
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolid-state NMR spectroscopy: an advancing tool to analyse the structure and properties of metal–organic frameworks
Eike
Brunner
 * and
Marcus
Rauche
* and
Marcus
Rauche
Chair of Bioanalytical Chemistry, Faculty of Chemistry and Food Chemistry, TU Dresden, 01062 Dresden, Germany. E-mail: eike.brunner@tu-dresden.de
First published on 6th April 2020
Abstract
Metal–organic frameworks (MOFs) gain increasing interest due to their outstanding properties like extremely high porosity, structural variability, and various possibilities for functionalization. Their overall structure is usually determined by diffraction techniques. However, diffraction is often not sensitive for subtle local structural changes and ordering effects as well as dynamics and flexibility effects. Solid-state nuclear magnetic resonance (ssNMR) spectroscopy is sensitive for short range interactions and thus complementary to diffraction techniques. Novel methodical advances make ssNMR experiments increasingly suitable to tackle the above mentioned problems and challenges. NMR spectroscopy also allows study of host–guest interactions between the MOF lattice and adsorbed guest species. Understanding the underlying mechanisms and interactions is particularly important with respect to applications such as gas and liquid separation processes, gas storage, and others. Special in situ NMR experiments allow investigation of properties and functions of MOFs under controlled and application-relevant conditions. The present minireview explains the potential of various solid-state and in situ NMR techniques and illustrates their application to MOFs by highlighting selected examples from recent literature.
 Eike Brunner | Eike Brunner received a diploma in physics (1987) and a Ph.D. (1989) from the University of Leipzig. Following a postdoctoral research stay at the Fritz Haber Institute of the Max Planck Society in 1990/1991, he was habilitated in 1996 (University of Leipzig) and received a Heisenberg fellowship from the DFG. After a research stay at the University of California at Berkeley in 1997/1998, he became Professor of Biophysics at the University of Regensburg (2001). Since 2007, he has been Professor for Bioanalytical Chemistry at TU Dresden. His research focuses on the NMR spectroscopic investigation of materials such as biominerals and MOFs. |
 Marcus Rauche | Marcus Rauche studied chemistry and received his Bachelor's (2013) and Master's (2015) degrees from TU Dresden. In 2016, he started working on his PhD thesis at TU Dresden dealing with the NMR spectroscopic characterisation of flexible MOFs. |
Metal–organic frameworks: general aspects and recent analytical challenges
Metal–organic frameworks (MOFs)1–5 are built from inorganic secondary building units (SBUs) and organic ligands (cf.Fig. 1). These components are linked together by coordination bonds thus forming highly porous and functional crystalline solids with specific surface areas up to 7800 m2 g−1.5 Various novel MOFs have been synthesized employing a multitude of different SBUs and linkers during the last few years.4 MOFs stand out due to their extremely high porosity compared with other porous materials. Linker functionalization offers the possibility to introduce special functionalities into the structure and to vary the pore size/geometry in a controlled fashion. For these reasons, MOFs are intensively studied as one of the most promising platforms for gas storage, separation, and purification and also for catalysis.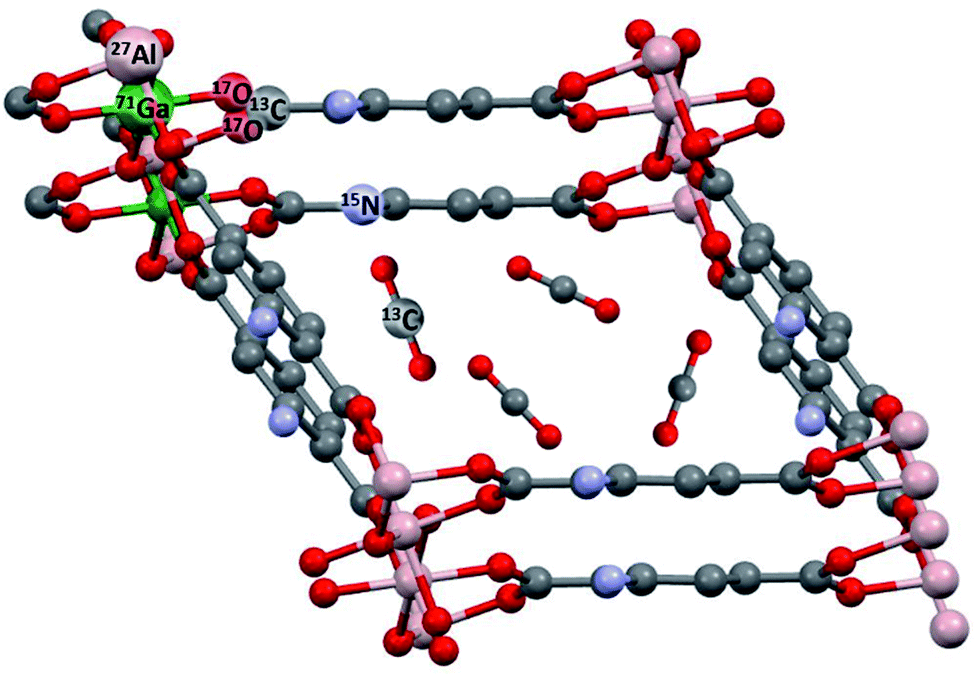 | ||
| Fig. 1 Typical NMR-active nuclei found in MOFs like MIL-53 (ref. 6) and their modifications. NH2-functionalized linkers are depicted as used in MIL-53-NH2(as).7 This topology was also prepared as a mixed-metal compound MIL-53(Al/Ga),8 see below. In situ NMR can detect adsorbed molecules like 13CO2 and their interactions with the MOF. | ||
The crystal structure of MOFs is commonly determined by diffraction techniques. However, MOF lattices exhibit various important properties which cannot or only partly be addressed by diffraction methods. The following challenges for MOF characterization can for example be tackled by NMR:
Local structure and order
As an example, MOFs with mixed metal sites or mixed linkers often pose problems because the distribution of the different metals or linkers in the framework is hardly accessible for diffraction methods. Incorporation and distribution of functionalities like linker side chains or nanoparticle catalysts in MOF crystallites is another example.Local and global flexibility
MOF lattices can exhibit different types of flexibility which can be subdivided into local and global effects. Globally “rigid” MOF structures can exhibit local motions, e.g., of the linkers, linker side chains, or functional groups. Such motions can be important for properties like adsorption behaviour. Furthermore, guest adsorption can result in subtle local structural changes even in “rigid” MOFs. A small subgroup of MOFs exhibits very pronounced global porosity switching in the crystalline solid state. These compounds are denoted as flexible, switchable, or “3rd generation” MOFs.3 Selected examples are MIL-53 (MIL: Materials of Institute Lavoisier6) and DUT-8 (DUT: Dresden University of Technology9). DUT-8 exhibits an adsorption-induced gating transition accompanied by a tremendous unit cell volume change of more than 100%! The mechanisms underlying these pronounced transitions are still insufficiently understood and need further investigation.Host–guest interactions and functional behaviour
