
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConcerted proton-electron transfer oxidation of phenols and hydrocarbons by a high-valent nickel complex†
Katherine J.
Fisher
,
Margalit L.
Feuer
,
Hannah M. C.
Lant
,
Brandon Q.
Mercado
,
Robert H.
Crabtree
 * and
Gary W.
Brudvig
*
* and
Gary W.
Brudvig
*
Department of Chemistry, Yale University, New Haven, CT 06520, USA. E-mail: robert.crabtree@yale.edu; gary.brudvig@yale.edu
First published on 6th January 2020
Abstract
The high-valent nickel(III) complex Ni(pyalk)2+ (2) was prepared by oxidation of a nickel(II) complex, Ni(pyalk)2 (1) (pyalk = 2-pyridyl-2-propanoate). 2 and derivatives were fully characterized by mass spectrometry and X-ray crystallography. Electron paramagnetic resonance spectroscopy and X-ray photoelectron spectroscopy confirm that the oxidation is metal-centered. 2 was found to react with a variety of phenolic and hydrocarbon substrates. A linear correlation between the measured rate constant and the substrate bond dissociation enthalpy (BDE) was found for both phenolic and hydrocarbon substrates. Large H/D kinetic isotope effects were also observed for both sets of substrates. These results suggest that 2 reacts through concerted proton-electron transfer (CPET). Analysis of measured thermodynamic parameters allows us to calculate a bond dissociation free energy (BDFE) of ∼91 kcal mol−1 for the O–H bond of the bound pyalk ligand. These findings may shed light onto CPET steps in oxidative catalysis and have implications for ligand design in catalytic systems.
Introduction
Reactions in which protons and electrons move in a single, concerted step (concerted proton-electron transfer, or CPET) play a significant role in many organic, inorganic, and bioinorganic catalytic systems. CPET at high-valent metal centers has been proposed or observed in the catalytic mechanisms ranging from enzymatic reactions1,2 to water-oxidation catalysis3,4 to organic synthesis.5,6 In many of these systems, the proton and electron are transferred to a high-valent metal–oxo species; however, another strategy involves the transfer of the proton to the ligand scaffold instead. This approach is also relevant to some bioinorganic systems. For instance, in nickel superoxide dismutase (NiSOD), a CPET step is proposed to occur at a highly oxidized nickel(III) intermediate in which a coordinated thiol or amide moiety acts as a proton donor.7–9Understanding the CPET reactivity of high-valent metal centers may give some insight into their reactivity in catalytic systems. In particular, understanding CPET steps in systems in which the proton is transferred to the ligand may provide insight into catalytic systems which do not or cannot go through metal–oxo intermediates, such as copper- or nickel-containing water-oxidation catalysts or NiSOD.
In our previous studies, we found that the strongly donating ligand 2-pyridinyl-2-propanoate (pyalk) can stabilize metal centers in high oxidation states, including Ir(V).10 In addition, the pyalk alkoxide has been suggested to act as a proton shuttle in catalytic water oxidation.11,12 We reasoned that a high-valent metal compound stabilized by the pyalk ligand was a good candidate for fast CPET. We now describe the preparation of a stable Ni(III) species capable of reacting with a variety of O–H and C–H bonds via CPET.
Results and discussion
Characterization of Ni3+
We prepared 1, a new square-planar nickel(II) complex with two pyalk ligands arranged in a trans orientation (Scheme 1), which was characterized by X-ray crystallography, 1H NMR spectroscopy, and cyclic voltammetry (CV). The CV of 1 shows a reversible redox feature at 0.15 V vs. Fc/Fc+, suggesting that oxidation generates a stable Ni(III) species (Fig. 1).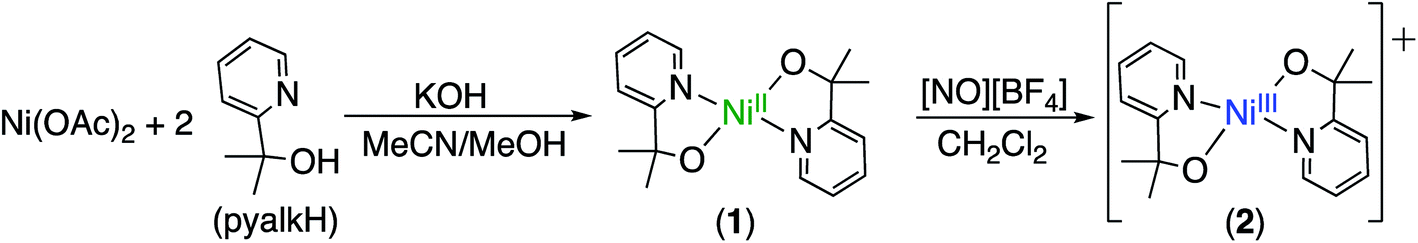 | ||
| Scheme 1 Synthetic route for preparation of NiII(pyalk)2 (1) and NiIII(pyalk)2+ (2). | ||
 | ||
| Fig. 1 (Left) X-ray crystal structure of Ni(pyalk)2 (1). Atoms are shown at the 50% probability level (1). (Right) Cyclic voltammogram of 1 in MeCN. The reversible wave at 0.15 V vs. Fc/Fc+ is assigned as a Ni(II/III) couple. | ||
In CH2Cl2, 1 was treated with [NO][BF4] or [NO][PF6](E0 = 0.6 V vs. Fc/Fc+) (Scheme 1). The solution immediately changed from light green to deep blue. UV-visible spectroscopy indicated the presence of two intense features in the absorption spectrum (λmax = 340 nm and 610 nm, Fig. S1†). This change suggested the formation of an oxidized species. Such intense absorption features in the visible and NIR regions are consistent with the data from other Ni(III) compounds,13 which led us to suspect that the Ni center had been oxidized. In addition, 1H NMR of 2 gave a broad paramagnetic spectrum, in contrast to the diamagnetic spectrum of 1 (Fig. S3†). Measurement of the solution magnetic susceptibility by the Evans method14 at room temperature gave a μeff of 1.87 for 2, consistent with the presence of one unpaired electron, as expected for a square-planar Ni(III) compound.
