. Li, L. R. Malins, J. T. Edwards, S. Kawamura, B. D. Maxwell, M. D. Eastgate and P. S. Baran, Science, 2016, 352, 801–805 CrossRef CAS PubMed.
- A. García-Domínguez, Z. Li and C. Nevado, J. Am. Chem. Soc., 2017, 139, 6835–6838 CrossRef PubMed.
- W. Shu, A. García-Domínguez, M. T. Quiróz, R. Mondal, D. J. Cárdenas and C. Nevado, J. Am. Chem. Soc., 2019, 141, 13812–13821 CrossRef CAS PubMed.
- J.-W. Gu, Q.-Q. Min, L.-C. Yu and X. Zhang, Angew. Chem., Int. Ed., 2016, 55, 12270–12274 CrossRef CAS PubMed.
- Z.-F. Yang, C. Xu, X. Zheng and X. Zhang, Chem. Commun., 2020, 56, 2642–2645 RSC.
- C. Xu, Z. F. Yang, L. An and X. Zhang, ACS Catal., 2019, 9, 8224–8229 CrossRef CAS.
- M. Chierchia, P. Xu, G. J. Lovinger and J. P. Morken, Angew. Chem., Int. Ed., 2019, 58, 14245–14249 CrossRef CAS PubMed.
- G. J. Lovinger and J. P. Morken, Eur. J. Org. Chem., 2019 DOI:10.1002/ejoc.201901600.
- (a) M. W. Campbell, J. S. Compton, C. B. Kelly and G. A. Molander, J. Am. Chem. Soc., 2019, 141, 20069–20078 CrossRef CAS PubMed; (b) S.-Z. Sun, Y. Duan, R. S. Mega, R. J. Somerville and R. Martin, Angew. Chem., Int. Ed., 2020, 59, 4370–4374 CrossRef CAS PubMed; (c) R. S. Mega, V. K. Duong, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2020, 59, 4375–4379 CrossRef CAS PubMed.
- (a) G. J. Lovinger and J. P. Morken, J. Am. Chem. Soc., 2017, 139, 17293–17296 CrossRef CAS PubMed; (b) S. M. Koo, A. J. Vendola, S. N. Momm and J. P. Morken, Org. Lett., 2020, 22, 666–669 CrossRef CAS PubMed.
- B. Shrestha, P. Basnet, R. K. Dhungana, S. KC, S. Thapa, J. M. Sears and R. Giri, J. Am. Chem. Soc., 2017, 139, 10653–10656 CrossRef CAS PubMed.
- S. Thapa, R. K. Dhungana, R. T. Magar, B. Shrestha, S. KC and R. Giri, Chem. Sci., 2018, 9, 904–909 RSC.
- S. KC, R. K. Dhungana, B. Shrestha, S. Thapa, N. Khanal, P. Basnet, R. W. Lebrun and R. Giri, J. Am. Chem. Soc., 2018, 140, 9801–9805 CrossRef CAS PubMed.
- S. KC, R. K. Dhungana, V. Aryal and R. Giri, Org. Process Res. Dev., 2019, 23, 1686–1694 CrossRef CAS.
- P. Gao, L.-A. Chen and M. K. Brown, J. Am. Chem. Soc., 2018, 140, 10653–10657 CrossRef CAS PubMed.
- D. Anthony, Q. Lin, J. Baudet and T. Diao, Angew. Chem., Int. Ed., 2019, 58, 3198–3202 CrossRef CAS PubMed.
- A. A. Kadam, T. L. Metz, Y. Qian and L. M. Stanley, ACS Catal., 2019, 9, 5651–5656 CrossRef CAS.
- J. Derosa, V. T. Tran, M. N. Boulous, J. S. Chen and K. M. Engle, J. Am. Chem. Soc., 2017, 139, 10657–10660 CrossRef CAS PubMed.
- J. Derosa, V. A. van der Puyl, V. T. Tran, M. Liu and K. M. Engle, Chem. Sci., 2018, 9, 5278–5283 RSC.
- V. A. van der Puyl, J. Derosa and K. M. Engle, ACS Catal., 2019, 9, 224–229 CrossRef CAS.
- R. Pan, C. Shi, D. Zhang, Y. Tian, S. Guo, H. Yao and A. Lin, Org. Lett., 2019, 21, 8915–8920 CrossRef CAS PubMed.
- W. Li, J. K. Boon and Y. Zhao, Chem. Sci., 2018, 9, 600–607 RSC.
- P. Basnet, R. K. Dhungana, S. Thapa, B. Shrestha, S. KC, J. M. Sears and R. Giri, J. Am. Chem. Soc., 2018, 140, 7782–7786 CrossRef CAS PubMed.
- P. Basnet, S. KC, R. K. Dhungana, B. Shrestha, T. J. Boyle and R. Giri, J. Am. Chem. Soc., 2018, 140, 15586–15590 CrossRef CAS PubMed.
- R. K. Dhungana, S. KC, P. Basnet, V. Aryal, L. J. Chesley and R. Giri, ACS Catal., 2019, 9, 10887–10893 CrossRef CAS.
- X. Zhao, H.-Y. Tu, L. Guo, S. Zhu, F.-L. Qing and L. Chu, Nat. Commun., 2018, 9, 3488 CrossRef PubMed.
- J. Derosa, R. Kleinmans, V. T. Tran, M. K. Karunananda, S. R. Wisniewski, M. D. Eastgate and K. M. Engle, J. Am. Chem. Soc., 2018, 140, 17878–17883 CrossRef CAS PubMed.
- V. T. Tran, Z. Li, T. J. Gallagher, J. Derosa, P. Liu and K. M. Engle, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie201915454.
- J. Derosa, T. Kang, V. T. Tran, S. R. Wisniewski, M. K. Karunananda, T. C. Jankins, K. L. Xu and K. M. Engle, Angew. Chem., Int. Ed., 2020, 58, 1202–1205 Search PubMed.
- L. Guo, H.-Y. Tu, S. Zhu and L. Chu, Org. Lett., 2019, 21, 4771–4776 CrossRef CAS PubMed.
- A. García-Domínguez, R. Mondal and C. Nevado, Angew. Chem., Int. Ed., 2019, 58, 12286–12290 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2020 |