In situ techniques10 are important for the investigation of MOFs under conditions relevant for later applications like adsorption and storage, separation, and catalysis, or for use as sensors. Such studies often require special experimental setups for in situ experiments on MOFs, for example under controlled loading, external gas pressure, educt flow in catalytic reactions, external electric fields, or mechanical pressure.Principles of solid-state NMR spectroscopy
Solid-state nuclear magnetic resonance (ssNMR) spectroscopy provides short-range, i.e., local structural information and is sensitive for local dynamics/thermal motions.Short-range internal interactions
NMR in condensed matter was reported for the first time in 1946 (ref. 11 and 12) (Nobel prize 1952). In 1948, Pake13 described the very broad and characteristic 1H NMR line shape of crystal water molecules in gypsum, which is now called the Pake doublet. It is dominated by the homo-nuclear magnetic dipole–dipole interaction (dipole coupling) between the two neighbouring 1H spins in crystal water. In this special case, straightforward line shape analysis allows inter-nuclear distance determination. Heteronuclear dipole coupling between different nuclear spins (e.g., 13C and 15N) is another distance-dependent interaction. This means dipole coupling makes distance information accessible for ssNMR experiments without the need of long-range order in contrast to diffraction-based crystallography. Further important interactions are the chemical shift anisotropy (CSA), the electric quadrupole interaction for nuclei with spin quantum numbers I > ½, or electron–nucleus interactions, i.e., hyperfine couplings in compounds with paramagnetic sites (see below).14–16 CSA reflects the spatial anisotropy of the chemical shift in solids (cf.Fig. 4 and related text below). The electric quadrupole interaction depends on the electric field gradient tensor at the site of the detected nucleus and dominates, e.g., the 2H NMR line shape in deuterated solids (cf.Fig. 3). These favourable properties verify ssNMR spectroscopy to be an excellent tool for structural analyses of solids.14–16High-resolution ssNMR spectroscopy
Typically, the simultaneous presence of various line-broadening interactions of comparable strength leads to broad but featureless Gaussian lines in solids. The invention of the magic angle spinning (MAS) technique17 was a major breakthrough for ssNMR spectroscopy. It relies on rapid sample spinning around an axis tilted against the external magnetic field B0 by the “magic angle” of 54.74°. This averages anisotropic interactions in analogy to the influence of isotropic molecular re-orientation in liquids and allows resolution of lines with different chemical shifts in solids. Continuous methodical development over decades now allows ultrafast MAS experiments at rates beyond 120 kHz.18 Ultrafast spinning is particularly important for systems with strong homo-nuclear dipole couplings between more than two spins19 like 1H-rich solids.20 Combination of MAS with cross-polarization (CP)21 enables sensitive detection of well-resolved CP MAS NMR spectra22 of intrinsically insensitive nuclei like 13C, 15N, or others. Cross-polarization is often used in two-dimensional (2D) hetero-nuclear correlation (HETCOR) spectroscopy. Application of MAS, however, removes anisotropic interactions like the dipole couplings which bear the distance information. Special experiments allow selective re-introduction of desired interactions under MAS. An example is rotational echo double resonance (REDOR).23 It allows measurement of inter-nuclear distances in hetero-nuclear spin pairs like 13C–15N.Solid-state NMR spectroscopy of MOFs
Solid-state NMR spectroscopy is increasingly applied to MOFs24–26 based on the detection of NMR-active nuclei in their framework (cf.Fig. 1). Detection of metal ions is possible for SBUs containing NMR-active nuclei. Examples are 27Al or 51V which exhibit high natural abundance and a moderately high magnetic moment.27 Various commonly used metal ions in MOFs exhibit NMR-active isotopes with low natural abundance and small magnetic moments, for example 25Mg and 67Zn.28–30 Special pulse sequences like CPMG (Carr–Purcell–Meiboom–Gill) can enhance the NMR signal thus enabling detection of such spectra. In addition, quadrupolar nuclei often exhibit very broad lines. Broad-band pulses or sequences like WURST (wideband uniform rate smooth truncation) must then be used to excite a sufficiently large frequency range.31Direct ssNMR detection of the nuclei in paramagnetic ions like Cr, Mn, Fe, Co, Ni, or Cu is often difficult or impossible. But it is sometimes feasible to study signals of nuclei close to the SBU. Examples are 13C or 17O in coordinating carboxylates of the linker molecules. These neighbouring nuclei act as “reporters” via their interactions with the paramagnetic centres in the SBU. It is possible to exploit characteristic effects like the Fermi contact and pseudo-contact shifts caused by paramagnetic centres, which are observable in the NMR signals of neighbouring nuclear spins.16 This is also true for SBUs with di-metal sites, e.g., typical paddle wheel units with antiferromagnetic coupling in their ground state32 because excited states may nevertheless be paramagnetic and can influence the NMR signals.33 In any case, the NMR signals of nuclei in the SBUs as well as neighbouring nuclei in the linkers like 1H/2H, 13C, or 15N contain valuable information as illustrated for selected examples in the following.