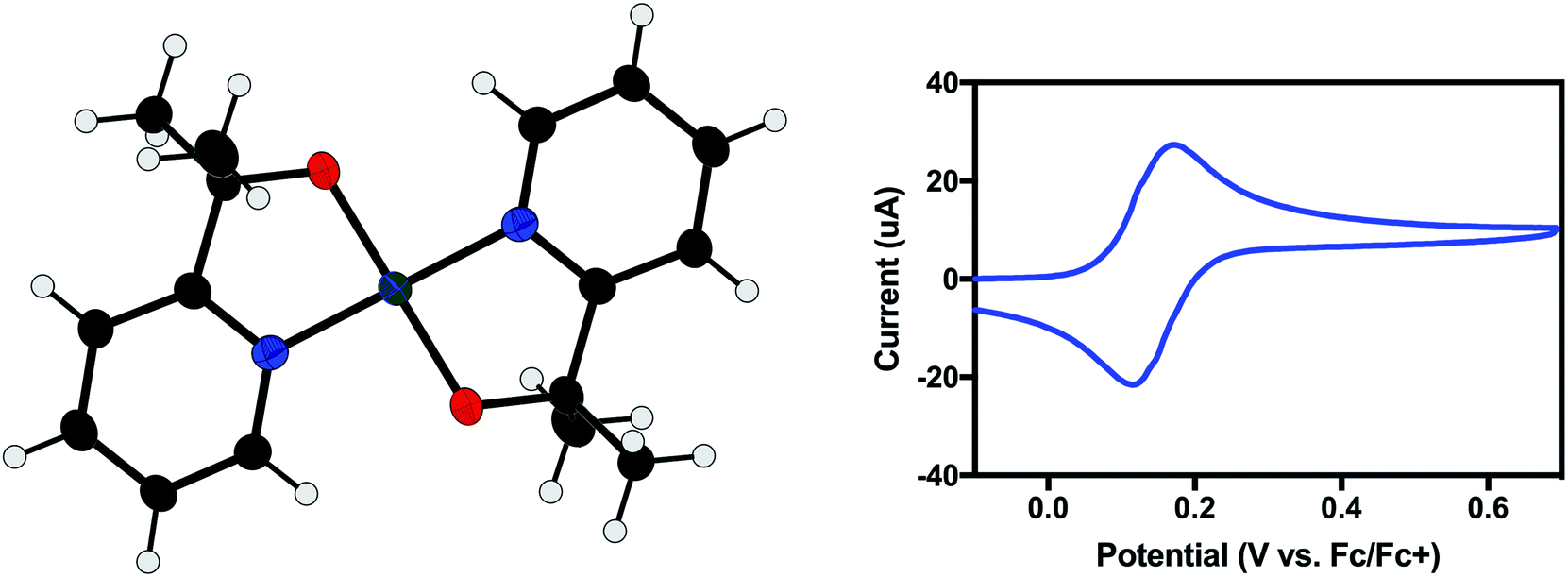
Electron paramagnetic resonance (EPR) spectroscopy showed the presence of a single S = ½ species (Fig. 2). A ligand centered oxidation would be expected to show an isotropic signal near g = 2.0. However, the observed rhombic spectrum had g values of gx = 2.077, gy = 2.091, gz = 2.274 (Fig. 2, left). These g values, as well as gave = 2.145, are consistent with a low-spin square-planar d7 Ni(III) complex.15
 | ||
| Fig. 2 (Left) Experimental (blue) and simulated (red) EPR spectra of 2 taken in CH2Cl2/toluene at 77 K. (Right) XPS spectrum of 2 (top) and 1 (bottom). The ∼1 eV shift is consistent with a one-electron oxidation of the nickel center. | ||
As demonstrated for a variety of iridium complexes,16–19 X-ray photoelectron spectroscopy (XPS) provides another method for identifying a metal-centered oxidation. For metal-centered redox events with no change to the ligand set, the binding energy is expected to increase with oxidation state. Between 1 and 2, the Ni 2p binding energy increases by 1.4 eV, consistent with a metal-centered oxidation from Ni(II) to Ni(III) (Fig. 2, right).
Electrospray ionization (ESI) mass spectrometry confirmed the molecular formula of 2. A solution of 2 in CH2Cl2 gave a peak at m/z = 330.09 with the expected isotopic pattern for nickel (Fig. S2a†). A mass spectrum of the parent compound, 1, was also taken, which showed a peak at m/z = 331.09 (Fig. S2b†), corresponding to [1 + H+]. Despite having the same elemental formula, 1 is uncharged, and thus is observed as the positively charged protonated species, whereas 2 bears a positive charge already, and so is observed without any associated ions. This result confirms that 2 is the one-electron oxidation product of 1. The ESI mass spectrum of 2 does show a small signal at m/z = 331.09, which may indicate that some reduction of 2 occurs during the mass spectral analysis or over time in CH2Cl2.
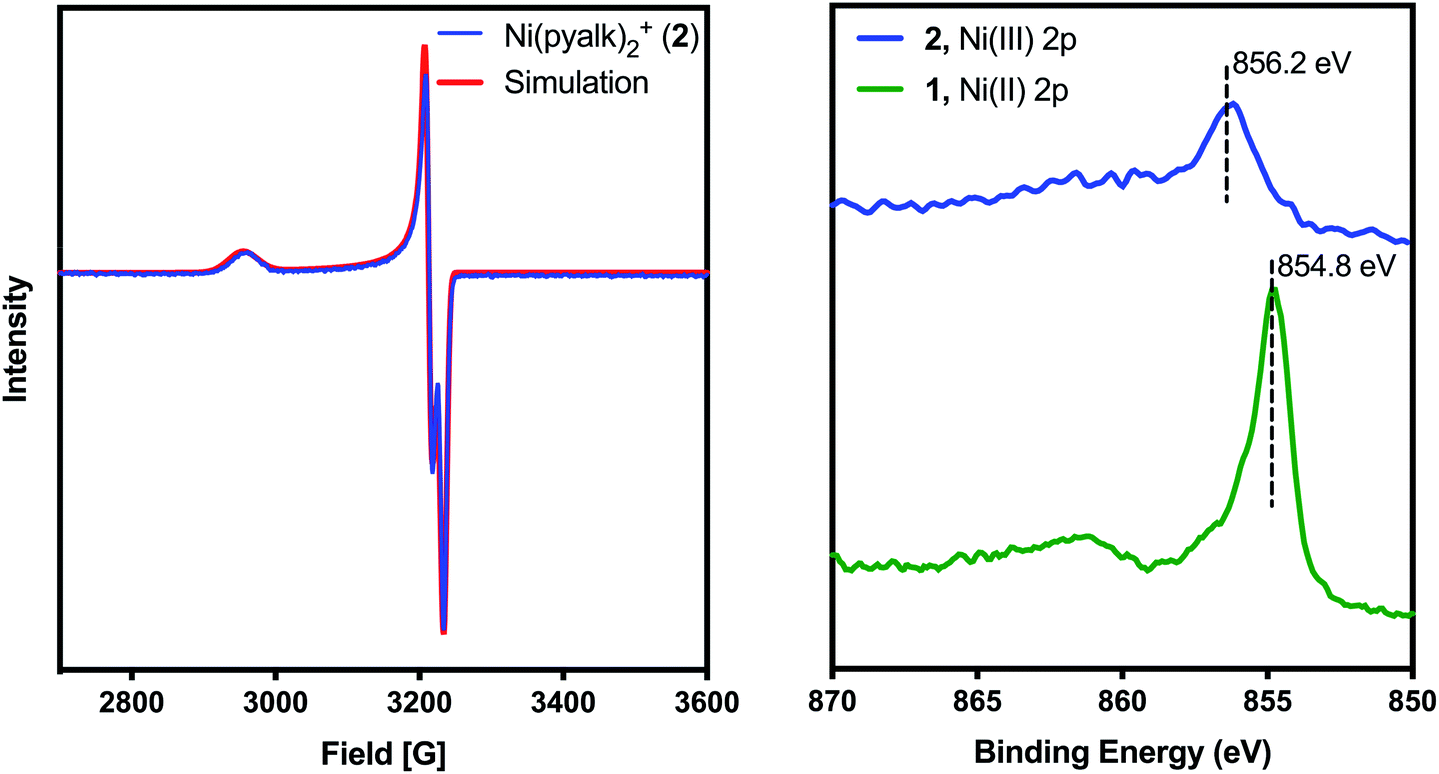
Despite our best efforts, we were unable to obtain a crystal structure of 2. While attempting to use pyridine as a co-solvent for crystallization, however, we observed that the solution changed from deep blue to bright orange (λmax = 420 nm, Fig. S1†). Crystals of this new complex, 3, were successfully grown from CH2Cl2/pentanes (Fig. 3, left). 3 proved to be an octahedral complex, with two equatorial pyalk ligands and two axial pyridines. 3 contains a PF6− anion, and a comparison of Ni–O bond lengths (Table S3†) indicates that the alkoxide arms of the pyalk ligands on 3 remain deprotonated, suggesting that 3 remains in the Ni(III) oxidation state. The EPR spectrum of 3 (Fig. 3, right) exhibits an axial signal with gx = 2.202, gy = 2.163, and gz = 2.030. A five-line hyperfine pattern is observed on the gz turning point, indicating coupling of the unpaired electron to the pyridyl nitrogens.20 We found that the pyridine ligands do not remain bound in solution unless excess pyridine is present; when crystals of 3 were dissolved in CH2Cl2, 3 immediately reverted to 2, as monitored by UV-visible spectroscopy (Fig. S1†). Upon addition of excess pyridine, the absorbance spectrum of 3 was re-established.