Local structure and order
Benzaqui et al.34 visualized subtle structural differences in the Al environment between hydrated and dehydrated MIL-96(Al) which were not found by X-ray diffraction. 27Al MAS NMR and 2D double-quantum–single-quantum (DQ–SQ) MAS NMR spectroscopy were applied. As can be seen in Fig. 2, dehydration of the sample results in a structural change of the Al3 site as reflected by a splitting into two signals denoted by Al3 and Al3′. Density functional theory (DFT) calculations provided a structural model for the responsible interactions between water molecules and the SBUs. The calculations agree well with the measured 27Al chemical shifts and quadrupole coupling constants. These observations emphasize the high sensitivity of ssNMR spectroscopy for subtle local structural changes in SBUs and highlight the usefulness of DFT combined with ssNMR spectroscopy.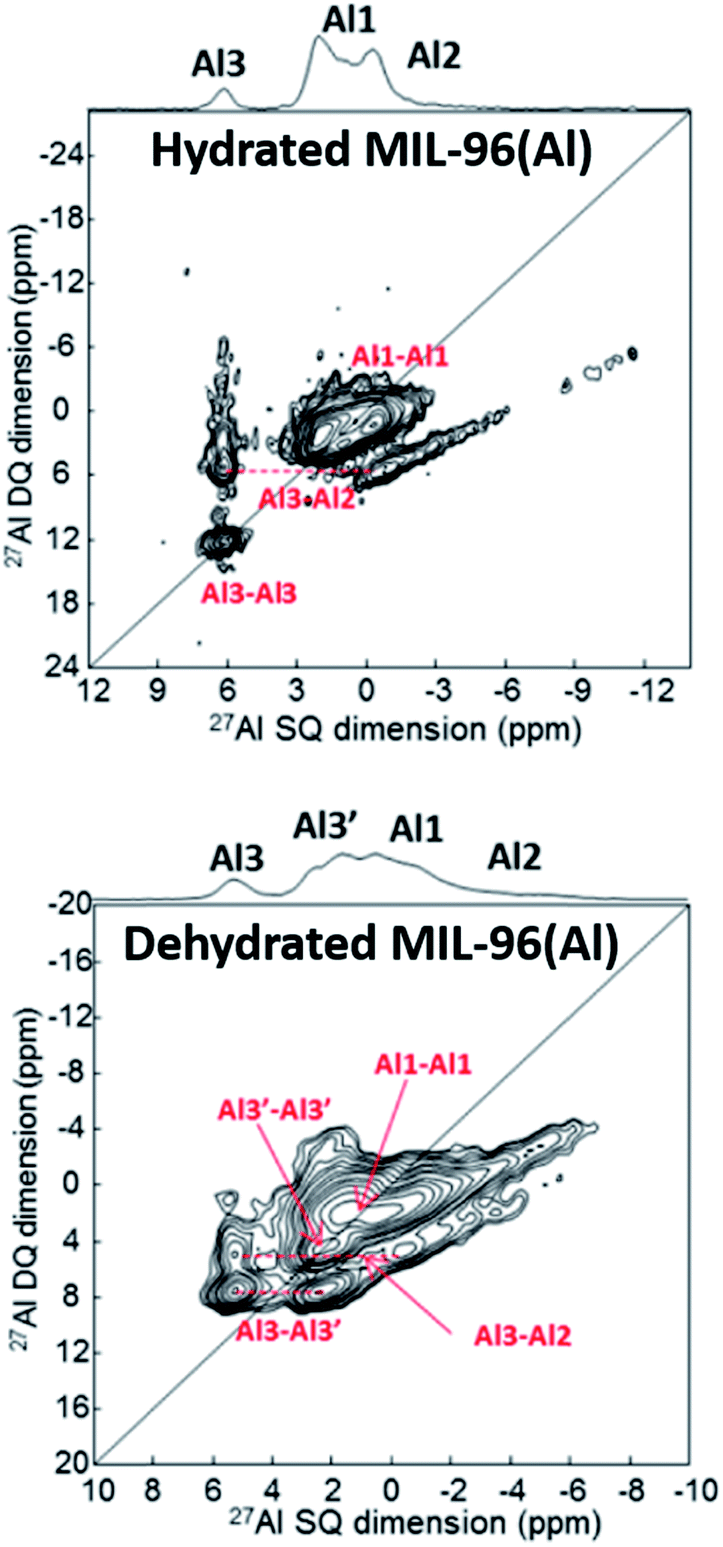 | ||
| Fig. 2 27Al DQ–SQ spectra of MIL-96(Al) showing the influence of the hydration state upon the environment of Al. Adapted with permission.34 Copyright 2017, American Chemical Society. | ||
Mali et al.35 studied the ion distribution in the metal oxo trimers of mixed-metal MIL-100(Fe/Al) by combining 27Al MAS NMR, EPR (electron paramagnetic resonance) and Mössbauer spectroscopy as well as EXAFS (extended X-ray absorption fine structure) and DFT. Paramagnetic shifts of 27Al occurred due to hyperfine interactions with the paramagnetic Fe species. These shifts are proportional to the hyperfine coupling constant and could be well reproduced by first-principles calculations. Random distribution of Al and Fe in the metal oxo trimers of MIL-100(Fe/Al) could be inferred from this study.
Oxygen, e.g., in carboxylate-based linkers, is located next to the metal centres and is sensitive to changes in the SBU. 17O NMR spectroscopy usually requires isotope enrichment because the natural abundance of this isotope only amounts to 0.05%. Bignami et al.8 described a cost-effective route to 17O-enriched terephthalic acid linkers in mixed-metal MIL-53(Al/Ga) by the so-called dry gel conversion (DGC) method. A pre-dried gel containing linker molecules and metal salt is treated in an autoclave with vapor generated by a small amount of water at elevated temperature. The cation distribution in this mixed metal MOF could be studied by 17O multiple-quantum (MQ) MAS and DOR (double rotation) NMR spectroscopy. The presence of Al and Ga bridged by one hydroxyl species was observed. It is thus concluded that the two metals do not separate and MIL-53(Al/Ga) is a true mixed-metal material. The discussed publications nicely demonstrate that ssNMR spectroscopy can elucidate the metal ion distribution in mixed-metal MOFs.
The linker distribution in MOFs with mixed linkers was studied by the Reimer group36 using the REDOR technique. The two linkers exhibit different functional groups selectively labelled with either 13C or 15N. The 13C–15N REDOR experiments performed allowed measurement of the average distance between the 13C and 15N nuclei located in the two different linkers. Based on molecular dynamics (MD) simulations, it became possible to derive a linker distribution model consistent with the measured distance.
Local thermal motions
Zhu et al.37 described the incorporation of a so-called “molecular shuttle” performing translational motions into a MOF denoted as UWDM-4 (UWDM: University of Windsor Dynamic Material). The motions of this molecular shuttle within the framework were demonstrated by 13C CP MAS NMR and 2D exchange spectroscopy (EXSY, see below).Linker molecules or certain parts of them like phenyl rings and attached side groups often exhibit thermal motions such as rotations and vibrations. 2H NMR spectroscopy is a powerful method to visualize and study such local motions.38 In favourable cases, not only can the specific motional mode of the linker be deduced, but it may also be possible to measure the motional frequency/exchange rate by line shape analysis of the 2H NMR signal.38 For example, Khudozhitkov et al.39 investigated 180° phenyl ring flips about the C2 axis of the linkers in [M2(bdc)2(dabco)] (M = Zn, Ni, Co, Cu; bdc = 1,4-benzene dicarboxylate, terephthalate; dabco = 1,4 diazabicyclo[2.2.2]octane). As indicated in Fig. 3 (right), the phenyl ring flip rate k slows down from 6000 kHz to 20 kHz after loading the MOF with dimethylformamide (DMF). For slow flips (20 kHz), the line shape almost perfectly corresponds to a Pake doublet with ca. 120 kHz frequency distance between the two intensity maxima. This is characteristic for rigid 2H–C groups. The line shape changes significantly for fast 180° flips (6000 kHz) present in the DMF-free sample. Line shape analysis allows determination of the exchange rate quantitatively.
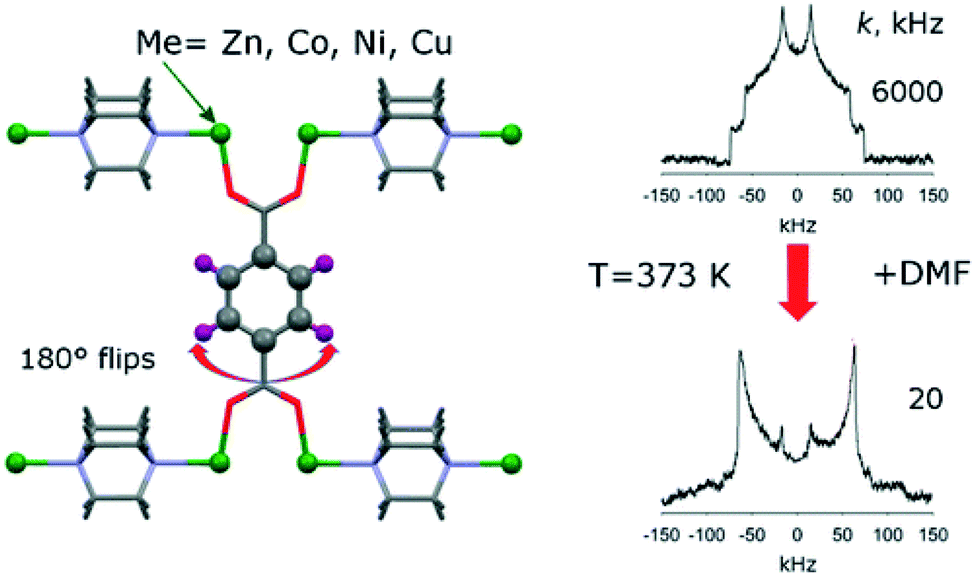 | ||
| Fig. 3 Illustration of typical 2H NMR line shapes and the influence of 180° flips of linker phenyl groups. Reproduced39 with permission. Copyright 2015, American Chemical Society. | ||
In situ NMR and host–guest interaction studies
Experiments under MAS
MAS NMR studies of host–guest interactions are usually performed on sealed samples loaded with liquids or gases. They sometimes require high pressures and/or temperatures. Standard MAS NMR probes operate at temperatures between ca. 120 and 370 K. Common MAS rotors do not allow high pressures because the rotor caps are not pressure tight. Therefore, increasing efforts are made to construct special in situ MAS probes and rotors. A sealed MAS NMR rotor system allowing temperatures and pressures up to 520 K and 100 bar was developed by Hu et al.40 Even higher temperatures at lower pressures are feasible, e.g., using high-frequency (hf)-heated rotor inserts.41 The synthesis of various materials including MOFs can be monitored by in situ liquid-state and solid-state NMR spectroscopy as reviewed by Haouas.42 An elegant combination of liquid- and solid-state NMR spectroscopy is denoted as CLASSIC (combined liquid- and solid-state in situ crystallization). It is performed on identical samples under MAS by interleaved detection of directly excited and cross-polarization spectra.43Gas loaded samples must be kept in tight containers like sealed glass ampoules. Such ampoules can even be inserted into MAS rotors if manufactured properly.44 Experiments are then performed at constant loading with a defined number of molecules or atoms before sample sealing. Temperature variation causes pressure changes because the adsorption equilibrium is temperature-dependent. However, exact pressure determination is then difficult.
Experiments under controlled pressure
It is thus often advantageous to run in situ gas adsorption measurements in a fashion where both thermodynamic variables, pressure and temperature, are under external control. Such in situ NMR data can directly be correlated with the results of volumetric adsorption experiments. On the other hand, this approach is hardly compatible with MAS because it requires the connection of the sample container with an outside gas reservoir for equilibration after temperature variation. However, in situ NMR without MAS can nevertheless deliver valuable information because thermal motions of the adsorbed species often result in sufficiently narrow signals. A corresponding experimental setup was described in ref. 45.Examples for host–guest interaction studies
Carboxylate 13C nuclei in MOFs with linkers coordinated with the SBUs via carboxylate groups are another probe close to the SBU. The natural isotopic abundance of 13C amounts to only 1.1%. Isotope enrichment of carboxylate carbon positions in 2,6-ndc (ndc: naphthalene dicarboxylate) was established and used to study the flexible compound DUT-8(Ni).46 The labelled carboxylates are then sensitively and selectively detected by 13C CP MAS NMR and a striking influence of solvent polarity upon the chemical shift was observed. This might indicate a specific interaction between the paddle wheel units and polar solvent molecules in contrast to non-polar solvents.The Senker group functionalized terephthalate linkers in MIL-101(Al) and MIL-101(Cr) by incorporation of 2-pyridyl urea (URPy) side groups resulting in the formation of so-called hydrogen bond donor–donor–acceptor patterns.47 Their influence upon the sorption selectivity was investigated by competitive adsorption of 2-aminopyridine (2-AP) and 3-aminopyridine (3-AP). 15N ssNMR spectroscopy on 15N-labeled samples was applied to elucidate structural reasons for the significantly higher selectivity coefficients for 3-AP observed on MIL-101-URPy compared with MIL-101-NH2. This highlights the potential of ssNMR spectroscopy to elucidate the structure of adsorption complexes in MOFs.