 | ||
| Fig. 3 (Left) X-ray crystal structure of 3. Atoms are shown at the 50% probability level. A non-coordinated pyridine has been omitted for clarity. (Right) Experimental (black) and simulated (red) EPR spectra of 3 in CH2Cl2/pyridine at −80 °C. | ||
Remarkably, 2 and 3 are stable at room temperature, both as solids and in solution; this is rare for Ni(III) compounds, which tend to be stable only between −80 °C and −20 °C.13,21,22
Oxidation of phenols
With the oxidation state of 2 established, we sought to test its oxidative reactivity. We had shown in previous studies that the pyalk ligand could be reversibly protonated while coordinated to first row transition metals;23 therefore, we hypothesized that 2 could undergo proton-coupled electron transfer (PCET) through the reaction proposed in Scheme 2.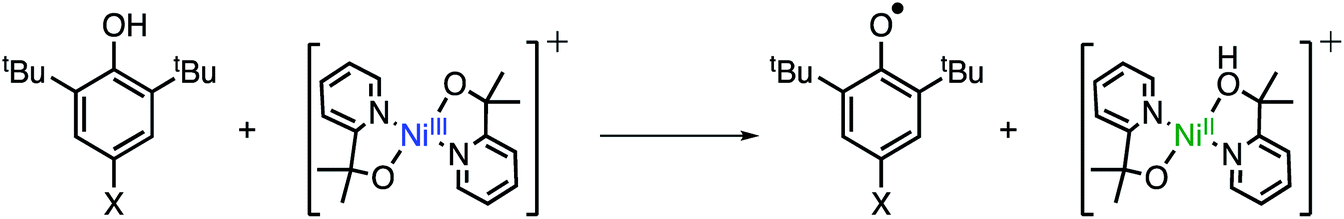 | ||
| Scheme 2 Proposed PCET pathway for oxidation of phenols. | ||
To test 2 for PCET reactivity, 2 was treated with 100 equivalents of 2,4,6-tri-tert-butylphenol (TTBP), a common substrate for PCET reactions. When the colorless solution of TTBP was added to the deep blue solution of 2 in CH2Cl2, the color changed immediately to bright blue. The appearance of the characteristic peaks at 383 nm and 400 nm in the absorbance spectrum of this solution (Fig. 4) indicated that the tri-tert-butylphenoxyl radical had been formed, causing the color change,24 and the disappearance of the features at 340 nm and 610 nm indicated that 2 had been consumed. In a similar reaction between 2 and 2,6-di-tert-butyl-phenol (DTBP), the radical coupling product 3,3′,5,5′-tetra-tert-butyl-[1,1′-bi(cyclohexylidene)]-2,2′,5,5′-tetraene-4,4′-dione was detected by gas chromatography-mass spectrometry (GC-MS, Fig. S4†). These results suggested that a PCET process was occurring.
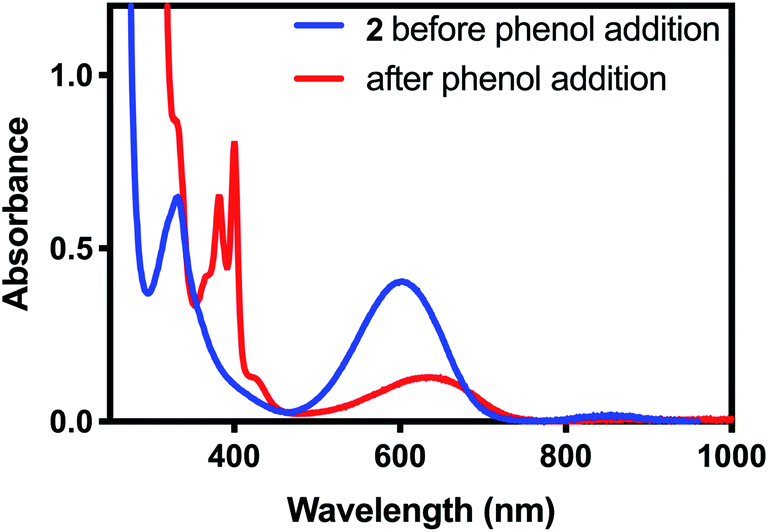 | ||
| Fig. 4 UV-visible spectrum of 2 before (blue) and after (red) addition of excess tri-tert-butylphenol. The red trace is consistent with the published UV-visible spectrum of the tri-tert-butylphenoxyl radical.24 | ||
For reactions of highly oxidized metal species with phenols, proton transfer followed by electron transfer (PT–ET), electron transfer followed by proton transfer (ET–PT), and concerted proton-electron transfer (CPET)/hydrogen atom transfer (HAT) mechanisms have all been observed.25–27 Analysis of kinetic measurements can be used to differentiate between these mechanisms. Therefore, the kinetics of the reaction of 2 with a series of para-substituted 2,6-di-t-butylphenols (4-X-2,6-DTBP) was studied by stopped-flow spectrophotometry in order to further investigate the mechanism of oxidation by 2. For these reactions, excess substrate was used to ensure pseudo-first order conditions (10–100 equivalents 4-X-2,6-DTBP). Representative UV-visible spectra as a function of time can be found in Fig. 5 and representative time traces can be found in Fig. S5 and S6.† Reactions with all substrates under these conditions fit well to a single exponential decay at a single wavelength (λ = 610 nm), for which global analysis gave kobs values. Plots of kobsvs. initial substrate concentration displayed a linear dependence (Fig. S7–S13†), allowing us to determine the second-order rate constant (k2) for each substrate (Table S1†).