Adsorption of CO2 on MOFs is increasingly studied because CO2 capture and storage is an environmentally important problem. MOFs are potent materials not only for gas storage but also for separation, e.g., removal of CO2 from other gases like methane. Various 13C NMR investigations of CO2 adsorbed on MOFs were reported meanwhile as can be found in the recent review by Witherspoon et al.48 Interaction of CO2 with open Mg sites in Mg-MOF-74 revealed a line shape characteristic of chemical shift anisotropy but with a reduced width compared to that of rigid CO2.49 It was concluded that adsorbed CO2 jumps between different open metal sites. The resulting partial averaging of the chemical shift anisotropy gives rise to the observed residual chemical shift anisotropy. Similar line shapes are also observed in other MOFs with and without open metal sites.50–54Fig. 4 shows the 1D in situ13C NMR and the 2D EXSY spectrum of adsorbed 13CO2 in a MOF. Isotropically reorienting free CO2 gas in the inter-particle space gives rise to a narrow signal at ca. 126 ppm. Adsorbed CO2 shows a characteristic broad signal of ca. 30 ppm overall line width due to the aforementioned residual chemical shift anisotropy. Rigid 13CO2 would give a similarly shaped but much broader line of 335 ppm width. The 2D EXSY experiment visualizes slow exchange processes on a large time scale compared with (2πΔν)−1 where Δν is the resonance frequency difference between the involved sites. At short mixing times, only diagonal signals are observed. The strong off-diagonal intensity at 1 s mixing time shows the presence of slow CO2 exchange between MOF crystallites within tmix. This means the chosen mixing time defines the time scale of exchange.
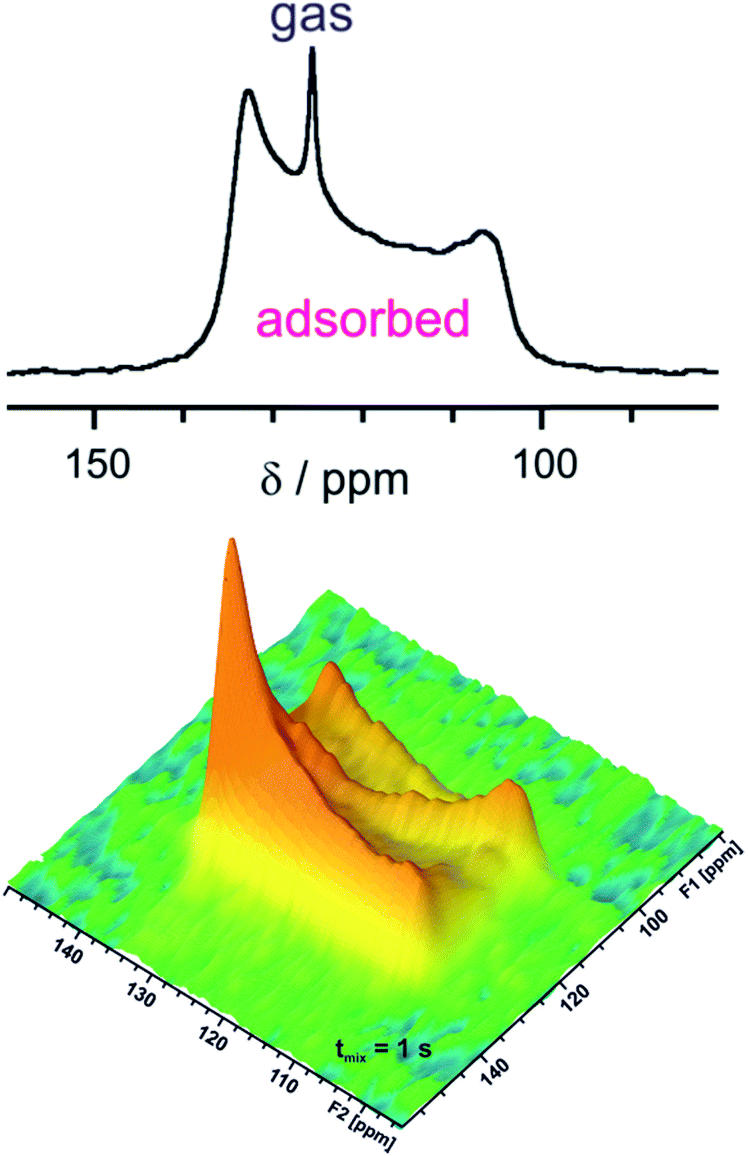 | ||
| Fig. 4 In situ 13C NMR spectrum (5 bar pressure, top) and 2D EXSY spectrum (7 bar pressure, bottom) for CO2 adsorbed on [Zn2(bdc)2dabco] measured at 232 K with 1 s mixing time tmix. Data adapted50 with permission. Copyright 2015, Elsevier. | ||
Adsorption of methane by MOFs is also of great interest because natural gas is a promising carrier for mobile applications. Yan et al.55 used 2H NMR spectroscopy of samples with deuterated linkers combined with neutron powder diffraction on samples loaded with deuterated methane to investigate methane adsorption in the MOFs MFM-112a, MFM-115a, and MFM-132a (MFM: Manchester Framework Material). It was shown that an optimal combination of linker dynamics, pore geometry, and favorable binding sites is responsible for the favorable methane uptake by these materials.
The selectivity of carbon dioxide adsorption from 13CO2/13CH4 mixtures by flexible MOFs was investigated by in situ13C NMR spectroscopy.56 This is possible since the characteristic residual chemical shift anisotropy of adsorbed CO2 and the narrow signal of the gas phase (cf.Fig. 4) allow quantitative determination of the ratio between adsorbed and gaseous CO2. Adsorbed and gas phase CH4 molecules can be discriminated by their different isotropic chemical shifts. Microscopic diffusion of pure and mixed methane and carbon dioxide in ZIF-11 (ZIF: zeolitic imidazolate framework) was studied by high field diffusion NMR spectroscopy.57
129Xe NMR spectroscopy58 delivers information on surface chemical properties and the porosity of materials. It is frequently used to characterize host–guest interactions and dynamic phenomena in MOFs. Note that MOFs are also considered for noble gas mixture separations, e.g. of Xe/Kr mixtures.
The influence of linker functionalization upon the noble gas adsorption properties of CAU-1 (CAU: Christian Albrecht University, Kiel) could be visualized and studied by in situ129Xe NMR spectroscopy using hyperpolarized 129Xe.59 The chemical shift sensitively reflects even minor changes in the accessible pore volume caused by functionalisation. This highlights the potential of 129Xe NMR spectroscopy for porosity characterization.
Flexibility studied by in situ129Xe NMR spectroscopy
Hyperpolarized 129Xe NMR spectroscopy was also used to characterize the adsorption-induced structural changes in MIL-53(Al).60 Further studies on MIL-53(Al) were carried out with and without MAS on sealed samples loaded with defined amounts of xenon.61 This allowed characterizing the exchange processes between adsorbed xenon in the large-pore form and in the narrow-pore form of the MOF at temperatures where both states co-exist. High-pressure in situ129Xe NMR spectroscopy was used to study adsorption-induced structural changes in flexible MOFs. It was, for example, shown that the gating transition in DUT-8(Ni) is an immediate and complete structural switching process accompanied by complete pore filling at the gate pressure.45Recently, the counterintuitive negative gas adsorption (NGA) phenomenon62 in DUT-49 and its homologs was studied by in situ129Xe NMR spectroscopy (cf.Fig. 5).63,64 NGA is caused by sudden mesopore contraction at a certain pressure (here: p/p0 = 0.18; p0 denotes the boiling pressure at the given temperature). Some of the gas adsorbed before in the open-pore state is released (NGA) during pore contraction which results in an increased density and intimate contact of xenon with the walls in the contracted-pore state. This causes a tremendous chemical shift jump of ca. 100 ppm. Re-opening of the pore system at higher relative pressure then gives rise to an increasingly intense signal at a lower chemical shift (Fig. 5).
 | ||
| Fig. 5 Top: 129Xe NMR signals of adsorbed xenon on DUT-49 measured at 200 K for various relative pressures during the adsorption experiment (op: open-pore phase, cp: contracted-pore phase). Bottom: Chemical shift of adsorbed xenon in DUT-49 at 200 K measured during the adsorption experiment (red symbols denote signals of DUT-49op; blue symbols denote those of DUT-49cp). Data adapted63 with permission. Copyright 2017, American Chemical Society. | ||
Finally, MOFs are also considered for catalysis and electrode applications. In situ NMR spectroscopic approaches for monitoring catalytic reactions65 and electro-adsorption in electrochemical devices under operating conditions66 already exist and are used for other materials.
Conclusions and outlook
Driven by continuous methodical development over decades, ssNMR spectroscopy is nowadays one of the most powerful methods for the determination of the local structure and dynamics of various materials. To date, NMR spectroscopy offers a wide variety of advanced experiments which are increasingly applied to characterize MOFs and their interactions with guest species. On one hand, ssNMR spectroscopy, usually in combination with MAS, allows studying of the local framework structure and adsorption-induced changes of the framework. In the future, these studies will substantially gain from ongoing developments such as the construction of high-field spectrometers beyond 1 GHz 1H resonance frequency, ultrafast MAS probes, and increasing application of novel spin hyperpolarization techniques like DNP.67 The first DNP-enhanced ssNMR experiments on MOFs have already been reported in the literature.68–70On the other hand, in situ NMR approaches with and without MAS allow the characterization of processes like gas adsorption, catalytic reactions, electro-adsorption, and others. In situ NMR techniques will find increasing interest in the future. This will in turn stimulate further methodical developments such as novel MAS probes allowing even higher temperatures and pressures. Miniaturized coil design in combination with spin hyperpolarization experiments may allow in situ NMR studies in microfluidic applications. Such future in situ NMR experiments will be particularly valuable for the characterization of properties and functions of MOFs with respect to envisioned applications. This will improve the knowledge base and help in designing tailored MOF syntheses in the future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support from the Deutsche Forschungsgemeinschaft within the research unit FOR 2433 “MOF-Switches”.Notes and references
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 974 CrossRef CAS PubMed.
- S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334 CrossRef CAS PubMed.
- S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695 CrossRef CAS PubMed.
- The Chemistry of Metal-Organic Frameworks, ed. S. Kaskel, Wiley-VCH, Weinheim, 2016 Search PubMed.
- I. M. Hönicke, I. Senkovska, V. Bon, I. A. Baburin, N. Bönisch, S. Raschke, J. D. Evans and S. Kaskel, Angew. Chem., Int. Ed., 2018, 57, 13780 CrossRef PubMed.
- C. Serre, F. Millange, C. Thouvenot, M. Noguès, G. Marsolier, D. Louër and G. Férey, J. Am. Chem. Soc., 2002, 124, 13519 CrossRef CAS PubMed.
- T. Ahnfeldt, D. Gunzelmann, T. Loiseau, D. Hirsemann, J. Senker, G. Férey and N. Stock, Inorg. Chem., 2009, 48, 3057 CrossRef CAS PubMed.
- G. P. M. Bignami, Z. H. Davis, D. M. Dawson, S. A. Morris, S. E. Russell, D. McKay, R. E. Parke, D. Iuga, R. E. Morris and S. E. Ashbrook, Chem. Sci., 2018, 9, 850 RSC.
- N. Kavoosi, V. Bon, I. Senkovska, S. Krause, C. Atzori, F. Bonino, J. Pallmann, S. Paasch, E. Brunner and S. Kaskel, Dalton Trans., 2017, 49, 4685 RSC.
- V. Bon, E. Brunner, A. Pöppl and S. Kaskel, Adv. Funct. Mater., 2020, 1907847 CrossRef.