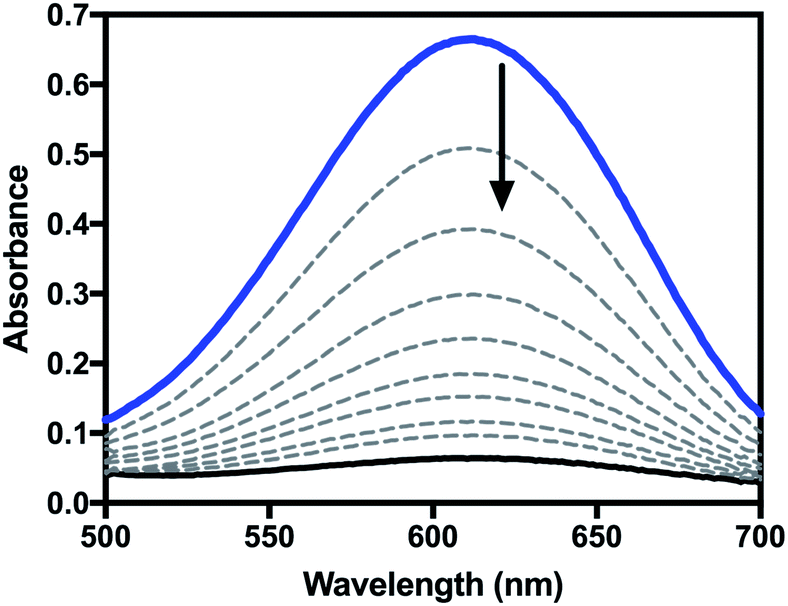 | ||
| Fig. 5 Representative UV-visible spectra as a function of time for the reaction of 2 with 4-X-2,6-DTBP in CH2Cl2. | ||
Measured k2 values were plotted against the Hammett parameter σp+ (Fig. 6, left). The σp+ constant was chosen rather than σp because σp+ is suggested to more accurately represent the electronic structure of the transition state for CPET from phenols28 and a strong negative correlation between σp+ and reaction rate is well known for this class of reactions.29 Hammett analysis showed that k2 decreases with the electron-withdrawing capabilities of the para-substituent of the substrate (ρ = −2.19). This result is inconsistent with a rate-limiting proton transfer followed by electron transfer (PT–ET) mechanism, which would show a positive linear correlation in the Hammett plot.30 The observed strong negative association is consistent with the formation of the electron deficient phenoxyl radical intermediate.31,32 These results suggest a CPET mechanism for the reaction of 4-X-2,6-DTBP substrates with 2. Similarly, the plot of log(k2) vs. BDEO–H of the phenol substrates also demonstrated a strong linear correlation (Fig. 6, right).33 In contrast, the plots of k2vs. substrate pKa or E1/2 showed a much poorer correlation (Fig. S14†). This linear dependence also suggests a CPET mechanism and is consistent with similar plots constructed for other metal-based oxidants.21,34,35 It should be noted that this mechanism could also be described as hydrogen atom transfer (HAT), which also operates through a concerted mechanism, and is frequently referred to as such in the literature.36
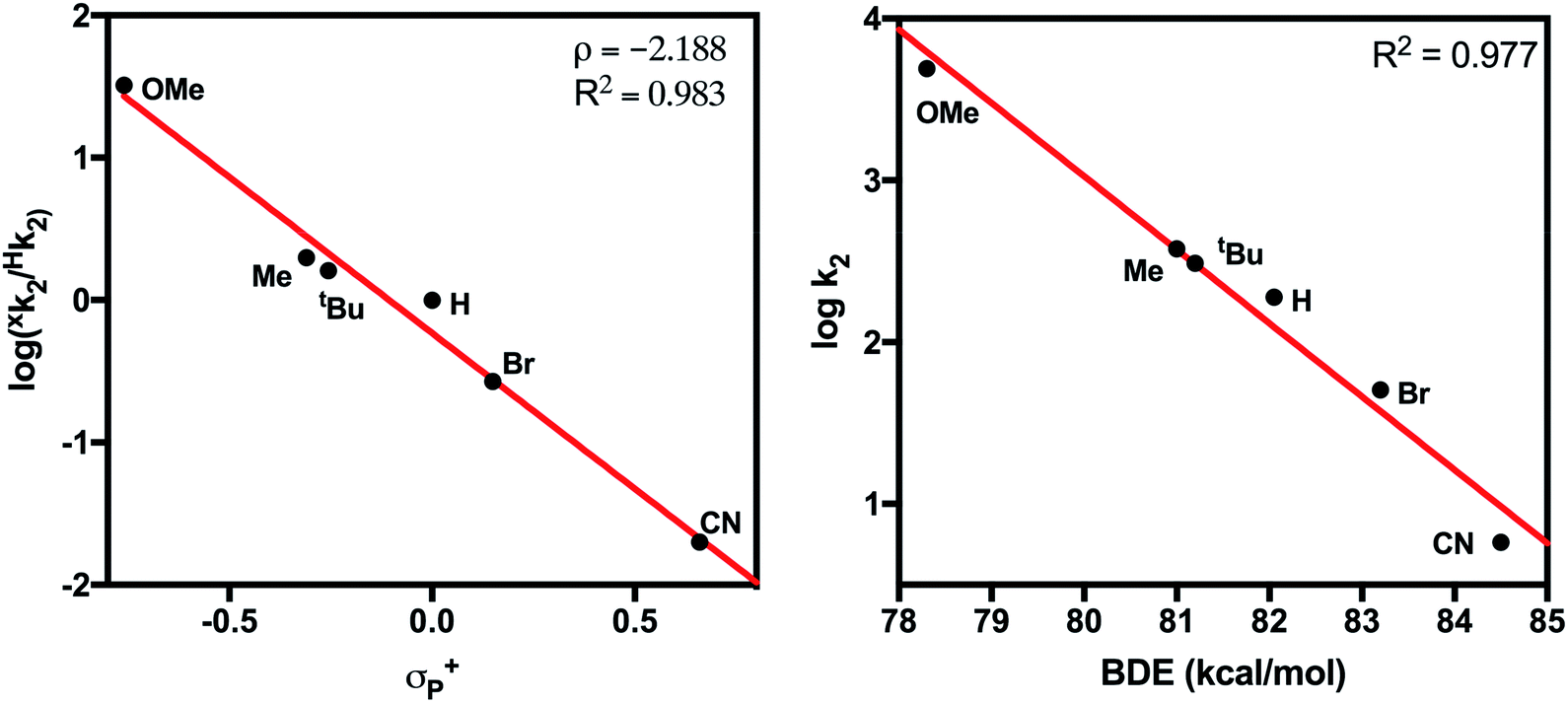 | ||
| Fig. 6 (Left) Hammett plot for the reaction of 2 with 4-X-2,6-DTBP substrates. (Right) Plot of log(k2) vs. BDE for 4-X-2,6-DTBP substrates. | ||
H/D kinetic isotope effects (k2(H)/k2(D), KIE) were also used to differentiate between the possible mechanisms. When the phenolic proton of 2,6-DTBP was replaced with a deuterium atom, the measured k2 value decreased dramatically, and a KIE of ∼4 was calculated (Fig. 7). A primary KIE of this magnitude implies that the cleavage of an O–H bond occurs in the rate-determining step, ruling out pathways involving rate-limiting electron transfer. This value for the KIE is also in good agreement with KIEs reported for the reaction of other metal-based oxidants with 2,6-DTBP that react via CPET.22,31
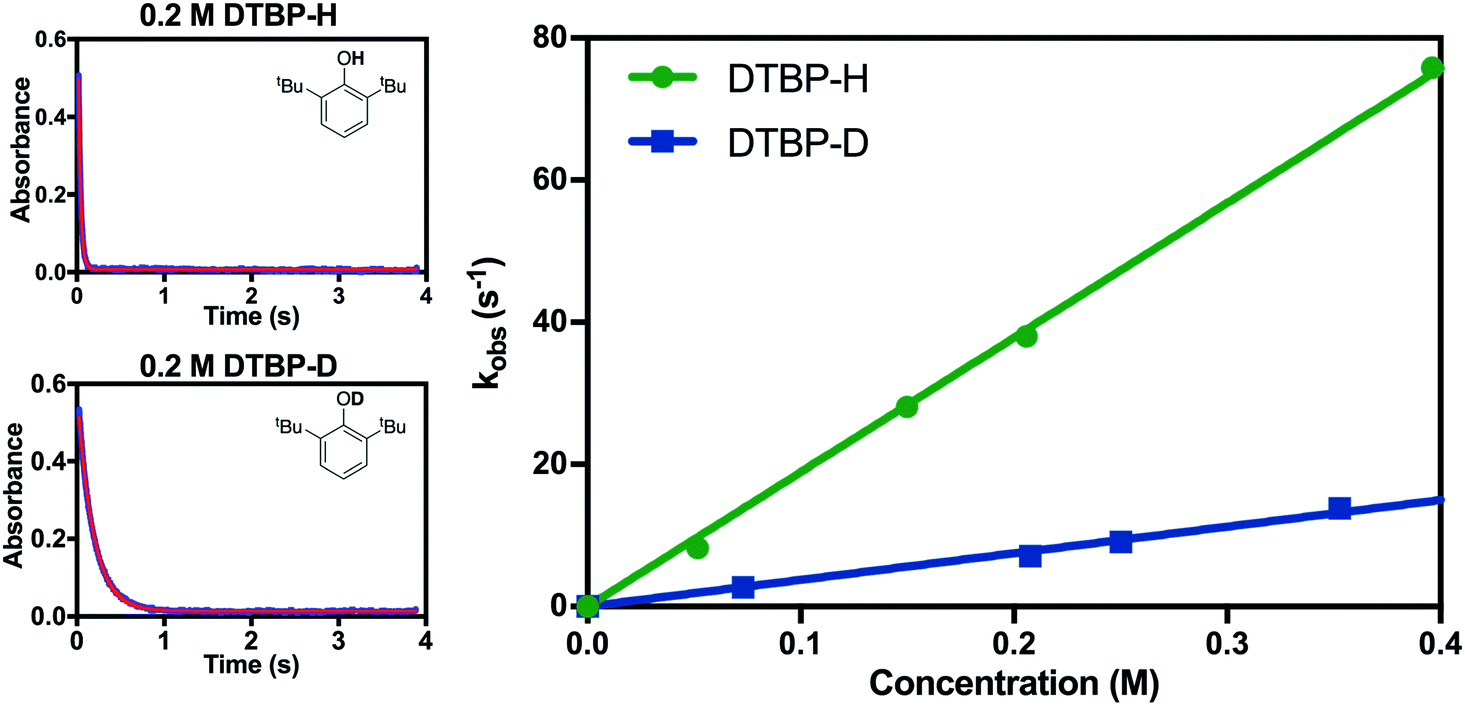 | ||
| Fig. 7 (Left) Representative time trace of the absorbance at λ = 610 nm for the reaction of 2 with 2-6-DTBP-H (top) and 2,6-DTBP-D (bottom). (Right) Plot of kobsvs. concentration for 2,6-DTBP-H and 2,6-DTBP-D substrates. | ||
It should be noted that pKa and BDE values used in our analysis were measured in dimethylsulfoxide (DMSO) rather than CH2Cl2, due to the lack of an absolute pKa scale in CH2Cl2.37 The general linear trend, however, is expected to remain the same regardless of solvent, as relative BDEs and BDFEs (bond dissociation free energies) do not change significantly with solvent. To test this assumption, the BDFEs in CH2Cl2 of several phenol substrates were estimated by converting from DMSO values using Abraham's empirical model.38–40 Calculating BDFEs in this manner relies on a number of assumptions, which are discussed in more detail in the ESI (p. S20†), so the BDFE values should be treated as estimates only. Even so, we still see a strong linear correlation between log(k2) and BDFECH2Cl2 (Fig. S15†). In a similar fashion, we also used Ingold's kinetic solvent effect relationship to calculate the expected rate constants in DMSO,41 which we then plotted against known BDFEs and BDEs measured in DMSO (Fig. S16†). Once again, we saw a strong linear correlation, further supporting our conclusion of a CPET mechanism.
Oxidation of hydrocarbons
To further probe its CPET reactivity, 2 was also treated with a number of hydrocarbon substrates. Initial reactions of 2 with 1,4-cyclohexadiene and 9,10-dihydroanthracene produced the expected products of benzene and anthracene, as determined by gas chromatography and 1H-NMR (Fig. S17a†). To determine the extent of reactivity of 2 with hydrocarbons, 2 was treated with a number of substrates having a range of C–H bond strengths.As with phenols, the reactions were monitored by stopped-flow UV-visible spectroscopy or by UV-visible spectroscopy for slowly reacting substrates. All reactions were carried out under pseudo-first order conditions (10–100 equivalents of substrate). Reactions with all substrates under these conditions gave a good fit to a single exponential decay at a single wavelength (λ = 610 nm), except for reactions with THF, and global analysis was used to find kobs values. For reactions with THF, the method of initial rates was used to find kobs values. Plots of kobsvs. substrate concentration gave good linear fits for all substrates, and the slopes of these fits were used to extract k2 values for each substrate (Fig. S19–S25†). When appropriate, the data were statistically corrected to account for the number of hydrogen atoms susceptible to oxidation. For kinetic analysis of multi-proton/multi-electron reactions, such as the oxidation of 9,10-dihydroanthracene to anthracene (Fig. S17b†), the first concerted proton-electron transfer step was considered to be the rate-determining step, since the resulting radical species tend to have significantly lower BDEs than the parent compounds.38,42,43
2 was found to react with hydrocarbons with C–H bond strengths that ranged from 77–92 kcal mol−1. For substrates with low C–H bond strengths, 2 reacted at appreciable rates – log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) k2 values of 0.89 and 0.74 were found for reactions of 2 with 1,4-cyclohexadiene and dihydroanthracene (DHA), respectively. A plot of log(k2) vs. C–H BDE showed a strong linear correlation (Fig. 8). In contrast, plots of log(k2) vs. E01/2 or pKa showed a very poor correlation (Fig. S26†). This result strongly suggests a CPET mechanism for hydrocarbon oxidation.
k2 values of 0.89 and 0.74 were found for reactions of 2 with 1,4-cyclohexadiene and dihydroanthracene (DHA), respectively. A plot of log(k2) vs. C–H BDE showed a strong linear correlation (Fig. 8). In contrast, plots of log(k2) vs. E01/2 or pKa showed a very poor correlation (Fig. S26†). This result strongly suggests a CPET mechanism for hydrocarbon oxidation.
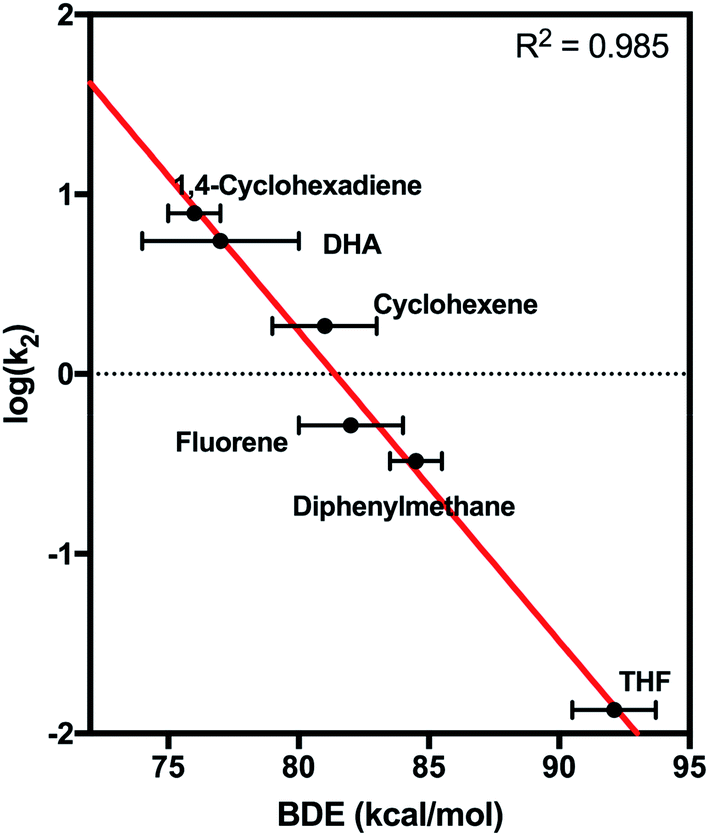 | ||
| Fig. 8 Plot of log(k2) vs. bond dissociation energy for the reaction of 2 with hydrocarbon substrates. | ||
To further investigate the mechanism, dihydroanthracene-d4 was prepared, and an H/D kinetic isotope effect was measured. A large H/D KIE of ∼11 was observed, indicating the involvement of a proton in the rate-determining step (Fig. S27†). This result, in combination with the linear correlation between log(k2) and C–H bond strength, further supports our assignment of a CPET mechanism.