- F. Bloch, W. W. Hansen and M. Packard, Phys. Rev., 1946, 69, 127 CrossRef.
- E. M. Purcell, H. C. Torrey and R. V. Pound, Phys. Rev., 1946, 69, 37 CrossRef CAS.
- G. E. Pake, J. Chem. Phys., 1948, 16, 327 CrossRef CAS.
- M. J. Duer, Solid-State NMR Spectroscopy, Principles and Applications, Blackwell Science, 2002 Search PubMed.
- K. J. D. MacKenzie and M. E. Smith, Multinuclear Solid-State NMR of Inorganic Materials, Pergamon Materials Series, Elsevier Science 2002 Search PubMed.
- A. J. Pell, G. Pintacuda and C. P. Grey, Prog. Nucl. Magn. Reson. Spectrosc., 2019, 111, 1 CrossRef CAS PubMed.
- E. R. Andrew, A. Bradbury and R. G. Eades, Nature, 1958, 182, 1659 CrossRef CAS.
- S. Penzel, A. Oss, M.-L. Org, A. Samoson, A. Böckmann, M. Ernst and B. H. Meier, J. Biomol. NMR, 2019, 73, 19 CrossRef CAS PubMed.
- E. Brunner, D. Freude, B. C. Gerstein and H. Pfeifer, J. Magn. Reson., 1990, 90, 90 CAS.
- U. Sternberg, R. Witter, I. Kuprov, J. M. Lamley, A. Oss, J. R. Lewandowski and A. Samoson, J. Magn. Reson., 2018, 291, 32 CrossRef CAS PubMed.
- A. Pines, M. G. Gibby and J. S. Waugh, J. Chem. Phys., 1973, 59, 569 CrossRef CAS.
- J. Schaefer and E. O. Stejskal, J. Am. Chem. Soc., 1976, 98, 1031 CrossRef CAS.
- T. Gullion and J. Schaefer, J. Magn. Reson., 1989, 81, 196 CAS.
- H. C. Hoffmann, M. Debowski, P. Müller, S. Paasch, I. Senkovska, S. Kaskel and E. Brunner, Materials, 2012, 5, 2537 CrossRef CAS.
- B. E. G. Lucier, S. Chen and Y. Huang, Acc. Chem. Res., 2018, 51, 319 CrossRef CAS PubMed.
- Y. T. A. Wong, V. Martins, B. E. G. Lucier and Y. Huang, Chem.–Eur. J., 2019, 25, 1848 CrossRef CAS PubMed.
- M. de Oliveira Jr, D. Seeburg, J. Weiß, S. Wohlrab, G. Buntkowsky, U. Bentrup and T. Gutmann, Catal. Sci. Technol., 2019, 9, 6180 RSC.
- V. Stavila, M. E. Foster, J. W. Brown, R. W. Davis, J. Edgington, A. I. Benin, R. A. Zarkesh, R. Parthasarathi, D. W. Hoyt, E. D. Walter, A. Andersen, N. M. Washton, A. S. Lipton and M. D. Allendorf, Chem. Sci., 2019, 10, 9880 RSC.
- J. Xu, V. V. Terskikh and Y. Huang, Chem.–Eur. J., 2013, 19, 4432 CrossRef CAS PubMed.
- A. Sutrisno, V. V. Terskikh, Q. Shi, Z. Song, J. Dong, S. Y. Ding, W. Wang, B. R. Provost, T. D. Daff, T. K. Woo and Y. Huang, Chem.–Eur. J., 2012, 18, 12251 CrossRef CAS PubMed.
- L. A. O'Dell and R. W. Schurko, Chem. Phys. Lett., 2008, 464, 97 CrossRef.
- K. Trepte, S. Schwalbe and G. Seifert, Phys. Chem. Chem. Phys., 2015, 17, 17122 RSC.
- Z. Ke, L. E. Jamieson, D. M. Dawson, S. E. Ashbrook and M. Bühl, Solid State Nucl. Magn. Reson., 2019, 101, 31 CrossRef CAS PubMed.
- M. Benzaqui, R. S. Pillai, A. Sabetghadam, V. Benoit, P. Normand, J. Marrot, N. Menguy, D. Montero, W. Shepard, A. Tissot, C. Martineau-Corcos, C. Sicard, M. Mihaylov, F. Carn, I. Beurroies, P. L. Llewellyn, G. De Weireld, K. Hadjiivanov, J. Gascon, F. Kapteijn, G. Maurin, N. Steunou and C. Serre, Chem. Mater., 2017, 29, 10326 CrossRef CAS.
- G. Mali, M. Mazaj, I. Arčon, D. Hanžel, D. Arčon and Z. Jagličić, J. Phys. Chem. Lett., 2019, 10, 1464 CrossRef CAS PubMed.
- X. Kong, H. Deng, F. Yan, J. Kim, J. A. Swisher, B. Smit, O. M. Yaghi and J. A. Reimer, Science, 2013, 341, 882 CrossRef CAS PubMed.
- K. Zhu, C. A. O'Keefe, V. N. Vukotic, R. W. Schurko and S. J. Loeb, Nat. Chem., 2015, 7, 514 CrossRef CAS PubMed.
- M. R. Hansen, R. Graf and H. W. Spiess, Acc. Chem. Res., 2013, 46, 1996 CrossRef CAS PubMed.
- A. E. Khudozhitkov, D. I. Kolokolov, A. G. Stepanov, V. A. Bolotov and D. N. Dybtsev, J. Phys. Chem. C, 2015, 119, 28038 CrossRef CAS.
- J. Z. Hu, M. Y. Hu, Z. Zhao, S. Xu, A. Vjunov, H. Shi, D. M. Camaioni, C. H. F. Peden and J. A. Lercher, Chem. Commun., 2015, 51, 13458 RSC.
- H. Kirchhain, J. Holzinger, A. Mainka, A. Spörhase, S. Venkatachalam, A. Wixforth and L. van Wüllen, Solid State Nucl. Magn. Reson., 2016, 78, 37 CrossRef CAS PubMed.
- M. Haouas, Materials, 2018, 11, 1416 CrossRef PubMed.
- C. E. Hughes, P. A. Williams and K. D. M. Harris, Angew. Chem., Int. Ed., 2014, 53, 8939 CrossRef CAS PubMed.