Thermodynamic analysis
We sought to identify the nickel-containing products of the reaction of 2 with phenol and hydrocarbon substrates. Based on our results suggesting that a CPET mechanism was at play, we suspected that the nickel center was being reduced and that the pyalk ligand was accepting a proton and transforming the alkoxide ligand to an alcohol, resulting in the formation [Ni(pyalk)(pyalkH)]+. The 1H NMR spectrum of reaction products of 2 with 2,6-DTBP, however, showed only the presence of the fully deprotonated 1 in solution. A blue precipitate was also identified. This precipitate was dissolved in water and extracted into CH2Cl2 using NaBArF (BArF = [B[3,5-(CF3)2C6H3]4]−). 1H NMR analysis of the resulting product indicated the presence of [Ni(pyalkH)2][2(BArF)] (4) (Fig. 9a and S28†). This result suggests that, once the nickel metal center has been reduced, the pyalkH proton is labile enough to rearrange. Attempts to crystallize 4 were unsuccessful; however, when treating Ni(OAc)2 with the pyalkH ligand, Ni(pyalkH)2(OAc)2 (5) was crystallized, demonstrating the binding of the pyalkH ligand to a nickel center (Fig. 9).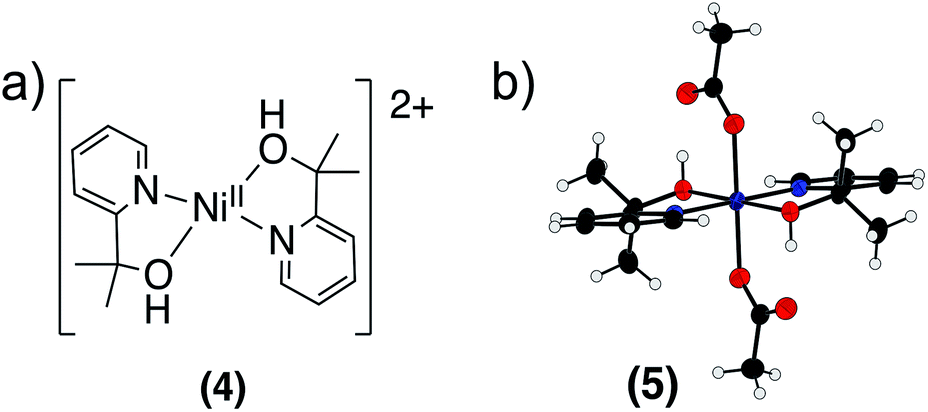 | ||
| Fig. 9 (a) Structure of [Ni(pyalkH)2]2+ (4). (b) X-ray crystal structure of Ni(pyalkH)2(OAc)2 (5). Atoms are shown at the 50% probability level. | ||
With the reaction products more fully understood, we constructed the square scheme in Scheme 3 to determine the thermodynamics of the reaction. The pKa for the first deprotonation of the pyalk ligand of 4 (pKa1, Fig. S29†) was estimated spectroscopically to be ∼25 in MeCN using data from the deprotonation of 4 to 1. Using this value, along with the E01/2 value of 0.15 V vs. Fc/Fc+ in MeCN, we were able to determine a BDFE for the bound pyalk O–H bond of ∼91 kcal mol−1 in MeCN using the square scheme shown in Scheme 3 and the following relationship:38
| BDFE = 13.7pKa + 23.06E0 + CG,sol | (1) |
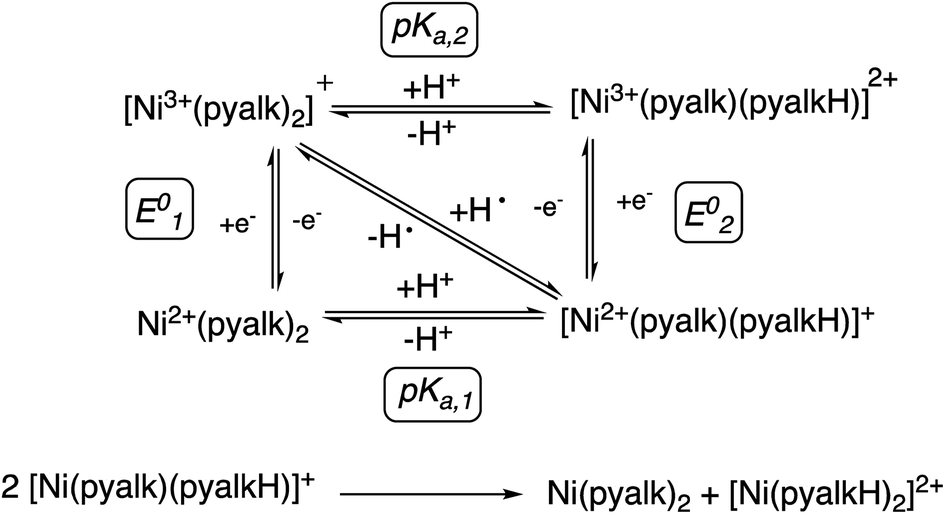 | ||
| Scheme 3 Thermochemical square scheme for stepwise vs. concerted proton and electron transfer to the complexes described in this work. The lower equation shows the proposed proton exchange, explaining the observed products of reactions of 2 with substrates. | ||
It is generally considered more appropriate to use BDFEs to describe the thermodynamics of PCET by transition metal complexes due to non-negligible entropic contributions.38 However, many reported high-valent metal–oxo or metal–hydroxo oxidants report only the BDE of the O–H bond formed upon the reaction with substrate. Therefore, in order to facilitate a comparison between 2 and reported high-valent CPET/HAT reagents, the BDE of the pyalk O–H bond was also calculated. The BDE of the pyalk O–H bond for the CPET product of 2 was calculated using same thermochemical parameters described above and the following equation:
| BDE = 13.7pKa + 23.06E0 + CH,sol | (2) |
For 2, a BDE of ∼94 kcal mol−1 in MeCN was calculated. This can be compared with a value of 105 kcal mol−1 for tBuO–H,44 taken as a model compound for free pyalkH, suggesting a modest O–H bond weakening on binding.
By cyclic voltammetry, 4 also showed a quasi-reversible redox feature in MeCN at E01/2 = 0.58 V vs. Fc/Fc+ (Fig. 10). Using the BDFE calculated from eqn (1) and the square-scheme relationship, we can estimate the value for pKa2 to be ∼18, 7 pKa units below the calculated pKa1 of 25.
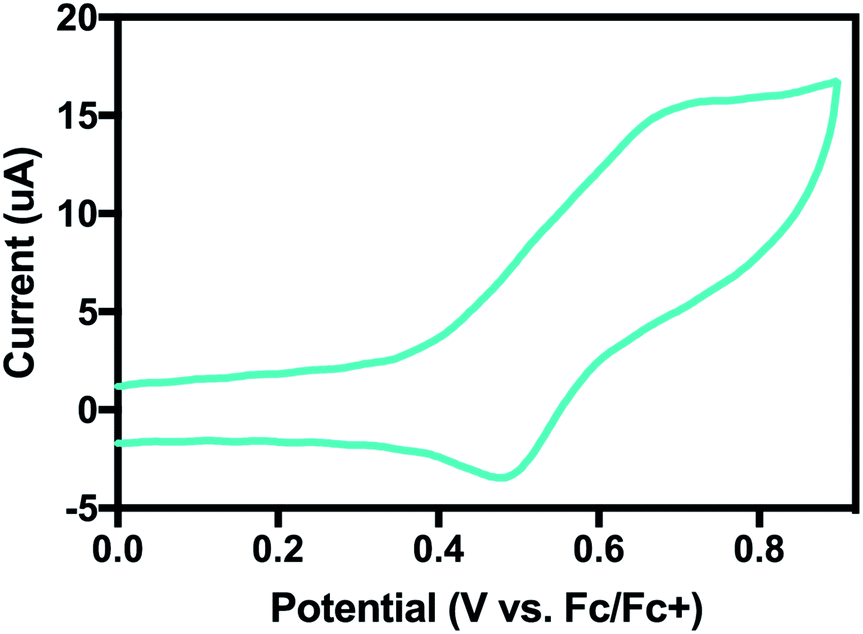 | ||
| Fig. 10 Cyclic voltammogram of 4 in MeCN. The quasi-reversible couple at E0 = 0.58 V vs. Fc/Fc+ is assigned as a Ni(II/III) couple. | ||
The reactivity of 2 with 2,6-DTBP and DHA compares favorably with other reported high-valent metal–oxo and metal–hydroxo complexes capable of CPET or HAT (Table 1). 2 reacts with DHA at a faster rate than several manganese– and iron–oxo complexes. 2 also compares extremely well with high-valent metal alkoxide and carboxylate compounds, including the only other reported Ni(III) systems, which tend to react with C–H bonds at slower rates than their metal–oxo counterparts. We hypothesize that the particularly strong BDE of the ligand O–H bond formed and the strong oxidizing power of 2 contribute to its high reactivity toward O–H and C–H bonds. This result demonstrates that fast CPET can be achieved in high-valent metal systems without necessarily going through a metal–oxo or metal–hydroxo intermediate, a result relevant to several proposed water-oxidation mechanisms in artificial photosynthetic systems of cobalt,49 copper,11 and nickel.50 This result also demonstrates that high-valent metal-alkoxide systems are capable of attacking strong C–H and O–H bonds, which is particularly relevant to water-oxidation catalysis. The pyalk ligand thus proves particularly useful for catalytic oxidations of this type.
| Compound | E01 (V vs. Fc/Fc+) | BDE (kcal mol−1) | log(k2) DHAa,b | log(k2) TTBPa,b | Ref. |
|---|---|---|---|---|---|
| a k 2 = second-order rate constant for reaction with the designated substrate, measured at 25 °C unless otherwise noted. b DHA = 9,10-diyhdroanthracene, TTBP = tri-tert-butylphenol (4-tBu-2,6-DTPB). c Vs. SCE. d Measured at −40 °C. e Measured at −50 °C. f MepyN2 = N,N′-(2,6-dimethylphenyl)-2,6-pyridinedicarboxamide. g MeAN = N,N,N′,N′,N′-pentamethyl-dipropylenetriamine and MeNacnac = [HC(CMeNC6H3(iPr)2)2]. h H3buea = tris[(N′-tert-butylureaylato)-N-ethylene]amine. i PY5 = 2,6-bis(bis(2-pyridyl)methoxymethane)pyridine. j iPrpyN2 = N,N′-bis(2,6-diisopropylphenyl)-2,6-pyridinedicarboxamide. | |||||
| [NiIII(pyalk)2]+ (2) | 0.15 | 94 | 0.74 | 2.48 | This work |
| NiIII(MepyN2)(ONO2)f | 0.43 | — | 0.91 | 21 | |
| NiIII(MepyN2)(Cl)f | 0.56 | — | — | 0.394d | 22 |
| [(MeAN)CuIII(μ-O2)NiIII(MeNacnac)]+g | — | — | — | −0.82 | 45 |
| [MnIII(H3buea)(O)]2−h | −2.0 | 77 | −0.53 | — | 25 |
| [MnIV(H3buea)(O)]−h | −1.0 | 89 | −1.59 | — | 25 |
| [FeIII(PY5)(OCH3)]2+i | 0.73 | 84 | −2.25 | −0.22e | 46 |
| CuIII(iPrpyN2)(OH)j | −0.074 | 90 | 2.27 | — | 34 |
| RuIV(bpy)2(py)(O) | 0.48c | 84 | 2.09 | — | 35, 47, and 48 |
Conclusions
We have synthesized and characterized a Ni(III)-alkoxide compound capable of reacting with strong C–H and O–H bonds at appreciable rates. A strong linear dependence between the second-order rate constant, k2, and substrate bond dissociation enthalpy indicates a CPET mechanism. Large H/D kinetic isotope effects also support this assignment. We attribute the fast reactivity of 2 to the strong O–H BDE of the pyalk/pyalkH supporting ligand and the high oxidizing power of the complex. This result demonstrates that fast PCET can occur in high-valent metal oxidants without a metal–oxo unit, which may be relevant to certain nickel-containing enzymes or water-oxidation catalysts of cobalt, copper, or nickel. This report also provides the first full thermodynamic analysis of CPET by a high-valent nickel complex. The value of pyalk as a ligand for catalytic oxidations is further supported.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences, and Biosciences as part of the Center for Light Energy Activated Redox Processes (LEAP) Energy Frontier Research Center under Award Number DE-SC0001059. K. J. F. acknowledges the National Science Foundation Graduate Research Fellowship Program (Grant No. DGE1122492). We thank the Mass Spectrometry & Proteomics Resource and Dr Fabian Menges for assistance with HRMS. We would also like to thank Dr Min Li and Dr Liam Sharninghausen for collection of XPS data. We would also like to thank Professor Jim Mayer and Catherine Wise for their helpful comments.References
- B. Meunier, S. P. de Visser and S. Shaik, Chem. Rev., 2004, 104, 3947–3980 CrossRef CAS PubMed.
- P. R. Ortiz de Montellano and J. J. De Voss, Nat. Prod. Rep., 2002, 19, 477–493 RSC.
- D. R. Weinberg, C. J. Gagliardi, J. F. Hull, C. F. Murphy, C. A. Kent, B. C. Westlake, A. Paul, D. H. Ess, D. G. McCafferty and T. J. Meyer, Chem. Rev., 2012, 112, 4016–4093 CrossRef CAS PubMed.
- Y. Zhang, H. Zhang, H. Ji, W. Ma, C. Chen and J. Zhao, J. Am. Chem. Soc., 2016, 138, 2705–2711 CrossRef CAS PubMed.
- D. C. Miller, K. T. Tarantino and R. R. Knowles, Top. Curr. Chem., 2016, 374, 30 CrossRef PubMed.
- S. W. M. Crossley, C. Obradors, R. M. Martinez and R. A. Shenvi, Chem. Rev., 2016, 116, 8912–9000 CrossRef CAS PubMed.
- D. P. Barondeau, C. J. Kassmann, C. K. Bruns, J. A. Tainer and E. D. Getzoff, Biochemistry, 2004, 43, 8038–8047 CrossRef CAS PubMed.
- J. Shearer, Angew. Chem., Int. Ed., 2013, 52, 2569–2572 CrossRef CAS PubMed.
- J. Shearer, Acc. Chem. Res., 2014, 47, 2332–2341 CrossRef CAS PubMed.
- T. K. Michaelos, D. Y. Shopov, S. B. Sinha, L. S. Sharninghausen, K. J. Fisher, H. M. C. Lant, R. H. Crabtree and G. W. Brudvig, Acc. Chem. Res., 2017, 50, 952–959 CrossRef CAS PubMed.
- B. Rudshteyn, K. J. Fisher, H. M. C. Lant, K. R. Yang, B. Q. Mercado, G. W. Brudvig, R. H. Crabtree and V. S. Batista, ACS Catal., 2018, 8, 7952–7960 CrossRef CAS.
- F. Chen, N. Wang, H. Lei, D. Guo, H. Liu, Z. Zhang, W. Zhang, W. Lai and R. Cao, Inorg. Chem., 2017, 56, 13368–13375 CrossRef CAS PubMed.
- P. Pirovano, E. R. Farquhar, M. Swart, A. J. Fitzpatrick, G. G. Morgan and A. R. McDonald, Chem.–Eur. J., 2015, 21, 3785–3790 CrossRef CAS PubMed.
- D. F. Evans and D. A. Jakubovic, J. Chem. Soc., Dalton Trans., 1988, 2927–2933 RSC.
- J. R. Lancaster, The Bioinorganic Chemistry of Nickel, VCH Publishers, 1988 Search PubMed.
- D. Y. Shopov, L. S. Sharninghausen, S. B. Sinha, B. Q. Mercado, D. Balcells, G. W. Brudvig and R. H. Crabtree, Inorg. Chem., 2018, 57, 5684–5691 CrossRef CAS PubMed.
- L. S. Sharninghausen, S. B. Sinha, D. Y. Shopov, B. Choi, B. Q. Mercado, X. Roy, D. Balcells, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 2016, 138, 15917–15926 CrossRef CAS PubMed.
- S. B. Sinha, D. Y. Shopov, L. S. Sharninghausen, C. J. Stein, B. Q. Mercado, D. Balcells, T. B. Pedersen, M. Reiher, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 2017, 139, 9672–9683 CrossRef CAS PubMed.
- L. S. Sharninghausen, S. B. Sinha, D. Y. Shopov, B. Q. Mercado, D. Balcells, G. W. Brudvig and R. H. Crabtree, Angew. Chem., 2017, 129, 13227–13231 CrossRef.
- B. De Castro and C. Freire, Inorg. Chem., 1990, 29, 5113–5119 CrossRef CAS.
- P. Pirovano, E. R. Farquhar, M. Swart and A. R. McDonald, J. Am. Chem. Soc., 2016, 138, 14362–14370 CrossRef CAS PubMed.
- P. Mondal, P. Pirovano, A. Das, E. R. Farquhar and A. R. McDonald, J. Am. Chem. Soc., 2018, 140, 1834–1841 CrossRef CAS PubMed.
- K. J. Fisher, K. L. Materna, B. Q. Mercado, R. H. Crabtree and G. W. Brudvig, ACS Catal., 2017, 7, 3384–3387 CrossRef CAS.
- V. W. Manner, T. F. Markle, J. H. Freudenthal, J. P. Roth and J. M. Mayer, Chem. Commun., 2008, 256–258 RSC.
- T. H. Parsell, M.-Y. Yang and A. S. Borovik, J. Am. Chem. Soc., 2009, 131, 2762–2763 CrossRef CAS PubMed.
- R. Gupta and A. S. Borovik, J. Am. Chem. Soc., 2003, 125, 13234–13242 CrossRef CAS PubMed.
- B. Chiavarino, R. Cipollini, M. E. Crestoni, S. Fornarini, F. Lanucara and A. Lapi, J. Am. Chem. Soc., 2008, 130, 3208–3217 CrossRef CAS PubMed.
- T. Yoshida, K. Hirozumi, M. Harada, S. Hitaoka and H. Chuman, J. Org. Chem., 2011, 76, 4564–4570 CrossRef CAS PubMed.
- D. A. Pratt, G. A. DiLabio, P. Mulder and K. U. Ingold, Acc. Chem. Res., 2004, 37, 334–340 CrossRef CAS PubMed.
- D. Dhar, G. M. Yee, T. F. Markle, J. M. Mayer and W. B. Tolman, Chem. Sci., 2017, 8, 1075–1085 RSC.
- D. E. Lansky and D. P. Goldberg, Inorg. Chem., 2006, 45, 5119–5125 CrossRef CAS PubMed.
- P. Mulder, O. W. Saastad and D. Griller, J. Am. Chem. Soc., 1988, 110, 4090–4092 CrossRef CAS.
- While the thermodynamics of this reaction are perhaps more accurately captured by the use of bond dissociation free energies (BDFEs), we chose to use BDEs in this analysis because of limited BDFE data for these compounds. A plot of log(k2) vs. BDFE is provided in the ESI† for substrates with reported BDFEs.
- D. Dhar and W. B. Tolman, J. Am. Chem. Soc., 2015, 137, 1322–1329 CrossRef CAS PubMed.
- J. R. Bryant and J. M. Mayer, J. Am. Chem. Soc., 2003, 125, 10351–10361 CrossRef CAS PubMed.
- J. W. Darcy, B. Koronkiewicz, G. A. Parada and J. M. Mayer, Acc. Chem. Res., 2018, 51, 2391–2399 CrossRef CAS PubMed.
- A. Kütt, S. Selberg, I. Kaljurand, S. Tshepelevitsh, A. Heering, A. Darnell, K. Kaupmees, M. Piirsalu and I. Leito, Tetrahedron Lett., 2018, 59, 3738–3748 CrossRef.
- J. J. Warren, T. A. Tronic and J. M. Mayer, Chem. Rev., 2010, 110, 6961–7001 CrossRef CAS PubMed.
- T. R. Porter and J. M. Mayer, Chem. Sci., 2014, 5, 372–380 RSC.
- M. H. Abraham, P. L. Grellier, D. V. Prior, R. W. Taft, J. J. Morris, P. J. Taylor, C. Laurence, M. Berthelot and R. M. Doherty, et al. , J. Am. Chem. Soc., 1988, 110, 8534–8536 CrossRef CAS.
- J. J. Warren and J. M. Mayer, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 5282 CrossRef CAS PubMed.
- Y. Gao, N. J. DeYonker, E. C. Garrett, A. K. Wilson, T. R. Cundari and P. Marshall, J. Phys. Chem. A, 2009, 113, 6955–6963 CrossRef CAS PubMed.
- A. J. Johansson, M. R. A. Blomberg and P. E. M. Siegbahn, J. Phys. Chem. C, 2007, 111, 12397–12406 CrossRef CAS.
- M. Finn, R. Friedline, N. K. Suleman, C. J. Wohl and J. M. Tanko, J. Am. Chem. Soc., 2004, 126, 7578–7584 CrossRef CAS PubMed.
- S. Kundu, E. Miceli, E. R. Farquhar and K. Ray, Dalton Trans., 2014, 43, 4264–4267 RSC.
- C. R. Goldsmith, R. T. Jonas and T. D. P. Stack, J. Am. Chem. Soc., 2002, 124, 83–96 CrossRef CAS PubMed.
- B. A. Moyer and T. J. Meyer, Inorg. Chem., 1981, 20, 436–444 CrossRef CAS.
- E. L. Lebeau, R. A. Binstead and T. J. Meyer, J. Am. Chem. Soc., 2001, 123, 10535–10544 CrossRef CAS PubMed.
- D. Das, S. Pattanayak, K. K. Singh, B. Garai and S. Sen Gupta, Chem. Commun., 2016, 52, 11787–11790 RSC.
- M. Zhang, M.-T. Zhang, C. Hou, Z.-F. Ke and T.-B. Lu, Angew. Chem., Int. Ed., 2014, 53, 13042–13048 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, supplementary figures, and crystallographic data. CCDC 1954011–1954013. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc05565g |
| This journal is © The Royal Society of Chemistry 2020 |