- D. Freude, M. Hunger and H. Pfeifer, Chem. Phys. Lett., 1982, 91, 307 CrossRef CAS.
- H. C. Hoffmann, B. Assfour, F. Epperlein, N. Klein, S. Paasch, I. Senkovska, S. Kaskel, G. Seifert and E. Brunner, J. Am. Chem. Soc., 2011, 133, 8681 CrossRef CAS PubMed.
- M. Rauche, S. Ehrling, S. Krause, I. Senkovska, S. Kaskel and E. Brunner, Chem. Commun., 2019, 55, 9140 RSC.
- T. Wittmann, C. B. L. Tschense, L. Zappe, C. Koschnick, R. Siegel, R. Stäglich, B. V. Lotsch and J. Senker, J. Mater. Chem. A, 2019, 7, 10379 RSC.
- V. J. Witherspoon, J. Xu and J. A. Reimer, Chem. Rev., 2018, 118, 10033 CrossRef CAS PubMed.
- X. Kong, E. Scott, W. Ding, J. A. Mason, J. R. Long and J. A. Reimer, J. Am. Chem. Soc., 2012, 134, 14341 CrossRef CAS PubMed.
- V. Bon, J. Pallmann, E. Eisbein, H. C. Hoffmann, I. Senkovska, I. Schwedler, A. Schneemann, S. Henke, D. Wallacher, R. A. Fischer, G. Seifert, E. Brunner and S. Kaskel, Microporous Mesoporous Mater., 2015, 216, 64 CrossRef CAS.
- R. M. Marti, J. Howe, C. R. Morelock, M. S. Conradi, K. Walton, D. S. Sholl and S. E. Hayes, J. Phys. Chem. C, 2017, 121, 25778 CrossRef CAS.
- Y. Zhang, B. E. G. Lucier and Y. Huang, Phys. Chem. Chem. Phys., 2016, 18, 8327 RSC.
- Y. Lu, B. E. G. Lucier, Y. Zhang, P. Ren, A. Zheng and Y. Huang, Phys. Chem. Chem. Phys., 2017, 19, 6130 RSC.
- A. C. Forse, M. I. Gonzalez, R. L. Siegelman, V. J. Witherspoon, S. Jawahery, R. Mercado, P. J. Milner, J. D. Martell, B. Smit, B. Blümich, J. R. Long and J. A. Reimer, J. Am. Chem. Soc., 2018, 140, 1663 CrossRef CAS PubMed.
- Y. Yan, D. I. Kolokolov, I. da Silva, A. G. Stepanov, A. J. Blake, A. Dailly, P. Manuel, C. C. Tang, S. Yang and M. Schröder, J. Am. Chem. Soc., 2017, 139, 13349 CrossRef CAS PubMed.
- M. Sin, N. Kavoosi, M. Rauche, J. Pallmann, S. Paasch, I. Senkovska, S. Kaskel and E. Brunner, Langmuir, 2019, 35, 3162 CrossRef CAS PubMed.
- E. M. Forman, B. R. Pimentel, K. J. Ziegler and R. P. Lively, Microporous Mesoporous Mater., 2017, 248, 158 CrossRef CAS.
- T. Ito and J. Fraissard, J. Chem. Phys., 1982, 76, 5225 CrossRef CAS.
- T. W. Kemnitzer, C. B. L. Tschense, T. Wittmann, E. A. Rössler and J. Senker, Langmuir, 2018, 34, 12538 CrossRef CAS PubMed.
- M.-A. Springuel-Huet, A. Nossov, Z. Adem, F. Guenneau, C. Volkringer, T. Loiseau, G. Férey and A. Gédéon, J. Am. Chem. Soc., 2010, 132, 11599 CrossRef CAS PubMed.
- R. Giovine, C. Volkringer, M.-A. Springuel-Huet, A. Nossov, F. Blanc, J. Trébosc, T. Loiseau, J.-P. Amoureux, O. Lafon and F. Pourpoint, J. Phys. Chem. C, 2017, 121, 19262 CrossRef CAS.
- S. Krause, V. Bon, I. Senkovska, U. Stoeck, D. Wallacher, D. M. Többens, S. Zander, R. S. Pillai, G. Maurin, F.-X. Coudert and S. Kaskel, Nature, 2016, 532, 348 CrossRef CAS PubMed.
- J. Schaber, S. Krause, S. Paasch, I. Senkovska, V. Bon, D. M. Többens, D. Wallacher, S. Kaskel and E. Brunner, J. Phys. Chem. C, 2017, 121, 5195 CrossRef CAS.
- F. Kolbe, S. Krause, V. Bon, I. Senkovska, S. Kaskel and E. Brunner, Chem. Mater., 2019, 31, 6193 CrossRef CAS.
- W. Zhang, S. Xu, X. Han and X. Bao, Chem. Soc. Rev., 2012, 41, 192 RSC.
- F. Blanc, M. Leskes and C. P. Grey, Acc. Chem. Res., 2013, 46, 1952 CrossRef CAS PubMed.
- A. B. Barnes, G. De Paëpe, P. C. A. van der Wel, K.-N. Hu, C. G. Joo, V. S. Bajaj, M. L. Mak-Jurkauskas, J. R. Sirigiri, J. Herzfeld, R. J. Temkin and R. G. Griffin, Appl. Magn. Reson., 2008, 34, 237 CrossRef CAS PubMed.
- A. J. Rossini, A. Zagdoun, M. Lelli, J. Canivet, S. Aguado, O. Ouari, P. Tordo, M. Rosay, W. E. Maas, C. Copéret, D. Farrusseng, L. Emsley and A. Lesage, Angew. Chem., Int. Ed., 2012, 51, 123 CrossRef CAS PubMed.
- F. Pourpoint, A. S. L. Thankamony, C. Volkringer, T. Loiseau, J. Trébosc, F. Aussenac, D. Carnevale, G. Bodenhausen, H. Vezin, O. Lafon and J.-P. Amoureux, Chem. Commun., 2014, 50, 933 RSC.
- T. Kobayashi, F. A. Perras, T. W. Goh, T. L. Metz, W. Huang and M. Pruski, J. Phys. Chem. Lett., 2016, 7, 2322 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |